 Brendan Antiochos*
Brendan Antiochos* Livia Casciola-Rosen
Livia Casciola-Rosen- Division of Rheumatology, Johns Hopkins University, Baltimore, MD, United States
Interferon (IFN) is a key component of the innate immune response. For reasons that remain incompletely understood, the IFN system is upregulated in several rheumatic diseases, particularly those that feature autoantibody production, such as SLE, Sjögren’s syndrome, myositis and systemic sclerosis. Interestingly, many of the autoantigens targeted in these diseases are components of the IFN system, representing IFN-stimulated genes (ISGs), pattern recognition receptors (PRRs), and modulators of the IFN response. In this review, we describe features of these IFN-linked proteins that may underlie their status as autoantigens. Note is also made of anti-IFN autoantibodies that have been described in immunodeficiency states.
Introduction
Autoantibodies arise in a wide array of immune-mediated diseases, including both organ-limited and systemic forms of autoimmunity (1, 2). Some autoantigens are organ-specific molecules that are expressed preferentially or even uniquely in the affected organ [e.g., thyroid-specific proteins in autoimmune thyroid disease (3)]. In contrast, antigens targeted in systemic autoimmune rheumatic diseases are frequently ubiquitously expressed, and perform a variety of essential cellular functions (2). The antigens most commonly targeted by antibodies in systemic rheumatic diseases are nuclear antigens, including proteins, nucleic acids, and nucleoprotein complexes (2). The mechanisms responsible for targeting these broadly distributed autoantigens are incompletely characterized, but are likely numerous and overlapping. Here, we will review the relationship between autoantibodies and the IFN system, highlighting the enrichment of IFN-linked antigens in systemic autoimmune rheumatic diseases, and potential explanations for their targeting by autoantibodies.
The IFN system (Figure 1) is a molecular network that perform host defense functions. Three types of IFNs are found in humans: type I IFNs are expressed by and act on nearly every cell type, type II IFN is more specific for immune cells, and type III IFNs mainly act on epithelial and endothelial cells at mucosal surfaces (4–6). Cell-intrinsic IFN signaling constitutes a primordial layer of innate immunity, enabling resident tissue cells to recognize and respond to a variety of microbial pathogens and nonmicrobial threats. Thus IFN induction within activated cells leads to IFN signaling in neighboring cells via IFNs and second messengers, and in both cases ISG induction occurs. IFNs also perform important cell-extrinsic functions, and are able to shape the immune response by influencing the behavior of immune cells (5).
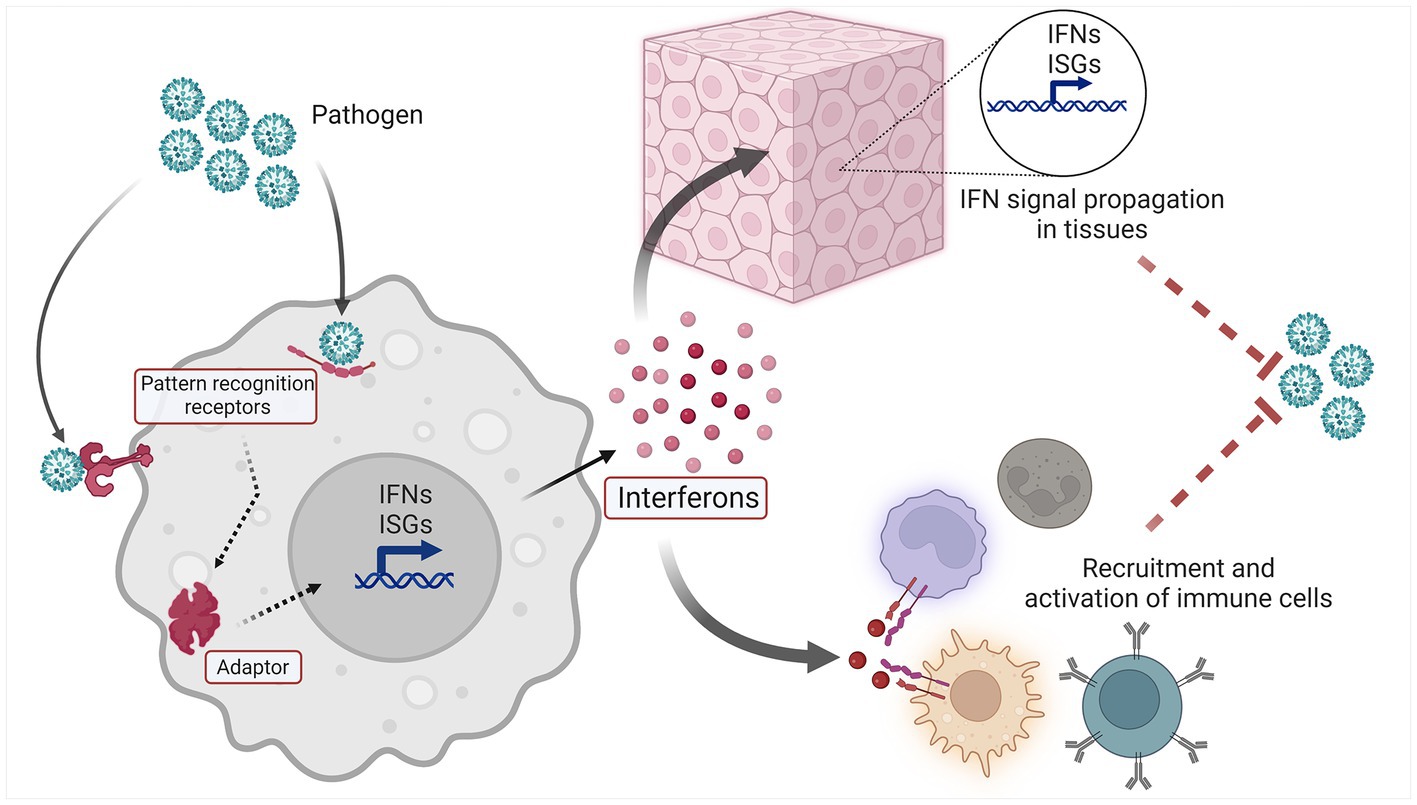
Figure 1. Schematic overview of the IFN system.
