 Shuqiao Yang1,2
Shuqiao Yang1,2 Qiao Ye
Qiao Ye- 1Clinical Center for Interstitial Lung Diseases, Beijing Institute of Respiratory Medicine, Beijing Chao-Yang Hospital, Capital Medical University, Beijing, China
- 2Department of Respiratory and Critical Care Medicine, Beijing Chao-Yang Hospital, Capital Medical University, Beijing, China
- 3Department of Occupational Medicine and Toxicology, Beijing Chao-Yang Hospital, Capital Medical University, Beijing, China
Background: Usual interstitial pneumonia (UIP) is a pattern of interstitial pneumonia that is caused by different etiologies. This study aimed to investigate the transplant-free survival (TFS) and the decline in forced vital capacity (FVC) of the patients with UIP and probable UIP patterns on CT caused by various underlying conditions.
Methods: A retrospective cohort study was conducted, enrolling patients with interstitial lung disease exhibiting a CT pattern consistent with UIP or probable UIP. Clinical and prognostic data of patients categorized by the etiology were compared.
Results: A total of 591 patients were included and classified into the following groups: idiopathic pulmonary fibrosis (IPF) (n = 320), connective tissue disease (CTD)-UIP (n = 229), asbestosis-UIP (n = 28), and hypersensitivity pneumonitis (HP)-UIP (n = 14). Advanced age, elevated levels of serum cytokeratin fraction 21-1 and percentage of neutrophils in bronchoalveolar lavage were observed in all groups. IPF patients showed a more rapid decline in FVC (133.9 mL/year) compared to CTD-UIP (24.5 mL/year, p = 0.001) and asbestosis-UIP (61.0 mL/year, p = 0.008) respectively. Sub-analysis of CTD-UIP revealed that patients with rheumatoid arthritis (RA)-UIP (88.1 mL/year) or antineutrophil cytoplasmic antibody-associated vasculitis (AAV)-UIP (72.9 mL/year) experienced a faster deterioration in FVC compared to those with primary Sjögren’s syndrome (pSS)-UIP (25.9 mL/year, p < 0.05). Kaplan–Meier curves showed that IPF had the poorest TFS (median 55.9 months), followed by HP-UIP (57.5 months), CTD-UIP (66.7 months), and asbestosis-UIP (TFS not reached). RA-UIP or AAV-UIP did not exhibit any prognostic advantages compared to IPF, while asbestosis-UIP and pSS-UIP showed better survival rates.
Conclusion: Patients with UIP caused by different underlying conditions share certain common features, but the trajectories of disease progression and survival outcomes differ.
Introduction
Usual interstitial pneumonia (UIP) is a commonly observed pattern of interstitial pneumonia characterized by heterogeneous fibrosis in the lung parenchyma, along with architectural distortion, fibroblast foci, and honeycombing. While UIP is diagnosed through pathology, extensive evidence supports the high specificity of typical and probable UIP patterns on high-resolution computed tomography (HRCT) for histopathologic UIP, eliminating the need for surgical lung biopsy. Idiopathic pulmonary fibrosis (IPF) represents the primary form of UIP with an unknown cause, but other interstitial lung diseases (ILDs), including connective tissue disease (CTD)-related, hypersensitivity pneumonitis (HP)-related, and asbestosis-related ILDs, can also exhibit a UIP pattern (1). UIP stands out from other ILD patterns, such as nonspecific interstitial pneumonia (NSIP), organizing pneumonia (OP), and lymphocytic interstitial pneumonia (LIP), due to its predominant fibrosis and minimal inflammation, which contribute to higher mortality rates, irrespective of whether it is idiopathic or secondary (2, 3).
Recent discussions have proposed the unification of UIP as a single diagnostic entity due to the presence of similar disease progression behaviors and underlying mechanisms. This unified approach has the potential to expedite antifibrotic trials for secondary UIP and trials of novel therapeutic agents (4). However, there is a scarcity of data on secondary UIP types, apart from CTD-UIP. Furthermore, previous analyses examining the disparities in survival rates between IPF and CTD-UIP have yielded inconsistent findings (5, 6). In this study, we conducted a comprehensive review of patients with UIP and probable UIP patterns on HRCT over a 10 years period at our hospital. The objective was to determine the transplant-free survival (TFS) and the decline in forced vital capacity (FVC) of the patients with different etiologies of UIP patterns and probable UIP on HRCT.
Materials and methods
Study design and patient selection
This study was an observational, retrospective cohort study conducted at Beijing Chao-Yang Hospital, China, which serves as a regional medical center specializing in ILD. We screened all the in-patients consecutively admitted to our hospital between January 2012 and December 2021 with newly diagnosed ILDs. Considering the limitations imposed by the local medical insurance policy and taking into account the patient’s individual preferences, hospitalized patients provide a more comprehensive set of clinical data for evaluating diseases, especially in terms of systemic organ involvement. The inclusion criterion was the presence of a UIP or probable UIP pattern on HRCT. Exclusion criteria included: (1) prior treatment with immunosuppressive or antifibrotic agents, (2) unclear diagnosis, (3) active infections, (4) malignancy or suspected malignancy at the time of diagnosis or within 1 year of diagnosis, (5) pneumothorax or pulmonary embolism at the time of diagnosis, (6) inadequate sample size for analysis, and (7) incomplete data or loss to follow-up. The study adhered to the principles of the Helsinki Declaration and its subsequent amendments and was approved by the Ethics Committee of Beijing Chao-Yang Hospital.
Diagnosis and image evaluation
All patients included in the final analysis underwent reevaluation and discussion by a multidisciplinary team comprising a pulmonologist, rheumatologist, and radiologist. The diagnosis of IPF was based on the 2018 ATS/ERS/JRS/ALAT guideline (7). CTD was diagnosed if the patients fulfilled the established classification criteria for each specific type of CTD (8–13). Asbestosis was diagnosed using the International Labor Organization classification criteria (14), the patients included all had a history of exposure to chrysotile asbestos. HP was diagnosed following the 2020 ATS/JRS/ALAT guideline (15).
All patients underwent chest HRCT with a 1 s scan time, 1.25 mm slice thickness, and 5 mm interval from the lung apex to the base, encompassing both lungs within the field of view. The CT patterns and signs of the patients were assessed by a radiologist and pulmonologist who were blinded to the clinical data. In patients of disagreement between the pulmonologist and radiologist after the initial assessment, consultation was sought from a ILD specialist. The presence of all the following features, as outlined in the 2018 ATS/ERS/JRS/ALAT guidelines (7), defined the UIP and probable UIP patterns: heterogeneous subpleural/basal predominance, reticular abnormalities, honeycombing (reticular pattern for probable UIP), with or without traction bronchiectasis, and the absence of features inconsistent with the UIP pattern (mild ground-glass opacity may be present in probable UIP). Positive emphysema findings were visually defined as the presence of areas of low attenuation indicating a lack of a distinct alveolar wall threshold comprising more than 10% of the lung volume (16). Pulmonary hypertension (PHT) was defined as a pulmonary artery systolic pressure exceeding 35 mmHg on echocardiography (17).
Data collection and follow-up
Demographics, smoking history, parameters of pulmonary function tests, and laboratory findings at the time of ILD diagnosis were extracted from the medical records. Prebronchodilator FVC, forced expiratory volume in the first second (FEV1), and single-breath diffusing capacity of the lung for carbon monoxide (DLCO SB) were measured according to the criteria set by the ATS/ERS (18, 19), and the percentages (%) of the normal predicted values for these parameters were recorded. The gender-age-physiology (GAP) score was calculated using sex, age, percent predicted FVC, and percent predicted DLCO, and patients were classified into GAP stages I, II, and III as previously described (20). The composite physiological index (CPI) was calculated using the formula (21): CPI = 91 − (0.65 × percent predicted DLCO) − (0.53 × percent predicted FVC) + (0.34 × percent predicted FEV1).
