 Fagui Yue1,2
Fagui Yue1,2 Yuting Jiang
Yuting Jiang Ruizhi Liu
Ruizhi Liu Hongguo Zhang
Hongguo Zhang- 1Center for Reproductive Medicine and Center for Prenatal Diagnosis, First Hospital, Jilin University, Changchun, China
- 2Jilin Engineering Research Center for Reproductive Medicine and Genetics, Jilin University, Changchun, China
- 3Department of Pediatrics, University of Oklahoma Health Sciences Center, Oklahoma City, OK, United States
Objective: Chromosomal 1q21.1 deletions and duplications are genomic disorders that are usually diagnosed postnatally. However, the genotype–phenotype correlations of 1q21.1 copy number variants (CNVs) during the prenatal period are still not clear. This study aimed to provide a systematic summary of prenatal phenotypes for such genomic disorders.
Methods: In total, 26 prenatal amniotic fluid samples diagnosed with 1q21.1 microdeletions/microduplications were obtained from pregnant women who opted for invasive prenatal testing. Karyotypic analysis and chromosomal microarray analysis (CMA) were performed for all cases simultaneously. The pregnancy outcomes and health conditions after birth in all cases were followed up. Meanwhile, prenatal cases with 1q21.1 microdeletions or microduplications in the literature were retrospectively collected.
Results: In total, 11 pregnancies (11/8,252, 0.13%) with 1q21.1 microdeletions and 15 (15/8,252, 0.18%) with 1q21.1 microduplications were identified. Among these 1q21.1 CNVs, 4 cases covered the thrombocytopenia-absent radius (TAR) region, 16 cases covered the 1q21.1 recurrent microdeletion/microduplication region, and 6 cases covered all regions mentioned above. The prenatal abnormal ultrasound findings were recorded in four participants with 1q21.1 deletions and seven participants with 1q21.1 duplications. Finally, three cases with 1q21.1 deletions and five with 1q21.1 duplications terminated their pregnancies.
Conclusion: In the prenatal setting, 1q21.1 microdeletions were associated with increased nuchal translucency (NT), anomalies of the urinary system, and cardiovascular abnormalities, while 1q21.1 microduplications were correlated with cardiovascular malformations, nasal bone dysplasia, and increased NT. In addition, cerebral ventriculomegaly might be correlated with 1q21.1 microduplications. Considering the variable expressivity and incomplete penetrance of 1q21.1 CNVs, long-term follow-up after birth should be carried out in these cases.
1. Introduction
Chromosomal rearrangements involving the 1q21.1 region are hotspot loci that are frequently discovered in patients with different clinical manifestations. The multiple low-copy repeats located in chromosome 1q21.1 could make this region susceptible to non-allelic homologous recombination (NAHR), which would cause recurrent deletions and duplications (1–3). Four segmental duplication blocks, referred to as breakpoints (BPs) BP1–BP4, were specified within the 1q21.1 region from centromere to telomere (4). The chromosomal 1q21.1 region is usually subdivided into two distinctive regions: the proximal region extends from BP2 to BP3, spanning ~0.2 Mb (chr1: 145.4–145.6 Mb, GRCh37/hg19), and the distal region extends from BP3 to BP4, spanning ~1.35 Mb (chr1: 146.5–147.9 Mb, GRCh37/h19) (1, 2). In addition, two classes, 1q21.1 deletions and duplications, were defined at the molecular level: class I located between BP3 and BP4 (~1.8 Mb) and class II located between BP1/BP2 and BP4 (~2.7 Mb) (3, 4).
Three clinic disorders with diverse copy number variants (CNVs) within the 1q21.1 region were described: chromosome 1q21.1 deletion syndrome (OMIM 612474), chromosome 1q21.1 duplication syndrome (OMIM 612475), and thrombocytopenia-absent radius (TAR) syndrome (OMIM 274000) (Figures 1A, 2A). Chromosome 1q21.1 deletions have been associated with developmental delay, intellectual disability, autism spectrum disorders, attention deficit hyperactivity disorder (ADHD), schizophrenia, cataracts, dysmorphic features (microcephaly, frontal bossing, deep-set eyes, epicanthic folds, large nasal bridge, long philtrum, highly arched palate, and trigonocephaly), and congenital anomalies (congenital heart disease, eye abnormalities, skeletal, and genitourinary malformations). Chromosomal 1q21.1 duplications exhibit a wide spectrum of anomalies, including developmental delay, intellectual disability, autism spectrum disorder, macrocephaly, congenital heart anomalies, and dysmorphic features (e.g., frontal bossing and hypertelorism) (5–7). The TAR syndrome is recognized as a congenital malformation syndrome characterized by bilateral absence of the radii and thrombocytopenia, musculoskeletal and gastrointestinal abnormalities, renal and cardiac anomalies, and intolerance to cow's milk (8, 9). It is evident that the clinical manifestations of CNVs at the 1q21.1 region are diverse and complicated, some of which could not be identified even by advanced machines and experienced clinicians in ultrasound.
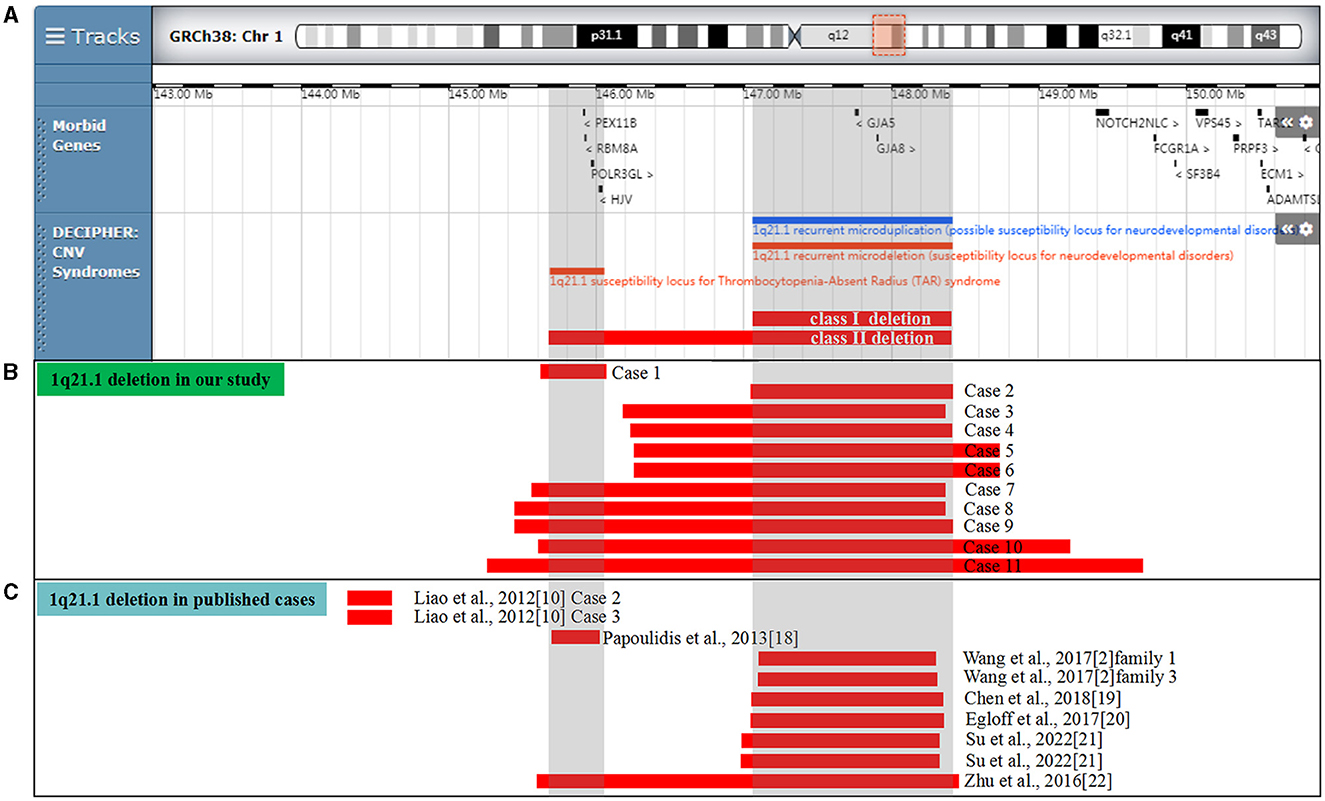
Figure 1. Scale representation of the deleted region in the 1q21.1 region (https://decipher.sanger.ac.uk/): (A) location of genes and genomic syndromes in the region; (B) deleted fragments in the present cases; and (C) previously described 1q21.1 deletions in the prenatal period. Genomic parameters are from GRCh38/hg38.