The upstream elements of the IFN system are innate pattern recognition receptors (PRRs), which recognize an array of Damage and Pathogen Associated Molecular Patterns (DAMPs and PAMPs, respectively) (7). Innate sensors are found in various compartments of the cell, including the endosome (e.g., TLR7), cell surface (e.g., TLR4), and cytoplasm [e.g., cyclic GMP-AMP synthase (cGAS) (8)]. The interaction of ligand and sensor leads to subsequent activation of downstream signaling adaptors, which include molecules such as stimulator of interferon genes (STING), mitochondrial antiviral-signaling protein (MAVS) and MyD88 (8). Activated adaptors then promote signaling through various kinases and transcription factors, which ultimately trigger the expression of Interferon Stimulated Genes (ISGs) and IFNs themselves. This expression of IFNs and ISGs results in both autocrine and paracrine cellular effects. Secreted IFNs bind their cognate receptors on the cell surface, leading to intracellular signaling via the JAK/STAT pathway and expression of ISGs and IFNs in IFN-activated cells (9). In this manner, the IFN system propagates a danger signal rapidly throughout an affected tissue or organ, readying resident parenchymal cells for host defense functions, and influencing the cellular immune response that follows.
Dysregulation of the IFN system has long been recognized as a feature of many autoimmune rheumatic diseases, most notably systemic lupus erythematosus (SLE), Sjögren’s syndrome (pSS), dermatomyositis (DM), and systemic sclerosis (SSc) (10–12). Upregulation of IFNs and ISGs has been observed both in the circulation and the target organs of patients with these diseases. In SLE, IFN I upregulation has been associated with increased markers of serologic and clinical disease activity. Interestingly, longitudinal studies have demonstrated that IFN expression is relatively stable despite changes in disease activity over time (13–17). In pSS, IFN expression has been linked to higher prevalence of autoantibodies and hypergammaglobulinemia, and increased lymphocytic infiltration of salivary tissues (18). Upregulation of IFN has been observed in many types of inflammatory myopathy, with IFN I upregulation particularly notable in DM muscle biopsies (19). Dysregulation of type I, II and III IFNs have all been observed, although the relative degree to which a specific IFN type is activated compared to others varies among individuals (19–23). In addition to the idiopathic rheumatic diseases, dysregulation of IFN has been identified as the driver of genetically-derived interferonopathy syndromes such as Aicardi–Goutières syndrome (AGS) and STING-associated vasculopathy with onset in infancy (SAVI); it is noteworthy that these genetic syndromes present with clinical features that often overlap with those of idiopathic rheumatic diseases (24). Taken together, these observations suggest that IFNs play a key role in the pathogenesis of the autoimmune rheumatic diseases. Consistent with this, therapeutic targeting of IFN has already shown promise in some patients, and is an area of ongoing research (25).
IFN-induced expression of autoantigens
An ISG is any gene whose expression is increased in response to IFN signaling; there are hundreds of such genes in human cells (26). Many autoantigens are included among the ISGs, suggesting that IFN-responsiveness may be involved in the development of this autoantibody subset. Notable among the IFN-induced autoantigens is Ro52 (encoded by the TRIM21 gene), which is targeted by autoantibodies in many rheumatic diseases, including pSS, SLE, DM, SSc and overlap syndromes (27). An important pathologic function of these antibodies has been defined - maternal anti-Ro52 antibodies demonstrate pathogenic function by mediating congenital heart block (28). In SLE, antibodies against Ro52 are associated with higher levels of circulating IFN I (29, 30). Ro52 is an IFN-induced E3 ligase that targets various substrates for removal via proteasomal degradation. In response to viral infection, Ro52 downregulates the innate immune response by enhancing clearance of the key transcription factor IRF3 (31, 32). Ro52 also promotes antiviral function by serving as a sensor of cytoplasmic IgG, marking intracellular viral-IgG immune complexes for proteasomal clearance (33, 34). Ro52 is therefore both a key regulator of the innate immune response and a functional component of the IFN pathway. It provides an important example of an antigen against which tolerance may be lost due to dysregulated IFN signaling. In this scenario, upregulated antigen expression in the setting of inflammation likely promotes the frequency with which the induced protein is displayed by antigen presenting cells, thereby increasing the likelihood that autoreactivity might occur. Continued expression of IFN in the affected organs would ensure sustained elevated levels of ISG antigens, fueling the propagation phase of such an autoimmune response. It is noteworthy that ISG upregulation caused by interferogenic stimuli (e.g., viral infection) facilitates additional intermolecular interactions that may also lead to breaking of tolerance against Ro52 or other relevant ISGs.
