 Rajalakshmy Ayilam Ramachandran†
Rajalakshmy Ayilam Ramachandran† Danielle M. Robertson
Danielle M. Robertson- Department of Ophthalmology, The University of Texas Southwestern Medical Center, Dallas, TX, United States
The cornea is the clear dome that covers the front portion of the globe. The primary functions of the cornea are to promote the refraction of light and to protect the eye from invading pathogens, both of which are essential for the preservation of vision. Homeostasis of each cellular layer of the cornea requires the orchestration of multiple processes, including the ability to respond to stress. One mechanism whereby cells respond to stress is autophagy, or the process of “self-eating.” Autophagy functions to clear damaged proteins and organelles. During nutrient deprivation, amino acids released from protein breakdown via autophagy are used as a fuel source. Mitophagy, a selective form of autophagy, functions to clear damaged mitochondria. Thus, autophagy and mitophagy are important intracellular degradative processes that sustain tissue homeostasis. Importantly, the inhibition or excessive activation of these processes result in deleterious effects on the cell. In the eye, impairment or inhibition of these mechanisms have been associated with corneal disease, degenerations, and dystrophies. This review summarizes the current body of knowledge on autophagy and mitophagy at all layers in the cornea in both non-infectious and infectious corneal disease, dystrophies, and degenerations. It further highlights the critical gaps in our understanding of mitochondrial dysfunction, with implications for novel therapeutics in clinical practice.
1. Introduction
The cornea is a five layered tissue that functions to refract light as it enters the eye and to protect the eye from invasion by pathogens (1). Of the five layers, Bowman’s layer is a modified basement membrane just beneath the corneal epithelium, while Descemet’s membrane is posterior to the stroma, functioning as a basement membrane for the corneal endothelium. The anterior-most cell layer of the cornea is composed of a five to seven cell layered stratified epithelium in various stages of differentiation. This includes basal cells that are able to undergo mitosis, wing or intermediate cells, and squamous cells that are contiguous with the conjunctival epithelium, forming the ocular surface. The next cellular layer is the collagenous stroma, the thickest layer of the cornea. The stroma is composed of predominantly type I collagen fibrils. It is the organization of these fibrils that contribute to corneal strength and transparency. The collagen is maintained by keratocytes localized within the corneal stroma. These cells have essential roles in corneal wound healing and fibrosis. Finally, the most posterior cellular layer in the cornea is the endothelium. This monolayer of cells functions to maintain corneal hydration. In humans, the corneal endothelium does not undergo cell division, but instead exhibits gross morphological changes following cell loss.
Autophagy or macroautophagy is a cannibalistic process whereby the cell “eats” misfolded proteins and damaged organelles in order to maintain homeostasis and generate energy for the cell (2). During the initial stages of autophagy, the isolation membrane or phagophore forms around cytoplasmic constituents that are destined for degradation, known as cargo. This double membrane structure continues to elongate until closing, encapsulating the cargo within the mature autophagosome. The autophagosome then docks and undergoes subsequent fusion with the lysosome where the cargo is subject to proteolysis by hydrolases. These raw materials are then recycled into macromolecules that are used in energy homeostasis. The microtubule-associated protein I light chain (LC3-I) is an integral protein involved in autophagy. LC3-I is an autophagosomal protein that is present within the inner and outer membranes. During autophagy, LC3-I undergoes lipidation to LC3-II. This shifts LC3 from the cytoplasm to the autophagosomal membrane during elongation, where it plays a key role in the conjugation of other autophagy proteins.
Mitophagy is a form of selective autophagy that degrades and recycles damaged mitochondria (3). There are multiple pathways through which mitophagy proceeds. In the presence of mitochondrial damage that results in mitochondrial depolarization, presenilins-associated rhomboid-like protein (PARL) activity is impaired, leading to stabilization of the PTEN induced kinase 1 (PINK1) at the outer mitochondrial membrane. This triggers recruitment of the E3 ligase PARKIN, which then ubiquitinates additional mitochondrial membrane proteins, targeting them for proteasomal degradation or the autophagy adapter p62. P62 in turn binds to LC3-II on the autophagosome. In non-PINK1 mediated mitophagy, mitophagy receptors that express LC3-II interacting region (LIR) are able to directly recruit LC3-II labeled autophagosomes. A schematic of the major autophagy and mitophagy pathways is detailed in Figure 1.
