 Maddalena Napolitano
Maddalena Napolitano Francesca di Vico2
Francesca di Vico2 Angelo Ruggiero
Angelo Ruggiero Gabriella Fabbrocini
Gabriella Fabbrocini
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 18 April 2023
Sec. Dermatology
Volume 10 - 2023 | https://doi.org/10.3389/fmed.2023.1165098
This article is part of the Research TopicCutaneous ImmunologyView all 8 articles
Recent evidence suggests that interleukin (IL)-13 is a crucial cytokine involved in the pathogenesis of atopic dermatitis (AD). It is a central driver of type-2 T-helper inflammation and is overexpressed in lesional skin of AD patients. Upon release in peripheral skin, IL-13 activates its receptors, recruits inflammatory cells, and modifies the skin microbiome. IL-13 also reduces the expression of epidermal barrier proteins and activates sensory nerve mediating the itch transmission signal. Novel therapeutics that target IL-13 seem to be efficacious and safe for the treatment of patients with moderate-to-severe AD. The aim of our manuscript is to review the role that IL-13 plays in AD immunopathogenesis.
Atopic dermatitis (AD) is a chronic inflammatory skin disease which has a complex and not yet fully understood pathophysiology (1, 2). AD is heterogeneous in its onset age, clinical phenotype, and severity of manifestations (1, 2). It has been reported that, in developed countries, AD affects in lifetime up to one fifth of the general population, resulting the most frequent among chronic inflammatory skin diseases (3–6). AD, especially in its moderate-to severe forms, results in a negative impact on the quality of life (QoL) of both patients, but and their families (6). Itch is the most troublesome symptom and has been linked with sleep disturbance, reduced work productivity, and poor mental health (7).
Atopic dermatitis pathophysiology implicates multiple interactions between altered type 2 immune responses, skin microbiome dysbiosis, and epidermal barrier disruption (EBD) (8). Recent research advancements, lead to a crucial change in AD pathogenic model, overcoming the preceding hypotheses based on (i) key role of the EBD (“outside-in” theory); (ii) immunoglobulin E (IgE)- response (type 1 hypersensitivity), or (iii) principal role of the abnormal systemic immune activation (“inside-out” theory) (9–11). Currently, however, AD is thought to be secondary to the complex interaction between several genetic defects, environmental stimuli and the activation of intricate inflammatory pathways that induce both the onset and chronicity of the disease (8–11). The discovery of the role for these multiple immune pathways and of related cytokines has led to the development of new drugs (8).
Atopic dermatitis is a disease related to type 2 immunity activation in response to environmental stimuli (2). Both adaptive and innate immune systems are involved in Type 2 immunity (2, 12). Indeed, in the innate immune system activation, it has been showed that key roles are played by Group 2 lymphoid cells (ILC2), mast cells, basophils, eosinophils, and macrophages activated by interleukin (IL)-4 and/or IL-13 (12–15). Furthermore, also keratinocyte is implicated in this mechanism by expressing an increased level of IL-25, IL-33, and thymic stromal lymphopoietin (TSLP) (12–15). These cytokines may act as alarmins, inducing the production of IL-4, IL-13, and IL-5 from both T helper (Th)2, and ILC2 cells, which in turn trigger the complex inflammatory cascade that underlies the clinical expression of AD (16).
Additionally, adaptive immune responses of T and B cells, including CD4 Th2 cells and Th17 cytokines, are required for the development and progression to systemic inflammation in the atopic march (16). The activation of Th2 and ILC2 pathways are at the center of type 2 inflammation (12–15). Conversely, although the expression of Th17-cytokines such as IL-17A has consistently been found to be increased in AD lesions, the role of these cytokines in AD pathogenesis is controversial (16). It has been supposed that they have a role in AD inflammation, rather, its presence is part of an effector response against Staphylococcus aureus (16).
In particular, IL-4 and IL-13 play a key role in AD pathogenesis, orchestrating effector Th2 immune responses (12). Even if IL-4 and IL-13 are encoded by adjacent genes and share a common receptor and signaling pathway, they are differently expressed in vivo by a variety of distinct cells that control both innate and adaptive immunity (12). Indeed, the follicular Th cells of the lymph node, regulating B-cell immunity, invariant natural killer T2 cells, and basophils express IL-4, while mucosal ILC2s mostly express IL-13 and little IL-4 (17). This difference suggests that IL-13 and IL-4 may have distinct actions in Th2 immunity (12). Indeed, it has been showed that IL-4, which drives the T cell differentiation, may have a role in the first steps of the pathogenesis of AD, while IL-13 effects appear to influence the peripheral tissue cells and the effector phase of the immune response (12–17). The aim of this narrative review is to provide an overview of the role that IL13 has in AD pathogenesis.
For this narrative review of literature, search of the English-language literature regarding the pathogenic role of IL-13 in AD was conducted. Different databases, namely Embase, PubMed, ResearchGate, Google Scholar and Scopus, have been consulted using the following terms: interleukin 13, IL-13, atopic dermatitis, atopic eczema, pathogenesis, pathogenic mechanism, Th2 inflammation.
IL-13 is an immunoregulatory cytokine with a structure characterized by a 4 α-helical hydrophobic bundle core (18). Although it is mainly secreted by Th2 cells, other cells, such as ILC2, mast cells and basophils, release IL-13 (4, 14).
A subgroup of ILCs, the ILC2, which are the only subset of lymphocytes with no antigen receptors rearranged, produce type 2 cytokines, promoting inflammation and hyperresponsiveness (18–21). In the skin, dermal ILC2s secrete IL-13 regardless of allergen exposure and independently of the alarmins (IL-2, TSLP, and IL-33), driving dendritic cells (DC)2 precursors differentiation to a CD11blow phenotype that fosters Th2 priming (12). Conversely, in the lung ILC2s require alarmins to secrete IL-13 and induce the expression of Th2 cells (12). However, TSLP may directly stimulate CD11c + DCs differentiation and activation, resulting in the stimulation of Th2-cell expression, inhibition of IL-12 secretion in the context of type-2 innate ligands, affecting B-cell development and survival (21). Recently, it has been demonstrated that the level of resident group ILC2 of the healthy human skin is notably increased in AD skin lesions (22). It has been showed that ILC2 in AD skin exhibits an elusive immunophenotype, and that among all the ILCs, inducible T-cell costimulatory (ICOS)-expressing cells, comprising both ILC2 and ILC3, are the main producer of IL-13 in the dermis (23). Moreover, ILCs present a higher level of IL-13Rα1 subunit of IL-13 receptor (IL-13R) than T cells (23).
