 Claire S. Whyte
Claire S. Whyte- Aberdeen Cardiovascular and Diabetes Centre, The Institute of Medical Sciences, School of Medicine, Medical Sciences and Nutrition, University of Aberdeen, Aberdeen, United Kingdom
The hemostatic and innate immune system are intertwined processes. Inflammation within the vasculature promotes thrombus development, whilst fibrin forms part of the innate immune response to trap invading pathogens. The awareness of these interlinked process has resulted in the coining of the terms “thromboinflammation” and “immunothrombosis.” Once a thrombus is formed it is up to the fibrinolytic system to resolve these clots and remove them from the vasculature. Immune cells contain an arsenal of fibrinolytic regulators and plasmin, the central fibrinolytic enzyme. The fibrinolytic proteins in turn have diverse roles in immunoregulation. Here, the intricate relationship between the fibrinolytic and innate immune system will be discussed.
Introduction
Over the last two decades there is increasing awareness of immunothrombosis, where components of the immune system promote coagulation to limit the action of invading pathogens (1). Whilst thromboinflammation describes the inflammatory process induced by pathogens leading to platelet-neutrophil and platelet-monocyte interactions and endothelial dysfunction that promote a prothrombotic environment (2, 3). Activation of monocytes and neutrophils induces release of tissue factor (TF) promoting the extrinsic coagulation pathway, whilst intrinsic coagulation is triggered by binding of factor XII (FXII) to neutrophils (4). Additionally, activated neutrophils degranulate and expel their nuclear and cytoplasmic content to form neutrophil extracellular traps (NETs) during the neutrophil death process, NETosis (4). NETs act as a surface for assembly of procoagulant proteins including TF, FXII and von Willebrand factor (5). Furthermore, released neutrophil elastase cleaves tissue factor pathway inhibitor, thereby dampening the anticoagulant effect and contributing to fibrin persistence (6). The fibrinolytic system is responsible for limiting ongoing fibrin formation and degrading the fibrin meshwork to resolve thrombi.
Fibrinolysis
Plasmin, the central enzyme responsible for fibrin degradation is formed after cleavage of Arg561-Val562 of the zymogen form, plasminogen, via plasminogen activators (Figure 1). The primary physiological activators are tissue plasminogen activator (tPA) and urokinase (uPA). Efficient tPA-mediated plasminogen activation requires binding of both proteins to fibrin or cellular surfaces. uPA-mediated activation can occur in solution, although it can be localized to cellular surfaces via urokinase plasminogen activator receptor (uPAR) (7). The fibrinolytic system is normally tightly regulated by various inhibitors. Plasminogen activation is primarily regulated by plasminogen activator inhibitor-1 (PAI-1) which forms a 1:1 complex with the activators (8). Plasminogen activator inhibitor-2 (PAI-2) is not as efficient an inhibitor as PAI-1 but does function in uPA-mediated extracellular activity (9). The principal plasmin inhibitor is the serine protease inhibitor (SERPIN), α2-antiplasmin (α2AP) which forms a non-covalent complex with the active enzyme (10). Crosslinking of α2AP to fibrin by active transglutaminase factor XIII (FXIIIa) enhances the ability of this SERPIN to inhibit plasmin (11). Thrombin activatable fibrinolysis inhibitor (TAFI) further acts as a fibrinolytic break by removing C-terminal lysines from fibrin which are required for the binding of plasminogen and tPA.
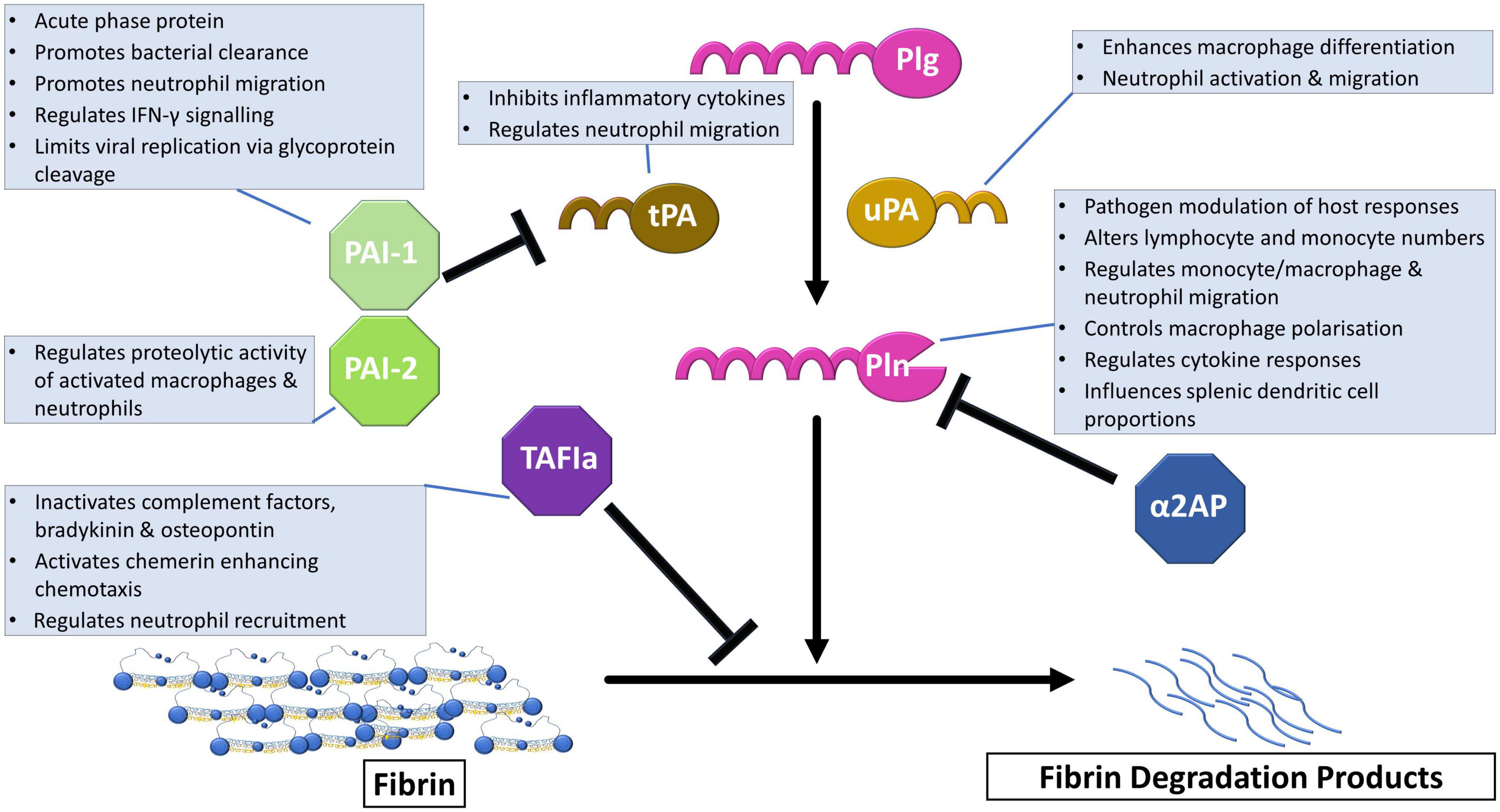
Figure 1. The fibrinolytic system in immune regulation. Plasminogen (Plg) is converted to the active enzyme plasmin (Pln) after cleavage by tissue plasminogen activator (tPA) or urokinase (uPA). This step is regulated by plasminogen activator inhibitor-1 (PAI-1), which is the primary physiological inhibitor, and plasminogen activator-2 (PAI-2). The active enzyme, plasmin, cleaves crosslinked fibrin into fibrin degradation products that can be cleared from the circulation. Alpha2-antiplasmin (α2AP) directly inhibits plasmin by forming a non-covalent complex. Activated thrombin activatable fibrinolysis inhibitor (TAFIa) exerts its effects by removal of C-terminal lysine required for plasminogen binding to fibrin. Boxes detail functions of the fibrinolytic proteins in immune regulation.
Pathogen hijacking of the fibrinolytic system
Invading pathogens take advantage of the fibrinolytic system, activating plasminogen in order to remove the confines of fibrin and extracellular matrix barriers and to evade the innate immune system (12). Indeed, certain strains of bacteria can produce plasminogen activators. Beta hemolytic strains of Streptococci possess streptokinase which induces non-proteolytic plasminogen activation by causing a conformational change that exposes the catalytic site and this complex can hydrolytically activate other plasminogen molecules (13). Staphylococcus aureus produces staphylokinase which also non-proteolytically activates plasminogen by forming a complex which generates plasmin (14). Staphylokinase is considered to be fibrin specific and in the absence of fibrin it is susceptible to inhibition by α2AP (15). Whilst Yersinia pestis, are able to proteolytically activate plasminogen and scuPA by the membrane protein Pla (16). Plasminogen contributes to lethality of Y. pestis, promoting spread of the bacteria and dampening immune cell recruitment to sites of infection [reviewed in (17)].