Blood leukocytes, neutrophils, lymphocytes, monocytes, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), immunoglobulin G (IgG), cytokeratin fraction 21-1 (CYFRA21-1), carcinoembryonic antigen (CEA), carbohydrate antigen (CA) 19-9, and CA 125 were recorded. The recommended normal ranges for each tumor marker are as follows: CYFRA21-1 ≤2.08 ng/mL, CEA ≤5 ng/mL, CA19-9 ≤37.00 U/mL, and CA125 ≤35.00 U/mL. Bronchoscopy with bronchoalveolar lavage (BAL) was performed following the guidelines of the ATS (22), and the results of the cellular analysis of the BAL fluid (BALF) were recorded. Normal BAL cell differential counts are as follows: macrophages ≥80%, lymphocytes ≤15%, neutrophils ≤3% and eosinophils ≤1%.
Longitudinal data on FVC, medication, and vital status were obtained by reviewing outpatient follow-up records and conducting phone interviews with patients or their family members. Patients were considered to have received immunosuppressive or antifibrotic therapy only if the medication was maintained for more than 6 months. For each patient, the decline in lung function was calculated as (last FVC − FVC at baseline)/duration of follow-up (in years) (23). The primary endpoint of the study was TFS, defined as the time from ILD diagnosis to the occurrence of either lung transplantation or death, up until August 2022.
Statistical analysis
Statistical analyses were conducted using the Statistical Package for the Social Sciences (SPSS) software, version 23 (IBM Corp., Armonk, NY, United States). Categorical baseline characteristics were presented as frequencies and percentages, while continuous characteristics were reported as mean ± standard deviation or median (interquartile range, IQR). Intergroup differences for continuous variables were assessed using one-way analysis of variance or the Mann–Whitney U test, while categorical data were analyzed using the chi-square test. Survival analysis was performed using Kaplan–Meier curves and log-rank test. When the log-rank test was significant, Bonferroni correction for multiple comparisons was applied and the alpha level was set at 0.0083 (0.05/6 tests). Univariate and Cox proportional hazard regression analyses were then conducted to determine the predictor variables for mortality or lung transplantation, presenting hazard ratios (HRs) and 95% confidence intervals (CIs). Statistical significance was set at a p-value <0.05.
Results
Study patients
Between 2012 and 2021, a total of 9,706 consecutive in-patients with chest HRCT detected ILD were initially screened. Among them, 1,395 patients exhibited a radiological UIP or probable UIP pattern on HRCT. Following the application of exclusion criteria, a total of 804 patients were excluded, resulting in 591 patients being included in the final analysis. The reasons for these patient’s hospitalization include dyspnea (45.5%), cough (31.6%), joint swelling or pain (7.4%), fever (4.3%), dry mouth (2.5%), abnormal chest imaging (2.0%), chest pain (1.9%), hemoptysis (1.9%), edema (1.4%), blood urine (0.8%) and fatigue (0.7%). Patients were classified into four groups: IPF (n = 320), CTD-UIP (n = 229), asbestosis associated UIP (asbestosis-UIP) (n = 28), and HP associated UIP (HP-UIP) (n = 14). Within the CTD-UIP group, there were 52 patients of rheumatoid arthritis (RA)-UIP, 71 patients of primary Sjögren’s syndrome (pSS)-UIP, 58 patients of antineutrophil cytoplasmic antibody-associated vasculitis (AAV)-UIP, and 48 patients of other CTD-UIP, which consisted of 10 patients of systemic sclerosis, 2 patients of dermatomyositis, 8 patients of mixed CTD, and 28 patients of interstitial pneumonia with autoimmune features. A detailed flow diagram illustrating the patient selection process is presented in Figure 1.
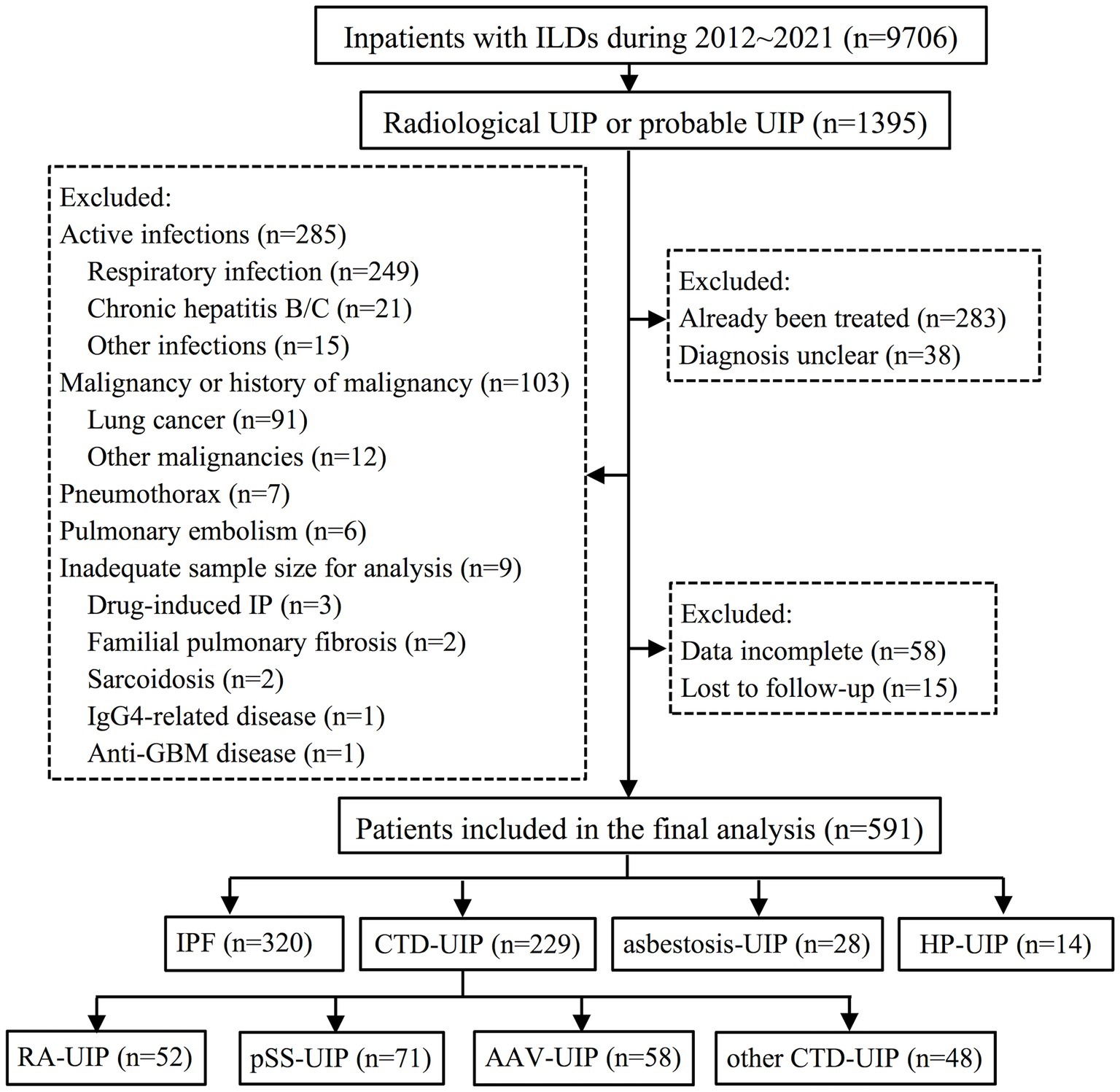
Figure 1. Flow diagram illustrating the patient identification process for the study. ILD, interstitial lung disease; UIP, usual interstitial pneumonia; IP, interstitial pneumonia; GBM, glomerular basement membrane; IPF, idiopathic pulmonary fibrosis; CTD, connective tissue disease; HP, hypersensitivity pneumonitis; RA, rheumatoid arthritis; pSS, primary Sjögren’s syndrome; AAV, anti-neutrophil cytoplasmic antibody-associated vasculitis.