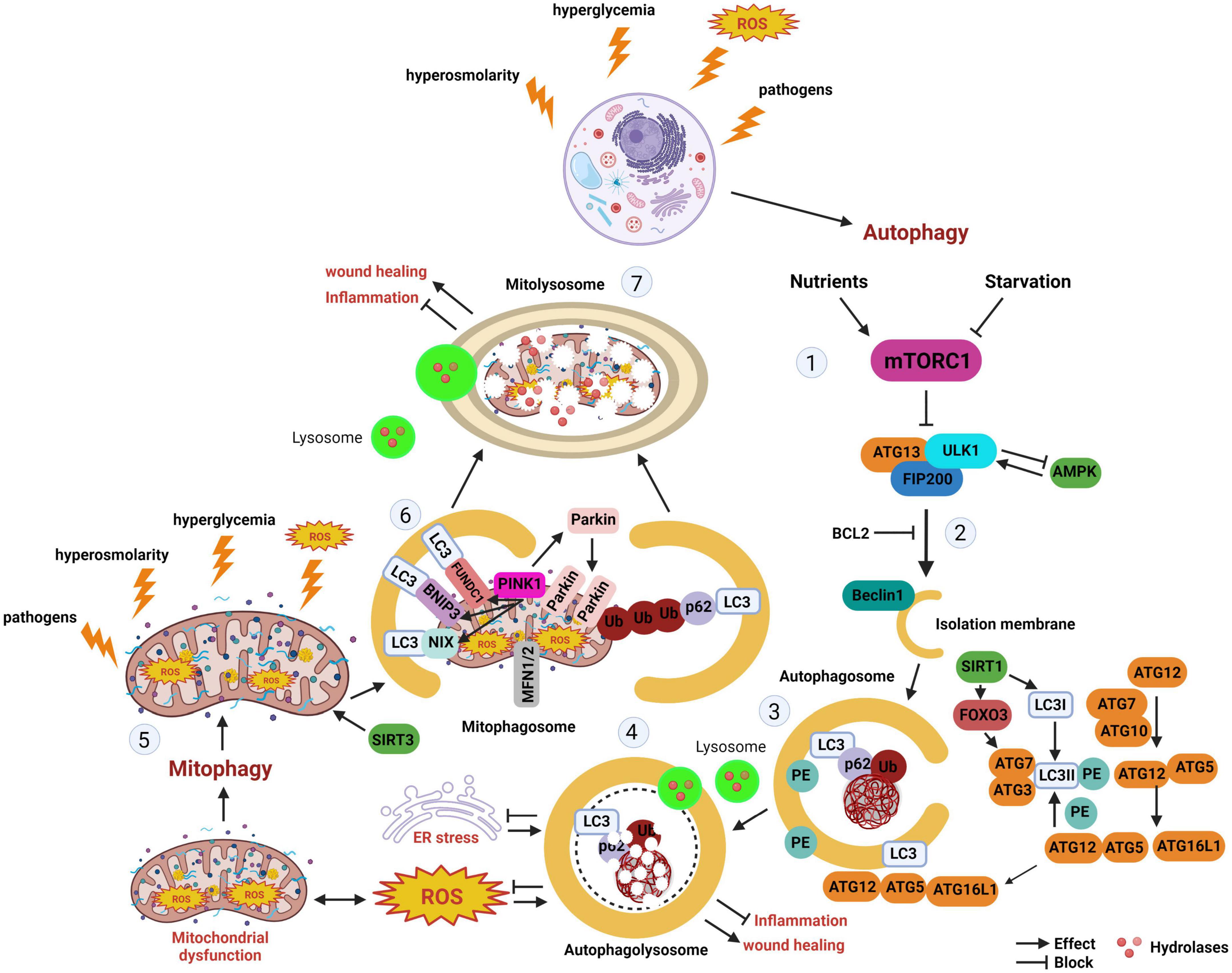
Figure 1. Autophagy and mitophagy. The stages of autophagy and mitophagy, a selective form of autophagy that clears damaged mitochondria. (Stage 1) Autophagy induction. Nutrient starvation and hyperglycemia differentially regulate mTORC1. During nutrient starvation, AMPK activates ULK1 to induce autophagy. In hyperglycemia, AMPK and SIRT1 inhibits autophagy. (Stage 2) Autophagy nucleation. Beclin 1 is liberated from BCL2, initiating autophagosome nucleation. (Stage 3) Autophagy expansion/completion. LC3II is formed from the association of LC3 with PE. SIRT1 interacts with autophagy proteins ATG5, ATG7, and LC3, as well as FOXO3. The autophagosome undergoes fusion with the lysosome. (Stage 4) Autophagy termination. Mature lysosomes are fused forming autophagolysosomes. Cargo undergoes proteolytic degradation by hydrolases. (Stage 5) Hyperglycemia, hypoxia, ROS, and other factors may activate mitophagy. (Stage 6) Depolarized mitochondria promote the recruitment of PINK1 to the mitochondria. PINK1 accumulation at the outer mitochondrial membrane leads to the recruitment of Parkin. As an E-3 ligase, Parkin promotes polyubiquitination of other proteins, including p62, leading to recognition of LC3 and mitophagosome formation. BNIP3, BNIP3L/NIX, and FUNDC1 are mitophagy receptors with LIR that can directly interact with LC3 on the autophagosome in the absence of PINK1 accumulation. (Stage 7) Like autophagy, mitophagosomes fuse with lysosomes, allowing for damaged mitochondria to undergo degradation by lysosomal enzymes. mTORC1, mechanistic target of rapamycin complex 1; ATG, autophagy related protein; ULK1, Unc-51-like kinase 1; FIP200, focal adhesion kinase family interacting protein; AMPK, adenosine monophosphate-activated protein kinase; SIRT1, sirtuin-silent mating type information regulation 2 homolog 1; SIRT3, sirtuin-silent mating type information regulation 2 homolog 3; BCL2, B-cell lymphoma; FOXO3, forkhead box O3; LC3, microtubule-associated protein 1A/1B-light chain 3; Ub, ubiquitin; p62, also known as p62/SQSTM1 (sequestosome 1); PE, phosphatidyl ethanolamine; ER, endoplasmic reticulum; ROS, reactive oxygen species; BNIP3, BCL2/adenovirus E1B interacting protein 3; BNIP3L/NIX, pro-apoptotic protein related to the BH3-only family; MFN1/2, mitofusin 1/2; PINK1, PTEN-induced kinase 1; FUNDC1, FUN14 domain containing 1; ATP, adenosine triphosphate.
While autophagy is a heavily conserved mechanism, mitophagy differs somewhat in mammals. Moreover, the role of autophagy and mitophagy in human disease appears to be cell and context specific. In this review, we summarized the current state of knowledge regarding autophagy and mitophagy in non-infectious and infectious corneal disease. We further highlight the major gaps in knowledge that exist with respect to these pathways in the cornea.
2. Non-infectious corneal diseases, dystrophies, and degenerations
2.1. The diabetic cornea
Diabetes mellitus (DM) is a complex and multifactorial disease characterized by progressive and chronic metabolic impairment due to the loss of insulin production and secretion from beta cells in the pancreas. This results in abnormal insulin signaling and, consequently, drives hyperglycemia and dyslipidemia (4). The prevalence of diabetes is increasing worldwide. Indeed, it has been estimated that by 2045, over 600 million adults will suffer from diabetes (5). Corneal complications from diabetes are common and occur in over two thirds of the diabetic population (6). Unlike diabetic retinopathy, corneal complications are painful in the early stages of disease, with pain levels decreasing in patients with fulminant nerve loss, a condition known as corneal neuropathy (7). The spectrum of diabetic corneal complications includes dry eye disease, corneal erosions, persistent epithelial defects and ulcerations, impaired wound healing, epithelial edema, alterations in limbal stem cells and in severe cases, blindness (Figure 2) (8–10).