Additionally, a plethora of plasminogen binding proteins (e.g. α-enolase, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and PAM) exist on bacteria, fungal pathogens, protozoan and helminth parasites (12, 18, 19). Bacteria utilize plasminogen to remove fibrin barriers and enable invasion through extracellular matrices both directly and indirectly by activating matrix metalloproteases (17, 19–21). Additionally, plasmin-mediated cleavage of members of the complement system and immunoglobulin facilitates immune evasion of some strains of bacteria (22, 23). Bacteria also use plasminogen as a molecular linker to enable interaction with host cells (23).
Binding of plasminogen to Cryptococcus neoformans may facilitate the ability of this fungal pathogen to cross the blood brain barrier (24). It has been suggested that the affinity for plasminogen binding could reflect the observed strain differences in virulence of C. neoformans (24). However, plasminogen may not function in promoting virulence of all fungal pathogens. Although Candida albicans binds plasminogen and can cleave fibrin when in the presence of exogenous plasminogen activators, this binding does not affect virulence or endothelial damage and therefore the in vivo significance is not known (18). Multiple species of helminth parasites possess plasminogen binding proteins that facilitate their invasion and immune evasion (19). Protozoans are also considered to use plasminogen to support their host invasion but binding varies with morphotype and age for Leishmania mexicana (25). As not all pathogens can endogenously activate the zymogen they therefore require interaction with host plasminogen activators (18).
Immune cells as sources of fibrinolytic proteins
Plasminogen activation is enhanced by assembly of plasminogen and its activators on fibrin or cellular surfaces (26–29) which also protect plasmin from inhibition by α2AP (30–32). Plasminogen receptors are found on endothelial cells, platelets, monocytes, macrophages and neutrophils [reviewed in (33)]. The multitude of plasminogen receptors have the common feature of availability of C-terminal basic residues (33). This includes binding proteins that lack a transmembrane protein (e.g., α-enolase and histone 2B), transmembrane proteins that require proteolysis to expose the C-terminal basic residue (e.g., integrins αIIbβ3 and αMβ2) and Plg-RKT a transmembrane protein synthesized with a C-terminal lysine residue (33, 34).
Plg-RKT was first identified on the surface of monocytes and macrophages and co-localizes with uPAR (35) and facilitates plasminogen activation by tPA (35) and uPA (36). Monocyte-derived uPA is required for incorporation of these cells into thrombi for efficient thrombus resolution (37). Although uPA is the predominant plasminogen activator in monocytes, stimulation with lipopolysaccharide (LPS), interferon-γ (IFN-γ) interleukin-4 (IL-4) all induce tPA secretion (38). Monocytes also express PAI-1 and are a major source of PAI-2 (39). Intracellular and secreted PAI-2 can be induced by stimulation of monocytes with thrombin and LPS (39, 40). Presence of PAI-2 in arterial and venous thrombi, presumed to be from monocytes, inhibits uPA-mediated lysis (41, 42). Both PAI-1 and PAI-2 are decreased by targeted upregulation of uPA which enhances fibrinolysis induced by monocyte-derived macrophages (43).
Thrombin activatable fibrinolysis inhibitor is also expressed by monocytes and macrophages with the level of expression being dependent on the activation status (44). Stimulation of macrophages with IL-4 downregulates TAFI expression whilst the proinflammatory stimuli IFN-γ and LPS has no effect (44). Additionally, monocytes and macrophages contain cellular FXIII-A (45, 46) which is trafficked to the membrane in association with golgi vesicles (47). IL-4 and IL-10-induced externalization of FXIII-A on monocytes stabilizes thrombi against degradation (48).
Polymorphonuclear leukocytes, assumed to be neutrophils, participate in endogenous thrombus lysis, mainly mediated by uPA with small contributions from tPA, elastase and cathepsin G (49). More recently neutrophils and their ability to form NETs have gained attention for their antifibrinolytic function. NETs consist of extruded nuclear and cytoplasmic content including histones, DNA strands and granular proteins including neutrophil elastase (4). The presence of DNA, histones and NETs inhibits plasminogen activation in vitro which can be reversed by degrading the chromatin with DNase (50, 51). Targeting DNA in vivo limits DVT growth in mice (52) and enhances tPA-mediated ex vivo thrombolysis of thrombi obtained from acute ischemic stroke patients (53, 54).
Alongside their role in promoting coagulation, platelets also regulate fibrinolysis and form part of the innate immune response. These anucleate cell fragments are packaged with granular content required for these multifaceted functions. Activated platelets expose P-selectin which facilitates interaction with the P-selectin glycoprotein ligand-1 (PSGL1) expressed on leukocytes and endothelial cells. Platelet-leukocyte interactions also occur via CD40-CD40L. These interactions allow platelets to direct leukocytes to sites of inflammation and propagate the inflammatory process (55, 56).
Platelet-rich thrombi are more resistant to lysis than erythrocyte-rich thrombi (57, 58) and platelets have largely been considered to be antifibrinolytic. Platelets are a major pool of circulating PAI-1 which is contained within the α-granules (59). Model thrombi formed at high shear rates contain elevated PAI-1 and lower tPA and plasminogen (60). This is consistent with the greater abundance of PAI-1 in platelet dense arterial thrombi compared to venous thrombi (61, 62). Platelet-derived PAI-1 is retained on activated platelet membranes, localizing to the platelet “cap” or “body” on phosphatidylserine (PS)-exposing procoagulant platelets or centrally over spread platelets (63, 64). This platelet-derived PAI-1 is functional in conferring resistance to lysis (63).
Additional anti-fibrinolytic factors contained within platelet α-granules include TAFI (65, 66), PN-1 (67), and α2AP (68, 69) which can downregulate fibrinolysis. The role of α2AP in maintaining thrombus stability may be limited as addition of circulating platelet concentration to α2AP-depeleted plasma does not protect against degradation (70). However, platelets contain a cytoplasmic pool of FXIII-A which crosslinks high molecular weight γ-dimers, α-polymers and α2AP-fibrin (71–75). Platelets retain externalized cellular FXIII-A in the “cap” region stabilizing thrombi against lysis due to crosslinking of α2AP (70). FXIII-A is also observed in platelet microparticles translocated via intracellular signaling that is calcium-independent (76).
In contrast to this, platelets support fibrinolytic activity through binding and exposure of plasminogen (28, 64, 77). Strong platelet stimulation facilitates plasminogen binding by fibrin-dependent and fibrin-independent mechanisms (64, 78). Plg-RKT accounts for binding of approximately 40% platelet-derived plasminogen (28). Plasminogen activators also localize to the platelet surface with tPA binding being fibrinogen-dependent (65). Single chain uPA is activated on the platelet surface in a mechanism of reciprocal activation with plasminogen (77).
Platelet dense granules contain polyphosphate (polyP), a biomolecule which functions in modulation of coagulation and inflammation (79). PolyP delays fibrin polymerization altering clot structure (80). The knotted fibrin structure downregulates tPA and plasminogen binding thereby inhibiting tPA-mediated fibrinolysis (81). The effect on uPA-mediated plasminogen activation may depend on the contribution of other proteins as polyP accelerates activation in a purified system (82) whilst inhibits it in a plasma-based system (83). FXII has close structural homology to tPA and uPA and as such can function as a plasminogen activator. PolyP auto activates FXII to active single chain FXII (84) which facilitates plasminogen activation (85). Platelet-derive polyP could therefore have differential roles in thrombus resolution and cellular proteolytic process depending on the surrounding environment.
During vascular insult, many of the innate cell immune responses require interaction with the endothelium. Endothelial cells are the main source of circulating tPA, and secretion occurs via both constitutive and regulated mechanisms (86). Both plasminogen and tPA can bind to endothelial cells and therefore have the potential to generate plasmin (87). Endothelial cells also secrete uPA which bind to the cell surface uPAR (88). Additionally, endothelial cells produce the fibrinolytic inhibitors PAI-1 (89, 90), PAI-2 (91), and TAFI (92) which are upregulated in response to inflammatory cytokines.
Interaction of innate immune cells within the thrombus environment could influence resolution and stability. In pulmonary thrombi, rolling neutrophils rip membrane fragments from PS-exposing platelets facilitating formation of neutrophil macroaggregates (93). It is interesting to speculate that this could act to deliver platelet-derived fibrinolytic proteins within these aggregates and may facilitate platelet-neutrophil fibrinolytic crosstalk.
