 Roberto Maglie
Roberto Maglie Farzan Solimani
Farzan Solimani Dario Didona4
Dario Didona4 Emiliano Antiga
Emiliano Antiga Giovanni Di Zenzo
Giovanni Di Zenzo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med. , 06 February 2023
Sec. Dermatology
Volume 10 - 2023 | https://doi.org/10.3389/fmed.2023.1128154
This article is part of the Research Topic Community Series - Cytokines and their Signaling in Chronic Inflammatory Diseases and Beyond: Volume II View all 8 articles
Bullous pemphigoid (BP) is the most common autoimmune bullous disease, characterized by severe pruritus and skin blistering. The loss of tolerance against Collagen XVII, also referred to as BP180, is the main pathogenic event of BP, leading to production of IgG autoantibodies which mainly target the juxtamembranous extracellular non-collagenous 16th A (NC16A) domain of BP180. A complex inflammatory network is activated upon autoantibody binding to the basement membrane zone; this inflammatory loop involves the complement cascade and the release of several inflammatory cytokines, chemokines and proteases from keratinocytes, lymphocytes, mast cells and granulocytes. Collectively, these events disrupt the integrity of the dermal-epidermal junction, leading to subepidermal blistering. Recent advances have led to identify novel therapeutic targets for BP, whose management is mainly based on the long-term use of topical and systemic corticosteroids. As an example, targeting type-2 T-helper cell-associated cytokines, such as Interleukin-4 and interleukin-13 has shown meaningful clinical efficacy in case series and studies; targeting IL-17 and IL-23 has also been tried, owing to an important role of these cytokines in the chronic maintenance phase of BP. In this review article, we discuss the complex cytokine milieu that characterized BP inflammation, highlighting molecules, which are currently investigated as present and future therapeutic targets for this life-threatening disease.
Bullous Pemphigoid (BP) represents the most common autoimmune bullous disorder and prevalently occurs in the elderly (1). The disease is characterized by circulating IgG autoantibodies which mainly target the non-collagenous (NC)16A domain of Collagen XVII, also referred to as BP180, a main component of the hemidesmosomes, which maintains the integrity of the dermal-epidermal junction (DEJ) (2). Patients with BP also develop antibodies against BP230, a cytoplasmic protein of the hemidesmosomal plaque that cross-link BP180 to keratin (K) 5 and K14 (3, 4).
Bullous Pemphigoid encompasses a heterogeneous spectrum of manifestations. The classic type is characterized by diffuse tense blisters arising on a background of erythematous-edematous skin (5). Pruritus is always present and, in some patients, may precede for years the appearance of manifest lesions (6). Further, several non-bullous forms have been described (7).
Over the recent years, the incidence of BP is raising significantly (2, 8). This phenomenon is partly explained by an increasing aging population in western countries, and easier access to serological diagnostic kits. Epidemiologic studies showed that BP is associated to some neurological disorders (9–11), particularly dementia and Parkinson, drugs, such as dipeptidyl peptidase IV inhibitors (DPP4i) and PD-1 inhibitors (12, 13) and malignancies (14, 15). The pandemic showed that BP might occur after either SARS-CoV-2 infection or related vaccines (16, 17).
Pathogenically, IgG binding to either BP180 or BP230 activates a cascade of inflammatory mediators resulting in the loss of dermal-epidermal adhesion (18). The increasing knowledge of this complex inflammatory cascade is pivotal for developing new therapeutic strategies for the disease, as its therapeutic management is still largely based on long-term immunosuppressive treatments (10). Indeed, the purpose of this review is to provide a concise overview of the cytokine milieu of BP, with a special focus on molecules currently under-investigation as potential therapeutic targets.
BP180-NC16A-reactive CD4+ T cells play a pivotal role in the pathogenesis of BP (19). Although Th1 and Th2 mixed profiles were considered in the past as the main mediators of the immune response in BP (20), BP is currently regarded as a prevalently Th2-cell skewed disease (Figure 1). Pickford et al. demonstrated strong IL-4 and IgE responses in peripheral blood mononuclear cell isolated from BP patients when exposed to NC16A peptides (21). A recent study on Chinese population identified two NC16A peptides that were associated with the induction of a Th2-type immune activation in BP. Specifically, the authors demonstrated that Th2 cell activation in BP occurred in an human leukocyte antigen-DR (HLA-DR) restricted fashion. IL-4 production by activated Th2 cells was associated with B-cell activation and autoantibody production (22). Th2 cytokines IL-4, IL-13, and IL-31, which play also a crucial role in eosinophil chemoattraction, maturation and activity, and induction of pruritus, have been shown to increase both in the peripheral blood and skin lesions of BP patients (23, 24) (Figure 1 and Table 1). Here, chemokines such as eotaxin and MCP-4, whose levels increase in the blister fluid, support the chemotaxis of Th2 cells (25), supporting a positive feedback loop between activated Th2 cells and eosinophils.
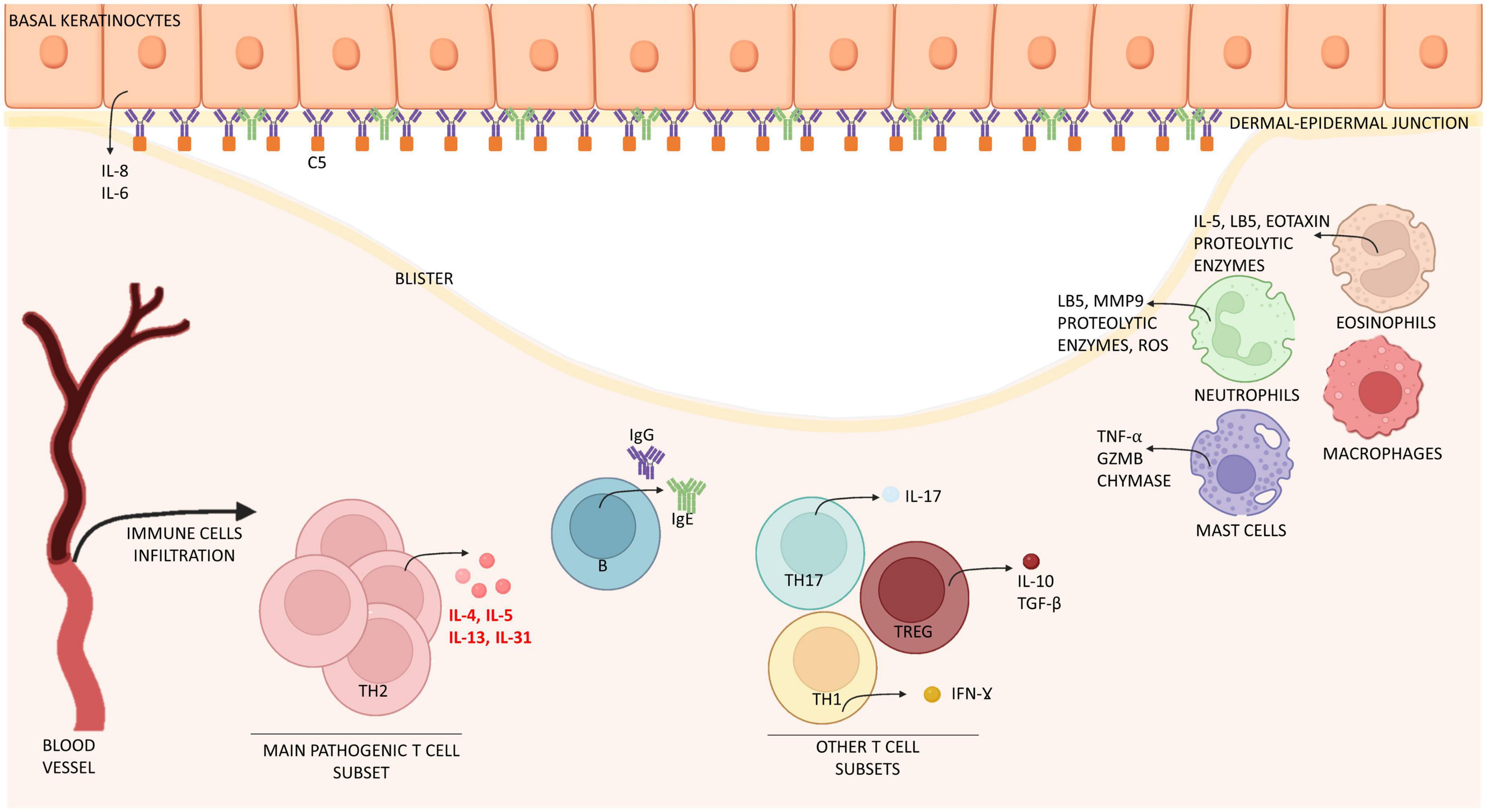
Figure 1. Schematic representation of immune cells and molecules involved in the pathogenesis of bullous pemphigoid. Bullous pemphigoid is determined by IgG, IgE attaching BP180 located in the dermal-epidermal junction. Epidermal cells react by releasing interleukin (IL) 6 and 8. Eventually, this process leads to the recruitment of immune cells (mast cells, macrophages, and eosinophils) which infiltrate the skin and release inflammatory interleukins (IL) and proteolytic enzymes. T cells contribute to this inflammatory process by releasing interleukins at both peripheral (blood) and lesional (skin) level. Especially IL-4, IL-13, IL-31 are crucially involved in B cell proliferation, antibody production and Ig-class switching, itch and eosinophils activation, while IL-17 support neutrophil recruitment. Together, immune cells induce expression of chemokines, thus increasing skin infiltration. The result of this process is the formation of erythematous urticarial plaques and, later, dermal-epidermal splitting causative of blistering. IL, interleukin; Ig, immunoglobulin; Th, T helper; TGF-β, tumor growth factor β; IFN-ɣ, interferon ɣ; GZMB, granzyme B; MMP9, matrix-metallopeptidase 9; LB5, leukotriene B 5; ROS, reactive oxygen species; C5, complement component 5.