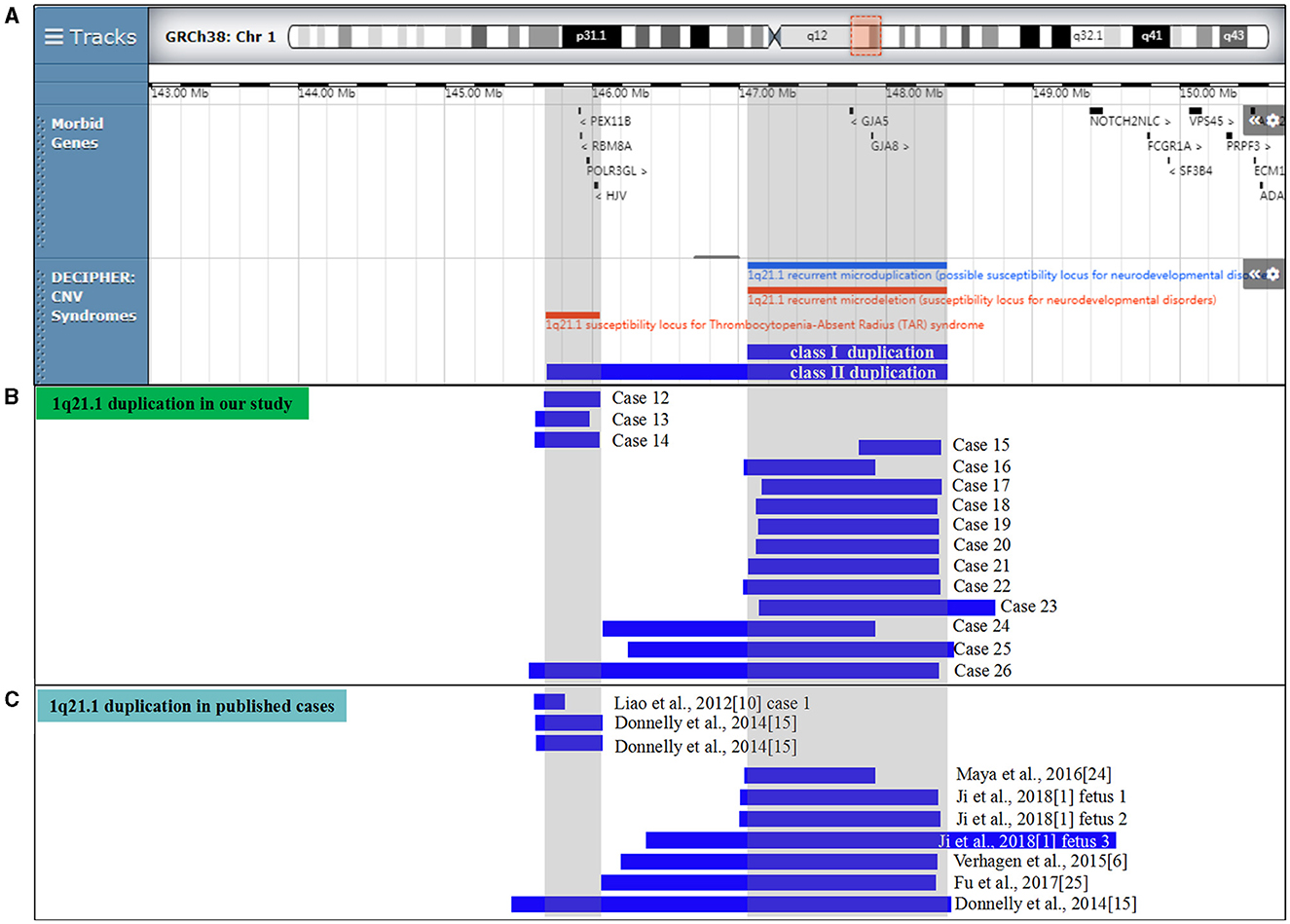
Figure 2. Scale representation of the duplicated region in the 1q21.1 region (https://decipher.sanger.ac.uk/): (A) location of genes and genomic syndromes in the region; (B) duplicated fragments in the present cases; and (C) previously described 1q21.1 duplications in the prenatal period. Genomic parameters are from GRCh38/hg38.
Till now, most studies on the CNV spectrum in the 1q21.1 region were diagnosed postnatally. Prenatal reports involving 1q21.1 duplications and deletions were limited (6, 10). To better our understanding of these prenatally detected chromosomal microscopic imbalances, we present the clinical and molecular findings of 26 cases with 1q21.1 microdeletions and microduplications in pregnant women undergoing prenatal invasive testing and provide a systematic summary of prenatal phenotypes for such genomic disorders.
2. Materials and methods
2.1. Clinical data
This retrospective study was performed from October 2018 to November 2022 and enrolled 26 cases carrying 1q21.1 microdeletions and microduplications, which were selected from 8,252 pregnant women. These women were referred to the First Hospital of Jilin University and underwent invasive diagnostic testing via amniocentesis. The main indications for prenatal diagnosis included non-invasive prenatal testing (NIPT) for aneuploidy, serum screening results for aneuploidy, ultrasound anomalies, and advanced maternal age. All pregnant women accepted routine prenatal ultrasound examinations during the gestation period, and abnormal ultrasound findings were included in the indications for prenatal diagnosis. All couples denied consanguineous marriage, and the pregnant women denied any exposure to teratogenic agents, irradiation, or infectious diseases during the pregnancy in question. All the prospective parents received detailed genetic counseling, and blood samples were collected after obtaining informed consent. The study protocol was approved by the Ethics Committee of the First Hospital of Jilin University (No. 2021-706), and written informed consent was obtained from all the couples.
2.2. Cytogenetic analysis
Pregnant women accepted amniocentesis for karyotyping analysis with written informed consent. A total of 30 ml of amniotic fluid cells were collected. Routine cytogenetic analysis was performed on G-band metaphases at 400–500 banding resolution, which was prepared from 20 ml of cultured amniotic fluid cells in accordance with standard protocols in our lab. In total, 20 metaphases were analyzed for all samples.
2.3. Chromosomal microarray analysis
The genomic DNA was extracted from the amniotic fluid cells and parental peripheral blood with the QIAamp® DNA Blood Mini Kit (Qiagen, Inc., Hilden, Germany) according to the manufacturer's protocol. Following written consent from all pregnant women, 10 ml of uncultured amniotic fluid cells were collected through amniocentesis. Then, the procedures were conducted through the CytoScan 750K array (Affymetrix, Santa Clara, CA, USA), in accordance with the manufacturer's protocol and our previous study (11). The procedure included genomic DNA extraction, digestion and ligation, PCR amplification, PCR product purification, quantification and fragmentation, labeling, array hybridization, washing, and scanning. Thresholds for genome-wide screening were set at ≥100 kb for gains and losses. The detected CNVs were comprehensively estimated by comparing them with the published literature and the public databases: (1) Database of Genomic Variants (DGVs; http://dgv.tcag.ca/dgv/app/home), (2) Database of Chromosomal Imbalance and Phenotype in Humans using Ensemble Resources (DECIPHERs, http://decipher.sanger.ac.uk/), (3) International Standards for Cytogenomic Arrays (ISCAs; https://www.iscaconsortium.org/), (4) Online Mendelian Inheritance in Man (OMIM, http://www.ncbi.nlm.nih.gov/omim), and (5) UCSC (http://genome.ucsc.edu/). All CNVs were classified as pathogenic (P), likely pathogenic (LP), variants of unknown significance (VOUS), likely benign (LB), and benign (B). Genomic positions refer to the Human Genome Assembly Dec. 2013 (GRCh38/hg38).