The role of the fibrinolytic system in immunomodulation
Fibrinolytic proteins have a multitude of roles outside of their primary function of fibrin degradation including regulating the immune response. PAI-1 is an acute phase protein that is upregulated in response to injury, infection and inflammation (90, 94, 95) (Figure 1). Upregulation of PAI-1 is considered to be a protective mechanism important for early immune responses against bacterial pathogens, including Haemophilus influenzae (96), Pseudomonas aeruginosa (97). PAI-1 promotes bacterial clearance and limits inflammation (96). Downregulation of PAI-1 by Streptococcus pneumoniae pneumolysin is associated with increased mortality which can be reversed by administering recombinant PAI-1, protecting against alveolar haemorrhage (98).
Plasminogen activator inhibitor-1 facilitates neutrophil migration and its inhibition or deletion reduces influx at the site of injury in response to Pseudomonas aeruginosa, Escherichia coli, and Klebsiella pneumoniae infections (97, 99, 100). PAI-1 regulates IFN- γ in response to LPS and Staphylococcal enterotoxin B (101) in a mechanism independent of the plasminogen activators. PAI-1 may also have a protective role in viral infections, due to inhibition of proteases required for glycoprotein cleavage, therefore limiting viral replication (102).
TAFIa modulates inflammation by removal of C-terminal arginine or lysine residues from C3a, C5a, bradykinin osteopontin and chenerin (103–105) (Figure 1). Cleavage of C5a by TAFI is protective in inflammatory models of LPS induced acute lung injury (106), bronchial asthma (107), and rheumatoid arthritis (108). The development of post-traumatic sepsis is associated with a reduction in TAFI and increased C5a (109). Additionally, TAFI-deficient mice display enhanced neutrophil recruitment and tumor necrosis factor-α (TNF-α) and IL-6 levels in the peritoneum after Escherichia coli induced abdominal sepsis (110). This was independent of its antifibrinolytic function (110). In contrast to this, in Pseudomonas aeruginosa-induced sepsis, TAFI inhibition potentiates the effects of the antibiotic, ceftazidime and reduces organ dysfunction (111).
Plasmin(ogen) has multifaceted roles in the regulation of proinflammatory processes [reviewed in (20)]. Plasminogen is required for efficient recruitment of monocytes and lymphocytes in response to inflammation (112) and promotes macrophage phagocytosis and migration (113, 114) (Figure 1). Deficiency of plasminogen alters the expression of phagocytic genes (113). Whilst the fibrinolytic activity of plasmin is required for macrophage migration in experimental peritonitis (114). Interestingly, the absence of fibrinogen or the integrin αMβ2 reverses the requirement for plasminogen suggesting fibrinolytic activity is required to remove the physical restraint of macrophages by fibrin(ogen) (114). Plg-RKT, is upregulated during differentiation of monocytes to macrophages (35) and drives polarization to an M2-like macrophage phenotype (115). Additionally, dendritic cell phagocytosis is enhanced by plasmin which maintains these cells in an immature phenotype an reduces migration to the lymph nodes (116).
Plasminogen activators modulate the innate immune response, in mechanisms both dependent and independent of their fibrinolytic action. In a Escherichia coli-induced sepsis model, tPA deficiency caused increased bacterial loads, reduced neutrophil migration and was associated with increased mortality by a plasmin-independent mechanism (117) (Figure 1). Consistent with this, enzymatically inactive tPA blocks LPS induced increase in proinflammatory cytokines such as TNF-α, and IL-6 via low density lipoprotein receptor-related protein-1 (LRP1) and N-methyl-D-aspartic acid receptor (NMDA-R) (118, 119). However, in an ischemia/reperfusion model, tPA-mediated plasmin activity was required for neutrophil transmigration and disruption of endothelial junctions which allows further recruitment of neutrophils (120). Plasmin does not directly activate neutrophils and recruitment of these cells requires mast cell activation and leukotriene generation (120). Whilst in a stroke model, tPA-mediated plasmin generation decreased lymphocyte and monocyte counts, elevated IL-10 and TNF-α and altered splenic dendritic cell proportions (121).
Urokinase enhances monocyte differentiation into macrophages (122) and promotes neutrophil activation and migration (123). The uPA receptor, uPAR facilitates neutrophil migration in response to LPS-induced peritonitis, but this was not observed with Escherichia coli or in a polymicrobial sepsis model suggesting a compensatory mechanism may occur (124, 125). The function of uPAR on neutrophil migration is independent of its role in plasminogen activation and requires toll-like receptor signaling (125). Deficiency of uPAR promotes proinflammatory cytokines and macrophage polarization towards M1 phenotype and reduced phagocytosis in an experimental colitis model (126).
The varying roles of the fibrinolytic system in immunomodulation highlights the complex interactions which must be carefully balanced so as not exacerbate the inflammatory response and promote a prothrombotic environment.
Dysregulation of fibrinolysis
Fibrinogen is an acute phase protein that dramatically increases during infection due to enhanced hepatic synthesis (127). Fibrin films form on the outside of blood clots which limit bacterial infiltration (128). However, aberrant fibrin accumulation contributes to development of a prothrombotic environment. During acute bacterial or viral infections, thrombotic complications can arise including deep vein thrombosis (DVT) and pulmonary embolisms (PE) (129, 130), acute myocardial infarction (AMI) (131, 132) and strokes (132). Thrombotic events occurring after infections affect various organ systems including respiratory, urinary and oral (133). The risk of thrombosis is higher in the first weeks succeeding infection and falls gradually after the initial infection (129). Consistent with a prothrombotic response to infection, seasonal variability in occurrence of AMI has been observed (134). The underlying mechanisms of the prothrombotic state are not fully understood. However, derailment of the fibrinolytic system is often a contributing factor to this.
Sepsis, a life-threatening response to infection, leads to tissue and organ damage and has a mortality rate of approximately 30%, although this is higher with older age or pre-existing conditions (135). As a result of the inflammatory state development of disseminated intravascular coagulation (DIC) can occur. This causes systemic dysregulation of coagulation and fibrinolysis resulting in depletion of coagulation factors and platelets and hemorrhaging. Platelet count is associated with severity, being significantly reduced with development of septic shock (136).
Plasmin(ogen) has a protective role in sepsis and levels are reduced with disease severity (137). However, a hypofibrinolytic state predominates in sepsis, largely due to elevated levels of PAI-1. Indeed, PAI-1 is a potential biomarker of disease severity and predictor of mortality (138). Initially, increased tPA and plasmin generation may predominate peaking at 2 h at which point TNF-α induces a steep increase in PAI-1 (139). Patients with the PAI-1 polymorphism 4G/5G, which is associated with elevated PAI-1 levels, are at increased risk of mortality from sepsis (140, 141). NETs may contribute to the elevated PAI-1 in sepsis as PAI-1 is downregulated in petidylarginine deiminase-4 (PAD-4) deficient mice which are unable to form NETs (142). NETs further contribute to a hypofibrinolytic state in sepsis due to the presence of cell-free DNA, an effect that can be overcome by DNase (143).
Hypofibrinolysis in sepsis may be further precipitated by other antifibrinolytic proteins. PAI-2 is not normally detected in healthy neutrophils but in patients with sepsis significant levels are present (144). Activation of TAFI could also be a contributing factor to the development of sepsis DIC (145). Interestingly, the TAFI Thr325 Ile/Ile single nucleotide polymorphism, which has increased antifibrinolytic potential, is associated with increased risk of contracting meningococcal disease and risk of mortality (146).
Acute respiratory distress syndrome (ARDS) is a hyperinflammatory condition that occurs in response to infection characterized by heightened alveolar-capillary permeability leading to extrusion of plasma proteins and inflammatory cytokines. This results in enhanced leukocytes and platelets recruitment to the lung microvasculature (147–149). Respiratory dysfunction and right heart failure develops, confounded by fibrin deposits which are observed in the air spaces and lung parenchyma due to the procoagulant environment along with hyaline-membranes and fibrosis (150–153).