The other major source of IL-13 is Th2 cells (4). In ex vivo coculture model, it has been demonstrated that a cutaneous lymphocyte-associated antigen (CLA)-dependent production of IL-13, upon activation with staphylococcal enterotoxin B (SEB), allows the differentiation of both Th2 high and low responder groups (24). SEB activation of the CLA T-cells resulted in a predominant IL-13 production among the Th2 cytokines (IL-5, IL-4, lL-13), and in stratification into the Th2 high and Th2 low groups, corresponding with disease activity (24). Additionally, in the Th2 high group, IL-13 response directly correlates with AD severity [measured by Eczema Area Severity Index (EASI)], anti-Staphylococcus aureus IgE plasma levels, sIL-2R, and CCL17 (24). Regardless of source, the increased IL-13 expression leads to the recruitment of eosinophils and activated T cells, resulting in an amplification of skin IL-13/IL-4 expression, driving the pathway of chronic inflammation in AD (25).
The signaling of IL-13 is regulated by a complex receptor system (4, 14). In non-hematopoietic cells, IL-13 engages a heterodimeric receptor composed of 2 subunits, the alpha chain of the IL-4 receptor (IL-4Rα) and the alpha-1 chain of IL-13 (IL-13 Rα1), binding IL-13 with low affinity; however, after forming a complex with IL-4Rα, it shows a higher affinity, resulting in the induction of the effector functions of IL-13 (4, 14). Another receptor, composed by IL-13-specific binding chain alpha 2, named IL-13 Rα2, is strictly related to IL-13 Rα1. IL-13 Rα2 binds IL-13 with high affinity, and it is considered for having a compensatory role (decoy receptor), as it would be able to remove excess IL-13 (4, 14). The IL-13 binding of the functional heterodimeric IL-4Rα /IL-13 Rα1 receptor results in the activation of downstream tyrosine kinase 2 (TYK2), and Janus kinases (JAK) activating the signal transducer and activator of transcription (STAT3, STAT1 and STAT6) (4, 14). Thus, the activation of JAK–STAT pathway is followed by the increased secretion of several chemokines for eosinophils, cytokines, angiogenic factors, and growth of IgE level binding to mast cell receptors, resulting in the exacerbation of the inflammation process of AD (25).
Previous studies showed the key role played by IL-13 in AD pathogenesis, regarding multiple aspects of disease pathogenesis such as skin barrier disruption, epidermal thickening, itch, inflammation, and infection (26). In biopsies AD skin, there is an overexpression of IL-13 in both lesional and non lesional skin compared to healthy controls (27). Additionally, AD severity is directly linked to the increased skin expression of IL-13, while a decrease in its concentration has been shown to correlate to improved clinical outcomes (27). Furthermore, a recent study confirms the dominance of the expression of IL-13 mRNA in both chronic and subacute lesions than non-lesional skin, and than healthy controls with near undetectable IL −4 expression (28–30). Recently, IL-13 has been identified in skin samples as a biomarker of AD, showing a strongest association with disease severity, circulating eosinophil levels, and total serum IgE (31). Furthermore, several atopic stigmata were associated with high levels of cutaneous IL13 such as thinning of the lateral eyebrow (Hertoghe sign), and maternal atopic rhinitis (31).
The STAT6 activation, by IL-4 and IL-13, reduce the expression of structural proteins like hornerin, desmoglein, loricrin, involucrin, desmocollin, filaggrin (FLG), keratin 1, and keratin 10, as well as the lipid composition (ceramides, free fatty acids, and cholesterol) important for normal skin barrier function (27, 32, 33). The overexpression of these cytokines, therefore, plays a key role in maintaining and increasing the impairment of the skin barrier in AD (34). Additionally, they induce a decreased production of AMP by keratinocytes, thus being important also in inducing skin dysbiosis, which is characterized by a significant colonization with Staphylococcus aureus, which may precede the onset of AD lesions (34).
OVOL1, an upstream transcription factor, regulates the expression of FLG (17); OVOL1 activation leads to its cytoplasmic-to-nuclear translocation, and resulting in the up-regulation of loricrin and FLG expression (35, 36). Interestingly, IL-13 and IL-4 inhibit FLG expression by interfering with OVOL1 signaling (16). Moreover, IL-13 inhibits the expression of involucrin in an OVOL1-independent way, exacerbating barrier dysfunction (35, 36). In case of barrier-disrupted skin, keratinocytes produce high levels of IL-33, IL-25 and TSLP, promoting Th2 cells differentiation, and ILC2s, resulting in the stimulation of IL-13 production (37). Thus, a vicious cycle is formed to develop atopic dry skin. These findings suggest that IL-13–OVOL1–FLG axis may play a central role in the pathogenesis of AD (17). Near this axis, IL-13-induced FLG down-regulation is in part mediated by the IL13–periostin–IL-24 axis (17, 38). It has been showed that, through STAT6 activation, IL-13 up-regulates the keratinocytes expression of periostin, stimulating keratinocyte production of IL-24, which down-regulates the FLG expression via STAT3 activation (38).
Furthermore, IL-13, acting on keratinocytes, reduces the expression of skin barrier proteins and lipids, regulating the expression of metalloproteinase (MMP)-9; MMP-9 mediates tissue remodeling and the migration of cells, through action of degradation on collagen IV of the basement membrane (39, 40). Moreover, IL-13 down-regulates the expression of MMP-13 in dermal fibroblasts, leading in a decrease of the degradation of collagen, resulting in fibrosis, as found in the thickened dermis of chronic lichenified AD manifestations (41).
