 Aitana Alonso-Gonzalez
Aitana Alonso-Gonzalez Eva Tosco-Herrera
Eva Tosco-Herrera Maria Molina-Molina
Maria Molina-Molina Carlos Flores
Carlos Flores- 1Unidad de Investigación, Hospital Universitario Nuestra Señora de Candelaria, Santa Cruz de Tenerife, Spain
- 2Universidad de Santiago de Compostela, Santiago de Compostela, Spain
- 3Servei de Pneumologia, Laboratori de Pneumologia Experimental, IDIBELL, Barcelona, Spain
- 4Campus de Bellvitge, Universitat de Barcelona, Barcelona, Spain
- 5CIBER de Enfermedades Respiratorias, Instituto de Salud Carlos III, Madrid, Spain
- 6Genomics Division, Instituto Tecnológico y de Energías Renovables (ITER), Santa Cruz de Tenerife, Spain
- 7Facultad de Ciencias de la Salud, Universidad Fernando Pessoa Canarias, Las Palmas de Gran Canaria, Spain
Idiopathic pulmonary fibrosis (IPF) is a chronic, rare progressive lung disease, characterized by lung scarring and the irreversible loss of lung function. Two anti-fibrotic drugs, nintedanib and pirfenidone, have been demonstrated to slow down disease progression, although IPF mortality remains a challenge and the patients die after a few years from diagnosis. Rare pathogenic variants in genes that are involved in the surfactant metabolism and telomere maintenance, among others, have a high penetrance and tend to co-segregate with the disease in families. Common recurrent variants in the population with modest effect sizes have been also associated with the disease risk and progression. Genome-wide association studies (GWAS) support at least 23 genetic risk loci, linking the disease pathogenesis with unexpected molecular pathways including cellular adhesion and signaling, wound healing, barrier function, airway clearance, and innate immunity and host defense, besides the surfactant metabolism and telomere biology. As the cost of high-throughput genomic technologies continuously decreases and new technologies and approaches arise, their widespread use by clinicians and researchers is efficiently contributing to a better understanding of the pathogenesis of progressive pulmonary fibrosis. Here we provide an overview of the genetic factors known to be involved in IPF pathogenesis and discuss how they will continue to further advance in this field. We also discuss how genomic technologies could help to further improve IPF diagnosis and prognosis as well as for assessing genetic risk in unaffected relatives. The development and validation of evidence-based guidelines for genetic-based screening of IPF will allow redefining and classifying this disease relying on molecular characteristics and contribute to the implementation of precision medicine approaches.
1. Introduction
Idiopathic pulmonary fibrosis (IPF), the most common type of interstitial pneumonia, is a chronic and progressive disease in which healthy tissue in the lung parenchyma is progressively replaced by an altered extracellular matrix leading to the loss of lung function (1, 2). This situation leads to dyspnea and abnormal gas exchange, restrictive physiology, hypoxemia, and finally, respiratory failure (3). Treatment options for IPF are limited, although many drugs targeting different pro-fibrotic pathways are being tested in clinical trials (4). Only two antifibrotic drugs, pirfenidone and nintedanib, have been approved for patient treatment by now. Both drugs reduce the decline in lung function, but none is curative (5, 6). Thus, although these drugs may slightly delay the 2–5 years estimated median survival time from diagnosis, disease lethality remains a challenge (7–9).
IPF often clusters in families where multiple members are affected. The term familial pulmonary fibrosis (FPF) is coined when at least two members from the same biological family are affected with IPF (10). In both familial and sporadic forms, common and rare genetic variants contribute to the disease architecture (11). Despite the genetic factors involved in both forms are incompletely understood, genetics is only partially overlapping. Rare variants affecting function of genes of the surfactant metabolism and telomere biology are commonly found in FPF cases when using sequencing studies in kindreds. Particularly, most of these deleterious variants affect telomere genes and they are associated with shorter telomere length (TL) (12). For that reason, obtaining TL measures are also recommended during the diagnostic process (11, 13, 14). The role of common genetic variants in IPF has been identified through genome-wide association studies (GWAS) in sporadic cases (15, 16). For now, nearly two dozen of genetic loci have been associated with IPF susceptibility, although the strongest common risk factor described is a single nucleotide polymorphism (SNP) in the promoter of MUC5B (17).
Despite the important recent advances in understanding the genetics underlying disease risks, the current IPF guidelines do not recommend the use of genetic testing for diagnosis (18). In this review, we outline the genetic architecture of IPF and discuss the clinical outcomes of specific variants within IPF genes. We also summarize the molecular technologies which could be applied to assist in the diagnosis of the patients and describe the situations in which it should be performed. Finally, we frame this scenario in the context of the fast developments occurring with the genomic technologies and in how they could further contribute to the implementation of precision medicine in IPF patients.
2. Genetics of IPF
Genetic factors play a significant role in the development of IPF. The known genetic variants associated with IPF are classified into two broad categories: common SNPs that are broadly found in the general population (allele frequency > 1%), and rare damaging variants in the spectrum of allele frequency below <1% which are typically not recurrent in the general population (19). Next-generation sequencing (NGS) technologies, where the order of nucleotides of the entire genome or of regions of interest are determined, are commonly applied to assess the contribution of rare variants to human phenotypes and have led to the identification of genes that are causal or associated with the risk of the disease. In IPF, most research and clinical sequencing studies have been performed in FPF patients and have identified rare variants in two distinct biological pathways: the surfactant metabolism and telomere maintenance (Figure 1). Collectively, they are found in about 25% of all patients with FPF and, in most of these cases, the disease displays an autosomal dominant (AD) inheritance pattern with incomplete penetrance. Since these rare variants only account for a small proportion of the population attributable risk of IPF, other approaches should be considered to define the role of common variants in the disease. In contrast to rare variants, common variants are present at a higher frequency in the general population although they individually associate with a smaller effect size (reduced penetrance). In GWAS studies, hundreds of thousands of common genetic variants are tested to find those that are statistically associated with a specific trait. If the trait of interest is dichotomous (presence/absence of IPF among the study participants) the study design involves the inclusion of cases (individuals with the trait) and controls (individuals without the trait) (21). This approach has highlighted new genes involved in IPF pathogenesis and possible new therapeutic targets. Thus, taken together, the results emerging from genetic studies support that IPF results from a complex interaction between genetic variants at variable frequency and environmental factors.
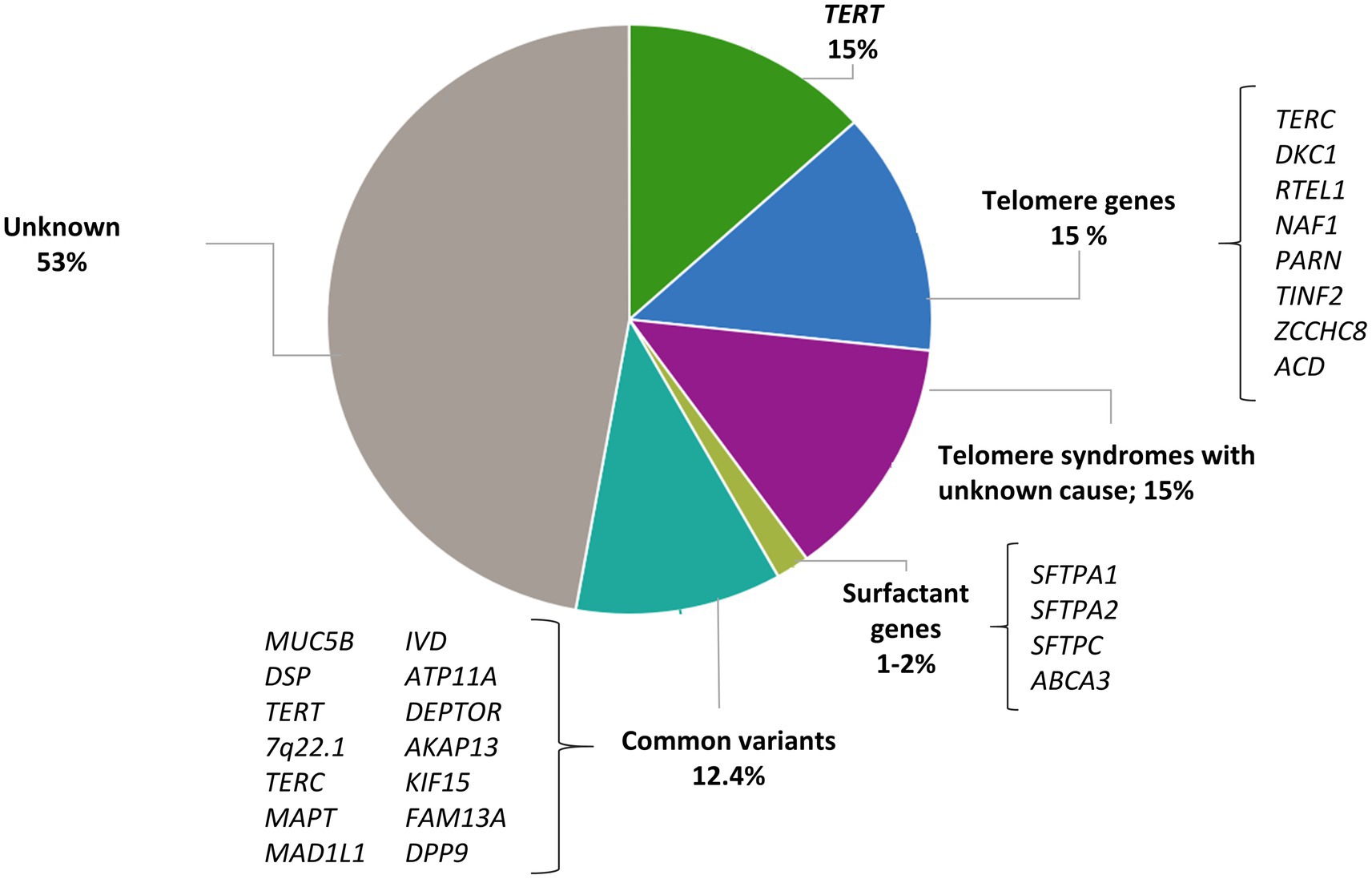
Figure 1. Contribution of rare and common variants to IPF risk. Proportion of risk explained by rare variants in telomere genes and surfactant genes; proportion of patients with short telomere length where a mutation has not been identified; proportion of risk explained by 14 IPF susceptibility variants estimated by Leavy et al. (20).
2.1. The genes involved in IPF development
The genetic studies in IPF have provided significant advances in the understanding of disease pathophysiology. The genes encoding proteins which fall in the same pathway are expected to disrupt the same mechanism and, therefore, can be targeted similarly. This fact reinforces the importance of disclosing the molecular causes underlying disease risk.
2.1.1. Telomere-related genes
Telomeres are specialized structures at the end of chromosomes. In humans, they typically consist of the tandemly repeated TTAGGG nucleotide sequence and the associated protective proteins known as the shelterin complex. Their main functions are to protect genome integrity and prevent degradation and chromosomal end-to-end fusion (22). As the DNA replication machinery is not capable of completely copying DNA at the extreme ends of linear chromosomes, telomeres shorten with each cell division. This situation is resolved in eukaryotes by the telomerase activity, which adds small (telomeric) repeat sequences to the end of chromosomes to compensate this attrition. This complex enzyme involves a catalytic subunit, the telomerase reverse transcriptase (TERT), the telomerase RNA component (TERC), and other relevant components, such as dyskerin (DKC1) and protein regulators (11, 23). However, the activity of the telomerase is limited, and telomeres inevitably shorten throughout the life span. When short telomeres reach a critical threshold across all chromosomes, cellular senescence and apoptosis are triggered by DNA damage responses (24).
Telomere biology disorders are a phenotypic group of diseases that share the molecular defect of short TL due to the presence of germline mutations that affect telomere maintenance (25). Dyskeratosis congenita was the first of these disorders to be described. Affected patients spontaneously develop pulmonary fibrosis at the second decade of life (26). Since then, multiple genes participating in the telomere maintenance have been linked to the pathogenesis of IPF. Gene defects affecting telomerase catalytic enzyme activity (TERT and TERC) were the first to be described in families with IPF and no history of dyskeratosis congenita (27, 28). With the advent of the NGS technologies, exome sequencing (ES) of patients with FPF have identified at first instance rare variants in two additional genes: PARN, which encodes for an enzyme that removes oligo(A) tails from the precursor RNA including that of TERC, and RTEL1, which encodes a helicase that unwinds the G-quadruplex and the T-loop secondary structures at the telomeric end (29, 30). Then, case reports have also identified mutations in DKC1 (31), ZCCHC8 (32), and NAF1 (33), all with a role in the biogenesis of telomerase, and TINF2 (34) and ACD (35), implicated in the telomere integrity (Table 1 and Figure 1). Pathogenic rare variants found in these genes lead to alterations in protein function, measured by decreased telomerase activity and telomere shortening in somatic cells, including blood leukocytes, oral mucosal epithelial cells, and lung epithelial cells (27–29, 33, 34, 36–38). Rare pathogenic variants in telomere-related genes are found in approximately one fourth of FPF cases and one tenth of sporadic IPF cases, the variants in TERT and TERC being the most frequent (8–15%) (27, 28). Rare pathogenic variants in PARN and RTEL1 comprise 5–10% of FPF cases (30). Mutations in other genes such as TINF2 and NAF1 collectively represent about 1% of the cases, and they have been identified in a few affected families so far (33, 34) (Figure 1).