DNA binding molecules
Several PRRs are included among the autoantigens targeted in rheumatic diseases. While some of these PRR antigens are also IFN-inducible, others are not. Among these non-IFN-induced antigens is Ku - a heterodimeric complex composed of Ku70 and Ku80 subunits that is targeted by autoantibodies in several autoimmune rheumatic diseases (35, 36). In SLE, anti-Ku antibodies have been reported at prevalence of 9.8–20.5% (35, 37). Anti-chromatin antibodies have been identified at greater prevalence among SLE patients with anti-Ku antibodies: anti-chromatin antibodies were found in 72.7% of anti-Ku positive versus 43.9% of anti-Ku negative patient sera in one study (p < 0.0001) (37). Anti-Ku antibodies have also been found in association with autoantibodies against additional DNA repair proteins, including DNA-PK, Mre11, WRN and PARP (35). While clearly implicated in DNA repair responses, the Ku complex has also been shown to serve as a cytoplasmic DNA sensor, translocating from the nucleus to the cytoplasm and binding dsDNA of various sorts (38, 39). Recently, Tao et al. demonstrated that cytoplasmic Ku interacts with cGAS to promote condensate formation and IFN signaling in response to cytoplasmic dsDNA (40). These findings raise the intriguing possibility that intermolecular interactions occurring in the context of DNA repair in the nucleus might underlie the targeting of Ku and related autoantigens in SLE, and that Ku may interact with other potential autoantigens in the cytoplasm.
A similar scenario has been observed in the case of poly(ADP-ribose) polymerase (PARP), an additional component of the cellular DNA damage response that is targeted as an autoantigen in SLE and other autoimmune conditions (41, 42). PARP1 translocates to the cytoplasm upon viral infection, where it PARylates cGAS. Interestingly, in contrast to the pro-IFN effect of Ku, this PARylation was reported to inhibit cGAS signaling (43). In addition, the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs), has also been identified as a sensor of cytoplasmic DNA (44), and recently demonstrated to negatively regulate cGAS via phosphorylation (45). It is noteworthy that DNA-PKcs, PARP and Ku are all translocated to the cytoplasm in the setting of dsDNA sensing, and together modulate the cell-intrinsic IFN I response generated by cGAS. Activation of the cytoplasmic dsDNA sensing pathway may therefore represent a stimulus that triggers antigenic changes in these proteins that are relevant to SLE pathogenesis.
RNA polymerase III (POL III) is a well-described autoantigen targeted in 15.3–26.6% of systemic sclerosis patients (46, 47). This enzyme transcribes a variety of noncoding RNA molecules required for routine cellular functions (48). However, its role in activating the IFN system is much less appreciated. A specific function for POL III in the innate immune response was reported by Chiu et al., who showed that POL III converts cytosolic dsDNA into 5′-ppp RNA, which is subsequently detected by RIG-like receptors (RLRs), generating a MAVS-dependent IFN response (49). Thus, Ku, PARP and POL III are all involved in the innate response to cytoplasmic dsDNA. The altered subcellular localization and interactions that occur in the setting of cytoplasmic dsDNA sensing therefore might represent additional mechanisms that could contribute to loss of tolerance against these antigens in autoimmune diseases characterized by an aberrant IFN I response.
Oligomerizing innate sensors
Several additional autoantigens combine the characteristics of IFN-induced expression, nucleic acid binding, and a third feature specific to their activation: oligomerization. Recent findings from this interesting autoantibody group are reviewed below.
Antibodies against a 140 kDa protein were first described in a Japanese cohort of patients with clinically amyopathic DM (50). The identity of this autoantigen was later demonstrated to be melanoma differentiation-associated protein 5 (MDA5) (51). The initial clinical phenotype described in association with MDA5 antibodies was that of mild muscle involvement, with severe pulmonary manifestations and a variety of cutaneous findings; additional cohort studies have yielded a broader spectrum of clinical manifestations (52). MDA5 is a member of the RIG-Like Receptor (RLR) group of cytoplasmic dsRNA sensors that promote antiviral IFN I production. Upon sensing long dsRNA, MDA5 assembles into filamentous oligomers that activate MAVS and trigger downstream IFN I signaling (53–55). Like Ro52, MDA5 expression is induced by IFN and interestingly, these two antibodies are often targeted together in this subset of DM patients. As MDA5 is both an IFN-inducible and an interferogenic protein, its dysregulation could readily contribute to sustained IFN signaling. Indeed, gain of function mutations in the gene encoding MDA5 (IFIH1) have been identified in patients with interferonopathy syndromes as well as SLE (56–58). Strong IFN I upregulation has been identified in anti-MDA5-associated DM (59), and some have proposed labeling this syndrome an acquired type I interferonopathy (60).