Intradermal injection of IL-13 has been reported to induce allokinesis and itching (42). The sensory neurons and keratinocytes express IL-4Rα/IL-13Rα1 and IL-13Rα2 (43). After binding to its receptor IL-13Rα1, IL-13 activates sensory neurons acting as an enhancer of other stimuli such as histamine (44). However, IL-13 is a potent neuroactive cytokine that potentiates also the responses of nonhistaminergic itch pathways (45). For example, it is involved in a histamine-independent direct stimulation of afferent nerve endings mediated by transient receptor potential ankyrin 1 (TRPA1) pathway (45).
Further, scratch injury enhanced the expression of IL-13Rα2, while no significant modification were found in the functional heterodimeric IL-13 receptor IL-13Rα1 expression (46). IL-13 Rα2, a decoy receptor binding with high affinity to IL-13, internalizes IL-13 and lowers the IL-13 level in the milieu; however, this receptor showed to have several other functions, especially in AD pathogenesis (47–51). IL-13 and scratch injury upregulate the expression of IL-13 Rα2 (47). which binds to IL-13 with high affinity, internalizes it, and decreases IL-13 levels in atopic skin (47).
Some authors also reported that IL-13 signals through IL-13 Rα2 is able to induce transforming growth factor beta (TGF-β) and promote fibrosis (48). It has been showed that even chitinase 3-like 1 (CHI3L1), also known as breast regression protein 39 (BRP-39) in mice and human homologue YKL-40, may act as an activator of IL-13 Rα2 (49, 50). The levels of both cutaneous and serum CHI3L1/YKL-40 are increased in AD patients (49). Notably, some variants of the CHI3L1 gene that codes for this protein have been associated with cases of severe AD with onset during late childhood, and a tendency to become chronic (51).
IL-13 is produced from ILC2s and Th2 cells and is increased in skin from AD patients (Figure 1) (27). IL-13 binds to subunit IL-13Rα1 of its heterodimeric receptor for signaling via JAK1 and JAK2 (4, 14). The binding of IL-13 to IL-13Rα2, lead to the association with YKL40; the exact signaling transducing machinery related to this complex has not been fully understood (27). IL-13 contributes to the start of AD and itching and, acting with IL-4, lead to the aggravation of EBD by downregulating involucrin and FLG, via inactivation of OVOL1 and stimulation of periostin and IL-24 (Figure 1) (17). This contributes to the increased trans epidermal water loss in AD patients (52, 53). Moreover, IL-13 showed to decrease antimicrobial peptides (AMPs) production by keratinocytes and to have a role in the dysbiosis of the skin, characterized by a prevalent Staphylococcus aureus colonization (Figure 1) (34, 54). Hence, the increased permeability of the skin lead to the entry of antigens that reach DC, stimulating the activation of naive T cells to Th2 lymphocytes, and finally resulting in the amplification of this loop mechanism (55). Both cytokines enhance the differentiation of B-cell and the production of IgE, Th2 development/differentiation, eosinophil recruitment, hence, the amplification of the inflammation mediated by Th2 cells (55). Furthermore, in the skin, dermal ILC2s secrete IL-13 regardless of allergen exposure and independently of the alarmins driving the differentiation to Th2 cells (12). IgE binding to both basophils and mast cells, lead to an increment of histamine and other inflammatory mediators release, increasing pruritus and vasodilation (55). Stimulating action on itching, however, mainly results from a histamine-independent direct stimulation of afferent nerve endings mediated by TRPA1 pathway (55). Indeed, IL-13 is an effective, neuroactive cytokine able to modulate human sensory neurons in their neuronal excitability, increasing the itch pathways response (56). Scratching also upregulates the expression of decoy receptor IL-13Rα2 that binds to IL-13 with high affinity, internalizes it, and lowers the IL-13 level in atopic skin. Thus, if on the one hand scratching exacerbates skin inflammation, on the other it triggers a reactive compensatory response against excess levels of IL-13 by upregulating the decoy IL-13 Rα2 (47). Moreover, IL-13 also plays a role in the maintenance of conjunctival well-being, hence, its inhibition may set the premises for dupilumab induced conjunctivitis. Interestingly, this appears to be associated with eosinophil fluctuations (which are on their turn the result of diminished tissue infiltration) (57).