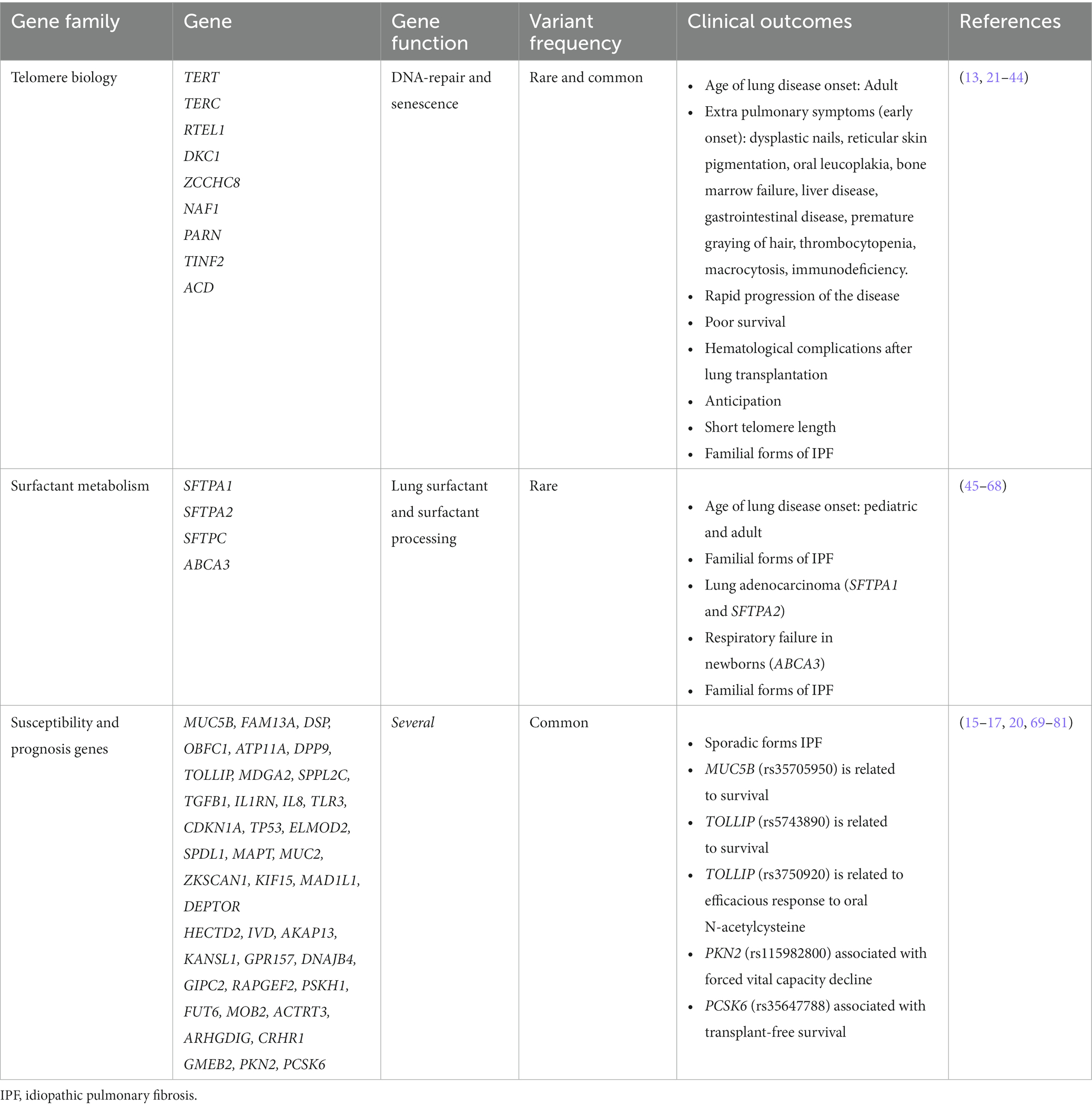
Table 1. Genes related to IPF and clinical outcomes from causal and associated variants.
Carriers of mutations in telomere-related genes have shortened telomeres, and the phenomenon of genetic anticipation is commonly described in these kindreds. This is defined as an earlier and severe onset of disease associated with the progressive shortening of telomeres of the kindred with each generation. Although approximately 25% of the sporadic IPF patients have telomeres shorter than the 10th percentile of the general population, rare pathogenic variants in genes of the telomere maintenance pathway are only found in 10% of these cases (37, 82). Thus, there are at least 15% of patients with clinical features of short telomere syndrome and short TL where a mutation is yet to be discovered (39) (Figure 1). This fact might be explained by, at least, three main reasons:
1. Additional genes that are critical for telomere maintenance might be involved in the pathophysiology of IPF. These genes may explain smaller fractions of the population risk and could be identified through sequencing additional cases.
2. The short telomeres can be inherited independently of the transmission of the mutations (40). This inherited telomere dysfunction might be sufficient to induce IPF due to persistent DNA damage, inducing cell senescence and alveolar type II cells (AEC2) apoptosis (83, 84).
3. Common variants associated with TL overlap with IPF genes identified through GWAS and might be responsible for the disease in a small fraction of patients where no rare variants were identified (85).
2.1.2. Surfactant-related genes
Pulmonary surfactant is a mixture of lipids and proteins produced by AEC2. Its main function is to reduce surface tension in the alveoli, thereby preventing the collapse of structures at end-expiration. Besides, it is also involved in host defense and the modulation of immune responses. Four surfactant proteins SP-A, SP-B, SP-C, and SP-D are encoded by the SFTPA1, SFTPA2, SFTPC, and SFTPD genes, respectively. They are translated within the endoplasmic reticulum (ER) in a pre-protein form and then transported to lamellar bodies where they are stored until they are secreted to the alveolar space. Rare pathogenic variants in SFTPC and SFTPA1-2 have been associated with both adult-onset and pediatric forms of IPF (45, 86) (Table 1 and Figure 1).
The first genetic association between the surfactant and IPF was obtained when Nogee and colleagues (46) reported a coding mutation in SFTPC gene in an infant and her mother, both affected with interstitial lung disease (ILD). Since then, numerous studies have identified more than 40 coding and non-coding rare pathogenic mutations in the gene. These mutations are described in pediatric and adult patients, predisposing to a range spectrum of fibrotic lung diseases which segregate in an AD pattern of inheritance (47–49, 87). One of these variants, predicting a Ile73Thr amino acid change, has been described in multiple cohorts, representing approximately 25–35% of the pathogenic alleles (47, 50, 87). Although multiple variants have been reported, there is a plausible common mechanism contributing to disease pathogenesis. Therefore, variants are mostly found in a BRICHOS domain localized in the SP-C pre-protein. Since this domain is critical for proper folding and trafficking within the secretory pathway, its mutations produce an abnormal pre-protein that accumulates within the ER. This results in ER stress and caspase pathway activation which leads to AEC2 injury (51–53). The frequency of rare pathogenic variants in SFTPC is low among cases, accounting for 1–2% of FPF patients (54, 55), although a higher prevalence (25%) was described in a Dutch cohort that could be explained by founder effects (56).
Surfactant protein A has two isoforms, SP-A1 and SP-A2, encoded by two adjacent genes, SFTPA1 and SFTPA2 (45). Both isoforms share a highly conserved carbohydrate recognition domain wherein all pathogenic heterozygous rare variants for IPF have been found (57, 58). Thus, pathogenic mutations in this domain result in an aberrant protein accumulation in the ER, producing ER stress (59). In contrast to SFTPC, which is only expressed in AEC2, SFTPA also expresses in Clara cells and submucosal glands from the respiratory airways, having important functions in host defense (60). Pathogenic mutations in SFTPA1 and SFTPA2 have been mainly linked to FPF cases and they usually also associate with lung cancer in adult patients (57, 58, 88) (Table 1).
In addition to those encoding the surfactant proteins, other genes encoding proteins involved in surfactant processing have been also involved in IPF pathogenesis. This is the case of the ATP-binding cassette transporter A3 (ABCA3), expressed in AEC2 lamellar bodies where it participates in lipid transportation (61, 62). Early studies reported homozygous or compound heterozygotic ABCA3 mutations causing fatal distress respiratory syndrome or childhood ILD (63–65) (Table 1). However, other studies describe pathogenic mutations in ABCA3 in adults with IPF. Moreover, heterozygous ABCA3 mutations have been described in combination with SFTPC mutations in infants with ILD, suggesting that the gene might be acting as a modifier of the disease severity in individuals with SFTPC mutations (66, 89).
2.1.3. Common genetic variants and other IPF genes
2.1.3.1. MUC5B variant is strongly associated to IPF
The genetic variant that most strongly associate with IPF susceptibility in the GWAS studies conducted to date is a SNP located in the promoter region of the MUC5B gene and identified as rs35705950. The variant was identified in 2011 for the first time in a seminal study that relied first on a linkage mapping approach followed by a fine mapping in familial and sporadic IPF cases (17). The same year, the genetic association was confirmed in another study with independent sporadic IPF cases (90). These initial studies firmly supported that the MUC5B variant is associated with a strong increase in IPF risk for both familial and sporadic forms. When measured in terms of odds ratios (OR), heterozygous and homozygous of the risk allele (T) of the polymorphism were estimated in as much as 6.8 (95% confidence interval [CI], 3.9–12) and 20.8 (95% CI, 3.8–113.7) for FPF, and 9.0 (95% CI, 6.2–13.1) and 21.8 (95% CI, 5.1–93.5) for sporadic IPF (Table 1).
MUC5B encodes mucin 5B, a major component of mucus which is found in various mucosal surfaces, including the lung (91). The promoter variant is in a critical regulatory domain of the gene (92). Consequently, rs35705950 is associated with significantly higher (37.4-fold increase for carriers of the T allele) MUC5B expression in lung tissue among unaffected subjects, although the expression is also higher in subjects with IPF (14-fold increase) than in unaffected controls irrespective of genotype (17).
After this seminal study, subsequent studies conducted in non-Hispanic White (NHW) cohorts have replicated the association of the variant with IPF risk (16, 17, 69–71, 93–95). rs35705950 has also proved to be a risk factor for IPF in Mexicans (96) and in Asian populations (97, 98), albeit the MUC5B variant is not as common in these populations as in NHW. One important piece of information in this context is that the prevalence of IPF is higher in NHW than in Hispanics or Asians. In contrast, the disease is extremely rare in African populations, where the T allele is also nearly absent.1 Although causation cannot be inferred because the presence of the variant alone is insufficient to cause the disease (T allele is also found in 9% of NHW subjects without disease), these observations illustrate an important positive correlation between the frequency of the MUC5B-rs35705950 T allele and the prevalence of IPF.
Besides being considered a dominant risk factor for IPF (69, 95), the MUC5B variant has been also associated with the presence of interstitial lung abnormalities (99). In addition, this variant associates with increased risk of ILD in patients with rheumatoid arthritis (100), although no associations have been found with asbestosis or sarcoidosis (95).
2.1.3.2. Other common variants associated to IPF
Other common variants have been associated with IPF risk through GWAS, providing further evidence that multiple common genetic variants have an important contribution to disease risk, with an overall risk likely higher than 12.4–17.7% explained by the common variation all together (20) (Figure 1). In the last decade, large-scale international collaborations have been established to combine independent genomic data across thousands of patients and controls to improve the statistical power and detect novel IPF genes. This strategy has allowed the identification of at least 23 independent genetic loci associated with IPF susceptibility and has revealed novel molecular processes involved in IPF pathogenesis including host defense, cell–cell adhesion, DNA repair, airway clearance, innate immunity, profibrotic signaling pathways, mTOR signaling, and mitotic signaling (15, 16, 70, 93, 94, 101).
The first two large GWAS were published in 2013. A GWAS of 1,616 IPF cases and 4,683 control subjects confirmed some previously identified associated loci such as TERT and MUC5B, and identified novel associations in or near FAM13A, DSP, OBFC1, ATP11A, DPP9, and in the 7q22 and 15q14-15 regions (70). The same year, a three-stage analysis, comprising 1,410 IPF cases and 2,934 controls in total, also identified associated loci in TOLLIP and SPPL2C (94). It should be noted that SPPL2C is found at 17q21.31. This region includes a well-known pleiotropic inversion polymorphism that is positively selected in Europeans (102). Intriguingly, this locus has been associated with many traits related to lung function such as chronic obstructive pulmonary disease (103), response to inhaled corticosteroids in asthma (104) and, more recently, to severe forms of coronavirus infection (COVID-19) (41). AKAP13, was later recognized as another IPF risk gene, therefore the profibrotic signaling pathway represents another potential target in IPF (93) (Table 1).
Recently, two GWAS meta-analyses of IPF have been conducted. The first one was carried out by Allen and colleagues, who confirmed mostly all previously reported signals in addition to identifying three novel signals implicating DEPTOR, KIF15, and MAD1L1 genes. These new findings further support the importance of mTOR signaling in IPF. It also suggested cell-cycle progression as another novel biological process, since KIF15 and MAD1L1 are both mitotic spindle assembly genes. Moreover, estimates of polygenic risk scores (PRS) for the first time in IPF showed that the disease is highly polygenic and there are potentially hundreds of, as yet unidentified, variants associated with IPF susceptibility (101). The first multi-ancestry meta-analysis of IPF included a total of 8,492 patients and nearly 1.36 million controls from 13 biobanks as part of the Global Biobank Meta-Analysis Initiative. This allowed to identify seven novel loci near GPR157, DNAJB4/GIPC2, RAPGEF2, FKBP5, RP11-286H14.4, PSKH1, and FUT6 (Table 1). Importantly, only one of these loci would have been associated under the stringent criteria of GWAS studies if the analysis had been restricted only to European populations (16). Thus, the study proves the advantage of increasing sample diversity, as new loci were identified since their index variants were more frequent in other genetic ancestries. The inclusion of understudied populations in GWAS studies is crucial as it allows to fully understand the genetic architecture of IPF while improves the ability to translate the genetic findings into clinical practice independently of ethnicity.
2.2. The clinical outcomes and the genetic discoveries
The clinical course of IPF is heterogeneous and yet unpredictable, and the molecular mechanisms underlying the disease are not completely understood. Specific genetic variants have clinical implications in terms of prognosis or associated comorbidities. Therefore, knowing the genetic causes could lead to patient stratification according to the expected prognosis or the treatment response, and contribute to the decision in patient prioritization for lung transplantation (Table 1).
2.2.1. Clinical outcomes of IPF patients with telomere gene mutations
Genetic mutations in telomere-related genes are often linked to the development of a telomeropathy syndrome, which is characterized by a heterogeneous phenotype that includes a wide range of pulmonary and extrapulmonary symptoms. Thus, although ILD seems to be the most predominant phenotype, others such as bone marrow dysfunction, liver cirrhosis, or early hair graying are also described when these mutations are present (23). Importantly, while the prevalence of IPF in carriers of telomere-related mutations increases with age, the extrapulmonary symptoms are often diagnosed at a younger age (105) (Table 1). The age at diagnosis of IPF has been proved to be different also among carriers of mutations: TERT mutation carriers are diagnosed at an earlier age (mean of 51 years) relative to mutation carriers in TERC (mean of 58 years), RTEL1 (mean of 60 years), and PARN (mean of 65 years) (105).
Clinical lung expression is very heterogeneous among telomerase mutation carriers; chronic hypersensitivity pneumonitis (7–12%), unclassifiable ILD (8–12%), and other idiopathic interstitial pneumonias (14–18%) are described (105, 106). However, the disease evolution is remarkably similar. Among carriers of mutations in TERT, RTEL1, PARN, and TERC, one consistent phenotype is progressive deterioration (as measured by absolute forced vital capacity or FVC), suggesting that telomere-related mutations are predictive of disease progression independently of the clinical diagnosis (105) (Table 1).