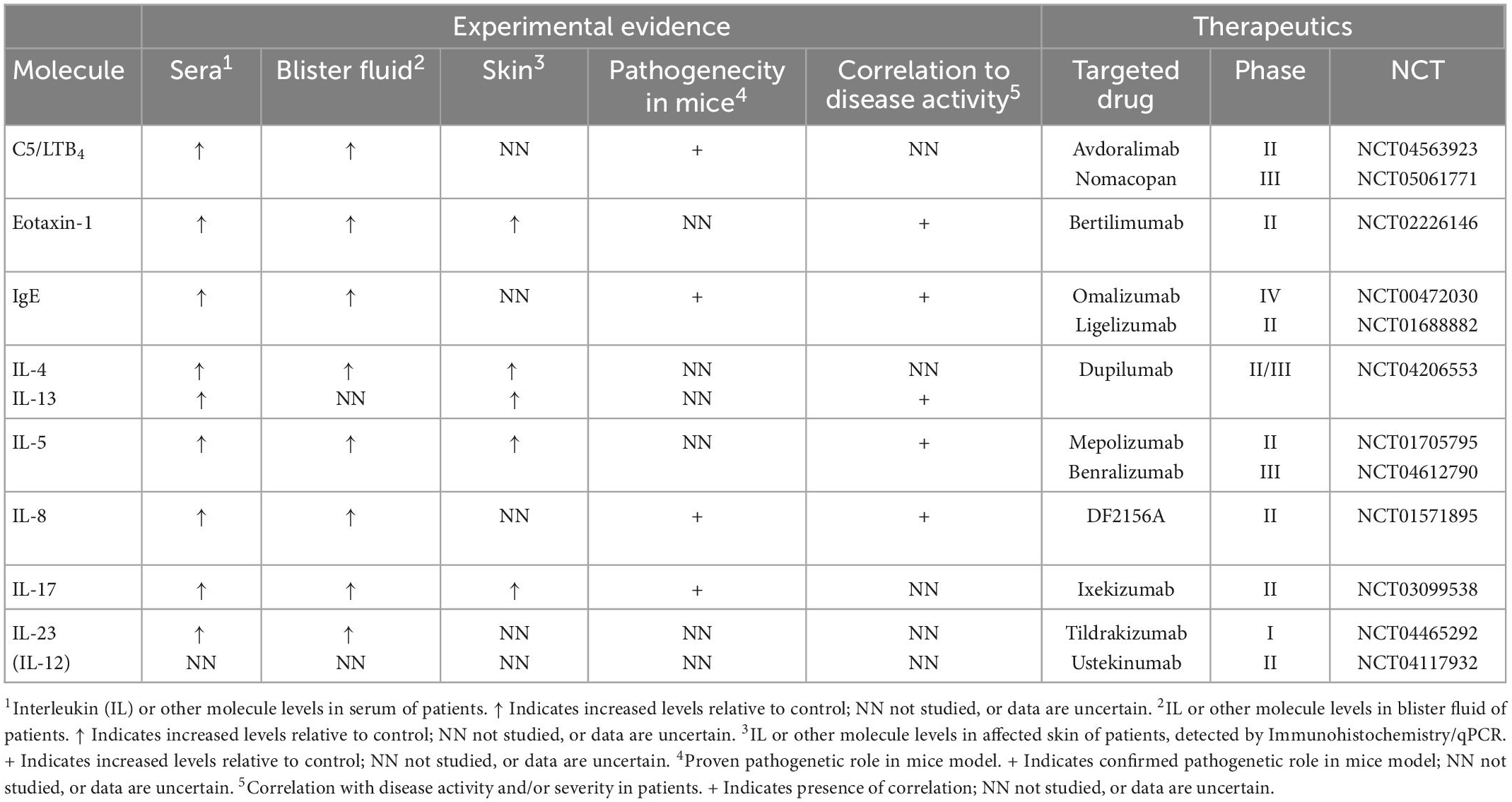
Table 1. Overview of immunological players involved in the pathogenesis of bullous pemphigoid according to findings from serological, blister fluid, skin samples, and mice models analysis as well as their current clinical relevance based on ongoing and terminated clinical trials (published or registered at www.clinicaltrials.gov, accessed on 15 December 2022).
B cell landscape of BP have been only poorly analyzed. Clinical evidence shows that B cells are pathogenically relevant, and their depletion with rituximab (RTX) is a viable therapeutic option, although less effective compared to pemphigus (26–31). This different qualitative effect exerted by RTX could be due to the persistency of IgE autoantibodies, which are still present also after RTX-treatment, and/or persistency of CD20- plasma cells (32). B-cells secreting autoantibodies were found also in BP lesions (33). Their trafficking into the skin seems to depend on the CXCR4/CXCL12 axis rather than cutaneous lymphocyte-associated antigen (CLA) expression, which is poorly expressed by B cells (34). Furthermore, CXCL12 activates C-Myc, which promotes B-cell differentiation into antibody-secreting cells and facilitate autoantibody production, and disruption of this axis results in altered antibody production in vitro (33).
B regulatory cells function in BP appears to be impaired. B regulatory cells are increased in the circulation of BP patients but show an inflammatory, rather than regulatory, phenotype secreting IFN-ɣ, IL-4, and TNF-α instead of IL-10 (35). Different studies reported that serum levels of B-cell activating factor (BAFF), a protein which regulates and stimulates B cell differentiation, is up-regulated in BP (36, 37).
T follicular helper (TFH) cells are a subset of T cells, characterized by the expression of CXCR5 and the capacity to migrate into germinal centers. TFH play a relevant role in autoimmune disorders by stimulating IgG switching and antibody production by activated B-cells (38). Inhibition of TFH cells represents a novel therapeutic approach in autoimmune diseases (39, 40). The signature cytokine of these cells is IL-21, which stimulate both TFH and B cell proliferation (41). Both TFH and IL-21 are increased in BP, positively correlating with anti-BP180 antibody levels; concordantly, treatment response in BP is accompanied by a decrease in TFH/IL-21 levels (42). Finally, absence of TFH cells or inhibition of IL-21 decreases autoantibody production by B-cells in in vitro T/B cell co-culture (42). In this context, also CXCL13, a chemokine which stimulate the migration of TFH cells is expressed in BP patients in both serum and skin (43).
T regulatory (T reg) cells are a crucial T cell subset in the pathogenesis of BP (Figure 1). Scurfy mice and patients affected by immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome have been reported to spontaneously generate antibodies against BP180 and BP230, suggesting that altered Treg function increases the risk of developing BP (44). In BP, Treg cells increase in lesional skin (45–47), while IL-10 serum levels are up-regulated during disease remission (45). These observational data question whether skin infiltrating T regs show undiminished suppression capacity in BP.
IL-17/IL-23 axis has a supportive role in the pathogenesis of BP. In a IL17–/– mice model, passive transfer of anti-BP180 antibodies led to reduced skin inflammation, whereas in wild-type mice levels of IL-17 production following anti-BP180 IgG passive transfer correlated to disease severity (48). Collectively, this findings suggest that IL-17 is pathogenically relevant in murine BP. In human BP IL-17 levels increase in lesional skin and blister fluid (49) and its production is mainly sustained by neutrophils (Figure 1). In the peripheral blood of BP patients, CD3+ lymphocytes appear to be the main source of IL-17 (24). Interestingly, longitudinal measurement of IL-17 and IL-23 serum concentration was found to predict relapse in BP patients, as relapsing patients were shown to have persisting increased levels of serum IL-17 as well as increasing serum concentration of IL-23 during the first month of treatment (50). The IL-17/IL23 axis promotes various pathological processes, including DNA extracellular trap formation (51), stimulation of IL-1β production in macrophages and production of matrix-metalloprotease (MMP)-9 and neutrophil elastase, enzymes involved in blister formation (52, 53).
Interestingly, only a subset of BP patients shows up-regulation of IL-17 at the baseline, without correlation with disease severity, suggesting that not all the patients could effectively benefit from therapeutic targeting of the IL-17/IL23 axis (30–32). Interestingly, targeting IL17/IL23 demonstrated efficacy in patients with coexisting BP and psoriasis, a prototype of IL17/IL23 driven disease (33, 54–57). Results from ongoing clinical trials are thereby necessary to understand the impact of this treatment approach also in BP patients without coexisting psoriasis (58). Inhibition of janus kinase may worth being investigated in BP, since this could allow simultaneous targeting of both IL-4 and IL-23 (59–62).