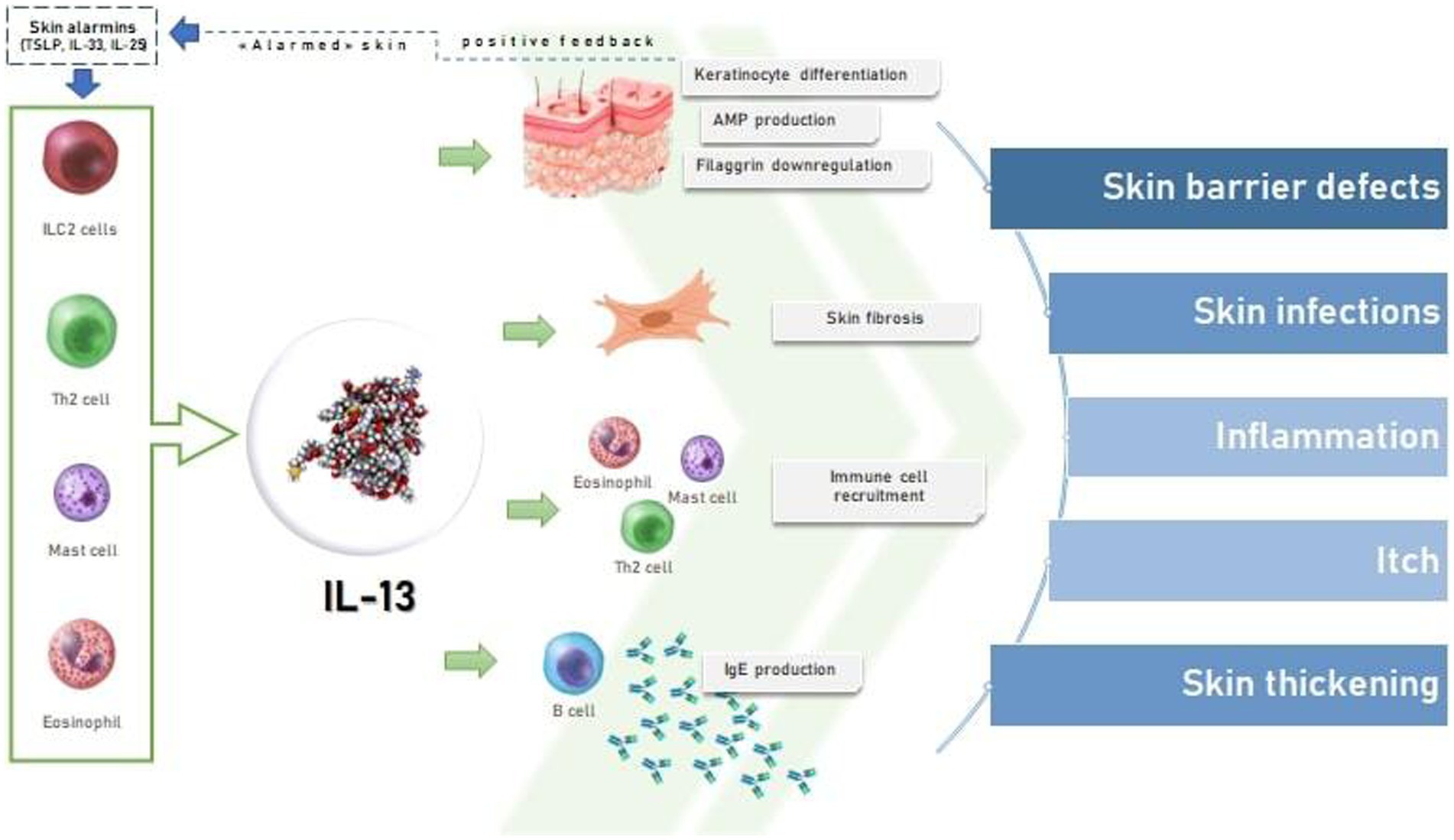
Figure 1. The effects of IL-13 on skin in atopic dermatitis.
The increased knowledge of the role played by IL-4 and IL-13 in the pathogenesis of AD, resulted in the development of new targeted therapies (58). Dupilumab, the first approved biological agent for the treatment of AD, which acts by blocking IL-4Rα and thus the activity of both IL-4 and IL-13, demonstrated the effectiveness of blocking type 2 cytokines, their receptors, or their intracellular signal transducers JAK/STAT pathway (8, 59–61).
Furthermore, IL-13 signaling alone is considered a potential therapeutic target for the management of AD (58). Tralokinumab, a fully human monoclonal antibody binding IL-13, acts by preventing the binding of IL-13 to both IL-13Rα1 and IL-13Rα2, resulting in the blockage of its signal transduction, showed to be a safe as and effective treatment option, even in monotherapy in adult AD patients (52, 58, 62, 63). A phase III, double-blind, placebo plus topical corticosteroids (TCS) controlled clinical trial, in which patients were randomized 2:1 to tralokinumab 300 mg or placebo every 2 weeks (Q2W) with TCS as needed over 16 weeks (63). At week 16, an higher rate of patients treated with tralokinumab than placebo achieved EASI 75: 56% vs. 35.7% [20.2% (9.8–30.6); p < 0.001], and Investigator Global Assessment (IGA) 0/1: 38.9% vs. 26.2% [12.4% (2.9–21.9); p = 0·015] (63). Of the patients who were tralokinumab responders at week 16, 89.6 and 92.5% of those treated with tralokinumab Q2W maintained an IGA 0/1 and EASI 75 response at week 32, respectively (63). Among patients not achieving EASI 75 and IGA 0/1 with tralokinumab Q2W after 16 weeks of treatment, 55.8 and 30.5% and achieved these endpoints, respectively, after 32 weeks of treatment (63). In the ECZTRA 6 trial, a phase III study, adolescents aged between 12 and 17 years old were randomized 1:1:1 to tralokinumab 150 mg or 300 mg Q2W, or placebo; after 16 weeks of treatment, a significantly greater rate of patients achieved EASI-75 and IGA0/1 on 150 mg/Q2W (IGA0/1: 21.4%; EASI75: 28.6%) and 300 mg/Q2W (IGA0/1: 17.5%; EASI-75: 27.8%) with no need of rescue therapy, versus placebo (IGA0/1: 4.3%, EASI-75: 6.4%) (64). Ongoing trials are evaluating the treatment of tralokinumab for AD pediatric patients [NCT05388760] (65).
Lebrikizumab, is a fully humanized IgG4 antibody, acts by binding IL-13 in a different non–receptor-binding domain, preventing IL-4Rα/IL13Rα1 heterodimerization and downstream signaling (58). In a double-blinded, placebo-controlled, multicenter, phase 3 clinical trial, 145 subjects have been randomized to subcutaneous lebrikizumab and 66 to placebo Q2W in combination with TCS. At week 16, IGA (0.1) was achieved by 60 (41.2%) patients in the lebrikizumab plus TCS group vs. 15 (22.1%) receiving placebo plus TCS (p = 0.01); corresponding proportions of patients achieving EASI-75 were 69.5% (101/145) vs. 42.2% (28/66) (p < 0.001) (66). The most frequently reported treatment-emergent adverse events were headache (7 [4.8%]), conjunctivitis (7 [4.8%]), injection site reactions (4 [2.8%]), hypertension (4 [2.8%]), and herpes infection (5 [3.4%]) (62). Full trial results on lebrikizumab are still lacking (67, 68).