Telomere shortening has also proven to be a predictor of survival (42), which may condition the stratification of patients with IPF before lung transplantation (43). Furthermore, IPF patients with short telomeres present an increase of specific morbidities after lung transplant such as infection, immunological and hematological dysfunction, potentially impacting on survival (13, 42, 44, 107) (Table 1).
Telomere syndromes are causally linked to the same underlying mechanism, short telomeres, which indicate that telomere-target therapies would be helpful in these patients. These therapeutic interventions are currently being investigated (108, 109). Danazol is a synthetic androgen that preserves TL and improves hematological response in 79% of the patients (110). However, the drug is poorly tolerated, especially due to liver adverse events, and its potential benefit in pulmonary fibrosis is being evaluated in clinical trials (TELO-SCOPE: NCT04638517). Meanwhile, the effect of currently used drugs have been also analyzed in these patients. While pirfenidone and nintedanib seem to be safe and beneficial in IPF patients with telomere related gene mutations and/or telomere shortening (111), a harmful effect of immunosuppression has been suggested (112).
2.2.2. Clinical outcomes of patients with mutations in surfactant biology genes
Surfactant-related gene expression is mainly related to the lung (113), although an increase of frequency in cancer has been also described. In general, the clinical expression related to surfactant genes is variable, but the age of disease onset usually ranges from the infancy to young adulthood (Table 1).
Disease causing SFTPC variants have been identified in different ILDs, including idiopathic interstitial pneumonias and pulmonary alveolar proteinosis (46, 47, 49, 87). ABCA3 gene variants are a common cause of hereditary respiratory failure in newborns (64). These cases are lethal and related to null mutations, leading to the absence of functional ABCA3 protein activity. Instead, milder phenotypes in children or even adults are associated to the absence of null mutations (65, 66, 72, 114). Mutations in SFTPA2 and SFTPA1 have been reported only in adults and they are considered a recognized cause of IPF and lung cancer (57). Precisely, this distinction has an impact in transplantation considerations (58).
To date, no specific drug therapy is determined for patients with mutations in surfactant genes.
2.2.3. Clinical outcomes and common gene polymorphisms
The common T allele of the promoter of MUC5B (rs35705950), which increases the IPF risk, has been associated with better IPF progression and survival (95, 115–118). Similarly, a G allele in rs5743890 near TOLLIP, associated with reduced susceptibility risk, was associated with increased mortality of IPF patients. The latter still lacks replication in independent studies (94).
These data could suggest that carriers of the MUC5B promoter variant are part of a subgroup of patients with a distinct prognosis (73, 94, 115). However, these paradoxical findings should be considered with extreme caution. A phenomenon called index event bias, in which a biased association can result from selection of subjects according to their disease status, could explain these results (119). A statistical approximation for adjusting such a bias has been developed and applied to this case. Despite the need for further replication of the findings in independent samples, the results of the application of the novel statistical method suggest that the association of the MUC5B variant with survival is biased and the risk allele may, in fact, be associated with decreased IPF survival (119). In this line, the existing evidence based on PRS model inferences suggest that genetic determinants of IPF susceptibility and progression may have limited overlap (101, 120).
Regarding therapies, the PANTHER-IPF clinical trial2 was conducted in carriers of the aforementioned MUC5B and TOLLIP variants to evaluate their modulatory effect on immunosuppressive and antioxidant treatments. The research revealed that, although N-Acetylcysteine did not show efficacy in treating IPF patients in general, a subset of the patients defined by the genotype at rs3750920 could benefit from the drug (121). Independent studies of this assessment are yet lacking from the literature.
Another relevant area of study in IPF involves the identification of genetic variants associated to disease progression rather than disease risk. The first GWAS of decline in lung function in individuals diagnosed with IPF was completed by Allen et al. In this study, the authors discovered a genetic locus that associated with a more rapid decline in the lung capacity, which lies in the RNA antisense gene PKN2 (101). Consistently, an inhibitor of this gene, fostamatinib, has been already suggested as a potential drug candidate in acute respiratory distress syndrome in patients with severe COVID-19 (122).
3. Genetic testing in IPF beyond TL
It is widely recognized that genetic testing could aid in the diagnosis, influence the clinical management, and assist in predicting the prognosis of some patients with IPF. However, there is no consensus recommendation about the implementation of genetic testing or how to identify those IPF patients that may benefit more from this practice (18). In fact, the diagnosis of IPF is still restricted to the identification of a pattern of interstitial pneumonia based on radiological or histological criteria. The American Thoracic Society (ATS), The European Respiratory Society (ERS), The Japanese Respiratory Society (JRS), and Asociación Latinoamericana de Tórax (ALAT) recently updated the clinical practice guidelines for IPF. However, no recommendations were offered regarding when to pursue genetic testing in patients or how to use these results in the clinical practice (18).
Recently, some initiatives developed by pulmonologists, patients, and patient families, confirmed the urgent necessity of guidelines, information, and equal access to patient testing (18, 123). To provide some guidance, the Pulmonary Fibrosis Foundation commissioned a genetic testing group which has reviewed the clinical scenarios in which genetic testing should be offered (125). At the same time, a multidisciplinary expert group of the ERS worked in a statement for better managing FPF, including a question regarding which patients could benefit more from genetic sequencing (11). All of them concur in a series of indications summarized in the following sections (Figure 2).
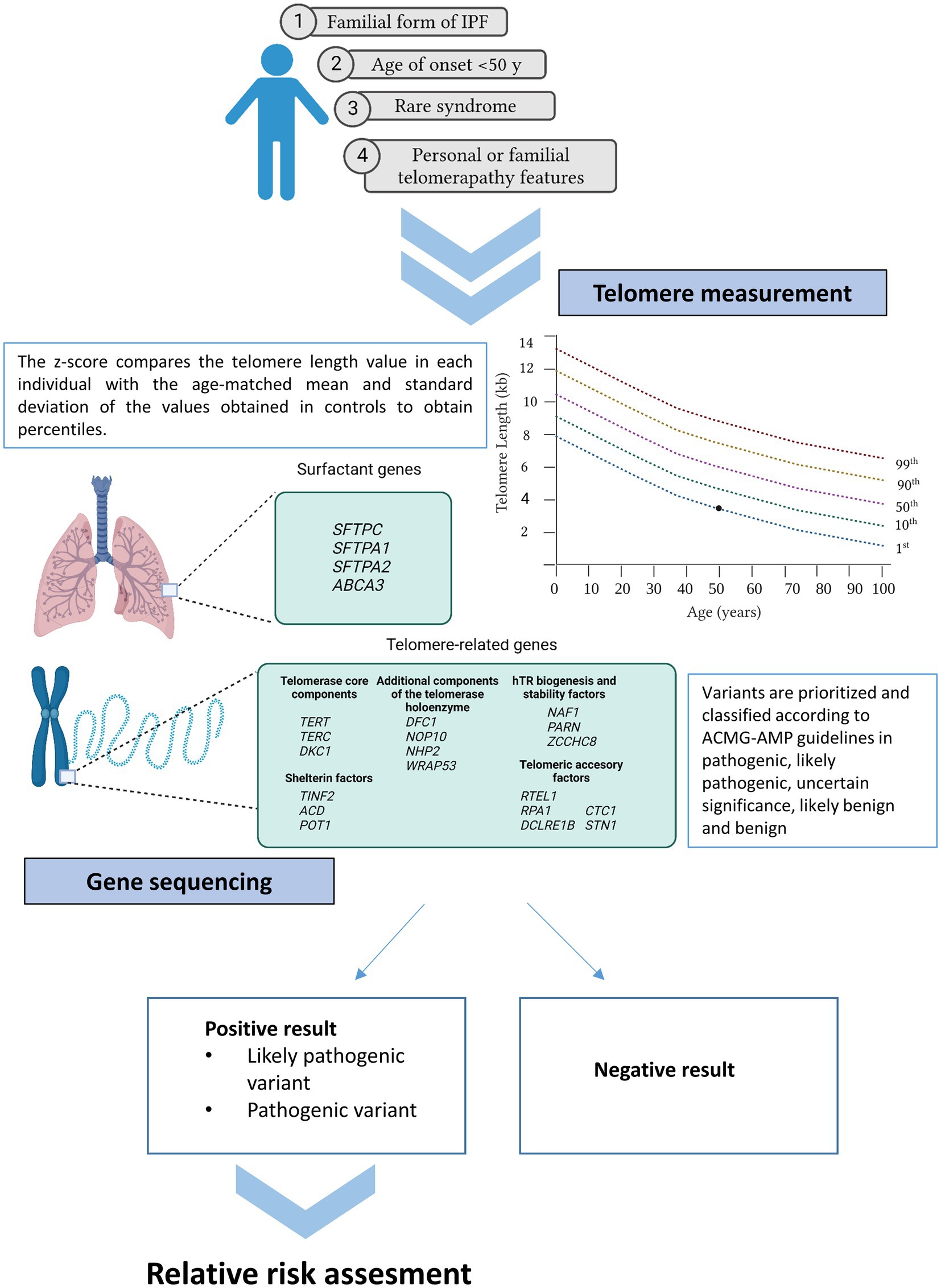
Figure 2. Current recommendations for genetic testing in IPF. Patients meeting at least one of the cited criteria may benefit from genetic testing (telomere length measurement and gene sequencing). If a positive result is obtained in the proband, first relatives can benefit from genetic analysis under request.
3.1. Methods for clinical genetic testing
The approach for identifying deleterious variants in IPF patients is DNA sequencing. In addition, cases with suspicion of telomere dysfunction can benefit from TL measurement.
3.1.1. TL determination
All methods developed to measure TL face advantages and disadvantages. The most validated methods include qPCR (quantitative Polymerase Chain Reaction) and flow-FISH (Fluorescence In Situ Hybridization). TeSLA (Telomere Shortest Length Assay) and TRF (Terminal Restriction Fragment) analyses have shown to be as reliable as other available options (126). More details about these techniques are provided in Table 2.
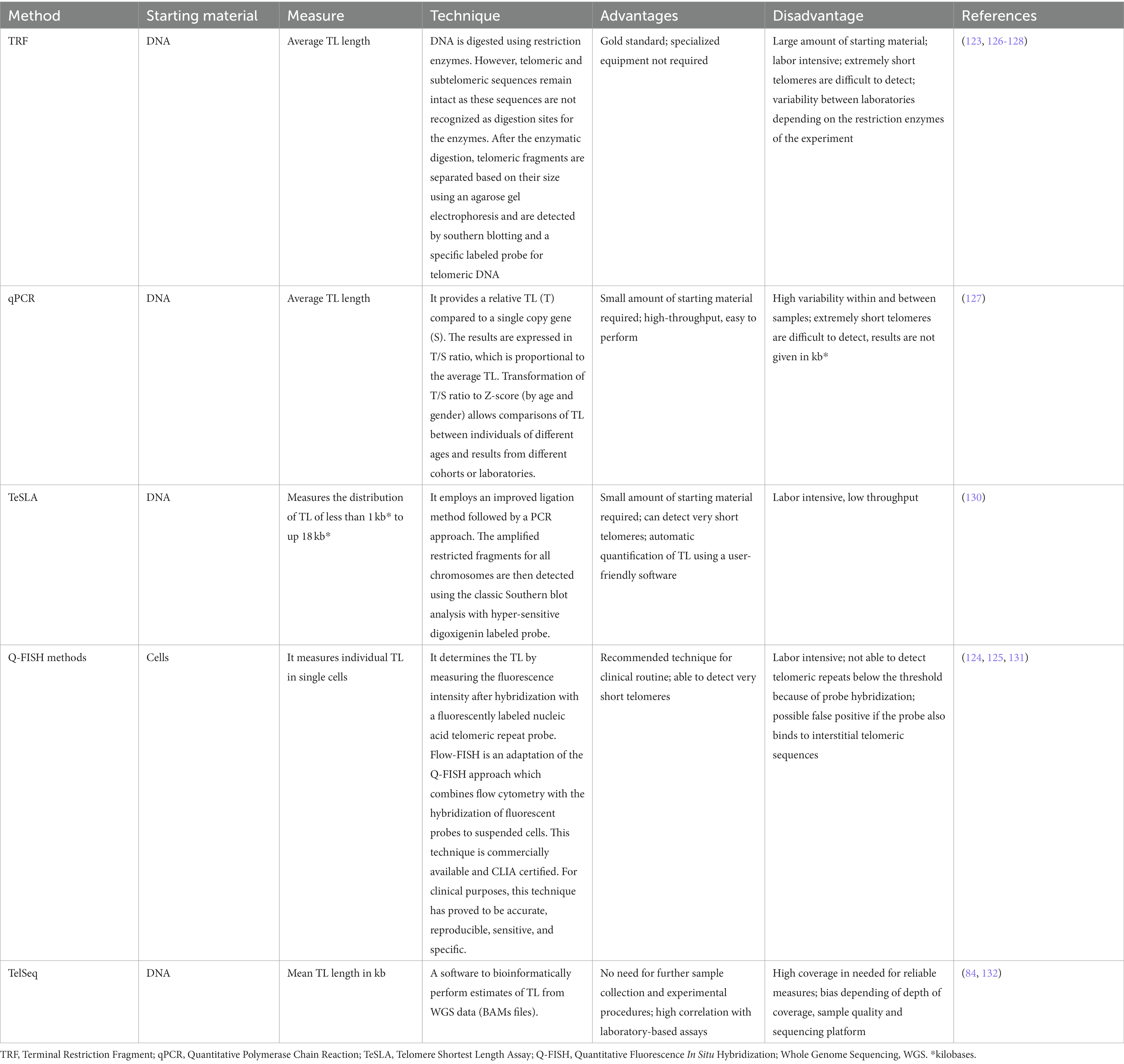
Table 2. Considerations of the available technologies for telomere length (TL) measurement.
Severe reduction is generally denoted when TL is below the 10th percentile of normal controls from the population. Clinical reporting of TL provides information about age-adjusted percentiles and are often represented graphically on telograms (Figure 2).
3.1.2. NGS methods and variant interpretation
3.1.2.1. The use of gene panels and ES
NGS allows sequencing of many genomic regions in a rapid and cost-effective way. Sequencing the whole genome, or a targeted portion of it, has proven to be extraordinarily useful for the identification of molecular causes of genetic diseases. Therefore, its use is widely accepted in the clinical routine (133). Two levels of analysis are commonly performed via NGS during the clinical diagnosis of IPF: gene panels and ES.