2.4. Selection of prenatally detected 1q21.1 microdeletion and microduplication
Given the lack of prenatal phenotypes of 1q21.1 deletions and duplications reported in the literature, we launched a search on PubMed (https://www.ncbi.nlm.nih.gov/pubmed/) for identifying relevant articles from inception to 2022. Criteria for case selection were defined as being in the English language, 1q21.1 deletions and duplications, and ultrasound phenotypes. Meanwhile, in order to investigate the candidate genes related to abnormal phenotypes, chromosomal microarray results for all reviewed cases should be guaranteed. A string of the following terms and their synonyms were used: 1q21.1 deletion/loss, 1q21.1 duplication/gain, prenatal diagnosis, chromosomal microarray analysis, and ultrasound findings. The combination of subject words and free words was also used for the search. The information collected included the general condition of the subjects (age, gravida and para, gestational age, and parental phenotypes), karyotype, inheritance, microarray results, indications for prenatal diagnosis (including ultrasound findings), and pregnancy outcomes.
2.5. Follow-up outcomes
The follow-up was mainly carried out through telephone interviews using a customized questionnaire by our center's follow-up staff after all the women had given birth. The specific follow-up contents contained pregnancy outcomes (miscarriages or birth), gestational ages of delivery, sex and birth weight/length of the neonate, ultrasound findings during the pregnancy period (nervous system, cardiovascular system, craniofacial growth, respiratory system, abdominal abnormalities, urinary system, alimentary system, musculoskeletal system, and others), and postnatal health conditions (congenital defects, craniofacial dysmorphisms, skeletal anomalies, and developmental details).
3. Results
3.1. Study population
Of 8,252 pregnant women opting for prenatal invasive testing in our center, 11 fetuses were identified with 1q21.1 microdeletions and 15 were diagnosed with 1q21.1 microduplications. The detection rate of CNVs involving the chromosomal 1q21.1 region was 0.31% (26/8,252). Tables 1, 2 summarize the clinical information, including gestational week at detection, indications for prenatal diagnosis, CMA results, maternal inheritance, and pregnancy outcomes for all the cases.
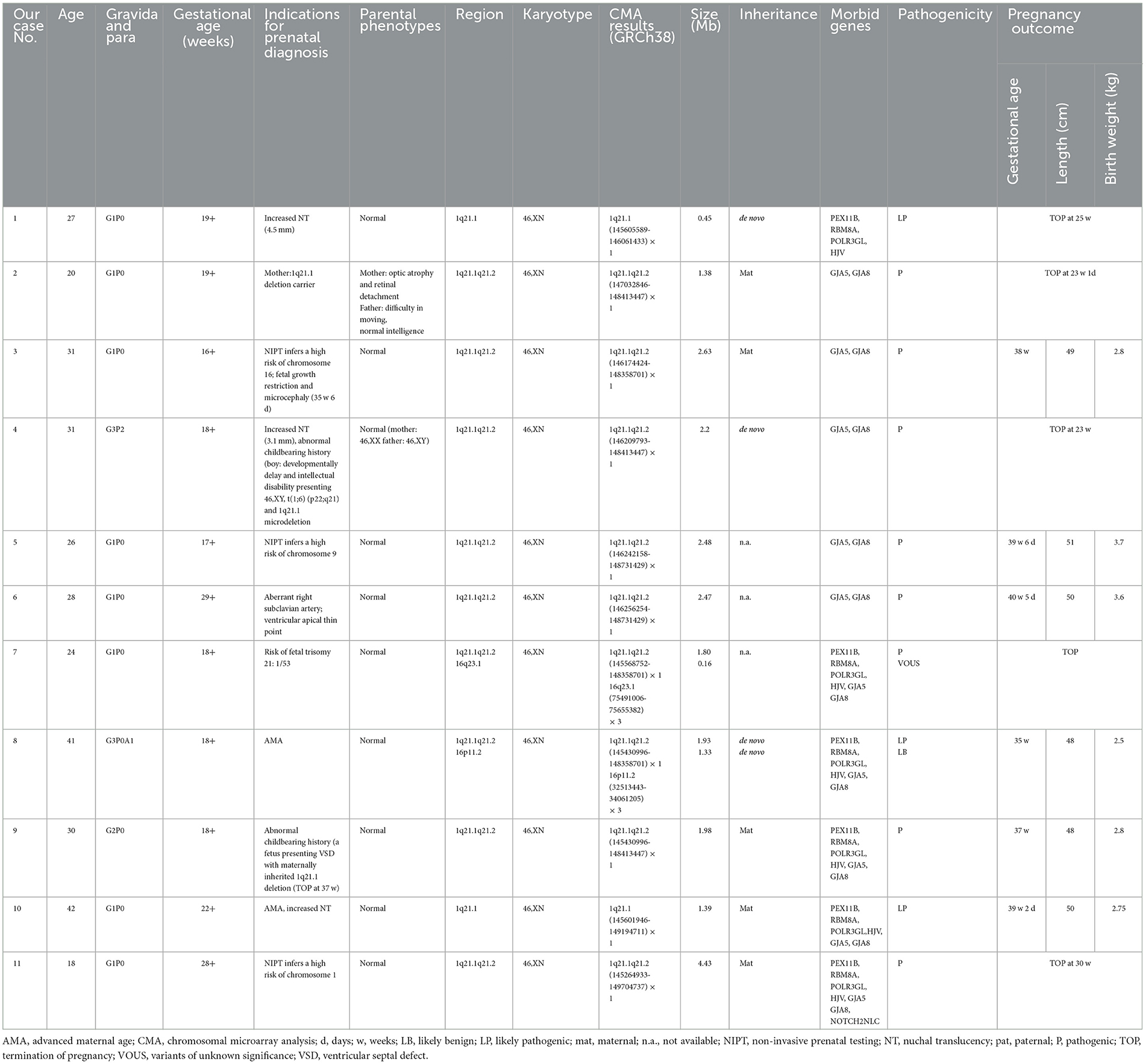
Table 1. Summary of clinical and molecular findings of fetuses presenting 1q21.1 microdeletion detected by CMA.
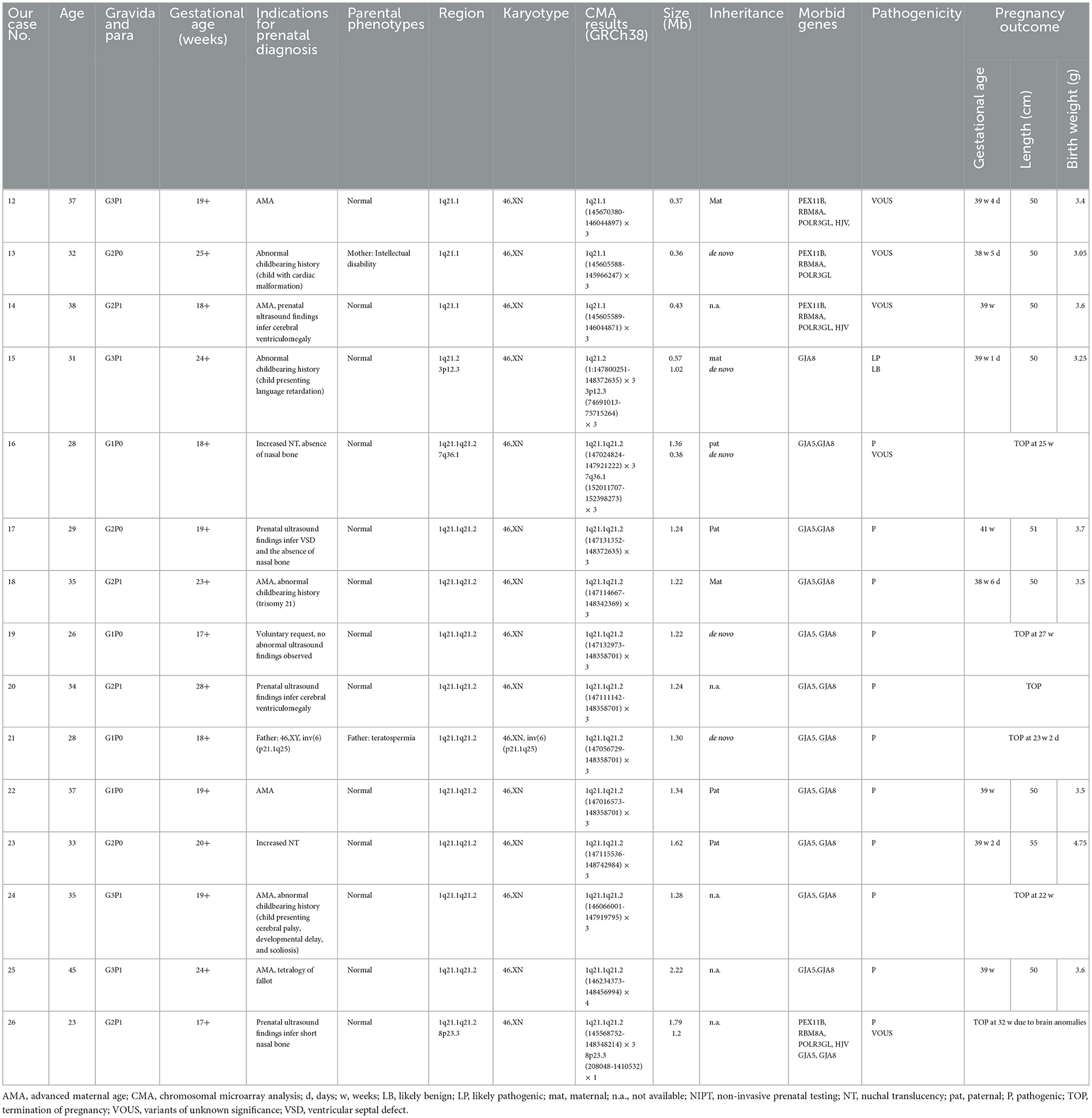
Table 2. Summary of clinical and molecular findings of fetuses presenting 1q21.1 microduplication detected by CMA.