Demographics and clinical characteristics
The average age of the patients was 65.5 years, with an average body mass index (BMI) of 24.2 kg/m2. Among the patients, 430 (72.8%) were male, and 351 (59.4%) had a smoking history.
A total of 436 patients (73.8%) had a UIP pattern, while 155 (26.2%) had a probable UIP pattern on HRCT. At the time of diagnosis, the average FVC was 2.8 L, with FVC% predicted, FEV1% predicted, and DLCO SB% predicted being 86.6%, 87.6%, and 53.6%, respectively. The median GAP score was 3.0 (IQR: 2.0–4.0), with 375 patients (63.5%) classified as stage I, 180 (30.4%) as stage II, and 36 (6.1%) as stage III. The mean composite physiological index (CPI) score was 40.0 (Table 1). Group comparisons showed that the CTD-UIP group had significantly fewer men (58.1% vs. 83.8%, p < 0.001), a lower BMI (23.5 kg/m2 versus 24.8 kg/m2, p < 0.001), and fewer ever-smokers (51.1% vs. 66.3%, p < 0.001) than the IPF group. Age, UIP/probable UIP ratio, pulmonary function, GAP score, and CPI score were comparable among the four groups at diagnosis.

Table 1. Baseline demographics of UIP patients in various etiology groups.
Regarding laboratory findings, the neutrophils, ESR, CRP, and IgG levels in the CTD-UIP patients were significantly higher than those in the other three patients (p < 0.001). Among the oncomarkers, the IPF patients had higher CEA levels than the CTD-UIP group (3.3 vs. 2.4, p < 0.001), whereas the CTD-UIP group had higher CA 19-9 and CA125 levels than the other three patients (p < 0.001). Bronchoscopy with BAL was performed in 307 patients (51.9%), and the median proportions of BALF macrophages, neutrophils, lymphocytes, and eosinophils were 60.0%, 28.0%, 6%, and 1%, respectively. The median proportion of neutrophils exceeded the normal level (3%) in patients with IPF (29.0%), CTD-UIP (30.0%), asbestosis-UIP (23.5%), and HP-UIP (22.0%) (Table 2).
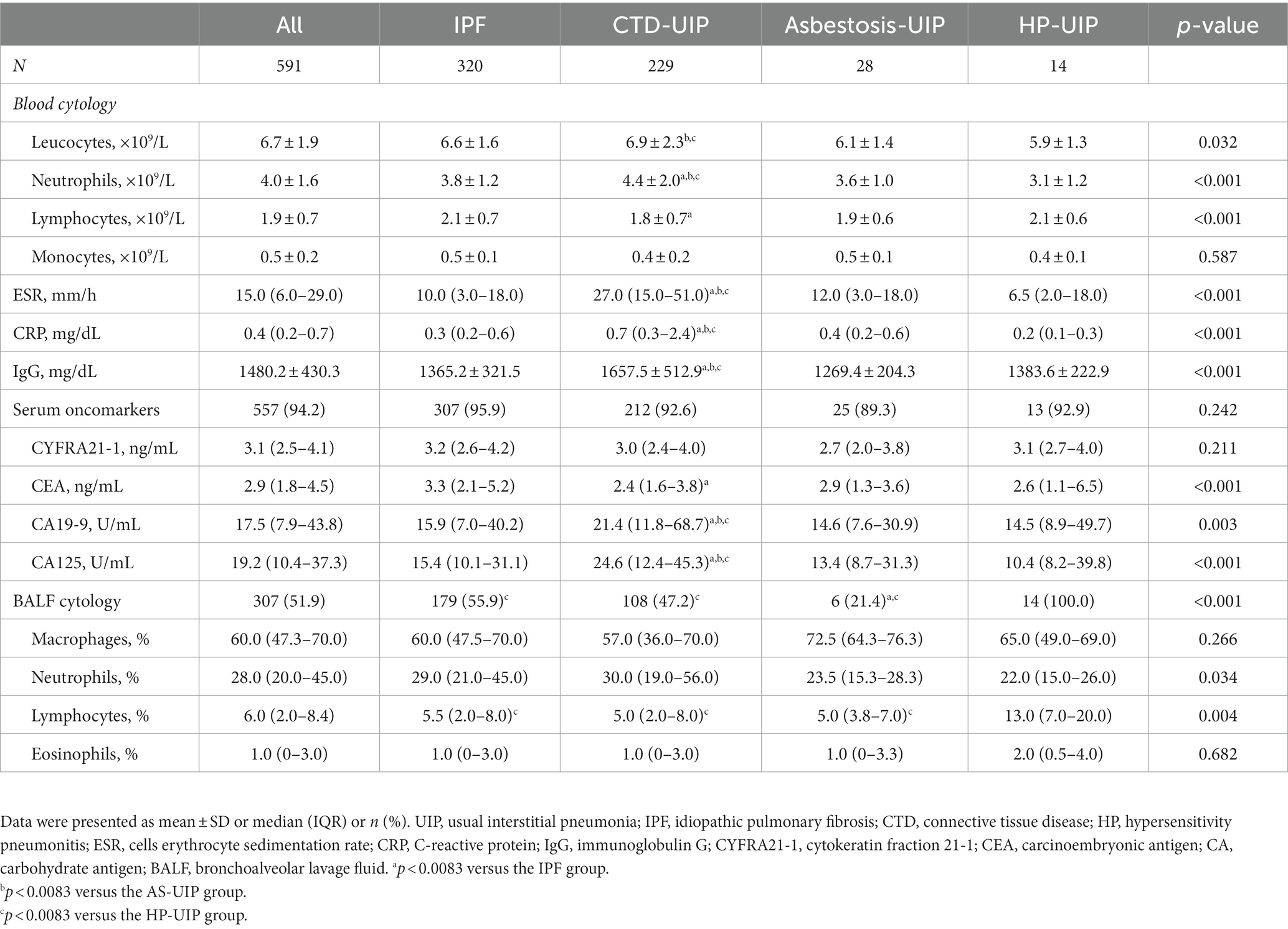
Table 2. Laboratory data of UIP patients in various etiology groups.
Follow-up data
Among all patients with UIP, the frequencies of complicated emphysema or pulmonary hypertension were 31.1% and 17.9%, respectively, with no significant differences between the patients. The median follow-up duration was 46.4 months, during which immunosuppressive medications were administered to 213 patients (36.0%), antifibrotic medications to 125 patients (21.2%), and lung transplants were performed in 9 patients (1.5%). The median annual decline in FVC was 76.0 mL. The 1 year, 3 years, and 5 years TFS rates were 90.0%, 71.0%, and 52.3%, respectively. Comparing the patients, antifibrotic therapy was more frequently used in patients with IPF compared to those with CTD-UIP (29.7% vs. 12.2%, p < 0.001) and asbestosis-UIP (29.7% vs. 0, p < 0.001). The median FVC decline in the IPF group (133.9 mL/year) was more rapid than the CTD-UIP (24.5 mL/year, p = 0.001) and the asbestosis-UIP groups (61.0 mL/year, p = 0.008) (Table 3).