Gene panels examine the sequence of a curated set of genes associated with the interrogated phenotype and they are considered the first-line test for multiple genetic disorders. By limiting the size of the target sequence, the results usually hold higher sensitivity and specificity for detecting pathogenic mutations (133). Moreover, if only genes with a proven established role in the disease are included, the ability to interpret the findings is greater. In the case of IPF, as in many other diseases, there is yet no consensus about which genes to be included in such a gene panel despite the existence of diverse commercial gene panels for it. IPF is a complex disease and there is still little knowledge about which genes might be involved in its pathogenesis. In addition, as larger cohorts are being sequenced, new disease genes are being discovered that may meet criteria for inclusion in the gene panels (133). This could result in different findings provided by different practitioners depending on the genetic solutions employed. One way to theoretically avoid this issue is by performing ES (133). ES focuses on determining the DNA sequence from nearly all protein coding genes (about 1–2%) and adjacent intronic regions across the genome. This region contains most disease-causing variants identified to date. Thus, one of the major advantages of this technique is the possibility to filter ES data and restrict the analysis to those genes specifically related to the disease. This approximation is equivalent to a virtual gene panel and provides the flexibility to expand the analysis by re-visiting the pre-existing ES data if new disease genes are identified, as it might be the case for IPF (134, 135).
3.1.2.2. Variant calling, filtering, and the challenge of variant prioritization
Independently of the technology of choice, following the steps of variant calling, filtering, and variant interpretation is critical to provide a precise result (Figure 3). Initially, bioinformatics analysis is required to align the target sequence (gene panel or exome) against the reference sequence of the human genome (136, 137). Thus, patient and reference sequences are compared, and any supported change detected by the variant calling algorithm is further subjected to additional filtering steps and added to the list of variants for the patient. In an ES analysis, however, this process can generate up to 20,000 variants hindering a manual examination (138). For that reason, variants are typically prioritized based on annotated data for relevant biological information such as genomic coordinates, coding sequence nomenclature, protein nomenclature and position relative to the gene. Additionally, relevant information from external resources such as population information or disease specific databases can also be included and used for prioritization and to facilitate the assessment of their clinical relevance (139). One of the first filters to apply uses information regarding the frequency of the variants in the general population from different ethnicities. Although common variants also contribute to the risk of IPF, they are recurrent in the population and their clinical implications are yet unknown. Because of this, genetic testing focuses on identifying rare, non-recurrent, and highly penetrant variants which can be causative of the disease. Therefore, specific thresholds of frequency can be set to restrict the search for rare variants of potential interest. Population databases such as gnomAD3 are used with that purpose (139). However, it cannot be assumed that only healthy individuals are represented in these public population databases, especially in the case of a late-onset disease such as IPF. Another useful information to assist in variant interpretation comes from disease-causing databases, for example the Human Gene Mutation Database (140) or ClinVar (141), which contain variants previously observed in affected individuals. Specifically for IPF, The Telomerase Database4 compile known mutations that cause human telomerase-deficiency diseases and their associated clinical phenotypes (141). In FPF, in which more than one family member is affected, is also relevant to sequence different family members to assess familial segregation. Thus, if a variant segregates with the phenotype in the family, it constitutes further evidence to support its pathogenicity (139). Finally, the functional consequence of the variant should be considered. In silico predictive pathogenicity tools, such as Sorting Intolerant from Tolerant (SIFT), PolyPhen2, Mutation Taster, Variant Effect Predictor (VEP), or the Combined Annotation Dependent Depletion (CADD) score can help in this step, especially for missense mutations (139). All these predictors, however, should be used with caution, as they usually use a fixed cutoff value, identical for all genes in order to filter out benign variants from the NGS data. To improve the use of some of these variant-level methods, the mutation significance cutoff (MSC)5 introduced gene-level and gene-specific phenotypic impact cutoff values. Thus, the MSC of a gene is defined as the lower limit of the 99, 95% or 90% CI for the CADD, Polyphen-2 or SIFT scores of all high-quality mutations described as pathogenic in the Human Gene Mutation Database or ClinVar database (142).
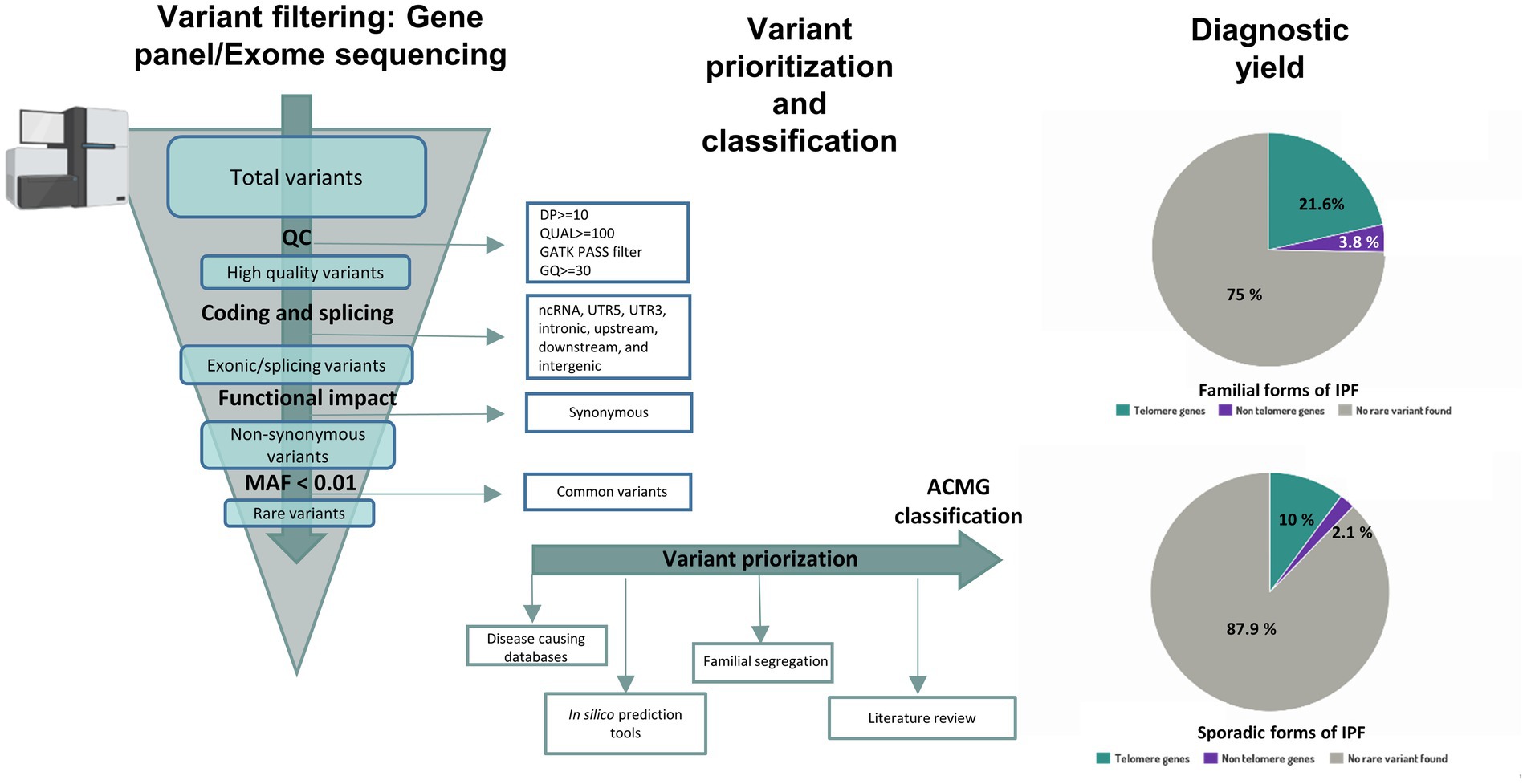
Figure 3. Workflow for variant interpretation in IPF. Annotated variants are filtered applying quality control metrics. High quality coding variants are filtered by predicted consequences on the protein level and non-coding RNA variants, intronic variants and variants inside 5’ or 3’ UTR are discarded. Population allele frequency filters are used to restrict following analysis of rare variants. For variant interpretation, disease causing databases, in-silico prediction tools, literature review and information about familial segregation can be used. Diagnostic yield in familial and sporadic forms of IPF from Zhang et al (12) is shown in pie charts.
After considering the above information, resulting variants are classified at the moment following recommendations from the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) in five categories: pathogenic, likely pathogenic, uncertain significance, likely benign, or benign (143). A positive result indicates that a patient harbors a pathogenic or likely pathogenic variant in a gene previously implicated with IPF, and therefore, it provides a molecular diagnosis. A negative result, however, means that the causative variant was not identified. Unfortunately, this is the most common result of a genetic testing. However, it should be noted that a negative result does not imply that there is no genetic cause associated to the condition in the patient. Despite efforts from the ACMG to provide criteria for classifying variants, the pieces of evidence are missing or are in conflict with each other in many cases. In these situations, the variant is reported as a variant of uncertain significance (VUS) (143). The advent of NGS technologies implies that more regions of the genome are sequenced, leading to a rapid increase in the number of VUS that can be identified (144). However, there is growing evidence that VUS reclassification is possible when several approaches are used: (1) functional in vitro studies at variant level that mimic the mutations and reproduce similar phenotype changes; (2) cumulative evidence from other cases sharing the same variant and phenotype, submitted to public databases; (3) segregation analysis of the variant between affected cases but within the same family; (4) existence of local population-level data to provide a more accurate allele frequency of the variant; and (5) improvement and updates in genetic variant annotation (145–148). Altogether, this evidence can contribute to new conclusions. The more data is collected for a variant, the higher probability will remain to reinterpret the result and refine its classification. For that reason, the reanalysis of NGS data after some time has improved the diagnostic yield in some settings (134, 149–151). Nowadays, 70% of IPF patients who undergo genetic testing have an unidentified genetic cause (Figure 1). However, diagnostic yield should improve as new genomic data is generated.
It should be noted that the workflow described above is specific for single nucleotide variants (SNVs) and indels (short insertions or deletions). Structural variants (SV) are defined as large regions (affecting more than 50 base pairs of sequence) that could involve changes in number of copies of a sequence (deletions, insertions, and duplications), orientation (inversions), or alterations of the chromosomal location (translocations) (152). They have been largely understudied across all diseases because they are more difficult to identify despite having high impact in the disease by altering gene dosage or regulatory elements that modulate gene expression (152). In the last years, many analytical tools for SV detection from NGS data have been developed and incorporated into the routine of clinical laboratories (152). Appropriate filtering strategies should be applied to identify causal SVs. To this aim, characteristics such as size and gene content, quality control metrics, allele frequency, and known association to the patient’s phenotype should be considered to classify and prioritize SVs out from genetic testing (153). However, SNPs and indels constitute the only types of variation that have been tested in relation to IPF for the moment.
3.2. IPF cases that most benefit from genetic testing
3.2.1. Patients with family history of IPF
Family history ascertainment is crucial for identifying familial cases of IPF. Therefore, the construction of a pedigree and the collection of the medical history from relatives within at least three generations are essential (125). Importantly, family history should be updated, as it has been described that up to 10% of sporadic forms will subsequently reclassify to familial forms as new relatives are diagnosed during the follow-up (154). The main reason of this procedure is that rare variants are enriched in families with IPF. These variants are expected to confer higher individual risk and tend to be eliminated from the population by purifying selection (155). These types of variants cosegregate with IPF in families as they alone can cause the disease. Consequently, the identification of the genetic cause in familial cases is higher in comparison with sporadic forms, their genetic diagnosis being estimated in 25% (95% [CI]: 21–30%) (12) (Figure 2).
3.2.2. Patients with extra pulmonary symptoms
Different extrapulmonary manifestations may be considered as part of telomere-related syndrome and suggestive of a telomere disorder. Accelerated telomere shortening manifests in a broad spectrum of symptoms involving tissues with high proliferation rates, such as epithelium or the hematological system (25). IPF patients with liver cirrhosis, bone narrow failure, myelodysplastic syndrome, acute myeloid leukemia, and/or premature hair graying present more frequently a pathogenic telomere-related gene mutation (154) (Figure 2).
3.2.3. Patients with early onset of disease
Classically, IPF is an aging disease that usually affects patients older than 60 years (105). Therefore, disease onset is rare before age of 50 and it might be suggestive of the existence of pathogenic mutations in surfactant or telomere related genes (86). Thus, even in sporadic forms of IPF, genetic testing may be useful in those patients in which clinical symptoms develops at a young age (Figure 2).
3.2.4. Monogenic syndromic diseases
Genetic testing should also be considered when there is a suspicion of a genetic syndrome such as Dyskeratosis congenita, Hermansky-Pudlak syndrome, Coats plus syndrome, Høyeraal–Hreidarsson syndrome, and Revesz syndrome (125). In all of them, pulmonary fibrosis is present along with other symptoms affecting multiple organs. Dyskeratosis congenita was the first recognized telomere syndrome, and it is clinically characterized by the triad of abnormal nails, reticular skin pigmentation, and oral leucoplakia. Affected patients are also at risk of experiencing bone marrow failure and cancer predisposition (156). A severe subset of Dyskeratosis congenita is the Høyeraal–Hreidarsson syndrome, which manifests with progressive bone marrow failure, cerebellar hypoplasia, immunodeficiency, and intrauterine growth retardation (156). The second subset of Dyskeratosis congenita is Revetz Syndrome, which also requires being diagnosed with the presence of bilateral exudative retinopathy and central nervous system calcifications. Hermansky-Pudlak syndrome manifests with oculocutaneous albinism and excessive bleeding of variable severity (156). Finally, Coats plus syndrome, a cerebroretinal microangiopathy with calcifications and cyst, is a rare disorder that mainly affects the eyes, brain, bone, and the gastrointestinal system (156) (Figure 2).
