 Rosa Sonja Alesci
Rosa Sonja Alesci Carola Hecking1
Carola Hecking1 Detlev Janssen
Detlev Janssen- 1IMD Blood Coagulation Centre, Bad Homburg, Germany
- 2Institute of Immunology and Genetics, Kaiserslautern, Germany
- 3Med-i-Scene Concept, Weisendorf, Germany
- 4IMD Blood Coagulation Centre, Mannheim, Germany
Background: The American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) have introduced an internationally shared framework for variant classification in genetic disorders. FVII deficiency is a rare inherited autosomal recessive bleeding disorder with sparse data concerning ACMG classification.
Methods: To develop an approach which may improve the utility of molecular genetic test results, 129 patients with FVII deficiency were retrospectively assigned to six subgroups for exploratory analysis: F7 gene wildtype (group 1), ACMG 1 (benign variant) or ACMG 2 (likely benign variant), only (group 2), ACMG 3 (variant of uncertain significance) ± ACMG 1–2 heterozygous or not classified variant (group 3), ACMG 4 (likely pathogenic variant), or ACMG 5 (pathogenic variant) single heterozygous ± ACMG 1–3 single heterozygous (group 4), ACMG 4–5 homozygous or ≥2 ACMG 4–5 heterozygous or ≥1 ACMG 4–5 heterozygous plus either ACMG 1 c.1238G>A modifying variant homozygous or ≥2 ACMG 1–3 (group 5), FVII deficiency and another bleeding disorder (group 6).
Results: Eleven of 31 patients (35.5%) in group 5 had abnormal ISTH-BS (n = 7) and/or history of substitution with recombinant factor VIIa (n = 5) versus 4 of 80 patients (5.0%, n = 1 abnormal ISTH-BS, n = 3 substitution) in groups 1 (n = 2/22), 2 (n = 1/29), 3 (n = 0/9), and 4 (n = 1/20). Four of 18 patients (22.2%) with FVII deficiency and another bleeding disorder (group 6) had an abnormal ISTH-BS (n = 2) and/or history of substitution with recombinant factor VIIa (n = 3).
Conclusion: Patients with a homozygous ACMG 4–5 variant or with specific combinations of heterozygous ACMG 4–5 ± ACMG 1–3 variants exhibited a high-risk bleeding phenotype in contrast to the remaining patients without another bleeding disorder. This result may serve as a basis to develop a genotype/phenotype prediction model in future studies.
Introduction
Congenital factor VII (FVII) deficiency is an autosomal recessively inherited rare bleeding disorder. The clinical phenotypes in FVII deficient patients show a wide variation from asymptomatic to severe with a poor correlation to FVII activity (1). Some patients with very low FVII activity had no bleeding symptoms whereas patients with partial FVII deficiency may have recurrent bleeding episodes (2). FVII levels above 20% are thought to be sufficient for prevention against spontaneous bleeding (1), possibly due to an enhanced effect of TF-FVII complex which activates coagulation even with small amounts of FVII (1, 3). However, previously asymptomatic patients with FVII levels above 20% may have an increased bleeding risk in situations such as major surgery or trauma. FVII deficiency may be caused by a quantitative FVII defect (type I, decreased FVII antigen) or qualitative FVII defect (type II, normal FVII antigen) (1).
Even though the prevalence of FVII deficiency was repeatedly reported to be 1:300,000–1:500,000 individuals (4–7) it is believed that mild, moderate or severe forms of FVII deficiency are by far more frequent reaching 1:59,000 individuals (8).
The coagulation factor VII gene (F7, HGNC ID: 3544) is located on chromosome 13 (13q34) with nine exons and eight introns, which compose a 12.8 kb gene locus near the telomeric region of the chromosome besides the gene promotor region (9). In the coagulation factor VII variant database of the European Association for Haemophilia and Allied Disorders (EAHAD) 271 unique variants of F7 gene in 1058 individuals with FVII activity below or above the normal range were recorded until May 2022 (10). Most F7 variants were small lesions with single nucleotide substitutions (point) in 86.7% of individuals followed by deletions (8.9%), duplications (2.6%), indel rearrangements (1.1%) and insertions (0.7%) (10). Missense variants represented 74% of single nucleotide substitutions (11).
The genotype/phenotype relationship in FVII deficient patients has extensively been studied in humans (2, 8, 12). FVII levels below 10% were seen in about 50% of patients who are homozygous or compound heterozygotes for pathogenic F7 gene variants compared to 7% of patients with heterozygous F7 pathogenic gene variants (6). Bleeding symptoms were discovered in 71% of patients with homozygous causative F7 gene variants versus 50% in compound heterozygotes and 19% in heterozygotes (2).
Whereas FVII:C shows a poor correlation to severity of bleeding phenotype the type and the site of a F7 variant may be helpful to predict hemorrhagic risks (13). For example, mutations affecting TFPI-binding exosites of FVII may markedly prolong clotting time (14). The utility of molecular genetic diagnostics depends on the validity of the pathogenicity classification for a specific variant detected. As a major step to establish an internationally shared framework for a systematic, objective and evidence-based variant classification in genetic disorders the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) published standards and guidelines for the interpretation of sequence variants in 2015 (15). However, these guidelines lacked specificity in several areas or resulted in contradictory or ambiguous interpretations. Therefore, validated “semiquantitative, hierarchical evidence-based rules for locus interpretation” (Sherloc) was developed by the ACMG-AMP (16).