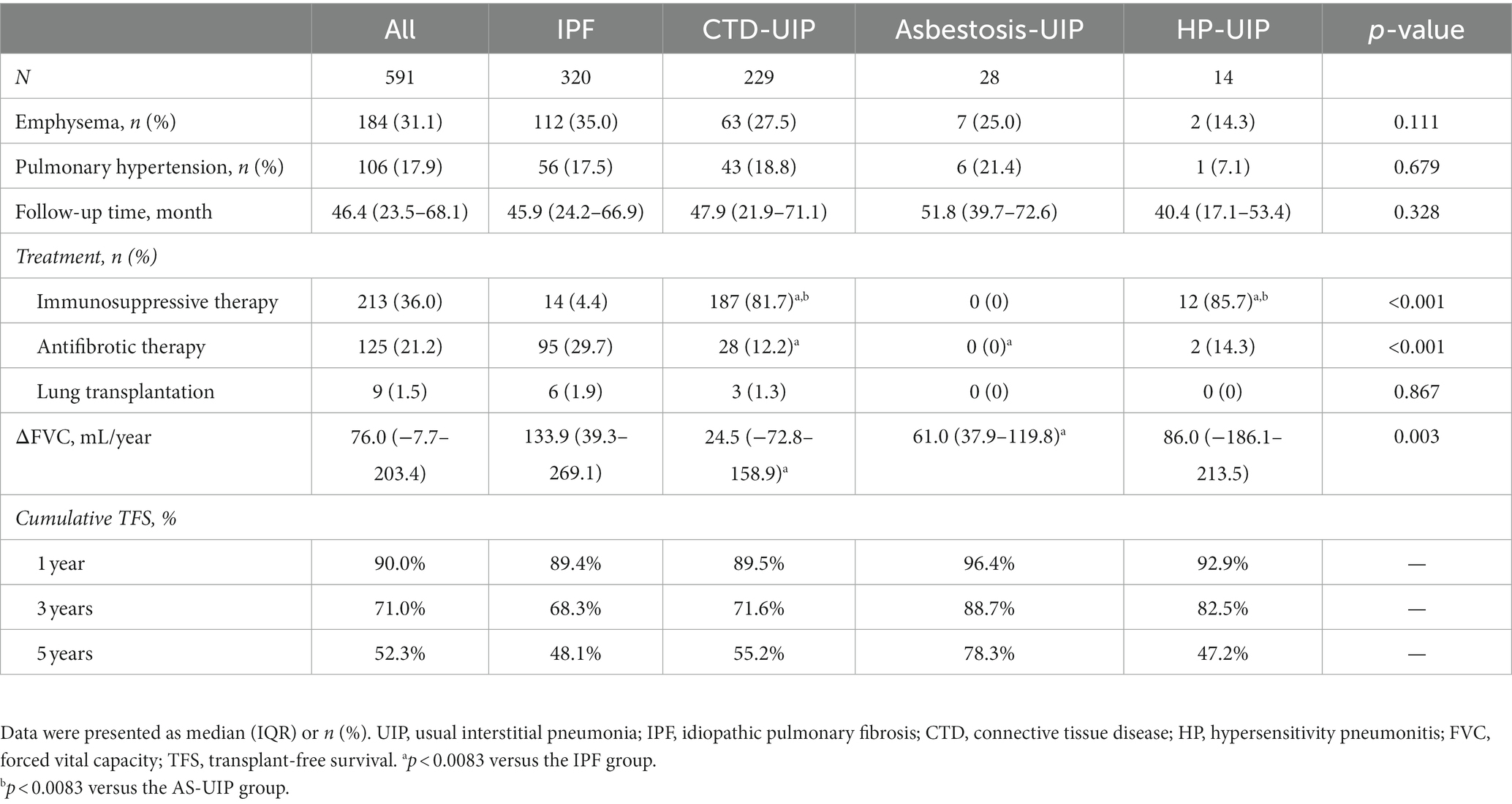
Table 3. Comorbidities, treatment, FVC decline and transplant-free survival of UIP patients in various etiology groups.
Subgroup analysis
A subgroup analysis was conducted specifically for patients with CTD-UIP. Most demographic and laboratory characteristics were comparable among patients with RA-UIP, pSS-UIP, AAV-UIP, and other CTD-UIP. However, there were notable differences in age and levels of blood inflammatory markers, including leukocytes, neutrophils, ESR, and CRP levels, which were higher in AAV-UIP patients. The rates of decline in FVC varied among the patients. Both RA-UIP (88.1 mL/year) and AAV-UIP (72.9 mL/year) patients exhibited significantly faster FVC decline compared to pSS-UIP (25.9 mL/year) and other CTD-UIP patients (20.7 mL/year), with a significance level of p < 0.05 (Supplementary Table S1).
Additional comparisons were conducted between patients with or without emphysema, and those with or without PHT in the IPF and CTD-UIP groups. Patients with emphysema presented slower median annual FVC decline than those without emphysema, both in the IPF group (50.7 mL/yr vs. 177.3 mL/yr, p = 0.047) and the CTD-UIP group (-35.6 mL/yr vs. 38.9 mL/yr, p = 0.024). Patients with PHT, however, presented significantly shorter TFS than those without PHT in the IPF group (35.9 vs. 65.2 month median TFS, p < 0.001) and the CTD-UIP group (28.5 vs. 80.9 month median TFS, p < 0.001) (see Supplementary Tables S2,S3).
Transplant-free survival and risk factors
The Kaplan–Meier curves in Figure 2 illustrate the differences in TFS among UIP patients with different etiologies. Statistically significant distinctions were observed among the IPF, CTD-UIP, asbestosis-UIP, and HP-UIP patients (p = 0.023) (Figure 2A). The IPF group exhibited the poorest prognosis (median TFS: 55.9 months), followed by the CTD-UIP group (median TFS: 66.7 months) and HP-UIP group (median TFS: 57.5 months). The asbestosis-UIP group demonstrated the best survival, with the median TFS not being reached. Pairwise comparisons revealed significant differences between the IPF and asbestosis-UIP patients (log-rank p < 0.001). While the difference did not reach statistical significance after Bonferroni correction, a marginal trend towards better survival was observed in the CTD-UIP group compared to the asbestosis-UIP group (p = 0.017).
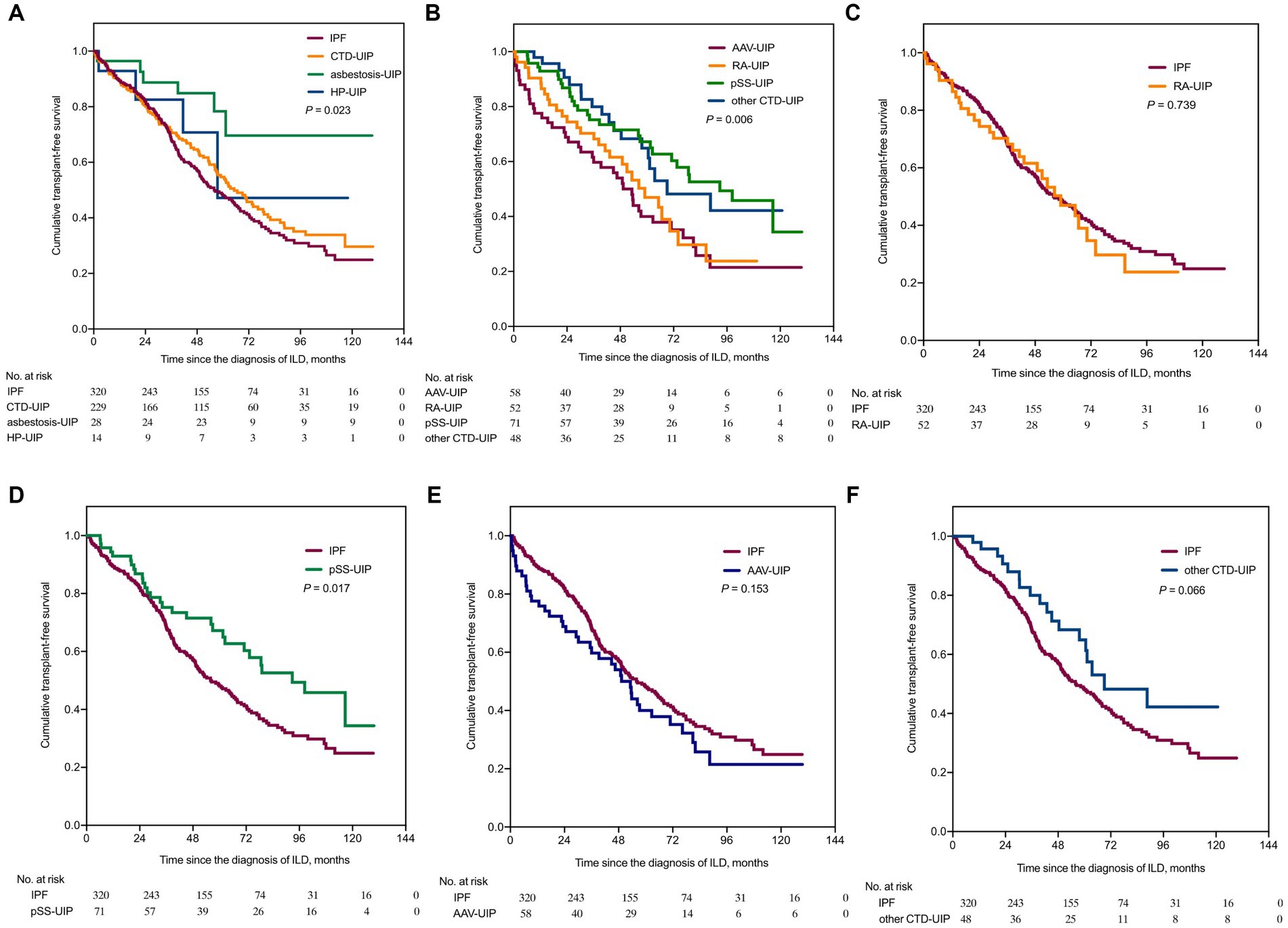