3.3. The main genes to test
It is expected that approximately up to 30% of the FPF cases are monogenic and that these cases are enriched with rare variants within genes related to two main pathways: surfactant metabolism and telomere biology (11). For that reason, genetic analysis is usually restricted to the genes from these pathways. Among the surfactant genes, SFTPC, SFTPA1, SFTPA2, and ABCA3 are included as consensus in the analysis (Figure 2), despite mutations in these genes explain a small percentage of the total number of cases (1–3%) (Figure 1) (86). Regarding the telomere biology genes, it still less clear which genes should be analyzed. In the past, most of the patients meeting the criteria for genetic testing only had TERT or TERC genes sequenced since they were the first genes to underlie AD forms of IPF (157, 158). Moreover, mutations in both genes explain up to 15% of the cases (159). Borie et al. and Kropski et al., based on their own experience, proposed the inclusion of at least TERT, TERC, RTEL1, PARN, NAF1, DKC1, and TINF2 genes in the analysis (11, 160). While others have extended the study to all 188 known genes that have been related to hematological disorders (84). More recently, additional telomere biology genes are being identified as causal of IPF via ES in affected families. Mutations in these genes are restricted to a few individuals. However, they contribute to better explain the spectrum of the disease causes and, for that reason, they should be also tested, especially in syndromic presentations. This is the case of NOP10, NHP2, WRAP53, ZCCHC8, ACD, POT1, RPA1, DCLRE1B, CTC1, and STN1. Rare variants in most of these genes are transmitted through various modes of inheritance (X-linked, autosomal recessive, and AD). Biallelic variants are usually related with syndromic forms such Dyskeratosis congenita, Høyeraal–Hreidarsson syndrome, and Coats plus syndrome (25) (Figure 2).
3.4. Utility of TL measurements
Telomere shortening is described in up to 50% of the familial forms (161, 162). In patients with telomerase complex mutations, a severe reduction of TL is observed in 80–90% of the cases. For that reason, some authors have proposed to limit gene sequencing to those patients with short telomeres (154), although some telomere related gene carriers and surfactant-related gene mutations would not be genetically diagnosed. Moreover, most of the techniques for telomere measurement offer an average on TL. However, it has been proved that it is not the average but the shortest telomeres of the cells the determinant factor of the responses. It implies that just a few short telomeres of a cell might trigger the DNA damage response (163). In addition, TL effects varies among telomere gene mutations (105).
Some studies suggest that TL testing should be performed in conjunction with gene sequencing, as it may provide information for variant interpretation and assist the clarification of the functional consequences of candidate variants. Alder et al. showed that age-adjusted TL below the 50th percentile has a 100% negative predicted value for clinically relevant mutations in telomere biology genes. Moreover, TL is also predictive of patient prognosis and the approximate timing of disease onset (131). First degree relatives may also benefit from this practice, even when no telomere biology gene mutation has been found, since they are still at risk of developing the disease if telomere shortening is observed in the kindred. In those cases, periodic clinical screening is recommended (40).
3.5. Genetic counseling
Briefly, the standard procedure for those patients who meet criteria for being tested is to provide them with relevant information about the method of testing and receive the required educational preparation and training about the benefits and risks of the process (164). In an optimal setting, this information is typically provided by genetic health care professionals (genetic counselors or clinical geneticists) which are members of a multi-disciplinary team (11, 132).
Pre-test counseling in a complex disease, such as IPF, is challenging but also necessary. Understanding the implication of genetic factors in the disease is a crucial step of the process which helps patients to understand the potential results and improve their decision-making. They must be provided with basic concepts of genetic inheritance. In addition, patients who receive genetic counseling should also understand complex concepts such as penetrance or the genetic anticipation (as could be the case for telomere disorders) because of the influence in the natural history of the disease in other family members (127). Thus, patients should understand that the potential results have consequences for them but also for their relatives. Identifying a pathogenic variant may influence the clinical management and may confer prognostic information. Despite this finding should prompt consideration of further genetic testing in asymptomatic relatives, the benefits of this are yet unclear in IPF (see next section). Another challenge arises when a VUS is identified instead of a pathogenic variant. This is, in fact, the most frequent scenario in FPF cases. Patients should be aware of this possibility and understand that VUS can be reclassified as either disease-causing or benign in the future (165). Finally, it should be clear that a negative result does not exclude the presence of a disease-causing variant in IPF genes that remain undiscovered. Thus, the findings of the genetic testing might be provisional results.
The current real situation is that there are wide differences between countries regarding aspects related to genetic counseling such as laws, health systems, or culture (166). Furthermore, in most centers, pulmonologists are responsible for providing information relative to the genetic tests and for communicating the results due to the lack of geneticists or genetic counselors that are integrated to ILD centers (132). A recent international survey revealed the urgent need for improving the genetic counseling in familial ILD by training ILD pulmonologists in genetics and/or including geneticists and genetic counselors in the diagnostic teams (123).
4. Future perspectives
In recent years, the application of high-throughput genomic technologies has led to the conclusion that a great inter-individual variation exists regarding the mechanisms that contribute to the disease. This variability is expected to impact its treatment, monitoring and diagnosis. In this review, we have summarized some important genetic aspects of IPF and how the current knowledge is starting to guide the molecular diagnosis of patients and relatives. However, this field is continuously evolving, and new approaches are becoming available and are expected to contribute to the implementation of precision medicine in the healthcare system.
Common and rare variants, as well as TL information can inform about disease risk and outcome of IPF patients. In addition, unexplored types of genetic variants of interest for IPF, such as SVs or variants affecting non-coding regions, are expected to be discovered as larger cohorts are included in genomic research studies (16). Despite the wide spectrum of discovered variants involved in IPF pathogenesis, genetic screening in clinical settings is not yet a common practice for the identification of rare variants with high effect sizes using gene panels or ES. Whole genome sequencing (WGS) has proven to increase the diagnostic yields in other diseases, thanks to the improvement of coding coverage consistency, increased power for detecting SVs, repeat variants, and sequencing of newly annotated coding-regions (167). Particularly in IPF, WGS offers an important advantage over ES due to its ability to identify rare and common variants, and to simultaneously estimate the TL (12). Thus, most rare deleterious qualifying variants in IPF patients are found in telomere biology genes (85%), while non-telomere biology variants are found in a smaller proportion (15%) (12). To determine the biological consequence of these variants, it is necessary to provide TL measures to verify that there is a correlation between the candidate variant effect and the TL. TL, however, is not usually provided by genetic testing laboratories and measures are usually given by alternative services which commonly apply Flow-FISH or qPCR approaches. Telseq and other approaches can bioinformatically estimate TL from WGS data (168) (Table 2). Some of these methods have shown a high correlation with Southern blot and Flow-FISH results (131, 168, 169). For that reason, this approach is becoming more widely used in large cohorts in which a reliable telomere measure is needed (85). However, bias in TL estimation may exist depending on depth of coverage, sample quality, and the sequencing platform used, and all these factors should be taken into account when candidate variants are prioritized (168). A further advantage of WGS is the possibility of detecting common variants beyond gene exons, which also contribute to the risk of IPF. However, effect sizes of individual common variants are very small. For that reason, they are usually discarded for genetic testing. One way to incorporate their information into the analysis is to measure their aggregated effect using PRS (170). The classic way to calculate the PRS is computing the sum of risk alleles that an individual harbors, weighted by the risk allele effect size as estimated by a GWAS on the phenotype (171). If a large number of variants are considered, a substantial greater predictive power is achieved (172). PRS calculated from risk variants previously linked to leukocyte TL (173) has been associated with short TL and disease progression (12). However, the estimation of PRS is not useful for all IPF patients and is not yet a standardized procedure despite its potential. Besides, both familial and sporadic forms might have a different genetic background, meaning that different approaches need to be applied for genetic screening purposes in these two situations. In agreement with this idea, the effect of common variant built PRS is notable only for sporadic cases (those who current guides do not recommend applying genetic screening). Instead, familial forms are five times more likely to be affected by an inherited rare pathogenic variant (12). An updated review of the personal and clinical utility of PRS in complex diseases has been published elsewhere (174).
Another important point of discussion regarding genetic testing is the possibility of offering it to asymptomatic family members. In general, if a monogenic disease is proven in a family, first-grade relatives may benefit from genetic screening if they can make their own informed decision. However, penetrance and expressivity (severity) in IPF is variable and should be carefully considered and explained when the subject receives the related information before being tested (125, 132, 160). The phenomenon of anticipation also complicates the decision. Genetic anticipation is observed under a progressively earlier age of onset and increase of severity of symptoms in each generation. Anticipation is frequently observed in families where IPF is caused by mutations in telomere-related genes and the mechanism that mostly explain it is believed to be telomere shortening (26, 105, 157). It has been suggested that the inheritance of short telomeres in non-mutation carriers of these kindreds might be a sufficient cause to induce the expression of the disease (40). In those cases, it is not the absence of telomerase but rather the short telomeres themselves that cause stem cell failure, as it has been shown in studies conducted in wild type mice with inherited short telomeres (26, 67). The latency before the accumulative effect of telomere shortening leading to IPF is still unknown, although a recent study of the frequently observed pathogenic TERT c.2005 T mutation in a Dutch cohort estimates the origin in a founder individual who lived in The Netherlands 300 years ago (68). For all of these reasons, there is no evidence-based practice about when and what tests (DNA sequencing and/or TL measures) should be included in the screening of asymptomatic family members. Further research is needed to assess the predictive power of genetic screening in IPF relatives.
Despite the important advances in the study of IPF genetics in recent years, there are still important caveats regarding molecular mechanisms and the genes involved in the disease. Large GWAS studies and exome or genome sequencing studies have been successful in identifying tens of loci associated with disease risk (15, 16, 82). However, it is expected that new risk genes will emerge when larger cohorts are analyzed, involving sequence data or other omics technologies that could be integrated to improve the predictive power of current studies. For example, an approach that has been used in other complex diseases, such as psychiatric disorders, is leveraging shared genetics between multiple phenotypes for discovering new genes. This is done through multi-trait association mapping (MTAG) studies (74). Another successful approach consists of integrating genetic signals into gene expression data and testing the association between the predicted gene expression and a trait through transcriptome-wide association studies (TWAS). In fact, the integration of both strategies in a cohort of IPF patients led to the discovery of two new candidate genes, MAFK and SMAD2. Both genes are transcription factors that are related to IPF by regulating target genes which are differentially expressed in specific IPF cell types of affected patients (75).
As new disease mechanisms are being unfolded, new target therapies could be formulated. Until now, the only approved drugs for IPF by the U.S Food and Drug Administration are nintedanib and pirfenidone. Nevertheless, multiple clinical trials are ongoing, and it is expected that target therapies for subsets of patients, defined by a particular genotype, will be developed in line with the principles of precision medicine.
5. Conclusion
IPF is a devastating disease with genetic predisposition to disease development and progression, already identified in sub-groups of patients such as those with family aggregation. Significant advances have been made in the field of genetics in the last decade. The ultimate goal of genomic research is to understand the complex genetic architecture of IPF. However, integrating all this knowledge into clinical practice is at reach despite the remaining challenges. In this study, we have attempted to provide detailed information of the known genetic basis of IPF and the potential impact of rare and common variants on disease outcomes. Although the lack of consensus guidelines about when to pursue genetic screening, here we expose current update derived from the perspective of different studies. Future research applying WGS or integrating approaches to combine multiple omics data will further help us to unravel the molecular mechanisms driving pulmonary fibrosis. Finally, advances in new compounds targeting identified gene dysfunctions will provide a better therapeutic approach for these patients.
Author contributions
AA-G and CF conceptualized the study and wrote the first draft of the manuscript. CF and MM-M obtained the funding. AA-G designed the tables and figures. AA-G, ET-H, MM-M, and CF contributed revising the manuscript. All authors approved the final version.
Funding
This research was funded by the Instituto de Salud Carlos III (PI20/00876; PMP22/00083) and co-financed by the European Regional Development Funds, “A way of making Europe” from the European Union; the agreement OA17/008 with Instituto Tecnológico y de Energías Renovables (ITER) to strengthen scientific and technological education, training, research, development and innovation in Genomics, Personalized Medicine and Biotechnology; and by Cabildo Insular de Tenerife (CGIEU0000219140). AA-G was supported by Ministerio de Universidades (modality Margarita Salas) and ET-H was supported by fellowship from Agencia Canaria de Investigación, Innovación y Sociedad de la Información del Gobierno de Canarias (TESIS2021010046).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
1. ^https://gnomad.broadinstitute.org/variant/11-1241221-G-T?dataset=gnomad_r2_1
2. ^https://clinicaltrials.gov: NCT00650091.
3. ^https://gnomad.broadinstitute.org
References
1. Meltzer, EB, and Noble, PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis. (2008) 3:8. doi: 10.1186/1750-1172-3-8
2. Richeldi, L, Collard, HR, and Jones, MG. Idiopathic pulmonary fibrosis. Lancet Lond Engl. (2017) 389:1941–52. doi: 10.1016/S0140-6736(17)30866-8
3. Mathai, SK, Schwartz, DA, and Borie, R. Genetic determinants of interstitial lung diseases In: RE Pyeritz, BR Korf, and WW Grody, editors. Emery and Rimoin’s principles and practice of medical genetics and genomics : Amsterdam, The Netherlands: Elsevier (2020). 405–37.