Complement activation plays a pivotal role in the pathogenesis of BP (63, 64). This assumption is supported by several evidences: (i) the complement component C3 is disposed in a linear fashion along the DEJ in perilesional BP skin, and is even stronger than IgG, or sometimes found in the absence of IgG, in direct immunofluorescence (65–68); (ii) the capacity of autoantibodies to activate complement ex vivo correlates with disease activity and levels of autoantibodies in BP patients (69); (iii) milder clinical phenotype of BP230-type seems to correlate with weaker complement deposition at DEJ (70) (iv) genetic deficiency and/or pharmacological depletion of various complement components reduce pathogenicity of anti-BP180 IgG and dampen skin inflammation in experimental mouse models of BP (71, 72).
In BP patients, the classical pathway activation, which occurs following antibody/antigen binding, is the major pathway (73). In vitro and in vivo evidence suggests that blockage of C1q prevents both complement activation and skin blistering. Likewise, genetic absence of C4 in experimental BP mouse models is enough to abolish mast cell (MC) degranulation and attraction of neutrophils (71) (Figure 1). The alternative pathway of complement activation plays a supportive role in the pathogenesis of BP (71). Accordingly, passive transfer of anti-BP180 antibodies in mice with genetic deficiency of alternative pathway component FB developed a delayed and mild disease phenotype (71).
The activation of complement generates the attachment of component C3 and release of anaphylatoxins C3a and C5a, of which C5a can mediate polymorphonuclear leukocytes chemotaxis and both C3a and C5a can mediate MC degranulation (74). Of relevance in the generation of experimental BP in mice is the interaction between C5a and C5aR1 (75). C5aR2 conversely plays an anti-inflammatory and protective role in BP (76, 77). C5a receptor 1 (C5aR1) on MC was shown to be critical for the formation of skin lesions (74). Further, C5a/C5aR1 interaction on the surface of neutrophils activates the release of LTB4 via 5-lipoxygenase (78). Collectively, these molecules are indispensable to an efficient recruitment of neutrophils into the interstitial skin tissue (79, 80) (Figure 1 and Table 1).
A last consequence of complement activation in BP is the formation of the terminal membrane attack complex, which exerts direct cytotoxic effects in the epidermal basal cells.
It is worth noting that loss of dermal-epidermal adhesion in BP may also occur via complement-independent mechanisms (76, 81, 82), which are thought to be preponderant during early and non-blistering phases of BP, where non-fixing complement IgG4 subclasses are predominant (83–85). These mechanisms include (i) internalization of BP180 from the surface of keratinocytes after IgG binding to BP180 (86); (ii) direct release of cytokines, e.g., IL-6 and IL-8, from keratinocytes (87); (iii) induction of MC degranulation and eosinophil activation by IgE autoantibodies (88).
It is thus possible that both complement-dependent and complement-independent mechanisms work together in inducing and perpetuating BP inflammation and blistering (85).
Loss of dermal-epidermal adhesion in BP is critically associated with skin infiltration of neutrophil and eosinophil granulocytes (89–91). In passive transfer models, pathogenicity of antibodies is significantly reduced in the absence of myeloid granulocytes (89, 92, 93). Moreover, BP models induced by genetic deletion of BP180 pathogenic domains are characterized by spontaneous infiltration of granulocytes (94). Granulocytes induce blistering by different mechanisms such as the release of proteases, e.g., MMP-9, reactive oxygen species (ROS) and either neutrophil or eosinophil extracellular traps (51, 92, 95–97) (Figure 1). In humans, neutrophils and eosinophils localize differently in BP skin, with the first predominating in the blister fluids and the second in the dermal skin (98). The blister fluid, as well as sera, of BP patients over-express several chemoattractant and pro-inflammatory molecules, including IL-1, IL-8, IL-10, IL-5, Tumor necrosis factor-alpha, IL-6, CCL17, CCL-1, galectin-9, periostin, CCL11, CCL26, thymic stromal lymphopoietin and eotaxins (99–107) (Table 1). Dermal-epidermal separation induced by eosinophils is mainly orchestrated by IL-5 and depends on adhesion and Fcγ receptor activation (108). Neutrophils and eosinophils also regulate different aspects of BP inflammation. As an example, NET release from neutrophils acts systemically by inducing B-cell differentiation into plasma cells via activation of the MAPK/p38 cascade (109). Eosinophils mediate specific IgE pathogenicity, release cytokines which enhance Th2 cell recruitment and stimulate peripheral nerve terminals, e.g., by releasing IL-31 (88, 110–112).
Cross talk between immune cells is likely to potentiate the effector functions of granulocytes. Accordingly, neutrophils in BP release significantly more ROS and MMP-9 when stimulated with monocyte supernatants in vitro (113). Recently, Granzyme B was shown to critically regulate monocyte-dependent neutrophil recruitment in BP, and its inhibition significantly ameliorated pemphigoid disease induced by immunization with anti-COL7 antibodies in mice (114).
Monocytes and neutrophils are also activated by CXCL10, whose levels are increased in early-relapsing patients and is produced by keratinocytes, fibroblasts, and infiltrating immune cells (115).
Tissue resident macrophages are increased in BP skin and are mainly polarized toward the M2 phenotype, which express CD163 and CD206 (116, 117). M2 macrophages produce large amounts of Th2-type cytokines and stimulate T-cell and eosinophil recruitment by releasing CCL18, CCL22, CCL24, and CCL2 (117, 118).
Furthermore, CD163+ M2 macrophages stimulated by LL37 in vitro produced CXCL10 and CCL20 as well CXCR5+, CXCL13+, which contribute to recruitment of TFH cells (43, 117).
Finally, BP skin is enriched in both basophils and MC. Basophils are implicated in BP-associated itch (119). The role of MCs is still matter of debate. Experimental evidence suggests that MC degranulation in mice with BP occur after different stimuli, including complement fractions and specific IgE antibodies (74). Finally, macrophage-mediated neutrophil infiltration depends on MC activation (120, 121) (Figure 1). However, while studies using KIT-dependent MC knock-out mice demonstrated that MC activation trigger BP (121), Kit-independent MC-deficient mice still develop the disease, without significant changes in immune cells infiltration. Collectively, these findings raise the hypothesis that MC activation could be a bystander effect of BP inflammation (122).
Until now, several targeted therapies for BP have been developed, including (i) cell-depleting therapies; (ii) autoantibody-targeting therapies and (iii) single cytokine/molecule-directed therapies (Table 1).
Rituximab, a B-cell depleting therapy, is still not approved for BP, but often applied off-label to patients who fail conventional therapies. BP patients receiving one or more RTX cycles experience high rate of complete remission (CR) (approximately 75–92% of CR and 40% of CR off therapy), with significant drop of autoantibody titer (27–29, 123). Notably, RTX was shown to halve the 5-year mortality rate of BP in one study (124). Although RTX is mostly applied according to either the lymphoma protocol (375 mg/m2 weekly for 4 weeks) or the rheumatoid arthritis protocol (2 infusions of 1,000 mg 2 weeks apart), increasing number of studies addressed the efficacy of low (2 infusions of 500 mg 2 weeks apart) or ultra low (100 mg weekly for 4 weeks) doses of RTX in BP (125, 126).
Intravenous immunoglobulins showed pleiotropic anti-inflammatory effects (26), including increased autoantibody catabolism, and demonstrated meaningful positive effects in several cases and studies in BP both as monotherapy or combined with RTX (127–130). More recently, efgartigimod, a monoclonal antibody targeting the neonatal Fc receptor and thereby hastening the internalization and degradation of immunoglobulins, has entered clinical trials in BP after promising results in pemphigus and myasthenia gravis patients (131, 132). Several studies reported disease improvement with omalizumab, a monoclonal antibody targeting the IgE-specific Fc epsilon receptor III (133–137) (Table 1). Omalizumab offers a favorable safety profile, making it suitable for patients with contraindication to prolonged corticosteroid/immunosuppressive regimens (138, 139). Intriguingly, it showed efficacy also in patients without detectable serum anti-BP180/BP230 IgE. Responder patients show a decrease in IgE skin deposition, circulating IgG autoantibody levels and circulating basophils, which suggests immune-modulatory effects beyond IgE inhibition (137, 138, 140).
Over the recent years, complement activation has served as an attractive target in BP, owing to the established role in BP pathogenesis demonstrated in animal studies.
In one study blockage of C1s by the specific inhibitor, TNT003, successfully blocked the complement activating capacity of BP sera. Likewise, Gutjahr et al. (141) found that tinzaparin sodium inhibited autoantibody-induced complement activation in BP sera.
More recently, Sadik et al. (142) reported the results of a phase IIa non-randomized clinical trial of BP patients treated with nomacopan (NCT05061771), an inhibitor of leukotriene B4 and complement C5. Seven of the nine patients recruited demonstrated remarkable reduction of Bullous Pemphigoid Disease Area Index (BPDAI) and pruritus after approximately 1.5 months. No serious adverse events were reported (Table 1).