IFI16 is an IFN-inducible dsDNA sensor in the AIM-like receptor (ALR) family (61). Similar to Ku, IFI16 translocates from the nucleus to the cytoplasm upon dsDNA sensing, where it promotes IFN signaling through STING (61). Similar to MDA5, IFI16 also assembles into filamentous oligomers when activated by dsDNA (62). Anti-IFI16 antibodies have been identified in SLE and pSS patients, and are associated with more severe disease features (63–66).
Absent in melanoma 2 (AIM2), another IFN-inducible dsDNA sensor in the ALR family, activates apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC) upon dsDNA sensing, triggering inflammasome assembly and IL-1/18 secretion (67). We recently identified anti-AIM2 autoantibodies in SLE. These frequently co-occurred with anti-IFI16 and anti-dsDNA antibodies, as well as disease activity markers (68). Autoantibodies targeting ASC (which is also IFN-inducible) have been identified in patients with inflammatory diseases, and anti-ASC antibodies demonstrated a pathogenic ability to enhance inflammasome activation in recipient phagocytes in mice (69).
A noteworthy feature common to the 3 autoantigens MDA5, IFI16 and AIM2 is that they are all IFN-inducible innate sensors of double stranded nucleic acids. Their activation leads to the generation of large, filamentous oligomers of protein and bound nucleic acid ligand. Sustained activation of these sensors at a disease site is one potential explanation for their targeting by autoantibodies. Indeed, our own observation of activated filamentous IFI16 present in the salivary tissues of some pSS patients supports this concept (70). In addition, cytoplasmic interaction of AIM2 and dsDNA has been detected in cell lines derived from pSS salivary tissue (71), and we observed both IFI16 and AIM2 bound to neutrophil extracellular trap DNA in SLE renal tissues (68). These findings provide compelling additional evidence that DNA-bound sensors are present at sites of disease activity.
The presence of oligomerized sensors coupled to nucleic acid ligands may lead to the generation of novel epitopes not found in the monomeric forms, or may increase the potential for autoreactivity via the increased valence present in oligomers that are conveyed to immune cells at sites of immune activation. These autoantigens may therefore represent key molecules whose activation causes pathogenic inflammatory signaling in affected organs, as well themselves being targets of the autoimmune response. Future studies are warranted to examine whether these and/or other autoantibodies serve as biomarkers that identify subsets of patients in whom such innate signaling pathways are especially relevant to disease initiation or propagation. Insights from such studies will likely inform the more effective use of IFN-specific therapies.
Interferons as autoantigens
In addition to the spectrum of intracellular autoantigens associated with systemic autoimmune rheumatic diseases, antibodies against extracellular antigens have also been described in a variety of scenarios. Anti-cytokine antibodies have been observed in patients with SLE and other rheumatic diseases, and also in viral infection and immunodeficiency states (72). In SLE, antibodies against type I, II and III IFNs have been observed (73). These authors found that antibodies against type I IFNs had a neutralizing function, and patients with blocking anti-type I IFN antibodies demonstrated normalized IFN expression levels. Conversely, SLE patients with anti-IFN II antibodies suffered from more severe disease manifestations, including upregulation of type I IFNs. Antibodies against IFNs were also measured in patients with pSS at a comparable prevalence, but were not observed as often in RA.
Nearly 20 years ago, neutralizing antibodies against type II IFN were recognized in patients suffering unusual, severe mycobacterial infections (74, 75). Since that time, several hundred cases of anti-IFN-gamma-autoantibodies (AIGA) have been reported in patients presenting with a variety of infections. In addition to mycobacterial disease, salmonella, varicella, and fungal species have also been recorded, making AIGA an antibody-mediated form of acquired immunodeficiency. Recent studies in SARS-CoV2 have strengthened the evidence that anti-IFN autoantibodies have functional consequences in the setting of infection, as antibodies directed against type I IFNs have been measured in patients who suffer severe disease outcomes from COVID19 (76–78). These observations suggest that, in the setting of infection, anti-IFN antibodies constitute a potentially treatable form of immunodeficiency that renders the host more susceptible to infection. Conversely, in the setting of autoimmune diseases such as SLE, it remains less clear whether anti-IFN antibodies contribute to disease pathogenesis, or serve as markers of aberrant IFN signaling.
Conclusion
Autoantibodies target a multitude of cellular antigens, and diverse mechanisms are likely responsible for their targeting through the humoral immune response. Several autoimmune rheumatic diseases feature upregulation of IFN signaling along with autoantibodies directed against components of the IFN system. These IFN-linked autoantigens include ISGs, DNA-binding proteins, and oligomerizing pattern recognition receptors. Pathogenic activation of these IFN system components may underlie their status as autoantigens, and these autoantibodies might therefore indicate patients in whom the antibody-targeted antigens play critical roles in driving IFN activation. Antibodies against IFNs themselves mediate increased susceptibility to some infections and represent a form of acquired immunodeficiency mediated by humoral autoimmunity.
Author contributions
BA and LC-R contributed to conceptualization and writing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
BA is supported by NIH K08AR077100.