4. Pitre, T, Mah, J, Helmeczi, W, Khalid, MF, Cui, S, Zhang, M, et al. Medical treatments for idiopathic pulmonary fibrosis: a systematic review and network meta-analysis. Thorax. (2022) 77:1243–50. doi: 10.1136/thoraxjnl-2021-217976
5. Sköld, CM, Bendstrup, E, Myllärniemi, M, Gudmundsson, G, Sjåheim, T, Hilberg, O, et al. Treatment of idiopathic pulmonary fibrosis: a position paper from a Nordic expert group. J Intern Med. (2017) 281:149–66. doi: 10.1111/joim.12571
6. Luppi, F, Spagnolo, P, Cerri, S, and Richeldi, L. The big clinical trials in idiopathic pulmonary fibrosis. Curr Opin Pulm Med. (2012) 18:428–32. doi: 10.1097/MCP.0b013e3283567ff9
7. Martinez, FJ, Safrin, S, Weycker, D, Starko, KM, Bradford, WZ, King, TE, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. (2005) 142:963–7. doi: 10.7326/0003-4819-142-12_Part_1-200506210-00005
8. Raghu, G, Collard, HR, Egan, JJ, Martinez, FJ, Behr, J, Brown, KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. (2011) 183:788–824. doi: 10.1164/rccm.2009-040GL
9. Fisher, M, Nathan, SD, Hill, C, Marshall, J, Dejonckheere, F, Thuresson, PO, et al. Predicting life expectancy for pirfenidone in idiopathic pulmonary fibrosis. J Manag Care Spec Pharm. (2017) 23:S17–24. doi: 10.18553/jmcp.2017.23.3-b.s17
10. Kropski, JA, Blackwell, TS, and Loyd, JE. The genetic basis of idiopathic pulmonary fibrosis. Eur Respir J. (2015) 45:1717–27. doi: 10.1183/09031936.00163814
11. Borie, R, Kannengiesser, C, Antoniou, K, Bonella, F, Crestani, B, Fabre, A, et al. European respiratory society statement on familial pulmonary fibrosis. Eur Respir J. (2022) 61:2201383. doi: 10.1183/13993003.01383-2022
12. Zhang, D, Povysil, G, Kobeissy, PH, Li, Q, Wang, B, Amelotte, M, et al. Rare and common variants in KIF15 contribute to genetic risk of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2022) 206:56–69. doi: 10.1164/rccm.202110-2439OC
13. Newton, CA, Kozlitina, J, Lines, JR, Kaza, V, Torres, F, and Garcia, CK. Telomere length in patients with pulmonary fibrosis associated with chronic lung allograft dysfunction and post–lung transplantation survival. J Heart Lung Transplant. (2017) 36:845–53. doi: 10.1016/j.healun.2017.02.005
14. Planas-Cerezales, L, Arias-Salgado, EG, Buendia-Roldán, I, Montes-Worboys, A, López, CE, Vicens-Zygmunt, V, et al. Predictive factors and prognostic effect of telomere shortening in pulmonary fibrosis. Respirol Carlton Vic. (2019) 24:146–53. doi: 10.1111/resp.13423
15. Allen, RJ, Guillen-Guio, B, Oldham, JM, Ma, SF, Dressen, A, Paynton, ML, et al. Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2020) 201:564–74. doi: 10.1164/rccm.201905-1017OC
16. Partanen, JJ, Häppölä, P, Zhou, W, Lehisto, AA, Ainola, M, Sutinen, E, et al. Leveraging global multi-ancestry meta-analysis in the study of idiopathic pulmonary fibrosis genetics. Cell Genomics. (2022) 2:100181. doi: 10.1016/j.xgen.2022.100181
17. Seibold, MA, Wise, AL, Speer, MC, Steele, MP, Brown, KK, Loyd, JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. (2011) 364:1503–12. doi: 10.1056/NEJMoa1013660
18. Raghu, G, Remy-Jardin, M, Richeldi, L, Thomson, CC, Inoue, Y, Johkoh, T, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2022) 205:e18–47. doi: 10.1164/rccm.202202-0399ST
19. Peljto, AL, Blumhagen, RZ, Walts, AD, Cardwell, J, Powers, J, Corte, TJ, et al. Idiopathic pulmonary fibrosis is associated with common genetic variants and limited rare variants. Am J Respir Crit Care Med. (2023). doi: 10.1164/rccm.202207-1331OC
20. Leavy, OC, Ma, SF, Molyneaux, PL, Maher, TM, Oldham, JM, Flores, C, et al. Proportion of idiopathic pulmonary fibrosis risk explained by known common genetic loci in European populations. Am J Respir Crit Care Med. (2020) 203:775–8. doi: 10.1164/rccm.202008-3211LE
21. Uffelmann, E, Huang, QQ, Munung, NS, de Vries, J, Okada, Y, Martin, AR, et al. Genome-wide association studies. Nat Rev Methods Primer. (2021) 1:59. doi: 10.1038/s43586-021-00056-9
22. Martínez, P, and Blasco, MA. Replicating through telomeres: a means to an end. Trends Biochem Sci. (2015) 40:504–15. doi: 10.1016/j.tibs.2015.06.003
23. Armanios, M, and Blackburn, EH. The telomere syndromes. Nat Rev Genet. (2012) 13:693–704. doi: 10.1038/nrg3246
24. Sperka, T, Song, Z, Morita, Y, Nalapareddy, K, Guachalla, LM, Lechel, A, et al. Author correction: puma and p21 represent cooperating checkpoints limiting self-renewal and chromosomal instability of somatic stem cells in response to telomere dysfunction. Nat Cell Biol. (2021) 23:292–2. doi: 10.1038/s41556-021-00633-w
25. Revy, P, Kannengiesser, C, and Bertuch, AA. Genetics of human telomere biology disorders. Nat Rev Genet. (2022) 24:86–108. doi: 10.1038/s41576-022-00527-z
26. Armanios, M, Chen, JL, Chang, YPC, Brodsky, RA, Hawkins, A, Griffin, CA, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci U S A. (2005) 102:15960–4. doi: 10.1073/pnas.0508124102
27. Armanios, MY, Chen, JJL, Cogan, JD, Alder, JK, Ingersoll, RG, Markin, C, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. (2007) 356:1317–26. doi: 10.1056/NEJMoa066157
28. Tsakiri, KD, Cronkhite, JT, Kuan, PJ, Xing, C, Raghu, G, Weissler, JC, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci. (2007) 104:7552–7. doi: 10.1073/pnas.0701009104
29. Cogan, JD, Kropski, JA, Zhao, M, Mitchell, DB, Rives, L, Markin, C, et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am J Respir Crit Care Med. (2015) 191:646–55. doi: 10.1164/rccm.201408-1510OC
30. Stuart, BD, Choi, J, Zaidi, S, Xing, C, Holohan, B, Chen, R, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet. (2015) 47:512–7. doi: 10.1038/ng.3278
31. Kropski, JA, Mitchell, DB, Markin, C, Polosukhin, VV, Choi, L, Johnson, JE, et al. A Novel dyskerin (DKC1) mutation is associated with familial interstitial pneumonia. Chest (2014);146:e1–e7. doi: 10.1378/chest.13-2224
32. Gable, DL, Gaysinskaya, V, Atik, CC, Talbot, CC Jr, Kang, B, Stanley, SE, et al. ZCCHC8, the nuclear exosome targeting component, is mutated in familial pulmonary fibrosis and is required for telomerase RNA maturation. Genes Dev. (2019) 33:1381–96. doi: 10.1101/gad.326785.119
33. Stanley, SE, Gable, DL, Wagner, CL, Carlile, TM, Hanumanthu, VS, Podlevsky, JD, et al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci Transl Med. (2016) 8:351ra107. doi: 10.1126/scitranslmed.aaf7837
34. Alder, JK, Stanley, SE, Wagner, CL, Hamilton, M, Hanumanthu, VS, and Armanios, M. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest. (2015) 147:1361–8. doi: 10.1378/chest.14-1947
35. Hoffman, TW, van der Vis, JJ, van der Smagt, JJ, Massink, MPG, Grutters, JC, and van Moorsel, CHM. Pulmonary fibrosis linked to variants in the ACD gene, encoding the telomere protein TPP1. Eur Respir J. (2019) 54:1900809. doi: 10.1183/13993003.00809-2019
36. Alder, JK, Chen, JJL, Lancaster, L, Danoff, S, Su, S, Chih, CJD, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci. (2008) 105:13051–6. doi: 10.1073/pnas.0804280105
37. Cronkhite, JT, Xing, C, Raghu, G, Chin, KM, Torres, F, Rosenblatt, RL, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med. (2008) 178:729–37. doi: 10.1164/rccm.200804-550OC
38. Kannengiesser, C, Borie, R, Ménard, C, Réocreux, M, Nitschké, P, Gazal, S, et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur Respir J. (2015) 46:474–85. doi: 10.1183/09031936.00040115
39. Schratz, KE, Haley, L, Danoff, SK, Blackford, AL, DeZern, AE, Gocke, CD, et al. Cancer spectrum and outcomes in the Mendelian short telomere syndromes. Blood. (2020) 135:1946–56. doi: 10.1182/blood.2019003264
40. van der Vis, JJ, van der Smagt, JJ, van Batenburg, AA, Goldschmeding, R, van Es, HW, Grutters, JC, et al. Pulmonary fibrosis in non-mutation carriers of families with short telomere syndrome gene mutations. Theatr Res Int. (2021) 26:1160–70. doi: 10.1111/resp.14145
41. Degenhardt, F, Ellinghaus, D, Juzenas, S, Lerga-Jaso, J, Wendorff, M, Maya-Miles, D, et al. Detailed stratified GWAS analysis for severe COVID-19 in four European populations. Hum Mol Genet. (2022) 31:3945–66. doi: 10.1093/hmg/ddac158
42. Planas-Cerezales, L, Arias-Salgado, EG, Berastegui, C, Montes-Worboys, A, González-Montelongo, R, Lorenzo-Salazar, JM, et al. Lung transplant improves survival and quality of life regardless of telomere dysfunction. Front Med. (2021) 8:695919. doi: 10.3389/fmed.2021.695919
43. Stuart, BD, Lee, JS, Kozlitina, J, Noth, I, Devine, MS, Glazer, CS, et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir Med. (2014) 2:557–65. doi: 10.1016/S2213-2600(14)70124-9
44. Borie, R, Kannengiesser, C, Hirschi, S, Le Pavec, J, Mal, H, Bergot, E, et al. Severe hematologic complications after lung transplantation in patients with telomerase complex mutations. J Heart Lung Transplant. (2015) 34:538–46. doi: 10.1016/j.healun.2014.11.010
45. Whitsett, JA, Wert, SE, and Weaver, TE. Diseases of pulmonary surfactant homeostasis. Annu Rev Pathol. (2015) 10:371–93. doi: 10.1146/annurev-pathol-012513-104644
46. Nogee, LM, Dunbar, AE, Wert, SE, Askin, F, Hamvas, A, and Whitsett, JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. (2001) 344:573–9. doi: 10.1056/NEJM200102223440805
47. Lawson, WE. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax. (2004) 59:977–80. doi: 10.1136/thx.2004.026336
48. Ono, S, Tanaka, T, Ishida, M, Kinoshita, A, Fukuoka, J, Takaki, M, et al. Surfactant protein C G100S mutation causes familial pulmonary fibrosis in Japanese kindred. Eur Respir J. (2011) 38:861–9. doi: 10.1183/09031936.00143610
49. Tredano, M, Griese, M, Brasch, F, Schumacher, S, de Blic, J, Marque, S, et al. Mutation of SFTPC in infantile pulmonary alveolar proteinosis with or without fibrosing lung disease. Am J Med Genet A. (2004) 126A:18–26. doi: 10.1002/ajmg.a.20670
50. Abou Taam, R, Jaubert, F, Emond, S, Le Bourgeois, M, Epaud, R, Karila, C, et al. Familial interstitial disease with I73T mutation: a mid- and long-term study. Pediatr Pulmonol. (2009) 44:167–75. doi: 10.1002/ppul.20970
51. Lawson, WE, Cheng, DS, Degryse, AL, Tanjore, H, Polosukhin, VV, Xu, XC, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci. (2011) 108:10562–7. doi: 10.1073/pnas.1107559108
52. Mulugeta, S, Nguyen, V, Russo, SJ, Muniswamy, M, and Beers, MF. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol. (2005) 32:521–30. doi: 10.1165/rcmb.2005-0009OC
53. Tanjore, H, Cheng, DS, Degryse, AL, Zoz, DF, Abdolrasulnia, R, Lawson, WE, et al. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. (2011) 286:30972–80. doi: 10.1074/jbc.M110.181164
54. Coghlan, MA, Shifren, A, Huang, HJ, Russell, TD, Mitra, RD, Zhang, Q, et al. Sequencing of idiopathic pulmonary fibrosis-related genes reveals independent single gene associations. BMJ Open Respir Res. (2014) 1:e000057. doi: 10.1136/bmjresp-2014-000057
55. Fernandez, BA, Fox, G, Bhatia, R, Sala, E, Noble, B, Denic, N, et al. A Newfoundland cohort of familial and sporadic idiopathic pulmonary fibrosis patients: clinical and genetic features. Respir Res. (2012) 13:64. doi: 10.1186/1465-9921-13-64
56. van Moorsel, CHM, van Oosterhout, MFM, Barlo, NP, de Jong, PA, van der Vis, JJ, Ruven, HJT, et al. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a Dutch cohort. Am J Respir Crit Care Med. (2010) 182:1419–25. doi: 10.1164/rccm.200906-0953OC
57. Wang, Y, Kuan, PJ, Xing, C, Cronkhite, JT, Torres, F, Rosenblatt, RL, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. (2009) 84:52–9. doi: 10.1016/j.ajhg.2008.11.010
58. van Moorsel, CHM, ten Klooster, L, van Oosterhout, MFM, de Jong, PA, Adams, H, Wouter van Es, H, et al. SFTPA2 mutations in familial and sporadic idiopathic interstitial pneumonia. Am J Respir Crit Care Med. (2015) 192:1249–52. doi: 10.1164/rccm.201504-0675LE
59. Maitra, M, Wang, Y, Gerard, RD, Mendelson, CR, and Garcia, CK. Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress. J Biol Chem. (2010) 285:22103–13. doi: 10.1074/jbc.M110.121467
60. Madsen, J, Tornøe, I, Nielsen, O, Koch, C, Steinhilber, W, and Holmskov, U. Expression and localization of lung surfactant protein a in human tissues. Am J Respir Cell Mol Biol. (2003) 29:591–7. doi: 10.1165/rcmb.2002-0274OC
61. Ban, N, Matsumura, Y, Sakai, H, Takanezawa, Y, Sasaki, M, Arai, H, et al. ABCA3 as a lipid transporter in pulmonary surfactant biogenesis. J Biol Chem. (2007) 282:9628–34. doi: 10.1074/jbc.M611767200
62. Yamano, G, Funahashi, H, Kawanami, O, Zhao, LX, Ban, N, Uchida, Y, et al. ABCA3 is a lamellar body membrane protein in human lung alveolar type II cells. FEBS Lett. (2001) 508:221–5. doi: 10.1016/S0014-5793(01)03056-3
63. Bullard, JE, Wert, SE, Whitsett, JA, Dean, M, and Nogee, LM. ABCA3 mutations associated with pediatric interstitial lung disease. Am J Respir Crit Care Med. (2005) 172:1026–31. doi: 10.1164/rccm.200503-504OC
64. Shulenin, S, Nogee, LM, Annilo, T, Wert, SE, Whitsett, JA, and Dean, M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med. (2004) 350:1296–303. doi: 10.1056/NEJMoa032178
65. Young, LR, Nogee, LM, Barnett, B, Panos, RJ, Colby, TV, and Deutsch, GH. Usual interstitial pneumonia in an adolescent with ABCA3 mutations*. Chest. (2008) 134:192–5. doi: 10.1378/chest.07-2652
66. Bullard, JE, and Nogee, LM. Heterozygosity for ABCA3 mutations modifies the severity of lung disease associated with a surfactant protein C gene (SFTPC) mutation. Pediatr Res. (2007) 62:176–9. doi: 10.1203/PDR.0b013e3180a72588
67. Armanios, M, Alder, JK, Parry, EM, Karim, B, Strong, MA, and Greider, CW. Short telomeres are sufficient to cause the degenerative defects associated with aging. Am J Hum Genet. (2009) 85:823–32. doi: 10.1016/j.ajhg.2009.10.028
68. van der Vis, JJ, van der Smagt, JJ, Hennekam, FAM, Grutters, JC, and van Moorsel, CHM. Pulmonary fibrosis and a TERT founder mutation with a latency period of 300 years. Chest. (2020) 158:612–9. doi: 10.1016/j.chest.2020.03.069