The ACMG classification uses a specific standard terminology to describe gene variants that are the cause of Mendelian disorders and is based on a complex process for assigning a variant to one of the five ACMG categories (15, 16): Variants classified as pathogenic (ACMG 5) or likely pathogenic (ACMG 4) have met specific criteria and may be used by the health-care provider for clinical decision making. A variant considered benign (ACMG 1) is not causative for the patient’s disorder or symptoms. ACMG 2 describes a variant which is likely benign (ACMG 2). Variants of uncertain significance (VUS; ACMG 3) should imply efforts for changing the classification to pathogenic or benign based on additional information.
In general, interpretation of gene variants classified as pathogenic is challenging (17). A pathogenic variant may be present in an individual without phenotypic correlate. In addition, classifications of specific variants may change over time or differ between databases such as Human Gene Mutation Database (HGMD) and ClinVar (17). To be useful as a contribution for therapeutic decisions the variants of the F7 gene should primarily be analyzed with respect to abnormal bleeding phenotype. There are several F7 gene variants associated with reduced FVII activity which are classified as benign (ACMG 1), for example the frequent missense F7 variant p.(Arg353Gln), also known as Arg353Gln polymorphism (R353Q; rs6046) or described by the single nucleotide substitution of Arginine by Glycine residue in position 413, p.(Arg413Gln) or by the complementary DNA (cDNA) change c.1238G>A. This variant leads to a mean decrease of FVII:C activity by about 20–30% in individuals with M1M2 genotype (heterozygous) (18–21), and by 43% in homozygotes (20). The minor allele frequency (MAF) of 0.1265 means that about 12.7% of the population are carriers of this variant. The variant c.1238G>A is known to diminish FVII secretion and is thought to be not directly pathogenic but modifying, i.e., contributing to the consequence of pathogenic variants (11, 22), for example by augmentation the FVII deficiency caused by the p.(Ala354Val) (c.1061C>T) variant (23).
The EAHAD F7 variant database provides tools to facilitate variant classification and has been prepared to list the pathogenicity of variants based on the ACMG classification (11). Unknown pathogenicity of a specific F7 gene variant represents only one of the difficulties in conjunction with interpretation of molecular genetic test results. Heterozygosity may be of clinical relevance if there are two or more pathogenic variants. To differentiate between compound heterozygous patients (both alleles affected) and double heterozygous patients (two variants in the same allele) molecular genetic testing of the mother or the father is usually needed. However, this is a hurdle in clinical practice. In addition, benign F7 gene variants may have a modifying effect in combination with pathogenic variants which increases the bleedings risk. Based on a large cohort of patients with FVII deficiency we explored an approach to increase the utility of ACMG classification which should facilitate interpretation of molecular genetic test results as a basis for successive advice and decision making in daily routine.
Materials and methods
Patients
Patients with FVII deficiency and genetic analyses prompted at the Coagulation Center Hochtaunus, Bad Homburg Germany, and the Coagulation Center Mannheim, Germany, between August 2012 and December 2021 were included in this retrospective exploratory analysis. According to center’s routine all patients with suspected bleeding disorder had completed the ISTH-SSC bleeding assessment tool (ISTH-BAT), a standardized questionnaire which can be used to generate a bleeding score (BS) in patients with bleedings disorders (24–26). Cut offs for an abnormal BS were ≥3 for children <12 years, ≥4 for males ≥12 years, and ≥6 for females ≥12 years. The higher score in females is due to the possibility of menorrhagia and postpartum bleeding (25). Besides standardized questionnaire the notes made by the physicians to document medical history in a non-standardized manner were used to identify and analyze bleeding symptoms. Treatment for substitution of FVII deficiency was documented.
Coagulation tests
As both centers belong to the same company the same procedures and reagents were used for coagulation tests (FVII:C, activated partial thromboplastin time, prothrombin time (PT) with Quick value) which were performed locally by routine. FVII:C was measured using Coagulation FVII deficient plasma (Siemens Healthineers, Erlangen, Germany) and the fully automated analyzer Sysmex® (Siemens Healthineers, Erlangen, Germany).
Molecular genetic analyses
Molecular genetic analyses were performed at the Office for Human Genetics, Wiesbaden, Germany, and at the Institute for Immunology and Genetics, Kaiserslautern, Germany. Before drawing peripheral blood for genetic testing all patients had to sign an informed consent form according to the German Gene Diagnostics Act. After isolation of genomic DNA the nine exons of the F7 gene on chromosome 13q34 and their flanking introns (since January 2015: RefSeqGene: NG_009262.1, transcript: GenBank accession number NM_000131.4) were analyzed by DNA sequencing. For exclusion of large deletions and duplications a variation of the multiplex polymerase chain reaction (PCR) named MLPA (Multiplex ligation-dependent probe amplification) analysis was performed using the MLPA-kit P207 from MRC Holland, Amsterdam, The Netherlands.
Assessment of pathogenicity of variants
Reports of individual molecular genetic testing in the cohort were screened for information about ACMG classification of specific variants. If a variant has not been classified, for example due to molecular genetic testing before the ACMG–AMP standards and guidelines were introduced, databases as follows were screened for information on pathogenicity: GnomAD,1 ClinVar,2 VarSome,3 Franklin by genoox,4 EAHAD-CFDB Factor VII Gene (F7) Variant Database.5 Additional information was available at the EAHAD F7 database (MAF, Grantham Score, PolyPhen-2 Prediction, SIFT Prediction, and PROVEAN Prediction) (11), and by case specific literature searches. If a variant could not be assigned to ACMG 1, 2, 4, or 5 it was classified as unknown or – if such classification was available – as variant of uncertain significance (VUS, ACMG 3).