Since a first report in 2018 (143), dupilumab, an IL-4 alpha subunit receptor inhibitor has been used increasingly in BP. Dupilumab mainly acts by suppressing IL-4- and IL-13-producing CD4+ T cells (144). In a case series of 13 patients published in 2020, dupilumab demonstrated a satisfactory response in 92.3% (145). BP patients receiving dupilumab in combination with conventional therapies achieve faster clinical response with a reduction of the cumulative steroid dose compared to those treated with conventional therapies (146, 147). Interestingly, combination of omalizumab and dupilumab demonstrated efficacy in one case of recalcitrant BP (148). Finally, the drug holds promise as a treatment for special settings of patients, including highly recalcitrant, rituximab-resistant, or immune checkpoint inhibitor-induced BP (149–151). So far, attempts to block IL-5 (mepolizumab) failed to show an impact on BP activity (NCT01705795), while a study with benralizumab, a monoclonal antibody targeting the IL-5 receptor is currently ongoing (NCT04612790) (Table 1).
Finally, it will be intriguing to evaluate the efficacy of nemolizumab, a monoclonal antibody targeting IL-31, on disease activity and pruritus of BP.
An intriguing aspect of the pathogenesis of BP is that antibody/antigen binding activates different pathways, which seem to act in parallel rather than as a single cascade. Hence, combining different target therapies will represent a feasible way to reduce the cumulative exposure of patients to systemic steroids. In a merely speculative manner, combination of rituximab and dupilumab might effectively target the T-B-cell cross-talk involved in the loss of tolerance against BP autoantigens; while, combination of anti-complement drugs and either neutrophil-or eosinophil-targeting therapies might be best suited to impair the effector phase of BP inflammation and pruritus. Indeed, with the number of available therapeutic options rapidly increasing, clinicians should focus on identifying comorbidities, clinical variables (e.g., bullous vs. non-bullous phenotypes and pruritus intensity), laboratory [e.g., neutrophil-rich vs. eosinophil-rich infiltrates at histopathology, or the intensity of complement deposition at direct immunofluorescence (DIF)] and serological findings (e.g., titer of IgG and IgE antibodies against BP180/BP230) or molecular factors (e.g., cytokine concentration) which may influence therapy-response and decision-making.
RM contributed to the design, writing, and speculation. FS, DD, and CP contributed to the writing and speculation. EA contributed to the design and writing. GD contributed to the design, writing, and supervision. All authors contributed to the article and approved the submitted version.
This study was supported by the “Progetto Ricerca Corrente” and “Ricerca Finalizzata” (N.12367807) of the Italian Ministry of Health, Rome, Italy.
IDI-IRCCS is healthcare provider of the European Reference Network for Rare and Undiagnosed Skin Diseases (ERN-Skin). FS is participant in the BIH Charité Clinician Scientist Program funded by the Charité – Universitätsmedizin Berlin, and the Berlin Institute of Health at Charité (BIH). The publication was made by a researcher (RM) with a research contract co-funded by the European Union–PON Research and Innovation 2014–2020 in accordance with Article 24, paragraph 3a, of Law No. 240 of December 30, 2010, as amended and Ministerial Decree No. 1062 of August 10, 2021. The Figure was created with the help of Biorender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ellebrecht C, Maseda D, Payne A. Pemphigus and pemphigoid: from disease mechanisms to druggable pathways. J Invest Dermatol. (2022) 142(Pt B. 3):907–14. doi: 10.1016/j.jid.2021.04.040
2. Opelka B, Schmidt E, Goletz S. Type XVII collagen: relevance of distinct epitopes, complement-independent effects, and association with neurological disorders in pemphigoid disorders. Front Immunol. (2022) 13:948108. doi: 10.3389/fimmu.2022.948108
3. Makita E, Matsuzaki Y, Fukui T, Matsui A, Minakawa S, Nakano H, et al. Autoantibodies to BPAG1e trigger experimental bullous pemphigoid in mice. J Invest Dermatol. (2021) 141:1167–76.e3. doi: 10.1016/j.jid.2020.08.031
4. Fontao L, Favre B, Riou S, Geerts D, Jaunin F, Saurat J, et al. Interaction of the bullous pemphigoid antigen 1 (BP230) and desmoplakin with intermediate filaments is mediated by distinct sequences within their COOH terminus. Mol Biol Cell. (2003) 14:1978–92. doi: 10.1091/mbc.e02-08-0548
5. Maglie R, Hertl M. Pharmacological advances in pemphigoid. Curr Opin Pharmacol. (2019) 46:34–43. doi: 10.1016/j.coph.2018.12.007
6. Didona D, Scarsella L, Fehresti M, Solimani F, Juratli H, Gobel M, et al. Autoreactive peripheral blood T helper cell responses in bullous pemphigoid and elderly patients with pruritic disorders. Front Immunol. (2021) 12:569287. doi: 10.3389/fimmu.2021.569287
7. Lamberts A, Meijer J, Pas H, Diercks G, Horvath B, Jonkman M. Nonbullous pemphigoid: insights in clinical and diagnostic findings, treatment responses, and prognosis. J Am Acad Dermatol. (2019) 81:355–63. doi: 10.1016/j.jaad.2019.04.029
8. Ahmed A, Anwar S, Reche P. Molecular basis for global incidence of pemphigoid diseases and differences in phenotypes. Front Immunol. (2022) 13:807173. doi: 10.3389/fimmu.2022.807173
9. Huang I, Wu P, Liu C, Huang Y. Association between bullous pemphigoid and psychiatric disorders: a systematic review and meta-analysis. J Dtsch Dermatol Ges. (2022) 20:1305–12. doi: 10.1111/ddg.14852
10. Borradori L, Van Beek N, Feliciani C, Tedbirt B, Antiga E, Bergman R, et al. Updated S2 K guidelines for the management of bullous pemphigoid initiated by the European academy of dermatology and venereology (EADV). J Eur Acad Dermatol Venereol. (2022) 36:1689–704. doi: 10.1111/jdv.18220
11. Moro F, Fania L, Sinagra J, Salemme A, Di Zenzo G. Bullous pemphigoid: trigger and predisposing factors. Biomolecules. (2020) 10:1432. doi: 10.3390/biom10101432
12. Tasanen K, Varpuluoma O, Nishie W. Dipeptidyl Peptidase-4 inhibitor-associated bullous pemphigoid. Front Immunol. (2019) 10:1238. doi: 10.3389/fimmu.2019.01238
13. Salemme A, Fania L, Scarabello A, Caproni M, Marzano A, Cozzani E, et al. Gliptin-associated bullous pemphigoid shows peculiar features of anti-BP180 and -BP230 humoral response: results of a multicenter study. J Am Acad Dermatol. (2022) 87:56–63. doi: 10.1016/j.jaad.2022.02.036
14. Maglie R, Antiga E, Vannucchi M, Del Bianco E, Bianchi B, Massi D, et al. Bullous eruption in a patient with B-cell chronic lymphocytic leukemia: a diagnostic challenge. Int J Dermatol. (2017) 56:1445–7. doi: 10.1111/ijd.13807
15. Atzmony L, Mimouni I, Reiter O, Leshem Y, Taha O, Gdalevich M, et al. Association of bullous pemphigoid with malignancy: a systematic review and meta-analysis. J Am Acad Dermatol. (2017) 77:691–9. doi: 10.1016/j.jaad.2017.05.006
16. Maronese C, Caproni M, Moltrasio C, Genovese G, Vezzoli P, Sena P, et al. Bullous pemphigoid associated with COVID-19 vaccines: an italian multicentre study. Front Med (Lausanne). (2022) 9:841506. doi: 10.3389/fmed.2022.841506
17. Kasperkiewicz M, Yale M, Strong R, Zillikens D, Woodley D, Recke A. COVID-19 pandemic and autoimmune bullous diseases: a cross-sectional study of the international pemphigus and pemphigoid foundation. J Eur Acad Dermatol Venereol. (2021) 35:e418–21. doi: 10.1111/jdv.17228
18. Ujiie H. What’s new in the pathogeneses and triggering factors of bullous pemphigoid. J Dermatol. (2022). [Epub ahead of print]. doi: 10.1111/1346-8138.16654
19. Ujiie H, Shibaki A, Nishie W, Shinkuma S, Moriuchi R, Qiao H, et al. Noncollagenous 16A domain of type XVII collagen-reactive CD4+ T cells play a pivotal role in the development of active disease in experimental bullous pemphigoid model. Clin Immunol. (2012) 142:167–75. doi: 10.1016/j.clim.2011.10.002
20. Hertl M, Eming R, Veldman CT. cell control in autoimmune bullous skin disorders. J Clin Invest. (2006) 116:1159–66. doi: 10.1172/JCI28547
21. Pickford W, Gudi V, Haggart A, Lewis B, Herriot R, Barker R, et al. T cell participation in autoreactivity to NC16a epitopes in bullous pemphigoid. Clin Exp Immunol. (2015) 180:189–200. doi: 10.1111/cei.12566
22. Zhang J, Fang H, Shen S, Dang E, Li Q, Qiao P, et al. Identification of Immunodominant Th2-Cell epitopes in chinese patients with bullous pemphigoid. J Invest Dermatol. (2018) 138:1917–24. doi: 10.1016/j.jid.2018.03.1515
23. Giomi B, Caproni M, Calzolari A, Bianchi B, Fabbri P. Th1, Th2 and Th3 cytokines in the pathogenesis of bullous pemphigoid. J Dermatol Sci. (2002) 30:116–28. doi: 10.1016/s0923-1811(02)00067-1
24. Feliciani C, Toto P, Mohammad Pour S, Coscione G, Amerio P, Amerio P. A Th2-like cytokine response is involved in bullous pemphigoid. the role of IL-4 and IL-5 in the pathogenesis of the disease. Int J Immunopathol Pharmacol. (1999) 12:55–61. doi: 10.1177/205873929901200202
25. Gounni Abdelilah S, Wellemans V, Agouli M, Guenounou M, Hamid Q, Beck L, et al. Increased expression of Th2-associated chemokines in bullous pemphigoid disease. role of eosinophils in the production and release of these chemokines. Clin Immunol. (2006) 120:220–31. doi: 10.1016/j.clim.2006.03.014
26. Amber K, Maglie R, Solimani F, Eming R, Hertl M. Targeted therapies for autoimmune bullous diseases: current status. Drugs. (2018) 78:1527–48. doi: 10.1007/s40265-018-0976-5
27. Tovanabutra N, Payne A. Clinical outcome and safety of rituximab therapy for pemphigoid diseases. J Am Acad Dermatol. (2020) 82:1237–9.