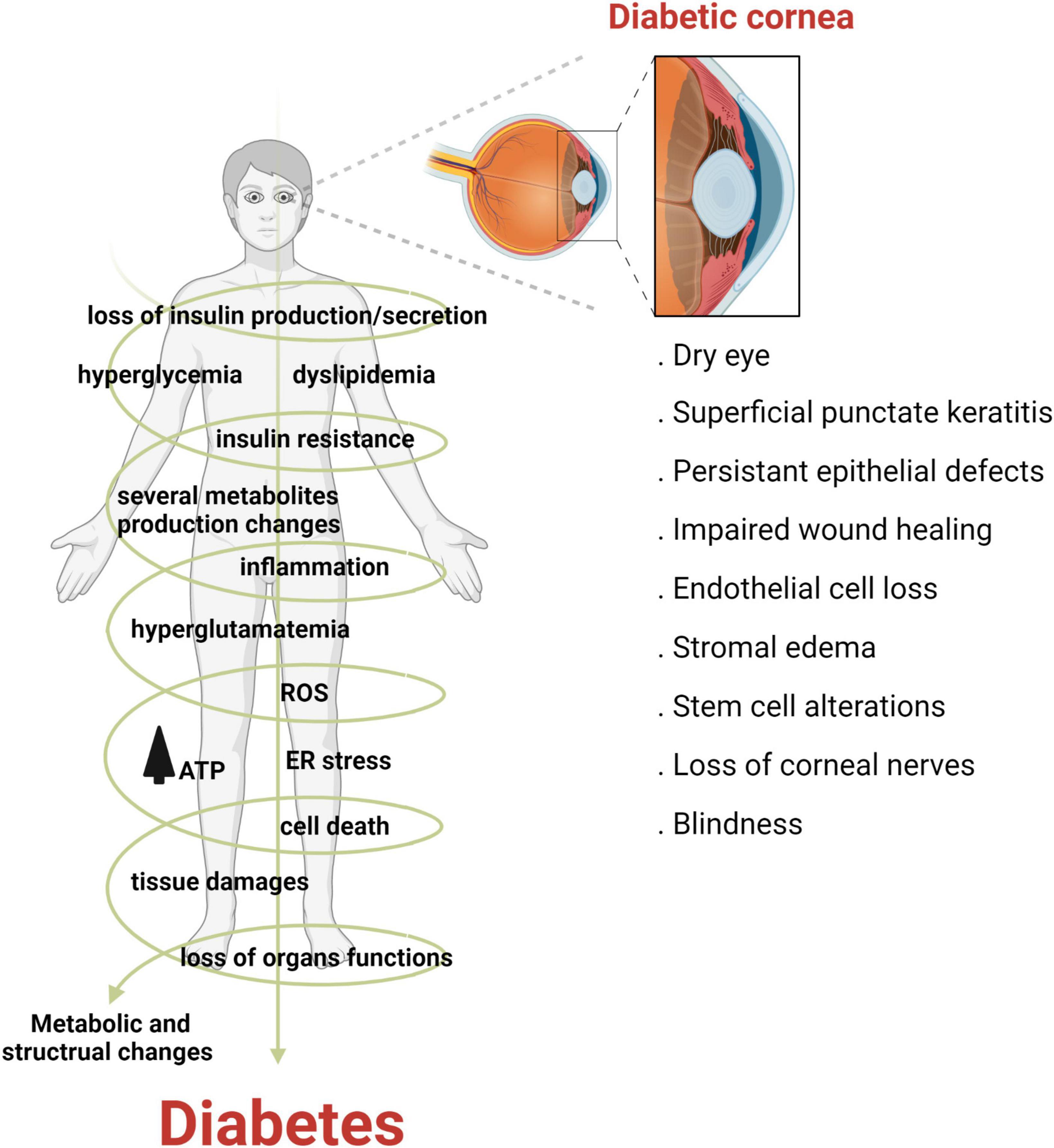
Figure 2. Diabetes adversely effects the cornea. Diabetes is a multifactorial disease characterized by metabolic dysfunction throughout the human body. In the cornea, diabetes can cause a host of complications ranging from dry eye disease to blindness, in severe cases.
As described earlier, autophagy is a vital cellular pathway that involves the recycling of misfolded proteins and damaged organelles to facilitate cell and tissue homeostasis. It is well established that too little or too much autophagy contributes to the development of many diseases. This is due to the cell being unable to recycled damaged components when autophagy is inhibited; whereas, excessive autophagy can lead to autophagic cell death. In the cornea, the dysregulation of canonical macro autophagy and mitophagy have been suggested as potential contributory mechanisms associated with the pathophysiology of diabetes (11–13). However, the relationship between autophagy and mitophagy with corneal complications in diabetes has yet to be fully elucidated.
Recent work evaluated the effects of hyperglycemia on the expression of the nicotinamide adenine dinucleotide (NAD+) dependent deacetylase sirtuin, the silent mating type information regulation 2 homolog 3 (SIRT3), and proteins involved in the forkhead box O-3A (FOXO3A)/PINK1-Parkin axis (14). SIRT3 localizes to the mitochondria and regulates key metabolic functions during stress (15). Using TKE2 mouse corneal epithelial progenitor cells in vitro and Ins2Akita/+ mice in vivo, Hu et al. reported a considerable decrease in SIRT3 expression, as well as LC3B, after culture in high glucose and in the diabetic cornea, suggesting the suppression of mitophagy in diabetes (14). They then used an adenoviral vector to over-express SIRT3, which led to an increase in LC3B in vitro and in vivo and augmented the FOXO3a/PINK1-Parkin pathway. This enhanced corneal epithelial cell migration in culture and promoted epithelial wound healing in the diabetic mouse cornea.