A phase 2, randomized, placebo-controlled, study evaluating the efficacy and safety of cendakimab (CC-93538) an anti- IL-13, in the treatment of moderate to severe AD, is still ongoing (69). Two hundred and fourteen patients have been enrolled in 3 different dosing regimens groups and in a placebo group (69). To date, no data are available (69).
Interestingly, it has been reported that the increased expression of IL-13 is linked with an optimal clinical response to dupilumab, while, on the other hand, non-responders patients showed to express less IL-13 and relatively greater levels of Type 1 and 3 cytokines. Hence, IL-13 levels may represent a predictor of response to IL-13 inhibitors (70).
IL-13 also showed to be a potential target even in other skin diseases, indirectly suggesting a pathogenetic role of its pathway. Indeed, dupilumab has been proposed as potentially safe and effective therapeutic option for other diseases, such as Grover disease. Evidence supporting this finding includes the fact that IL-4 infusions may induce Grover disease and that the immunophenotype of the immune cells infiltrating Grover disease demonstrate a TH2 cytokine phenotype (71).
Finally, some authors proposed the use of anti-IL-13 vaccination which may have the potential of outperforming monoclonal antibody-based approaches (72, 73).
Current evidence suggest IL-13 as a crucial cytokine AD pathogenesis, supporting its significant contribution in mediating several clinical features, including skin inflammation and pruritus. Therefore, IL-13 is considered a valid target for AD. Indeed, IL-13 inhibitors such as tralokinumab and lebrikizumab seem to be a possible treatment for patients with moderate-to-severe AD, with good safety and efficacy profiles. However, further investigations will be worthy to clarify the position of these treatments in the therapeutic ladder of AD.
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
MN, CP, FV, AR, and GF: conceptualization. CP, MN, FV, and AR: methodology. MN, FV, and AR: software. CP, MN, FV, AR, and GF: formal analysis. MN, CP, FV, and AR: data curation and writing–original draft preparation. CP: writing–review and editing. GF: visualization. GF and CP: supervision. All authors have read and agreed to the published version of the manuscript.
MN served as advisory board member and consultant, and has received fees and speaker’s honoraria for Sanofi, Abbvie, Leo Pharma, Lilly, Amgen. GF is a member of the journal’s Editorial Board, and has been principal investigator in clinical trials sponsored by and/or has received personal fees from AbbVie, Abiogen, Almirall, Celgene, Eli-Lilly, Leo Pharma, Novartis, Sanofi, and UCB. CP acted as investigator, speaker, consultant, and advisory board member for AbbVie, Eli Lilly, Novartis, Pfizer, and Sanofi.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Puar, N, Chovatiya, R, and Paller, AS. New treatments in atopic dermatitis. Ann Allergy Asthma Immunol. (2021) 126:21–31. doi: 10.1016/j.anai.2020.08.016
2. Moniaga, CS, Tominaga, M, and Takamori, K. The pathology of type 2 inflammation-associated itch in atopic dermatitis. Diagnostics (Basel). (2021) 11:2090. doi: 10.3390/diagnostics11112090
3. Kim, BE, and Leung, DYM. Significance of skin barrier dysfunction in atopic dermatitis. Allergy Asthma Immunol Res. (2018) 10:207–15. doi: 10.4168/aair.2018.10.3.207
4. Tubau, C, and Puig, L. Therapeutic targeting of the IL-13 pathway in skin inflammation. Expert Rev Clin Immunol. (2021) 17:15–25. doi: 10.1080/1744666X.2020.1858802
5. Abuabara, K, Magyari, A, McCulloch, CE, Linos, E, Margolis, DJ, and Langan, SM. Prevalence of atopic eczema among patients seen in primary Care: data from the health improvement network. Ann Intern Med. (2019) 170:354–6. doi: 10.7326/M18-2246
6. Silverberg, JI. Public health burden and epidemiology of atopic dermatitis. Dermatol Clin. (2017) 35:283–9. doi: 10.1016/j.det.2017.02.002
7. Chang, YS, and Chiang, BL. Sleep disorders and atopic dermatitis: a 2-way street? J Allergy Clin Immunol. (2018) 142:1033–40. doi: 10.1016/j.jaci.2018.08.005
8. Chiricozzi, A, Maurelli, M, Peris, K, and Girolomoni, G. Targeting IL-4 for the treatment of atopic dermatitis. Immunotargets Ther. (2020) 9:151–6. doi: 10.2147/ITT.S260370
9. Elias, PM, and Steinhoff, M. “Outside-to-inside” (and now back to “outside”) pathogenic mechanisms in atopic dermatitis. J Invest Dermatol. (2008) 128:1067–70. doi: 10.1038/jid.2008.88
10. Werfel, T, Allam, JP, Biedermann, T, Eyerich, K, Gilles, S, Guttman-Yassky, E, et al. Cellular and molecular immunologic mechanisms in patients with atopic dermatitis. J Allergy Clin Immunol. (2016) 138:336–49. doi: 10.1016/j.jaci.2016.06.010
11. Paller, AS, Kabashima, K, and Bieber, T. Therapeutic pipeline for atopic dermatitis: end of the drought? J Allergy Clin Immunol. (2017) 140:633–43. doi: 10.1016/j.jaci.2017.07.006