3.2. Chromosomal anomalies detected by karyotyping
Amniotic fluid samples from all subjects were subjected to conventional karyotyping to identify balanced chromosomal rearrangements that could not be detected through CMA. Of the 11 1q21.1 microdeletions, no karyotypic anomalies were observed. Among the 15 1q21.1 microduplications, the karyotype of case 21 was 46,XN,inv(6) (p21.1q25).
3.3. Chromosome 1q21.1 microdeletions in affected fetuses
In our report, 11 cases (0.13%, 11/8,252) with 1q21.1 microdeletions were identified by CMA, ranging from 0.45 Mb to 4.43 Mb (Table 1). In addition, CMA detected a 0.16 Mb duplication of 16q23.1 in case 6 and a 1.33 Mb duplication of 16p11.2 in case 8, the clinic pathogenicity of which was VOUS and LB, respectively. The distribution of indications for prenatal diagnosis was as follows: non-structural anomalies (5/11), NIPT inferring aneuploidy (3/11), abnormal childbearing history (2/11), advanced maternal age (2/11), risk of Down syndrome (1/11), and maternal abnormal karyotype (1/11). The abnormal ultrasound findings were recorded in five participants with 1q21.1 deletions, three of whom presented increased nuchal translucency (NT). Among the 5 of 11 cases with maternal inheritance, only the mother of case 2 presented optic atrophy and retinal detachment. Three of the 11 cases were de novo, and 3 of the 11 cases were not available. One case (case 1) encompassed the TAR region, five cases (cases 2–6) encompassed 1q21.1 recurrent microdeletion, and five cases (cases 7–11) covered both the TAR region and 1q21.1 recurrent microdeletion in common (Figure 1B).
3.4. Chromosome 1q21.1 microduplications in affected fetuses
CMA successfully identified 15 fetuses (0.18%, 15/8,252) with 1q21.1 microduplications ranging from 0.36 Mb to 1.79 Mb (Table 2). In addition, CMA detected a 0.57 Mb duplication of 3p12.3 in case 15, a 0.38 Mb duplication of 7q36.1 in case 16, and a 1.2 Mb deletion of 8p23.2 in case 26, with LB, VOUS, and VOUS clinic pathogenicity, respectively. The distribution of indications for prenatal diagnosis was as follows: advanced maternal age (6/15), nasal bone dysplasia (3/15), cerebral ventriculomegaly (2/15), cardiac malformation (2/15), increased NT (2/15), abnormal childbearing history (4/15), voluntary request (1/15), and parental chromosome anomaly (1/15). Prenatal abnormal ultrasound findings were recorded in seven cases, in which cerebral ventriculomegaly, nasal bone dysplasia, and heart malformations were observed. Among them, 7 of 15 cases were parentally inherited, 3 of 15 cases were de novo, and 5 of 15 cases were unavailable. Three cases (cases 12–14) encompassed the TAR locus, 11 cases (cases 15–25) shared 1q21.1 recurrent microduplication, and 1 case (case 26) covered the TAR region and 1q21.1 recurrent microduplication (Figure 2B).
3.5. Prenatal and postnatal follow-up assessment
Of the 11 1q21.1 deletion cases, five eventually terminated their pregnancies: two (cases 1 and 4) were de novo, two (cases 2 and 11) were maternally inherited, and one (case 7) was unavailable. Among the six cases that continued the pregnancies, three were maternal inheritance, one was de novo, and two were unavailable. It was noteworthy that the two pregnancies of case 9 carried the maternally inherited 1q21.1 deletion. However, the first pregnancy presented a ventricular septal defect (VSD), while no ultrasound findings were discovered for her second pregnancy, so the woman continued the pregnancy and delivered a healthy child at term. In the 15 1q21.1 duplication cases, 6 opted for the termination of pregnancy (TOP): 2 cases (cases 19 and 21) were de novo, 1 (case 16) was paternally inherited, and 3 (cases 20, 24, and 26) were unavailable. Among the nine cases opting for ongoing pregnancies, six were parentally inherited, one was de novo, and two were unavailable. The clinic pathogenicity of the duplicated TAR region was VOUS, which was probably the reason for ongoing pregnancies in cases 10–12.
We followed up on all neonates with 1q21.1 microdeletions and microduplications after birth, including congenital defects, craniofacial dysmorphisms, and skeletal anomalies developmental details. Overall, they were in healthy states, with no evident anomalies observed up until the writing of this article. However, since all subjects were of a young age, long-term follow-up should be guaranteed for them.
4. Discussion
In our study, we systematically described 26 prenatal cases referred to our center for prenatal invasive testing and found recurrent chromosomal 1q21.1 rearrangements. Among them, chromosomal 1q21.1 microdeletions were detected in 11 cases, five of which eventually chose TOP. Chromosomal 1q21.1 microduplications were identified in 15 cases, and six opted for TOP. Compared with postnatal phenotypes, prenatal phenotypes involving 1q21.1 deletions/duplications were limited in the clinic. To the best of our knowledge, this is the largest cohort study with a detailed follow-up for prenatally diagnosed CNVs at the 1q21.1 locus in China.
CMA has been adopted as an effective diagnostic tool in identifying new microdeletion and microduplication syndromes, such as Williams–Beuren syndrome, 17q21.31 microdeletion syndrome, Prader–Willi syndrome, and Angelman syndrome. As a hot spot region, CNVs at the 1q21 locus were frequently reported in postnatal settings and in populations with intellectual disabilities, developmental delays, schizophrenia, and autism (12). In a study involving 5,218 persons with idiopathic intellectual disabilities, autism, or congenital anomalies, Mefford et al. identified 25 unrelated probands with 1q21.1 deletions (0.5%) and nine persons with 1q21.1 duplications (0.2%), with no 1q21.1 microdeletions and only one microduplication found in 4,737 controls (13). Brunetti-Pierri et al. described 21 probands with 1q21.1 microdeletions and 15 probands with 1q21.1 microduplications in 16,557 affected individuals presenting intellectual disabilities, autism, and/or congenital anomalies, with detection frequencies of 0.13 and 0.09%, respectively (14). However, the detection rates of CNVs at the 1q21.1 locus in a prenatal setting were rarely described. It was reported that the frequencies of chromosomal 1q21.1 deletions/duplications with and without fetal anomalies were 4.9 and 9.6%, respectively (15, 16). In our 8,252 prenatal cases referred for genetic microarray testing, the detection rates of 1q21.1 microdeletions and microduplications were 0.13 and 0.18%, respectively.
As one of the most commonly detected structural aberrations, individuals carrying chromosomal 1q21.1 CNVs could exhibit diverse phenotypes, including intellectual disabilities, autism, schizophrenia, congenital anomalies, dysmorphic features, or normal phenotypes (17). Till now, most research involving 1q21.1 CNVs focused on postnatal cases, and the prenatal genotype–phenotype correlation was still unclear due to inadequate reports in the clinic. Considering the phenotypic diversity, the incomplete penetrance, and the lack of distinct prenatal features for 1q21.1 CNVs, it is challenging to offer genetic counseling for such prenatal cases. Hence, to provide a better understanding of 1q21.1 CNVs in the prenatal setting, we summarized the clinical data and molecular findings of prenatally detected 1q21.1 microdeletions and microduplications in the published literature (Tables 3, 4, Figures 1C, 2C).