Statistics
The data were analyzed descriptively with categories of patients expressed as numbers and percentages. For the other variables, mean values ± standard deviation (SD) as well as median (minimum, maximum) were calculated.
A combined endpoint consisting of an abnormal bleeding score and/or substitution history for FVII deficiency was used to identify high risk phenotypes in patients without another bleeding disorder, and to support the hypothesis that heterozygous ACMG 5 variants are of clinical relevance if combined with homozygous modifying ACMG 1–3 variants.
First, a subgroup of FVII deficient patients with homozygous modifying ACMG 1 variant c.1238G>A plus heterozygous ACMG 5 variant c.1061C>T was selected and compared with a subgroup of patients who had either a single c.1238G>A variant, only, or were heterozygous for c.1238G>A variant plus heterozygous for ACMG 5 variant c.1061C>T. Thereafter, the total cohort was divided into six genetic subgroups according to F7 genotype and presence of another bleeding disorder as shown in Figure 1. The combined endpoint mentioned above was used to compare group 5 (assumed high-risk phenotype) with pooled groups 1 to 4 (assumed low-risk phenotype).
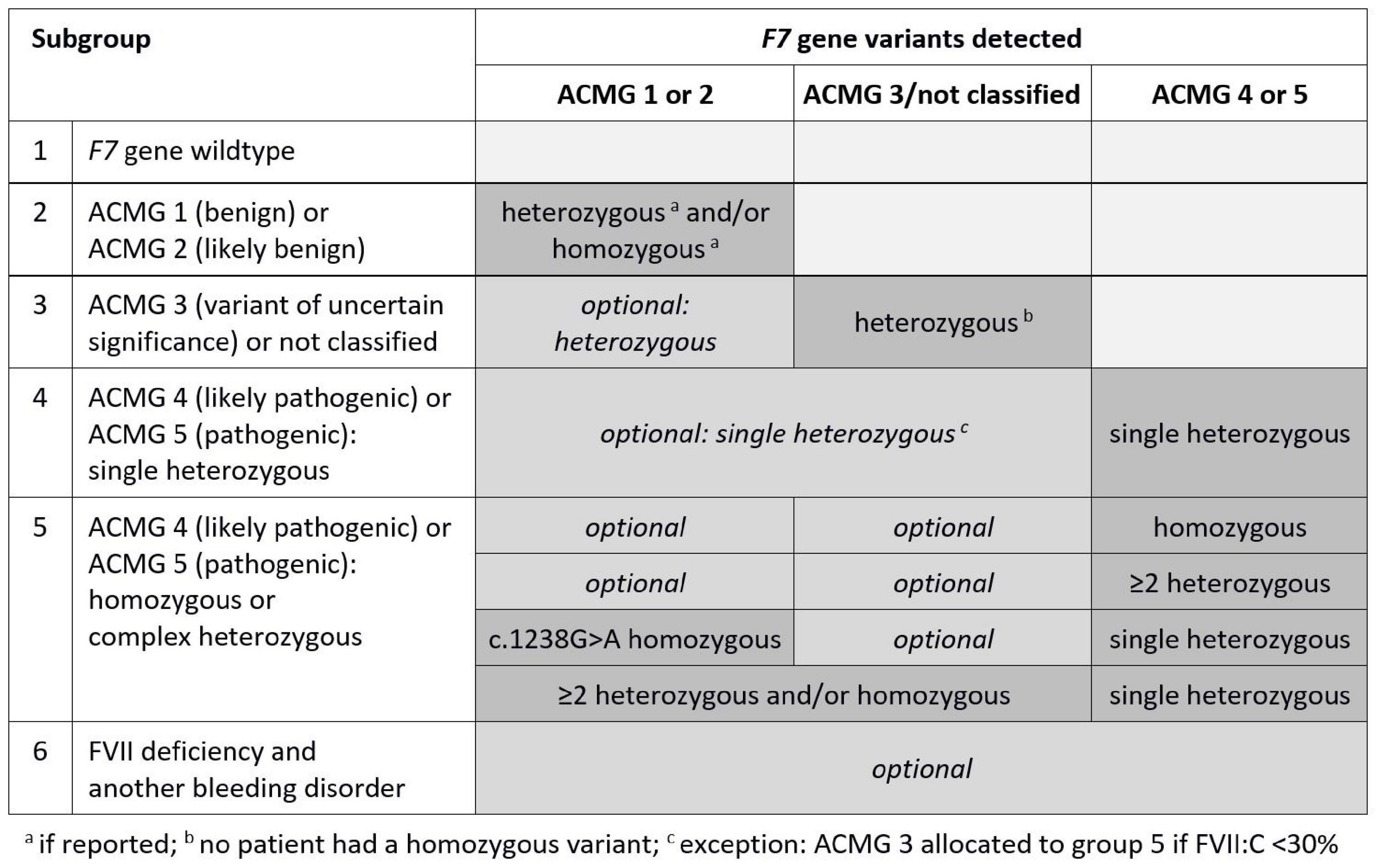
Figure 1. Genetic subgroups according to F7 genotype, ACMG classification and presence of another bleeding disorder.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent from the patients was not required to participate in this study in accordance with the local legislation and institutional requirements. The retrospective analysis complied with the principles of the Declaration of Helsinki and the Good Clinical Practice (GCP).