28. Polansky M, Eisenstadt R, DeGrazia T, Zhao X, Liu Y, Feldman R. Rituximab therapy in patients with bullous pemphigoid: a retrospective study of 20 patients. J Am Acad Dermatol. (2019) 81:179–86.
29. Cho Y, Chu C, Wang L. First-line combination therapy with rituximab and corticosteroids provides a high complete remission rate in moderate-to-severe bullous pemphigoid. Br J Dermatol. (2015) 173:302–4. doi: 10.1111/bjd.13633
30. Berkani N, Joly P, Golinski M, Colliou N, Lim A, Larbi A, et al. B-cell depletion induces a shift in self antigen specific B-cell repertoire and cytokine pattern in patients with bullous pemphigoid. Sci Rep. (2019) 9:3525.
31. Chee R, Nagendran V, Bansal A, Casie Chetty S, Harland C. B-cell targeted therapy alone may not be effective in bullous pemphigoid. Clin Exp Dermatol. (2007) 32:111–2.
32. Hall R III, Streilein R, Hannah D, McNair P, Fairley J, Ronaghy A, et al. Association of serum B-cell activating factor level and proportion of memory and transitional B cells with clinical response after rituximab treatment of bullous pemphigoid patients. J Invest Dermatol. (2013) 133:2786–8. doi: 10.1038/jid.2013.236
33. Fang, H, Xue K, Cao T, Li Q, Dang E, Liu Y, et al. CXCL12/CXCR4 axis drives the chemotaxis and differentiation of B cells in bullous pemphigoid. J Invest Dermatol. (2023) 143:197–208. doi: 10.1016/j.jid.2022.08.041
34. Leyendeckers H, Tasanen K, Bruckner-Tuderman L, Zillikens D, Sitaru C, Schmitz J, et al. Memory B cells specific for the NC16A domain of the 180 kDa bullous pemphigoid autoantigen can be detected in peripheral blood of bullous pemphigoid patients and induced in vitro to synthesize autoantibodies. J Invest Dermatol. (2003) 120:372–8. doi: 10.1046/j.1523-1747.2003.12071.x
35. Liu Z, Dang E, Li B, Qiao H, Jin L, Zhang J, et al. Dysfunction of CD19(+)CD24(hi)CD27(+) B regulatory cells in patients with bullous pemphigoid. Sci Rep. (2018) 8:703.
36. Asashima N, Fujimoto M, Watanabe R, Nakashima H, Yazawa N, Okochi H, et al. Serum levels of BAFF are increased in bullous pemphigoid but not in pemphigus vulgaris. Br J Dermatol. (2006) 155:330–6.
37. Qian H, Kusuhara M, Li X, Tsuruta D, Tsuchisaka A, Ishii N, et al. B-cell activating factor detected on both naive and memory B cells in bullous pemphigoid. Exp Dermatol. (2014) 23:596–605. doi: 10.1111/exd.12421
38. Craft J. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol. (2012) 8:337–47.
39. Kim A, Han D, Choi J, Seok J, Kim S, Seo S, et al. Targeting inducible costimulator expressed on CXCR5(+)PD-1(+) T(H) cells suppresses the progression of pemphigus vulgaris. J Allergy Clin Immunol. (2020) 146:1070–9.e8. doi: 10.1016/j.jaci.2020.03.036
40. Reighard S, Cranert S, Rangel K, Ali A, Gyurova I, de la Cruz-Lynch AT, et al. Therapeutic targeting of follicular T cells with chimeric antigen receptor-expressing natural killer cells. Cell Rep Med. (2020) 1:100003.
41. Vogelzang A, McGuire H, Yu D, Sprent J, Mackay C, King CA. fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity. (2008) 29:127–37.
42. Li Q, Liu Z, Dang E, Jin L, He Z, Yang L, et al. Follicular helper T Cells (Tfh) and IL-21 involvement in the pathogenesis of bullous pemphigoid. PLoS One. (2013) 8:e68145. doi: 10.1371/journal.pone.0068145
43. Ohuchi K, Fujimura T, Lyu C, Amagai R, Muto Y, Aiba S. Possible roles of CXCL13/CXCR5 axis in the development of bullous pemphigoid. J Dermatol. (2021) 48:353–9. doi: 10.1111/1346-8138.15713
44. Muramatsu K, Ujiie H, Kobayashi I, Nishie W, Izumi K, Ito T, et al. Regulatory T-cell dysfunction induces autoantibodies to bullous pemphigoid antigens in mice and human subjects. J Allergy Clin Immunol. (2018) 142:1818–30.e6. doi: 10.1016/j.jaci.2018.03.014
45. Antiga E, Quaglino P, Volpi W, Pierini I, Del Bianco E, Bianchi B, et al. Regulatory T cells in skin lesions and blood of patients with bullous pemphigoid. J Eur Acad Dermatol Venereol. (2014) 28:222–30.
46. Muramatsu K, Zheng M, Yoshimoto N, Ito T, Ujiie I, Iwata H, et al. Regulatory T cell subsets in bullous pemphigoid and dipeptidyl peptidase-4 inhibitor-associated bullous pemphigoid. J Dermatol Sci. (2020) 100:23–30. doi: 10.1016/j.jdermsci.2020.08.004
47. Gambichler T, Tsitlakidon A, Skrygan M, Hoxtermann S, Susok L, Hessam ST. regulatory cells and other lymphocyte subsets in patients with bullous pemphigoid. Clin Exp Dermatol. (2017) 42:632–7.
48. Chakievska L, Holtsche M, Kunstner A, Goletz S, Petersen B, Thaci D, et al. IL-17A is functionally relevant and a potential therapeutic target in bullous pemphigoid. J Autoimmun. (2019) 96:104–12. doi: 10.1016/j.jaut.2018.09.003
49. Arakawa M, Dainichi T, Ishii N, Hamada T, Karashima T, Nakama T, et al. Lesional Th17 cells and regulatory T cells in bullous pemphigoid. Exp Dermatol. (2011) 20:1022–4.
