 Clara Bernardelli
Clara Bernardelli Anna Caretti
Anna Caretti Elena Lesma
Elena Lesma- 1Laboratory of Pharmacology, Department of Health Sciences, Università degli Studi di Milano, Milan, Italy
- 2Laboratory of Biochemistry and Molecular Biology, Department of Health Sciences, Università degli Studi di Milano, Milan, Italy
A chronic inflammatory condition characterizes various lung diseases. Interestingly, a great contribution to inflammation is made by altered lipids metabolism, that can be caused by the deregulation of the mammalian target of rapamycin complex-1 (mTORC1) activity. There is evidence that one of mTOR downstream effectors, the sterol regulatory element-binding protein (SREBP), regulates the transcription of enzymes involved in the de novo fatty acid synthesis. Given its central role in cell metabolism, mTOR is involved in several biological processes. Among those, mTOR is a driver of senescence, a process that might contribute to the establishment of chronic lung disease because the characteristic irreversible inhibition of cell proliferation, associated to the acquisition of a pro-inflammatory senescence-associated secretory phenotype (SASP) supports the loss of lung parenchyma. The deregulation of mTORC1 is a hallmark of lymphangioleiomyomatosis (LAM), a rare pulmonary disease predominantly affecting women which causes cystic remodeling of the lung and progressive loss of lung function. LAM cells have senescent features and secrete SASP components, such as growth factors and pro-inflammatory molecules, like cancer cells. Using LAM as a paradigm of chronic and metastatic lung disease, here we review the published data that point out the role of dysregulated lipid metabolism in LAM pathogenesis. We will discuss lipids’ role in the development and progression of the disease, to hypothesize novel LAM biomarkers and to propose the pharmacological regulation of lipids metabolism as an innovative approach for the treatment of the disease.
Introduction
Lungs rely on a unique lipid biology that ensures respiratory function and is involved in the regulation of the immune response. The alveolar area sustains active lipid metabolism to maintain surfactant homeostasis thus ensuring optimal respiration cycle. Mostly composed of phospholipids with 5–10% of cholesterol and small amount of sphingolipids, surfactant is essential in reducing the surface tension in the alveolar walls (1). Sterol-regulatory element-binding proteins (SREBPs) regulate lung lipid biosynthesis and sterol homeostasis at transcriptional level (2). This family of transcription factors promotes the expression of cholesterogenic and lipogenic genes involved in fat storage (3). Growing evidence shows that SREBPs are involved in numerous pathogenic processes such as endoplasmic reticulum stress and inflammation. LPS-challenged macrophages from mice with a targeted deficiency in the gene encoding SREBP-1a, failed to activate lipogenesis, secretion of IL-1β and gene encoding Nlrp1a, which is a core inflammasome component (4). Oishi et al. demonstrated that in macrophages, SREBP1 reprograms lipid metabolism to produce the anti-inflammatory polyunsaturated fatty acids thus promoting the late resolution of TLR4-induced gene activation (5). SREBP1 induction in alveolar type 2 cells of Insig1/2Δ/Δ mice causes neutral lipid accumulation in type II cells and in the alveoli, resulting in pulmonary inflammation and airspace remodeling (6). Transcriptome network analysis reveals that SREBP1 activation promotes lipotoxicity that in turn induces inflammation and fibrosis in the lung (7). Recent data have shown that SREBP2 integrates cholesterol metabolism with NLRP3 inflammasome activation in macrophages (8). SREBP1 is regulated by the mammalian target of rapamycin (mTOR) (9) that is a regulator of lipid and nucleotide synthesis. Once activated, SREBP1 translocates from the endoplasmic reticulum to the nucleus to promote the transcription of the enzymes responsible for glugoneogenesis and lipogenesis (10).
Lymphangioleiomyomatosis (LAM) is a rare progressive lung disease characterized by lung cystic destruction, and the progressive loss of lung function (11). LAM occurs sporadically or associated to Tuberous Sclerosis Complex (TSC) (12). Loss of TSC gene function causes to dysregulated mTOR signaling (13). mTOR has a central role in cell metabolism, integrating environmental signals to promote anabolic processes (14). Recently, likely because of mTOR dysregulation, several studies indicated the alteration of lipid metabolism in LAM as an interesting target to uncover novel therapeutic approaches (15).
The minireview describes the current state of knowledge of emerging role of lipids in LAM considering their contribution in inflammation and in the pathogenesis and progression of chronic lung diseases.
Lipids in pulmonary diseases
Inflammation
Lung inflammatory diseases are tightly linked with aberrant SREBP activity in respiratory epithelial cells and with dyslipidemia. SREBP2-mediated lipid metabolism promotion correlates with (COVID-19)-induced cytokine storm (16). The increased expression of SREBP-1 after respiratory virus infection (i.e., SARS-CoV), regulates the accumulation of lipid droplets, cell organelles involved in the amplification of inflammatory mediators’ production (17). Dysregulation of free fatty acids, cholesterol, and ceramides homeostasis in the lungs is associated with the pathogenesis and the progression of chronic obstructive pulmonary disease (COPD). Cigarette smoke–induced alveolar accumulation of lipids is an important event that triggers inflammation in alveolar macrophages (18). Several studies have demonstrated that ceramides, the central hub of sphingolipids metabolism, are elevated in COPD and contribute to chronic inflammation (19). Idiopathic pulmonary fibrosis (IPF) is a progressive lung disease characterized by inflammation and fibrosis. As recently reviewed by Suryadevara et al. (20), bioactive lipid mediators derived from fatty acids, glycerolipids, phospholipids, and sphingolipids exhibit pro- or anti-fibrotic/inflammatory effects in lung tissues of IPF patients. Acute Respiratory Distress Syndrome (ARDS) is a severe form of clinical acute lung injury (ALI), both sharing severe inflammatory conditions. Phospholipase A2 enzymes (sPLA2s), that generate free fatty acids and lysophospholipids from glycerophospholipids, are upregulated in different cell types in the ALI/ARDS lung compartment. Significant evidence demonstrates that in these pathological settings, PLA2s are functionally important as regulators of inflammatory signaling (21). Caused by several factors, pneumonia is the inflammation localized to the terminal airways, the alveoli, and the interstitium of the lungs. A pilot study conducted in 2017 during community-acquired pneumonia (22), reported that monounsaturated and polyunsaturated fatty acid accumulation elicits an inflammatory response during pneumonia progression. Cystic Fibrosis (CF) is an inherited recessive disease. CF is caused by mutations in the CF transmembrane conductance regulator gene, with most of the mortality given by the lung dysfunction (23), being inflammation an independent risk factor for disease progression. We previously demonstrated that CF bronchial epithelial cells accumulate the sphingolipid ceramide (24), which contributes to CF airways inflammation (25), as well as glycerophospholipids and lyso-glycerophospholipids, the latest considered pro-inflammatory molecules (26), cholesterols, cholesterol esters and triacylglycerols (27), compared to normal cells.