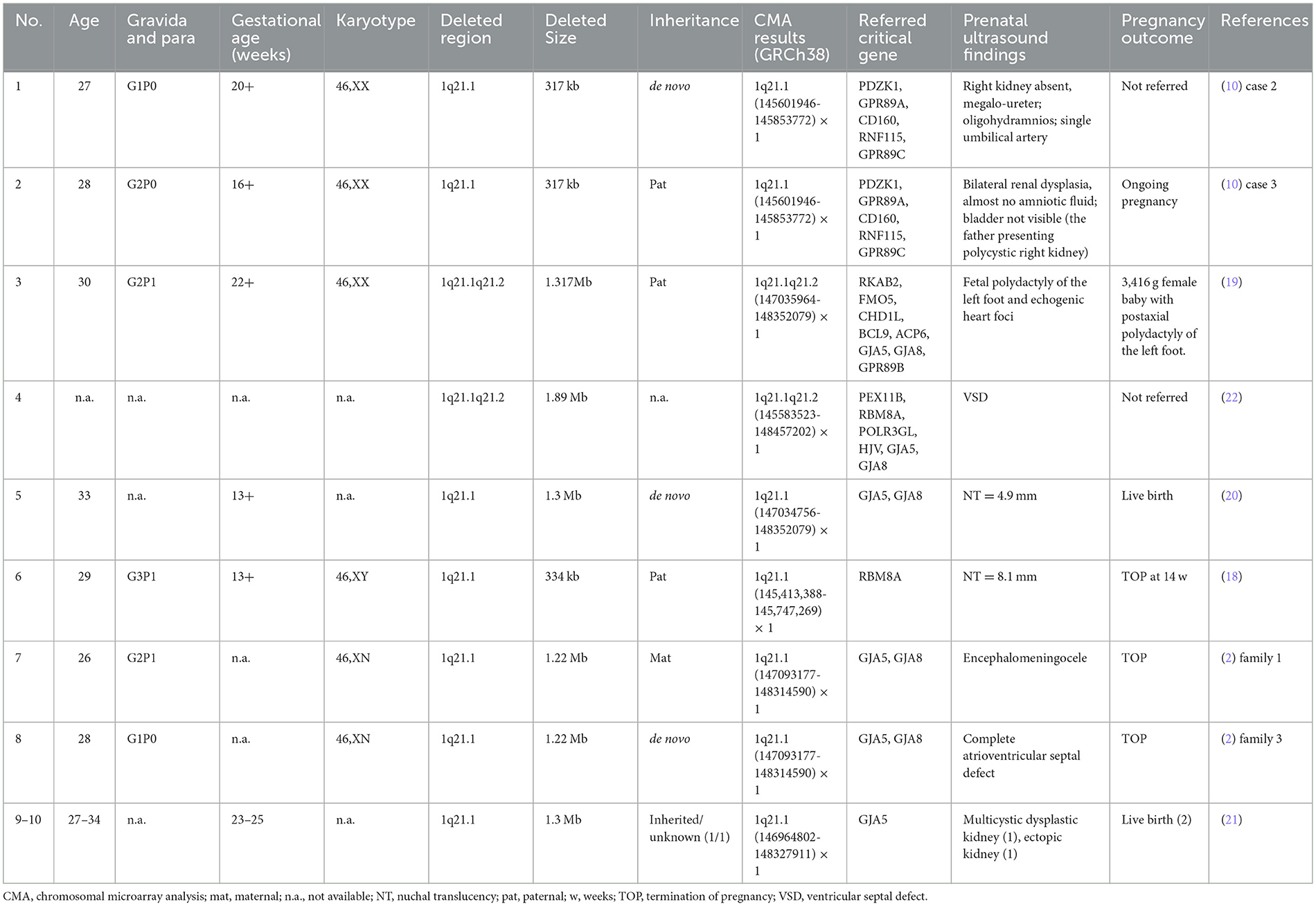
Table 3. Clinical data of fetuses presenting 1q21.1 microdeletion detected by CMA in the published literature.
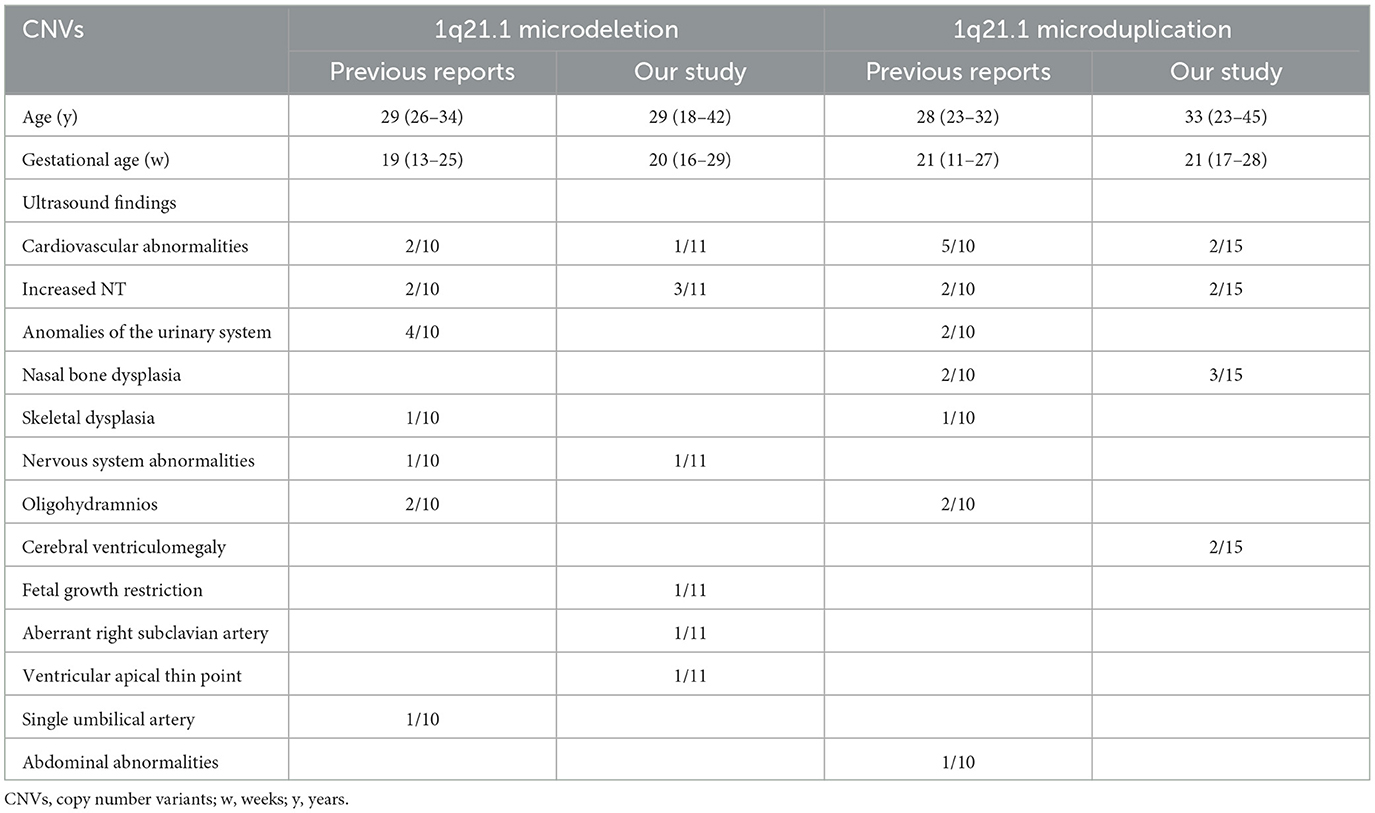
Table 4. Pooled data from all fetuses presenting 1q21.1 microdeletion and microduplication.