Acknowledgments
Figure 1 was created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mastrandrea, LD. An overview of organ-specific autoimmune diseases including immunotherapy. Immunol Investig. (2015) 44:803–16. doi: 10.3109/08820139.2015.1099409
2. Suurmond, J, and Diamond, B. Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest. (2015) 125:2194–202. doi: 10.1172/JCI78084
3. Ralli, M, Angeletti, D, Fiore, M, D’Aguanno, V, Lambiase, A, Artico, M, et al. Hashimoto’s thyroiditis: an update on pathogenic mechanisms, diagnostic protocols, therapeutic strategies, and potential malignant transformation. Autoimmun Rev. (2020) 19:102649. doi: 10.1016/j.autrev.2020.102649
4. Manivasagam, S, and Klein, RS. Type III interferons: emerging roles in autoimmunity. Front Immunol. (2021) 12:764062. doi: 10.3389/fimmu.2021.764062
5. Lee, AJ, and Ashkar, AA. The dual nature of type I and type II interferons. Front Immunol. (2018) 9:2061. doi: 10.3389/fimmu.2018.02061
6. Wack, A, Terczyńska-Dyla, E, and Hartmann, R. Guarding the frontiers: the biology of type III interferons. Nat Immunol. (2015) 16:802–9. doi: 10.1038/ni.3212
7. Gong, T, Liu, L, Jiang, W, and Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
8. Cao, X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol. (2016) 16:35–50. doi: 10.1038/nri.2015.8
9. Ivashkiv, LB, and Donlin, LT. Regulation of type I interferon responses. Nat Rev Immunol. (2014) 14:36–49. doi: 10.1038/nri3581
10. Crow, MK, Olferiev, M, and Kirou, KA. Type I interferons in autoimmune disease. Annu Rev Pathol. (2019) 14:369–93. doi: 10.1146/annurev-pathol-020117-043952
11. Goel, RR, Kotenko, SV, and Kaplan, MJ. Interferon lambda in inflammation and autoimmune rheumatic diseases. Nat Rev Rheumatol. (2021) 17:349–62. doi: 10.1038/s41584-021-00606-1
12. Hooks, JJ, Moutsopoulos, HM, Geis, SA, Stahl, NI, Decker, JL, and Notkins, AL. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. (1979) 301:5–8. doi: 10.1056/NEJM197907053010102
13. Kirou, KA, Lee, C, George, S, Louca, K, Peterson, MG, and Crow, MK. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. (2005) 52:1491–503. doi: 10.1002/art.21031
14. Feng, X, Wu, H, Grossman, JM, Hanvivadhanakul, P, FitzGerald, JD, Park, GS, et al. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum. (2006) 54:2951–62. doi: 10.1002/art.22044
15. Landolt-Marticorena, C, Bonventi, G, Lubovich, A, Ferguson, C, Unnithan, T, Su, J, et al. Lack of association between the interferon-alpha signature and longitudinal changes in disease activity in systemic lupus erythematosus. Ann Rheum Dis. (2009) 68:1440–6. doi: 10.1136/ard.2008.093146
16. Petri, M, Singh, S, Tesfasyone, H, Dedrick, R, Fry, K, Lal, P, et al. Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. Lupus. (2009) 18:980–9. doi: 10.1177/0961203309105529
17. Mai, L, Asaduzzaman, A, Noamani, B, Fortin, PR, Gladman, DD, Touma, Z, et al. The baseline interferon signature predicts disease severity over the subsequent 5 years in systemic lupus erythematosus. Arthritis Res Ther. (2021) 23:29. doi: 10.1186/s13075-021-02414-0
18. Hall, JC, Baer, AN, Shah, AA, Criswell, LA, Shiboski, CH, Rosen, A, et al. Molecular subsetting of interferon pathways in Sjogren’s syndrome. Arthritis Rheumatol. (2015) 67:2437–46. doi: 10.1002/art.39204
19. Pinal-Fernandez, I, Casal-Dominguez, M, Derfoul, A, Pak, K, Plotz, P, Miller, FW, et al. Identification of distinctive interferon gene signatures in different types of myositis. Neurology. (2019) 93:e1193–204. doi: 10.1212/WNL.0000000000008128
20. Hall, JC, Casciola-Rosen, L, Berger, AE, Kapsogeorgou, EK, Cheadle, C, Tzioufas, AG, et al. Precise probes of type II interferon activity define the origin of interferon signatures in target tissues in rheumatic diseases. Proc Natl Acad Sci U S A. (2012) 109:17609–14. doi: 10.1073/pnas.1209724109
21. Apostolou, E, Kapsogeorgou, EK, Konsta, OD, Giotakis, I, Saridaki, MI, Andreakos, E, et al. Expression of type III interferons (IFNλs) and their receptor in Sjögren’s syndrome. Clin Exp Immunol. (2016) 186:304–12. doi: 10.1111/cei.12865
22. Oke, V, Gunnarsson, I, Dorschner, J, Eketjäll, S, Zickert, A, Niewold, TB, et al. High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res Ther. (2019) 21:107. doi: 10.1186/s13075-019-1878-y