Results
In total, 129 patients with diagnosis of FVII deficiency (mean FVII:C 42.4% ± 15.1%) and availability of molecular genetic report were evaluated. Eighty-nine patients were females (69.0%). Mean age at molecular genetic testing was 34.1 ± 16.7 years (median 32.5 years, range 0–84 years). Reasons for consultation of the Coagulation Center were bleeding symptoms in 23 patients (17.8%), reduced Quick value in 31 patients (24.0%), testing of a family member in 12 patients (9.3%), known FVII deficiency in 17 patients (13.2%), recurrent abortions and/or wish for a child in 38 patients (29.5%), and miscellaneous in 8 patients (6.2%).
Variants
F7 gene wildtype (WT) was diagnosed in 25 patients (19.4%). In total, the genetic reports of 104 non-WT patients contained 182 variant findings (1.8 ± 1.2 variants per patient; median 1, maximum 10). Most findings in the molecular genetic reports were ACMG 1 or 2 variants (n = 102) followed by ACMG 4 or 5 variants (n = 65). About half of ACMG 1 or 2 findings were homozygous (n = 54; 53%), whereas 62 of 65 ACMG 4 or 5 findings (95.4%) were heterozygous. Further details of the variants are listed in the Supplementary Tables 1–4.
About half of the 53 different F7 gene variants identified were pathogenic (ACMG 5 n = 17; 32.1%) or likely pathogenic (ACMG 4 n = 11; 20.8%) (Supplementary Tables 1–3). Only 13 variants were benign (ACMG 1 n = 10; 18.9%) or likely benign (ACMG 2 n = 3; 5.7%). Ten variants were of uncertain significance (ACMG 3; 18.9%), and 2 variants remained unclassified (3.8%). Twenty variants (37.7%) seen in one (n = 16), two (n = 2) or three (n = 2) patients are not listed in the EAHAD database for F7 gene variants, mostly ACMG 1 or 2 variants (n = 8), and ACMG 3 or not classified variants (n = 8) (Supplementary Tables 1–3). For many variants with single nucleotide substitutions the EAHAD F7 database contains information concerning MAF, Grantham Score, PolyPhen-2 Prediction, SIFT Prediction, and PROVEAN Prediction (Supplementary Tables 1–3).
Frequent variants c.1238G>A ± c.1061C>T
The missense F7 gene variant c.1238G>A was detected in 81 of 104 non-WT patients (77.9%), 47 of 81 (58.0%) were homozygous (Supplementary Table 1). In 35 of 81 carriers of c.1238G>A variant (43.2%) no other F7 gene variant was found (Table 1). Mean FVII:C in 12 patients with single heterozygous c.1238G>A variant was 53.8 ± 14.9% with FVII:C below 50% in two patients (18 and 30%, respectively). Twenty-three patients with single homozygous c.1238G>A variant had mean FVII:C of 44.5 ± 12.1%.

Table 1. Characteristics, ISTH BS, and substitution in FVII deficient patients with c.1238G>A variant ± heterozygous c.1061C>T variant.
In 18 patients with c.1238G>A variant the ACMG 5 variant c.1061C>T was present (all heterozygous). One of these had F7 deficiency together with another bleeding disorder (von Willebrand syndrome). The c.1061C>T variant was not accompanied by the c.1238G>A variant in one patient, only. However, this male patient with FVII:C 33% and ISTH BS 5 had the c.1388delC variant (heterozygous, ACMG 4) which was also present in six other patients together with the c.1061C>T variant. As shown in Table 1, six of 12 patients (50.0%) with homozygous c.1238G>A variant and heterozygous c.1061C>T variant had an abnormal ISTH bleeding score (n = 4) and/or history of rFVIIa substitution (n = 2) versus only 1 of 41 patients (2.4%) in the other three subgroups of patients with c.1238G>A variant. This finding supports allocation of patients with a single heterozygous ACMG 4–5-variant plus the homozygous ACMG 1 c.1238G>A variant to group 5. However, 5 of 12 patients with homozygous c.1238G>A variant and c.1061C>T variant also had heterozygous c.1391delC variant. Of these, one patient had an abnormal ISTH BS and another patient needed substitution with rFVIIa.
Genotype/phenotype analyses
For genotype/phenotype analyses, 18 patients (14.7%) with another bleeding disorder besides FVII deficiency (WT n = 3, ACMG 1–3 variants, only, n = 9, ACMG 4 or 5 variants n = 6) were excluded from the assignment to the groups 1 to 5. Of the remaining 111 patients, 22 (19.8%) were F7 gene wildtype, 29 (26.1%) had ACMG 1 or 2 variants, only, 51 (45.9%) had at least one ACMG 4 or 5 variant, and 9 (8.1%) had to be assigned to group 3 due to ACMG 3 or unclassified variants (Table 2). Only one patient with at least two heterozygote ACMG 4 or 5 variants was known to be compound heterozygote.

Table 2. Characteristics, ISTH BS, and substitution in genetic subgroups of patients with FVII deficiency according to Figure 1.