Senescence-mediated inflammation
Senescence is a stress-response process characterized by the irreversible arrest of the cell cycle following DNA damage. Senescent cells develop a senescence-associated secretory phenotype (SASP) to maintain their status and to communicate within their microenvironment. SASP factors, both released as soluble molecules or within extracellular vesicles (EVs), have strong biological activities on neighboring cells, including the promotion of inflammation. Indeed, the study of SASP in all in vitro generated models demonstrated the presence of the proinflammatory interleukin-6 (IL-6), CXC chemokine ligand 8 (IL-8), and monocyte chemoattractant protein 1 (28). Even if senescence allows the maintaining of tissue homeostasis, an irreversible proliferation arrest might limit the regenerative capacity of the tissue, and the presence of a pro-inflammatory milieu might sustain the onset of pathological condition and age-related disorders. In pulmonary diseases, accelerated or premature aging emerged to be significant. The oxidative stress caused by cigarette smoke induces senescence in alveolar epithelial cells (AEC) and endothelial cells that accumulate in COPD lungs, while in IPF the shortening of the telomeres might be responsible for the senescent status of fibroblasts and AEC (29). Of note, AEC might be driven to senescence also after the serine/threonine kinase mTOR hyperactivation (30). The constitutive activation of mTOR in the lung epithelial cells of transgenic mice results in weakness tight junctions’ (TJ), that is a sign of injury in the lung epithelium. In parallel, also epithelial to mesenchymal transition (EMT) was enhanced, ultimately contributing to lung fibrosis. In this study, however, senescence was not observed, but the authors suggested that this might be due to differences in the timing of mTOR activation compared to previous studies (31). Moreover, the peculiar contribution of the AEC to local inflammation might be ascribed also to the release of EVs. Interestingly, it was recently demonstrated that AEC released EVs that differs on their miRNA content, depending on the apical or basolateral origin site of their release. EVs released from the apical side of epithelial cells are enriched in miRNAs enhancing the mTOR signaling pathways, probably being responsible for the TJ vulnerability and for the EMT, which can cause lung injury (32). Of note, senescence and lipid metabolism share many regulatory proteins, among which mTOR has a crucial role (33). Senescent cells have increased lipids uptake and accumulation in lipids droplets (34) and it has recently arisen that the regulation of specific lipid species plays a critical role in senescence contributing to the chronic inflammation associated with SASP (35). For example, the prostaglandin E2 (PGE2), which is synthesized from the arachidonic acid, is expressed at higher levels in COPD fibroblasts compared to healthy controls and is responsible for the autocrine and paracrine spreading of senescence in neighboring cells, as well as of the induction of inflammatory response through the up-regulation of its receptors EP2 and EP4 in a cyclooxygenase 2 -dependent reactive oxygen species response (36). Notably, PGE2 is released also by epithelial cells to relax airways smooth muscle cells, indicating that the roles of lipid molecules in the crosstalk between cells in the lung environment is complex and needs further investigation (37). Additionally, the signaling lipid leukotrienes, that are SASP components, are secreted by senescent fibroblasts and contribute to worsen IPF promoting fibrosis in lung (38). Finally, lysophosphatidylcholines (LPCs) have SASP activity, being expressed at high levels in senescent fibroblasts and being capable to induce the secretion of IL-8 and IL-6 (35).
LAM and mTOR
Lymphangioleiomyomatosis (LAM) primarily affects women of childbearing age. LAM cells are smooth muscle-like cells bearing a mutation in TSC1 or TSC2 tumor suppressor genes that causes the hyperactivation of Rheb with an increase of mTORC1 and adenosine 5′-monophosphate activated protein kinase (AMPK) activity (39, 40). AMPK control the energy levels by promoting the catabolic processes and by inhibiting the anabolic activities. Interestingly, tuberin-null cells show the AMPK hyperactivation which correlates with the cytoplasmic localization of p27Kip1 that negatively regulates the cyclin dependent kinase 2 (41). mTORC1 or mTORC2 induce the expression and the proteolytic process of SREBP1; therefore, mTORC1 hyperactivation, through SREBP1 and acetyl-CoA carboxylase, controls the lipid accumulation (10). Moreover, the synthesis of PC is controlled by mTORC1 through SREBP1 (9). As a consequence of the TSC mutations, the loss of heterozygosity in TSC1 or TSC2, which causes the lack of hamartin or tuberin, leads to the hyperactivation of mTOR that promotes cell growth and proliferation by stimulating anabolic metabolism with the increase of protein and lipid synthesis (42, 43). Furthermore, constitutive mTOR activation leads to an increased invasion of lymphatics and lungs where LAM cells and wild type stromal cells form cysts with the consequent pneumothorax and progressive loss of pulmonary function (44).
The proliferation of LAM cells in the lung parenchyma is associated with diffuse cystic lesions and their infiltration into the lymphatic walls causes lymphatic abnormalities leading to damage or obstruction of lymphatic vessels (45). Lymphangiogenesis plays a pathogenetic role whereby LAM cells invade and spread through the lymphatic, a process that is related to a high levels of vascular endothelial growth factor (VEGF)-D (46). As well, nearby and into the LAM lesions, the increased expression of VEGF-A stimulates angiogenesis that also might be a mechanism by which LAM cells invade the circulation (47). Clinical manifestations of LAM include progressive dyspnea, cough, chest pain, recurrent pneumothoraces, and chylous complications including chylothorax, chyloptysis, and chylous ascites (13). Angiomyolipomas, benign tumors composed of LAM cells, smooth muscle cells, adipose tissue, and vessels, can often occur in LAM patients.
The discovery of the genetic basis of LAM cells led to consider the inhibition of mTOR hyperactivation as a therapeutic approach. Rapamycin indeed inhibits LAM cell proliferation resulting in the stabilization of the lung function, in the reduction of lymphatic abnormalities, and of the angiomyolipoma volumes, with no efficacy in the regression of the existing pulmonary lesions. So far, the treatment with rapamycin is the only approved for LAM (48).
After being classified as an interstitial lung disease, LAM has been recently reconsidered as a low-grade, destructive, metastasizing neoplasm for several features as the invasive LAM cell properties, the recurrency in donor allografts of patients who have undergone lung transplantation, and the metabolic reprogramming. However, LAM differs from other neoplasms for the bilateral and symmetrical disruption of the lung without a clear primary tumor site and dominant mass lesions (49).
