 Yuning Chen1†
Yuning Chen1† Matthew Wilson
Matthew Wilson Yan-Ruide Li
Yan-Ruide Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 29 March 2023
Sec. Precision Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fmed.2023.1135468
This article is part of the Research Topic Engineered Medicines to Mitigate Resistance to Cancer Immunotherapy View all 7 articles
The realm of cell-based immunotherapy holds untapped potential for the development of next-generation cancer treatment through genetic engineering of chimeric antigen receptor (CAR)-engineered T (CAR-T) cell therapies for targeted eradication of cancerous malignancies. Such allogeneic “off-the-shelf” cell products can be advantageously manufactured in large quantities, stored for extended periods, and easily distributed to treat an exponential number of cancer patients. At current, patient risk of graft-versus-host disease (GvHD) and host-versus-graft (HvG) allorejection severely restrict the development of allogeneic CAR-T cell products. To address these limitations, a variety of genetic engineering strategies have been implemented to enhance antitumor efficacy, reduce GvHD and HvG onset, and improve the overall safety profile of T-cell based immunotherapies. In this review, we summarize these genetic engineering strategies and discuss the challenges and prospects these approaches provide to expedite progression of translational and clinical studies for adoption of a universal cell-based cancer immunotherapy.
Recent developments in cell-based engineering have revolutionized the field of cancer treatment in terms of enhancing both the specificity and efficacy of potential therapeutics (1–3). Delivered agents such as immune checkpoint inhibitors or engineered immune cells bolster immunoreactivity and are designed to specifically target tumor cells with minimal off-target effects (Table 1) (2). Currently, cancer vaccines, oncolytic viruses, T and natural killer (NK) cells, stem cells, monoclonal antibodies and recombinant proteins are major areas of investigation for the development of new cancer therapeutics (3). As a relatively novel field of medicine, cell-based approaches provide a “living therapy” distinct from other forms of treatment, attracting massive interest as a highly malleable and dynamic platform (4, 5). Furthermore, compared to autologous cell therapy, wherein therapeutic cells are collected directly from the intended patient, manufactured and engineered in vitro, and used to exclusively treat that patient, allogeneic cell therapy can be preemptively positioned at treatment centers, manufactured at large scale, and is less logistically challenging and costly (6, 7).

Table 1. Engineering strategies for allogeneic cell products.
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) has been widely recognized as the earliest form of allogeneic cell therapy, after its implementation to treat inherited anemias and immune deficiencies in the 1950s (8). The Allo-HSCT platform was found to be particularly effective against hematological malignancies with limited response in solid tumors, and could potentially address refractory malignancies (8). Despite powerful graft-versus-tumor (GvT) implications, allo-HSCT-induced immune cell activation and proliferation also produced harmful graft-versus-host disease (GvHD) restricting its delivery.
Among peripheral lymphocytes, T and NK cells are the most commonly studied immune cells for antitumor allogeneic cell therapy, in the form of chimeric antigen receptor (CAR)-engineered T and NK (CAR-T and CAR-NK) cells (5, 9). Currently, all CAR-T cell therapies approved by the United States Food and Drug Administration (FDA) remain autologous, targeting CD19-positive B-cell leukemia and lymphoma (i.e., Kymriah, Yescarta, Tecartus, and Breyanzi), as well as B-cell maturation antigen (BCMA)-positive multiple myeloma (MM) (i.e., Abecma and Carvykti). Though CAR-T cells have shown less potency against solid tumors, several investigations are underway to target solid tumors with CARs. Ongoing clinical trials are targeted against several tumors, including prostate stem cell antigen (PSCA)-CAR-T cells against advanced prostate cancer, IL13Ra2-CAR T cells and epidermal growth factor receptor variant III (EGFRvIII)-CAR-T cells against advanced glioblastoma, glypican 3 (GPC3)-CAR-T cells against hepatocellular carcinoma (HCC), CD70-CAR-T cells against renal cell carcinoma (RCC), GD2-CAR-T cells against neuroblastoma, and mesothelin (MSLN)-CAR-T cells against MSLN-positive solid tumors (e.g., ovarian, breast, lung, and pancreatic cancer). However, these therapies are limited in their autologous nature due to potentially low healthy T cell numbers in patients, lengthy manufacturing process, and high cost. Allogeneic therapies address these limitations by increasing availability as an off-the-shelf therapy and reducing manufacturing variability, thereby decreasing the production costs.
Pioneering clinical trials are investigating allogeneic cell therapy by collecting peripheral blood mononuclear cells (PBMCs) via leukapheresis from healthy donors for in vitro expansion (9, 10). Further engineering involves arming of these cells with tumor antigen-specific CARs and ablation of the TRAC locus to eliminate the risk of GvHD (9, 11, 12). Some trials also involve engineering cord blood-derived hematopoietic stem cells (HSCs) or induced pluripotent stem cells (iPSCs) to generate NK cells with CARs enhancing NK-mediated killing (9). The resulting genome-edited, donor-derived allogeneic CAR-T cell products can be distributed to treat multiple patients (10, 12). In addition to conventional T or NK cells, innate-like T cell subtypes including invariant natural killer T (iNKT) and mucosal-associated invariant T (MAIT) cells have been explored due to their limited TCR repertoires that evade MHC restriction, suggesting their potential as a highly efficient allogeneic cell product (10, 11).
Several sources have been investigated for the generation of allogeneic cell therapies, including PBMCs, iPSCs, and HSCs. PBMCs are the simplest cell product to derive by isolating the desired cell type from healthy donors via leukapheresis, engineering via retro-or lentiviral transduction, and culturing to high purity. However, stem cell-derived therapies provide a more beneficial and malleable platform for therapeutic cell production (13). These approaches, including HSC-and iPSC-differentiation, hold several advantages over PBMC-derived cell generation; ubiquitously, engineering nascent stem cells has lower cost and produces higher yields of therapeutic cells (13, 14). iPSCs can be derived from any mature cells, commonly skin cells, and can be differentiated into any cell type. HSCs are derived from bone marrow or cord blood donors and can be differentiated into any hematopoietic lineage cell including blood cells, immune cells, or platelets. Current pre-clinical and clinical research has focused on HSCs and iPSCs as the initial source for engineered allogeneic cell products. We have demonstrated that human CD34+ cord blood-derived HSCs could be cultured, engineered, and differentiated into CAR-engineered iNKT (CAR-iNKT) cells with high yield and purity. These cells show high tumor-killing specificity and efficacy in murine models (6, 15, 16). In addition, current research involving iPSCs show that these cells can be guided across human leukocyte antigen (HLA) barriers to differentiate into many therapeutic cell types including CD8 T cells and NK cells (17–19). Clinical trials for iPSC-derived CAR-NK cells show promising results against hematological and solid malignancies in humans (19). Thus, drawbacks in manufacturing throughput and efficacy characteristic of primary immune cell therapies could be overcome through incorporation of stem cells (6, 19).
Nevertheless, allogeneic cell-based cancer immunotherapy still has several noteworthy limitations. Most notably, graft immunosuppression challenges both off-the-shelf cell products and autologous cell-based therapies alike. For instance, the heterogeneous tumor microenvironment (TME) associated with solid tumors suppresses effector cell activation and antitumor capacity through anti-inflammatory agents including tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and anti-inflammatory cytokines (9, 20). Additionally, allogeneic cell products typically induce dual GvHD and host-versus-graft (HvG) effects from immunogenic HLA mismatch between donor and recipient (10, 19). TCR recognition of effector cells against healthy host cells by donor effector cells unleashes cytotoxic cascade through expression of FAS ligand, perforin, and granzymes (10). iPSC-dervied NK cells have demonstrated the capability to produce a hyperinflammatory environment, inducing cytokine release syndrome (CRS) in recipients (21). Additionally, HvG effect in host rejection of graft cells reduces the antitumor efficacy and persistence of allogeneic cells, precluding maximum therapeutic capacity (11).
To address these problems, diverse approaches have been explored and implemented in cancer research, leading to significant advancement in effector cell capabilities. The invention of genetic engineering techniques such as viral transduction, CRISPR-Cas9, and transcription activator-like effector nuclease (TALEN) largely expands the potential to artificially modulate genetic expression of therapeutic cells. In this review, we discuss the major obstacles that hinder cancer treatment and summarize current genetic engineering strategies to overcome these difficulties (Table 1). Enhancement of antitumor efficacy, amelioration of GvHD, reduction of HvG effect, and improvement of safety profiles are essential directions for the progression and adoption of next generation of allogeneic cell-based cancer immunotherapy.
CARs are an engineering milestone in synthetic biology responsible for unprecedented response rates in patients since its initial U.S. FDA approval in 2017 for CD19-targeting CAR-T cell therapy against relapsed or refractory B cell acute lymphoblastic leukemia (ALL) (22). CARs were first developed in T cells with four distinct functional domains: an extracellular antigen-recognition domain, a flexible hinge region, a transmembrane domain, and an intracellular T cell signaling domain (23). The extracellular antigen-binding domain could be comprised of various structures including single chain variable fragments of antibodies (scFv) (24), cell receptors (25), ligands (26), other derived peptides programmed for specific interactions (27), or nanobodies (28). This domain is constructed with a range of specificities to the specified target to provide ample stimulation while avoiding activation-induced cell death (23). Several studied targets for the extracellular domain in hematological malignancies include, but are not limited to, CD19, CD20, CD22, CD30, CD38, and BCMA. Targets in solid tumors include human epidermal growth factor 2 (HER2), epidermal growth factor receptor (EGFR), GD2, mucin 1, mesothelin, programmed death ligand 1 (PD-L1), and CD171, among others (29).
The hinge region connects the extracellular domain to the transmembrane domain, which then connects the structure to the intracellular signaling domain. First-generation CARs are composed of this basic structure, with the intracellular domain typically a CD3ζ domain for T cell activation (30). Second-and third-generation CARs consist of one or two additional costimulatory domains, respectively; most commonly, 4-1BB or CD28 costimulatory domains are can included to provide enhanced activation and expansion (31). Fourth-generation CARs include a cytokine-releasing cassette that is constitutive or induced, allowing for increased toxicity or proliferation by cytokine stimulation (e.g., IL-12, IL-15, and IL-18) (31, 32).
CARs are most commonly engineered on T cells harvested from the patient receiving treatment. So far, patient-derived T cells are the base of each FDA-approved CAR-T cell therapy, with allogeneic T cells posing the risk of graft-versus-host alloreaction due to histoincompatibility (29). CAR-T cells also pose increased risk due to cytokine release syndrome (CRS) and neurotoxicity (33). Alternative cells for CAR-engineering include NK (34) and unconventional innate T cells, such as iNKT, MAIT and gamma delta T (γδ T) cells (14). These alternative effector cells demonstrate reduced risk of allorejection and exhibit potential for off-the-shelf cell products derived from healthy donor PBMCs and stem cells (14). Furthermore, they exhibit a shorter duration of activity and cause less severe toxicities, while also utilizing multiple cytotoxic mechanisms that are not dependent on CAR signaling to enhance tumor targeting (35, 36).
One popular extracellular domain for CAR-NK cells is NKG2D, an NK activating receptor that recognizes stress-induced ligands including MICA, MICB, and ULBP that are expressed on DNA-damaged or transformed cells (37). NKG2D-CAR-NK cells exhibit enhanced killing and higher specificity than conventional NK cells, with the ability to augment persistence with IL-15 co-expression (37, 38). Furthermore, compared to CAR-T cells, NKG2D-CAR-NK cells exhibit lower expression of cytotoxic molecules such as granzyme B, perforin, and IFN-γ while showing better tumor-killing and survival in vivo, attributed to an increase in the expression of immunological activation genes (37). iNKT cells also exhibit benefits in CAR-engineering against hematological malignancies, targeting acute myeloid leukemia and B cell lymphoma more efficiently than CAR-T cells while ameliorating GvHD from allogeneic transplantation (14). GvHD ablation by iNKT cells was shown to be achieved by the rapid depletion of CD14+ myeloid cells responsible for exacerbated alloreaction in mice (14).
One barrier restricting cell-based cancer therapy is limited efficacy due to the downregulation of costimulatory molecules on tumor cells (39). The genes responsible for producing cytokines, which are crucial for the proper function and persistence of immune cells, have been modified and incorporated into engineered cells, resulting in an improved ability of these cells to specifically target tumors. IL-15 is an important cytokine in CD8+ T cell and NK cell stimulation and proliferation (40). It has been demonstrated the engineered ectopic expression of IL-15 in CD19-CAR-T cells results in 3-to 15-fold greater expansion, survival, antitumor efficacy, and decreased PD-1 expression in vivo (41). Membrane-bound IL-15 engineered on CD19-CAR-T with a platform to promote T-memory stem cells (TSCM) showed similar effects, increasing persistence in vivo and promoting a CD45ROnegCCR7+CD95+ phenotype akin to TSCM. T-cell persistence independent of CAR signaling was achieved by the membrane-bound IL-15 due to STAT5 signaling, enabling CAR-T cells to undergo long-term memory stem-cell differentiation (42). IL-2 has been studied as a key stimulant of T cells similar to IL-15, however its constitutive stimulation of immunosuppressive regulatory T cells limits its effectiveness in cancer therapies. IL-2 is also associated with activation-induced cell death, limiting long-term activation and durability in vivo (43).