Median number of F7 gene variants per patient was 2 in group 4 (range 1–2) as well as in group 5 (range 1–10) whereas median number was 1 in groups 2 (range 1–3), 3 (range 1 – 5) and 6 (range 0–3) (Table 2). Mean age ranged from 30.2 ± 15.4 years in group 3 (ACMG 3 or not classified) to 36.9 ± 14.1 years in group 1 (F7 gene wildtype).
The groups differed with respect to the proportion of patients with female sex between 54.8% in group 5 and 83.3% in group 6. Reasons for consultation of the Coagulation Center were also different. Group 6 reached the numerically highest rates in the consultation categories bleeding symptoms (44.4%), and abortions and/or wish for a child (50.0%), the latter together with group 1 (54.5%). Most patients in group 5 were admitted due to abnormal laboratory findings (45.2%) or known FVII deficiency (29.0%) (Table 2).
Mean FVII:C and Quick values were lowest in group 5 (29.5 and 54.4%, respectively), and ranged from 43.4 to 49.7%, and 68.2 to 76.2% in the other groups, respectively (Figure 2 and Table 2). In group 5, FVII:C was 5–6% in three patients with homozygous ACMG 4 or 5 variants, 26.4 ± 10.7% in 10 patients with ≥2 heterozygous ACMG 4 or 5 variants (n = 8) or ACMG 3–5 variants (n = 2), and 35.3 ± 11.1% in 18 patients with one heterozygous ACMG 4 or 5 variant combined with homozygous c.1238G>A or ≥2 ACMG 1–3 variants.

Figure 2. FVII activities stratified by six subgroups according to Figure 1.
In females ≥12 years, mean ISTH BS was numerically higher in group 5 (3.5 ± 2.7; median 3; range 0–9) versus 1.9 ± 1.6 (median 1, range 0–6) in groups 1 to 4 (pooled data of 57 females). In males ≥12 years, 13 patients of group 5 had mean ISTH BS of 1.6 ± 1.4 (median 2, range 0–5) versus 1.0 ± 1.0 (median 1, range 0–3) in 19 males ≥12 years included in groups 1 to 4. Both children included in group 5 had an abnormal ISTH BS versus none of 4 children in groups 1 to 4 (Table 2).
Eleven of 31 pts. (35.5%) in group 5 had an abnormal ISTH-BS (n = 7) and/or history of substitution with recombinant factor VIIa (n = 5) versus 4 of 80 pts. (5.0%, n = 1 abnormal ISTH-BS, n = 3 substitution) in groups 1 to 4 (Table 2). In group 5, the percentages of patients with abnormal ISTH-BS and/or history of substitution were similar in patients with homozygous ACMG 4 or 5 variants (1 of 3 patients, 33%), ≥2 heterozygous ACMG 4 or 5 variants (3 of 10 patients, 30%), one heterozygous ACMG 4 or 5 variant plus homozygous c.1238G>A variant (6 of 15 patients, 40%) and one heterozygous ACMG 4 or 5 variant plus ≥2 heterozygous ACMG 1 to 3 variants (1 of 3 patients, 33%). In group 6 (FVII deficiency and another bleeding disorder), 4 of 18 patients (22.2%) had an abnormal ISTH-BS (n = 2) and/or history of substitution with recombinant factor VIIa (n = 3).
Discussion
To the best of our knowledge, this exploratory analysis is the first investigation focusing on the usefulness of ACMG classification for interpretation of molecular genetic test results in patients with FVII deficiency. On one site, an abnormal ISTH-BS and/or a history of substitution was observed in few patients (4/80; 5.0%) with F7 gene wildtype or ACMG 1–3 variants or single heterozygous ACMG 4–5 variant with/without single heterozygous ACMG 1–3 variant. On the other site, 11 of 31 patients (35.5%) with four different scenarios of ACMG 4–5 ± ACMG 1–3 variant findings reached the combined endpoint of this exploratory analysis. Pooling patients with different constellations of zygosity and ACMG classifications in two genotypic categories was based on our preceding analysis of 44 patients with co-existence of heterozygous or homozygous c.1238G>A variant and heterozygous ACMG 5 c.1061C>T variant which was not biased by possible differences between variants of the same ACMG class such as truncating versus not-truncating variations (6) or large deletions versus others (27). This preceding analysis supports the hypothesis, that heterozygous ACMG 4–5 variants may become clinically relevant if combined with a modifying homozygous ACMG 1–3 variant or ≥2 heterozygous ACMG 1–3 variants. In the total cohort, 7 of 18 patients (38.9%) with this kind of molecular genetic finding reached the combined end point which is similar to 4 of 13 patients (30.8%) with homozygous or compound/double heterozygous ACMG 4 or 5 variants. Homozygous and compound heterozygous pathogenic F7 variants are known to be associated with an increased bleeding risk (2, 6, 28). Of note, differentiation between double heterozygosity affecting one allele, only, and compound heterozygosity was not available in 9 of 10 patients with two heterozygous ACMG 3–5 variants in our cohort.