Fibrin persistence is exacerbated by the inflammatory environment which promotes an imbalance in the fibrinolytic factors. Of note, PAI-1 synthesis is upregulated by several proinflammatory cytokines. Elevated levels of PAI-1 are observed with respiratory infections including influenza (154), severe acute respiratory syndrome coronavirus (SARS-CoV) (155) and SARS-CoV2 which downregulates fibrinolytic activity (156). Elevated PAI-1 is associated with worsening disease severity after SARS-CoV2 infection (156, 157). IL-6 induces an upregulation in PAI-1 gene expression and plasma levels of both PAI-1 and tPA (158–160). In endothelial cells, trans-signaling by IL-6 causes a circular amplification of IL-6 as well as IL-8, MCP-1 and PAI-1 synthesis (161). Additionally, endothelial cells release PAI-1 in response to the acute phase reactant, C-reactive protein (CRP) (150, 162, 163). Levels of uPA antigen are unaffected in ARDs but the heightened levels of PAI-1 cause a downregulation in fibrinolytic activity in the bronchoalveolar space (150).
Therapeutic potential of targeting the fibrinolytic pathway
The appropriation of fibrinolytic system by pathogens to evade the host immune response and the varied function of fibrinolytic proteins in immunomodulation makes them potential therapeutic targets. Plasmin(ogen) binding and subsequent proteolytic activity are inhibited by lysine analogues. Lysine analogues therefore have potential in modulating the proinflammatory and immunosuppressive properties of plasmin. One such lysine analogue, epsilon aminocaproic acid (εACA), has been shown to reduce experimental Group B streptococcus meningitis and neonatal mortality rates (164). Whilst tranexamic acid (TXA), has shown promise at reducing rates of post-surgical infection (165). Furthermore, plasmin inhibition by aprotinin, εACA or TXA reduces neutrophil recruitment and may have potential to ischemia-reperfusion reduced injury (166).
On the other hand, when aberrant fibrin(ogen) develops during infection, promoting fibrinolysis is desirable. The use of recombinant tPA as an adjuvant therapy in a small retrospective study of infective endocarditis facilitated clearance of fibrin rich vegetations that encase the bacteria (167). In trauma or sepsis induced ARDS, uPA and streptokinase, were beneficial producing a significant improvement in PaO2 (168, 169). Coronavirus disease-19 (COVID-19) is caused by infection with severe acute respiratory SARS-CoV2. Severely ill patients with COVID-19 are prone to thrombosis and can develop ARDS, sepsis and multiorgan failure. Thrombolytic therapy has therefore garnered interest for treatment in severely ill COVID patients (170, 171). Initial studies indicate that tPA improves PaO2/FiO2 ratio, however, larger studies are required to establish treatment regimens and the safety profile (172, 173). Targeting the inflammatory response also has potential to correct fibrinolytic dysregulation. Indeed, blocking IL-6 with Tocilizumab decreases PAI-1 levels and this was found to be beneficial in SARS-CoV2 infection and is a recommended therapy in ICU patients (161, 174).
Summary
The fibrinolytic and innate immune systems work in concert to protect from infection and inflammation and to regulate thrombus resolution. Derailment of one system therefore influences the other. Invading pathogens take advantage of plasminogen and its activators to evade protective immune responses. Whilst immune cells are a source of fibrinolytic proteins and act as a surface for their assembly and function in thrombus resolution. The fibrinolytic system participates in host immune responses, however, dysregulation can precipitate in aberrant fibrin distribution or impede immune cell function. There is much still to learn on the interplay between the fibrinolytic and innate immune systems. Improved understanding of these intricacies could lead to development of more targeted immunothrombolytic or immunomodulating therapies.
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
CW was supported by British Heart Foundation (PG/20/17/35050).
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. (2013) 13:34–45.
2. Martinod K, Deppermann C. Immunothrombosis and thromboinflammation in host defense and disease. Platelets. (2021) 32:314–24.
3. Bonaventura A, Vecchi A, Dagna L, Martinod K, Dixon D, Tassell BW, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. (2021) 21:319–29. doi: 10.1038/s41577-021-00536-9
4. Brinkmann V, Reichard U, Goosmann C. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5.
5. Stark K, Massberg S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat Rev Cardiol. (2021) 18:666–82.
6. Massberg S, Grahl L, Bruehl M, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. (2010) 16:887–96.
7. Stoppelli M, Corti A, Soffientini A, Cassani G, Blasi F, Assoian R. Differentiation-enhanced binding of the amino-terminal fragment of human urokinase plasminogen activator to a specific receptor on U937 monocytes. Proc Natl Acad Sci USA. (1985) 82:4939–43. doi: 10.1073/pnas.82.15.4939
8. Kruithof E, Tran-Thang C, Ransijn A, Bachmann F. Demonstration of a fast-acting inhibitor of plasminogen activators in human plasma. Blood. (1984) 64:907–13.
9. Thorsen S, Philips M, Selmer J, Lecander I, Astedt B. Kinetics of inhibition of tissue-type and urokinase-type plasminogen activator by plasminogen-activator inhibitor type 1 and type 2. Eur J Biochem. (1988) 175:33–9.
10. Lu B, Sofian T, Law R, Coughlin P, Horvath A. Contribution of conserved lysine residues in the alpha2-antiplasmin C terminus to plasmin binding and inhibition. J Biol Chem. (2011) 286:24544–52. doi: 10.1074/jbc.M111.229013
11. Sakata Y, Aoki N. Significance of cross-linking of alpha 2-plasmin inhibitor to fibrin in inhibition of fibrinolysis and in hemostasis. J Clin Invest. (1982) 69:536–42.
12. Peetermans M, Vanassche T, Liesenborghs L, Lijnen R, Verhamme P. Bacterial pathogens activate plasminogen to breach tissue barriers and escape from innate immunity. Crit Rev Microbiol. (2016) 42:866–82. doi: 10.3109/1040841X.2015.1080214
13. Wang X, Lin X, Loy J, Tang J, Zhang X. Crystal structure of the catalytic domain of human plasmin complexed with streptokinase. Science. (1998) 281:1662–5.
14. Collen D, Schlott B, Engelborghs Y, Hoef BV, Hartmann M, Lijnen HR, et al. On the mechanism of the activation of human plasminogen by recombinant staphylokinase. J Biol Chem. (1993) 268:8284–9.
15. Collen D. Staphylokinase: a potent, uniquely fibrin-selective thrombolytic agent. Nat Med. (1998) 4:279–84. doi: 10.1038/nm0398-279
16. Järvinen H, Laakkonen L, Haiko J, Johansson T, Juuti K, Suomalainen M, et al. Human single-chain urokinase is activated by the omptins PgtE of Salmonella enterica and Pla of Yersinia pestis despite mutations of active site residues. Mol Microbiol. (2013) 89:507–17. doi: 10.1111/mmi.12293
17. Degen J, Bugge T, Goguen J. Fibrin and fibrinolysis in infection and host defense. J Thromb Haemost. (2007) 5:24–31.
18. Crowe J, Sievwright I, Auld G, Moore N, Gow N, Booth N. Candida albicans binds human plasminogen: identification of eight plasminogen-binding proteins. Mol Microbiol. (2003) 47:1637–51. doi: 10.1046/j.1365-2958.2003.03390.x
19. Ayon-Nunez D, Fragoso G, Bobes R, Laclette J. Plasminogen-binding proteins as an evasion mechanism of the host’s innate immunity in infectious diseases. Biosci Rep. (2018) 38:5. doi: 10.1042/BSR20180705
20. Medcalf R, Keragala C. Fibrinolysis: a primordial system linked to the immune response. Int J Mol Sci. (2021) 22:7. doi: 10.3390/ijms22073406
21. Bhattacharya S, Ploplis V, Castellino F. Bacterial plasminogen receptors utilize host plasminogen system for effective invasion and dissemination. J Biomed Biotechnol. (2012) 2012:482096. doi: 10.1155/2012/482096
22. Castiblanco-Valencia MM, Fraga TR, Pagotto AH, Serrano SM, Abreu PA, Barbosa AS, et al. Plasmin cleaves fibrinogen and the human complement proteins C3b and C5 in the presence of Leptospira interrogans proteins: a new role of LigA and LigB in invasion and complement immune evasion. Immunobiology. (2016) 221:679–89. doi: 10.1016/j.imbio.2016.01.001
23. Sanderson-Smith M, De Oliveira D, Ranson M, McArthur J. Bacterial plasminogen receptors: mediators of a multifaceted relationship. J Biomed Biotechnol. (2012) 2012:272148. doi: 10.1155/2012/272148
24. Stie J, Bruni G, Fox D. Surface-associated plasminogen binding of Cryptococcus neoformans promotes extracellular matrix invasion. PLoS One. (2009) 4:e5780. doi: 10.1371/journal.pone.0005780
25. Calcagno M, Avilan L, Colasante C, Berrueta L, Salmen S. Interaction of different Leishmania mexicana morphotypes with plasminogen. Parasitol Res. (2002) 88:972–8. doi: 10.1007/s00436-002-0688-2
26. Hoylaerts M, Rijken D, Lijnen H, Collen D. Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role fibrin. J Biol Chem. (1982) 257:2912–9.