12. Lamiable, O, Brewerton, M, and Ronchese, F. IL-13 in dermal type-2 dendritic cell specialization: from function to therapeutic targeting. Eur J Immunol. (2022) 52:1047–57. doi: 10.1002/eji.202149677
13. Matsunaga, K, Katoh, N, Fujieda, S, Izuhara, K, and Oishi, K. Dupilumab: basic aspects and applications to allergic diseases. Allergol Int. (2020) 69:187–96. doi: 10.1016/j.alit.2020.01.002
14. Gandhi, NA, Bennett, BL, Graham, NM, Pirozzi, G, Stahl, N, and Yancopoulos, GD. Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discov. (2016) 15:35–50. doi: 10.1038/nrd4624
15. Tojima, I, Matsumoto, K, Kikuoka, H, Hara, S, Yamamoto, S, Shimizu, S, et al. Evidence for the induction of Th2 inflammation by group 2 innate lymphoid cells in response to prostaglandin D2 and cysteinyl leukotrienes in allergic rhinitis. Allergy. (2019) 74:2417–26. doi: 10.1111/all.13974
16. Ong, PY. Atopic dermatitis: is innate or adaptive immunity in control? A clinical perspective. Front Immunol. (2022) 13:943640. doi: 10.3389/fimmu.2022.943640
17. Furue, K, Ito, T, Tsuji, G, Ulzii, D, Vu, YH, Kido-Nakahara, M, et al. The IL-13-OVOL1-FLG axis in atopic dermatitis. Immunology. (2019) 158:281–6. doi: 10.1111/imm.13120
18. Leonard, WJ, and Lin, JX. Cytokine receptor signaling pathways. J Allergy Clin Immunol. (2000) 105:877–88. doi: 10.1067/mai.2000.106899
19. Pasha, MA, Patel, G, Hopp, R, and Yang, Q. Role of innate lymphoid cells in allergic diseases. Allergy Asthma Proc. (2019) 40:138–45. doi: 10.2500/aap.2019.40.4217
20. von Moltke, J, O'Leary, CE, Barrett, NA, Kanaoka, Y, Austen, KF, and Locksley, RM. Leukotrienes provide an NFAT-dependent signal that synergizes with IL-33 to activate ILC2s. J Exp Med. (2017) 214:27–37. doi: 10.1084/jem.20161274
21. Scanlon, ST, and McKenzie, AN. The messenger between worlds: the regulation of innate and adaptive type-2 immunity by innate lymphoid cells. Clin Exp Allergy. (2015) 45:9–20. doi: 10.1111/cea.12464
22. Kim, BS, Siracusa, MC, Saenz, SA, Noti, M, Monticelli, LA, Sonnenberg, GF, et al. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. (2013):170ra16. doi: 10.1126/scitranslmed.3005374
23. Alkon, N, Bauer, WM, Krausgruber, T, Goh, I, Griss, J, Nguyen, V, et al. Single-cell analysis reveals innate lymphoid cell lineage infidelity in atopic dermatitis. J Allergy Clin Immunol. (2022) 149:624–39. doi: 10.1016/j.jaci.2021.07.025
24. Sans-De San Nicolàs, L, Figueras-Nart, I, Bonfill-Ortí, M, De Jesús-Gil, C, García-Jiménez, I, Guilabert, A, et al. SEB-induced IL-13 production in CLA+ memory T cells defines Th2 high and Th2 low responders in atopic dermatitis. Allergy. (2022) 77:3448–51. doi: 10.1111/all.15424
25. Szalus, K, Trzeciak, M, and Nowicki, RJ. JAK-STAT inhibitors in atopic dermatitis from pathogenesis to clinical trials results. Microorganisms. (2020) 8:1743. doi: 10.3390/microorganisms8111743
26. Czarnowicki, T, He, H, Krueger, JG, and Guttman-Yassky, E. Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol. (2019) 143:1–11. doi: 10.1016/j.jaci.2018.10.032
27. Bieber, T. Interleukin-13: targeting an underestimated cytokine in atopic dermatitis. Allergy. (2020) 75:54–62. doi: 10.1111/all.13954
28. Tsoi, LC, Rodriguez, E, Degenhardt, F, Baurecht, H, Wehkamp, U, Volks, N, et al. Atopic dermatitis is an IL −13 dominant disease with greater molecular heterogeneity com-pared to psoriasis. J Invest Dermatol. (2019) 139:1480–9. doi: 10.1016/j.jid.2018.12.018
29. Koppes, SA, Brans, R, Ljubojevic Hadzavdic, S, Frings-Dresen, MH, Rustemeyer, T, and Kezic, S. Stratum corneum tape stripping: monitoring of inflammatory mediators in atopic dermatitis patients using topical therapy. Int Arch Allergy Immunol. (2016) 170:187–93. doi: 10.1159/000448400
30. Szegedi, K, Lutter, R, Res, PC, Bos, JD, Luiten, RM, Kezic, S, et al. Cytokine profiles in interstitial fluid from chronic atopic dermatitis skin. J Eur Acad Dermatol Venereol. (2015) 29:2136–44. doi: 10.1111/jdv.13160
31. Maintz, L, Welchowski, T, Herrmann, N, Brauer, J, Traidl-Hoffmann, C, Havenith, R, et al. IL-13, periostin and dipeptidyl-peptidase-4 reveal endotype-phenotype associations in atopic dermatitis. Allergy. (2023) 17. doi: 10.1111/all.15647
32. Kim, BE, Leung, DY, Boguniewicz, M, and Howell, MD. Loricrin and in-volucrin expression is down-regulated by Th2 cytokines through S TAT-6. Clin Immunol. (2008) 126:332–7. doi: 10.1016/j.clim.2007.11.006
33. Berdyshev, E, Goleva, E, Bronova, I, Dyjack, N, Rios, C, Jung, J, et al. Lipid abnormalities in atopic skin are driven by type 2 cytokines. JCI. Insight. (2018) 3:e98006. doi: 10.1172/jci.insight.98006
34. Howell, MD, Boguniewicz, M, Pastore, S, Novak, N, Bieber, T, Girolomoni, G, et al. Mechanism of HBD-3 deficiency in atopic dermatitis. Clin Immunol. (2006) 121:332–8. doi: 10.1016/j.clim.2006.08.008