Figure 2. Kaplan–Meier survival curves comparing transplant-free survival among patients with UIP of various etiologies. TFS, transplant-free survival. (A) TFS comparison between IPF, CTD-UIP, asbestosis-UIP, and HP-UIP (p = 0.023). (B) TFS comparison among RA-UIP, pSS-UIP, AAV-UIP, and other CTD-UIP (p = 0.006). (C) TFS comparison between IPF and RA-UIP [HR = 1.069 (95% CI: 0.715, 1.597), p = 0.739, compared to IPF]. (D) TFS comparison between IPF and pSS-UIP [HR = 0.624 (95% CI: 0.446, 0.872), p = 0.017, compared to IPF]. (E) TFS comparison between IPF and AAV-UIP [HR = 1.286 (95% CI: 0.882, 1.875), p = 0.153, compared to IPF]. (F) TFS comparison between IPF and other CTD-UIP, [HR = 0.638 (95% CI: 0.425, 0.958), p = 0.066, compared to IPF]. IPF, idiopathic pulmonary fibrosis; CTD, connective tissue disease; UIP, usual interstitial pneumonia; HP, hypersensitivity pneumonitis; RA, rheumatoid arthritis; pSS, primary Sjögren’s syndrome; AAV, anti-neutrophil cytoplasmic antibody-associated vasculitis; HR, hazard ratio; CI, confidence interval.
Furthermore, differences in TFS were observed within the CTD-UIP patients (p = 0.006) (Figure 2B). The AAV-UIP group (median TFS: 49.3 months) and RA-UIP group (median TFS: 59.0 months) had the poorest prognoses, while the pSS-UIP group (median TFS: 92.8 months) and other CTD-UIP group (median TFS: 69.1 months) exhibited better TFS. A significant difference was found between the AAV-UIP and pSS-UIP patients (log-rank p < 0.001), and a trend towards better survival was observed in the pSS-UIP group compared to the RA-UIP group (p = 0.017). When the IPF group was used as a control, neither the RA-UIP nor AAV-UIP patients showed a survival advantage, whereas a significant survival advantage was found in the pSS-UIP group (p = 0.017), and a marginal survival advantage was observed in the other CTD-UIP group (p = 0.066) (Figures 2C–F).
Univariate analysis indicated that age (HR 1.043, p < 0.001) and PHT (HR 1.769, p = 0.002) were risk factors, while female sex, probable UIP (HR 0.433, p < 0.001, compared with UIP), FVC% predicted (HR 0.971, p < 0.001), DLCO SB% predicted (HR 0.971, p < 0.001), pSS-UIP (HR 0.599, p = 0.011, compared with IPF), and asbestosis-UIP (HR 0.320, p = 0.006, compared with IPF) were protective factors for mortality or lung transplant. Cox hazard modeling demonstrated that the impact of etiology on survival persisted even after adjusting for age, sex, radiological patterns, pulmonary function, and PHT at the time of diagnosis (Table 4).
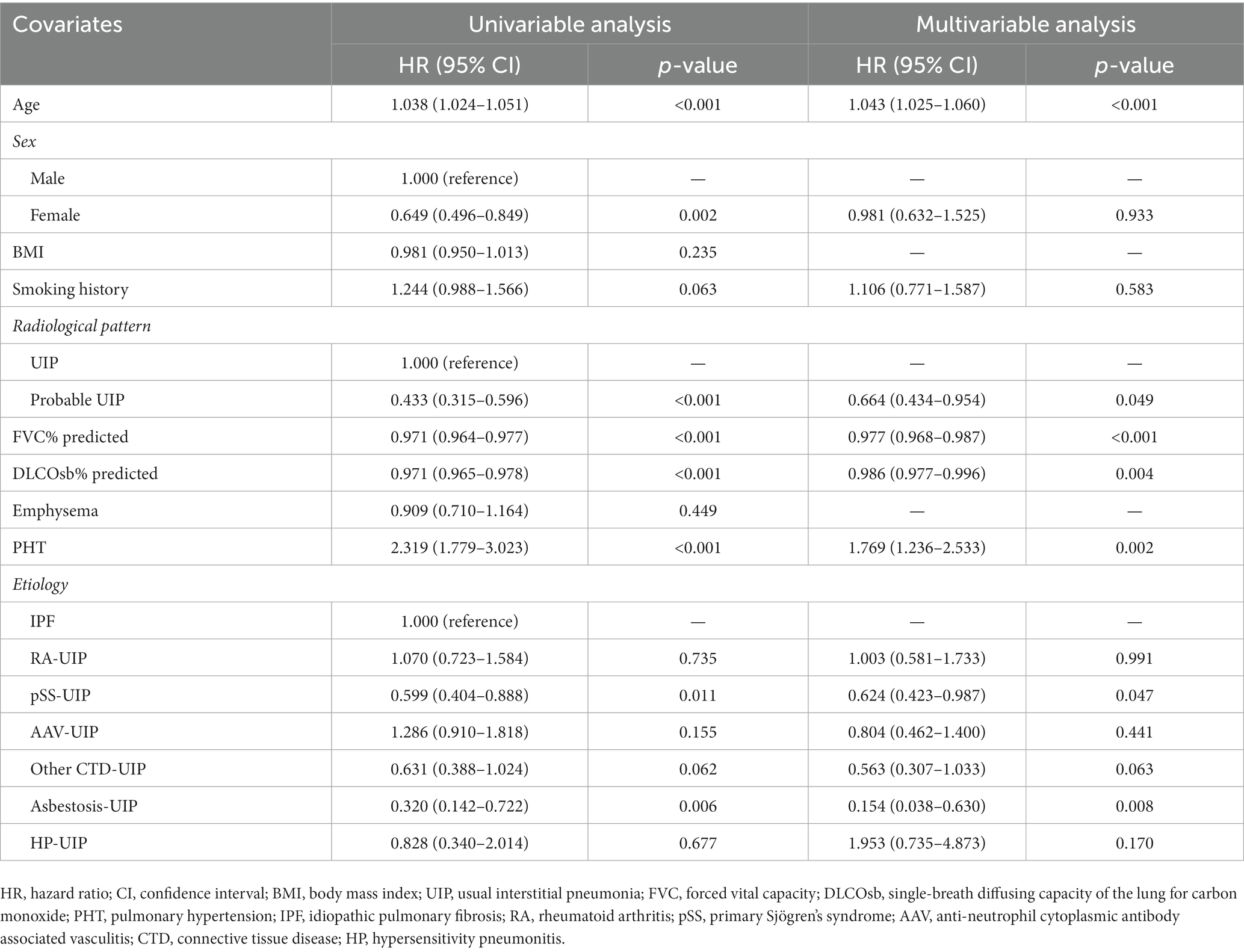
Table 4. Risk factors associated with death or lung transplantation in UIP patients.