IL-7 is an important cytokine for lymphoid differentiation from hematopoietic stem cells. Administration of IL-7 exhibits increases in CD4+ and CD8+ T cells in circulation dependent on dose (44). In comparison to IL-15, IL-7 promotes the expansion of naïve T cells and leads to a more potent tumor-killing response (44). Transgenic IL-7 expression in CAR-T cells has therefore been explored and proven to increase tumor-targeting and killing in the presence of regulatory T cells in hematological and solid tumors (45). Specifically, CD20-, CD19-, GD2-, and mesothelin-CAR-T cells showed enhanced proliferation and cytotoxicity. These were much improved with the co-expression of CCL19 or CCL21 to further improve chemotaxis and proliferation (45, 46).
IL-12 is associated with a pro-inflammatory immune response in both innate and adaptive immune cells. Genetically modifying CAR-T cells to express IL-12 upon CAR or TCR stimulation under the control of the NFAT promoter provides T cells with a steady and high level of IL-12 secretion. By controlling IL-12 expression with an NFAT response, IL-12 is restricted in tissues the effector cells pass through, limiting toxicity in healthy tissues (47). In tumor tissues IL-12-engineered CAR-T cells exhibited elevated IFN-γ secretion, greater antitumor efficacy, and greater innate immune response (47). By activating both the innate and adaptive immune response, IL-12 may enhance tumor-targeting against cells that have undergone antigen-escape to avoid killing by antigen-specific CAR-T cells. Innate response is evidenced by elevated levels of macrophages, NK, and NKp46+ cells following IL-12 expression within the tumor environment (48). Studies have also shown that IL-12 increases antigen processing and presentation to increase T cell stimulation indirectly (49). Engineering inducible IL-12 activation is being investigated in phase I/II clinical trials for an EGFR-specific CAR-T cell against metastatic colorectal cancer (43).
IL-21 is an essential cytokine for optimal proliferation, differentiation, and activation of T cells utilizing the STAT3 pathway (49). Batra et al. demonstrated that GPC3-CAR-T cells coexpressing IL-21 effectively target GPC3-positive tumor lines, with the most robust expansion and persistence in cells expressing combined IL-21 and IL-15 (50). IL-21-expressing cells exhibited greater cytotoxicity to tumor cells and greater survival in mice than IL-15 or IL-2 treated CAR-T cells despite higher levels of IFN-γ and Bcl-2 expression (43, 50, 51). Because the levels of circulating IL-15 and IL-21 in mouse peripheral blood were 100-1,000-fold lower than the maximum tolerable dose in humans, systemic toxicities are not anticipated with CAR-T cells coexpressing these cytokines (50, 51). The efficacy of these engineered T cells are being evaluated in two ongoing clinical trials (NCT02932956 and NCT02905188) for GPC3+ liver cancer and hepatocellular carcinoma (52).
Immune checkpoint blockade has rapidly become one of the most promising therapeutic strategies for triggering anti-tumor immunity. As a key component of the immune system, immune checkpoints are essential for preserving self-tolerance as well as regulating the intensity and duration of physiological immune response in peripheral tissues to protect cells from being attacked indiscriminately (53). Tumor cells are able to express immune checkpoint ligands interacting with receptors on immune cells to inhibit their activity, and thereby bypass the immune system. This has limited cell therapies by enabling tumor cells to mask themselves from immune cells, causing immune resistance. To address the immune suppression against tumors from this ligand-receptor interaction, antibodies for immune checkpoint blockade have shown promise in blocking the interaction to enable tumor-targeting by immune cells (Figure 1).
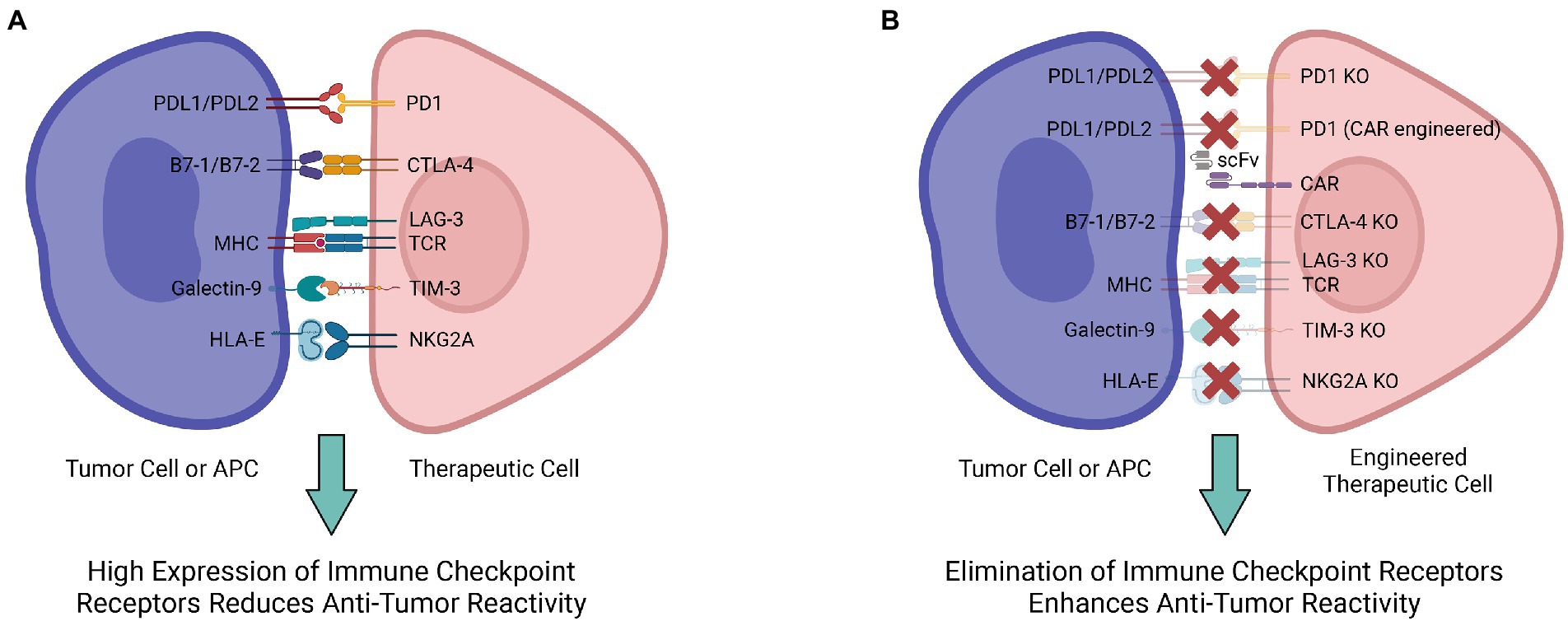
Figure 1. Multiple engineering strategies to mitigate the immune checkpoint receptor-mediated immunosuppression. (A) Various ligand-receptor interactions are involved in the immunosuppression and decrease antitumor reactivity of therapeutic cells. (B) Engineering therapeutic cells to ablate their immune checkpoint receptors and interrupt ligand-receptor interactions for enhanced antitumor reactivity.
Among the diverse immune checkpoint proteins, two of the most extensively studied are programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (54). Generally, PD-1 is responsible for maintaining peripheral tolerance during cancer, infection, or chronic inflammation (54, 55). PD-1 ligands PD-L1 and PD-L2 expressed in tumor cells bind to PD-1 expressed on the surface of T cells, leading to inhibitory checkpoint signaling that reduces cytotoxicity and T-cell exhaustion (56). The PD-1/PD-L1/2 interaction downregulates the immune responses and provides an opportunity for tumor cells to persist uninterrupted. In response to this immune resistance, anti-PD-1 treatment is aimed at enhancing T cell function by blocking the inhibitory signals (53). By identifying mutated peptides, such as neoantigens, tumor-infiltrating T cells can trigger a potent immune response (57). Specifically, nivolumab, an FDA-approved antibody targeting human PD-1, has shown clinical efficacy in 2010 and 2012 against mesothelioma and gastric cancer (58, 59). According to a phase I clinical study of nivolumab, treatment efficacy was correlated with the expression of PD-1 ligands in tumor cells (58). However, PD-1 ligands are differentially expressed across different tumors; it is predicted that the therapeutic effect is selective for PD-1high tumors (e.g., lung cancer, glioblastoma, melanoma) (55, 60).
CARs also provide a potent mechanism for PD-1 blockade. Because of the immunosuppressive TME and feeble natural immune responses against tumors, clinical trials utilizing checkpoint inhibitors have shown limited effectiveness regarding some types of tumors, such as pancreatic ductal adenocarcinoma (61, 62). Despite the promise of PD-1 blockadeas a form of cancer therapy, its efficacy may be limited for certain types of cancer and between patients due to tumor heterogeneity. CARs can disrupt the interaction between PD-1 and PD-L1 to effectively treat PD-L1+ cancers. For example, in pancreatic cancer (PaC) third-generation CARs with costimulatory domains such as CD28, CD137, and OX40, can effectively target PD-L1 on PaC cells. Furthermore, in xenograft/orthotopic mouse models, the targeting specificity of CARs largely determines the cytotoxic activity of the engineered CAR-T cells, with greater binding affinity of the scFv associated with greater cytotoxicity (62–64).
In contrast to PD-1, the CTLA-4 immune checkpoint regulates T-cell proliferation in lymph nodes during the early stages of an immune response (65). CTLA-4 maintains immune balance by downregulating the function of activated T cells through inhibitory signals from binding B7, a membrane protein expressed on activated antigen presenting cells (APCs) (65). Anti-CTLA-4 monoclonal antibodies such as ipilimumab bind CTLA-4 to shut down inhibitory signals, enabling CTLs to persist with their cytotoxic activities against cancer cells (53). Furthermore, preclinical data has indicated that CTLA-4-blockade has a significant effect on boosting the anti-tumor response, suggesting that targeting CTLA-4 could serve as a viable method for activating cytotoxic T-lymphocytes (CTLs) in cancer therapy (66–68). In metastatic melanoma, ipilimumab improved patients’ survival after established lines of treatments had failed (69, 70). CRISPR-Cas9 has been utilized to target CTLA-4 as well. To construct CTLA-4 knockout CTLs, lentiviral vectors containing guide RNA specific for CTLA-4 are used to create double-strand breaks and block CTLA-4 expression (71). Following CTLA-4 knockout, anti-tumor cytotoxicity and cytokine secretion by CTLs are notably increased, as well as the apoptosis and caspase activities of tumor cells. This is indicative of an overall improvement in antitumor efficacy following CTLA-4 knockout with CRISPR-Cas9 (71).
In addition to PD-1 and CTLA-4, several new immunological checkpoints have been discovered. Lymphocyte activation gene-3 (LAG-3), adjacent to the CD4 gene on chromosome 12, is a negative regulator for T cell activities (54). Structural similarities between LAG-3 and CD4 make MHC-II a high-affinity ligand of LAG-3 (72). CRISPR-Cas9 is also employed to understand the mechanisms behind LAG-3 checkpoint regulation. After using CRISPR-Cas9 with electroporation to knockout LAG-3 in both T cells and CAR-T cells, the immune phenotype and viability of the edited cells are not significantly different from the control cells in vitro. However, in vivo studies demonstrate that LAG-3 modulates pool size and downregulates T cell expansion, evident from increased T cell populations in LAG-3 knockout models (73, 74). Though further research is required to understand the functions of LAG-3, gene-editing provides a potential mechanism for enhancing lymphocyte recognition of tumor cells.
T cell immunoglobulin mucin family member 3 (TIM-3), also known as hepatitis A virus cellular receptor 2 (HAVCR2), has been increasingly studied because of its distinct characteristics as a suppressive marker. As a type I transmembrane protein, TIM-3 is essential for suppressing Th1 responses and the production of cytokines, including TNF and INF-γ. TIM-3-associated dysregulation is usually implicated in autoimmune disease. Generally, Tim-3 expression on T cells is inversely linked with the progression of autoimmune disorders (75). Furthermore, as the TIM-3 expression level increases, the level of T cell exhaustion increases as well, marking the gradual loss of T cell function during long-term viral infections and tumor growth. Studies on both human and mouse tumors indicate that TIM-3 controls T cell exhaustion in tumor-infiltrating leukocytes (TILs), acting as a checkpoint for tumor immunity (75). High TIM-3 expression on CD8+ T cells has been correlated with poor prognosis in cancer patients. Moreover, TIM-3 and PD-1 expression on CD8+ TILs are highly correlated (76). With increased research in TIM-3 and CRISPR knockouts, evidence is mounting that double-positive TIM-3+PD-1+ CD8+ T cells are linked to higher levels of T cell exhaustion (77). Further research utilizing TIM-3 knockouts will be able to uncover the mechanisms behind TIM3-mediated suppression and may evidence greater potential for therapeutics.