35. Hashimoto-Hachiya, A, Tsuji, G, Murai, M, Yan, X, and Furue, M. Upregulation of FLG, LOR, and IVL expression by Rhodiola crenulata root extract via aryl hydrocarbon receptor: differential involvement of OVOL1. Int J Mol Sci. (2018) 19:E1654. doi: 10.3390/ijms19061654
36. Tsuji, G, Hashimoto-Hachiya, A, Kiyomatsu-Oda, M, Takemura, M, Ohno, F, Ito, T, et al. Aryl hydrocarbon receptor activation restores filaggrin expression via OVOL1 in atopic dermatitis. Cell Death Dis. (2017) 8:e2931. doi: 10.1038/cddis.2017.322
37. Camelo, A, Rosignoli, G, Ohne, Y, Stewart, RA, Overed-Sayer, C, Sleeman, MA, et al. IL33, IL-25, and TSLP induce a distinct phenotypic and activation profile in human type 2 innate lymphoid cells. Blood Adv. (2017) 1:577–89. doi: 10.1182/bloodadvances.2016002352
38. Mitamura, Y, Nunomura, S, Nanri, Y, Ogawa, M, Yoshihara, T, Masuoka, M, et al. The IL13/periostin/IL-24 pathway causes epidermal barrier dysfunction in allergic skin inflammation. Allergy. (2018) 73:1881–91. doi: 10.1111/all.13437
39. Ungar, B, Garcet, S, Gonzalez, J, Dhingra, N, Correa da Rosa, J, Shemer, A, et al. An integrated model of atopic dermatitis biomarkers highlights the systemic nature of the dis-ease. J Invest Dermatol. (2017) 137:603–13. doi: 10.1016/j.jid.2016.09.037
40. Moriya, C, Jinnin, M, Yamane, K, Maruo, K, Muchemwa, FC, Igata, T, et al. Expression of matrix metallo-proteinase-13 is controlled by IL-13 via PI3K/Akt3 and PKC-d in nor-mal human dermal fibroblasts. J Invest Dermatol. (2011) 131:655–61. doi: 10.1038/jid.2010.361
41. Shin, JW, Choi, YJ, Choi, HR, Na, JI, Kim, KH, Park, IA, et al. Defective basement membrane in atopic dermatitis and possible role of IL-13. J Eur Acad Dermatol Venereol. (2015) 29:2060–2. doi: 10.1111/jdv.12596
42. van den Bogaard, EH, Bergboer, JG, Vonk-Bergers, M, van Vlijmen-Willems, IM, Hato, SV, van der Valk, PG, et al. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J Clin Invest. (2013) 123:917–27. doi: 10.1172/JCI65642
43. Xiao, S, Lu, Z, Steinhoff, M, Li, Y, Buhl, T, Fischer, M, et al. Innate immune regulates cutaneous sensory IL-13 receptor alpha 2 to promote atopic dermatitis. Brain Behav Immun. (2021) 98:28–39. doi: 10.1016/j.bbi.2021.08.211
44. Brightling, CE, Saha, S, and Hollins, F. Interleukin-13: prospects for new treatments. Clin Exp Allergy. (2010) 40:42–9. doi: 10.1111/j.1365-2222.2009.03383.x
45. Ratnarajah, K, Le, M, Muntyanu, A, Mathieu, S, Nigen, S, Litvinov, IV, et al. Inhibition of IL-13: a new pathway for atopic dermatitis [formula: see text]. J Cutan Med Surg. (2021) 25:315–28. doi: 10.1177/1203475420982553
46. Ulzii, D, Kido-Nakahara, M, Nakahara, T, Tsuji, G, Furue, K, Hashimoto-Hachiya, A, et al. Scratching counteracts IL-13 signaling by upregulating the decoy receptor IL-13Rα2 in keratinocytes. Int J Mol Sci. (2019) 20:3324. doi: 10.3390/ijms20133324
47. Furue, M, Ulzii, D, Nakahara, T, Tsuji, G, Furue, K, Hashimoto-Hachiya, A, et al. Implications of IL-13Rα2 in atopic skin inflammation. Allergol Int. (2020) 69:412–6. doi: 10.1016/j.alit.2020.01.005
48. Fichtner-Feigl, S, Strober, W, Kawakami, K, Puri, RK, and Kitani, A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. (2006) 12:99–106. doi: 10.1038/nm1332
49. Kwak, EJ, Hong, JY, Kim, MN, Kim, SY, Kim, SH, Park, CO, et al. Chitinase 3-like 1 drives allergic skin inflammation via Th2 immunity and M2 macrophage activation. Clin Exp Allergy. (2019) 49:1464–74. doi: 10.1111/cea.13478
50. Lee, CM, He, CH, Nour, AM, Zhou, Y, Ma, B, Park, JW, et al. Corrigendum: IL-13Rα2 uses TMEM219 in chitinase 3-like-1-induced signalling and effector responses. Nat Commun. (2016) 7:13541. doi: 10.1038/ncomms13541
51. Dežman, K, Korošec, P, Rupnik, H, and Rijavec, M. SPINK5 is associated with early-onset and CHI3L1 with late-onset atopic dermatitis. Int J Immunogenet. (2017) 44:212–8. doi: 10.1111/iji.12327
52. Kelleher, MM, Dunn-Galvin, A, Gray, C, Murray, DM, Kiely, M, Kenny, L, et al. Skin barrier impair-ment at birth predicts food allergy at 2 years of age. J Allergy Clin Immunol. (2016) 137:1111–1116.e8. doi: 10.1016/j.jaci.2015.12.1312
53. Horimukai, K, Morita, K, Narita, M, Kondo, M, Kabashima, S, Inoue, E, et al. Transepidermal water loss measurement during infancy can predict the subsequent develop-ment of atopic dermatitis regardless of filaggrin mutations. Allergol Int. (2016) 65:103–8. doi: 10.1016/j.alit.2015.09.004
54. Meylan, P, Lang, C, Mermoud, S, Johannsen, A, Norrenberg, S, Hohl, D, et al. Skin colonization by Staphylococcus aureus precedes the clinical diagnosis of atopic dermatitis in infancy. J Invest Dermatol. (2017) 137:2497–504. doi: 10.1016/j.jid.2017.07.834
55. Miron, Y, Miller, PE, Hughes, C, Indersmitten, T, Lerner, EA, and Cevikbas, F. Mechanistic insights into the antipruritic effects of lebrikizumab, an anti-IL-13 mAb. J Allergy Clin Immunol. (2022) 150:690–700. doi: 10.1016/j.jaci.2022.01.028