In Table 3, 10 prenatally detected 1q12.1 microdeletion cases with detailed microarray results and ultrasound findings were collected (Figure 1C) (2, 10, 18–22). The gestational age was between 13 and 25 weeks. All cases varied in size and ranged from 317 kb to 1.89 Mb: class I deletion (5/10), class II deletion (2/10), TAR syndrome (1/10), and atypical 1q21.1 deletion (2/10). Among these deletions, 5 of 10 were parentally inherited, 3 of 10 were de novo, and 2 of 10 cases were not available. Pregnancy outcomes were as follows: five gave birth to a child or opted for ongoing pregnancy, three chose TOP, and two were unavailable. The pooled data from all fetuses presenting 1q21.1 microdeletion in the published literature and our study are listed in Table 4. Maternal age at diagnosis ranged from 26 to 34 years in the published literature and ranged from 18 to 42 years in our cases, with a mean age of 29 years. The gestational age at diagnosis ranged from 13 to 25 weeks in the published cases and from 16 to 29 weeks in our cases, with the mean gestational age of 19 and 20 weeks, respectively. The summarized frequencies of abnormal prenatal phenotypes in the literature and our study were as follows: increased NT (5/21), anomalies of the urinary system (4/21), cardiovascular abnormalities (3/21), nervous system abnormalities (2/21), oligohydramnios (2/21), skeletal dysplasia (1/21), fetal growth restriction (1/21), aberrant right subclavian artery (1/21), ventricular apical thin point (1/21), and single umbilical artery (1/21). Based on the findings mentioned above, we assumed that 1q21.1 deletions were closely associated with increased NT, anomalies of the urinary system, and cardiovascular abnormalities in prenatal ultrasound findings. It was noteworthy that the asymptomatic pregnant woman in our case 9 transmitted the 1q21.1 microdeletion to her two pregnancies. However, the first fetus showed VSD, while the second fetus showed no anomalies in ultrasonography, which indicated the incomplete penetrance of the 1q21.1 deletion in the prenatal setting. Meanwhile, diverse prenatal phenotypes of 1q21.1 deletions may be presented in the same family. In addition, TAR syndrome could be prenatally diagnosed by abnormal examination, mainly including bilateral radial hypoplasia/agenesis with or without humeral shortness and the presence of thumbs on both hands (23). No limb anomalies were observed except for increased NT in prenatal ultrasound findings for our case 1, which might indicate the phenotypic diversity of TAR syndrome in the prenatal setting. However, whether TAR syndrome should be added to the long list of genetic syndromes associated with increased NT should require more clinical evidence.
In Table 5, 10 prenatal 1q21.1 microduplication cases with detailed microarray results and ultrasound findings are reviewed (Figure 2C) (1, 6, 10, 15, 24, 25). The gestational age was between 11 and 27 weeks. The duplicated size ranged from 258 kb to 2.7 Mb: class I duplication (6/10), class II duplication (1/10), and 1q21.1 duplication involving the TAR region (3/10). Among these duplications, 5 of 10 were maternally inherited, 4 of 10 were de novo, and 1 of 10 were unavailable. Four cases opted for TOP, and the pregnancy outcomes of the remaining cases were unavailable. The pooled data from all fetuses presenting 1q21.1 microduplication in the published literature and our study are listed in Table 4. Maternal age at diagnosis ranged from 23 to 32 years in the published literature and from 23 to 45 years in our cases, with mean ages of 28 and 33 years, respectively. The gestational age at diagnosis ranged from 11 to 27 weeks in the published cases and ranged from 17 to 28 weeks in our cases, with a mean gestational age of 21 weeks. The summarized frequencies of abnormal prenatal phenotypes in the literature and our study were as follows: cardiovascular abnormalities (7/25), nasal bone dysplasia (5/25), increased NT (4/25), anomalies of the urinary system (2/25), cerebral ventriculomegaly (2/25), oligohydramnios (2/25), skeletal dysplasia (1/25), and abdominal abnormalities (1/25). In addition, Ji et al. inferred that nasal bone loss might be related to 1q21.1 duplication (1). Based on the findings mentioned above, we assumed that 1q21.1 duplications were closely correlated with cardiovascular abnormalities, nasal bone dysplasia, and increased NT in prenatal settings. In addition, cerebral ventriculomegaly observed in our study was not reported in prenatally detected 1q21.1 microduplication before, which might be associated with 1q21.1 duplication, but more evidence should be collected.
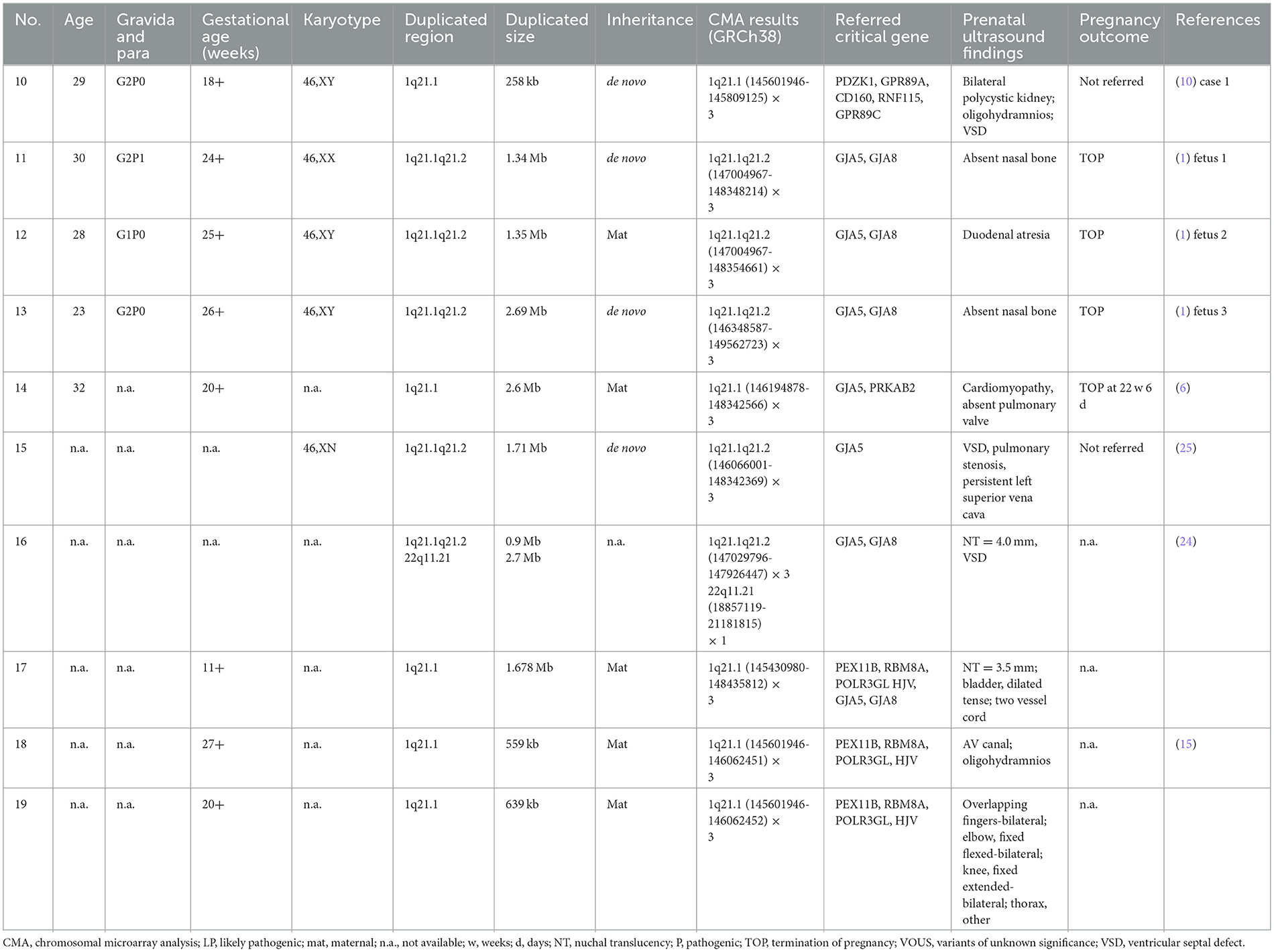
Table 5. Clinical data of fetuses presenting 1q21.1 microduplication detected by CMA in the published literature.