50. Plee J, Le Jan S, Giustiniani J, Barbe C, Joly P, Bedane C, et al. Integrating longitudinal serum IL-17 and IL-23 follow-up, along with autoantibodies variation, contributes to predict bullous pemphigoid outcome. Sci Rep. (2015) 5:18001. doi: 10.1038/srep18001
51. Giusti D, Bini E, Terryn C, Didier K, Le Jan S, Gatouillat G, et al. NET formation in bullous pemphigoid patients with relapse is modulated by IL-17 and IL-23 Interplay. Front Immunol. (2019) 10:701. doi: 10.3389/fimmu.2019.00701
52. Le Jan S, Plee J, Vallerand D, Dupont A, Delanez E, Durlach A, et al. Innate immune cell-produced IL-17 sustains inflammation in bullous pemphigoid. J Invest Dermatol. (2014) 134:2908–17. doi: 10.1038/jid.2014.263
53. Le Jan S, Muller C, Plee J, Durlach A, Bernard P, Antonicelli F. IL-23/IL-17 Axis Activates IL-1beta-Associated inflammasome in macrophages and generates an auto-inflammatory response in a subgroup of patients with bullous pemphigoid. Front Immunol. (2019) 10:1972. doi: 10.3389/fimmu.2019.01972
54. Holtsche M, Hammers C, Chakievska L, Ludwig R, Thaci D, Zillikens D, et al. Adjuvant treatment with secukinumab induced long term remission in a patient with severe bullous pemphigoid. J Dtsch Dermatol Ges. (2020) 18:1478–80. doi: 10.1111/ddg.14291
55. Yun J, Scardamaglia L, Tan C, McCormack C. Successful secukinumab treatment of active bullous pemphigoid and chronic severe psoriasis: a case report. Australas J Dermatol. (2022) 63:e155–8. doi: 10.1111/ajd.13803
56. Lu L, Yu Y, Zhang J, Fan X, Qi Y, Lin B. Incidental amelioration of bullous pemphigoid during ixekizumab treatment for psoriasis. J Dermatol. (2022) 49:e13–5. doi: 10.1111/1346-8138.16189
57. Xiao Y, Gu Y, Xia D, Zhou X, Li W. Ixekizumab successfully treated refractory psoriasis concurrent bullous pemphigoid. J Dermatol. (2022). [Epub ahead of print]. doi: 10.1111/1346-8138.16559
58. Hsieh C, Tsai T. Management of coexisting bullous pemphigoid and psoriasis: a review. Am J Clin Dermatol. (2022) 23:869–79. doi: 10.1007/s40257-022-00719-7
59. Solimani F, Meier K, Ghoreschi K. Emerging topical and systemic JAK inhibitors in dermatology. Front Immunol. (2019) 10:2847. doi: 10.3389/fimmu.2019.02847
60. Solimani F, Hilke F, Ghoreschi K. [Pharmacology of Janus kinase inhibitors]. Hautarzt. (2019) 70:934–41.
61. Ociepa K, Danilewicz M, Wagrowska-Danilewicz M, Peterson-Jeckowska R, Wojcicka-Rubin A, Lewkowicz N, et al. Expression of the selected proteins of JAK/STAT signaling pathway in diseases with oral mucosa involvement. Int J Mol Sci. (2022) 24:323. doi: 10.3390/ijms24010323
62. Xiao Y, Xiang H, Li W. Concurrent bullous pemphigoid and plaque psoriasis successfully treated with Janus kinase inhibitor Baricitinib. Dermatol Ther. (2022) 35:e15754. doi: 10.1111/dth.15754
63. Kushner C, Payne A. Increasing the complement of therapeutic options in bullous pemphigoid. J Invest Dermatol. (2018) 138:246–8.
64. Iwata H, Kitajima Y. Bullous pemphigoid: role of complement and mechanisms for blister formation within the lamina lucida. Exp Dermatol. (2013) 22:381–5. doi: 10.1111/exd.12146
65. Romeijn T, Jonkman M, Knoppers C, Pas H, Diercks G. Complement in bullous pemphigoid: results from a large observational study. Br J Dermatol. (2017) 176:517–9. doi: 10.1111/bjd.14822
66. Jordon R, Sams W Jr., Beutner E. Complement immunofluorescent staining in bullous pemphigoid. J Lab Clin Med. (1969) 74:548–56.
67. Black M, Bhogal B, Willsteed E. Immunopathological techniques in the diagnosis of bullous disorders. Acta Derm Venereol Suppl (Stockh). (1989) 151:96–105; discussion 106–10.
68. Hashimoto T, Kanazawa N, Inoue N. Anticomplement therapy in bullous pemphigoid. Br J Dermatol. (2019) 181:448–9. doi: 10.1111/bjd.18229
69. Chiorean R, Baican A, Mustafa M, Lischka A, Leucuta D, Feldrihan V, et al. Complement-Activating capacity of autoantibodies correlates with disease activity in bullous pemphigoid patients. Front Immunol. (2018) 9:2687. doi: 10.3389/fimmu.2018.02687
70. Zheng M, Ujiie H, Iwata H, Muramatsu K, Yoshimoto N, Ito T, et al. Characteristics of IgG subclasses and complement deposition in BP230-type bullous pemphigoid. J Eur Acad Dermatol Venereol. (2019) 33:595–600. doi: 10.1111/jdv.15325
71. Nelson K, Zhao M, Schroeder P, Li N, Wetsel R, Diaz L, et al. Role of different pathways of the complement cascade in experimental bullous pemphigoid. J Clin Invest. (2006) 116:2892–900. doi: 10.1172/JCI17891
72. Liu Z, Giudice G, Swartz S, Fairley J, Till G, Troy J, et al. The role of complement in experimental bullous pemphigoid. J Clin Invest. (1995) 95:1539–44. doi: 10.1172/JCI117826
73. Freire P, Munoz C, Derhaschnig U, Schoergenhofer C, Firbas C, Parry G, et al. Specific inhibition of the classical complement pathway prevents C3 deposition along the dermal-epidermal junction in bullous pemphigoid. J Invest Dermatol. (2019) 139:2417–24.e2. doi: 10.1016/j.jid.2019.04.025
74. Heimbach L, Li Z, Berkowitz P, Zhao M, Li N, Rubenstein D, et al. The C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J Biol Chem. (2011) 286:15003–9. doi: 10.1074/jbc.M111.221036
75. Mihai S, Hirose M, Wang Y, Thurman J, Holers V, Morgan B, et al. Specific inhibition of complement activation significantly ameliorates autoimmune blistering disease in mice. Front Immunol. (2018) 9:535. doi: 10.3389/fimmu.2018.00535
76. Karsten C, Beckmann T, Holtsche M, Tillmann J, Tofern S, Schulze F, et al. Tissue destruction in bullous pemphigoid can be complement independent and may be mitigated by C5aR2. Front Immunol. (2018) 9:488. doi: 10.3389/fimmu.2018.00488
77. Emtenani S, Holtsche M, Stahlkopf R, Seiler D, Burn T, Liu H, et al. Differential expression of C5aR1 and C5aR2 in innate and adaptive immune cells located in early skin lesions of bullous pemphigoid patients. Front Immunol. (2022) 13:942493. doi: 10.3389/fimmu.2022.942493
78. Sezin T, Krajewski M, Wutkowski A, Mousavi S, Chakievska L, Bieber K, et al. The Leukotriene B(4) and its receptor BLT1 act as critical drivers of neutrophil recruitment in murine bullous pemphigoid-like epidermolysis bullosa acquisita. J Invest Dermatol. (2017) 137:1104–13. doi: 10.1016/j.jid.2016.12.021
79. Sezin T, Murthy S, Attah C, Seutter M, Holtsche M, Hammers C, et al. Dual inhibition of complement factor 5 and leukotriene B4 synergistically suppresses murine pemphigoid disease. JCI Insight. (2019) 4:e128239. doi: 10.1172/jci.insight.128239
80. Sadik C, Miyabe Y, Sezin T, Luster A. The critical role of C5a as an initiator of neutrophil-mediated autoimmune inflammation of the joint and skin. Semin Immunol. (2018) 37:21–9. doi: 10.1016/j.smim.2018.03.002
81. Ujiie H, Sasaoka T, Izumi K, Nishie W, Shinkuma S, Natsuga K, et al. Bullous pemphigoid autoantibodies directly induce blister formation without complement activation. J Immunol. (2014) 193:4415–28.
82. Natsuga K, Nishie W, Shinkuma S, Ujiie H, Nishimura M, Sawamura D, et al. Antibodies to pathogenic epitopes on type XVII collagen cause skin fragility in a complement-dependent and -independent manner. J Immunol. (2012) 188:5792–9. doi: 10.4049/jimmunol.1003402
83. Lamb P, Patton T, Deng J. The predominance of IgG4 in prodromal bullous pemphigoid. Int J Dermatol. (2008) 47:150–3. doi: 10.1111/j.1365-4632.2008.03361.x
84. Zuo Y, Evangelista F, Culton D, Guilabert A, Lin L, Li N, et al. IgG4 autoantibodies are inhibitory in the autoimmune disease bullous pemphigoid. J Autoimmun. (2016) 73:111–9.
85. Papara C, Karsten C, Ujiie H, Schmidt E, Schmidt-Jimenez L, Baican A, et al. The relevance of complement in pemphigoid diseases: a critical appraisal. Front Immunol. (2022) 13:973702. doi: 10.3389/fimmu.2022.973702
86. Iwata H, Kamio N, Aoyama Y, Yamamoto Y, Hirako Y, Owaribe K, et al. IgG from patients with bullous pemphigoid depletes cultured keratinocytes of the 180-kDa bullous pemphigoid antigen (type XVII collagen) and weakens cell attachment. J Invest Dermatol. (2009) 129:919–26. doi: 10.1038/jid.2008.305
87. Schmidt E, Reimer S, Kruse N, Jainta S, Brocker E, Marinkovich M, et al. Autoantibodies to BP180 associated with bullous pemphigoid release interleukin-6 and interleukin-8 from cultured human keratinocytes. J Invest Dermatol. (2000) 115:842–8.