We recently demonstrated that LAM/TSC cells, derived by chylous thorax of a LAM/TSC patient, have senescent features dependent from mTOR hyperactivation and the capability to induce senescence in neighboring cells (50). As demonstrated in other respiratory disease such as IPF, senescence might drive the progressive loss of parenchymal structure in LAM which ultimately causes the impairment of lung function (51). It has been demonstrated that LAM cells secrete molecules known to be SASP components comprised metalloproteinases (52), proinflammatory cytokines IL-6 (53) and IL-8 (50), kathepsin K (54), and VEGF-D (39) reinforcing the hypothesis of a LAM cell communication with the microenvironment and a SASP modulation of the remodeling of the lung parenchyma. Interestingly, IL-8, a potent neutrophil chemotactic factor that triggers chemotaxis and neutrophil activation through a phosphorylation cascade in the inflammatory response (55), has been demonstrated to be involved in the pathogenesis and progression of lung diseases such as ARDS and SARS CoV-2, suggesting the possibility to use IL-8 as a biomarker or as a therapeutic target (56). In LAM, besides its role in senescence, IL-8 might reinforce the senescence/inflammatory milieu driving the progression of the disease.
Furthermore, mTOR role in LAM senescence can be supported by the senolitic effect of rapamycin and the relapse following the suspension of rapamycin treatment caused by an irreversible senescent state, called geroconversion, sustained by mTOR hyperactivation (57).
Lipids as novel potential therapeutic targets and biomarkers
Pulmonary diseases
In view of the deep interplay between dysregulated lipid metabolism and chronic inflammatory condition that characterizes several lung diseases, restoring lipid homeostasis could represent a novel therapeutic approach. Herein, there are examples of preclinical studies focusing on the potential anti-inflammatory efficacy of compounds targeting lipid pathway in pulmonary diseases (Table 1). Treatment with A922500, a pharmacological inhibitor of acyl-CoA:diacylglycerol acyltransferase-1, inhibits lipid droplets biogenesis triggered by SARS-CoV-2 infection in A549 human epithelial cells and in primary human monocytes. Thereon, the synthesis of pro-inflammatory lipid production and cytokinesis is downregulated (58). Interestingly, we were able to also reduce glycerol- and cholesterol-based lipids, to promote fatty acids oxidation, to reduce inflammation in in vitro model of CF by reducing ceramide accumulation with Myriocin, the inhibitor of the rate-limiting step in sphingolipid biosynthesis (27). Finally, in an in vivo model, the bleomycin-induced lung fibrosis and inflammation in rats was attenuated by AK106-001616 that inhibits the cytosolic PLA2 responsible for the generation of pro-inflammatory eicosanoids (59).
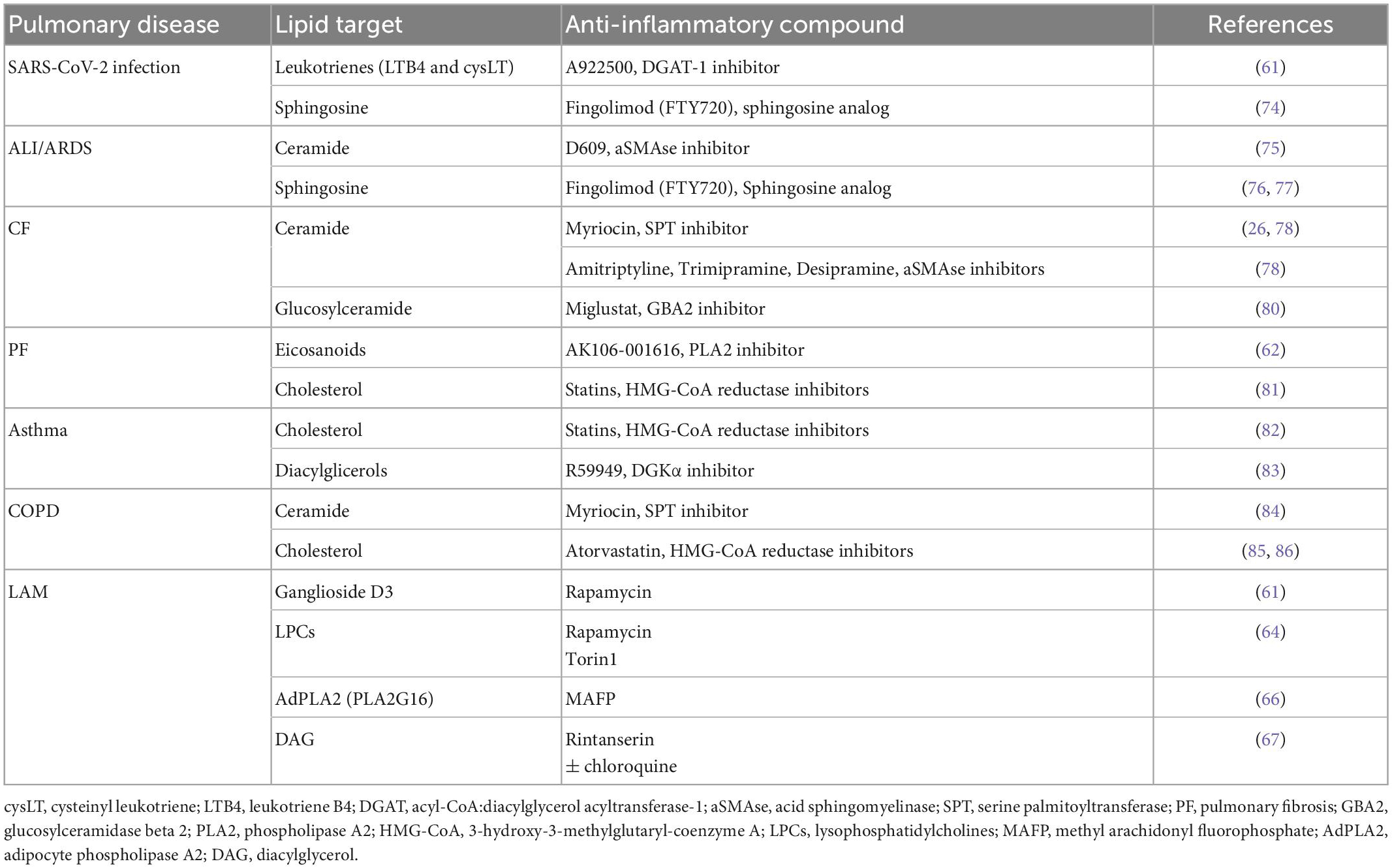
Table 1. Anti-inflammatory efficient compounds targeting lipid pathways in pulmonary diseases.
LAM
Lymphangioleiomyomatosis (LAM) cells have a metabolic signature of increased fatty acid uptake and synthesis, a characteristic in common with cancer cells (60), making altered lipid species in LAM both disease relevant biomarkers and potential therapeutic targets.