23. Chiche, L, Jourde-Chiche, N, Whalen, E, Presnell, S, Gersuk, V, Dang, K, et al. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol. (2014) 66:1583–95. doi: 10.1002/art.38628
24. Crow, YJ, and Stetson, DB. The type I interferonopathies: 10 years on. Nat Rev Immunol. (2022) 22:471–83. doi: 10.1038/s41577-021-00633-9
25. De Ceuninck, F, Duguet, F, Aussy, A, Laigle, L, and Moingeon, P. IFN-α: a key therapeutic target for multiple autoimmune rheumatic diseases. Drug Discov Today. (2021) 26:2465–73. doi: 10.1016/j.drudis.2021.06.010
26. Schoggins, JW. Interferon-stimulated genes: what do they all do? Ann Rev Virol. (2019) 6:567–84. doi: 10.1146/annurev-virology-092818-015756
27. Jones, EL, Laidlaw, SM, and Dustin, LB. TRIM21/Ro52 - roles in innate immunity and autoimmune disease. Front Immunol. (2021) 12:738473. doi: 10.3389/fimmu.2021.738473
28. Salomonsson, S, Sonesson, SE, Ottosson, L, Muhallab, S, Olsson, T, Sunnerhagen, M, et al. Ro/SSA autoantibodies directly bind cardiomyocytes, disturb calcium homeostasis, and mediate congenital heart block. J Exp Med. (2005) 201:11–7. doi: 10.1084/jem.20041859
29. Andraos, R, Ahmad, A, Eriksson, P, Dahlström, Ö, Wirestam, L, Dahle, C, et al. Autoantibodies associated with systemic sclerosis in three autoimmune diseases imprinted by type I interferon gene dysregulation: a comparison across SLE, primary Sjögren’s syndrome and systemic sclerosis. Lupus Sci Med. (2022) 9:e000732. doi: 10.1136/lupus-2022-000732
30. Weckerle, CE, Franek, BS, Kelly, JA, Kumabe, M, Mikolaitis, RA, Green, SL, et al. Network analysis of associations between serum interferon-α activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum. (2011) 63:1044–53. doi: 10.1002/art.30187
31. Higgs, R, Gabhann, JŃ, Larbi, NB, Breen, EP, Fitzgerald, KA, and Jefferies, CA. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J Immunol. (2008) 181:1780–6. doi: 10.4049/jimmunol.181.3.1780
32. Kimura, T, Jain, A, Choi, SW, Mandell, MA, Schroder, K, Johansen, T, et al. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol. (2015) 210:973–89. doi: 10.1083/jcb.201503023
33. Mallery, DL, McEwan, WA, Bidgood, SR, Towers, GJ, Johnson, CM, and James, LC. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc Natl Acad Sci U S A. (2010) 107:19985–90. doi: 10.1073/pnas.1014074107
34. Keeble, AH, Khan, Z, Forster, A, and James, LC. TRIM21 is an IgG receptor that is structurally, thermodynamically, and kinetically conserved. Proc Natl Acad Sci U S A. (2008) 105:6045–50. doi: 10.1073/pnas.0800159105
35. Schild-Poulter, C, Su, A, Shih, A, Kelly, OP, Fritzler, MJ, Goldstein, R, et al. Association of autoantibodies with Ku and DNA repair proteins in connective tissue diseases. Rheumatology (Oxford). (2008) 47:165–71. doi: 10.1093/rheumatology/kem338
36. Cavazzana, I, Fredi, M, Taraborelli, M, Quinzanini, M, Tincani, A, and Franceschini, F. A subset of systemic sclerosis but not of systemic lupus erythematosus is defined by isolated anti-Ku autoantibodies. Clin Exp Rheumatol. (2013) 31:118–21.
37. Mahler, M, Swart, A, Wu, J, Szmyrka-Kaczmarek, M, Senécal, JL, Troyanov, Y, et al. Clinical and serological associations of autoantibodies to the Ku70/Ku80 heterodimer determined by a novel chemiluminescent immunoassay. Lupus. (2016) 25:889–96. doi: 10.1177/0961203316640918
38. Sui, H, Chen, Q, and Imamichi, T. Cytoplasmic-translocated Ku70 senses intracellular DNA and mediates interferon-lambda1 induction. Immunology. (2021) 163:323–37. doi: 10.1111/imm.13318
39. Sui, H, Zhou, M, Imamichi, H, Jiao, X, Sherman, BT, Lane, HC, et al. STING is an essential mediator of the Ku70-mediated production of IFN-λ1 in response to exogenous DNA. Sci Signal. (2017) 10:aah5054. doi: 10.1126/scisignal.aah5054
40. Tao, X, Song, J, Song, Y, Zhang, Y, Yang, J, Zhang, P, et al. Ku proteins promote DNA binding and condensation of cyclic GMP-AMP synthase. Cell Rep. (2022) 40:111310. doi: 10.1016/j.celrep.2022.111310
41. Muller, S, Briand, JP, Barakat, S, Lagueux, J, Poirier, GG, de Murcia, G, et al. Autoantibodies reacting with poly(ADP-ribose) and with a zinc-finger functional domain of poly(ADP-ribose) polymerase involved in the recognition of damaged DNA. Clin Immunol Immunopathol. (1994) 73:187–96. doi: 10.1006/clin.1994.1187