88. Freire P, Munoz C, Stingl G. IgE autoreactivity in bullous pemphigoid: eosinophils and mast cells as major targets of pathogenic immune reactants. Br J Dermatol. (2017) 177:1644–53. doi: 10.1111/bjd.15924
89. Liu Z, Giudice G, Zhou X, Swartz S, Troy J, Fairley J, et al. A major role for neutrophils in experimental bullous pemphigoid. J Clin Invest. (1997) 100:1256–63. doi: 10.1172/JCI119639
90. Jones V, Patel P, Amber K. Eosinophils in bullous pemphigoid. Panminerva Med. (2021) 63:368–78. doi: 10.23736/S0031-0808.20.03997-X
91. Park S, Lee S, Kim J, Kim S. Circulating eosinophil and neutrophil counts correlate with disease severity in bullous pemphigoid. Ann Dermatol. (2018) 30:544–9. doi: 10.5021/ad.2018.30.5.544
92. Liu Z, Shapiro S, Zhou X, Twining S, Senior R, Giudice G, et al. A critical role for neutrophil elastase in experimental bullous pemphigoid. J Clin Invest. (2000) 105:113–23. doi: 10.1172/JCI3693
93. Liu Z. Bullous pemphigoid: using animal models to study the immunopathology. J Investig Dermatol Symp Proc. (2004) 9:41–6. doi: 10.1111/j.1087-0024.2004.00841.x
94. Lindgren O, Le Menn G, Tuusa J, Chen Z, Tasanen K, Kokkonen N. Absence of NC14A domain of COLXVII/BP180 in mice results in IL-17?associated skin inflammation. J Invest Dermatol. (2022). 143:48–56.e7 doi: 10.1016/j.jid.2022.07.019
95. Lin L, Betsuyaku T, Heimbach L, Li N, Rubenstein D, Shapiro S, et al. Neutrophil elastase cleaves the murine hemidesmosomal protein BP180/type XVII collagen and generates degradation products that modulate experimental bullous pemphigoid. Matrix Biol. (2012) 31:38–44. doi: 10.1016/j.matbio.2011.09.003
96. Verraes S, Hornebeck W, Polette M, Borradori L, Bernard P. Respective contribution of neutrophil elastase and matrix metalloproteinase 9 in the degradation of BP180 (type XVII collagen) in human bullous pemphigoid. J Invest Dermatol. (2001) 117:1091–6. doi: 10.1046/j.0022-202x.2001.01521.x
97. Murthy S, Schilf P, Patzelt S, Thieme M, Becker M, Kroger L, et al. Dapsone suppresses disease in preclinical murine models of pemphigoid diseases. J Invest Dermatol. (2021) 141:2587–95.e2. doi: 10.1016/j.jid.2021.04.009
98. Margaroli C, Bradley B, Thompson C, Brown M, Giacalone V, Bhatt L, et al. Distinct compartmentalization of immune cells and mediators characterizes bullous pemphigoid disease. Exp Dermatol. (2020) 29:1191–8. doi: 10.1111/exd.14209
99. Pruessmann J, Pruessmann W, Holtsche M, Linnemann B, Hammers C, van Beek N, et al. Immunomodulator Galectin-9 is increased in blood and skin of patients with bullous pemphigoid. Acta Derm Venereol. (2021) 101:adv00419. doi: 10.2340/00015555-3771
100. Fujimura T, Kakizaki A, Furudate S, Aiba S. A possible interaction between periostin and CD163(+) skin-resident macrophages in pemphigus vulgaris and bullous pemphigoid. Exp Dermatol. (2017) 26:1193–8. doi: 10.1111/exd.13157
101. Kagami S, Kai H, Kakinuma T, Miyagaki T, Kamata M, Sugaya M, et al. High levels of CCL26 in blister fluid and sera of patients with bullous pemphigoid. J Invest Dermatol. (2012) 132:249–51. doi: 10.1038/jid.2011.251
102. Kowalski E, Kneibner D, Kridin K, Amber K. Serum and blister fluid levels of cytokines and chemokines in pemphigus and bullous pemphigoid. Autoimmun Rev. (2019) 18:526–34.
103. Solimani F, Didona D, Li J, Bao L, Patel P, Gasparini G, et al. Characterizing the proteome of bullous pemphigoid blister fluid utilizing tandem mass tag labeling coupled with LC-MS/MS. Arch Dermatol Res. (2022) 314:921–8. doi: 10.1007/s00403-021-02253-8
104. Wakugawa M, Nakamura K, Hino H, Toyama K, Hattori N, Okochi H, et al. Elevated levels of eotaxin and interleukin-5 in blister fluid of bullous pemphigoid: correlation with tissue eosinophilia. Br J Dermatol. (2000) 143:112–6. doi: 10.1046/j.1365-2133.2000.03599.x
105. Sun C, Wu J, Wong T, Wang L, Chuan M. High levels of interleukin-8, soluble CD4 and soluble CD8 in bullous pemphigoid blister fluid. The relationship between local cytokine production and lesional T-cell activities. Br J Dermatol. (2000) 143:1235–40. doi: 10.1046/j.1365-2133.2000.03894.x
106. Miyagaki T, Sugaya M, Kagami S, Nakashima H, Ishiura N, Watanabe R, et al. Increased CCL1 levels in the sera and blister fluid of patients with bullous pemphigoid. J Dermatol Sci. (2009) 54:45–7. doi: 10.1016/j.jdermsci.2008.10.012
107. Gunther C, Wozel G, Meurer M, Pfeiffer C. Up-regulation of CCL11 and CCL26 is associated with activated eosinophils in bullous pemphigoid. Clin Exp Immunol. (2011) 166:145–53. doi: 10.1111/j.1365-2249.2011.04464.x
108. de Graauw E, Sitaru C, Horn M, Borradori L, Yousefi S, Simon H, et al. Evidence for a role of eosinophils in blister formation in bullous pemphigoid. Allergy. (2017) 72:1105–13. doi: 10.1111/all.13131
109. Fang H, Shao S, Xue K, Yuan X, Qiao P, Zhang J, et al. Neutrophil extracellular traps contribute to immune dysregulation in bullous pemphigoid via inducing B-cell differentiation and antibody production. FASEB J. (2021) 35:e21746. doi: 10.1096/fj.202100145R
110. Rudrich U, Gehring M, Papakonstantinou E, Illerhaus A, Engmann J, Kapp A, et al. Eosinophils are a major source of interleukin-31 in bullous pemphigoid. Acta Derm Venereol. (2018) 98:766–71. doi: 10.2340/00015555-2951
111. Amber K, Valdebran M, Kridin K, Grando S. The role of eosinophils in bullous pemphigoid: a developing model of eosinophil pathogenicity in mucocutaneous disease. Front Med (Lausanne). (2018) 5:201. doi: 10.3389/fmed.2018.00201
112. Lin L, Hwang B, Culton D, Li N, Burette S, Koller B, et al. Eosinophils mediate tissue injury in the autoimmune skin disease bullous pemphigoid. J Invest Dermatol. (2018) 138:1032–43. doi: 10.1016/j.jid.2017.11.031
113. de Graauw E, Sitaru C, Horn M, Borradori L, Yousefi S, Simon D, et al. Monocytes enhance neutrophil-induced blister formation in an ex vivo model of bullous pemphigoid. Allergy. (2018) 73:1119–30. doi: 10.1111/all.13376
114. Hiroyasu S, Zeglinski M, Zhao H, Pawluk M, Turner C, Kasprick A, et al. Granzyme B inhibition reduces disease severity in autoimmune blistering diseases. Nat Commun. (2021) 12:302. doi: 10.1038/s41467-020-20604-3
115. Riani M, Le Jan S, Plee J, Durlach A, Le Naour R, Haegeman G, et al. Bullous pemphigoid outcome is associated with CXCL10-induced matrix metalloproteinase 9 secretion from monocytes and neutrophils but not lymphocytes. J Allergy Clin Immunol. (2017) 139:863–72.e3. doi: 10.1016/j.jaci.2016.08.012
116. Furudate S, Fujimura T, Kambayashi Y, Kakizaki A, Aiba S. Comparison of CD163+ CD206+ M2 macrophages in the lesional skin of bullous pemphigoid and pemphigus vulgaris: the possible pathogenesis of bullous pemphigoid. Dermatology. (2014) 229:369–78. doi: 10.1159/000365946
117. Riani M, Muller C, Bour C, Bernard P, Antonicelli F, Le Jan S. Blister fluid induces MMP-9-Associated M2-Type macrophages in bullous pemphigoid. Front Immunol. (2019) 10:1858. doi: 10.3389/fimmu.2019.01858
118. Tanita K, Fujimura T, Sato Y, Lyu C, Aiba S. Minocycline decreases Th2 chemokines from M2 macrophages: possible mechanisms for the suppression of bullous pemphigoid by traditional bullous disease drugs. Exp Dermatol. (2018) 27:1268–72. doi: 10.1111/exd.13779
119. Hashimoto T, Kursewicz C, Fayne R, Nanda S, Shah S, Nattkemper L, et al. Pathophysiologic mechanisms of itch in bullous pemphigoid. J Am Acad Dermatol. (2020) 83:53–62. doi: 10.1016/j.jaad.2019.07.060
120. Chen R, Fairley J, Zhao M, Giudice G, Zillikens D, Diaz L, et al. Macrophages, but not T and B lymphocytes, are critical for subepidermal blister formation in experimental bullous pemphigoid: macrophage-mediated neutrophil infiltration depends on mast cell activation. J Immunol. (2002) 169:3987–92. doi: 10.4049/jimmunol.169.7.3987
121. Chen R, Ning G, Zhao M, Fleming M, Diaz L, Werb Z, et al. Mast cells play a key role in neutrophil recruitment in experimental bullous pemphigoid. J Clin Invest. (2001) 108:1151–8. doi: 10.1172/JCI11494
122. Nsiah-Dosu S, Scholz C, Orinska Z, Sadik C, Ludwig R, Schmidt E, et al. Mast cell-deficient mice Mcpt5Cre/Dicer (fl/fl) redefine the role of mast cells in experimental bullous pemphigoid. Skin Health Dis. (2022) 2:e70. doi: 10.1002/ski2.70
124. Yoo D, Lee J, Kim S, Kim J. Mortality and clinical response of patients with bullous pemphigoid treated with rituximab. Br J Dermatol. (2021) 185:210–2.