For instance, in the perspective of a LAM immunotherapy, it was demonstrated that LAM lung sections of both patients and Tsc2–/– mice have a strong positivity to the ganglioside D3 (GD3), whose expression is a characteristic that LAM cells maintain also after multiple passages in vitro (61). Interestingly, LAM patients have lower anti-GD3 antibodies in their serum compared to healthy controls and the in vitro treatment of LAM cells with commercial anti-GD3 antibodies plus human complement induce death in the 12–42% of the population. This might suggest an approach similar to melanoma, in which the targeting of GD3 by antibodies reduces tumor growth through the activation of the natural killer T cells (62).
Even if GD3 expression in LAM cells and lipids accumulation in the renal angiomyolipomas of LAM patients can be considered a consequence of mTOR deregulation (63), emerging researches demonstrate the existence of uncovered TSC1/2-dependent-mTORC1-independent regulation of lipids metabolism that may contribute to the LAM pathogenesis and progression.
In the first systematic study of the LAM lipidome, high levels of four LPCs (C16:0, C18:0, C18:1, and C20:4) were found in the plasma of women with LAM compared with healthy controls, suggesting novel potential biomarkers (64). LPCs are bioactive lipids generated by the activity of phospholipase A (PLA) on the precursor PC (9) and preclinical models of lung cancer showed that those molecules might have a direct role in tumor angiogenesis (65), a condition that, together with lymphangiogenesis, is characteristic also in pulmonary and extrapulmonary LAM (49). Interestingly, the increase of LPCs were also observed in Tsc2–/– mouse embryonic fibroblasts (MEFs), consistently with the hypothesis that tuberin loss might enhance several phospholipid and neutral lipid species. The treatment with the mTOR inhibitors rapamycin or torin1, or the down-regulation of SREBP1 does not suppress LPCs, while the down-regulation of specific PLAs decreases selectively the proliferation of Tsc2–/– MEFs compared with Tsc2+/+ MEFs (58). Among PLAs, the accumulation of the PLA2G16 isoform was observed in the lung nodules and in the renal angiomyolipomas of LAM patients, and it was demonstrated that the presence of tuberin negatively regulates PLA2G16 overexpression both in vitro and in vivo, affecting also the production of prostaglandins, that are critical mediators of chronic inflammation and cancer progression. In the same study, Tsc2–/– MEFs treated with rapamycin or torin1 did not change the expression of PLA2G16, while the PLA inhibitor methyl arachidonyl fluorophosphonate (MAFP) reduced the growth and induced apoptosis on tuberin-deficient cells derived from LAM patient. Interestingly, the tuberin induced-expression in these cells prevents the effects of MAFP treatment (66).
Since mTORC1 hyperactivation causes the reduction of autophagy and induces a metabolic reprogramming of tuberin-deficient cells, a novel therapeutic approach was proposed in a recent study which demonstrates that Tsc2–/– MEFs are sensitive to the treatment with rintaserin in combination with chloroquine, while the impact of those two drugs on Tsc2+/+ MEFs is minimal (67). Rintaserin is an inhibitor of the diacylglycerol kinase alpha (DGKA), that induces the formation of the phosphatidic acid from diacylglycerol, ultimately regulating the homeostasis of cell membranes (68). Tsc2–/– MEFs increased the expression and the activity of DGKA, with a consequent 5-fold higher accumulation of phosphatidic acid compared to Tsc2+/+ MEFs and a higher capability to uptake nutrients from the extracellular space through macropinocytosis. The treatment with rintaserin blocks macropinocytosis and leads to the accumulation of diacylglycerol, reprogramming the phospholipid metabolism by reducing the storage of lipids in droplets and enhancing the synthesis of phospholipids (67). Interestingly, high levels of DGKA, DGKD, DGKQ, and DGKZ were found in the angiomyolipomas of TSC patients, that often develop LAM as pulmonary manifestation (69). In vivo, the injection of tuberin-deficient cells with the downregulation of DGKA does not cause the enlargement of lung alveoli observed in preclinical models of LAM (67, 70).
Taken together, these studies indicate that targeting of key metabolic pathways in lipids synthesis might be novel therapeutic approaches for LAM.
Moreover, lipids can be relevant biomarkers to understand LAM pathogenesis and progression. Indeed, the first evaluation of the LAM serum metabolome showed that there are differences in the metabolic profile when LAM patients were stratified according to menopausal status, lung function, disease burden and disease activity (15). These metabolic abnormalities involved almost exclusively sphingolipids, phospholipids and acylcarnitine fatty acids, reflecting metabolic processes downstream of mTOR. Remarkably, the observation that FEV1 loss, used as measure of airflow limitation, was related to sphingolipid and acylcarnitine fatty acid metabolism, might reflect the extent of lung parenchyma disruption, resulting in the loss of elastic recoil. In the same way, the airflow obstruction in patients with COPD is associated with glycerophospholipids and sphingolipids metabolites, suggesting a common mechanism involving mTOR hyperactivation (71). Moreover, uncovered changes in glycerophospholipids were observed in patients with sporadic LAM compared with LAM/TSC ones after rapamycin treatment (15).
Finally, the altered lipid metabolism in LAM cells can be exploited to identify novel metabolic imaging biomarkers for the disease. Indeed, the previously reported enhancing in PC levels and neutral lipids in LAM cells (64) allowed to test the in vivo uptake of [18F]fluorocholine (FCH) and [18F]fluoroacetate (FACE) to detect tuberin-deficient cells in solid tumors and to monitor the response to rapamycin through dynamic PET (72). [18F]FCH uptake, but not [18F]FACE uptake in tuberin-deficient xenografts was rapamycin-sensitive and in vitro study on tuberin-deficient cells indicated that this difference might be due to an accumulation of these two compounds in two different cells compartment. In fact, [18F]FCH is mainly incorporated into lipids, while [18F]FACE enters into mitochondria and can be used as marker of mitochondrial activity, which is not suppressed by rapamycin, confirming a specific metabolic feature of tuberin-deficiency in LAM cells (73).
Conclusion
Regulation of precise lipid species plays a critical role in senescence and senescence-mediated inflammation associated to chronic pulmonary diseases. Aberrant activity of SREBP and its upstream regulator, mTOR, is highly involved in lipotoxicity that induces inflammation and fibrosis in several lung diseases including LAM (Figure 1). The evidence of an altered lipid metabolism in tuberin-null cells, e.g., involving LPCs and GD3, and the demonstration of metabolic abnormalities in LAM patient serum, mainly LPCs, sphingolipids, phospholipids and acylcarnitine fatty acids, provides the need to promote the comprehension of lipid dysmetabolism in LAM as novel biomarkers for a metabolic signature for stratifying LAM patients and to suggest useful therapeutic targets, also to associate to mTOR inhibitor rapamycin, for the treatment of LAM.