69. Borie, R, Crestani, B, Dieude, P, Nunes, H, Allanore, Y, Kannengiesser, C, et al. The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the European Caucasian population. PLoS One. (2013) 8:e70621. doi: 10.1371/journal.pone.0070621
70. Fingerlin, TE, Murphy, E, Zhang, W, Peljto, AL, Brown, KK, Steele, MP, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. (2013) 45:613–20. doi: 10.1038/ng.2609
71. Wei, R, Li, C, Zhang, M, Jones-Hall, YL, Myers, JL, Noth, I, et al. Association between MUC5B and TERT polymorphisms and different interstitial lung disease phenotypes. Transl Res J Lab Clin Med. (2014) 163:494–502. doi: 10.1016/j.trsl.2013.12.006
72. Wambach, JA, Casey, AM, Fishman, MP, Wegner, DJ, Wert, SE, Cole, FS, et al. Genotype-phenotype correlations for infants and children with ABCA3 deficiency. Am J Respir Crit Care Med. (2014) 189:1538–43. doi: 10.1164/rccm.201402-0342OC
73. van der Vis, JJ, Snetselaar, R, Kazemier, KM, ten Klooster, L, Grutters, JC, and van Moorsel, CHM. Effect of Muc5b promoter polymorphism on disease predisposition and survival in idiopathic interstitial pneumonias. Respirol Carlton Vic. (2016) 21:712–7. doi: 10.1111/resp.12728
74. Turley, P, Walters, RK, Maghzian, O, Okbay, A, Lee, JJ, Fontana, MA, et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat Genet. (2018) 50:229–37. doi: 10.1038/s41588-017-0009-4
75. Chen, M, Zhang, Y, Adams, T, Ji, D, Jiang, W, Wain, LV, et al. Integrative analyses for the identification of idiopathic pulmonary fibrosis-associated genes and shared loci with other diseases. Thorax. (2022) 10:thoraxjnl-2021-217703. doi: 10.1136/thorax-2021-217703
76. Hodgson, U, Pulkkinen, V, Dixon, M, Peyrard-Janvid, M, Rehn, M, Lahermo, P, et al. ELMOD2 Is a Candidate Gene for Familial Idiopathic Pulmonary Fibrosis. Am J Hum Genet [Internet]. (2006) 79:149–54. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0002929707600093
77. Korthagen, NM, CHM, van Moorsel, Barlo, NP, Kazemier, KM, HJT, Ruven, and Grutters, JC. Association between Variations in Cell Cycle Genes and Idiopathic Pulmonary Fibrosis. G Zissel, editor. PLoS ONE [Internet]. 2012 ;7:e30442. Available from: https://dx.plos.org/10.1371/journal.pone.0030442
78. Korthagen, NM, van Moorsel, CHM, Kazemier, KM, Ruven, HJT, and Grutters, JC. IL1RN genetic variations and risk of IPF: a meta-analysis and mRNA expression study. Immunogenetics [Internet]. (2012) 64:371–7. Available from: http://link.springer.com/10.1007/s00251-012-0604-6
79. Koskela, JT, Häppölä, P, Liu, A, FinnGen, PJ, Genovese, G, et al. Genetic variant in SPDL1 reveals novel mechanism linking pulmonary fibrosis risk and cancer protection [Internet]. Genetic and Genomic Medicine. (2021) Available from: http://medrxiv.org/lookup/doi/10.1101/2021.05.07.21255988
80. O’Dwyer, DN, Armstrong, ME, Trujillo, G, Cooke, G, Keane, MP, Fallon, PG, et al. The Toll-like Receptor 3 L412F Polymorphism and Disease Progression in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med [Internet]. 2013;188:1442–50. Available from: doi: 10.1164/rccm.201304-0760OC
81. Son, JY, Kim, SY, Cho, SH, Shim, HS, Jung, JY, Kim, EY, et al. TGF-β1 T869C Polymorphism May Affect Susceptibility to Idiopathic Pulmonary Fibrosis and Disease Severity. Lung [Internet]. (2013) 191:199–205. Available from: http://link.springer.com/10.1007/s00408-012-9447-z
82. Petrovski, S, Todd, JL, Durheim, MT, Wang, Q, Chien, JW, Kelly, FL, et al. An exome sequencing study to assess the role of rare genetic variation in pulmonary fibrosis. Am J Respir Crit Care Med. (2017) 196:82–93. doi: 10.1164/rccm.201610-2088OC
83. Povedano, JM, Martinez, P, Flores, JM, Mulero, F, and Blasco, MA. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep. (2015) 12:286–99. doi: 10.1016/j.celrep.2015.06.028
84. Arias-Salgado, EG, Galvez, E, Planas-Cerezales, L, Pintado-Berninches, L, Vallespin, E, Martinez, P, et al. Genetic analyses of aplastic anemia and idiopathic pulmonary fibrosis patients with short telomeres, possible implication of DNA-repair genes. Orphanet J Rare Dis. (2019) 14:82. doi: 10.1186/s13023-019-1046-0
85. Taub, MA, Conomos, MP, Keener, R, Iyer, KR, Weinstock, JS, Yanek, LR, et al. Genetic determinants of telomere length from 109,122 ancestrally diverse whole-genome sequences in TOPMed. Cell Genomics. (2022) 2:100084. doi: 10.1016/j.xgen.2021.100084
86. Sutton, RM, Bittar, HT, Sullivan, DI, Silva, AG, Bahudhanapati, H, Parikh, AH, et al. Rare surfactant-related variants in familial and sporadic pulmonary fibrosis. Hum Mutat. (2022) 43:2091–101. doi: 10.1002/humu.24476
87. Cameron, HS, Somaschini, M, Carrera, P, Hamvas, A, Whitsett, JA, Wert, SE, et al. A common mutation in the surfactant protein C gene associated with lung disease. J Pediatr. (2005) 146:370–5. doi: 10.1016/j.jpeds.2004.10.028
88. Nathan, N, Giraud, V, Picard, C, Nunes, H, Dastot-Le Moal, F, Copin, B, et al. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum Mol Genet. (2016) 25:1457–67. doi: 10.1093/hmg/ddw014
89. Crossno, PF, Polosukhin, VV, Blackwell, TS, Johnson, JE, Markin, C, Moore, PE, et al. Identification of early interstitial lung disease in an individual with genetic variations in ABCA3 and SFTPC. Chest. (2010) 137:969–73. doi: 10.1378/chest.09-0790
90. Zhang, Y, Noth, I, Garcia, JGN, and Kaminski, N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med. (2011) 364:1576–7. doi: 10.1056/NEJMc1013504
91. Roy, MG, Livraghi-Butrico, A, Fletcher, AA, McElwee, MM, Evans, SE, Boerner, RM, et al. Muc5b is required for airway defence. Nature. (2014) 505:412–6. doi: 10.1038/nature12807
92. Helling, BA, Gerber, AN, Kadiyala, V, Sasse, SK, Pedersen, BS, Sparks, L, et al. Regulation of MUC5B expression in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. (2017) 57:91–9. doi: 10.1165/rcmb.2017-0046OC
93. Allen, RJ, Porte, J, Braybrooke, R, Flores, C, Fingerlin, TE, Oldham, JM, et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genome-wide association study. Lancet Respir Med. (2017) 5:869–80. doi: 10.1016/S2213-2600(17)30387-9
94. Noth, I, Zhang, Y, Ma, SF, Flores, C, Barber, M, Huang, Y, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. (2013) 1:309–17. doi: 10.1016/S2213-2600(13)70045-6
95. Stock, CJ, Sato, H, Fonseca, C, Banya, WAS, Molyneaux, PL, Adamali, H, et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax. (2013) 68:436–41. doi: 10.1136/thoraxjnl-2012-201786
96. Peljto, AL, Selman, M, Kim, DS, Murphy, E, Tucker, L, Pardo, A, et al. The MUC5B promoter polymorphism is associated with idiopathic pulmonary fibrosis in a Mexican cohort but is rare among Asian ancestries. Chest. (2015) 147:460–4. doi: 10.1378/chest.14-0867
97. Horimasu, Y, Ohshimo, S, Bonella, F, Tanaka, S, Ishikawa, N, Hattori, N, et al. MUC5B promoter polymorphism in Japanese patients with idiopathic pulmonary fibrosis. Respirol Carlton Vic. (2015) 20:439–44. doi: 10.1111/resp.12466
98. Wang, C, Zhuang, Y, Guo, W, Cao, L, Zhang, H, Xu, L, et al. Mucin 5B promoter polymorphism is associated with susceptibility to interstitial lung diseases in Chinese males. PLoS One. (2014) 9:e104919. doi: 10.1371/journal.pone.0104919
99. Hunninghake, GM, Hatabu, H, Okajima, Y, Gao, W, Dupuis, J, Latourelle, JC, et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N Engl J Med. (2013) 368:2192–200. doi: 10.1056/NEJMoa1216076
100. Juge, PA, Lee, JS, Ebstein, E, Furukawa, H, Dobrinskikh, E, Gazal, S, et al. MUC5B promoter variant and rheumatoid arthritis with interstitial lung disease. N Engl J Med. (2018) 379:2209–19. doi: 10.1056/NEJMoa1801562
101. Allen, RJ, Oldham, JM, Jenkins, DA, Leavy, OC, Guillen-Guio, B, Melbourne, CA, et al. Longitudinal lung function and gas transfer in individuals with idiopathic pulmonary fibrosis: a genome-wide association study. Lancet Respir Med. (2022) 11:65–73. doi: 10.1016/S2213-2600(22)00251-X
102. Boettger, LM, Handsaker, RE, Zody, MC, and McCarroll, SA. Structural haplotypes and recent evolution of the human 17q21.31 region. Nat Genet. (2012) 44:881–5. doi: 10.1038/ng.2334
103. Sakornsakolpat, P, Prokopenko, D, Lamontagne, M, Reeve, NF, Guyatt, AL, Jackson, VE, et al. Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat Genet. (2019) 51:494–505. doi: 10.1038/s41588-018-0342-2
104. Tantisira, KG, Lazarus, R, Litonjua, AA, Klanderman, B, and Weiss, ST. Chromosome 17: association of a large inversion polymorphism with corticosteroid response in asthma. Pharmacogenet Genomics. (2008) 18:733–7. doi: 10.1097/FPC.0b013e3282fe6ebf
105. Newton, CA, Batra, K, Torrealba, J, Kozlitina, J, Glazer, CS, Aravena, C, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. (2016) 48:1710–20. doi: 10.1183/13993003.00308-2016
106. Hoffman, TW, van Moorsel, CHM, Borie, R, and Crestani, B. Pulmonary phenotypes associated with genetic variation in telomere-related genes. Curr Opin Pulm Med [Internet]. (2018) 24:269–80. doi: 10.1097/MCP.0000000000000475
107. Silhan, LL, Shah, PD, Chambers, DC, Snyder, LD, Riise, GC, Wagner, CL, et al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur Respir J. (2014) 44:178–87. doi: 10.1183/09031936.00060014
108. Nagpal, N, Wang, J, Zeng, J, Lo, E, Moon, DH, Luk, K, et al. Small-molecule PAPD5 inhibitors restore telomerase activity in patient stem cells. Cell Stem Cell. (2020) 26:896–909.e8. doi: 10.1016/j.stem.2020.03.016
109. Pintado-Berninches, L, Montes-Worboys, A, Manguan-García, C, Arias-Salgado, EG, Serrano, A, Fernandez-Varas, B, et al. GSE4-loaded nanoparticles a potential therapy for lung fibrosis that enhances pneumocyte growth, reduces apoptosis and DNA damage. FASEB J Off Publ Fed Am Soc Exp Biol. (2021) 35:e21422. doi: 10.1096/fj.202001160RR
110. Townsley, DM, Dumitriu, B, Liu, D, Biancotto, A, Weinstein, B, Chen, C, et al. Danazol treatment for telomere diseases. N Engl J Med. (2016) 374:1922–31. doi: 10.1056/NEJMoa1515319
111. Justet, A, Klay, D, Porcher, R, Cottin, V, Ahmad, K, Molina Molina, M, et al. Safety and efficacy of pirfenidone and nintedanib in patients with idiopathic pulmonary fibrosis and carrying a telomere-related gene mutation. Eur Respir J. (2021) 57:2003198. doi: 10.1183/13993003.03198-2020
112. Newton, CA, Molyneaux, PL, and Oldham, JM. Clinical genetics in interstitial lung disease. Front Med. (2018) 5:116. doi: 10.3389/fmed.2018.00116
113. Han, S, and Mallampalli, RK. The role of surfactant in lung disease and host defense against pulmonary infections. Ann Am Thorac Soc. (2015) 12:765–74. doi: 10.1513/AnnalsATS.201411-507FR
114. Epaud, R, Delestrain, C, Louha, M, Simon, S, Fanen, P, and Tazi, A. Combined pulmonary fibrosis and emphysema syndrome associated with ABCA3 mutations. Eur Respir J. (2014) 43:638–41. doi: 10.1183/09031936.00145213
115. Peljto, AL, Zhang, Y, Fingerlin, TE, Ma, SF, Garcia, JGN, Richards, TJ, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. (2013) 309:2232. doi: 10.1001/jama.2013.5827
116. Cosgrove, GP, Groshong, SD, Peljto, AL, Talbert, J, McKean, D, Markin, C, et al. The MUC5B promoter polymorphism is associated with a less severe pathological form of familial interstitial pneumonia (FIP). Am J Respir Crit Care Med. (2012) 185:A6865. doi: 10.1164/ajrccm-conference.2012.185.1_MeetingAbstracts.A6865
117. Biondini, D, Cocconcelli, E, Bernardinello, N, Lorenzoni, G, Rigobello, C, Lococo, S, et al. Prognostic role of MUC5B rs35705950 genotype in patients with idiopathic pulmonary fibrosis (IPF) on antifibrotic treatment. Respir Res. (2021) 22:98. doi: 10.1186/s12931-021-01694-z
118. van der Vis, JJ, Prasse, A, Renzoni, EA, Stock, CJW, Caliskan, C, Maher, TM, et al. MUC5B rs35705950 minor allele associates with older age and better survival in idiopathic pulmonary fibrosis. Theatr Res Int. (2022). doi: 10.1111/resp.14440
119. Dudbridge, F, Allen, RJ, Sheehan, NA, Schmidt, AF, Lee, JC, Jenkins, RG, et al. Adjustment for index event bias in genome-wide association studies of subsequent events. Nat Commun. (2019) 10:1561. doi: 10.1038/s41467-019-09381-w
120. Oldham, JM, Allen, RJ, Lorenzo-Salazar, JM, Molyneaux, PL, Ma, SF, Joseph, C, et al. PCSK6 and survival in idiopathic pulmonary fibrosis. medRxiv (2022). Available at: https://www.medrxiv.org/content/early/2022/05/07/2022.05.06.22274705
121. Oldham, JM, Ma, SF, Martinez, FJ, Anstrom, KJ, Raghu, G, Schwartz, DA, et al. TOLLIP, MUC5B, and the response to N-acetylcysteine among individuals with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2015) 192:1475–82. doi: 10.1164/rccm.201505-1010OC
122. Kost-Alimova, M, Sidhom, EH, Satyam, A, Chamberlain, BT, Dvela-Levitt, M, Melanson, M, et al. A high-content screen for Mucin-1-reducing compounds identifies Fostamatinib as a candidate for rapid repurposing for acute lung injury. Cell Rep Med. (2020) 1:100137. doi: 10.1016/j.xcrm.2020.100137
123. Terwiel, M, Borie, R, Crestani, B, Galvin, L, Bonella, F, Fabre, A, et al. Genetic testing in interstitial lung disease: an international survey. Respirol Carlton Vic. (2022) 27:747–57. doi: 10.1111/resp.14303
124. Gutierrez-Rodrigues, F, Santana-Lemos, BA, Scheucher, PS, Alves-Paiva, RM, and Calado, RT. Direct comparison of flow-FISH and qPCR as diagnostic tests for telomere length measurement in humans. PloS One. (2014) 9:e113747
125. Newton, CA, Oldham, JM, Applegate, C, Carmichael, N, Powell, K, Dilling, D, et al. The role of genetic testing in pulmonary fibrosis. Chest. (2022) 162:394–405. doi: 10.1016/j.chest.2022.03.023
126. Lai, TP, Wright, WE, and Shay, JW. Comparison of telomere length measurement methods. Philos Trans R Soc B Biol Sci. (2018) 373:20160451. doi: 10.1098/rstb.2016.0451
127. Shelton, CA, and Whitcomb, DC. Evolving roles for physicians and genetic counselors in managing complex genetic disorders. Clin Transl Gastroenterol. (2015) 6:e124. doi: 10.1038/ctg.2015.46
128. Aubert, G, Hills, M, and Lansdorp, PM. Telomere length measurement-caveats and a critical assessment of the available technologies and tools. Mutat Res. (2012) 730:59–67.