1q21.1 recurrent microduplication/microdeletion shares the same coordinates within the 1q21.1 region. Of the 26 fetuses in our study, 22 cases (10 1q21.1 deletions and 12 1q21.1 duplications) covered partial or the whole 1q21.1 recurrent microduplication/microdeletion region, in which 9 OMIM genes (PRKAB2, FMO5, CHD1L, BCL9, ACP6, GJA5, GJA8, GPR89B, and NBPF24) were located. Among them, the GJA5 and GJA8 genes are morbid genes associated with clinical diseases. As a critical candidate gene for the cardiac phenotype, GJA5 encodes the gap junction protein connexin 40 (CX40), and its heterozygous mutations would lead to familial atrial fibrillation 11 (OMIM 614049) and atrial standstill (OMIM 108770) (3). GJA5 might be responsible for the phenotypic specificity in congenital heart disease (CHD) resulting from 1q21.1 CNVs (26, 27). Its flanking gene, GJA8 (OMIM 600897), encodes the gap junction protein connexin 50 (CX50), and its heterozygous mutation would result in autosomal dominant cataract 1, multiple types (OMIM 116200) (28). The abnormal expression of GJA5 and GJA8 has been closely associated with CHD (10). Cases 17, 25, and the first pregnancy of case 9 in our study presented VSD, which might be attributed to the losses and gains of GJA5 and GJA8 genes. The pooled data from all reviewed studies indicate that the losses and gains of the GJA5 and GJA8 genes were probably the most common genetic causes associated with 1q21.1 deletions or duplications. The PRKAB2 gene, highly expressed in the right ventricular outflow tract and skeletal muscles, has been associated with schizophrenia (29–31). The CHD1L gene, implicated in chromatin remodeling, relaxation, and decatenation, plays a role in DNA damage response. Overexpression of CHD1L was discovered in the tetralogy of Fallot (TOF), double-outlet right ventricle, and infundibular pulmonary stenosis (32). In addition, CHD1L is regarded as a candidate gene for autism, ADHD, and congenital anomalies of the kidney and urinary tract (19, 31). Our case 25 presented TOF, which might be correlated with CHD1L to some degree, but the correlation between CHD1L and TOF still needs further investigation. The BCL9 gene is proposed as a candidate gene for schizophrenia (33). Meanwhile, it is also involved in language deficits in patients with the 1q21.1 duplication (31). With current knowledge, there is not enough evidence to show that the functions and effects of the remaining OMIM genes have close correlations with the abnormal phenotypes observed in the prenatal setting.
Four fetuses carrying 1q21.1 deletions (case 1) and 1q21.1 duplications (cases 12-14) involved the TAR region. A total of 10 OMIM genes are located in the TAR region, including CD160, RNF115, POLR3C, PIAS3, ITGA10, PEX11B, RBM8A, POLR3GL, TXNIP, and HJV. The RBM8A gene encodes the exon-junction complex subunit member Y14, one of the four components of the exon-junction splicing complex, which plays a critical role in cellular functions (34). The first study revealing the correlation between heterozygous 1q21.1 microdeletion involving the RBM8A gene and TAR syndrome was reported in 2007 (35). Along with further study on TAR syndrome, it has been observed that 1q21.1 deletion is regarded as necessary but not sufficient to result in TAR syndrome. Other genetic alterations could be involved in the process. Most patients with TAR syndrome had compound heterozygous mutations, including a proximal 1q21.1 deletion spanning at least 200 kb involving the RBM8A gene and one of the two low-frequency SNPs in regulatory regions of the RBM8A gene (18). There is currently insufficient evidence that other genes are associated with TAR syndrome and the prenatal phenotypes observed in our study.
There are some limitations to our study. First, the enrolled subjects with 1q21.1 deletions and duplications were collected in a single center, and the total number was relatively small. In order to establish a more clear correlation between 1q21.1 deletions/duplications and prenatal phenotypes, multi-center collaboration should be adopted to enlarge the sample size in the future. Second, the current follow-up outcomes were acquired after birth, and all participants were still young. Although all subjects with 1q21.1 deletions and duplications were in a healthy state with no obvious abnormalities observed until the writing of this article, it is difficult to predict whether abnormal phenotypes will appear in the future. Thus, long-term follow-up should be guaranteed, including for autism, intellectual disability, ADHD, hearing impairments, seizures, cardiac disease, and motor difficulties (12). In addition, some cases carried VOUS CNVs in addition to 1q21.1 microdeletions or duplications, and whether these CNVs would have potential impacts still needed further investigation.
5. Conclusion
In conclusion, we described the clinical data and molecular findings in 26 prenatal cases aiming to investigate the correlation between 1q21.1 CNVs and prenatal phenotypes. Our study mainly focused on the prenatal phenotypes of 1q21.1 CNVs based on our findings and the published cases. In the prenatal setting, 1q21.1 microdeletions were associated with increased nuchal translucency (NT), anomalies of the urinary system, and cardiovascular abnormalities, while 1q21.1 microduplications were correlated with cardiovascular malformations, nasal bone dysplasia, and increased NT. In addition, cerebral ventriculomegaly might be correlated with 1q21.1 microduplications. More relevant studies are necessary for a better understanding of the prenatal phenotype–genotype correlation of 1q21.1 CNVs. For postnatal cases with 1q21.1 CNVs, regardless of whether ultrasound anomalies were observed or not during the pregnancy period, regular follow-up should be guaranteed till adulthood in case abnormal developmental behaviors may emerge.
Data availability statement
The data presented in the study are deposited in the Gene Expression Omnibus repository, accession number GSE240611.
Ethics statement
The study was approved by the Ethics Committee of the First Hospital of Jilin University. Written informed consent to participate was obtained for all the couples before collecting samples of amniotic fluid.
Author contributions
FY obtained the clinical information, collected data from the literature, and wrote the manuscript. XY, YJ, and HZ performed the cytogenetic study and chromosomal microarray analysis on the amino fluid samples. SL, RL, and HZ conceived and designed the study, performed the final review, and editing of the manuscript. All authors have read and approved the final manuscript.
Funding
This study was supported by the Science and Technology Department of Jilin Province, China (grant number: 20210101302JC).