27. Urano T, Castellino F, Suzuki Y. Regulation of plasminogen activation on cell surfaces and fibrin. J Thromb Haemost. (2018) 16:1487–97.
28. Whyte CS, Morrow GB, Baik N, Booth NA, Jalal MM, Parmer RJ, et al. Exposure of plasminogen and a novel plasminogen receptor, Plg-RKT, on activated human and murine platelets. Blood. (2021) 137:248–57. doi: 10.1182/blood.2020007263
29. Stricker R, Wong D, Shiu D, Reyes P, Shuman M. Activation of plasminogen by tissue plasminogen activator on normal and thrombasthenic platelets: effects on surface proteins and platelet aggregation. Blood. (1986) 68:275–80.
30. Hall S, Humphries J, Gonias S. Inhibition of cell surface receptor-bound plasmin by alpha 2-antiplasmin and alpha 2-macroglobulin. J Biol Chem. (1991) 266:12329–36.
31. Lucas M, Fretto L, McKee P. The binding of human plasminogen to fibrin and fibrinogen. J Biol Chem. (1983) 258:4249–56.
32. Plow E, Freaney D, Plescia J, Miles L. The plasminogen system and cell surfaces: evidence for plasminogen and urokinase receptors on the same cell type. J Cell Biol. (1986) 103:2411–20.
33. Plow E, Doeuvre L, Das R. So many plasminogen receptors: why? J Biomed Biotechnol. (2012) 2012:141806.
34. Miles L, Ny L, Wilczynska M, Shen Y, Ny T, Parmer R. Plasminogen receptors and fibrinolysis. Int J Mol Sci. (2021) 22:4.
35. Andronicos NM, Chen EI, Baik N, Bai H, Parmer C, Kiosses WB, et al. Proteomics-based discovery of a novel, structurally unique, and developmentally regulated plasminogen receptor, Plg-RKT, a major regulator of cell surface plasminogen activation. Blood. (2010) 115:1319–30. doi: 10.1182/blood-2008-11-188938
36. Lighvani S, Baik N, Diggs J, Khaldoyanidi S, Parmer R, Miles L. Regulation of macrophage migration by a novel plasminogen receptor Plg-R KT. Blood. (2011) 118:5622–30. doi: 10.1182/blood-2011-03-344242
37. Singh I, Burnand KG, Collins M, Luttun A, Collen D, Boelhouwer B, et al. Failure of thrombus to resolve in urokinase-type plasminogen activator gene-knockout mice: rescue by normal bone marrow-derived cells. Circulation. (2003) 107:869–75. doi: 10.1161/01.cir.0000050149.22928.39
38. Hart P, Burgess D, Vitti G, Hamilton J. Interleukin-4 stimulates human monocytes to produce tissue-type plasminogen activator. Blood. (1989) 74:1222–5.
39. Ritchie H, Jamieson A, Booth N. Thrombin modulates synthesis of plasminogen activator inhibitor type 2 by human peripheral blood monocytes. Blood. (1995) 86:3428–35.
40. Ritchie H, Booth N. Secretion of plasminogen activator inhibitor 2 by human peripheral blood monocytes occurs via an endoplasmic reticulum-golgi-independent pathway. Exp Cell Res. (1998) 242:439–50. doi: 10.1006/excr.1998.4118
41. Ritchie H, Robbie L, Kinghorn S, Exley R, Booth N. Monocyte plasminogen activator inhibitor 2 (PAI-2) inhibits u-PA-mediated fibrin clot lysis and is cross-linked to fibrin. Thromb Haemost. (1999) 81:96–103.
42. Siefert SA, Chabasse C, Mukhopadhyay S, Hoofnagle MH, Strickland DK, Sarkar R, et al. Enhanced venous thrombus resolution in plasminogen activator inhibitor type-2 deficient mice. J Thromb Haemost. (2014) 12:1706–16. doi: 10.1111/jth.12657
43. Humphries J, Gossage JA, Modarai B, Burnand KG, Sisson TH, Murdoch C, et al. Monocyte urokinase-type plasminogen activator up-regulation reduces thrombus size in a model of venous thrombosis. J Vasc Surg. (2009) 50:1127–34. doi: 10.1016/j.jvs.2009.06.047
44. Claesen K, De Loose J, Van Wielendaele P. ProCPU is expressed by (primary) human monocytes and macrophages and expression differs between states of differentiation and activation. Int J Mol Sci. (2023) 24:4. doi: 10.3390/ijms24043725
45. Henriksson P, Becker S, Lynch G, McDonagh J. Identification of intracellular factor XIII in human monocytes and macrophages. J Clin Invest. (1985) 76:528–34.
46. Muszbek L, Adany R, Szegedi G, Polgar J, Kavai M. Factor XIII of blood coagulation in human monocytes. Thromb Res. (1985) 37:401–10.
47. Cordell P, Kile B, Standeven K, Josefsson E, Pease R, Grant P. Association of coagulation factor XIII-a with golgi proteins within monocyte-macrophages: implications for subcellular trafficking and secretion. Blood. (2010) 115:2674–81. doi: 10.1182/blood-2009-08-231316
48. Alshehri F, Whyte C, Tuncay A, Williams M, Wilson H, Mutch N. Monocytes expose factor XIII-a and stabilize thrombi against fibrinolytic degradation. Int J Mol Sci. (2021) 22:12. doi: 10.3390/ijms22126591
49. Moir E, Booth N, Bennett B, Robbie L. Polymorphonuclear leucocytes mediate endogenous thrombus lysis via a u-PA-dependent mechanism. Br J Haematol. (2001) 113:72–80. doi: 10.1046/j.1365-2141.2001.02696.x
50. Varj I, Longstaff C, Szab L, Farkas ÁZ, Varga-Szab V, Tanka-Salamon A, et al. DNA, histones and neutrophil extracellular traps exert anti-fibrinolytic effects in a plasma environment. Thromb Haemost. (2015) 113:1289–98. doi: 10.1160/TH14-08-0669
51. Longstaff C, Varj I, Sótonyi P, Szab L, Krumrey M, Hoell A, et al. Mechanical stability and fibrinolytic resistance of clots containing fibrin. DNA, and histones. J Biol Chem. (2013) 288:6946–56. doi: 10.1074/jbc.M112.404301
52. Brühl M-, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
53. Ducroux C, Meglio L, Loyau S, Delbosc S, Boisseau W, Deschildre C, et al. Thrombus neutrophil extracellular traps content impair tPA-induced thrombolysis in acute ischemic stroke. Stroke. (2018) 49:754–7.
54. Laridan E, Denorme F, Desender L, François O, Andersson T, Deckmyn H, et al. Neutrophil extracellular traps in ischemic stroke thrombi. Ann Neurol. (2017) 82:223–32.
55. Sreeramkumar V, Adrover J, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. (2014) 346:1234–8.
56. Zuchtriegel G, Uhl B, Puhr-Westerheide D, Pörnbacher M, Lauber K, Krombach F, et al. Platelets guide leukocytes to their sites of extravasation. PLoS Biol. (2016) 14:e1002459. doi: 10.1371/journal.pbio.1002459
57. Jang IK, Gold HK, Ziskind AA, Fallon JT, Holt RE, Leinbach RC, et al. Differential sensitivity of erythrocyte-rich and platelet-rich arterial thrombi to lysis with recombinant tissue-type plasminogen activator. A possible explanation for resistance to coronary thrombolysis. Circulation. (1989) 79:920–8. doi: 10.1161/01.cir.79.4.920
58. Tomkins A, Schleicher N, Murtha L, Kaps M, Levi C, Nedelmann M, et al. Platelet rich clots are resistant to lysis by thrombolytic therapy in a rat model of embolic stroke. Exp Transl Stroke Med. (2015) 7:2. doi: 10.1186/s13231-014-0014-y
59. Booth N, Simpson A, Croll A, Bennett B, MacGregor I. Plasminogen activator inhibitor (PAI-1) in plasma and platelets. Br J Haematol. (1988) 70:327–33.
60. Whyte C, Mostefai H, Baeten K. Role of shear stress and tPA concentration in the fibrinolytic potential of thrombi. Int J Mol Sci. (2021) 22:4. doi: 10.3390/ijms22042115
61. Booth N, Robbie L, Croll A, Bennett B. Lysis of platelet-rich thrombi: the role of PAI-1. Ann NY Acad Sci. (1992) 667:70–80.
62. Robbie L, Bennett B, Croll A, Brown P, Booth N. Proteins of the fibrinolytic system in human thrombi. Thromb Haemost. (1996) 75:127–33.