129. Montpetit, AJ, Alhareeri, AA, Montpetit, M, Starkweather, AR, Elmore, LW, Filler, K, et al. Telomere length: a review of methods for measurement. Nurs Res. (2014) 63:289–99.
130. Lai, TP, Zhang, N, Noh, J, Mender, I, Tedone, E, Huang, E, et al. A method for measuring the distribution of the shortest telomeres in cells and tissues. Nat Commun [Internet]. 2017;8:1356. Available from: doi: 10.1038/s41467-017-01291-z
131. Alder, JK, Hanumanthu, VS, Strong, MA, DeZern, AE, Stanley, SE, Takemoto, CM, et al. Diagnostic utility of telomere length testing in a hospital-based setting. Proc Natl Acad Sci. (2018) 115:E2358–65. doi: 10.1073/pnas.1720427115
132. Borie, R, Kannengiesser, C, Sicre de Fontbrune, F, Gouya, L, Nathan, N, and Crestani, B. Management of suspected monogenic lung fibrosis in a specialised centre. Eur Respir Rev. (2017) 26:160122. doi: 10.1183/16000617.0122-2016
133. Khincha, PP, Dagnall, CL, Hicks, B, Jones, K, Aviv, A, Kimura, M, et al. Correlation of leukocyte telomere length measurement methods in patients with dyskeratosis congenita and in their unaffected relatives. Int J Mol Sci. (2017) 18:1765. doi: 10.3390/ijms18081765
134. Fung, JLF, Yu, MHC, Huang, S, Chung, CCY, Chan, MCY, Pajusalu, S, et al. A three-year follow-up study evaluating clinical utility of exome sequencing and diagnostic potential of reanalysis. NPJ Genomic Med. (2020) 5:37. doi: 10.1038/s41525-020-00144-x
135. Molina-Ramírez, LP, Kyle, C, Ellingford, JM, Wright, R, Taylor, A, Bhaskar, SS, et al. Personalised virtual gene panels reduce interpretation workload and maintain diagnostic rates of proband-only clinical exome sequencing for rare disorders. J Med Genet. (2022) 59:393–8. doi: 10.1136/jmedgenet-2020-107303
136. Rehder, C, Bean, LJH, Bick, D, Chao, E, Chung, W, Das, S, et al. Next-generation sequencing for constitutional variants in the clinical laboratory, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med Off J Am Coll Med Genet. (2021) 23:1399–415. doi: 10.1038/s41436-021-01139-4
137. Rexach, J, Lee, H, Martinez-Agosto, JA, Németh, AH, and Fogel, BL. Clinical application of next-generation sequencing to the practice of neurology. Lancet Neurol. (2019) 18:492–503. doi: 10.1016/S1474-4422(19)30033-X
138. Auton, A, Abecasis, GR, Altshuler, DM, Durbin, RM, Abecasis, GR, Bentley, DR, et al. A global reference for human genetic variation. Nature. (2015) 526:68–74. doi: 10.1038/nature15393
139. Zhang, J, Yao, Y, He, H, and Shen, J. Clinical interpretation of sequence variants. Curr Protoc Hum Genet. (2020) 106:e98. doi: 10.1002/cphg.98
140. Stenson, PD, Mort, M, Ball, EV, Evans, K, Hayden, M, Heywood, S, et al. The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. (2017) 136:665–77. doi: 10.1007/s00439-017-1779-6
141. Landrum, MJ, Lee, JM, Benson, M, Brown, G, Chao, C, Chitipiralla, S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. (2016) 44:D862–8. doi: 10.1093/nar/gkv1222
142. Itan, Y, Shang, L, Boisson, B, Ciancanelli, MJ, Markle, JG, Martinez-Barricarte, R, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods. (2016) 13:109–10. doi: 10.1038/nmeth.3739
143. Richards, S, Bale, S, Bick, D, Das, S, Gastier-Foster, J, Grody, WW, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–23. doi: 10.1038/gim.2015.30
144. Hoffman-Andrews, L. The known unknown: the challenges of genetic variants of uncertain significance in clinical practice. J Law Biosci. (2017) 4:648–57. doi: 10.1093/jlb/lsx038
145. Mesman, RLS, Calléja, FMGR, Hendriks, G, Morolli, B, Misovic, B, Devilee, P, et al. The functional impact of variants of uncertain significance in BRCA2. Genet Med Off J Am Coll Med Genet. (2019) 21:293–302. doi: 10.1038/s41436-018-0052-2
146. Chong, JX, Yu, JH, Lorentzen, P, Park, KM, Jamal, SM, Tabor, HK, et al. Gene discovery for Mendelian conditions via social networking: de novo variants in KDM1A cause developmental delay and distinctive facial features. Genet Med Off J Am Coll Med Genet. (2016) 18:788–95. doi: 10.1038/gim.2015.161
147. Atzeni, R, Massidda, M, Fotia, G, and Uva, P. VariantAlert: a web-based tool to notify updates in genetic variant annotations. Hum Mutat. (2022) 43:1808–15. doi: 10.1002/humu.24495
148. Iancu, IF, Avila-Fernandez, A, Arteche, A, Trujillo-Tiebas, MJ, Riveiro-Alvarez, R, Almoguera, B, et al. Prioritizing variants of uncertain significance for reclassification using a rule-based algorithm in inherited retinal dystrophies. Npj Genomic Med. (2021) 6:18. doi: 10.1038/s41525-021-00182-z
149. Deignan, JL, Chung, WK, Kearney, HM, Monaghan, KG, Rehder, CW, Chao, EC, et al. Points to consider in the reevaluation and reanalysis of genomic test results: a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2019) 21:1267–70. doi: 10.1038/s41436-019-0478-1
150. Schobers, G, Schieving, JH, Yntema, HG, Pennings, M, Pfundt, R, Derks, R, et al. Reanalysis of exome negative patients with rare disease: a pragmatic workflow for diagnostic applications. Genome Med. (2022) 14:66. doi: 10.1186/s13073-022-01069-z
151. Sun, Y, Xiang, J, Liu, Y, Chen, S, Yu, J, Peng, J, et al. Increased diagnostic yield by reanalysis of data from a hearing loss gene panel. BMC Med Genomics. (2019) 12:76. doi: 10.1186/s12920-019-0531-6
152. Ho, SS, Urban, AE, and Mills, RE. Structural variation in the sequencing era. Nat Rev Genet. (2020) 21:171–89. doi: 10.1038/s41576-019-0180-9
153. Raca, G, Astbury, C, Behlmann, A, De Castro, MJ, Hickey, SE, Karaca, E, et al. Points to consider in the detection of germline structural variants using next-generation sequencing: a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2022) 25:100316. doi: 10.1016/j.gim.2022.09.017
154. Kropski, JA, Young, LR, Cogan, JD, Mitchell, DB, Lancaster, LH, Worrell, JA, et al. Genetic evaluation and testing of patients and families with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2017) 195:1423–8. doi: 10.1164/rccm.201609-1820PP
155. Quintana-Murci, L. Understanding rare and common diseases in the context of human evolution. Genome Biol [Internet]. (2016) 17:225. doi: 10.1186/s13059-016-1093-y
156. Savage, SA, and Alter, BP. Dyskeratosis congenita. Hematol Oncol Clin North Am. (2009) 23:215–31. doi: 10.1016/j.hoc.2009.01.003
157. Diaz de Leon, A, Cronkhite, JT, Katzenstein, ALA, Godwin, JD, Raghu, G, Glazer, CS, et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS One. (2010) 5:e10680. doi: 10.1371/journal.pone.0010680
158. Vulliamy, T, Marrone, A, Goldman, F, Dearlove, A, Bessler, M, Mason, PJ, et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. (2001) 413:432–5. doi: 10.1038/35096585
159. Borie, R, Tabèze, L, Thabut, G, Nunes, H, Cottin, V, Marchand-Adam, S, et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur Respir J. (2016) 48:1721–31. doi: 10.1183/13993003.02115-2015
160. Kropski, JA, Lawson, WE, Young, LR, and Blackwell, TS. Genetic studies provide clues on the pathogenesis of idiopathic pulmonary fibrosis. Dis Model Mech. (2013) 6:9–17. doi: 10.1242/dmm.010736
161. Courtwright, AM, and El-Chemaly, S. Telomeres in interstitial lung disease: the short and the long of it. Ann Am Thorac Soc. (2019) 16:175–81. doi: 10.1513/AnnalsATS.201808-508CME
162. Opresko, PL, and Shay, JW. Telomere-associated aging disorders. Ageing Res Rev. (2017) 33:52–66. doi: 10.1016/j.arr.2016.05.009
163. Hemann, MT, Strong, MA, Hao, LY, and Greider, CW. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cells. (2001) 107:67–77. doi: 10.1016/S0092-8674(01)00504-9
164. Resta, R, Biesecker, BB, Bennett, RL, Blum, S, Estabrooks Hahn, S, Strecker, MN, et al. A new definition of genetic counseling: National Society of genetic counselors’ task force report. J Genet Couns. (2006) 15:77–83. doi: 10.1007/s10897-005-9014-3
165. Clarke, AJ, and Wallgren-Pettersson, C. Ethics in genetic counselling. J Community Genet [Internet]. (2019) 10:3–33. doi: 10.1007/s12687-018-0371-7
166. Abacan, M, Alsubaie, L, Barlow-Stewart, K, Caanen, B, Cordier, C, Courtney, E, et al. The global state of the genetic counseling profession. Eur J Hum Genet. (2019) 27:183–97. doi: 10.1038/s41431-018-0252-x
167. Belkadi, A, Bolze, A, Itan, Y, Cobat, A, Vincent, QB, Antipenko, A, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A. (2015) 112:5473–8. doi: 10.1073/pnas.1418631112
168. Ding, Z, Mangino, M, Aviv, A, Spector, T, Durbin, R, and Consortium, U. Estimating telomere length from whole genome sequence data. Nucleic Acids Res. (2014) 42:e75. doi: 10.1093/nar/gku181
169. Kimura, M, Stone, RC, Hunt, SC, Skurnick, J, Lu, X, Cao, X, et al. Measurement of telomere length by the southern blot analysis of terminal restriction fragment lengths. Nat Protoc. (2010) 5:1596–607. doi: 10.1038/nprot.2010.124
170. Purcell, SM, Wray, NR, Stone, JL, Visscher, PM, O’Donovan, MC, Sullivan, PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. (2009) 460:748–52. doi: 10.1038/nature08185
171. Choi, SW, Mak, TSH, and O’Reilly, PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc. (2020) 15:2759–72. doi: 10.1038/s41596-020-0353-1
172. Dudbridge, F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. (2013) 9:e1003348. doi: 10.1371/journal.pgen.1003348
173. Li, C, Stoma, S, Lotta, LA, Warner, S, Albrecht, E, Allione, A, et al. Genome-wide association analysis in humans links nucleotide metabolism to leukocyte telomere length. Am J Hum Genet. (2020) 106:389–404. doi: 10.1016/j.ajhg.2020.02.006
Keywords: Idiopathic pulmonary fibrosis, genetic testing, precision medicine, exome sequencing, telomere length
Citation: Alonso-Gonzalez A, Tosco-Herrera E, Molina-Molina M and Flores C (2023) Idiopathic pulmonary fibrosis and the role of genetics in the era of precision medicine. Front. Med. 10:1152211. doi: 10.3389/fmed.2023.1152211
Edited by:
Alice Chen, Consultant, Potomac, MD, United StatesReviewed by:
Martin Petrek, Palacký University Olomouc, CzechiaYuFeng Zhang, Nanjing University of Chinese Medicine, China
Copyright © 2023 Alonso-Gonzalez, Tosco-Herrera, Molina-Molina and Flores. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos Flores, Y2Zsb3Jlc0B1bGwuZWR1LmVz