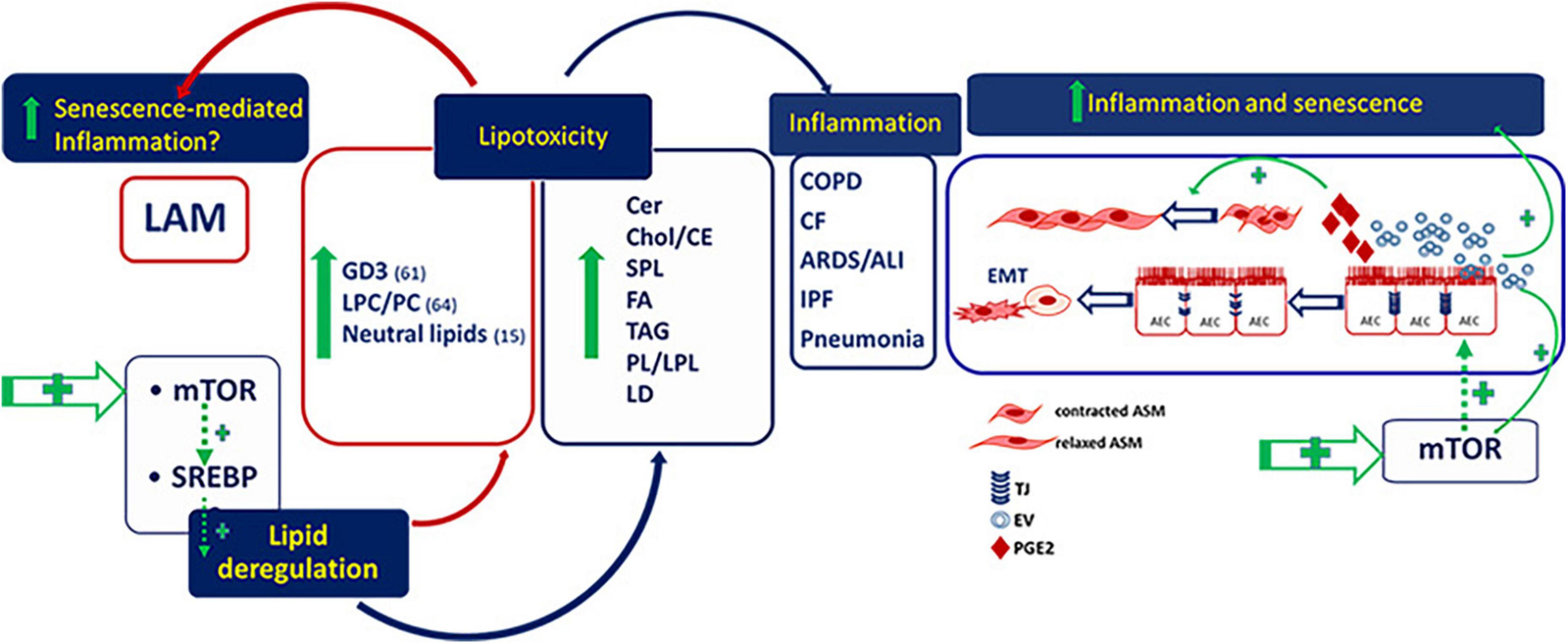
Figure 1. Deregulated lipids in lymphangioleiomyomatosis (LAM) and pulmonary diseases. GD3, ganglioside D3; LPC/PC, lysophosphatidylcholine/phosphatidylcholine; Cer, ceramide; Chol/CE, cholesterol/cholesterol esthers; SPL, sphingolipids; FA, fatty acids; TAG, riacylglycerols; PL/LPL, phospholipids/lysophospholipids; LD, lipid droplets; AEC, alveolar epithelial cells; ASM, airway smooth muscle.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Acknowledgments
The authors acknowledge support from the University of Milan through the APC initiative.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Agudelo C, Samaha G, Garcia-Arcos I. Alveolar lipids in pulmonary disease. A review. Lipids Health Dis. (2020) 19:122. doi: 10.1186/s12944-020-01278-8
2. Horton J, Goldstein J, Brown M. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. (2002) 109:1125–31. doi: 10.1172/JCI0215593
3. Walker A, Yang F, Jiang K, Ji J, Watts J, Purushotham A, et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. (2010) 24:1403–17. doi: 10.1101/gad.1901210
4. Im S, Yousef L, Blaschitz C, Liu J, Edwards R, Young S, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. (2011) 13:540–9. doi: 10.1016/j.cmet.2011.04.001
5. Oishi Y, Spann N, Link V, Muse ED, Strid T, Edillor C, et al. SREBP1 contributes to resolution of pro-inflammatory TLR4 signaling by reprogramming fatty acid metabolism. Cell Metab. (2017) 25:412–27. doi: 10.1016/j.cmet.2016.11.009
6. Plantier L, Besnard V, Xu Y, Ikegami M, Wert S, Hunt A, et al. Activation of sterol-response element-binding proteins (SREBP) in alveolar type II cells enhances lipogenesis causing pulmonary lipotoxicity. J Biol Chem. (2012) 287:10099–114. doi: 10.1074/jbc.M111.303669
7. Shichino S, Ueha S, Hashimoto S, Otsuji M, Abe J, Tsukui T, et al. Transcriptome network analysis identifies protective role of the LXR/SREBP-1c axis in murine pulmonary fibrosis. JCI Insight. (2019) 4:e122163. doi: 10.1172/jci.insight.122163
8. Guo C, Chi Z, Jiang D, Xu T, Yu W, Wang Z, et al. Cholesterol homeostatic regulator SCAP-SREBP2 integrates NLRP3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity. (2018) 49:842.e–56.e. doi: 10.1016/j.immuni.2018.08.021
9. Quinn W, Wan M, Shewale S, Gelfer R, Rader D, Birnbaum M, et al. mTORC1 stimulates phosphatidylcholine synthesis to promote triglyceride secretion. J Clin Invest. (2017) 127:4207–15. doi: 10.1172/JCI96036
10. Yi J, Zhu J, Wu J, Thompson C, Jiang X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci. (2020) 117:31189–97. doi: 10.1073/pnas.2017152117
11. Meraj R, Wikenheiser-Brokamp K, Young L, McCormack F. Lymphangioleiomyomatosis: new concepts in pathogenesis, diagnosis, and treatment. Semin Respir Crit Care Med. (2012) 33:486–97. doi: 10.1055/s-0032-1325159
12. Volpi A, Sala G, Lesma E, Labriola F, Righetti M, Alfano R, et al. Tuberous sclerosis complex: new insights into clinical and therapeutic approach. J Nephrol. (2019) 32:355–63. doi: 10.1007/s40620-018-0547-6
13. McCarthy C, Gupta N, Johnson S, Yu J, McCormack F. Lymphangioleiomyomatosis: pathogenesis, clinical features, diagnosis, and management. Lancet Respir Med. (2021) 9:1313–27. doi: 10.1016/S2213-2600(21)00228-9
14. Kennedy B, Lamming D. The mechanistic target of rapamycin: the grand conductor of metabolism and aging. Cell Metab. (2016) 23:990–1003. doi: 10.1016/j.cmet.2016.05.009
15. Bottolo L, Miller S, Johnson S. Sphingolipid, fatty acid and phospholipid metabolites are associated with disease severity and mTOR inhibition in lymphangioleiomyomatosis. Thorax. (2020) 75:679–88. doi: 10.1136/thoraxjnl-2019-214241
16. Sohrabi Y, Reinecke H, Godfrey R. Altered cholesterol and lipid synthesis mediates hyperinflammation in COVID-19. Trends Endocrinol Metab. (2021) 32:132–4. doi: 10.1016/j.tem.2021.01.001
17. Yuan S, Chu H, Chan J, Ye Z, Wen L, Yan B, et al. SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat Commun. (2019) 10:120. doi: 10.1038/s41467-018-08015-x
18. Lugg S, Scott A, Parekh D, Naidu B, Thickett D. Cigarette smoke exposure and alveolar macrophages: mechanisms for lung disease. Thorax. (2022) 77:94–101. doi: 10.1136/thoraxjnl-2020-216296