NKG2A, an inhibitory regulator in the NKG2 family of proteins, is an important immune checkpoint for both CD8+ T cells and NK cells. Similar to other immune checkpoints, NKG2A binds to its ligand, peptide-presenting human leukocyte antigen-E (HLA-E), to activate inhibitory signals (78). The expression pattern of NKG2A in CD8+ T cells is highly regulated. In healthy individuals, NKG2A expression is minimal in CD8+ T cells, whereas during tumor development and persistent viral infection, the expression level increases (79, 80). Cytokines (e.g., IL-2, IL-4, and IL-6) can effectively modulate NKG2A expression on CD8+ T cells. NKG2/CD94 receptor interaction conducts inhibitory signals in CD8+ T cells, leading to a decrease in cytotoxic activity (81). NKG2A is present in about half of the human peripheral blood NK cells. Although NKG2/CD94 activation has a restrictive effect on the cytotoxicity of NK cells, interrupting the receptor-ligand interaction with HLA-E reinvigorates NK killing (78). Tumors often overexpress HLA-E and are thus able to suppress NK antitumor activity. In fact, HLA-E overexpression is associated with poor prognoses in several cancers, including glioblastoma and breast cancer (82, 83). NKG2A-mediated immune suppression in NK cells has been alleviated via NKG2A protein expression blocker (PEBL) transduction and anti-NKG2A antibody interference. PEBL transduction has been shown to induce a higher level of NK cell cytotoxicity without risking the relapse of NKG2A expression (84). Based on data from murine models, downregulating NKG2A effectively enhances anti-tumor immune response in both CD8+ T cells and NK cells (84).
Antigen spreading, a result of sequential immune response, must be taken into consideration during immune checkpoint blockade (85). This phenomenon involves broadening the immune response from the initial targeted response against a particular epitope to include other subdominant or previously unrecognized epitopes. Such a process may occur in response to self and foreign peptides, and it can lead to a more diverse an `11d robust immune response against the target antigen (86). Although past clinical trials did not investigate antigen spreading in immune checkpoint inhibition, there have been reports demonstrating that patients responding to the treatment experienced the recruitment and expansion of tumor-specific T cells that were not detectable before the therapy (87–90). Additionally, there has been experimental evidence indicating that checkpoint inhibition therapy was able to induce antigen spreading (85). Further studies are needed to analyze how the occurrence of antigen spreading could enhance the efficiency of immune checkpoint inhibition therapy.
Transcription factors and epigenetic enzyme modulation play vital roles in immune cell activation, differentiation, and effector function (91). One frequently studied enzyme is the histone methyltransferase EZH1, an antagonistic lymphoid potential regulator during embryonic hematopoiesis. Human induced pluripotent stem cells (iPSCs) are essential resources for cell therapies, but deriving mature cell types can be difficult (92). Studies have shown that using iPSCs derived from T cells that have productively altered TCRs or incorporating organoid or thymic culture systems results in improved T cell maturation in vitro (93–95). To further improve the outcomes, EZH1 programming is included during cell derivation. By coupling a stroma-free T cell differentiation system with EZH1-knockdown-mediated epigenetic engineering, iPSC-derived T (EZ-T) cells can be produced. These artificially generated cells possess mature molecular markers similar to those found on peripheral blood-derived TCRαβ T cells and exhibit a very diverse TCR repertoire (92). Effector and memory T cell subsets are produced as a result of EZ-T cell activation. EZ-T cells display robust anti-tumor responses both in vitro and in vivo in xenograft models after being transduced with CARs. Developmentally mature T cells can be efficiently produced from iPSCs and employed in adoptive cell therapy due to epigenetic remodeling of EZH1 repression (92).
Another crucial reprogramming method for increasing anti-tumor efficacy is via DNA methyltransferase 3A (DNMT3A), an enzyme that catalyzes 5-methylcytosine methylation (96). DNMT3A is an essential factor in both embryonic and hematopoietic stem cell (HSC) differentiation (96). The loss of DNMT3A causes long-term HSC differentiation impairment in vivo (97). Mutations of DNMT3A can be found in human HSCs and are often associated with hematological malignancies (96). Due to this characteristic of DNMT3A, reprogramming has become a research target and therapeutic method for both hematological and solid malignancies. Epigenetically altered CAR-T cells with modulated DNMT3A have been studied to further understand the role of DNMT3A in anti-tumor activities. One significant drawback of traditional CAR-T cell therapy is exhaustion during prolonged antigen exposure. Under antigen stimulation, the expression of DNMT3A is upregulated by T cells. While the DNMT3A-regulated epigenetic changes in CAR-T cells induce exhaustion, removing DNMT3A from CAR-T cells improves the anti-tumor activities and avoids possible dysfunction in an IL-10-dependent manner (98). CRISPR-Cas9 can be used to generate CAR-T cells with DNMT3A knocked out with consequent constant proliferation and successful control of tumor development in vivo, exhibiting the benefits of targeting DNMT3A in immunotherapies (99).
One mechanism ADCC is mediated is through immunoglobulin G (IgG) Fc receptor FcγRIIIa, otherwise known as CD16a. On NK cells CD16a is associated with CD3ζ or Fcɛ receptor I (FcɛRI) γ chains. When it binds to the Fc portion of IgG, CD16a causes the cell to undergo biochemical events similar to T cell activation and lead to cytotoxicity (100, 101). Following NK cell activation, a disintegrin and metalloproteinase-17 (ADAM17) cleaves CD16a, reducing ADCC capacity and leading to NK cell dysfunction (101). FDA-approved monoclonal antibodies such as rituximab (anti-CD20) and trastuzumab (anti-CD19) are aimed at bringing NK cells within proximity of tumor cells and activating ADCC function to kill the tumor (102).
Zhu et al. have engineered “off-the-shelf” NK cells from induced pluripotent stem cells (iPSC) and engineered high-affinity non-cleavable CD16a (hnCD16a) to maintain NK cytotoxicity (102). These cells expressed similar NK activation and maturation markers but showed much higher CD16a expression and persistence. This was achieved by replacing a serine residue with a proline in the membrane proximal region of the CD16 receptor (103). In vitro and in vivo assays demonstrate hnCD16a-NK cells with a single dose of monoclonal antibody mediate ADCC against lymphoma and ovarian cancer, with better survival than PBMC-NK cells (102). Engineered hnCD16-NK cells from iPSCs serve as a potential allogeneic cell therapy because of the lack of HLA specificity in NK cells. Cichocki et al. (104) reported engineered CD16-CAR-NK cells with membrane-bound IL-15 from iPSCs capable of targeting multiple myeloma and being mass produced. These data showed enhanced durability without supplemention with stimulatiory cytokines against NSG models of multiple myeloma. Because of their potential for adoptive transfer without inducing GvHD, several clinical trials are in progress to induce ADCC against B cell lymphoma, pancreatic and other advanced solid tumors, and multiple myeloma via monoclonal antibody/CD16-NK cell combined treatment (105).
Expression of HLA molecules largely mediates the clinical manifestation of GvHD for adoptive cell therapy recipients on account of alloreactivity against host-mismatch recognition. Prior studies have shown the capacity to suppress GvHD while maintaining the desired GvL reaction through depletion of mature alloreactive graft cells (106–109), dosage titration (110), and allo-anergization through exposure to host cells ex vivo (111, 112). The standardization of genetic editing techniques has since drawn attention towards the viability of employing genome-based methods to preclude GvHD onset. In particular, disrupting expression of a functional TCR at the constituent TCR receptor α constant (TRAC) and β constant (TRBC) loci effectively curtails graft cell capacity for immunoresponse, including alloreaction (113, 114). In order to produce a viable graft, insertion of a chimeric antigen receptor (CAR) gene at the TRAC/TRBC loci can effectively substitute a functional TCR. A wide variety of differentially expressed tumor-associated antigens have been identified within malignancies (e.g., BCMA, CD19, GD2, and Mesothelin) which can be selectively targeted by the CAR depending on tumor expression profile (115–117). Clinical trials administering CD19-targeting CAR-T cell therapy that lacked endogenous TCR demmonstrated enhanced tumor recognition despite its missing receptor, suggesting the feasibility of adopting a universal CAR-T therapy for cancer treatment (118–120).
However, limitations including antigen escape and graft impurity limit the safety of adopting a “universal” CAR-T cell-based therapy (121). Other promising methods employ a transgene encoding a non-conventional TCR that does not engage in alloreactivity while retaining TCR-mediated antigen recognition; so far this strategy has been most prominently adopted using NY-ESO-1 TCR transgene. The NY-ESO-1 TCR displays enhanced affinity for the antigen NY-ESO-1 when complexed with HLA-A2, both of which are highly expressed on the surface of multiple myeloma MM and testicular cancer cells (122, 123). A study performed by Mastaglio et al. employed zinc finger nuclease (ZFN) to disrupt the TRAC locus with a copy of the NY-ESO-1 TCR gene produce NY-ESO-1 T cells; compared to a non-TCR knockout graft, NY-ESO-1 T cells maintained similar reduction of NY-ESO-1+ MM tumor burden while abating GvHD response (124). Single TRAC knockout was sufficient to abolish endogenous TCR formation and suppress alloreactivity, attributed to non-productive mispairing of the remaining TCR β chain with NY-ESO-1 TCR α chain.
Another viable alternative lies in using unconventional T cell subsets, such as iNKT cells, that recognize antigens via an invariant TCR, independent of HLA. The human iNKT TCR is composed of a Vα24-Jα17 chain paired with a heavily restricted repertoire of Vβ chains, primarily Vβ11 (125, 126). Unlike conventional TCRs, the iNKT TCR recognizes antigens in an HLA-independent manner, restricted to glycolipids presented by CD1d, a non-classical MHC molecule (127). By virtue of their inability to recognize HLA antigen complexes, iNKT cells possess powerful antitumor capacity without the risk of inducing GvHD response (6, 128). Additionally, iNKT expression of NK activating receptors (i.e., DNAM-1, NKG2D, NKp33) permit alternative, TCR-independent recognition of tumors (129, 130). Clinical engraftment of iNKT cells against a variety of malignancies, including MM (130), lung cancer (131), and head and neck cancer (132), demonstrated significant reduction in tumor burden with greater safety profile compared to checkpoint inhibitor and CAR-T cell therapies. The improved safety of iNKT cell adoptive transfer, as well as its dual employment of innate and adaptive immune response position iNKT cells as a prime candidate for allogeneic “off-the-shelf” cancer immunotherapy.
Beta-2 microglobulin (B2M) monomers form a functional complex with major histocompatibility class I (MHC I) molecules that are universally expressed on the surface of nucleated cells (133). The mature MHC I complex organizes presentation of intracellular antigens towards its cognate CD8 co-receptor expressed by single positive CD8 cytotoxic T cells. Cytosolic proteins are degraded into antigen peptides and loaded onto MHC Class I molecule (134). This complex translocates to the cell surface for TCR-mediated recognition of loaded antigens, inducing activation of CD8+ cytotoxic lymphocytes (Figure 2A). Expression of MHC I restricts the implementation of allogeneic therapy twofold. First, MHC I mismatch on graft cells activates endogenous lymphocytes producing a HvG effect that limits the survival and persistence of therapeutic donor cells (135). Secondly, cell therapies that function using TCR-mediated pathways recognize MHC I mismatch on host cells and induce onset of GvHD. Because proper MHC I loading and recognition requires complexing with B2M, methods to deplete its expression abrogate the HvG effect and augment engraftment success rate. In conjunction with using cell types that employ MHC-independent recognition pathways, such as iNKT and NY-ESO TCRs as mentioned above, successful adoption of an allogeneic “off-the-shelf” cell therapy becomes feasible.
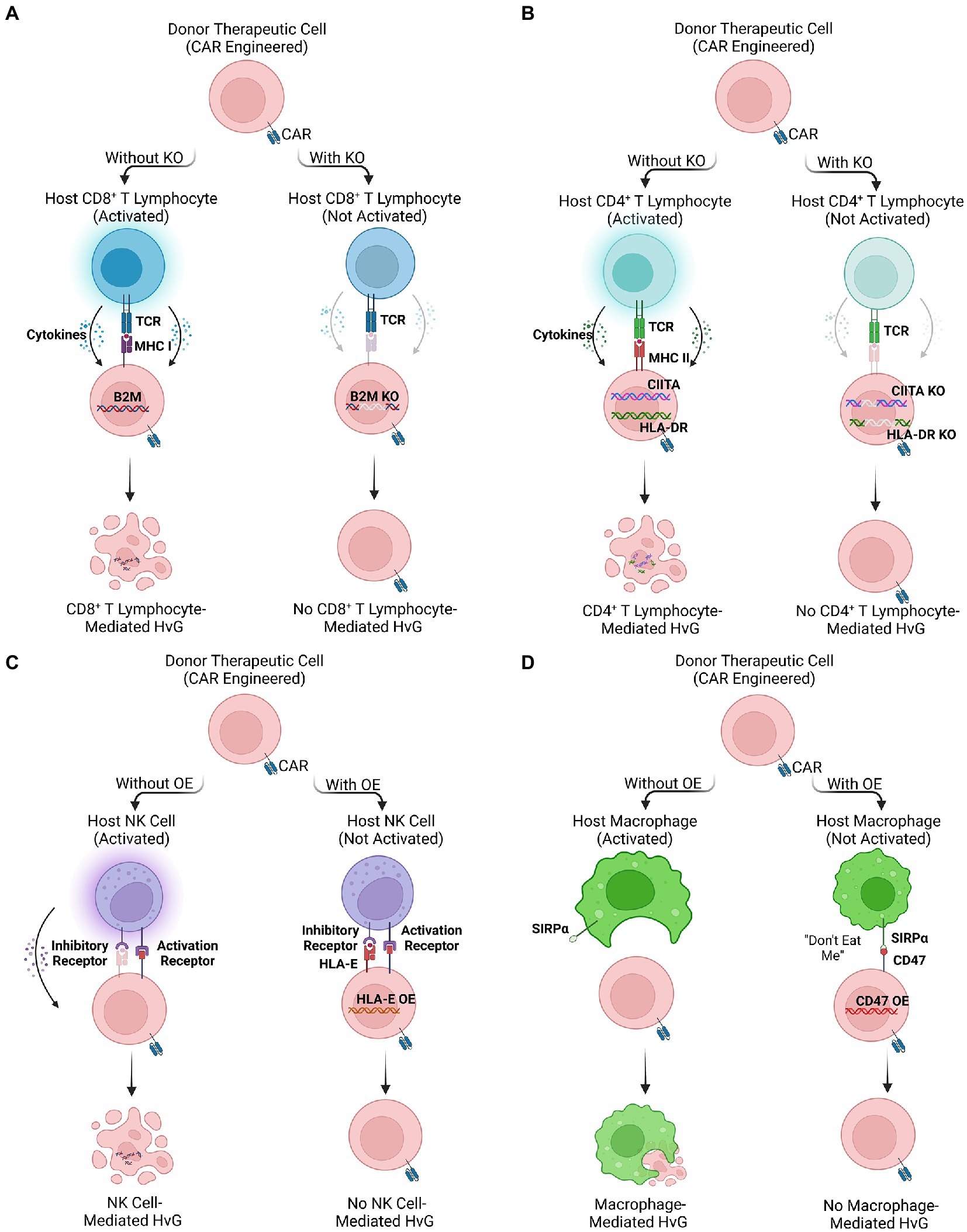
Figure 2. Multiple engineering strategies to reduce host-versus-graft (HvG) effect. (A,B) The ablation of HLA molecules expressed on therapeutic cells by knocking out HLA-related genes (e.g., B2M, HLA-DR, and CIITA), avoiding CD8+ and CD4+ T lymphocyte-mediated HvG effect. (C,D) The overexpression of inhibitory ligands (e.g., HLA-E and CD47) of therapeutic cells. The binding of inhibitory ligands prevents NK signaling pathway and macrophage phagocytosis, reducing NK cell-and macrophage-mediated HvG effect.