Discussion
This study focused on patients with ILD exhibiting UIP and probable UIP patterns on HRCT. Our findings revealed significant heterogeneity in disease progression and some shared characteristics among UIP patients with different etiologies.
In the current study, we observed significant heterogeneity in FVC decline rates and TFS among patients with UIP of different etiologies. Asbestosis, characterized by irregular and/or linear opacities with basal preponderance, ground-glass opacities, and mosaic attenuation accompanied by pleural abnormalities, is caused by repeated asbestos inhalation (24). Our recently published data showed that 9.8% (20/204) of patients with asbestosis manifested a UIP pattern on HRCT (25). Limited data are available regarding the survival of asbestosis-UIP patients. In our study, asbestosis-UIP patients demonstrated a slowly progressive disease course and better survival outcomes compared to IPF patients, despite the two groups being indistinguishable in terms of clinical and laboratory features. Another disease entity in our cohort, UIP associated with pSS also exhibited a prognostic advantage over IPF. Pathological comparisons revealed that fibroblast foci, a characteristic feature of IPF, are infrequent and sporadically observed in pSS-UIP and asbestosis patients, whereas interstitial inflammation, plasma cell infiltration, lymphoid follicles with germinal centers, cysts, bronchiolitis, and pleuritis were significantly more pronounced in the lungs of pSS-UIP patients (26, 27). Recent studies utilizing artificial intelligence to quantify fibroblast foci have demonstrated that a large area occupied by fibroblast foci predicts a poor prognosis in IPF (28). Similarly, data from patients with chronic HP have indicated that the presence of fibroblast foci is an independent predictor of time to death or transplantation (29). Thus, we consider the number of fibroblastic foci to be at least one of the crucial factors contributing to the differences in fibrosis progression and mortality among patients with UIP. However, the factors influencing the presence and number of fibroblastic foci remain unknown.
In contrast to pSS-UIP, both the decline rate in FVC and TFS in patients with RA-UIP and AAV-UIP were similar to those observed in IPF. This finding could help explain the conflicting evidence regarding prognostic differences between CTD-UIP and IPF, as the composition of CTD patients varies across studies (5, 6). Studies comparing survival between RA-UIP and IPF, as well as AAV-UIP and IPF, have reported results consistent with our findings (30, 31). Interestingly, in a study by Watanabe et al. (32), while the decline in lung volume and median survival time were similar between AAV-UIP and IPF patients, the causes of death differed. AAV-UIP patients more frequently experienced deaths related to anti-immune therapy (infectious complications) or vasculitis itself (alveolar hemorrhage and potentially cardiovascular disease), whereas in IPF, most deaths were related to respiratory system issues. Nevertheless, considering the potential benefits, early initiation of antifibrotic therapy may be considered in patients with RA and AAV exhibiting a UIP pattern. Moreover, due to the evident heterogeneity, it may be inappropriate to treat CTD-ILD as a unified entity when conducting analyses related to ILD progression and survival. Furthermore, we recommend considering the specific etiology before conducting clinical trials, as therapeutic effects may diverge among patients with different progression patterns.
It is worth to mention that the annual decline rate of FVC observed in IPF patients from the current study appears to be slower than that reported in several large-scale clinical trials. In the TOMORROW trial, the annual decline rate of FVC was 60 mL in the 150 mg twice daily nintedanib group and 190 mL in the placebo group (33). In the INPULSIS trial, adjusted rate of decline in FVC was 112.4 mL/year with nintedanib and 223.3 mL/year with placebo (34). We noticed that the baseline FVC% (85.4%) of the patients in this cohort was higher than that in the TOMORROW (80.2%) and INPULSIS trials (80%). Besides, a portion of patients in the current study had received antifibrotic treatments during the study period. These could be the possible reasons for the different FVC decline rate in our study.
Despite the heterogeneities, the clinical and laboratory data of patients with UIP from various causes display remarkable similarities, shedding light on the shared underlying mechanisms of this pattern. The findings of our current study demonstrate that the median age of patients in each group exceeded 60 years, indicating a probable correlation between the UIP pattern and aging. Advanced age has long been recognized as a risk factor for IPF. Epidemiological evidence consistently reveals a higher prevalence and incidence of IPF with increasing age (35), while mechanistic investigations have underscored the crucial role of age-related processes, such as telomere attrition, cellular senescence, stem cell exhaustion, and disrupted intercellular communication, in the development of IPF (36). Similarly, in secondary ILDs, studies have reported older age in ILDs associated with CTD compared to CTD without ILD (37, 38), as well as in chronic HP compared to acute HP (39). In a recent study by Laurent et al. (40), a significant positive correlation was found between definite or possible UIP patterns and age in individuals exposed to asbestos. However, previous studies have yielded inconsistent findings regarding age differences between patients with UIP and NSIP (30, 41). Thus, it is plausible that the association with aging might be more closely related to the fibrosis behavior itself rather than specific ILD patterns. Another noteworthy shared characteristic among patients with UIP is the presence of elevated levels of CYFRA21-1 in the serum and neutrophils in BALF. The significance of CYFRA21-1 in ILDs has been increasingly recognized in recent years. CYFRA21-1 is a cytoskeletal protein expressed in type 1 and type 2 pneumocytes, as well as respiratory bronchiolar epithelial cells, and its release is triggered by lung injury (42). Notably, Molyneaux et al. (43) discovered that baseline concentrations of CYFRA21-1 could potentially identify individuals at risk of disease progression within 12 months and predict overall mortality in patients with IPF. In our study, we observed comparably elevated levels of CYFRA21-1 in both IPF and secondary UIP patients. Similarly, previous investigations have underscored the importance of neutrophils in the immune pathogenesis of IPF (44, 45), and our data revealed that BALF neutrophils were equally elevated in IPF and secondary UIP, indicating analogous immune pathogenesis in UIP caused by various underlying conditions. However, the elevation in BALF neutrophils does not appear to be exclusive to UIP (46). One possible explanation is that BALF neutrophils are associated with lung fibrosis in general rather than specifically with the UIP pattern, which is supported by the increase in neutrophils and decrease in lymphocytes observed in the BALF during the transition from acute to chronic hypersensitivity pneumonitis (39).
The similarities between IPF and other ILDs with “IPF-like” behaviors gave birth to the concept of progressive pulmonary fibrosis (PPF), which indeed, has its clinical values, especially on drug development and application. However, precise and personalized medication should not be neglected. Thus, “smarter lumping and smarter splitting” maybe advocated for these patients (47, 48). As for UIP patients, on the one hand, exploring the common biological pathways during UIP development may provide broader directions for therapy; on the other hand, tailored therapies based on precision medicine urge deeper investigations for the causes of heterogeneity in disease progression and prognosis of these patients.
The current study had several limitations that should be mentioned. Firstly, being a retrospective single-center study, it may be prone to selection bias, and the number of asbestosis-UIP and HP-UIP patients was relatively small. In the real world, the cases of HP-UIP and asbestosis are limited, a previous published study by our team showed that 9.8% (20/204) of patients with asbestosis and 10.8% (8/74) of patients with fibrotic HP manifested a UIP pattern on HRCT (25). However, this limitation may affect the accuracy of our assessments and result in an insufficient statistical power to detect existing differences, thus, some of the results must be interpretated with caution. Secondly, not all patients underwent a multidisciplinary team (MDT) assessment at the time of ILD diagnosis, although we conducted retrospective MDT discussions for each patient and excluded those with unclear diagnoses, there remains a possibility of misjudgments regarding the etiology in a few patients. Thirdly, the majority of the pathological results were derived from transbronchial lung biopsies, which prevented the comprehensive analysis of pathological features in this study. Fourthly, the assessment of the likelihood of developing PHT relied on echocardiography rather than right heart catheterization (RHC), which may have compromised accuracy. However, it is important to note that echocardiography offers the advantage of being non-invasive. RHC is an invasive procedure associated with potential risks for patients, and its benefits for patients with ILD are limited. Studies have demonstrated that in patients with PHT, the measurement of tricuspid regurgitation velocity (TRV) correlates significantly with pulmonary artery systolic pressure (PASP) obtained through RHC. Echocardiography has consistently proven to be a reliable method for diagnosing PHT (49). Last, the cause of death was not analyzed due to the fact that most deaths occurred outside the hospital, thus the specific reasons for the observed survival disparities among patients remain unknown.