SIRT1 is a NAD-dependent deacetylase with pivotal roles in fat and glucose metabolism (16, 17). SIRT1 has been shown to modulate insulin sensitivity and apoptosis through the deacetylation of p53 (18, 19). This post-translational modification of p53 exerts a protective role in corneal wound healing via the insulin-like growth factor (IGF)-1/AKT pathway. The IGF pathway is dysregulated in the diabetic cornea due in part to an increase in expression of the IGF binding protein (BP)-3 in tear fluid (20–23). Wang et al. showed that hyperglycemia reduced SIRT1 expression in corneal epithelial cells in culture and in the Ins2Akita/+ mouse model. The decrease in SIRT1 expression was associated with increased p53 acetylation and suppressed AKT signaling in corneal epithelial cells. In contrast, rescue of SIRT1 expression promoted cell migration. This corresponded to a reduction in acetylated p53 and expression levels of IGFBP-3, while AKT signaling was restored. While this study did not directly investigate autophagy, given the established role for IGFBP-3 in the inhibition of autophagy in corneal epithelial cells, we hypothesize that there would be an increase in autophagy in cells expressing high levels of SIRT1 (24). This would be consistent with the relationship between SIRT3 and mitophagy.
Similar to the diabetic corneal epithelium, hyperglycemia has also been shown to downregulate mitophagy in corneal endothelial cells. Using a cell culture model, human corneal endothelial cells (HCECs) grown in hyperglycemic conditions exhibited altered mitochondrial morphology and a decrease in certain mitophagy proteins including LC3B, Parkin, PINK1, and the mitophagy receptor BNIP3L/Nix (25). This was associated with a decrease in Na+/K+ ATPase and zonula occludens-1 (ZO-1). The decrease in the Na+/K+ ATPase was further associated with an increase in corneal thickness due to stromal edema in a streptozotocin-induced type 1 diabetic mouse model. Exposing HCECs to hyperglycemic conditions while co-treating with carbonyl cyanide m-chlorophenyl hydrazine (CCCP), a compound that robustly depolarizes mitochondria thereby inducing PINK-mediated mitophagy, increased LC3B and PINK1 expression and decreased reactive oxygen species (ROS) production. The upregulation of mitophagy was also associated with increased levels of Na+/K+ ATPase and ZO-1. In vivo, treatment with CCCP restored corneal thickness values to control levels.
While insulin has not been studied as a mediator of autophagy or mitophagy in the corneal epithelium in the setting of hyperglycemia, insulin has been shown to inhibit mitophagy in corneal epithelial cells (26). Titone et al. used telomerase-immortalized human corneal epithelial (hTCEpi) cells and primary cultured human corneal epithelial cells to investigate the effects of insulin on metabolic activity and mitochondrial quality control in cells exposed to growth factor withdrawal (26). They reported that insulin blocks PINK-mediated mitophagy by preventing mitochondrial depolarization during prolonged stress. This occurred through activation of the insulin receptor. Surprisingly, however, the authors reported on the translocation of insulin receptor, insulin-like growth factor type receptor, and epidermal growth factor receptor to the mitochondria. The ability of these mitochondrial-localized receptors to stabilize the mitochondria and mediate mitophagy remains unknown.
In addition to metabolic changes in the corneal epithelium, chronic hyperglycemia is associated with loss of the corneal subbasal nerve plexus (27–30). Together these changes cause cell and tissue damage (6, 7). Corneal nociceptors are derived from the trigeminal ganglion. Assessment of the trigeminal ganglion in hyperglycemic culture and in STZ-induced type 1 diabetic mice has shown an increase in the microRNA, miR-34c, that paralleled a reduction in the expression of key autophagy proteins, ATG4B and LC3-II (31). The authors further showed that local inhibition of miR-34c via a subconjunctival injection with a miR-34c antagomir, promoted corneal wound healing. Importantly, the downregulation of miR-34c was also associated with an increase in autophagy (31). A subsequent study by this same group showed similar results with miR-181a. Specifically, they found that miR-181a was also highly expressed in STZ-treated diabetic mice and in neurons cultured in high glucose in vitro (32). The upregulation of miR-181 similarly led to a down regulation of autophagy-related proteins ATG5, LC3B-II, and B-cell lymphoma-2 (BCL-2). Again, the inhibition of miR-181a restored autophagy, promoted the expansion of neuronal axons in vitro, and showed evidence of nerve repair in vivo. These findings, summarized in Table 1, suggest that the modulation of autophagy to increase autophagic flux may be useful in the prevention and treatment of diabetic keratopathy.

Table 1. The role of autophagy/mitophagy in corneal diseases, dystrophies, and degenerations.
2.2. Dry eye disease
Dry eye disease (DED) is a multifactorial disease that affects the ocular surface and is characterized by deficient aqueous tear production and/or an increase in tear evaporation. The instability of the precorneal tear film and resultant hyperosmolarity triggers inflammation, leading to tissue damage and neurosensory abnormalities (33). In both corneal epithelial cells exposed to hyperosmolar stress and in animal models of DED, an increase in oxidative stress has been well documented (34). It is well established that oxidative stress activates autophagy in many cell types (35). This is a key mechanism to maintain cell viability and prevent apoptosis. Thus, in the corneal epithelium, autophagy may function to protect against corneal epithelial tissue damage and maintain ocular surface homeostasis (Figure 3).