Engineered MHC I deficient cells may be employed to successfully target tumors through a combination of CRISPR-Cas 9 knockdown of endogenous TCR and transduction of a CAR moiety to provide MHC I-independent tumor recognition capacity. Using lentivirus transduction, Ren et al. produced allogeneic CD19-targeting CAR-T (CAR19-T) cells possessing abrogated expression of the TCR, HLA class I molecules, and immune checkpoint PD-1 (136). When CAR19-T cells were co-cultured with allogeneic PBMCs, alloreactivity was largely diminished indicating their favorable profile for allogeneic engraftment. This was similarly applicable within our studies BMCA-targeting CAR iNKT (BCAR-iNKT) cells that were depleted for B2M (11). Within an in vivo multiple myeloma mouse xenograft model, depleted BCAR-iNKT cells achieved longer survival than non-depleted counterparts while still maintaining total tumor clearance. For both CAR-T and CAR-iNKT based therapies, ablation of B2M or HLA I expression renders a potent strategy to abrogate HvG effects and promote long-term graft persistence.
Human Leukocyte Antigen-DR isotype (HLA-DR) is an MHC class II surface antigen-presenting molecule, with expression restricted to professional APCs, including B cells, macrophages, and dendritic cells. Morphologically, HLA-DR is a heterodimer composed of α and β chains then complex to recognize its cognate CD4 co-receptor (137). In contrast to MHC I which presents cytosolic antigens, MHC II molecules present exogenous antigens for recognition by single positive CD4 helper T cells (Figure 2B). Expression of HLA-DR mediates both direct and indirect HvG effect through respective recognition of either graft-derived or endogenous APCs (138). The direct pathway holds more relevance for acute allograft rejection, while the indirect pathway remains a causal factor for chronic graft rejection (139). Through depletion of HLA-DR expression on effector cell transplants, alloreactive host lymphocytes cannot be primed for both direct and indirect HvG effect.
As a relatively early technology, zinc finger nucleases (ZFNs) were successfully utilized to ablate HLA expression in allogeneic hematopoietic stem cells (HSCs) while maintaining pluripotency (140). More recently, CRISPR-Cas 9 multiplex gene editing has provided an avenue for more specific disruption of HLA II α genes, including HLA-DRA, DQA, and DPA (141). After 13 days of quadruple gRNAs transfection, Lee et al. observed that the gene-edited CD3+ T cells composed 62.1% of HLA-I/II-double-negative cells, while retaining surface phenotype and functionality. However, for generation of universally compatible iPSCs (142), results concluded that TALEN restriction enzymes had the greatest consistency; they were more maneuverable than ZFNs and had fewer off-target events than CRISPR/Cas 9. Even when stimulated with high concentrations of IFN-γ, TALEN-treated fibroblasts lacked HLA-DR expression, which was maintained within iPSCs generation (142). HLA-DR knockout remained effective and safe in these therapies, suggesting their potential to modify effector cells to prevent HvG effect in cancer immunotherapy (143).
Class II transactivator (CIITA) is a transcriptional coactivator that regulates IFN-γ-activated MHC gene expression. CIITA primarily controls expression and production of MHC class II molecules but also plays a role in supporting constitutive expression of MHC I expression (144). Thus, CIITA is a viable target to induce knockout of MHC II, thus reducing HvG incidence mediated by HLA class II molecules. Through multiplex gene editing, researchers disrupted the CIITA gene in human PSCs (145). In vitro and in vivo immunoassay suggested that alloreactive response against gene-edited PSCs was minimized, suggesting this strategy’s potential to develop a universal cell therapy (146). Our own experiments to develop allogeneic HSC-derived iNKT cells, have produced CIITA-KO and B2M-KO universal BCAR iNKT cells that could completely abrogate HvG effect (6), emphasizing effectiveness within fully differentiated T cell subsets.
HLA class I histocompatibility antigen, alpha chain E (HLA-E) and alpha chain G (HLA-G) are non-classical MHC class I molecules that act as inhibitory ligands for NK cells (147). Recognition of HLA-E by CD94/NKG cell receptors, protects targeted cells from NK cell-mediated lysis (148). Although HLA ablation may abate targeting from host lymphocytes, these cell therapies alone remain vulnerable to NK cell recognition pathways (148, 149). Engineering HLA-E/HLA-G overexpression may provide protective effects within effector cells, thus safeguarding them from innate targeting to enhance in vivo persistence (150, 151). Zhao et al. successfully generated human embryonic stem cells (hESCs) with high expression of HLA-G (152), providing protective effects against NK cell-mediated cytotoxicity. In a similar vein, Torikai et al. utilized a Sleeping Beauty (CD19RCD28) transposon system to homogenously induce overexpression of HLA-E in CAR19-T cells, consequently reducing measured cell lysis in vitro (153). Using AAV-mediated gene editing to insert the gene for HLA-E at the B2M locus in hPSCs, Gornalusse et al. generated cells overexpressing HLA-E single-chain dimers or trimers while disrupting HLA-A/B/C production. These gene-edited human PSCs demonstrated strong resistance against NK cell killing as well as minimal recognition by CD8+ cytotoxic lymphocytes (154). Regardless of strategy, insertion of HLA-E/G genes provided sufficient protection against NK cell-mediated killing enhancing effector cell survival and persistence (Figure 2C).
CD47 is a transmembrane glycoprotein expressed universally among human cells (155, 156), and is upregulated within tumors to promote survival (157–159). Through interaction with signal receptor protein-alpha (SIRPα) expressed on macrophages, targeted cells inhibit phagocytosis and cell digestion to evade eradication (155, 156, 159). Macrophages largely contribute to HvG effect through both direct killing as well as their pro-inflammatory function in activating markers, cytokine profile, and engagement in ADCC (160–162). To prevent macrophage killing, exploitation of CD47/SIRPα pathway provides a potential remedy; akin to CD47hi tumors, graft cells engineered to overexpress CD47 evade macrophage-mediated phagocytosis (163). By combining lentivirus transduction of CD47 with gRNA guided CRISPR-Cas 9 knockdown of B2M and CIITA, it is feasible to engineer HLA−CD47hi effector cells that are immune to both T cell and macrophage mediated HvG effect (163, 164). Compared to non-engineered allogeneic grafts, CD47 modified cells showed enhanced survival and persistence within in vivo mouse xenograft models (163). By incorporating CD47 to evade macrophage killing, engrafted effector cells promote low HvG response, enhancing their long-term ability to suppress tumor growth (Figure 2D).
Lymphodepletion is a crucial step in cell therapies to reduce suppressive immune cells and decrease immunogenicity of the host immune system prior to cell transfer. One method for temporary lymphodepletion is to use alemtuzumab, a monoclonal antibody that targets CD52 and induces ADCC against mature lymphocytes but not naïve lymphocyte progenitors (165); however, this method cannot be used for adoptive transfer due to indiscriminate targeting of healthy and cancerous lymphocytes (165). One genetic engineering approach to address this challenge is the disruption of CD52 expression with TALEN mRNA to eliminate the binding cite of alemtuzumab in order to maintain antitumor efficacy. This approach is used in CD19-CAR-T cells against lymphoma, generating a CAR-T cell product capable of allogeneic engraftment (165). Engineering CD52 deficient CAR-T cells allows for concurrent treatment of alemtuzumab and CD19-CAR-T cells for combined host cell depletion and donor cell engraftment. Several clinical trials utilize this TALEN-mediated CD52-knockout for successful selective lymphodepletion in allogeneic CD19-CAR-T cell therapy (166). Genetically engineering a mechanism for selective lymphodepletion provides an opportune avenue for allogeneic transfer of CAR-T cells without the risk of GvHD.
Despite the aforementioned engineering strategies to ameliorate GvHD, safety remains an issue for allogeneic cell products, which may cause unpredictable effects post therapeutic cell infusion (2, 3). Engineered cell products such as CAR-T cells might produce lifetime sustainment in patients as well as elicit adverse effects related to permanent gene transfer (2). CRS and neurotoxicity are also common safety risks for cell-based immunotherapy (3, 4). To achieve more desirable safety profile and avoid harm to patients, the introduction of suicide genes has provided an effective approach to allow selective destruction of modified cells at a designated time point (2, 5).
Suicide gene technology could be classified into three groups (5). First, gene-directed enzyme prodrug therapy (GDEPT) converts a nontoxic drug into a toxic compound through a genetically encoded molecule, usually the herpes simplex virus thymidine kinase (HSV-TK) (1, 2, 5). Second, apoptotic genes such as human caspase-9 could be linked to a drug binding domain for conditional dimerization (5), and administration of a non-therapeutic small molecule dimerizer would induce apoptosis (5). The third category utilizes monoclonal antibody-mediated cell removal mechanisms to eliminate allogeneic cells with a specific membrane protein expression (e.g., CD20) (5).
Among the three categories, HSV-TK is the most extensively tested suicide gene therapy utilized in human cells (2). The HSV-TK gene has been inserted into donor cells using retroviral or lentiviral transduction, which are then selected for purity (3, 5). The transduced cells constantly express the TK gene, which could phosphorylate pyrimidine and guanosine analogs (e.g., acyclovir, ACV; ganciclovir, GCV; penciclovir, PCV) (6). The phosphorylation leads to DNA misconformation through integration of DNA polymerase, eventually resulting in cell death (6). Thus, administration of guanosine analogs could eliminate more than 90% of circulating TK gene-engineered cells in most cases (5).
Our previous studies demonstrated the use of TK suicide gene in allogeneic cell-based cancer immunotherapy, where the lentivector was constructed through integration of an iNKT TCR gene with an sr39TK gene (3). The humanized SR39 (SR39h) was tested to be the most sensitive TK variant to suicide induction (6). Human HSCs were transduced with this Lenti/iNKT-sr39TK vector and further differentiated into allogeneic iNKT cells (3). The yield and functions of iNKT cell products were not affected by the incorporation of a suicide gene, displaying strong antitumor killing efficacy to multiple caners including melanoma, myelogenous leukemia, lung cancer, prostate cancer, and multiple myeloma (3). Therefore, TCR engineering, as well as CAR engineering and HLA ablation, could be combined with suicide gene engineering at the same time (3). Furthermore, Qin et al. established a safety-cell system through linking HSV-TK gene and a cell-division gene (CDK1), protecting the suicide system from inactivation in dividing cells (3). They found that the safety-cell system with GCV could eliminate all mitotically active cells and thus represented a safety measure before cell infusion into a patient (167). The incorporation of suicide genes provides additional safety measures for allogeneic cell-based immunotherapy, minimizing the potential risks for cancer patients for clinical development of future drugs.