In conclusion, this respective cohort study demonstrates that patients with UIP stemming from various etiologies exhibit common features such as advanced age, elevated serum levels of CYFRA21-1, and increased neutrophil counts in BALF. However, there are notable differences in the decline rate of FVC and TFS among these patients. Specifically, UIP associated with asbestosis and pSS-UIP exhibit slower disease progression and improved survival rates compared to IPF.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by The Ethics Committee of Beijing Chao-Yang Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
QY: conceptualization and methodology. SY: data curation, formal analysis, and writing-original draft preparation. JW: writing-reviewing and editing. DS: software and validation. YW: visualization and investigation. CX: project administration and supervision. All authors contributed to the article and approved the submitted version.
Funding
The work was sponsored by High Level Public Health Technology Talent Construction Project (DL-02-21) and Reform and Development Program of Beijing Institute of Respiratory Medicine (ysrh2022013).
Acknowledgments
The authors extend our sincere gratitude to all the patients and investigators who participated in this study. The authors would also like to express our thanks to Luling Li and Xiang Gong from Beijing Chaoyang Hospital for their invaluable contributions in diagnosing connective tissue diseases (CTD) and evaluating computed tomography (CT) scans. Their assistance greatly contributed to the quality and accuracy of our research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2023.1246767/full#supplementary-material
References
1. Raghu, G, Remy-Jardin, M, Richeldi, L, Thomson, CC, Inoue, Y, Johkoh, T, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2022) 205:e18–47. doi: 10.1164/rccm.202202-0399ST
2. Nagai, S, Kitaichi, M, Itoh, H, Nishimura, K, Izumi, T, and Colby, TV. Idiopathic nonspecific interstitial pneumonia/fibrosis: comparison with idiopathic pulmonary fibrosis and BOOP. Eur Respir J. (1998) 12:1010–9. doi: 10.1183/09031936.98.12051010
3. Kim, HC, Lee, JS, Lee, EY, Ha, Y-J, Chae, EJ, Han, M, et al. Risk prediction model in rheumatoid arthritis-associated interstitial lung disease. Respirology. (2020) 25:1257–64. doi: 10.1111/resp.13848
4. Selman, M, Pardo, A, and Wells, AU. Usual interstitial pneumonia as a stand-alone diagnostic entity: the case for a paradigm shift? Lancet Respir Med. (2023) 11:188–96. doi: 10.1016/S2213-2600(22)00475-1
5. Park, JH, Kim, DS, Park, I-N, Jang, SJ, Kitaichi, M, Nicholson, AG, et al. Prognosis of fibrotic interstitial pneumonia: idiopathic versus collagen vascular disease-related subtypes. Am J Respir Crit Care Med. (2007) 175:705–11. doi: 10.1164/rccm.200607-912OC
6. Alhamad, EH. Clinical characteristics and survival in idiopathic pulmonary fibrosis and connective tissue disease-associated usual interstitial pneumonia. J Thorac Dis. (2015) 7:386–93. doi: 10.3978/j.issn.2072-1439.2014.12.40
7. Raghu, G, Remy-Jardin, M, Myers, JL, Richeldi, L, Ryerson, CJ, Lederer, DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2018) 198:e44–68. doi: 10.1164/rccm.201807-1255ST
8. Aletaha, D, Neogi, T, Silman, AJ, Funovits, J, Felson, DT, Bingham, CO, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. (2010) 62:2569–81. doi: 10.1002/art.27584
9. van den Hoogen, F, Khanna, D, Fransen, J, Johnson, SR, Baron, M, Tyndall, A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. (2013) 65:2737–47. doi: 10.1002/art.38098
10. Bohan, A, and Peter, JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. (1975) 292:344–7. doi: 10.1056/NEJM197502132920706
11. Vitali, C, Bombardieri, S, Jonsson, R, Moutsopoulos, HM, Alexander, EL, Carsons, SE, et al. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis. (2002) 61:554–8. doi: 10.1136/ard.61.6.554
12. Sharp, GC, Irvin, WS, Tan, EM, Gould, RG, and Holman, HR. Mixed connective tissue disease-an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med. (1972) 52:148–59. doi: 10.1016/0002-9343(72)90064-2
13. Fischer, A, Antoniou, KM, Brown, KK, Cadranel, J, Corte, TJ, du Bois, RM, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J. (2015) 46:976–87. doi: 10.1183/13993003.00150-2015
14. American Thoracic Society. Diagnosis and initial management of nonmalignant diseases related to asbestos. Am J Respir Crit Care Med. (2004) 170:691–715. doi: 10.1164/rccm.200310-1436ST
15. Raghu, G, Remy-Jardin, M, Ryerson, CJ, Myers, JL, Kreuter, M, Vasakova, M, et al. Diagnosis of hypersensitivity pneumonitis in adults. An official ATS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2020) 202:e36–69. doi: 10.1164/rccm.202005-2032ST
16. Ryerson, CJ, Hartman, T, Elicker, BM, Ley, B, Lee, JS, Abbritti, M, et al. Clinical features and outcomes in combined pulmonary fibrosis and emphysema in idiopathic pulmonary fibrosis. Chest. (2013) 144:234–40. doi: 10.1378/chest.12-2403
17. Ponikowski, P, Voors, AA, Anker, SD, Bueno, H, Cleland, JGF, Coats, AJS, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. (2016) 37:2129–200. doi: 10.1093/eurheartj/ehw128
18. Miller, MR, Hankinson, J, Brusasco, V, Burgos, F, Casaburi, R, Coates, A, et al. Standardisation of spirometry. Eur Respir J. (2005) 26:319–38. doi: 10.1183/09031936.05.00034805
19. Macintyre, N, Crapo, RO, Viegi, G, Johnson, DC, van der Grinten, CPM, Brusasco, V, et al. Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur Respir J. (2005) 26:720–35. doi: 10.1183/09031936.05.00034905
20. Ryerson, CJ, Vittinghoff, E, Ley, B, Lee, JS, Mooney, JJ, Jones, KD, et al. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest. (2014) 145:723–8. doi: 10.1378/chest.13-1474
21. Wells, AU, Desai, SR, Rubens, MB, Goh, NSL, Cramer, D, Nicholson, AG, et al. Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med. (2003) 167:962–9. doi: 10.1164/rccm.2111053
22. Meyer, KC, Raghu, G, Baughman, RP, Brown, KK, Costabel, U, du Bois, RM, et al. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. (2012) 185:1004–14. doi: 10.1164/rccm.201202-0320ST
23. Lange, P, Celli, B, Agustí, A, Boje Jensen, G, Divo, M, Faner, R, et al. Lung-function trajectories leading to chronic obstructive pulmonary disease. N Engl J Med. (2015) 373:111–22. doi: 10.1056/NEJMoa1411532
24. Keskitalo, E, Salonen, J, Vähänikkilä, H, and Kaarteenaho, R. Survival of patients with asbestosis can be assessed by risk-predicting models. Occup Environ Med. (2021) 78:516–21. doi: 10.1136/oemed-2020-106819
25. Ma, R, Li, S, Wang, Y, Yang, S, Bao, N, and Ye, Q. High-resolution computed tomography features of asbestosis versus fibrotic hypersensitivity pneumonitis: an observational study. BMC Pulm Med. (2022) 22:207. doi: 10.1186/s12890-022-01967-3