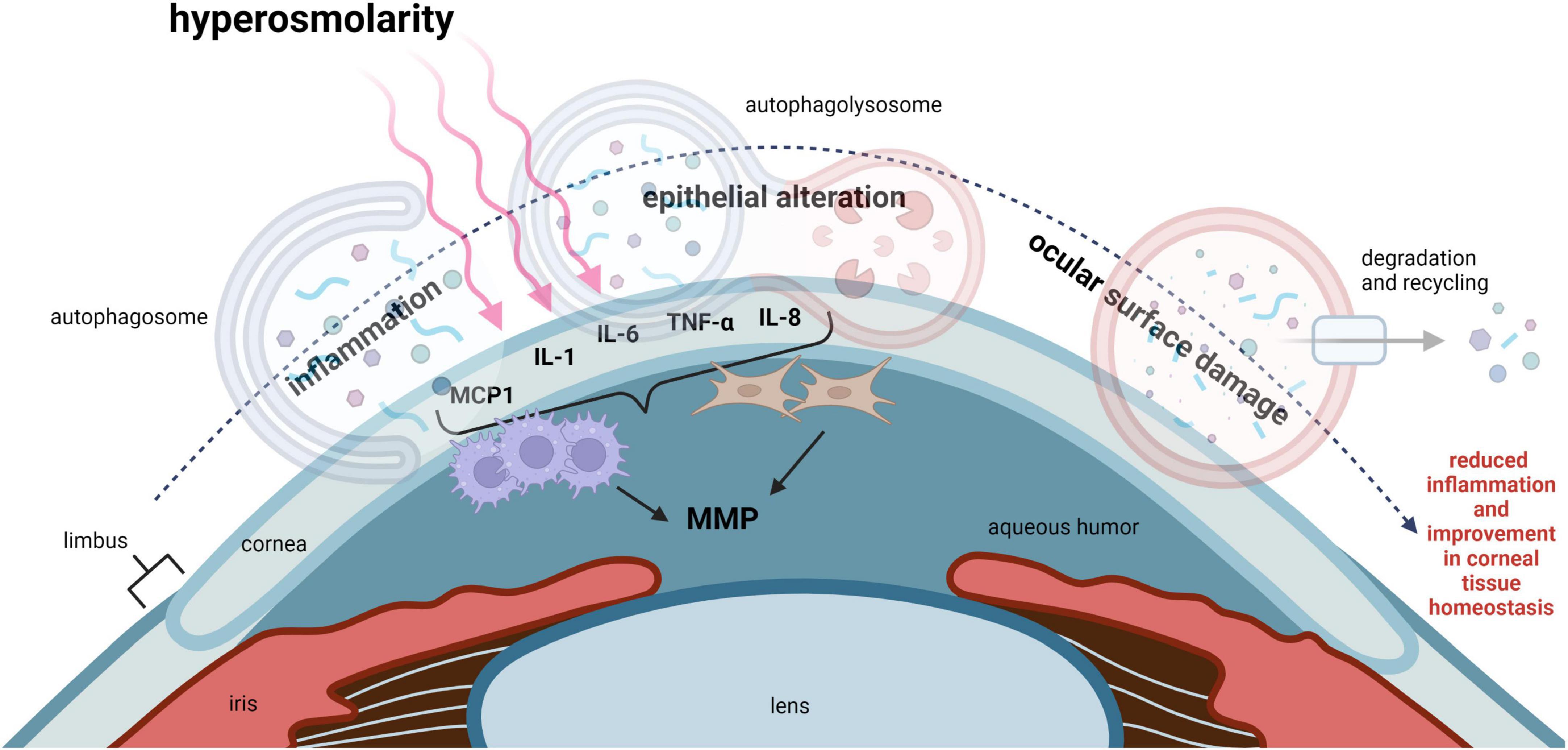
Figure 3. Autophagy regulates inflammation in DED and improves corneal epithelial homeostasis. Hyperosmolarity, desiccating stress and chronic mechanical irritation are common stressors that trigger inflammation in DED. This leads to ocular surface damage. IL-1, IL-6, IL-8, TNF-α, and MCP1, have all been shown to be elevated in DED. These pro-inflammatory cytokines function to recruit leukocytes such as macrophages to the cornea. Inflammation-induced ocular surface damage is associated with activation of MMPs, where MMP3 and MMP9 play an important role in degenerative tissue remodeling. Available evidence suggests that autophagy may act as a key regulator of inflammation and restore corneal epithelial homeostasis in DED. MCP1, monocyte chemoattractant protein-1; IL-1, interleukin 1; IL-6, interleukin 6; IL-8, interleukin 8; TNF-α, tumor necrosis factor alpha; MMP, matrix metalloproteinase.
Several studies have investigated the role of autophagy in DED using the desiccating stress mouse model. In a study by Ma et al., the authors reported increased expression of LC3B in the corneal epithelium after 5, 10, and 20 days of desiccating stress based upon immunofluorescence and an increase in the LC3B II/I ratio by immunoblotting (36). Autophagosome accumulation was also noted. Autophagy was further increased in mouse corneas treated topically with LYN-1604, a molecule known to induce autophagy through activation of the unc-51-like kinase (ULK) and then blocked using 3-methyladenine (3-MA), which inhibits phosphoinositide 3-phosphokinase (PI3K) (36, 37). Interestingly, LYN-1604 appeared to improve tear production in mice with DED and decreased the inflammatory markers tumor necrosis factor-alpha (TNF-α), matrix metalloproteinase (MMP)-3, and MMP-9 (36). In contrast, the inhibition of autophagy worsened all parameters, consistent with greater disease pathology in DED.