As a promising immunotherapy, allogeneic cell-based therapy has been increasingly studied for cancer treatment. This review comprehensively introduced current strategies of genetically engineering cells, especially allogeneic cell products, for strengthening the anti-tumor reactivity and reducing alloreactivity, thereby improving treatment efficacy. Cell engineering provides a platform to enhance anti-tumor efficacy of engrafted cells. Patient-derived T cells genetically engineered to express CAR are the typical base for CAR-T cell therapy but still carry a potential risk for GvHD and HvG. In response to this problem, alternative cells, such as NK and unconventional innate T cells, are utilized for safer CAR engineering. Another engineering method is the incorporation of cytokine armoring genes, which leads to the production of cytokines required for T cell growth, including IL-15, IL-2, IL-7, IL-12, and IL-21, hence strengthening tumor-killing abilities. Moreover, the blockade of several immune checkpoints, such as PD-1, CTLA-4, and LAG-3, represents an alternative way of inducing anti-tumor immunity. Eliminating these crucial ligand-receptor interactions prevents tumor cells from bypassing eradication from the immune system, as these greatly confer immune resistance to cancerous cells. Additionally, immune checkpoint blockade therapies have been utilized in combination with CAR T cell therapy. One study has shown that for human triple-negative breast cancer cells with mesothelin overexpression that can be targeted by CAR T cells, by counteracting the inhibitory effect of PD-1 on CAR-T cells through CRISPR-Cas9 ribonucleoprotein-mediated editing, PD-1hi population was significantly reduced with a minimal impact on the proliferation of T cells. In contrast, CAR-T cells with PD-1 disruption demonstrated improved tumor control and relapse prevention compared to CAR T cells with or without αPD-1 antibody blockade. These findings support the integration of immune checkpoint blockade with CAR-T cells to advantageously manipulate solid tumors and offer an alternative method to CAR-T cell therapy (168). Graft anti-tumor responses can also be improved through the modulation of transcription factors and epigenetic enzymes, two widely studied methods being the knockdown of EZH1 and DNTM3A. In addition, maintaining the cytotoxicity of NK cells through the induction of CD16 has shown promise in suppressing tumor cells as well.
To ensure the safety of allogeneic cell-based therapy, strategies have been designed to reduce post-treatment alloreactivity. One way to ameliorate potential GvHD is to ablate TCR expression, which can be achieved through disruption of endogenous TCR and substitution with a CAR moiety to maintain tumor recognition. Alternatively, incorporating the NY-ESO-1 TCR transgene enables the encoding of a non-conventional TCR, which is not implicated in alloreactive pathways. In addition, unconventional T cell subsets, including iNKT cells, provide possibilities of limiting alloreactivity through endogenous, HLA-independent recognition pathways. From the perspective of the HvG effect, studies have indicated that ablating HLAs, including B2M, HLA-DR, and CIITA could successfully augment graft success rate. Moreover, overexpressing inhibitory ligands, such as HLA-E, protects effector cells from NK cell-mediated killing, whereas CD47 incorporation enables effector cells to evade macrophages activity. Finally, disrupting the expression of CD52 plays a vital role in successful lymphodepletion, an essential step for preventing GvHD. The combination of various strategies for enhancing anti-tumor efficacy and suppressing alloreactivity suggests a promising future for allogeneic cell products in cancer immunotherapy.
One type of current allogeneic CAR cell therapy, ALLO-715, utilizing BCMA-targeting CAR T cells, has been tested in part A of in-human phase 1 trial against multiple myeloma (MM) named UNIVERSAL (169). TALEN technology enables two additional alterations in this therapy: TRAC knockout and CD52 knockout. Removing TRAC downregulates the expression of the TCRαβ complex, thereby preventing the recognition of histocompatibility antigens mediated by TCRαβ, lowering the risk of GvHD consequently. In addition, CD52 is expressed on multiple immune cell types whose implications with HvG effect would diminish ALLO-715 persistence. Therefore, to ensure the cell expansion and persistence of ALLO-715, patients undergo lymphodepletion in conjunction with ALLO-647 engraftment, using anti-CD52 blockade. Favorable outcomes, such as lower rates of CRS and neurotoxicity, reflect the feasibility and overall safety of ALLO-715 administration.
Another existing type of off-the-shelf allogeneic CAR-T cells is UCART19, whose mRNA encoding TALENs knocks out the TCR-α-constant-chain-encoding and CD52 genes (12). Such a function reduces the risk of GvHD in utilizing allogeneic UCART19. Therapeutic effects of UCART19 have been tested in two clinical trials, PALL trial for children and CALM trial for adults, for treating pediatric and adult B-cell acute lymphoblastic leukemia, where UCART19 cells generated through recombinant lentiviral transduction were infused after the 7-day lymphodepletion treatment. Despite the occurrence of adverse events, such as CRS, satisfying outcomes include in vivo expansion of UCART19 and antileukemic response in a relatively safer manner. These trials demonstrate the feasibility of employing allogeneic UCART19 to treat B-cell acute lymphoblastic leukemia, especially when facing the rapid progression of the disease and the unavailability of autologous CAR-T-cell treatment. In addition, such trials provide a template for practically incorporating allogeneic cell products with multiple genetic engineering strategies, heralding the promise of allogeneic cell-based cancer therapy. In spite of the prospect of allogeneic CAR-T-cell products like UCART19, one accompanying limitation is manufacturing complexity (6). Since the risk of GvHD resides in HLA incompatibility for conventional TCRαβ T cells, endogenous TCR expression attenuation is required, which guarantees safety but simultaneously increases production difficulty.
In response to this problem, generating stem cell-derived products to express CARs and TCRs for tumor suppression offer possible alternatives (13). In addition to conventional CAR-T cells, CAR-engineered stem cells like CAR-NK and CAR-myeloid cells lead to a more extensive anti-tumor response. One study has demonstrated the performance of CAR-NK cell therapy against CD19-positive lymphoid tumors (170). Researchers derived the HLA-mismatched anti-CD19 CAR-NK cells from cord blood. Retroviral vector for transduction encoded anti-CD19 CAR, IL-15, and inducible caspase 9. In phase 1 and 2 trials, most patients experiencing engineered CAR-NK cell infusion showed quick responses to the therapy without accompanying side effects such as CRS, neurotoxicity, and hemophagocytic lymphohistiocytosis, except for hematologic toxic events related to lymphodepletion chemotherapy. In addition, GvHD did not occur after CAR-NK cell administration, indicating the viability and safety of allogeneic CAR-NK cell therapy.
Despite the promising features of current allogeneic CAR cell therapies mentioned above, a few drawbacks should be considered. In the application of BCMA-targeting CAR-T cells and UCART19 cells, the ablation of TCR and CD52 might lead to increased manufacturing difficulties and high costs (6, 12, 118, 171, 172). In contrast, although the cord blood-derived CAR-NK cells effectively avoid the risk of GvHD, the sensitive and complex process of thawing blood units could potentially lower the treatment’s efficacy. Furthermore, the availability of allogeneic CAR cell therapies depends on several factors. For example, due to the limited length of lentiviral/retroviral vectors required for transduction (169), transgene should be optimized before practical administration. Development of new gene delivery systems might overcome this problem. Although the occurrence of side effects is controlled at a relatively low level, such adverse problems remain unsolved. As seen in CRS, since IL-6, IL-1, and nitric oxide (NO) secreted by recipient macrophages mediate the severity of the syndrome rather than cytokines obtained from CAR T cells, blocking IL-6 and IL-1 and inhibiting inducible NO synthase (iNOS) provides an ameliorating effect (173). Moreover, as human microglia generate iNOS and proinflammatory cytokines via IL-1 activation, IL-1 blockade also plays an essential role in alleviating CAR-T cell-mediated neurotoxicity (173). Further studies should focus on discovering alternatives for improving the safety control of allogeneic CAR cell therapies.
Similar to the UCART19 therapy mentioned above, genetically engineered stem cells can also be utilized in allogeneic treatment. Specifically, in HSC transplantation (HSCT), it is possible to produce immune cells that target specific tumors over an extended period using genetically modified cells. On the contrary, PSCs, including ESCs and iPSCs, can self-renew while preserving pluripotency and provide endless supplies for target cells. Furthermore, the accessible source PBMCs allows for convenient iPSC reprogramming. By starting the engineering process with a small number of stem cells, the cost can be controlled while efficiency is enhanced.
However, while various engineering strategies and high accessibility of cell sources indicate the potential application of stem cell-based cancer therapy, several limitations are worth mentioning. For example, incorporating additional genes such as the sr39TK/GCV suicide switch may increase the immunogenicity and dependence on the cell cycle (6). Moreover, during the induction of T cells from iPSC and T-iPSC, uncharacterized serum and feeder cells of murine origin occur despite the successful generation, which is incompatible with the production at a clinical level (19). Therefore, future investigations should focus on improving the overall quality of genetically engineered cells and optimizing the generation process, thereby further improving the practicality of deriving immune cells from undifferentiated stem cells and advancing the development of allogeneic cell-based immunotherapy for cancer.
Y-RL and LY: conceptualization. YC, AK, YZ, YF, and MW: writing-original draft preparation. YC, AK, MW, YF, and Y-RL: writing-review and editing. YC and YF: visualization. Y-RL and LY: supervision. All authors have read and agreed to the published version of the manuscript.
This work was supported by an Ablon Scholars Award (to LY), a UCLA BSCRC Innovation Award (to LY), and an M. John Pickett Post-Doctoral Fellow Award (to Y-RL).
The authors would like to acknowledge the members of the Yang Lab for contributing insights and discussion surrounding the topic. Figures were created in biorender.com.
Y-RL and LY are inventors on patents relating to this article filed by UCLA. LY is a scientific advisor to AlzChem and Amberstone Biosciences, and a co-founder, stockholder, and advisory board member of Appia Bio.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mellman, I, Coukos, G, and Dranoff, G. Cancer immunotherapy comes of age. Nature. (2011) 480:480–9. doi: 10.1038/nature10673
2. Riley, RS, June, CH, Langer, R, and Mitchell, MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov. (2019) 18:175–96. doi: 10.1038/s41573-018-0006-z
3. Farkona, S, Diamandis, EP, and Blasutig, IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. (2016) 14:1–18. doi: 10.1186/s12916-016-0623-5
4. Weber, EW, Maus, MV, and Mackall, CL. The emerging landscape of immune cell therapies. Cells. (2020) 181:46–62. doi: 10.1016/j.cell.2020.03.001
5. Guedan, S, Ruella, M, and June, CH. Emerging cellular therapies for cancer. Annu Rev Immunol. (2019) 37:145–71. doi: 10.1146/annurev-immunol-042718-041407
6. Li, Y-R, Zhou, Y, Kim, YJ, Zhu, Y, Ma, F, Yu, J, et al. Development of allogeneic HSC-engineered iNKT cells for off-the-shelf cancer immunotherapy. Cell Rep Med. (2021) 2:100449. doi: 10.1016/j.xcrm.2021.100449
7. DiNofia, AM, and Grupp, SA. Will allogeneic CAR T cells for CD19+ malignancies take autologous CAR T cells ‘off the shelf’? Nat Rev Clin Oncol. (2021) 18:195–6. doi: 10.1038/s41571-021-00485-1
8. Jenq, RR, and Van den Brink, MR. Allogeneic haematopoietic stem cell transplantation: individualized stem cell and immune therapy of cancer. Nat Rev Cancer. (2010) 10:213–21. doi: 10.1038/nrc2804