63. Morrow G, Whyte C, Mutch N. Functional plasminogen activator inhibitor 1 is retained on the activated platelet membrane following platelet activation. Haematologica. (2020) 105:2824–33. doi: 10.3324/haematol.2019.230367
64. Whyte CS, Swieringa F, Mastenbroek TG, Lionikiene AS, Lanc MD, Meijden PE, et al. Plasminogen associates with phosphatidylserine-exposing platelets and contributes to thrombus lysis under flow. Blood. (2015) 125:2568–78. doi: 10.1182/blood-2014-09-599480
65. Ni R, Neves M, Wu C. Activated thrombin-activatable fibrinolysis inhibitor (TAFIa) attenuates fibrin-dependent plasmin generation on thrombin-activated platelets. J Thromb Haemost. (2020) 18:2364–76. doi: 10.1111/jth.14950
66. Schadinger S, Lin J, Garand M, Boffa M. Secretion and antifibrinolytic function of thrombin-activatable fibrinolysis inhibitor from human platelets. J Thromb Haemost. (2010) 8:2523–9.
67. Boulaftali Y, Ho-Tin-Noe B, Pena A, Loyau S, Venisse L, François D, et al. Platelet protease nexin-1, a serpin that strongly influences fibrinolysis and thrombolysis. Circulation. (2011) 123:1326–34. doi: 10.1161/CIRCULATIONAHA.110.000885
68. Plow E, Collen D. The presence and release of alpha 2-antiplasmin from human platelets. Blood. (1981) 58:1069–74.
69. Gogstad G, Stormorken H, Solum N. Platelet alpha 2-antiplasmin is located in the platelet alpha-granules. Thromb Res. (1983) 31:387–90. doi: 10.1016/0049-3848(83)90339-0
70. Mitchell J, Lionikiene A, Fraser S, Whyte C, Booth N, Mutch N. Functional factor XIII-a is exposed on the stimulated platelet surface. Blood. (2014) 124:3982–90. doi: 10.1182/blood-2014-06-583070
71. Reed G, Matsueda G, Haber E. Fibrin-fibrin and alpha 2-antiplasmin-fibrin cross-linking by platelet factor XIII increases the resistance of platelet clots to fibrinolysis. Trans Assoc Am Physicians. (1991) 104:21–8.
72. Reed G, Matsueda G, Haber E. Platelet factor XIII increases the fibrinolytic resistance of platelet-rich clots by accelerating the crosslinking of alpha 2-antiplasmin to fibrin. Thromb Haemost. (1992) 68:315–20.
73. Hevessy Z, Haramura G, Boda Z, Udvardy M, Muszbek L. Promotion of the crosslinking of fibrin and alpha 2-antiplasmin by platelets. Thromb Haemost. (1996) 75:161–7.
74. Francis C, Marder V. Rapid formation of large molecular weight alpha-polymers in cross-linked fibrin induced by high factor XIII concentrations. Role of platelet factor XIII. J Clin Invest. (1987) 80:1459–65. doi: 10.1172/JCI113226
75. Rubens F, Perry D, Hatton M, Bishop P, Packham M, Kinlough-Rathbone R. Platelet accumulation on fibrin-coated polyethylene: role of platelet activation and factor XIII. Thromb Haemost. (1995) 73:850–6.
76. Somodi L, Beke Debreceni I, Kis G. Activation mechanism dependent surface exposure of cellular factor XIII on activated platelets and platelet microparticles. J Thromb Haemost. (2022) 20:1223–35. doi: 10.1111/jth.15668
77. Baeten K, Richard M, Kanse S, Mutch N, Degen J, Booth N. Activation of single-chain urokinase-type plasminogen activator by platelet-associated plasminogen: a mechanism for stimulation of fibrinolysis by platelets. J Thromb Haemost. (2010) 8:1313–22.
78. Miles L, Plow E. Binding and activation of plasminogen on the platelet surface. J Biol Chem. (1985) 260:4303–11.
79. Muller F, Mutch N, Schenk W. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. (2009) 139:1143–56.
80. Whyte C, Chernysh I, Domingues M. Polyphosphate delays fibrin polymerisation and alters the mechanical properties of the fibrin network. Thromb Haemost. (2016) 116:897–903. doi: 10.1160/TH16-01-0062
81. Mutch N, Engel R, de Willige S, Philippou H, Ariens R. Polyphosphate modifies the fibrin network and down-regulates fibrinolysis by attenuating binding of tPA and plasminogen to fibrin. Blood. (2010) 115:3980–8. doi: 10.1182/blood-2009-11-254029
82. Whyte C, Mutch N. uPA-mediated plasminogen activation is enhanced by polyphosphate. Haematologica. (2021) 106:522–31. doi: 10.3324/haematol.2019.237966
83. Smith S, Mutch N, Baskar D, Rohloff P, Docampo R, Morrissey J. Polyphosphate modulates blood coagulation and fibrinolysis. Proc Natl Acad Sci USA. (2006) 103:903–8.
84. Engel R, Brain C, Paget J, Lionikiene A, Mutch N. Single-chain factor XII exhibits activity when complexed to polyphosphate. J Thromb Haemost. (2014) 12:1513–22. doi: 10.1111/jth.12663
85. Mitchell J, Lionikiene A, Georgiev G. Polyphosphate colocalizes with factor XII on platelet-bound fibrin and augments its plasminogen activator activity. Blood. (2016) 128:2834–45. doi: 10.1182/blood-2015-10-673285
86. Emeis J, Eijnden-Schrauwen Y, Hoogen C, de Priester W, Westmuckett A, Lupu F. An endothelial storage granule for tissue-type plasminogen activator. J Cell Biol. (1997) 139:245–56.
87. Miles L, Parmer R. Plasminogen receptors: the first quarter century. Semin Thromb Hemost. (2013) 39:329–37. doi: 10.1055/s-0033-1334483
88. Quax P, Grimbergen J, Lansink M. Binding of human urokinase-type plasminogen activator to its receptor: residues involved in species specificity and binding. Arterioscler Thromb Vasc Biol. (1998) 18:693–701.
89. van Mourik J, Lawrence D, Loskutoff D. Purification of an inhibitor of plasminogen activator (antiactivator) synthesized by endothelial cells. J Biol Chem. (1984) 259:14914–21.
90. Morrow G, Whyte C, Mutch NJ. A serpin with a finger in many PAIs: PAI-1’s central function in thromboinflammation and cardiovascular disease. Front Cardiovasc Med. (2021) 8:653655. doi: 10.3389/fcvm.2021.653655
91. Boncela J, Przygodzka P, Papiewska-Pajak I, Wyroba E, Cierniewski C. Association of plasminogen activator inhibitor type 2 (PAI-2) with proteasome within endothelial cells activated with inflammatory stimuli. J Biol Chem. (2011) 286:43164–71. doi: 10.1074/jbc.M111.245647
92. Lin J, Garand M, Zagorac B. Identification of human thrombin-activatable fibrinolysis inhibitor in vascular and inflammatory cells. Thromb Haemost. (2011) 105:999–1009.
93. Yuan Y, Alwis I, Wu M. Neutrophil macroaggregates promote widespread pulmonary thrombosis after gut ischemia. Sci Transl Med. (2017) 9:409. doi: 10.1126/scitranslmed.aam5861
94. Shetty S, Padijnayayveetil J, Tucker T, Stankowska D, Idell S. The fibrinolytic system and the regulation of lung epithelial cell proteolysis, signaling, and cellular viability. Am J Physiol Lung Cell Mol Physiol. (2008) 295:L967–75. doi: 10.1152/ajplung.90349.2008