42. Decker, P, Briand, JP, de Murcia, G, Pero, RW, Isenberg, DA, and Muller, S. Zinc is an essential cofactor for recognition of the DNA binding domain of poly(ADP-ribose) polymerase by antibodies in autoimmune rheumatic and bowel diseases. Arthritis Rheum. (1998) 41:918–26. doi: 10.1002/1529-0131(199805)41:5<918::AID-ART20>3.0.CO;2-W
43. Wang, F, Zhao, M, Chang, B, Zhou, Y, Wu, X, Ma, M, et al. Cytoplasmic PARP1 links the genome instability to the inhibition of antiviral immunity through PARylating cGAS. Mol Cell. (2022) 82:2032–2049.e7. doi: 10.1016/j.molcel.2022.03.034
44. Ferguson, BJ, Mansur, DS, Peters, NE, Ren, H, and Smith, GL. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. elife. (2012) 1:e00047. doi: 10.7554/eLife.00047
45. Sun, X, Liu, T, Zhao, J, Xia, H, Xie, J, Guo, Y, et al. DNA-PK deficiency potentiates cGAS-mediated antiviral innate immunity. Nat Commun. (2020) 11:6182. doi: 10.1038/s41467-020-19941-0
46. Nikpour, M, Hissaria, P, Byron, J, Sahhar, J, Micallef, M, Paspaliaris, W, et al. Prevalence, correlates and clinical usefulness of antibodies to RNA polymerase III in systemic sclerosis: a cross-sectional analysis of data from an Australian cohort. Arthritis Res Ther. (2011) 13:R211. doi: 10.1186/ar3544
47. Moinzadeh, P, Fonseca, C, Hellmich, M, Shah, AA, Chighizola, C, Denton, CP, et al. Association of anti-RNA polymerase III autoantibodies and cancer in scleroderma. Arthritis Res Ther. (2014) 16:R53. doi: 10.1186/ar4486
48. Yeganeh, M, and Hernandez, N. RNA polymerase III transcription as a disease factor. Genes Dev. (2020) 34:865–82. doi: 10.1101/gad.333989.119
49. Chiu, YH, Macmillan, JB, and Chen, ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cells. (2009) 138:576–91. doi: 10.1016/j.cell.2009.06.015
50. Sato, S, Hirakata, M, Kuwana, M, Suwa, A, Inada, S, Mimori, T, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum. (2005) 52:1571–6. doi: 10.1002/art.21023
51. Sato, S, Hoshino, K, Satoh, T, Fujita, T, Kawakami, Y, Fujita, T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. (2009) 60:2193–200. doi: 10.1002/art.24621
52. Fuzzi, E, Gatto, M, Zen, M, Franco, C, Zanatta, E, Ghirardello, A, et al. Anti-MDA5 dermatomyositis: an update from bench to bedside. Curr Opin Rheumatol. (2022) 34:365–73. doi: 10.1097/BOR.0000000000000908
53. Peisley, A, Lin, C, Wu, B, Orme-Johnson, M, Liu, M, Walz, T, et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci U S A. (2011) 108:21010–5. doi: 10.1073/pnas.1113651108
54. Berke, IC, and Modis, Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. (2012) 31:1714–26. doi: 10.1038/emboj.2012.19
55. Berke, IC, Yu, X, Modis, Y, and Egelman, EH. MDA5 assembles into a polar helical filament on dsRNA. Proc Natl Acad Sci U S A. (2012) 109:18437–41. doi: 10.1073/pnas.1212186109
56. Rutsch, F, MacDougall, M, Lu, C, Buers, I, Mamaeva, O, Nitschke, Y, et al. A specific IFIH1 gain-of-function mutation causes singleton-Merten syndrome. Am J Hum Genet. (2015) 96:275–82. doi: 10.1016/j.ajhg.2014.12.014
57. Rice, GI, del Toro Duany, Y, Jenkinson, EM, Forte, GM, Anderson, BH, Ariaudo, G, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. (2014) 46:503–9. doi: 10.1038/ng.2933
58. van Eyck, L, de Somer, L, Pombal, D, Bornschein, S, Frans, G, Humblet-Baron, S, et al. Brief report: IFIH1 mutation causes systemic lupus erythematosus with selective IgA deficiency. Arthritis Rheumatol. (2015) 67:1592–7. doi: 10.1002/art.39110
59. Nombel, A, Fabien, N, and Coutant, F. Dermatomyositis with anti-MDA5 antibodies: bioclinical features, pathogenesis and emerging therapies. Front Immunol. (2021) 12:773352. doi: 10.3389/fimmu.2021.773352
60. Mehta, P, Machado, PM, and Gupta, L. Understanding and managing anti-MDA 5 dermatomyositis, including potential COVID-19 mimicry. Rheumatol Int. (2021) 41:1021–36. doi: 10.1007/s00296-021-04819-1
61. Unterholzner, L, Keating, SE, Baran, M, Horan, KA, Jensen, SB, Sharma, S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. (2010) 11:997–1004. doi: 10.1038/ni.1932
62. Morrone, SR, Wang, T, Constantoulakis, LM, Hooy, RM, Delannoy, MJ, and Sohn, J. Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc Natl Acad Sci U S A. (2014) 111:E62–71. doi: 10.1073/pnas.1313577111