19. Chakinala R, Khatri A, Gupta K, Koike K, Epelbaum O. Sphingolipids in COPD. Eur Respir Rev. (2019) 28:190047.
20. Suryadevara V, Ramchandran R, Kamp D, Natarajan V. Lipid mediators regulate pulmonary fibrosis: potential mechanisms and signaling pathways. Int J Mol Sci. (2020) 21:4257. doi: 10.3390/ijms21124257
21. Letsiou E, Htwe Y, Dudek S. Secretory phospholipase A2 enzymes in acute lung injury. Cell Biochem Biophys. (2021) 79:609–17. doi: 10.1007/s12013-021-01003-x
22. Zheng Y, Ning P, Luo Q, He Y, Yu X, Liu X, et al. Inflammatory responses relate to distinct bronchoalveolar lavage lipidome in community-acquired pneumonia patients: a pilot study. Respir Res. (2019) 20:82. doi: 10.1186/s12931-019-1028-8
24. Zulueta A, Peli V, Dei Cas M, Colombo M, Paroni R, Falleni M, et al. Inflammatory role of extracellular sphingolipids in cystic fibrosis. Int J Biochem Cell Biol. (2019) 116:105622. doi: 10.1016/j.biocel.2019.105622
25. Teichgräber V, Ulrich M, Endlich N, Riethmüller J, Wilker B, de Oliveira–Munding C, et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med. (2008) 14:382–91. doi: 10.1038/nm1748
26. Chiurchiù V, Leuti A, Maccarrone M. Bioactive lipids and chronic inflammation: managing the fire within. Front Immunol. (2018) 9:38. doi: 10.3389/fimmu.2018.00038
27. Mingione A, Dei Cas M, Bonezzi F, Caretti A, Piccoli M, Anastasia L, et al. Inhibition of sphingolipid synthesis as a phenotype-modifying therapy in cystic fibrosis. Cell Physiol Biochem. (2020) 54:110–25. doi: 10.33594/000000208
28. di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. (2021) 22:75–95. doi: 10.1038/s41580-020-00314-w
29. Barnes P, Baker J, Donnelly L. Cellular senescence as a mechanism and target in chronic lung diseases. Am J Respir Crit Care Med. (2019) 200:556–64. doi: 10.1164/rccm.201810-1975TR
30. Summer R, Shaghaghi H, Schriner D, Roque W, Sales D, Cuevas-Mora K, et al. Activation of the mTORC1/PGC-1 axis promotes mitochondrial biogenesis and induces cellular senescence in the lung epithelium. Am J Physiol Lung Cell Mol Physiol. (2019) 316:L1049–60. doi: 10.1152/ajplung.00244.2018
31. Saito M, Mitani A, Ishimori T, Miyashita N, Isago H, Mikami Y, et al. Active mTOR in lung epithelium promotes epithelial–mesenchymal transition and enhances lung fibrosis. Am J Respir Cell Mol Biol. (2020) 62:699–708. doi: 10.1165/rcmb.2019-0255OC
32. Schindler V, Alhamdan F, Preußer C, Hintz L, Alashkar Alhamwe B, Nist A, et al. Side-directed release of differential extracellular vesicle-associated microRNA profiles from bronchial epithelial cells of healthy and asthmatic subjects. Biomedicines. (2022) 10:622. doi: 10.3390/biomedicines10030622
33. Iglesias-Bartolome R, Patel V, Cotrim A, Leelahavanichkul K, Molinolo A, Mitchell J, et al. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. (2012) 11:401–14. doi: 10.1016/j.stem.2012.06.007
34. Flor A, Wolfgeher D, Wu D, Kron SJ. A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence. Cell Death Discov. (2017) 3:17075. doi: 10.1038/cddiscovery.2017.75
35. Hamsanathan S, Gurkar A. Lipids as regulators of cellular senescence. Front Physiol. (2022) 13:796850. doi: 10.3389/fphys.2022.796850
36. Dagouassat M, Gagliolo J, Chrusciel S, Bourin M, Duprez C, Caramelle P, et al. The cyclooxygenase-2–prostaglandin E 2 pathway maintains senescence of chronic obstructive pulmonary disease fibroblasts. Am J Respir Crit Care Med. (2013) 187:703–14. doi: 10.1164/rccm.201208-1361OC
37. Ruan Y, Zhou W, Chan H. Regulation of smooth muscle contraction by the epithelium: role of prostaglandins. Physiology. (2011) 26:156–70. doi: 10.1152/physiol.00036.2010
38. Wiley C, Brumwell A, Davis S, Jackson J, Valdovinos A, Calhoun C, et al. Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight. (2019) 4:e130056. doi: 10.1172/jci.insight.130056
39. Johnson S, Taveira-DaSilva A, Moss J. Lymphangioleiomyomatosis. Clin Chest Med. (2016) 37:389–403. doi: 10.1016/j.ccm.2016.04.002
40. Short J, Houston K, Dere R, Cai S, Kim J, Johnson C, et al. AMP-Activated protein kinase signaling results in cytoplasmic sequestration of p27. Cancer Res. (2008) 68:6496–506. doi: 10.1158/0008-5472.CAN-07-5756
41. Lacher M, Pincheira R, Zhu Z, Camoretti-Mercado B, Matli M, Warren R, et al. Rheb activates AMPK and reduces p27Kip1 levels in Tsc2-null cells via mTORC1-independent mechanisms: implications for cell proliferation and tumorigenesis. Oncogene. (2010) 29:6543–56. doi: 10.1038/onc.2010.393
42. Ben-Sahra I, Hoxhaj G, Ricoult S, Asara J, Manning B. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. (2016) 351:728–33. doi: 10.1126/science.aad0489
43. Ricoult S, Yecies J, Ben-Sahra I, Manning B. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene. (2016) 35:1250–60. doi: 10.1038/onc.2015.179
44. Clements D, Dongre A, Krymskaya V, Johnson S. Wild type mesenchymal cells contribute to the lung pathology of lymphangioleiomyomatosis. PLoS One. (2015) 10:e0126025. doi: 10.1371/journal.pone.0126025