Trehalose is another known inducer of autophagy with fewer cytotoxic effects in mammalian cells than other chemical inducers (38). A recent study investigated the effects of trehalose on primary cultured human corneal fibroblasts and SV-40 immortalized corneal epithelial cells exposed to desiccating or inflammatory stress (39). They further examined the effects of trehalose when topically applied to the human cornea in patients with DED. In their cell culture models, neither TNF-α nor desiccating stress induced autophagy. The simultaneous treatment of cells with TNF-α and trehalose increased autophagy, evidenced by an increase in the LC3 II/I ratio, but this did not occur when trehalose was used in conjunction with desiccating stress. While trehalose was effective in the reduction of TNF-α-induced inflammation, it only reduced monocyte chemotactic protein-1 (MCP-1) and had no effect on IL-6, IL-8, and MMP-9 in the desiccating stress model. Similarly, in human studies, the administration of trehalose topically to the eye reduced the ocular surface disease index score, indicating reduced symptomology, and reduced MMP9. Consistent with their desiccating stress findings, IL-6 and IL-8 were unchanged in tears following trehalose treatment for DED (39).
Chloroquine is an antimalarial drug that has been shown to block autophagy by inhibiting lysosomal fusion with the autophagosome (40). In their in vitro desiccating stress model, Shivakumar et al. similarly found that autophagy was upregulated in corneal epithelial cells, evidenced by an increase in LC3B and the lysosomal marker LAMP1 (41). This was associated with an increase in NF-κB activity. Interestingly, rather than activate autophagy, they blocked autophagy in corneal epithelial cells using chloroquine. While this was effective in reducing NF-κB, it had no effect on autophagy induced by desiccating stress.
Liu et al. used primary human corneal epithelial cells (HCECs) to assess the effects of hyperosmolar stress on pro-inflammatory mediators (42). These included TNF-α, IL-1b, IL-6, and IL-8. They found that there was an increase in all four pro-inflammatory cytokines. They also reported an increase in autophagy markers ULK1, ATG5, Beclin 1, and LC3. Paradoxically, treatment of cells exposed to hyperosmolar stress with rapamycin, a known inducer of autophagy, suppressed inflammation and increased cell viability, albeit this latter effect was small in magnitude. The authors concluded that while autophagy is increased in cells exposed to hyperosmolar stress, further activation through rapamycin attenuated inflammation and promoted cell survival, without the induction of autophagic cell death.
Consistent with these prior studies, Wang et al. also showed that LC3B-II was increased in a desiccating stress mouse model of DED (43). They further demonstrated this in vitro using immortalized SV40 human corneal epithelial cells and primary cultured corneal epithelial cells exposed to hyperosmolar stress. Using electron microscopy, their data suggested a potential increase in autophagosome formation. They further showed evidence of accumulation of p62 in both in vitro and in vivo models, while Beclin 1 and ATG5 were unchanged. Similar to the increase in LC3II and p62, there was a parallel increase in the DNA damage-inducible transcript 4 (DDIT4). Previous work has found that the upregulation of DDIT4 is associated with DED and increased ROS production (44). In support of this view, Wang et al. found that siRNA-mediated knockdown of DDIT4 in corneal epithelial cells showed decreased levels of ROS in response to hyperosmolar stress (43). To evaluate whether the DDIT4-ROS axis mediates autophagy during hyperosmolar stress, N-acetylcysteine (NAC) was used. The addition of NAC restored levels of LC3-II and p62 to normal levels in HCECs in vitro. Likewise, in the DED mouse model, NAC exerted a protective effect on the ocular surface. The authors concluded that DDIT4 may function as an endogenous regulator of autophagy and mediate ROS-induced autophagy in DED.
Lacritin is a multifactorial tear glycoprotein that is produced by the lacrimal and meibomian glands, in addition to the corneal and conjunctival epithelium (45). Lacritin is an epithelial mitogen. Consistent with this role, lacritin has been shown to be an important molecule in corneal wound healing (46). A deficiency of lacritin in tear fluid has also been associated with DED (46–49). In a separate study by Wang et al., cells co-treated with inflammatory cytokines and tears from non-DED patients demonstrated cytosolic localization of the transcription factor FOXO3 (45). In contrast to this, treatment of cornea epithelial cells with pro-inflammatory cytokines and tears from patients with DED revealed a nuclear localized FOXO3, suggesting an impairment in corneal epithelial homeostasis in the dry eye condition. Interestingly, co-treatment of cells incubated with tears from dry eyes and lacritin reversed the nuclear localization of FOXO3, sequestering it in the cytosol. The authors went on to show that the lacritin-dependent improvement in corneal epithelial homeostasis appeared to be dependent on the activation of autophagy. This effect was mediated by the acetylation of FOXO3, suggesting that it functions as a novel Atg101 ligand.
Melatonin-loaded polymer polyvinyl caprolactam–polyvinyl acetate–polyethylene glycol graft copolymer micelles (Mel-Mic) have similarly been reported to ameliorate hyperosmolarity-induced ocular surface damage by augmenting autophagic flux and mitophagy via PINK1 (50). In this study, treatment of corneal epithelial cells subject to hyperosmolar stress with melatonin or melatonin-loaded micelles (Mel-Mic) led to an enhancement in LC3B and Beclin 1 expression, while p62 levels were decreased (50). When they assessed mitophagy, they found that expression of both PINK1 and Parkin were increased by treatment with Mel-Mic, while translocase of outer membrane 20 (TOM20) was downregulated. Similarly, the topical administration of Mel-Mic in mice with DED attenuated corneal epithelial cell surface damage and restored tear production. There was also an increase in the number of conjunctival goblet cells. Similar to their in vitro findings, LC3B was increased in the cornea of dry eye mice treated with Mel-Mic and there was a corresponding decrease in the expression of TOM20 (50). Autophagosomes were also observed by transmission electron microscopy of corneal epithelial tissue treated with Mel-Mic, along with an increase in the expression of PINK1 and Parkin. Collectively, Mel-Mic administration showed an improvement in autophagic flux and mitophagy and this was associated with the alleviation of ocular surface damage in DED (50).