63. Baer, AN, Petri, M, Sohn, J, Rosen, A, and Casciola-Rosen, L. Association of Antibodies to interferon-inducible Protein-16 with markers of more severe disease in primary Sjogren’s syndrome. Arthritis Care Res. (2016) 68:254–60. doi: 10.1002/acr.22632
64. Caneparo, V, Cena, T, De Andrea, M, Dell’oste, V, Stratta, P, Quaglia, M, et al. Anti-IFI16 antibodies and their relation to disease characteristics in systemic lupus erythematosus. Lupus. (2013) 22:607–13. doi: 10.1177/0961203313484978
65. Mondini, M, Vidali, M, De Andrea, M, Azzimonti, B, Airo, P, D’Ambrosio, R, et al. A novel autoantigen to differentiate limited cutaneous systemic sclerosis from diffuse cutaneous systemic sclerosis: the interferon-inducible gene IFI16. Arthritis Rheum. (2006) 54:3939–44. doi: 10.1002/art.22266
66. Uchida, K, Akita, Y, Matsuo, K, Fujiwara, S, Nakagawa, A, Kazaoka, Y, et al. Identification of specific autoantigens in Sjogren’s syndrome by SEREX. Immunology. (2005) 116:53–63. doi: 10.1111/j.1365-2567.2005.02197.x
67. Hornung, V, Ablasser, A, Charrel-Dennis, M, Bauernfeind, F, Horvath, G, Caffrey, DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. (2009) 458:514–8. doi: 10.1038/nature07725
68. Antiochos, B, Trejo-Zambrano, D, Fenaroli, P, Rosenberg, A, Baer, A, Garg, A, et al. The DNA sensors AIM2 and IFI16 are SLE autoantigens that bind neutrophil extracellular traps. elife. (2022) 11:e72103. doi: 10.7554/eLife.72103
69. Franklin, BS, Bossaller, L, De Nardo, D, Ratter, JM, Stutz, A, Engels, G, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol. (2014) 15:727–37. doi: 10.1038/ni.2913
70. Antiochos, B, Matyszewski, M, Sohn, J, Casciola-Rosen, L, and Rosen, A. IFI16 filament formation in salivary epithelial cells shapes the anti-IFI16 immune response in Sjögren’s syndrome. JCI Insight. (2018) 3:e120179. doi: 10.1172/jci.insight.120179
71. Vakrakou, AG, Svolaki, IP, Evangelou, K, Gorgoulis, VG, and Manoussakis, MN. Cell-autonomous epithelial activation of AIM2 (absent in melanoma-2) inflammasome by cytoplasmic DNA accumulations in primary Sjogren’s syndrome. J Autoimmun. (2020) 108:102381. doi: 10.1016/j.jaut.2019.102381
72. Merkel, PA, Lebo, T, and Knight, V. Functional analysis of anti-cytokine autoantibodies using flow cytometry. Front Immunol. (2019) 10:1517. doi: 10.3389/fimmu.2019.01517
73. Gupta, S, Tatouli, IP, Rosen, LB, Hasni, S, Alevizos, I, Manna, ZG, et al. Distinct functions of autoantibodies against interferon in systemic lupus erythematosus: a comprehensive analysis of Anticytokine autoantibodies in common rheumatic diseases. Arthritis Rheumatol. (2016) 68:1677–87. doi: 10.1002/art.39607
74. Döffinger, R, Helbert, MR, Barcenas-Morales, G, Yang, K, Dupuis, S, Ceron-Gutierrez, L, et al. Autoantibodies to interferon-gamma in a patient with selective susceptibility to mycobacterial infection and organ-specific autoimmunity. Clin Infect Dis. (2004) 38:e10–4. doi: 10.1086/380453
75. Höflich, C, Sabat, R, Rosseau, S, Temmesfeld, B, Slevogt, H, Döcke, WD, et al. Naturally occurring anti-IFN-gamma autoantibody and severe infections with Mycobacterium cheloneae and Burkholderia cocovenenans. Blood. (2004) 103:673–5. doi: 10.1182/blood-2003-04-1065
76. Bastard, P, Vazquez, S, Liu, J, Laurie, MT, Wang, CY, Gervais, A, et al. Vaccine breakthrough hypoxemic COVID-19 pneumonia in patients with auto-abs neutralizing type I IFNs. Science immunology. (2022):eabp8966. doi: 10.1126/sciimmunol.abp8966
77. Bastard, P, Rosen, LB, Zhang, Q, Michailidis, E, Hoffmann, HH, Zhang, Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. (2020) 370:eabd4585. doi: 10.1126/science.abd4585
Keywords: autoantibody, autoimmunity, interferon, innate immunity, autoantigen
Citation: Antiochos B and Casciola-Rosen L (2023) Interferon and autoantigens: intersection in autoimmunity. Front. Med. 10:1165225. doi: 10.3389/fmed.2023.1165225
Edited by:
Helena Enocsson, Linköping University, SwedenReviewed by:
Maija-Leena Eloranta, Uppsala University, SwedenCopyright © 2023 Antiochos and Casciola-Rosen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brendan Antiochos, YmFudGlvYzFAamguZWR1