45. Kumasaka T, Seyama K, Mitani K, Sato T, Souma S, Kondo T, et al. Lymphangiogenesis in lymphangioleiomyomatosis. Am J Surg Pathol. (2004) 28:1007–16. doi: 10.1097/01.pas.0000126859.70814.6d
46. Terraneo S, Lesma E, Ancona S, Imeri G, Palumbo G, Torre O, et al. Exploring the role of matrix metalloproteinases as biomarkers in sporadic lymphangioleiomyomatosis and tuberous sclerosis complex. A pilot study. Front Med. (2021) 8:605909. doi: 10.3389/fmed.2021.605909
47. Dodd K, Yang J, Shen M, Sampson J, Tee A. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene. (2015) 34:2239–50. doi: 10.1038/onc.2014.164
48. Yao J, Taveira-DaSilva A, Jones A, Julien-Williams P, Stylianou M, Moss J. sustained effects of sirolimus on lung function and cystic lung lesions in lymphangioleiomyomatosis. Am J Respir Crit Care Med. (2014) 190:1273–82. doi: 10.1164/rccm.201405-0918OC
49. McCormack F, Travis W, Colby T, Henske E, Moss J. Lymphangioleiomyomatosis. Am J Respir Crit Care Med. (2012) 186:1210–2. doi: 10.1164/rccm.201205-0848OE
50. Bernardelli C, Ancona S, Lazzari M, Lettieri A, Selvaggio P, Massa V, et al. LAM Cells as potential drivers of senescence in lymphangioleiomyomatosis microenvironment. Int J Mol Sci. (2022) 23:7040. doi: 10.3390/ijms23137040
51. Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, et al. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am J Respir Crit Care Med. (2021) 203:707–17. doi: 10.1164/rccm.202004-1274OC
52. Ancona S, Orpianesi E, Bernardelli C, Chiaramonte E, Chiaramonte R, Terraneo S, et al. Differential modulation of matrix metalloproteinases-2 and-7 in lam/tsc cells. Biomedicines. (2021) 9:1760. doi: 10.3390/biomedicines9121760
53. Wang J, Filippakis H, Hougard T, Du H, Ye C, Liu H, et al. Interleukin-6 mediates PSAT1 expression and serine metabolism in TSC2-deficient cells. Proc Natl Acad Sci. (2021) 118:e2101268118. doi: 10.1073/pnas.2101268118
54. Dongre A, Clements D, Fisher A, Johnson S. Cathepsin K in lymphangioleiomyomatosis. Am J Pathol. (2017) 187:1750–62. doi: 10.1016/j.ajpath.2017.04.014
55. Ortiz-Montero P, Londoño-Vallejo A, Vernot J. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal. (2017) 15:17. doi: 10.1186/s12964-017-0172-3
56. Cesta M, Zippoli M, Marsiglia C, Gavioli E, Mantelli F, Allegretti M, et al. The role of interleukin-8 in lung inflammation and injury: implications for the management of COVID-19 and hyperinflammatory acute respiratory distress syndrome. Front Pharmacol. (2022) 12:808797. doi: 10.3389/fphar.2021.808797
57. Walters H, Cox L. mTORC inhibitors as broad-spectrum therapeutics for age-related diseases. Int J Mol Sci. (2018) 19:2325. doi: 10.3390/ijms19082325
58. Dias S, Soares V, Ferreira A, Sacramento C, Fintelman-Rodrigues N, Temerozo J, et al. Lipid droplets fuel SARS-CoV-2 replication and production of inflammatory mediators. PLoS Pathog. (2020) 16:e1009127. doi: 10.1371/journal.ppat.1009127
59. Shimizu H, Ito A, Sakurada K, Nakamura J, Tanaka K, Komatsu M, et al. AK106-001616, a potent and selective inhibitor of cytosolic phospholipase A 2: in vivo efficacy for inflammation, neuropathic pain, and pulmonary fibrosis. J Pharmacol Exp Ther. (2019) 369:511–22. doi: 10.1124/jpet.118.255034
60. Mossmann D, Park S, Hall M. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer. (2018) 18:744–57. doi: 10.1038/s41568-018-0074-8
61. Gilbert E, Eby J, Hammer A, Klarquist J, Christensen D, Barfuss A, et al. Positioning ganglioside D3 as an immunotherapeutic target in lymphangioleiomyomatosis. Am J Pathol. (2013) 183:226–34. doi: 10.1016/j.ajpath.2013.04.002
62. Park J, Wu D, Prendes M, Lu S, Ragupathi G, Schrantz N, et al. Fine specificity of natural killer T cells against GD3 ganglioside and identification of GM3 as an inhibitory natural killer T-cell ligand. Immunology. (2008) 123:145–55. doi: 10.1111/j.1365-2567.2007.02760.x
63. Zhang H, Huang J, Düvel K, Boback B, Wu S, Squillace R, et al. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS One. (2009) 4:e6189. doi: 10.1371/journal.pone.0006189
64. Priolo C, Ricoult S, Khabibullin D, Filippakis H, Yu J, Manning B, et al. Tuberous sclerosis complex 2 loss increases lysophosphatidylcholine synthesis in lymphangioleiomyomatosis. Am J Respir Cell Mol Biol. (2015) 53:33–41. doi: 10.1165/rcmb.2014-0379RC
65. Linkous A, Yazlovitskaya E, Hallahan D. Cytosolic phospholipase A2 and lysophospholipids in tumor angiogenesis. JNCI J Natl Cancer Inst. (2010) 102:1398–412. doi: 10.1093/jnci/djq290
66. Li C, Zhang E, Sun Y, Lee P, Zhan Y, Guo Y, et al. Rapamycin-insensitive up-regulation of adipocyte phospholipase A2 in tuberous sclerosis and lymphangioleiomyomatosis. PLoS One. (2014) 9:e104809. doi: 10.1371/journal.pone.0104809
67. Kovalenko A, Sanin A, Kosmas K, Zhang L, Wang J, Akl E, et al. Therapeutic targeting of DGKA-mediated macropinocytosis leads to phospholipid reprogramming in tuberous sclerosis complex. Cancer Res. (2021) 81:2086–100. doi: 10.1158/0008-5472.CAN-20-2218