Acknowledgments
We thank all the children and their respective families for participating in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ji X, Pan Q, Wang Y, Wu Y, Zhou J, Liu A, et al. Prenatal diagnosis of recurrent distal 1q211 duplication in three fetuses with ultrasound anomalies. Front Genet. (2018) 9:275. doi: 10.3389/fgene.2018.00275
2. Wang HD, Liu L, Wu D, Li T, Cui CY, Zhang LZ, et al. Clinical and molecular cytogenetic analyses of four families with 1q21.1 microdeletion or microduplication. J Gene Med. (2017) 19:2948. doi: 10.1002/jgm.2948
3. Dolcetti A, Silversides CK, Marshall CR, Lionel AC, Stavropoulos DJ, Scherer SW, et al. 1q211 Microduplication expression in adults. Genet Med. (2013) 15:282–9. doi: 10.1038/gim.2012.129
4. Pang H, Yu X, Kim YM, Wang X, Jinkins JK, Yin J, et al. Disorders associated with diverse, recurrent deletions and duplications at 1q211. Front Genet. (2020) 11:577. doi: 10.3389/fgene.2020.00577
5. Van Dijck A, van der Werf IM, Reyniers E, Scheers S, Azage M, Siefkas K, et al. Five patients with a chromosome 1q211 triplication show macrocephaly, increased weight and facial similarities. Eur J Med Genet. (2015) 58:503–8. doi: 10.1016/j.ejmg.2015.08.004
6. Verhagen JM, de Leeuw N, Papatsonis DN, Grijseels EW, de Krijger RR, Wessels MW. Phenotypic variability associated with a large recurrent 1q211 microduplication in a three-generation family. Mol Syndromol. (2015) 6:71–6. doi: 10.1159/000431274
7. Busè M, Cuttaia HC, Palazzo D, Mazara MV, Lauricella SA, Malacarne M, et al. Expanding the phenotype of reciprocal 1q211 deletions and duplications: a case series. Ital J Pediatr. (2017) 43:61. doi: 10.1186/s13052-017-0380-x
8. Albers CA, Newbury-Ecob R, Ouwehand WH, Ghevaert C. New insights into the genetic basis of TAR (thrombocytopenia-absent radii) syndrome. Curr Opin Genet Dev. (2013) 23:316–23. doi: 10.1016/j.gde.2013.02.015
9. Greenhalgh KL, Howell RT, Bottani A, Ancliff PJ, Brunner HG, Verschuuren-Bemelmans CC, et al. Thrombocytopenia-absent radius syndrome: a clinical genetic study. J Med Genet. (2002) 39:876–81. doi: 10.1136/jmg.39.12.876
10. Liao C, Fu F, Yi CX, Li R, Yang X, Xu Q, et al. Prenatal diagnosis of an atypical 1q211 microdeletion and duplication associated with foetal urogenital abnormalities. Gene. (2012) 507:92–4. doi: 10.1016/j.gene.2012.07.008
11. Zhang H, Yue F, Zhang X, He J, Jiang Y, Liu R, et al. Prenatal detection of distal 1q211q212 microduplication with abnormal ultrasound findings: two cases report and literature review. Medicine. (2021) 100:e24227. doi: 10.1097/MD.0000000000024227
12. Bernier R, Steinman KJ, Reilly B, Wallace AS, Sherr EH, Pojman N, et al. Clinical phenotype of the recurrent 1q211 copy-number variant. Genet Med. (2016) 18:341–9. doi: 10.1038/gim.2015.78
13. Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, et al. Recurrent rearrangements of chromosome 1q211 and variable pediatric phenotypes. N Engl J Med. (2008) 359:1685–99. doi: 10.1056/NEJMoa0805384
14. Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, et al. Recurrent reciprocal 1q211 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. (2008) 40:1466–71. doi: 10.1038/ng.279
15. Donnelly JC, Platt LD, Rebarber A, Zachary J, Grobman WA, Wapner RJ. Association of copy number variants with specific ultrasonographically detected fetal anomalies. Obstet Gynecol. (2014) 124:83–90. doi: 10.1097/AOG.0000000000000336
16. Levy B, Wapner R. Prenatal diagnosis by chromosomal microarray analysis. Fertil Steril. (2018) 109:201–12. doi: 10.1016/j.fertnstert.2018.01.005
17. Harvard C, Strong E, Mercier E, Colnaghi R, Alcantara D, Chow E, et al. Understanding the impact of 1q211 copy number variant. Orphanet J Rare Dis. (2011) 6:54. doi: 10.1186/1750-1172-6-54
18. Papoulidis I, Oikonomidou E, Orru S, Siomou E, Kontodiou M, Eleftheriades M, et al. Prenatal detection of TAR syndrome in a fetus with compound inheritance of an RBM8A SNP and a 334-kb deletion: a case report. Mol Med Rep. (2014) 9:163–5. doi: 10.3892/mmr.2013.1788
19. Chen CP, Chang SY, Chen YN, Chern SR, Wu PS, Chen SW, et al. Prenatal diagnosis of a familial 1q211-q212 microdeletion in a fetus with polydactyly of left foot on prenatal ultrasound. Taiwan J Obstet Gynecol. (2018) 57:739–44. doi: 10.1016/j.tjog.2018.08.024
20. Egloff M, Hervé B, Quibel T, Jaillard S, Le Bouar G, Uguen K, et al. Diagnostic yield of chromosomal microarray analysis in fetuses with isolated increased nuchal translucency: a French multicenter study. Ultrasound Obstet Gynecol. (2018) 52:715–21. doi: 10.1002/uog.18928
21. Su J, Qin Z, Fu H, Luo J, Huang Y, Huang P, et al. Association of prenatal renal ultrasound abnormalities with pathogenic copy number variants in a large Chinese cohort. Ultrasound Obstet Gynecol. (2022) 59:226–33. doi: 10.1002/uog.23702
22. Zhu X, Li J, Ru T, Wang Y, Xu Y, Yang Y, et al. Identification of copy number variations associated with congenital heart disease by chromosomal microarray analysis and next-generation sequencing. Prenat Diagn. (2016) 36:321–7. doi: 10.1002/pd.4782
23. Bottillo I, Castori M, De Bernardo C, Fabbri R, Grammatico B, Preziosi N, et al. Prenatal diagnosis and post-mortem examination in a fetus with thrombocytopenia-absent radius (TAR) syndrome due to compound heterozygosity for a 1q211 microdeletion and a RBM8A hypomorphic allele: a case report. BMC Res Notes. (2013) 6:376. doi: 10.1186/1756-0500-6-376
24. Maya I, Kahana S, Yeshaya J, Tenne T, Yacobson S, Agmon-Fishman I, et al. Chromosomal microarray analysis in fetuses with aberrant right subclavian artery. Ultrasound Obstet Gynecol. (2017) 49:337–41. doi: 10.1002/uog.15935
25. Fu F, Deng Q, Lei TY, Li R, Jing XY, Yang X, et al. Clinical application of SNP array analysis in fetuses with ventricular septal defects and normal karyotypes. Arch Gynecol Obstet. (2017) 296:929–40. doi: 10.1007/s00404-017-4518-2
26. Soemedi R, Topf A, Wilson IJ, Darlay R, Rahman T, Glen E, et al. Phenotype-specific effect of chromosome 1q21.1 rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Hum Mol Genet. (2012) 21:1513–20. doi: 10.1093/hmg/ddr589
27. Guida V, Ferese R, Rocchetti M, Bonetti M, Sarkozy A, Cecchetti S, et al. A variant in the carboxyl-terminus of connexin 40 alters GAP junctions and increases risk for tetralogy of Fallot. Eur J Hum Genet. (2013) 21:69–75. doi: 10.1038/ejhg.2012.109
28. Arora A, Minogue PJ, Liu X, Addison PK, Russel-Eggitt I, Webster AR, et al. A novel connexin50 mutation associated with congenital nuclear pulverulent cataracts. J Med Genet. (2008) 45:155–60. doi: 10.1136/jmg.2007.051029
29. Oliveira SM, Ehtisham J, Redwood CS, Ostman-Smith I, Blair EM, Watkins H. Mutation analysis of AMP-activated protein kinase subunits in inherited cardiomyopathies: implications for kinase function and disease pathogenesis. J Mol Cell Cardiol. (2003) 35:1251–5. doi: 10.1016/S0022-282800237-2
30. Thornton C, Snowden MA, Carling D. Identification of a novel AMP-activated protein kinase beta subunit isoform that is highly expressed in skeletal muscle. J Biol Chem. (1998) 273:12443–50. doi: 10.1074/jbc.273.20.12443
31. Benítez-Burraco A, Barcos-Martínez M, Espejo-Portero I, Fernández-Urquiza M, Torres-Ruiz R, Rodríguez-Perales S, et al. Narrowing the genetic causes of language dysfunction in the 1q211 microduplication syndrome. Front Pediatr. (2018) 6:163. doi: 10.3389/fped.2018.00163
32. Morano M, Zacharzowski U, Maier M, Lange PE, Alexi-Meskishvili V, Haase H, et al. Regulation of human heart contractility by essential myosin light chain isoforms. J Clin Invest. (1996) 98:467–73. doi: 10.1172/JCI118813
33. Uhrig S, Schlembach D, Waldispuehl-Geigl J, Schaffer W, Geigl J, Klopocki E, et al. Impact of array comparative genomic hybridization-derived information on genetic counseling demonstrated by prenatal diagnosis of the TAR (thrombocytopenia-absent-radius) syndrome-associated microdeletion 1q211. Am J Hum Genet. (2007) 81:866–8. doi: 10.1086/521338
34. Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet. (2012) 44:435–9. doi: 10.1038/ng.1083
Keywords: chromosomal 1q21.1 microdeletions and microduplications, chromosomal microarray analysis, prenatal phenotypes, pregnancy outcomes, cerebral ventriculomegaly
Citation: Yue F, Yang X, Jiang Y, Li S, Liu R and Zhang H (2023) Prenatal phenotypes and pregnancy outcomes of fetuses with recurrent 1q21.1 microdeletions and microduplications. Front. Med. 10:1207891. doi: 10.3389/fmed.2023.1207891
Received: 18 April 2023; Accepted: 19 July 2023;
Published: 24 August 2023.
Edited by:
Raigam Jafet Martinez-Portilla, Instituto Nacional de Perinatología (INPER), MexicoReviewed by:
Tao Wang, Sichuan University, ChinaSimona Boito, IRCCS Ca 'Granda Foundation Maggiore Policlinico Hospital, Italy
Copyright © 2023 Yue, Yang, Jiang, Li, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongguo Zhang, emhhbmdoZ3VvQGpsdS5lZHUuY24=