9. Dahlberg, CI, Sarhan, D, Chrobok, M, Duru, AD, and Alici, E. Natural killer cell-based therapies targeting cancer: possible strategies to gain and sustain anti-tumor activity. Front Immunol. (2015) 6:605. doi: 10.3389/fimmu.2015.00605
10. Caldwell, KJ, Gottschalk, S, and Talleur, AC. Allogeneic CAR cell therapy—more than a pipe dream. Front Immunol. (2021) 11:618427. doi: 10.3389/fimmu.2020.618427
11. Perez, C, Gruber, I, and Arber, C. Off-the-shelf allogeneic T cell therapies for cancer: opportunities and challenges using naturally occurring universal donor T cells. Front Immunol. (2020) 11:583716. doi: 10.3389/fimmu.2020.583716
12. Benjamin, R, Graham, C, Yallop, D, Jozwik, A, Mirci-Danicar, OC, Lucchini, G, et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1 studies. Lancet. (2020) 396:1885–94. doi: 10.1016/S0140-6736(20)32334-5
13. Li, Y-R, Zhou, Y, Kramer, A, and Yang, L. Engineering stem cells for cancer immunotherapy. Trends Cancer. (2021) 7:1059–73. doi: 10.1016/j.trecan.2021.08.004
14. Li, Y-R, Zeng, S, Dunn, ZS, Zhou, Y, Li, Z, Yu, J, et al. Off-the-shelf third-party HSC-engineered iNKT cells for ameliorating GvHD while preserving GvL effect in the treatment of blood cancers. Iscience. (2022) 25:104859. doi: 10.1016/j.isci.2022.104859
15. Smith, DJ, Liu, S, Ji, S, Li, B, McLaughlin, J, Cheng, D, et al. Genetic engineering of hematopoietic stem cells to generate invariant natural killer T cells. Proc Natl Acad Sci U S A. (2015) 112:1523–8. doi: 10.1073/pnas.1424877112
16. Gschweng, E, De Oliveira, S, and Kohn, DB. Hematopoietic stem cells for cancer immunotherapy. Immunol Rev. (2014) 257:237–49. doi: 10.1111/imr.12128
17. Jiang, Z, Han, Y, and Cao, X. Induced pluripotent stem cell (iPSCs) and their application in immunotherapy. Cell Mol Immunol. (2014) 11:17–24. doi: 10.1038/cmi.2013.62
18. Potdar, P, and Chaudhary, S. Current challenges in the therapeutic use of induced pluripotent stem cells (iPSCs) in cancer therapy. Appl Cancer Res. (2017) 37:1–8. doi: 10.1186/s41241-017-0015-y
19. Nianias, A, and Themeli, M. Induced pluripotent stem cell (iPSC)–derived lymphocytes for adoptive cell immunotherapy: recent advances and challenges. Curr Hematol Malig Rep. (2019) 14:261–8. doi: 10.1007/s11899-019-00528-6
20. Zhu, Y, Kang, E, Wilson, M, Basso, T, Chen, E, Yu, Y, et al. 3D tumor spheroid and Organoid to model tumor microenvironment for cancer immunotherapy. Organ. (2022) 1:149–67. doi: 10.3390/organoids1020012
21. Shankar, K, Capitini, CM, and Saha, K. Genome engineering of induced pluripotent stem cells to manufacture natural killer cell therapies. Stem Cell Res Ther. (2020) 11:1–14. doi: 10.1186/s13287-020-01741-4
22. Yip, A, and Webster, RM. The market for chimeric antigen receptor T cell therapies. Nat Rev Drug Discov. (2018) 17:161–2. doi: 10.1038/nrd.2017.266
23. Zhang, C, Zhuang, Q, Liu, J, and Liu, X. Synthetic biology in chimeric antigen receptor T (CAR T) cell engineering. ACS Synth Biol. (2022) 11:1–15. doi: 10.1021/acssynbio.1c00256
24. Moritz, D, Wels, W, Mattern, J, and Groner, B. Cytotoxic T lymphocytes with a grafted recognition specificity for ERBB2-expressing tumor cells. Proc Natl Acad Sci U S A. (1994) 91:4318–22. doi: 10.1073/pnas.91.10.4318
25. Rafiq, S, Purdon, TJ, Daniyan, AF, Koneru, M, Dao, T, Liu, C, et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms tumor 1 antigen. Leukemia. (2017) 31:1788–97. doi: 10.1038/leu.2016.373
26. Ramírez-Chacón, A, Betriu-Méndez, S, Bartoló-Ibars, A, González, A, Martí, M, and Juan, M. Ligand-based CAR-T cell: different strategies to drive T cells in future new treatments. Front Immunol. (2022) 13:932559. doi: 10.3389/fimmu.2022.932559
27. Elazar, A, Chandler, NJ, Davey, AS, Weinstein, JY, Nguyen, JV, Trenker, R, et al. De novo-designed transmembrane domains tune engineered receptor functions. elife. (2022) 11:e75660. doi: 10.7554/eLife.75660
28. Li, H, Zhong, D, Luo, H, Shi, W, Xie, S, Qiang, H, et al. Nanobody-based CAR T cells targeting intracellular tumor antigens. Biomed Pharmacother. (2022) 156:113919. doi: 10.1016/j.biopha.2022.113919
29. Keshavarz, A, Salehi, A, Khosravi, S, Shariati, Y, Nasrabadi, N, Kahrizi, MS, et al. Recent findings on chimeric antigen receptor (CAR)-engineered immune cell therapy in solid tumors and hematological malignancies. Stem Cell Res Ther. (2022) 13:1–22. doi: 10.1186/s13287-022-03163-w
30. Van Schandevyl, S, and Kerre, T. Chimeric antigen receptor T-cell therapy: design improvements and therapeutic strategies in cancer treatment. Acta Clin Belg. (2018) 75:26–32. doi: 10.1080/17843286.2018.1545373
31. Savoldo, B, Ramos, CA, Liu, E, Mims, MP, Keating, MJ, Carrum, G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. J Clin Invest. (2011) 121:1822–6. doi: 10.1172/JCI46110
32. Chmielewski, M, and Abken, H. CAR T cells releasing IL-18 convert to T-Bethigh FoxO1low effectors that exhibit augmented activity against advanced solid tumors. Cell Rep. (2017) 21:3205–19. doi: 10.1016/j.celrep.2017.11.063
33. Hay, KA. Cytokine release syndrome and neurotoxicity after CD 19 chimeric antigen receptor-modified (CAR-) T cell therapy. Br J Haematol. (2018) 183:364–74. doi: 10.1111/bjh.15644
34. Yilmaz, A, Cui, H, Caligiuri, MA, and Yu, J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J Hematol Oncol. (2020) 13:1–22. doi: 10.1186/s13045-020-00998-9
35. Rezvani, K, Rouce, R, Liu, E, and Shpall, E. Engineering natural killer cells for cancer immunotherapy. Mol Ther. (2017) 25:1769–81. doi: 10.1016/j.ymthe.2017.06.012
36. Smyth, MJ, Hayakawa, Y, Takeda, K, and Yagita, H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat Rev Cancer. (2002) 2:850–61. doi: 10.1038/nrc928
37. Leivas, A, Valeri, A, Córdoba, L, García-Ortiz, A, Ortiz, A, Sánchez-Vega, L, et al. NKG2D-CAR-transduced natural killer cells efficiently target multiple myeloma. Blood Cancer J. (2021) 11:1–11. doi: 10.1038/s41408-021-00537-w
38. du, Z, Ng, YY, Zha, S, and Wang, S. piggyBac system to co-express NKG2D CAR and IL-15 to augment the in vivo persistence and anti-AML activity of human peripheral blood NK cells. Mol Ther Methods Clin Dev. (2021) 23:582–96. doi: 10.1016/j.omtm.2021.10.014
39. Sadelain, M, Brentjens, R, and Rivière, I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. (2009) 21:215–23. doi: 10.1016/j.coi.2009.02.009
40. Santana Carrero, RM, Beceren-Braun, F, Rivas, SC, Hegde, SM, Gangadharan, A, Plote, D, et al. IL-15 is a component of the inflammatory milieu in the tumor microenvironment promoting antitumor responses. Proc Natl Acad Sci U S A. (2019) 116:599–608. doi: 10.1073/pnas.1814642116
41. Hoyos, V, Savoldo, B, Quintarelli, C, Mahendravada, A, Zhang, M, Vera, J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. (2010) 24:1160–70. doi: 10.1038/leu.2010.75
42. Hurton, LV, Singh, H, Najjar, AM, Switzer, KC, Mi, T, Maiti, S, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A. (2016) 113:E7788–97. doi: 10.1073/pnas.1610544113
43. Bell, M, and Gottschalk, S. Engineered cytokine signaling to improve CAR T cell effector function. Front Immunol. (2021) 12:684642. doi: 10.3389/fimmu.2021.684642
44. Sportès, C, Babb, RR, Krumlauf, MC, Hakim, FT, Steinberg, SM, Chow, CK, et al. Phase I study of recombinant human Interleukin-7 Administration in Subjects with refractory malignancy. Clin Cancer Res. (2010) 16:727–35. doi: 10.1158/1078-0432.CCR-09-1303
45. Perna, SK, Pagliara, D, Mahendravada, A, Liu, H, Brenner, MK, Savoldo, B, et al. Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin Cancer Res. (2014) 20:131–9. doi: 10.1158/1078-0432.CCR-13-1016
46. Luo, H, Su, J, Sun, R, Sun, Y, Wang, Y, Dong, Y, et al. Coexpression of IL7 and CCL21 increases efficacy of CAR-T cells in solid tumors without requiring preconditioned lymphodepletion. Clin Cancer Res. (2020) 26:5494–505. doi: 10.1158/1078-0432.CCR-20-0777
47. Chmielewski, M, and Abken, H. CAR T cells transform to trucks: chimeric antigen receptor–redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol Immunother. (2012) 61:1269–77. doi: 10.1007/s00262-012-1202-z
48. Dowell, AC, Oldham, KA, Bhatt, RI, Lee, SP, and Searle, PF. Long-term proliferation of functional human NK cells, with conversion of CD56dim NK cells to a CD56bright phenotype, induced by carcinoma cells co-expressing 4-1BBL and IL-12. Cancer Immunol Immunother. (2012) 61:615–28. doi: 10.1007/s00262-011-1122-3
49. Kerkar, SP, Goldszmid, RS, Muranski, P, Chinnasamy, D, Yu, Z, Reger, RN, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. (2011) 121:4746–57. doi: 10.1172/JCI58814
50. Batra, SA, Rathi, P, Guo, L, Courtney, AN, Fleurence, J, Balzeau, J, et al. Glypican-3–specific CAR T cells Coexpressing IL15 and IL21 have superior expansion and antitumor activity against hepatocellular carcinoma. Cancer Immunol Res. (2020) 8:309–20. doi: 10.1158/2326-6066.CIR-19-0293
51. Moroz, A, Eppolito, C, Li, Q, Tao, J, Clegg, CH, and Shrikant, PA. IL-21 enhances and sustains CD8+ T cell responses to achieve durable tumor immunity: comparative evaluation of IL-2, IL-15, and IL-21. J Immunol. (2004) 173:900–9. doi: 10.4049/jimmunol.173.2.900
52. Ortiz, MV, Roberts, SS, Glade Bender, J, Shukla, N, and Wexler, LH. Immunotherapeutic targeting of GPC3 in pediatric solid embryonal tumors. Front Oncol. (2019) 9:108. doi: 10.3389/fonc.2019.00108
53. Lee, L, Gupta, M, and Sahasranaman, S. Immune checkpoint inhibitors: an introduction to the next-generation cancer immunotherapy. J Clin Pharmacol. (2016) 56:157–69. doi: 10.1002/jcph.591
54. Qin, S, Xu, L, Yi, M, Yu, S, Wu, K, and Luo, S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer. (2019) 18:155:1–14. doi: 10.1186/s12943-019-1091-2
55. Zitvogel, L., and Kroemer, G., Targeting PD-1/PD-L1 interactions for cancer immunotherapy. (2012), Abingdon, UK: Taylor & Francis. p. 1223–1225.
56. Goodman, A, Patel, SP, and Kurzrock, R. PD-1–PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat Rev Clin Oncol. (2017) 14:203–20. doi: 10.1038/nrclinonc.2016.168
57. Chamoto, K, Hatae, R, and Honjo, T. Current issues and perspectives in PD-1 blockade cancer immunotherapy. Int J Clin Oncol. (2020) 25:790–800. doi: 10.1007/s10147-019-01588-7
58. Brahmer, JR, Drake, CG, Wollner, I, Powderly, JD, Picus, J, Sharfman, WH, et al. Phase I study of single-agent anti–programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. (2010) 28:3167–75. doi: 10.1200/JCO.2009.26.7609
59. Topalian, SL, Hodi, FS, Brahmer, JR, Gettinger, SN, Smith, DC, McDermott, DF, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
60. Motzer, RJ, Escudier, B, McDermott, DF, George, S, Hammers, HJ, Srinivas, S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. (2015) 373:1803–13. doi: 10.1056/NEJMoa1510665
61. Yu, S, Yi, M, Qin, S, and Wu, K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol Cancer. (2019) 18:1–13. doi: 10.1186/s12943-019-1057-4
62. Yang, C-Y, Fan, MH, Miao, CH, Liao, YJ, Yuan, RH, and Liu, CL. Engineering chimeric antigen receptor T cells against immune checkpoint inhibitors PD-1/PD-L1 for treating pancreatic cancer. Mol Ther Oncolytics. (2020) 17:571–85. doi: 10.1016/j.omto.2020.05.009
63. Slaney, CY, Wang, P, Darcy, PK, and Kershaw, MH. CARs versus BiTEs: a comparison between T cell–redirection strategies for cancer TreatmentCAR T-cell and BiTE therapies for cancers. Cancer Discov. (2018) 8:924–34. doi: 10.1158/2159-8290.CD-18-0297
64. Kershaw, MH, Westwood, JA, and Darcy, PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer. (2013) 13:525–41. doi: 10.1038/nrc3565
65. Buchbinder, EI, and Desai, A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98. doi: 10.1097/COC.0000000000000239
66. Yang, Y-F, Zou, JP, Mu, J, Wijesuriya, R, Ono, S, Walunas, T, et al. Enhanced induction of antitumor T-cell responses by cytotoxic T lymphocyte-associated molecule-4 blockade: the effect is manifested only at the restricted tumor-bearing stages. Cancer Res. (1997) 57:4036–41.
67. Mangsbo, SM, Sandin, LC, Anger, K, Korman, AJ, Loskog, A, and Tötterman, TH. Enhanced tumor eradication by combining CTLA-4 or PD-1 blockade with CpG therapy. J Immunother. (2010) 33:225–35. doi: 10.1097/CJI.0b013e3181c01fcb
68. Espenschied, J, Lamont, J, Longmate, J, Pendas, S, Wang, Z, Diamond, DJ, et al. CTLA-4 blockade enhances the therapeutic effect of an attenuated poxvirus vaccine targeting p53 in an established murine tumor model. J Immunol. (2003) 170:3401–7. doi: 10.4049/jimmunol.170.6.3401
69. Robert, C, Thomas, L, Bondarenko, I, O'Day, S, Weber, J, Garbe, C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. (2011) 364:2517–26. doi: 10.1056/NEJMoa1104621
70. Ribas, A. “Tumor immunotherapy directed at PD-1,” in The New England Journal of Medicine. (Massachusetts Medical Society) (2012) 2517–2519.