26. Enomoto, Y, Takemura, T, Hagiwara, E, Iwasawa, T, Okudela, K, Yanagawa, N, et al. Features of usual interstitial pneumonia in patients with primary Sjögren’s syndrome compared with idiopathic pulmonary fibrosis. Respir Investig. (2014) 52:227–35. doi: 10.1016/j.resinv.2014.02.003
27. Roggli, VL, Gibbs, AR, Attanoos, R, Churg, A, Popper, H, Cagle, P, et al. Pathology of asbestosis-an update of the diagnostic criteria: report of the asbestosis committee of the college of American Pathologists and Pulmonary Pathology Society. Arch Pathol Lab Med. (2010) 134:462–80. doi: 10.5858/134.3.462
28. Mäkelä, K, Mäyränpää, MI, Sihvo, H-K, Bergman, P, Sutinen, E, Ollila, H, et al. Artificial intelligence identifies inflammation and confirms fibroblast foci as prognostic tissue biomarkers in idiopathic pulmonary fibrosis. Hum Pathol. (2021) 107:58–68. doi: 10.1016/j.humpath.2020.10.008
29. Wang, P, Jones, KD, Urisman, A, Elicker, BM, Urbania, T, Johannson, KA, et al. Pathologic findings and prognosis in a large prospective cohort of chronic hypersensitivity pneumonitis. Chest. (2017) 152:502–9. doi: 10.1016/j.chest.2017.02.011
30. Maillet, T, Goletto, T, Beltramo, G, Dupuy, H, Jouneau, S, Borie, R, et al. Usual interstitial pneumonia in ANCA-associated vasculitis: a poor prognostic factor. J Autoimmun. (2020) 106:102338. doi: 10.1016/j.jaut.2019.102338
31. Kim, EJ, Elicker, BM, Maldonado, F, Webb, WR, Ryu, JH, Van Uden, JH, et al. Usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J. (2010) 35:1322–8. doi: 10.1183/09031936.00092309
32. Watanabe, T, Minezawa, T, Hasegawa, M, Goto, Y, Okamura, T, Sakakibara, Y, et al. Prognosis of pulmonary fibrosis presenting with a usual interstitial pneumonia pattern on computed tomography in patients with myeloperoxidase anti-neutrophil cytoplasmic antibody-related nephritis: a retrospective single-center study. BMC Pulm Med. (2019) 19:194. doi: 10.1186/s12890-019-0969-5
33. Richeldi, L, Costabel, U, Selman, M, Kim, DS, Hansell, DM, Nicholson, AG, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. (2011) 365:1079–87. doi: 10.1056/NEJMoa1103690
34. Richeldi, L, Cottin, V, du Bois, RM, Selman, M, Kimura, T, Bailes, Z, et al. Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS® trials. Respir Med. (2016) 113:74–9. doi: 10.1016/j.rmed.2016.02.001
35. Raghu, G, Weycker, D, Edelsberg, J, Bradford, WZ, and Oster, G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2006) 174:810–6. doi: 10.1164/rccm.200602-163OC
36. Lu, Y, Chen, J, Wang, S, Tian, Z, Fan, Y, Wang, M, et al. Identification of genetic signature associated with aging in pulmonary fibrosis. Front Med. (2021) 8:744239. doi: 10.3389/fmed.2021.744239
37. Kim, H, Cho, S-K, Song, Y-J, Kang, J, Jeong, S-A, Kim, HW, et al. Clinical characteristics of rheumatoid arthritis patients with interstitial lung disease: baseline data of a single-center prospective cohort. Arthritis Res Ther. (2023) 25:43. doi: 10.1186/s13075-023-03024-8
38. Toyoda, Y, Koyama, K, Kawano, H, Nishimura, H, Kagawa, K, Morizumi, S, et al. Clinical features of interstitial pneumonia associated with systemic lupus erythematosus. Respir Investig. (2019) 57:435–43. doi: 10.1016/j.resinv.2019.04.005
39. Wang, L-J, Cai, H-R, Xiao, Y-L, Wang, Y, and Cao, M-S. Clinical characteristics and outcomes of hypersensitivity pneumonitis: a population-based study in China. Chin Med J. (2019) 132:1283–92. doi: 10.1097/CM9.0000000000000256
40. Laurent, F, Benlala, I, Dournes, G, Gramond, C, Thaon, I, Clin, B, et al. Interstitial lung abnormalities detected by CT in asbestos-exposed subjects are more likely associated to age. J Clin Med. (2021) 10:3130. doi: 10.3390/jcm10143130
41. Yunt, ZX, Chung, JH, Hobbs, S, Fernandez-Perez, ER, Olson, AL, Huie, TJ, et al. High resolution computed tomography pattern of usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease: relationship to survival. Respir Med. (2017) 126:100–4. doi: 10.1016/j.rmed.2017.03.027
42. Moll, R, Franke, WW, Schiller, DL, Geiger, B, and Krepler, R. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cells. (1982) 31:11–24. doi: 10.1016/0092-8674(82)90400-7
43. Molyneaux, PL, Fahy, WA, Byrne, AJ, Braybrooke, R, Saunders, P, Toshner, R, et al. CYFRA 21-1 predicts progression in idiopathic pulmonary fibrosis: a prospective longitudinal analysis of the PROFILE cohort. Am J Respir Crit Care Med. (2022) 205:1440–8. doi: 10.1164/rccm.202107-1769OC
44. Warheit-Niemi, HI, Huizinga, GP, Edwards, SJ, Wang, Y, Murray, SK, O’Dwyer, DN, et al. Fibrotic lung disease alters neutrophil trafficking and promotes neutrophil elastase and extracellular trap release. Immunohorizons. (2022) 6:817–34. doi: 10.4049/immunohorizons.2200083
45. Gregory, AD, Kliment, CR, Metz, HE, Kim, K-H, Kargl, J, Agostini, BA, et al. Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J Leukoc Biol. (2015) 98:143–52. doi: 10.1189/jlb.3HI1014-493R
46. Veeraraghavan, S, Latsi, PI, Wells, AU, Pantelidis, P, Nicholson, AG, Colby, TV, et al. BAL findings in idiopathic nonspecific interstitial pneumonia and usual interstitial pneumonia. Eur Respir J. (2003) 22:239–44. doi: 10.1183/09031936.03.00105202
47. Wells, AU, Brown, KK, Flaherty, KR, Kolb, M, and Thannickal, VJ. What’s in a name? That which we call IPF, by any other name would act the same. Eur Respir J. (2018) 51:1800692. doi: 10.1183/13993003.00692-2018
48. Karampitsakos, T, Juan-Guardela, BM, Tzouvelekis, A, and Herazo-Maya, JD. Precision medicine advances in idiopathic pulmonary fibrosis. EBioMedicine. (2023) 95:104766. doi: 10.1016/j.ebiom.2023.104766
49. Greiner, S, Jud, A, Aurich, M, Hess, A, Hilbel, T, Hardt, S, et al. Reliability of noninvasive assessment of systolic pulmonary artery pressure by Doppler echocardiography compared to right heart catheterization: analysis in a large patient population. J Am Heart Assoc. (2014) 3:e001103. doi: 10.1161/JAHA.114.001103
Glossary
Keywords: usual interstitial pneumonia, idiopathic pulmonary fibrosis, etiology, pulmonary function, transplant-free survival
Citation: Yang S, Wang J, Sun D, Wang Y, Xue C and Ye Q (2023) Disease progression in patients with usual interstitial pneumonia and probable UIP patterns on computed tomography with various underlying etiologies: a retrospective cohort study. Front. Med. 10:1246767. doi: 10.3389/fmed.2023.1246767
Edited by:
Eirini Vasarmidi, University of Crete, GreeceReviewed by:
Theodoros Karampitsakos, University of South Florida, United StatesMaria Otaola, University of Buenos Aires, Argentina
Copyright © 2023 Yang, Wang, Sun, Wang, Xue and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiao Ye, eWVxaWFvX2NoYW95YW5nQHNpbmEuY29t