2.3. Limbal stem cells
Limbal stem cells are essential for the maintenance of the corneal epithelium. Deficiencies in the limbal stem compartment are associated with conjunctivalization of the corneal surface. Several studies have implicated autophagy as a critical mediator of the limbal stem cell compartment (51, 52). Specifically, autophagy has been shown to exert a protective effect on the limbal epithelium (51, 53). In Beclin 1 deficient (+/-) mice, RNA sequencing studies have shown a reduction in genes that drive proliferation. One of the most highly affected genes in this study was the PDZ-binding kinase (PBK). Importantly, siRNA knockdown of PBK in hTCEpi cells triggered cell cycle arrest (54). The authors speculated that the change in expression of PBK likely explains the 50% decrease in stem and early transient amplifying cells in Beclin 1 +/- mice.
H2A histone family member X (H2AX) expression was also much lower in the corneal epithelium of Beclin 1 +/- compared to wild type mice, suggesting the presence of an impaired stress response due to cells not being able to undergo proper DNA damage repair (54). Likewise, the activating transcription factor 3 (ATF3) was upregulated in the limbal stem cell compartment. When hTCEpi cells were interrogated after siRNA knockdown of ATF3, there was a significant increase in cell growth. Thus, increased expression of ATF3 in Beclin 1 +/- mice may attenuate epithelial renewal. This would also be consistent with the reported decrease in corneal epithelial wound healing.
miR-103/107 is a miRNA that is predominantly expressed in the basal cells of the limbal epithelium. A prior study has reported that miR-103/107 regulates genes that function in stem cell maintenance (52). Subsequent work by this same group explored the relationship between miR103/107 and autophagy. Using antagomirs to silence miR-103/107 and bafilomycin to block autophagosomal-lysosomal fusion, they found that late-stage autophagy was impaired. This occurred through an increase in protein kinase C (PKC) activity, phosphorylation of dynamin, and an attenuation in end stage autophagy. Since autophagy has been proposed as a potential regulator of the stem cell niche, this same group went on to further show that blocking autophagy by treating cells with bafilomycin decreased holoclone formation. Consistent with this, corneal epithelial wound healing was impaired in Beclin +/- deficient mice.
As discussed previously, the induction of autophagy has been linked to oxidative stress and ROS production (55). Thus, increased autophagy has been suggested to reduce oxidative stress and mitigate tissue damage (56). Given the crucial role for autophagy in the protection of limbal stem cells, Zhou et al. investigated the effects of H2O2 on murine corneal epithelial progenitor cells (TKE2) compared to mature murine corneal epithelial (MCE) cells (57). Importantly, the authors reported that TKE2 cells treated with H2O2 increased expression of superoxide dismutase (SOD) and glutathione S-transferase P (GSTP) and autophagy, while suppressing levels of ROS. In contrast, the opposite effects were seen in MCE cells. In addition, Beclin 1 was upregulated in TKE2 cells and downregulated in MCE cells. This paralleled changes in the ratio of LC3-II to LC3-I. Together, these data suggest that the TKE2 progenitor cells resist oxidative stress by enhancing autophagy, whereas oxidative stress inhibits autophagy in mature MCE cells.
2.4. Keratoconus
Keratoconus is a progressive ectatic corneal disease where stromal thinning leads to the formation of a cone due to the abnormal protrusion of the weakened cornea. This results in irregular astigmatism and visual compromise. Many of these patients require surgical intervention at some point in the disease process, whether it be corneal cross-linking to prevent further thinning or penetrating keratoplasty to restore vision. Increased levels of oxidative stress have been reported in keratoconic corneas. Due to the relationship between oxidative stress and autophagy, several studies have attempted to link the increase in oxidative stress with changes in autophagy in keratoconus.
Yildiz et al. (58) investigated PINK1-mediated mitophagy in human corneal epithelium collected from patients undergoing photorefractive keratectomy or corneal cross-linking for myopia or keratoconus, respectively (59). Evaluation of both mRNA and protein revealed a significant decrease in PINK1 in the epithelium of keratoconic patients. There were no changes in expression levels of ATG5, LC3B, or Beclin 1. In contrast to this work, Iqbal et al. (60) investigated LC3 expression using immunohistochemistry in 13 keratoconic corneas compared to controls and reported higher expression levels in three of the keratoconus samples (59). Finally, Shetty et al. (61) investigated the expression of autophagy markers in corneal epithelial tissue from 78 patients with keratoconus that were undergoing corneal cross-linking (62). They reported differential expression of LC3 and p62 in keratoconic tissue compared to controls, with a clear decrease in LC3 expression at all grades of severity. Collectively, these findings are somewhat equivocal and highlight the need for further studies to investigate the role of autophagy and mitophagy in keratoconus.
2.5. Fuchs endothelial corneal dystrophy
Fuchs endothelial corneal dystrophy (FECD) is one of the most common corneal endothelial dystrophies worldwide. FECD is characterized by a bilateral heterogeneous progressive degeneration in corneal endothelial cells (62, 63). The pathophysiology of FECD is marked by the loss of corneal endothelial cells and the presence of extracellular matrix excrescences in Descemet’s membrane, known as guttae. Guttae are encircled by cells forming rosette structures. This leads to endothelial dysfunction, corneal edema, and vision loss (64). Changes in the intracellular environment of corneal endothelial cells in FECD are associated with macromolecular alterations and senescence, where cellular stress stimulates ROS production, an imbalance in the oxidant-antioxidant response, and mitochondrial damage (Figure 4) (65–67). Given the abundance of mitochondria within the corneal endothelium, it is not surprising that mitochondrial dysfunction plays a central role in FECD (68).