68. Boroda S, Niccum M, Raje V, Purow B, Harris T. Dual activities of ritanserin and R59022 as DGKα inhibitors and serotonin receptor antagonists. Biochem Pharmacol. (2017) 123:29–39. doi: 10.1016/j.bcp.2016.10.011
69. Martin K, Zhou W, Bowman M, Shih J, Au K, Dittenhafer-Reed K, et al. The genomic landscape of tuberous sclerosis complex. Nat Commun. (2017) 8:15816. doi: 10.1038/ncomms15816
70. Bernardelli C, Chiaramonte E, Ancona S, Sirchia S, Cerri A, Lesma E. Primary TSC2-/meth cells induce follicular neogenesis in an innovative TSC mouse model. Int J Mol Sci. (2022) 23:9713. doi: 10.3390/ijms23179713
71. Bowler R, Jacobson S, Cruickshank C, Hughes G, Siska C, Ory D, et al. Plasma sphingolipids associated with chronic obstructive pulmonary disease phenotypes. Am J Respir Crit Care Med. (2015) 191:275–84. doi: 10.1164/rccm.201410-1771OC
72. Verwer E, Kavanagh T, Mischler W, Feng Y, Takahashi K, Wang S, et al. [18F]Fluorocholine and [18F]Fluoroacetate PET as imaging biomarkers to assess phosphatidylcholine and mitochondrial metabolism in preclinical models of TSC and LAM. Clin Cancer Res. (2018) 24:5925–38. doi: 10.1158/1078-0432.CCR-17-3693
73. Parkhitko A, Priolo C, Coloff J, Yun J, Wu J, Mizumura K, et al. Autophagy-dependent metabolic reprogramming sensitizes TSC2-deficient cells to the antimetabolite 6-aminonicotinamide. Mol Cancer Res. (2014) 12:48–57. doi: 10.1158/1541-7786.MCR-13-0258-T
74. Tasat D, Yakisich J. Rationale for the use of sphingosine analogues in COVID-19 patients. Clin Med. (2021) 21:e84–7. doi: 10.7861/clinmed.2020-0309
75. Pandolfi R, Barreira B, Moreno E, Lara-Acedo V, Morales-Cano D, Martínez-Ramas A, et al. Role of acid sphingomyelinase and IL-6 as mediators of endotoxin-induced pulmonary vascular dysfunction. Thorax. (2017) 72:460–71. doi: 10.1136/thoraxjnl-2015-208067
76. Huang Z, Liu H, Zhang X, Wen G, Zhu C, Zhao Y, et al. Transcriptomic analysis of lung tissues after hUC-MSCs and FTY720 treatment of lipopolysaccharide-induced acute lung injury in mouse models. Int Immunopharmacol. (2018) 63:26–34. doi: 10.1016/j.intimp.2018.06.036
77. Zhang Z, Li W, Heng Z, Zheng J, Li P, Yuan X, et al. Combination therapy of human umbilical cord mesenchymal stem cells and FTY720 attenuates acute lung injury induced by lipopolysaccharide in a murine model. Oncotarget. (2017) 8:77407–14. doi: 10.18632/oncotarget.20491
78. Caretti A, Bragonzi A, Facchini M, de Fino I, Riva C, Gasco P, et al. Anti-inflammatory action of lipid nanocarrier-delivered myriocin: therapeutic potential in cystic fibrosis. Biochim Biophys Acta Gen Subj. (2014) 1840:586–94. doi: 10.1016/j.bbagen.2013.10.018
79. Becker K, Riethmüller J, Lüth A, Döring G, Kleuser B, Gulbins E. Acid sphingomyelinase inhibitors normalize pulmonary ceramide and inflammation in cystic fibrosis. Am J Respir Cell Mol Biol. (2010) 42:716–24. doi: 10.1165/rcmb.2009-0174OC
80. Dechecchi M, Nicolis E, Mazzi P, Cioffi F, Bezzerri V, Lampronti I, et al. Modulators of sphingolipid metabolism reduce lung inflammation. Am J Respir Cell Mol Biol. (2011) 45:825–33. doi: 10.1165/rcmb.2010-0457OC
81. Kou L, Kou P, Luo G, Wei S. Progress of statin therapy in the treatment of idiopathic pulmonary fibrosis. Oxid Med Cell Longev. (2022) 2022:1–18. doi: 10.1155/2022/6197219
82. Han W, Li J, Tang H, Sun L. Treatment of obese asthma in a mouse model by simvastatin is associated with improving dyslipidemia and decreasing leptin level. Biochem Biophys Res Commun. (2017) 484:396–402. doi: 10.1016/j.bbrc.2017.01.135
83. Kambayashi T, Deshpande D. The role of diacylglycerol kinases in allergic airway disease. Curr Opin Pharmacol. (2020) 51:50–8. doi: 10.1016/j.coph.2020.07.008
84. Karandashova S, Kummarapurugu A, Zheng S, Chalfant C, Voynow J. Neutrophil elastase increases airway ceramide levels via upregulation of serine palmitoyltransferase. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L206–14. doi: 10.1152/ajplung.00322.2017
85. Mroz R, Lisowski P, Tycinska A, Bierla J, Trzeciak P, Minarowski L, et al. Anti-inflammatory effects of atorvastatin treatment in chronic obstructive pulmonary disease. A controlled pilot study. J Physiol Pharmacol. (2015) 66:111–28.
Keywords: chronic lung diseases, inflammation, senescence-mediated inflammation, mTOR, lymphangioleiomyomatosis, lipids
Citation: Bernardelli C, Caretti A and Lesma E (2023) Dysregulated lipid metabolism in lymphangioleiomyomatosis pathogenesis as a paradigm of chronic lung diseases. Front. Med. 10:1124008. doi: 10.3389/fmed.2023.1124008
Received: 14 December 2022; Accepted: 06 January 2023;
Published: 19 January 2023.
Edited by:
Shuibang Wang, National Institutes of Health, United StatesReviewed by:
Stephen C. Land, University of Dundee, United KingdomCopyright © 2023 Bernardelli, Caretti and Lesma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Lesma,  ZWxlbmEubGVzbWFAdW5pbWkuaXQ=
ZWxlbmEubGVzbWFAdW5pbWkuaXQ=