Since 2015, the ACMG classification has been introduced as the internationally accepted standard for the interpretation of gene variants in heritable diseases (15, 16). For example, novel F8 and F9 gene variants in patients with hemophilia A or B included in the PedNet Registry were classified according to ACMG/AMP guidelines (29). In children with suspected inherited bleeding disorder genetic screening based on the ACMG guidelines may serve as a tool for early diagnosis, especially in patients with inherited thrombocytopenia (30). In the EAHAD blood coagulation factor VII variant database the assignment of F7 gene variants to ACMG classes is already foreseen (11). Based on the ACMG classification a simplification may be possible by assigning patients with FVII deficiency and no other bleeding disorder to the categories “low risk” and “high risk” for abnormal bleeding tendency.
Our investigation may serve as a kick-off to promote the use of ACMG classification in FVII deficiency. One major question to be answered in patients with FVII deficiency refers to prophylaxis with procoagulants for surgery (1) or for delivery (31). In the past, the prediction of bleeding risk to decide on substitution therapy has been mainly based on FVII:C levels, the personal clinical history and the first bleeding symptom (1, 32). In a larger cohort and preferably prospective study, the hypothesis may be tested that the two categories identified in our investigation (“low risk” versus “high risk” for abnormal bleeding tendency) are appropriate to support ACMG classification-based interpretation of molecular genetic test results in FVII deficient patients as a contribution to successive advice and decision making in daily routine.
Even in the “high risk” group the median FVII:C was rather high (30.0%, range 2–61%) if compared to an Italian investigation which showed FVII:C > 25% in 7.1% of homozygous or compound heterozygous patients, only (6). Whereas the Italian study included patients with FVII:C < 50% diagnosed because of bleeding tendency or prolonged prothrombin time, only, maximum FVII:C was higher in our retrospective study and other reasons for consultation such as family screening or abortions/wish for a child were allowed.
As already shown by the Greifswald FVII deficiency study, the most frequent pathogenic variant in the German population is the ACMG 5 missense mutation p.(Ala294Val) (c.1061C>T) (33), also frequently combined with the ACMG 5 variant c.1391delC p.(Pro464Hisfs*32) (34) which was the same in Polish and Italian cohorts (35, 36). In our cohort the c.1061C>T variant was present in 19 of 129 patients (14.7%). Six of these patients had a combination with the c.1391delC variant and 18 patients with the c.1238G>A modifying variant. As published by Fromovich-Amit et al. (23) FVII secretion was not further reduced by the variant c.1238G>A in the presence of the c.1061C>T variant. In our cohort there was a small difference with respect to FVII activity in patients heterozygous for c.1061C>T variant together with homozygous c.1238G>A variant (FVII:C 32.6 ± 5.8%, n = 12) versus combination with heterozygote c.1238G>A variant (FVII:C 38.3 ± 11.1%, n = 6) (Table 1).
We observed a difference in zygosity of ACMG 1 or 2 variants versus ACMG 4 or 5 variants (homozygous: 52.9% versus 4.6%). This may reflect the typically higher minor allele frequency (MAF) of ACMG 1 or 2 variants versus ACMG 4 or 5 variants in conjunction with the higher probability of decreased FVII:C in homozygous versus heterozygous variants.
Key limitations of this investigation are the retrospective design based on patient datasets and some ACMG findings derived from partially outdated rules for classification of pathogenicity and older literature searches. It should be noted that the ACMG classification is a learning system with recent or upcoming new recommendations (37). A disease specific expert panel has been founded for coagulation factor deficiencies (38). However, until now, published experiences to adapt criteria for ACMG classification are available for other diseases such as recessively inherited hemoglobinopathies, only (39). Further limitations of our analyses are the exclusion of patients without molecular genetic testing, the baseline differences between groups, no FVII antigen testing to evaluate possible FVII:C discrepancies (40), and restriction to descriptive statistics without p-values as no formal hypothesis was defined in advance.
In conclusion, ACMG classification is a promising tool to improve interpretation of molecular genetic test results in patients with FVII deficiency. The results of this exploratory analysis suggest phenotype differences between patients with a homozygous ACMG 4–5 variant or specific combinations of heterozygous ACMG 4–5 ± ACMG 1–3 variants, on one side, and patients with F7 gene wildtype, ACMG 1–3 variants, only, or single heterozygous ACMG 4–5 ± single heterozygous ACMG 1–3 variants, on the other side. This finding may serve as a basis to develop a genotype/phenotype prediction model in future studies.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article and Supplementary Tables 1–3.
Author contributions
RA had the idea and concept for the study and did the preparing work as well as manuscript outline. CH and C-ED helped to detect the patients, gave their input to the idea, and did proofreading. BR worked in the lab and did essential work to find out the ACMG classification. DJ performed the statistical analysis, literature and F7 variant database reviews, and was involved in the preparation of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study received funding from Novo Nordisk Pharma GmbH, Mainz, Germany. The funder was not involved in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
DJ is the owner of Med-i-Scene Concept GmbH, Weisendorf, Germany, which received the funding for this study from Novo Nordisk Pharma GmbH, Mainz, Germany, in conjunction with an existing basic agreement for scientific services.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor FS declared a past co-authorship with the author RA.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2023.1220813/full#supplementary-material
Footnotes
- ^ https://gnomad.broadinstitute.org
- ^ https://www.ncbi.nlm.nih.gov/clinvar/
- ^ https://varsome.com
- ^ https://franklin.genoox.com
- ^ https://f7-db.eahad.org
References
1. Napolitano M, Siragusa S, Mariani G. Factor FVII deficiency: clinical phenotype, genotype and therapy. J Clin Med. (2017) 6:38. doi: 10.3390/jcm6040038
2. Herrmann FH, Wulff K, Auerswald G, Schulman S, Astermark J, Batorova A, et al. Greifswald Factor FVII Deficiency Study Group. Factor VII deficiency: clinical manifestations of 717 subjects from Europe and Latin America with mutations in the factor 7 gene. Haemophilia. (2009) 15:267–80. doi: 10.1111/j.1365-2516.2008.01910.x
3. Monroe DM, Key NS. The tissue-factor VIIa complex: procoagulant activity, regulation, and multitasking. J Thromb Haemost. (2007) 5:1097–105. doi: 10.1111/j.1538-7836.2007.02435.x
4. Franchini M, Marano G, Pupella S, Vaglio S, Masiello F, Veropalumbo E, et al. Rare congenital bleeding disorders. Ann Transl Med. (2018) 6:331.