71. Shi, L, Meng, T, Zhao, Z, Han, J, Zhang, W, Gao, F, et al. CRISPR knock out CTLA-4 enhances the anti-tumor activity of cytotoxic T lymphocytes. Gene. (2017) 636:36–41. doi: 10.1016/j.gene.2017.09.010
72. Workman, CJ, Dugger, KJ, and Vignali, DA. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J Immunol. (2002) 169:5392–5. doi: 10.4049/jimmunol.169.10.5392
73. Zhang, Y, Zhang, X, Cheng, C, Mu, W, Liu, X, Li, N, et al. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front Med. (2017) 11:554–62. doi: 10.1007/s11684-017-0543-6
74. Workman, CJ, Cauley, LS, Kim, IJ, Blackman, MA, Woodland, DL, and Vignali, DAA. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol. (2004) 172:5450–5. doi: 10.4049/jimmunol.172.9.5450
75. Das, M, Zhu, C, and Kuchroo, VK. Tim-3 and its role in regulating anti-tumor immunity. J Immunol. (2008) 181:7660. doi: 10.1111/imr.12520
76. Morimoto, T, Nakazawa, T, Matsuda, R, Nishimura, F, Nakamura, M, Yamada, S, et al. CRISPR-Cas9–mediated TIM3 knockout in human natural killer cells enhances growth inhibitory effects on human Glioma cells. Int J Mol Sci. (2021) 22:3489. doi: 10.3390/ijms22073489
77. Sakuishi, K, Apetoh, L, Sullivan, JM, Blazar, BR, Kuchroo, VK, and Anderson, AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. (2010) 207:2187–94. doi: 10.1084/jem.20100643
78. Wang, X, Xiong, H, and Ning, Z. Implications of NKG2A in immunity and immune-mediated diseases. Front Immunol. (2022) 13:960852. doi: 10.3389/fimmu.2022.1076546
79. Borst, L, van der Burg, SH, and van Hall, T. The NKG2A–HLA-E Axis as a novel checkpoint in the tumor MicroenvironmentNKG2A as a novel checkpoint in the tumor microenvironment. Clin Cancer Res. (2020) 26:5549–56. doi: 10.1158/1078-0432.CCR-19-2095
80. van Hall, T, André, P, Horowitz, A, Ruan, DF, Borst, L, Zerbib, R, et al. Monalizumab: inhibiting the novel immune checkpoint NKG2A. J Immunother Cancer. (2019) 7:1–8. doi: 10.1186/s40425-019-0761-3
81. van Montfoort, N, Borst, L, Korrer, MJ, Sluijter, M, Marijt, KA, Santegoets, SJ, et al. NKG2A blockade potentiates CD8 T cell immunity induced by cancer vaccines. Cells. (2018) 175:1744–1755.e15. e15. doi: 10.1016/j.cell.2018.10.028
82. Kren, L, Slaby, O, Muckova, K, Lzicarova, E, Sova, M, Vybihal, V, et al. Expression of immune-modulatory molecules HLA-G and HLA-E by tumor cells in glioblastomas: an unexpected prognostic significance? Neuropathology. (2011) 31:129–34. doi: 10.1111/j.1440-1789.2010.01149.x
83. de Kruijf, EM, Sajet, A, van Nes, JGH, Natanov, R, Putter, H, Smit, VTHBM, et al. HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J Immunol. (2010) 185:7452–9. doi: 10.4049/jimmunol.1002629
84. Kamiya, T, Seow, SV, Wong, D, Robinson, M, and Campana, D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J Clin Invest. (2019) 129:2094–106. doi: 10.1172/JCI123955
85. Brossart, P. The role of antigen spreading in the efficacy of immunotherapies. Clin Cancer Res. (2020) 26:4442–7. doi: 10.1158/1078-0432.CCR-20-0305
86. Vanderlugt, CL, and Miller, SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. (2002) 2:85–95. doi: 10.1038/nri724
87. Kvistborg, P, Philips, D, Kelderman, S, Hageman, L, Ottensmeier, C, Joseph-Pietras, D, et al. Anti–CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med. (2014) 6:254ra128–8. doi: 10.1126/scitranslmed.3008918
88. Yuan, J, Gnjatic, S, Li, H, Powel, S, Gallardo, HF, Ritter, E, et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc Natl Acad Sci. (2008) 105:20410–5. doi: 10.1073/pnas.0810114105
89. Yuan, J, Adamow, M, Ginsberg, BA, Rasalan, TS, Ritter, E, Gallardo, HF, et al. Integrated NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc Natl Acad Sci. (2011) 108:16723–8. doi: 10.1073/pnas.1110814108
90. Robert, L, Tsoi, J, Wang, X, Emerson, R, Homet, B, Chodon, T, et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin Cancer Res. (2014) 20:2424–32. doi: 10.1158/1078-0432.CCR-13-2648
91. Dai, E, Zhu, Z, Wahed, S, Qu, Z, Storkus, WJ, and Guo, ZS. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol Cancer. (2021) 20:1–27. doi: 10.1186/s12943-021-01464-x
92. Jing, R, Scarfo, I, Najia, MA, Lummertz da Rocha, E, Han, A, Sanborn, M, et al. EZH1 repression generates mature iPSC-derived CAR T cells with enhanced antitumor activity. Cell Stem Cell. (2022) 29:1181–1196.e6. doi: 10.1016/j.stem.2022.06.014
93. Iriguchi, S, Yasui, Y, Kawai, Y, Arima, S, Kunitomo, M, Sato, T, et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat Commun. (2021) 12:1–15. doi: 10.1038/s41467-020-20658-3
94. Vizcardo, R, Klemen, ND, Islam, SMR, Gurusamy, D, Tamaoki, N, Yamada, D, et al. Generation of tumor antigen-specific iPSC-derived thymic emigrants using a 3D thymic culture system. Cell Rep. (2018) 22:3175–90. doi: 10.1016/j.celrep.2018.02.087
95. Montel-Hagen, A, Seet, CS, Li, S, Chick, B, Zhu, Y, Chang, P, et al. Organoid-induced differentiation of conventional T cells from human pluripotent stem cells. Cell Stem Cell. (2019) 24:376–389.e8. doi: 10.1016/j.stem.2018.12.011
96. Yang, L, Rau, R, and Goodell, MA. DNMT3A in haematological malignancies. Nat Rev Cancer. (2015) 15:152–65. doi: 10.1038/nrc3895
97. Challen, GA, Sun, D, Jeong, M, Luo, M, Jelinek, J, Berg, JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. (2012) 44:23–31. doi: 10.1038/ng.1009
98. Prinzing, B, Zebley, CC, Petersen, CT, Fan, Y, Anido, AA, Yi, Z, et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci Transl Med. (2021) 13:eabh0272. doi: 10.1126/scitranslmed.abh0272
99. Ito, Y, and Kagoya, Y. Epigenetic engineering for optimal chimeric antigen receptor T cell therapy. Cancer Sci. (2022) 113:3664. doi: 10.1111/cas.15541
100. Ravetch, J. Fcgamma receptors: old friends and new family members. Immunity. (2006) 24:19–28. doi: 10.1016/j.immuni.2005.11.010
101. Mandelboim, O, Malik, P, Davis, DM, Jo, CH, Boyson, JE, and Strominger, JL. Human CD16 as a lysis receptor mediating direct natural killer cell cytotoxicity. Proc Natl Acad Sci. (1999) 96:5640–4. doi: 10.1073/pnas.96.10.5640
102. Zhu, H, Blum, RH, Bjordahl, R, Gaidarova, S, Rogers, P, Lee, TT, et al. Pluripotent stem cell–derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood. (2020) 135:399–410. doi: 10.1182/blood.2019000621
103. Jing, Y, Ni, Z, Wu, J, Higgins, LA, Markowski, TW, Kaufman, DS, et al. Identification of an ADAM17 cleavage region in human CD16 (FcγRIII) and the engineering of a non-cleavable version of the receptor in NK cells. PLoS One. (2015) 10:e0121788. doi: 10.1371/journal.pone.0121788
104. Cichocki, F, Bjordahl, R, Goodridge, JP, Mahmood, S, Gaidarova, S, Abujarour, R, et al. Quadruple gene-engineered natural killer cells enable multi-antigen targeting for durable antitumor activity against multiple myeloma. Nat Commun. (2022) 13:1–15. doi: 10.1038/s41467-022-35127-2
105. Lamers-Kok, N, Panella, D, Georgoudaki, AM, Liu, H, Özkazanc, D, Kučerová, L, et al. Natural killer cells in clinical development as non-engineered, engineered, and combination therapies. J Hematol Oncol. (2022) 15:1–55. doi: 10.1186/s13045-022-01382-5
106. Wehler, TC, Nonn, M, Brandt, B, Britten, CM, Gröne, M, Todorova, M, et al. Targeting the activation-induced antigen CD137 can selectively deplete alloreactive T cells from antileukemic and antitumor donor T-cell lines. Blood. (2007) 109:365–73. doi: 10.1182/blood-2006-04-014100
107. Hartwig, U, Nonn, M, Khan, S, Meyer, RG, Huber, C, and Herr, W. Depletion of alloreactive T cells via CD69: implications on antiviral, antileukemic and immunoregulatory T lymphocytes. Bone Marrow Transplant. (2006) 37:297–305. doi: 10.1038/sj.bmt.1705238
108. Amrolia, PJ, Muccioli-Casadei, G, Huls, H, Adams, S, Durett, A, Gee, A, et al. Adoptive immunotherapy with allodepleted donor T-cells improves immune reconstitution after haploidentical stem cell transplantation. Blood. (2006) 108:1797–808. doi: 10.1182/blood-2006-02-001909
109. Solomon, SR, Mielke, S, Savani, BN, Montero, A, Wisch, L, Childs, R, et al. Selective depletion of alloreactive donor lymphocytes: a novel method to reduce the severity of graft-versus-host disease in older patients undergoing matched sibling donor stem cell transplantation. Blood. (2005) 106:1123–9. doi: 10.1182/blood-2005-01-0393
110. Mackinnon, S, Papadopoulos, EB, Carabasi, MH, Reich, L, Collins, NH, Boulad, F, et al. Adoptive immunotherapy evaluating escalating doses of donor leukocytes for relapse of chronic myeloid leukemia after bone marrow transplantation: separation of graft-versus-leukemia responses from graft-versus-host disease. Blood. (1995) 86:1261–8. doi: 10.1182/blood.V86.4.1261.bloodjournal8641261
111. Davies, JK, Singh, H, Huls, H, Yuk, D, Lee, DA, Kebriaei, P, et al. Combining CD19 redirection and Alloanergization to generate tumor-specific human T cells for allogeneic cell therapy of B-cell malignancies. Cancer Res. (2010) 70:3915–24. doi: 10.1158/0008-5472.CAN-09-3845
112. Barbon, CM, Janec, KJ, Kelner, RH, Norton, JE, and Guinan, EC. Alloanergization method for inducing allospecific hyporesponsiveness in PBMC exposed to allostimulation in vitro in the context of costimulatory molecule blockade In:. Immunological tolerance: Methods and protocols. New York, NY: Springer (2019). 103–18.
113. Kagoya, Y, Guo, T, Yeung, B, Saso, K, Anczurowski, M, Wang, CH, et al. Genetic ablation of HLA class I, class II, and the T-cell receptor enables allogeneic T cells to be used for adoptive T-cell therapy. Cancer Immunol Res. (2020) 8:926–36. doi: 10.1158/2326-6066.CIR-18-0508
114. MacLeod, DT, Antony, J, Martin, AJ, Moser, RJ, Hekele, A, Wetzel, KJ, et al. Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T cells. Mol Ther. (2017) 25:949–61. doi: 10.1016/j.ymthe.2017.02.005
115. June, CH, and Sadelain, M. Chimeric antigen receptor therapy. N Engl J Med. (2018) 379:64–73. doi: 10.1056/NEJMra1706169
116. Straathof, K, Flutter, B, Wallace, R, Jain, N, Loka, T, Depani, S, et al. Antitumor activity without on-target off-tumor toxicity of GD2–chimeric antigen receptor T cells in patients with neuroblastoma. Sci Transl Med. (2020) 12:eabd6169. doi: 10.1126/scitranslmed.abd6169
117. Morello, A, Sadelain, M, and Adusumilli, PS. Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov. (2016) 6:133–46. doi: 10.1158/2159-8290.CD-15-0583
118. Qasim, W, Zhan, H, Samarasinghe, S, Adams, S, Amrolia, P, Stafford, S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. (2017) 9:eaaj2013. doi: 10.1126/scitranslmed.aaj2013
119. Abramson, JS. Anti-CD19 CAR T-cell therapy for B-cell non-Hodgkin lymphoma. Transfus Med Rev. (2020) 34:29–33. doi: 10.1016/j.tmrv.2019.08.003
120. Ying, Z, Huang, XF, Xiang, X, Liu, Y, Kang, X, Song, Y, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. (2019) 25:947–53. doi: 10.1038/s41591-019-0421-7
121. Majzner, RG, and Mackall, CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. (2018) 8:1219–26. doi: 10.1158/2159-8290.CD-18-0442
122. Robbins, PF, Li, YF, el-Gamil, M, Zhao, Y, Wargo, JA, Zheng, Z, et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol. (2008) 180:6116–31. doi: 10.4049/jimmunol.180.9.6116
123. Rapoport, AP, Stadtmauer, EA, Binder-Scholl, GK, Goloubeva, O, Vogl, DT, Lacey, SF, et al. NY-ESO-1–specific TCR–engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. (2015) 21:914–21. doi: 10.1038/nm.3910
124. Mastaglio, S, Genovese, P, Magnani, Z, Ruggiero, E, Landoni, E, Camisa, B, et al. NY-ESO-1 TCR single edited stem and central memory T cells to treat multiple myeloma without graft-versus-host disease. Blood. (2017) 130:606–18. doi: 10.1182/blood-2016-08-732636
125. Baranek, T, de Amat Herbozo, C, Mallevaey, T, and Paget, C. Deconstructing iNKT cell development at single-cell resolution. Trends Immunol. (2022) 43:503–12. doi: 10.1016/j.it.2022.04.012
126. Zhou, Y, Li, YR, Zeng, S, and Yang, L. Methods for studying mouse and human invariant natural killer T cells In:. Invariant natural killer T-cells: Methods and protocols. New York, NY: Springer (2021). 35–57.