95. Levi M, Poll T. Coagulation in patients with severe sepsis. Semin Thromb Hemost. (2015) 41:9–15.
96. Lim J, Woo C, Li J. Critical role of type 1 plasminogen activator inhibitor (PAI-1) in early host defense against nontypeable Haemophilus influenzae (NTHi) infection. Biochem Biophys Res Commun. (2011) 414:67–72. doi: 10.1016/j.bbrc.2011.09.023
97. Goolaerts A, Lafargue M, Song Y. PAI-1 is an essential component of the pulmonary host response during Pseudomonas aeruginosa pneumonia in mice. Thorax. (2011) 66:788–96. doi: 10.1136/thx.2010.155788
98. Lim J, Stirling B, Derry J. Tumor suppressor CYLD regulates acute lung injury in lethal Streptococcus pneumoniae infections. Immunity. (2007) 27:349–60. doi: 10.1016/j.immuni.2007.07.011
99. Roelofs J, Teske G, Bonta P. Plasminogen activator inhibitor-1 regulates neutrophil influx during acute pyelonephritis. Kidney Int. (2009) 75:52–9. doi: 10.1038/ki.2008.454
100. Renckens R, Roelofs J, Bonta P. Plasminogen activator inhibitor type 1 is protective during severe Gram-negative pneumonia. Blood. (2007) 109:1593–601. doi: 10.1182/blood-2006-05-025197
101. Renckens R, Pater J, van der Poll T. Plasminogen activator inhibitor type-1-deficient mice have an enhanced IFN-gamma response to lipopolysaccharide and staphylococcal enterotoxin B. J Immunol. (2006) 177:8171–6. doi: 10.4049/jimmunol.177.11.8171
102. Dittmann M, Hoffmann H, Scull M. A serpin shapes the extracellular environment to prevent influenza a virus maturation. Cell. (2015) 160:631–43. doi: 10.1016/j.cell.2015.01.040
103. Campbell W, Lazoura E, Okada N, Okada H. Inactivation of C3a and C5a octapeptides by carboxypeptidase R and carboxypeptidase N. Microbiol Immunol. (2002) 46:131–4. doi: 10.1111/j.1348-0421.2002.tb02669.x
104. Plug T, Meijers J. Structure-function relationships in thrombin-activatable fibrinolysis inhibitor. J Thromb Haemost. (2016) 14:633–44.
105. Shinohara T, Sakurada C, Suzuki T. Pro-carboxypeptidase R cleaves bradykinin following activation. Int Arch Allergy Immunol. (1994) 103:400–4. doi: 10.1159/000236661
106. Naito M, Taguchi O, Kobayashi T. Thrombin-activatable fibrinolysis inhibitor protects against acute lung injury by inhibiting the complement system. Am J Respir Cell Mol Biol. (2013) 49:646–53. doi: 10.1165/rcmb.2012-0454OC
107. Fujiwara A, Taguchi O, Takagi T. Role of thrombin-activatable fibrinolysis inhibitor in allergic bronchial asthma. Lung. (2012) 190:189–98. doi: 10.1007/s00408-011-9337-9
108. Song J, Hwang I, Cho K. Plasma carboxypeptidase B downregulates inflammatory responses in autoimmune arthritis. J Clin Invest. (2011) 121:3517–27. doi: 10.1172/JCI46387
109. Vollrath J, Marzi I, Herminghaus A, Lustenberger T, Relja B. Post-traumatic sepsis is associated with increased C5a and decreased TAFI levels. J Clin Med. (2020) 9:4. doi: 10.3390/jcm9041230
110. Renckens R, Roelofs J, Horst S. Absence of thrombin-activatable fibrinolysis inhibitor protects against sepsis-induced liver injury in mice. J Immunol. (2005) 175:6764–71. doi: 10.4049/jimmunol.175.10.6764
111. Muto Y, Suzuki K, Iida H. EF6265, a novel inhibitor of activated thrombin-activatable fibrinolysis inhibitor, protects against sepsis-induced organ dysfunction in rats. Crit Care Med. (2009) 37:1744–9. doi: 10.1097/CCM.0b013e31819ffc14
112. Ploplis V, French E, Carmeliet P, Collen D, Plow E. Plasminogen deficiency differentially affects recruitment of inflammatory cell populations in mice. Blood. (1998) 91:2005–9.
113. Das R, Ganapathy S, Settle M, Plow E. Plasminogen promotes macrophage phagocytosis in mice. Blood. (2014) 124:679–88.
114. Silva L, Lum A, Tran C. Plasmin-mediated fibrinolysis enables macrophage migration in a murine model of inflammation. Blood. (2019) 134:291–303. doi: 10.1182/blood.2018874859
115. Vago J, Sugimoto M, Lima K. Plasminogen and the plasminogen receptor, Plg-R(KT), regulate macrophage phenotypic, and functional changes. Front Immunol. (2019) 10:1458. doi: 10.3389/fimmu.2019.01458
116. Borg R, Samson A, Au A. Dendritic cell-mediated phagocytosis but not immune activation is enhanced by plasmin. PLoS One. (2015) 10:7. doi: 10.1371/journal.pone.0131216
117. Renckens R, Roelofs J, Florquin S. Endogenous tissue-type plasminogen activator is protective during Escherichia coli-induced abdominal sepsis in mice. J Immunol. (2006) 177:1189–96. doi: 10.4049/jimmunol.177.2.1189
118. Mantuano E, Azmoon P, Brifault C. Tissue-type plasminogen activator regulates macrophage activation and innate immunity. Blood. (2017) 130:1364–74.
119. Mantuano E, Brifault C, Lam M, Azmoon P, Gilder A, Gonias SL. LDL receptor-related protein-1 regulates NFkappaB and microRNA-155 in macrophages to control the inflammatory response. Proc Natl Acad Sci USA. (2016) 113:1369–74. doi: 10.1073/pnas.1515480113
120. Uhl B, Zuchtriegel G, Puhr-Westerheide D. Tissue plasminogen activator promotes postischemic neutrophil recruitment via its proteolytic and nonproteolytic properties. Arterioscler Thromb Vasc Biol. (2014) 34:1495–504. doi: 10.1161/ATVBAHA.114.303721
121. Draxler D, Lee F, Ho H, Keragala C, Medcalf R, Niego B. t-PA suppresses the immune response and aggravates neurological deficit in a murine model of ischemic stroke. Front Immunol. (2019) 10:591. doi: 10.3389/fimmu.2019.00591
122. Paland N, Aharoni S, Fuhrman B. Urokinase-type plasminogen activator (uPA) modulates monocyte-to-macrophage differentiation and prevents Ox-LDL-induced macrophage apoptosis. Atherosclerosis. (2013) 231:29–38. doi: 10.1016/j.atherosclerosis.2013.08.016
123. Abraham E, Gyetko M, Kuhn K. Urokinase-type plasminogen activator potentiates lipopolysaccharide-induced neutrophil activation. J Immunol. (2003) 170:5644–51.
124. Renckens R, Roelofs J, Florquin S, van der Poll T. Urokinase-type plasminogen activator receptor plays a role in neutrophil migration during lipopolysaccharide-induced peritoneal inflammation but not during Escherichia coli-induced peritonitis. J Infect Dis. (2006) 193:522–30. doi: 10.1086/499601
125. Kiyan Y, Tkachuk S, Rong S. TLR4 response to LPS is reinforced by urokinase receptor. Front Immunol. (2020) 11:573550. doi: 10.3389/fimmu.2020.573550
126. Genua M, D’Alessio S, Cibella J. The urokinase plasminogen activator receptor (uPAR) controls macrophage phagocytosis in intestinal inflammation. Gut. (2015) 64:589–600. doi: 10.1136/gutjnl-2013-305933
127. Dowton S, Colten H. Acute phase reactants in inflammation and infection. Semin Hematol. (1988) 25:84–90.
128. Macrae F, Duval C, Papareddy P. A fibrin biofilm covers blood clots and protects from microbial invasion. J Clin Invest. (2018) 128:3356–68. doi: 10.1172/JCI98734
129. Smeeth L, Cook C, Thomas S, Hall A, Hubbard R, Vallance P. Risk of deep vein thrombosis and pulmonary embolism after acute infection in a community setting. Lancet. (2006) 367:1075–9. doi: 10.1016/S0140-6736(06)68474-2
130. Lozinguez O, Arnaud E, Belec L. Demonstration of an association between Chlamydia pneumoniae infection and venous thromboembolic disease. Thromb Haemost. (2000) 83:887–91.
131. Meier C, Jick S, Derby L, Vasilakis C, Jick H. Acute respiratory-tract infections and risk of first-time acute myocardial infarction. Lancet. (1998) 351: 1467–71.
132. Smeeth L, Thomas S, Hall A, Hubbard R, Farrington P, Vallance P. Risk of myocardial infarction and stroke after acute infection or vaccination. N Engl J Med. (2004) 351:2611–8.
133. Beristain-Covarrubias N, Perez-Toledo M, Thomas M, Henderson I, Watson S, Cunningham A. Understanding infection-induced thrombosis: lessons learned from animal models. Front Immunol. (2019) 10:2569. doi: 10.3389/fimmu.2019.02569
134. Meier C. Seasonal variations in the incidence of acute myocardial infarction. J Am Coll Cardiol. (1998) 32:2103–4.
135. Angus D, Linde-Zwirble W, Lidicker J, Clermont G, Carcillo J, Pinsky M. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. (2001) 29:1303–10.