5. Peyvandi F, Menegatti M. Treatment of rare factor deficiency in 2016. Hematol Am Soc Hematol Educ Prog. (2016) 2016:663–9. doi: 10.1182/asheducation-2016.1.663
6. Quintavalle G, Riccardi F, Rivolta GF, Martorana D, Di Perna C, Percesepe A, et al. F7 gene variants modulate protein levels in a large cohort of patients with factor VII deficiency. Results from a genotype–phenotype study. Thromb Haemost. (2017) 117:1455–64. doi: 10.1160/TH17-02-0085
7. Sevenet PO, Kaczor DA, Depasse F. Factor VII deficiency: from basics ot clinical laboratory diagnosis in patient management. Clin Appl Thromb Haemostasis. (2017) 23:703–10. doi: 10.1177/1076029616670257
8. Bernardi F, Mariani G. Biochemical, molecular and clinical aspects of coagulation factor VII and its role in hemostasis and thrombosis. Haematologica. (2021) 106:351–62. doi: 10.3324/haematol.2020.248542
9. O’Hara PJ, Grant FJ, Haldeman BA, Gray CL, Insley MY, Hagen FS, et al. Nucleotide sequence of the gene coding for human factor VII, a vitamin K-dependent protein participating in blood coagulation. Proc Natl Acad Sci USA. (1987) 84:5158–62. doi: 10.1073/pnas.84.15.5158
10. EAHAD. EAHAD Coagulation Factor Variant Databases. Factor FVII Gene (F7) Variant Database. (2022). Available online at: https://f7-db.eahad.org (accessed December 20, 2022).
11. Giansily Blaizot M, Rallapalli PM, Perkins SJ, Kemball-Cook G, Hampshire DJ, Gomez K, et al. The EAHAD blood coagulation factor VII variant database. Hum Mut. (2020) 41:1209–19.
12. Giansily Blaizot M, Aguilar-Martinez P, Biron-Andreani C, Jeanjean P, Igual H, Schved J-F. Analysis of the genotypes and phenotypes of 37 unrelated patients with inherited factor VII deficiency. Eur J Hum Genet. (2001) 9:105–12. doi: 10.1038/sj.ejhg.5200593
13. Zhang X, Wang S, Leng S, Feng Q, Zhang Y, Xu S, et al. Novel factor VII gene mutations in six families with hereditary coagulation factor VII deficiency. J Clin Lab Anal. (2021) 35:e23905. doi: 10.1002/jcla.23905
14. Seanoon K, Payongsri P, Vivithanaporn P, Sirachainan N, Chuansumrit A, Hongeng S, et al. Mutations of TFPI-binding exosites on factor VII cause bleeding phenotypes in factor VII deficiency. Blood Adv. (2022) 6:5887–97. doi: 10.1182/bloodadvances.2022007560
15. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association of Molecular Pathology. Genet Med. (2015) 17:405–24.
16. Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Yuan-Yuan H, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. (2017) 19:1105–17.
17. Van Rooij J, Arp P, Broer L, Verlouw J, van Rooij F, Kraaij R, et al. Reduced penetrance of pathogenic ACMG variants in a deeply phenotyped cohort study and evaluation of ClinVar classification. Genet Med. (2020) 22:1812–20. doi: 10.1038/s41436-020-0900-8
18. Green F, Kelleher C, Wilkes H, Temple A, Meade T, Humphries S. A common genetic polymorphism associated with lower coagulation factor VII levels in healthy individuals. Aterioscler Thromb. (1991) 11:540–6.
19. Humphries S, Temple A, Lane A, Green J, Cooper J, Miller G. Low plasma levels of factor VIIc and antigen are more strongly associated with the 10 base pair promoter (-323) insertion than the glutamine 353 variant. Thromb Haemost. (1996) 75:567–72.
20. Mtiraoui N, Aboud N, Bouraoui H, Haizem S, Gris JC, Busson M, et al. Reduction of coagulation factor VII plasma levels by R353Q but not the -323P0/10 promotor polymorphism in healthy Tunisians. Am J Hematol. (2005) 79:11–6.