56. Dubin, C, Del Duca, E, and Guttman-Yassky, E. The IL-4, IL-13 and IL-31 pathways in atopic dermatitis. Expert Rev Clin Immunol. (2021) 17:835–52. doi: 10.1080/1744666X.2021.1940962
57. Ferrucci, S, Angileri, L, Tavecchio, S, Fumagalli, S, Iurlo, A, Cattaneo, D, et al. Elevation of peripheral blood eosinophils during dupilumab treatment for atopic dermatitis is associated with baseline comorbidities and development of facial redness dermatitis and ocular surface disease. J Dermatolog Treat. (2022) 33:2587–92. doi: 10.1080/09546634.2022.2049588
58. Le Floc’h, A, Allinne, J, Nagashima, K, Scott, G, Birchard, D, Asrat, S, et al. Dual blockade of IL-4 and IL13 with dupilumab, an IL-4Rα antibody, is required to broadly inhibit type 2 inflammation. Allergy. (2020) 75:1188–204. doi: 10.1111/all.14151
59. Napolitano, M, Fabbrocini, G, Neri, I, Stingeni, L, Boccaletti, V, Piccolo, V, et al. Dupilumab treatment in children aged 6-11 years with atopic dermatitis: a multicentre, real-life study. Paediatr Drugs. (2022) 24:671–8. doi: 10.1007/s40272-022-00531-0
60. Stingeni, L, Bianchi, L, Antonelli, E, Caroppo, ES, Ferrucci, SM, Gurioli, C, et al. DADA – Dupilumab for atopic dermatitis of the adolescence study group. A 52-week update of a multicentre Italian real-world experience on effectiveness and safety of dupilumab in adolescents with moderate-to-severe atopic dermatitis. J Eur Acad Dermatol Venereol. (2022) 36:1292–9. doi: 10.1111/jdv.18648
61. Patruno, C, Fabbrocini, G, Longo, G, Argenziano, G, Ferrucci, SM, Stingeni, L, et al. Effectiveness and safety of long-term dupilumab treatment in elderly patients with atopic dermatitis: a Multicenter real-life observational study. Am J Clin Dermatol. (2021) 22:581–6. doi: 10.1007/s40257-021-00597-5
62. Wollenberg, A, Blauvelt, A, Guttman-Yassky, E, Worm, M, Lynde, C, Lacour, JP, et al. Tralokinumab for moderate-to-severe atopic dermatitis: results from two 52- week, randomized, double-blind, multicentre, placebo-controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br J Dermatol. (2021) 184:437–49. doi: 10.1111/bjd.19574
63. Silverberg, JI, Toth, D, Bieber, T, Alexis, AF, Elewski, BE, Pink, AE, et al. Tralokinumab plus topical corticosteroids for the treatment of moderate-to-severe atopic dermatitis: results from the double-blind, randomized, multi-Centre, placebo-controlled phase III ECZTRA 3 trial. Br J Dermatol. (2021) 184:450–63. doi: 10.1111/bjd.19573
64. Paller, AS, Blauvelt, A, Soong, W, Imafuku, S, Hong, CH, Schuttelaar, M, et al. Efficacy and safety of traloki-numab in adolescents with moderate-to-severe atopic dermati-tis: results of the phase 3 ECZTRA 6 trial. SKIN J Cutaneous Med. (2022) 6:s29. doi: 10.25251/skin.6.supp.s29
65. Available at: https://clinicaltrials.gov/ct2/results?cond=&term=NCT05388760&cntry=&state=&city=&dist (Accessed February 10, 2023)
66. Simpson, EL, Gooderham, M, Wollenberg, A, Weidinger, S, Armstrong, A, Soung, J, et al. Efficacy and safety of lebrikizumab in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis: a randomized clinical trial (ADhere). JAMA Dermatol. (2023):e225534. doi: 10.1001/jamadermatol.2022.5534
67. Available at: https://clinicaltrials.gov/ct2/results?cond=&term=NCT04146363&cntry=&state=&city=&dist (Accessed February 10, 2023)
68. Available at: https://clinicaltrials.gov/ct2/results?cond=&term=NCT04178967&cntry=&state=&city=&dist (Accessed February 10, 2023)
69. Available at: https://clinicaltrials.gov/ct2/show/NCT04800315?term=NCT04800315&draw=2&rank=1 (Accessed February 10, 2023)
70. Singh, K, Valido, K, Swallow, M, Okifo, KO, Wang, A, Cohen, JM, et al. Baseline skin cytokine profiles determined by RNA in situ hybridization correlate with response to dupilumab in patients with eczematous dermatitis. J Am Acad Dermatol. (2023) S0190-9622:00179–2. doi: 10.1016/j.jaad.2022.12.052
71. Butler, DC, Kollhoff, A, and Berger, T. Treatment of Grover disease with dupilumab. JAMA Dermatol. (2021) 157:353–6. doi: 10.1001/jamadermatol.2020.5097
72. Foerster, J, and Molęda, A. Virus-like particle-mediated vaccination against Interleukin-13 May harbour general anti-allergic potential beyond atopic dermatitis. Viruses. (2020) 12:438. doi: 10.3390/v12040438
Keywords: atopic dermatitis (AD), interleukin-13, pathogenesis, type 2 inflammation, review
Citation: Napolitano M, di Vico F, Ruggiero A, Fabbrocini G and Patruno C (2023) The hidden sentinel of the skin: An overview on the role of interleukin-13 in atopic dermatitis. Front. Med. 10:1165098. doi: 10.3389/fmed.2023.1165098
Edited by:
Angelo Valerio Marzano, University of Milan, ItalyReviewed by:
Carlo Alberto Maronese, IRCCS Ca 'Granda Foundation Maggiore Policlinico Hospital, ItalyCopyright © 2023 Napolitano, di Vico, Ruggiero, Fabbrocini and Patruno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maddalena Napolitano, bWFkZHkubmFwb2xpdGFub0BnbWFpbC5jb20=; Cataldo Patruno, Y2F0YWxkby5wYXRydW5vQHVuaWN6Lml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.