127. Van Kaer, L, Parekh, VV, and Wu, L. Invariant natural killer T cells: bridging innate and adaptive immunity. Cell Tissue Res. (2011) 343:43–55. doi: 10.1007/s00441-010-1023-3
128. Bae, E-A, Seo, H, Kim, IK, Jeon, I, Kang, CY, et al. Roles of NKT cells in cancer immunotherapy. Arch Pharm Res. (2019) 42:543–8. doi: 10.1007/s12272-019-01139-8
129. Li, Y-R, Brown, J, Yu, Y, Lee, D, Zhou, K, Dunn, ZS, et al. Targeting immunosuppressive tumor-associated macrophages using innate T cells for enhanced antitumor reactivity. Cancers. (2022) 14:2749. doi: 10.3390/cancers14112749
130. Lanier, LL. NKG2D receptor and its ligands in host defense. Cancer Immunol Res. (2015) 3:575–82. doi: 10.1158/2326-6066.CIR-15-0098
131. Exley, MA, Friedlander, P, Alatrakchi, N, Vriend, L, Yue, S, Sasada, T, et al. Adoptive transfer of invariant NKT cells as immunotherapy for advanced melanoma: a phase I clinical TrialPhase I trial of invariant NKT cells for advanced melanoma. Clin Cancer Res. (2017) 23:3510–9. doi: 10.1158/1078-0432.CCR-16-0600
132. Cheng, X, Wang, J, Qiu, C, Jin, Y, Xia, B, Qin, R, et al. Feasibility of iNKT cell and PD-1+ CD8+ T cell-based immunotherapy in patients with lung adenocarcinoma: preliminary results of a phase I/II clinical trial. Clin Immunol. (2022) 238:108992:108992. doi: 10.1016/j.clim.2022.108992
133. Güssow, D, Rein, R, Ginjaar, I, Hochstenbach, F, Seemann, G, Kottman, A, et al. The human beta 2-microglobulin gene. Primary structure and definition of the transcriptional unit. J Immunol. (1987) 139:3132–8. doi: 10.4049/jimmunol.139.9.3132
134. Wang, H, Liu, B, and Wei, J. Beta2-microglobulin (B2M) in cancer immunotherapies: biological function, resistance and remedy. Cancer Lett. (2021) 517:96–104. doi: 10.1016/j.canlet.2021.06.008
135. Halle, S, Halle, O, and Förster, R. Mechanisms and dynamics of T cell-mediated cytotoxicity in vivo. Trends Immunol. (2017) 38:432–43. doi: 10.1016/j.it.2017.04.002
136. Ren, J, Liu, X, Fang, C, Jiang, S, June, CH, and Zhao, Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 InhibitionMultiplex genome editing to generate universal CAR T cells. Clin Cancer Res. (2017) 23:2255–66. doi: 10.1158/1078-0432.CCR-16-1300
137. Mosaad, Y. Clinical role of human leukocyte antigen in health and disease. Scand J Immunol. (2015) 82:283–306. doi: 10.1111/sji.12329
138. Heeger, PS. T-cell allorecognition and transplant rejection: a summary and update. Am J Transplant. (2003) 3:525–33. doi: 10.1034/j.1600-6143.2003.00123.x
139. Angaswamy, N, Tiriveedhi, V, Sarma, NJ, Subramanian, V, Klein, C, Wellen, J, et al. Interplay between immune responses to HLA and non-HLA self-antigens in allograft rejection. Hum Immunol. (2013) 74:1478–85. doi: 10.1016/j.humimm.2013.07.002
140. Torikai, H, Mi, T, Gragert, L, Maiers, M, Najjar, A, Ang, S, et al. Genetic editing of HLA expression in hematopoietic stem cells to broaden their human application. Sci Rep. (2016) 6:1–11. doi: 10.1038/srep21757
141. Lee, J, Sheen, JH, Lim, O, Lee, Y, Ryu, J, Shin, D, et al. Abrogation of HLA surface expression using CRISPR/Cas9 genome editing: a step toward universal T cell therapy. Sci Rep. (2020) 10:1–10. doi: 10.1038/s41598-020-74772-9
142. Kwon, Y-W, Ahn, HS, Lee, JW, Yang, HM, Cho, HJ, Kim, SJ, et al. HLA DR genome editing with TALENs in human iPSCs produced immune-tolerant dendritic cells. Stem Cells Int. (2021) 2021:1–14. doi: 10.1155/2021/8873383
143. Depil, S, Duchateau, P, Grupp, SA, Mufti, G, and Poirot, L. ‘Off-the-shelf’allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
144. Devaiah, BN, and Singer, DS. CIITA and its dual roles in MHC gene transcription. Front Immunol. (2013) 4:476. doi: 10.3389/fimmu.2013.00476
145. Han, X, Wang, M, Duan, S, Franco, PJ, Kenty, JHR, Hedrick, P, et al. Generation of hypoimmunogenic human pluripotent stem cells. Proc Natl Acad Sci U S A. (2019) 116:10441–6. doi: 10.1073/pnas.1902566116
146. Mattapally, S, Pawlik, KM, Fast, VG, Zumaquero, E, Lund, FE, Randall, TD, et al. Human leukocyte antigen class I and II knockout human induced pluripotent stem cell–derived cells: universal donor for cell therapy. J Am Heart Assoc. (2018) 7:e010239. doi: 10.1161/JAHA.118.010239
147. Lee, N, Llano, M, Carretero, M, Ishitani, A, Navarro, F, López-Botet, M, et al. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci. (1998) 95:5199–204. doi: 10.1073/pnas.95.9.5199
148. Braud, VM, Allan, DSJ, O'Callaghan, CA, Söderström, K, D'Andrea, A, Ogg, GS, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. (1998) 391:795–9. doi: 10.1038/35869
149. Riteau, B, Menier, C, Khalil-Daher, I, Martinozzi, S, Pla, M, Dausset, J, et al. HLA-G1 co-expression boosts the HLA class I-mediated NK lysis inhibition. Int Immunol. (2001) 13:193–201. doi: 10.1093/intimm/13.2.193
150. Lanza, R, Russell, DW, and Nagy, A. Engineering universal cells that evade immune detection. Nat Rev Immunol. (2019) 19:723–33. doi: 10.1038/s41577-019-0200-1
151. Pontrelli, P, Rascio, F, Castellano, G, Grandaliano, G, Gesualdo, L, and Stallone, G. The role of natural killer cells in the immune response in kidney transplantation. Front Immunol. (2020) 11:1454. doi: 10.3389/fimmu.2020.01454
152. Zhao, H-X, Jiang, F, Zhu, YJ, Wang, L, Li, K, Li, Y, et al. Enhanced immunological tolerance by HLA-G1 from neural progenitor cells (NPCs) derived from human embryonic stem cells (hESCs). Cell Physiol Biochem. (2017) 44:1435–44. doi: 10.1159/000485539
153. Torikai, H, Reik, A, Soldner, F, Warren, EH, Yuen, C, Zhou, Y, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. (2013) 122:1341–9. doi: 10.1182/blood-2013-03-478255
154. Gornalusse, GG, Hirata, RK, Funk, SE, Riolobos, L, Lopes, VS, Manske, G, et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol. (2017) 35:765–72. doi: 10.1038/nbt.3860
155. Lindberg, FP, Gresham, HD, Schwarz, E, and Brown, EJ. Molecular cloning of integrin-associated protein: an immunoglobulin family member with multiple membrane-spanning domains implicated in alpha v beta 3-dependent ligand binding. J Cell Biol. (1993) 123:485–96. doi: 10.1083/jcb.123.2.485
156. Takimoto, C, Chao, MP, Gibbs, C, McCamish, MA, Liu, J, Chen, JY, et al. The macrophage ‘do not eat me’ signal, CD47, is a clinically validated cancer immunotherapy target. Ann Oncol. (2019) 30:486–9. doi: 10.1093/annonc/mdz006
157. Giatromanolaki, A, Mitrakas, A, Anestopoulos, I, Kontosis, A, Koukourakis, IM, Pappa, A, et al. Expression of CD47 and SIRPα macrophage immune-checkpoint pathway in non-small-cell lung cancer. Cancers. (2022) 14:1801. doi: 10.3390/cancers14071801
158. Shi, M, Gu, Y, Jin, K, Fang, H, Chen, Y, Cao, Y, et al. CD47 expression in gastric cancer clinical correlates and association with macrophage infiltration. Cancer Immunol Immunother. (2021) 70:1831–40. doi: 10.1007/s00262-020-02806-2
159. Jalil, AR, Andrechak, JC, and Discher, DE. Macrophage checkpoint blockade: results from initial clinical trials, binding analyses, and CD47-SIRPα structure–function. Antibody Ther. (2020) 3:80–94. doi: 10.1093/abt/tbaa006
160. Salehi, S, and Reed, EF. The divergent roles of macrophages in solid organ transplantation. Curr Opin Organ Transplant. (2015) 20:446. doi: 10.1097/MOT.0000000000000209
161. Magil, A.B., Monocytes/macrophages in renal allograft rejection. Transplant Rev, (2009). 23: p. 199–208. doi: 10.1016/j.trre.2009.06.005 0955-470X.
162. Jiang, X, Tian, W, Sung, YK, Qian, J, and Nicolls, MR. Macrophages in solid organ transplantation. Vascular Cell. (2014) 6:5–9. doi: 10.1186/2045-824X-6-5
163. Deuse, T, Hu, X, Gravina, A, Wang, D, Tediashvili, G, de, C, et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat Biotechnol. (2019) 37:252–8. doi: 10.1038/s41587-019-0016-3
164. Hu, X, Dao, M, White, K, Clarke, R, Landry, S, Basco, R, et al. Abstract LB144: overexpression of CD47 protects hypoimmune CAR T cells from innate immune cell killing. Cancer Res. (2021) 81:LB144–4. doi: 10.1158/1538-7445.AM2021-LB144
165. Poirot, L, Schiffer-Mannioui, C, le Clerre, D, Chion-Sotinel, I, Derniame, S, Potrel, P, et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell ImmunotherapiesGenome editing for Allogenic adoptive T-cell immunotherapy. Cancer Res. (2015) 75:3853–64. doi: 10.1158/0008-5472.CAN-14-3321
166. Smirnov, S, Petukhov, A, Levchuk, K, Kulemzin, S, Staliarova, A, Lepik, K, et al. Strategies to circumvent the side-effects of immunotherapy using allogeneic CAR-T cells and boost its efficacy: results of recent clinical trials. Front Immunol. (2021) 12:780145. doi: 10.3389/fimmu.2021.780145
167. Perica, K, Varela, JC, Oelke, M, and Schneck, J. Adoptive T cell immunotherapy for cancer. Rambam Maimonides Med J. (2015) 6:e0004. doi: 10.5041/RMMJ.10179
168. Hu, W, Zi, Z, Jin, Y, Li, G, Shao, K, Cai, Q, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. (2019) 68:365–77. doi: 10.1007/s00262-018-2281-2
169. Mailankody, S, Matous, JV, Chhabra, S, Liedtke, M, Sidana, S, Oluwole, OO, et al. Allogeneic BCMA-targeting CAR T cells in relapsed/refractory multiple myeloma: phase 1 UNIVERSAL trial interim results. Nat Med. (2023) 29:422–9. doi: 10.1038/s41591-022-02182-7
170. Liu, E, Marin, D, Banerjee, P, Macapinlac, HA, Thompson, P, Basar, R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
171. Stadtmauer, EA, Fraietta, JA, Davis, MM, Cohen, AD, Weber, KL, Lancaster, E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. (2020) 367:eaba7365. doi: 10.1126/science.aba7365
172. Benjamin, R, Graham, C, Yallop, D, Jozwik, A, Ciocarlie, O, Jain, N, et al. Preliminary data on safety, cellular kinetics and anti-leukemic activity of UCART19, an allogeneic anti-CD19 CAR T-cell product, in a pool of adult and pediatric patients with high-risk CD19+ relapsed/refractory B-cell acute lymphoblastic leukemia. Blood. (2018) 132:896. doi: 10.1182/blood-2018-99-111356
Keywords: gene engineering, cancer immunotherapy, allogeneic cell products, CRISPR-Cas9, chimeric antigen receptor-engineered T (CAR-T) cells, immune checkpoints, graft-versus-host disease, alloreactivity
Citation: Chen Y, Zhu Y, Kramer A, Fang Y, Wilson M, Li Y-R and Yang L (2023) Genetic engineering strategies to enhance antitumor reactivity and reduce alloreactivity for allogeneic cell-based cancer therapy. Front. Med. 10:1135468. doi: 10.3389/fmed.2023.1135468
Edited by:
Kishu Ranjan, Yale University, United StatesReviewed by:
Cristina Maccalli, Sidra Medicine, QatarCopyright © 2023 Chen, Zhu, Kramer, Fang, Wilson, Li and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan-Ruide Li, Y2hhcmxpZS5saUB1Y2xhLmVkdQ==; Lili Yang, bGlsaXlhbmdAdWNsYS5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.