21. Girolami A, Paoletti M, Ferrari S, Garcia D. Peculiar congenital factor VII defect with the proposita and her mother showing the same compound heterozygosity for Thr384Met and Arg413Gln. Acta Haematol. (2021) 144:100–4. doi: 10.1159/000507071
22. Miolo G, Caggiari L, De Zorzi M, Tedeschi M, Tessitori G, Percesepe A, et al. Impact of a new genetic variant on FVII:C activity. Integr Mol Med. (2019) 6:2–4. doi: 10.15761/IMM.1000381
23. Fromovich-Amit Y, Zivelin A, Rosenberg N, Tamary H, Landau M, Seligsohn U. Characterization of mutations causing factor VII deficiency in 61 unrelated Israeli patients. J Thromb Haemost. (2004) 2:1774–81. doi: 10.1111/j.1538-7836.2004.00921.x
24. Rodeghiero F, Tosetto A, Abshire T, Arnold DM, Coller B, James P, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. (2010) 8:2063–5. doi: 10.1111/j.1538-7836.2010.03975.x
25. Elbatarny M, Mollah S, Grabell J, Bae S, Deforest M, Tuttle A, et al. Normal range of bleeding scores for the ISTH–BAT: adult and pediatric data from the merging project. Haemophilia. (2014) 20:831–5. doi: 10.1111/hae.12503
26. ISTH-SSC. ISTH-SSC Bleeding Assessment Tool. (2022). Available online at: https://bleedingscore.certe.nl (accessed December 20, 2022).
27. Rath M, Najm J, Sirb H, Kentouche K, Dufke A, Pauli S, et al. Large deletions play a minor but essential role in congenital coagulation factor VII and X deficiencies. Hamostaseologie. (2015) 35(Suppl. 1):S36–42.
28. Mariani G, Herrmann FH, Dolce A, Batorova A, Etro D, Peyvandi F, et al. Clinical phenotypes and factor VII genotype in congenital factor VII deficiency. Thromb Haemost. (2005) 93:481–7.
29. Andersson NG, Labarque V, Letelier A, Mancuso ME, Bührlen M, Fischer K, et al. Novel F8 and F9 gene variants from the PedNet hemophilia registry classified according to ACMG/AMP guidelines. Hum Mutat. (2020) 41:2058–72. doi: 10.1002/humu.24117
30. Andersson NG, Rossing M, Ferrari MF, Gabrielaite M, Leinoe E, Ljung R, et al. Genetic screening of children with suspected inherited bleeding disorders. Haemophilia. (2020) 26:314–24.
31. Baumann Kreuziger LM, Morton CT, Reding MT. Is prophylaxis required for delivery in women with factor VII deficiency? Haemophilia. (2013) 19:827–32.
32. Di Minno MN, Dolce A, Mariani G, Ster Study Group. Bleeding symptoms at disease presentation and prediction of ensuing bleeding in inherited FVII deficiency. Thromb Haemost. (2013) 109:1051–9.
33. Herrmann FH, Wulff K, Strey R, Siegemund A, Astermark J, Schulmann S. Variability of clinical manifestations of factor VII-deficiency in homozygous and heterozygous subjects of the European F7 gene mutation A294V. Haematologica. (2008) 93:1273–5. doi: 10.3324/haematol.12567
34. Wulff K, Herrmann FH. Twenty two novel mutations of the factor VII gene in factor VII deficicency. Hum Mutat. (2000) 15:489–96. doi: 10.1002/1098-1004(200006)15:6<489::AID-HUMU1>3.0.CO;2-J
35. Arbini AA, Bodkin D, Lopaciuk S, Bauer KA. Molecular analysis of Polish patients with factor VII deficiency. Blood. (1994) 84:2214–20.
36. Bernardi F, Liney DL, Patracchini P, Gemmati D, Legnani D, Arcieri P, et al. Molecular defects in CRM+ factor VII deficiencies: modelling of missense mutations in the catalytic domain of FVII. Br J Haematol. (1994) 86:610–8. doi: 10.1111/j.1365-2141.1994.tb04793.x
37. Pejaver V, Byrne AB, Feng B-J, Pagel KA, Mooney SD, Karchin R, et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet. (2022) 109:2163–77. doi: 10.1016/j.ajhg.2022.10.013
38. Mohan S, Lee K, Carcao M, Doshi BS, Downes K, Kemball-Cook G, et al. ClinGen coagulation factor deficiency variant curation expert panel: meeting the need for recommendations to curate variants in the coagulation factor genes. Blood. (2019) 134(Suppl 1):5794.
39. Kountouris P, Stephanou C, Lederer CW, Traeger-Synodinos J, Bento C, Harteveld CL, et al. Adapting the ACMG/AMP variant classification framework: a perspective from the ClinGen hemoglobinopathy variant curation expert panel. Hum Mutat. (2022) 43:108. doi: 10.1002/humu.24280
Keywords: FVII deficiency, genotype, phenotype, ACMG classification, genetic variations
Citation: Alesci RS, Hecking C, Racké B, Janssen D and Dempfle C-E (2023) Utility of ACMG classification to support interpretation of molecular genetic test results in patients with factor VII deficiency. Front. Med. 10:1220813. doi: 10.3389/fmed.2023.1220813
Received: 11 May 2023; Accepted: 16 June 2023;
Published: 14 July 2023.
Edited by:
Felix Carl Fabian Schmitt, University Hospital Heidelberg, GermanyReviewed by:
Daniel Gruneberg, University Hospital Heidelberg, GermanyJohannes Zipperle, Ludwig Boltzmann Institute for Traumatology, Austria
Copyright © 2023 Alesci, Hecking, Racké, Janssen and Dempfle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosa Sonja Alesci, cy5hbGVzY2lAZ2VyaW5udW5nc3plbnRydW0taG9jaHRhdW51cy5kZQ==