125. Suarez-Carantona C, Jimenez-Cauhe J, Gonzalez-Garcia A, Fernandez-Guarino M, Asuncion Ballester M. Low-Dose rituximab for bullous pemphigoid. protocol and single-center experience. Actas Dermosifiliogr. (2023) 114:T62–8.
126. Chen Q, Hu B, Wan L, Hu L, Zhou X, Chen L, et al. Three cases of refractory bullous pemphigoid in the elderly treated successfully with ultra-low-dose rituximab. J Dermatol. (2022). [Epub ahead of print]. doi: 10.1111/1346-8138.16668
127. Li N, Culton D, Diaz L, Liu Z. Modes of action of intravenous immunoglobulin in bullous pemphigoid. J Invest Dermatol. (2018) 138:1249–51. doi: 10.1016/j.jid.2018.02.020
128. Amagai M, Ikeda S, Hashimoto T, Mizuashi M, Fujisawa A, Ihn H, et al. A randomized double-blind trial of intravenous immunoglobulin for bullous pemphigoid. J Dermatol Sci. (2017) 85:77–84. doi: 10.1016/j.jdermsci.2016.11.003
129. Sami N, Ali S, Bhol K, Ahmed A. Influence of intravenous immunoglobulin therapy on autoantibody titres to BP Ag1 and BP Ag2 in patients with bullous pemphigoid. J Eur Acad Dermatol Venereol. (2003) 17:641–5. doi: 10.1046/j.1468-3083.2003.00714.x
130. Engineer L, Ahmed A. Role of intravenous immunoglobulin in the treatment of bullous pemphigoid: analysis of current data. J Am Acad Dermatol. (2001) 44:83–8.
131. Micevic G, Lo S, Rajan N. Targeting the neonatal Fc receptor in pemphigus: safety first. Br J Dermatol. (2022) 186:389–90. doi: 10.1111/bjd.20939
132. Goebeler M, Bata-Csorgo Z, De Simone C, Didona B, Remenyik E, Reznichenko N, et al. Treatment of pemphigus vulgaris and foliaceus with efgartigimod, a neonatal Fc receptor inhibitor: a phase II multicentre, open-label feasibility trial. Br J Dermatol. (2022) 186:429–39. doi: 10.1111/bjd.20782
133. Vassallo C, Somenzi A, De Amici M, Barruscotti S, Brazzelli V. Omalizumab as a corticosteroid-sparing agent in the treatment of bullous pemphigoid. Dermatol Ther. (2022) 35:e15946.
134. Gonul M, Keseroglu H, Ergin C, Ozcan I, Erdem O. Bullous pemphigoid successfully treated with omalizumab. Indian J Dermatol Venereol Leprol. (2016) 82:577–9.
135. D’Aguanno K, Gabrielli S, Ouchene L, Muntyanu A, Ben-Shoshan M, Zhang X, et al. Omalizumab for the treatment of bullous pemphigoid: a systematic review of efficacy and safety. J Cutan Med Surg. (2022) 26:404–13.
136. Navarro-Trivino F, Llamas-Molina J, Ayen-Rodriguez A, Cancela-Diez B, Ruiz-Villaverde R. Dramatic improvement of bullous pemphigoid with omalizumab in an elderly patient. Eur J Hosp Pharm. (2021) 28:350–2. doi: 10.1136/ejhpharm-2020-002418
137. Maglie R, Antiga E, Quintarelli L, Verdelli A, Caproni M. Dramatic exacerbation of bullous pemphigoid following rituximab and successful treatment with omalizumab. Eur J Dermatol. (2019) 29:213–5. doi: 10.1684/ejd.2019.3499
138. De D, Kaushik A, Handa S, Mahajan R, Schmidt E. Omalizumab: an underutilized treatment option in bullous pemphigoid patients with co-morbidities. J Eur Acad Dermatol Venereol. (2021) 35:e469–72. doi: 10.1111/jdv.17229
139. Cao P, Xu W, Zhang L. Rituximab, omalizumab, and dupilumab treatment outcomes in bullous pemphigoid: a systematic review. Front Immunol. (2022) 13:928621. doi: 10.3389/fimmu.2022.928621
140. Seyed Jafari S, Gadaldi K, Feldmeyer L, Yawalkar N, Borradori L, Schlapbach C. Effects of omalizumab on fcepsilonRI and IgE expression in lesional skin of bullous pemphigoid. Front Immunol. (2019) 10:1919. doi: 10.3389/fimmu.2019.01919
141. Gutjahr A, Heck F, Emtenani S, Hammers A, Hundt J, Muck P, et al. Bullous pemphigoid autoantibody-mediated complement fixation is abolished by the low-molecular-weight heparin tinzaparin sodium. Br J Dermatol. (2019) 181:593–4.
142. Sadik C, Rashid H, Hammers C, Diercks G, Weidinger A, Beissert S, et al. Evaluation of nomacopan for treatment of bullous pemphigoid: a phase 2a nonrandomized controlled trial. JAMA Dermatol. (2022) 158:641–9. doi: 10.1001/jamadermatol.2022.1156
143. Kaye A, Gordon S, Deverapalli S, Her M, Rosmarin D. Dupilumab for the treatment of recalcitrant bullous pemphigoid. JAMA Dermatol. (2018) 154:1225–6.
144. Takamura S, Teraki Y. Treatment of bullous pemphigoid with dupilumab: dupilumab exerts its effect by primarily suppressing T-helper 2 cytokines. J Dermatol. (2022) 49:845–50. doi: 10.1111/1346-8138.16428
145. Abdat R, Waldman R, de Bedout V, Czernik A, McLeod M, King B, et al. Dupilumab as a novel therapy for bullous pemphigoid: a multicenter case series. J Am Acad Dermatol. (2020) 83:46–52. doi: 10.1016/j.jaad.2020.01.089
146. Yang J, Gao H, Zhang Z, Tang C, Chen Z, Wang L, et al. Dupilumab combined with low-dose systemic steroid therapy improves efficacy and safety for bullous pemphigoid. Dermatol Ther. (2022) 35:e15648. doi: 10.1111/dth.15648
147. Zhang Y, Xu Q, Chen L, Chen J, Zhang J, Zou Y, et al. Efficacy and safety of dupilumab in moderate-to-severe bullous pemphigoid. Front Immunol. (2021) 12:738907. doi: 10.3389/fimmu.2021.738907
148. Seyed Jafari S, Feldmeyer L, Bossart S, Simon D, Schlapbach C, Borradori L. Case report: combination of omalizumab and dupilumab for recalcitrant bullous pemphigoid. Front Immunol. (2020) 11:611549. doi: 10.3389/fimmu.2020.611549
149. Baffa M, Maglie R, Montefusco F, Pipito C, Senatore S, Antiga E. Severe bullous pemphigoid following Covid-19 vaccination resistant to rituximab and successfully treated with dupilumab. J Eur Acad Dermatol Venereol. (2022). 37:e135–7. doi: 10.1111/jdv.18673
150. Bruni, M, Moar A, Schena D, Girolomoni G. A case of nivolumab-induced bullous pemphigoid successfully treated with dupilumab. Dermatol Online J. (2022) 15:28. doi: 10.5070/D328257396
Keywords: cytokine, bullous pemphigoid, target therapy, interleukin, Th2, lymphocyte, eosinophil, neutrophil
Citation: Maglie R, Solimani F, Didona D, Pipitò C, Antiga E and Di Zenzo G (2023) The cytokine milieu of bullous pemphigoid: Current and novel therapeutic targets. Front. Med. 10:1128154. doi: 10.3389/fmed.2023.1128154
Received: 20 December 2022; Accepted: 23 January 2023;
Published: 06 February 2023.
Edited by:
Ralf J. Ludwig, University of Lübeck, GermanyReviewed by:
Hiroaki Iwata, Gifu University, JapanCopyright © 2023 Maglie, Solimani, Didona, Pipitò, Antiga and Di Zenzo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanni Di Zenzo,  Zy5kaXplbnpvQGlkaS5pdA==
Zy5kaXplbnpvQGlkaS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.