136. Mavrommatis A, Theodoridis T, Orfanidou A, Roussos C, Christopoulou-Kokkinou V, Zakynthinos S. Coagulation system and platelets are fully activated in uncomplicated sepsis. Crit Care Med. (2000) 28:451–7. doi: 10.1097/00003246-200002000-00027
137. Vago J, Brito L, Teixeira L. The role of plasmin/plasminogen in experimental polymicrobial sepsis. Blood. (2020) 136:16–7.
138. Tipoe T, Wu W, Chung L. Plasminogen activator inhibitor 1 for predicting sepsis severity and mortality outcomes: a systematic review and meta-analysis. Front Immunol. (2018) 9:1218. doi: 10.3389/fimmu.2018.01218
139. Biemond B, Levi M, Ten Cate H. Plasminogen activator and plasminogen activator inhibitor I release during experimental endotoxaemia in chimpanzees: effect of interventions in the cytokine and coagulation cascades. Clin Sci. (1995) 88:587–94. doi: 10.1042/cs0880587
140. Li L, Nie W, Zhou H, Yuan W, Li W, Huang W. Association between plasminogen activator inhibitor-1 -675 4G/5G polymorphism and sepsis: a meta-analysis. PLoS One. (2013) 8:e54883. doi: 10.1080/15513815.2020.1775736
141. Hermans P, Hibberd M, Booy R. 4G/5G promoter polymorphism in the plasminogen-activator-inhibitor-1 gene and outcome of meningococcal disease. Meningococcal Res Group Lancet. (1999) 354:556–60.
142. McDonald B, Davis R, Kim S. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. (2017) 129:1357–67.
143. Gould T, Vu T, Stafford A. Cell-free DNA modulates clot structure and impairs fibrinolysis in sepsis. Arterioscler Thromb Vasc Biol. (2015) 35:2544–53. doi: 10.1161/ATVBAHA.115.306035
144. Haj M, Robbie L, Adey G, Bennett B. Inhibitors of plasminogen activator in neutrophils and mononuclear cells from septic patients. Thromb Haemost. (1995) 74:1528–32.
145. Emonts M, de Bruijne E, Guimaraes A. Thrombin-activatable fibrinolysis inhibitor is associated with severity and outcome of severe meningococcal infection in children. J Thromb Haemost. (2008) 6:268–76. doi: 10.1111/j.1538-7836.2007.02841.x
146. Kremer Hovinga J, Franco R, Zago M, Ten Cate H, Westendorp R, Reitsma PH. A functional single nucleotide polymorphism in the thrombin-activatable fibrinolysis inhibitor (TAFI) gene associates with outcome of meningococcal disease. J Thromb Haemost. (2004) 2:54–7.
147. Martin C, Papazian L, Payan M, Saux P, Gouin F. Pulmonary fibrosis correlates with outcome in adult respiratory distress syndrome. A study in mechanically ventilated patients. Chest. (1995) 107:196–200.
148. Moine P, McIntyre R, Schwartz M. NF-kappaB regulatory mechanisms in alveolar macrophages from patients with acute respiratory distress syndrome. Shock. (2000) 13:85–91.
149. Huang X, Xiu H, Zhang S, Zhang G. The role of macrophages in the pathogenesis of ALI/ARDS. Mediators Inflamm. (2018) 2018:1264913.
150. Bertozzi P, Astedt B, Zenzius L. Depressed bronchoalveolar urokinase activity in patients with adult respiratory distress syndrome. N Engl J Med. (1990) 322:890–7.
151. Bone R, Francis P, Pierce A. Intravascular coagulation associated with the adult respiratory distress syndrome. Am J Med. (1976) 61:585–9.
152. Ware L. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med. (2006) 27:337–49.
154. Keller T, van der Sluijs K, de Kruif M. Effects on coagulation and fibrinolysis induced by influenza in mice with a reduced capacity to generate activated protein C and a deficiency in plasminogen activator inhibitor type 1. Circ Res. (2006) 99:1261–9. doi: 10.1161/01.RES.0000250834.29108.1a
155. Gralinski L, Bankhead A, Jeng S. Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. mBio. (2013) 4:4.
156. Whyte C, Simpson M, Morrow G. The suboptimal fibrinolytic response in COVID-19 is dictated by high PAI-1. J Thromb Haemost. (2022) 20:2394–406.
157. Juneja G, Castelo M, Yeh C. Biomarkers of coagulation, endothelial function, and fibrinolysis in critically ill patients with COVID-19: a single-center prospective longitudinal study. J Thromb Haemost. (2021) 19:1546–57. doi: 10.1111/jth.15327
158. Mestries J, Kruithof E, Gascon M, Herodin F, Agay D, Ythier A. In vivo modulation of coagulation and fibrinolysis by recombinant glycosylated human interleukin-6 in baboons. Eur Cytokine Netw. (1994) 5:275–81.
159. Kruithof E. Regulation of plasminogen activator inhibitor type 1 gene expression by inflammatory mediators and statins. Thromb Haemost. (2008) 100:969–75.
160. Kruithof E, Mestries J, Gascon M, Ythier A. The coagulation and fibrinolytic responses of baboons after in vivo thrombin generation–effect of interleukin 6. Thromb Haemost. (1997) 77:905–10.
161. Kang S, Tanaka T, Inoue H. IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci USA. (2020) 117:22351–6. doi: 10.1073/pnas.2010229117
162. Prabhakaran P, Ware L, White K, Cross M, Matthay M, Olman M. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am J Physiol Lung Cell Mol Physiol. (2003) 285:L20–8.
163. Ware L, Matthay M, Parsons P. Pathogenetic and prognostic significance of altered coagulation and fibrinolysis in acute lung injury/acute respiratory distress syndrome. Crit Care Med. (2007) 35:1821–8. doi: 10.1097/01.CCM.0000221922.08878.49
164. Magalhaes V, Andrade E, Alves J. Group B Streptococcus hijacks the host plasminogen system to promote brain endothelial cell invasion. PLoS One. (2013) 8:e63244. doi: 10.1371/journal.pone.0063244
165. Draxler D, Yep K, Hanafi G. Tranexamic acid modulates the immune response and reduces postsurgical infection rates. Blood Adv. (2019) 3:1598–609. doi: 10.1182/bloodadvances.2019000092
166. Reichel C, Lerchenberger M, Uhl B. Plasmin inhibitors prevent leukocyte accumulation and remodeling events in the postischemic microvasculature. PLoS One. (2011) 6:e17229. doi: 10.1371/journal.pone.0017229
167. Levitas A, Krymko H, Richardson J, Zalzstein E, Ioffe V. Recombinant tissue plasminogen activator as a novel treatment option for infective endocarditis: a retrospective clinical study in 32 children. Cardiol Young. (2016) 26:110–5. doi: 10.1017/S104795111400273X
168. Hardaway R. A brief overview of acute respiratory distress syndrome. World J Surg. (2006) 30:1829–34.
169. Hardaway R, Harke H, Tyroch A, Williams C, Vazquez Y, Krause G. Treatment of severe acute respiratory distress syndrome: a final report on a phase I study. Am Surg. (2001) 67:377–82.
170. Whyte C, Morrow G, Mitchell J, Chowdary P, Mutch N. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J Thromb Haemost. (2020) 18:1548–55. doi: 10.1111/jth.14872
171. Moore H, Barrett C, Moore E. Is there a role for tissue plasminogen activator as a novel treatment for refractory COVID-19 associated acute respiratory distress syndrome? J Trauma Acute Care Surg. (2020) 88:713–4. doi: 10.1097/TA.0000000000002694
172. Wang J, Hajizadeh N, Moore E. Tissue plasminogen activator (tPA) treatment for COVID-19 associated acute respiratory distress syndrome (ARDS): a case series. J Thromb Haemost. (2020) 18:1752–5.
173. Barrett C, Moore H, Moore E, Wang J, Hajizadeh N, Biffl W, et al. Study of alteplase for respiratory failure in SARS-CoV-2 COVID-19: a vanguard multicenter, rapidly adaptive, pragmatic, randomized controlled trial. Chest. (2022) 161:710–27. doi: 10.1016/j.chest.2021.09.024
Keywords: fibrinolysis, innate immune, infection, plasminogen, thrombosis
Citation: Whyte CS (2023) All tangled up: interactions of the fibrinolytic and innate immune systems. Front. Med. 10:1212201. doi: 10.3389/fmed.2023.1212201
Received: 25 April 2023; Accepted: 17 May 2023;
Published: 02 June 2023.
Edited by:
Eleni Gavriilaki, G. Papanikolaou General Hospital, GreeceReviewed by:
Oonagh Shannon, Malmö University, SwedenCopyright © 2023 Whyte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claire S. Whyte, Yy5zLndoeXRlQGFiZG4uYWMudWs=