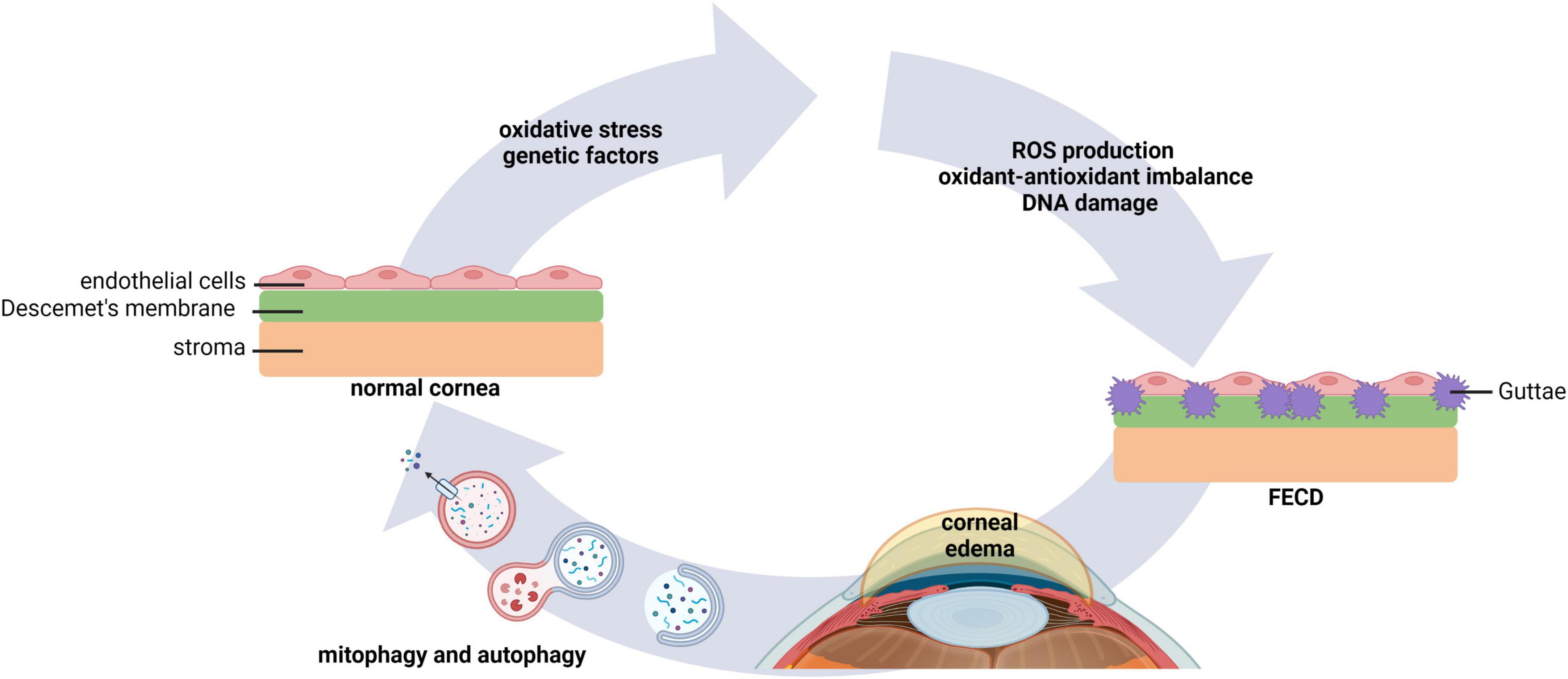
Figure 4. Autophagy and mitophagy play key roles in FECD. Environmental factors that lead to oxidative stress, as well as genetic factors, underlie morphological changes in corneal endothelial cells. This leads to an increase in ROS production, oxidant-antioxidant impairments, mitochondrial fragmentation, and DNA damage. Extracellular matrix excrescences form in Descemet’s membrane. Dysregulated pump function in the compromised corneal endothelium causes stromal edema and vision loss. Evidence suggests that modulation of mitophagy and autophagy may improve corneal homeostasis in FECD. FECD, Fuchs endothelial corneal dystrophy.
Mitochondrial dynamics, fusion and fission, are associated with mitochondrial quality control in response to damaged mitochondria (65). Under conditions of cellular stress, unsalvageable mitochondria undergo fission and are then targeted for degradation by mitophagy. It is well-established that the loss of mitochondrial membrane potential is a key trigger for PINK/Parkin-mediated mitophagy (65, 69). One of the ubiquitination targets for Parkin is the fusion protein mitofusin 2 (MFN2). The degradation of MFN2 in turn promotes mitochondrial fragmentation, allowing for the removal of damaged mitochondria (65). Indeed, in corneal tissue collected from patients with FECD, there is a reduction in MFN2 expression (70, 71). This decrease in MFN2 is associated with a fission dominant morphology and mitochondrial fragmentation. This was further associated with a decrease in mitochondrial mass, and the activation of fission-induced mitophagy, evidenced by an increase in levels of LC3-II and the lysosome associated membrane protein 1 (LAMP1).
Based upon these findings, a subsequent study also evaluated mitochondrial changes and mitophagy in post-surgical FECD tissues compared to normal corneas (72). Here, the authors demonstrated excessive mitochondrial clearance through mitophagy in FECD. The association of PINK1, phosphorylated Parkin and autophagosome formation were shown in conjunction with down-regulation of dynamin related protein 1 (DRP1) (72). While DRP1 is another key protein that regulates mitochondrial fission, DRP1 is not necessarily required for mitophagy (73, 74). To complement their ex vivo tissue studies, the authors used menadione (2-methyl-1,4-naphthoquinone) to induce robust oxidative damage of nuclear and mitochondrial DNA in cultured human corneal endothelial cells (HCEnC)-21T cells. Importantly, Parkin-dependent mitophagy was found to be a key regulator in this system (72). Moreover, the ROS-dependent translocation of YFP-Parkin was observed in fragmented mitochondria, together with upregulation of phosphorylated Parkin, suggesting the induction of mitophagy in a PINK1-independent manner. Collectively, these data support that increased oxidative stress in corneal endothelial cells mediates mitophagy through Parkin-mediated mitochondrial fragmentation in FECD, while DRP1 and PINK1 are degraded during the process of degenerative cell loss in the corneal endothelium.
In addition to mitophagy, the role of autophagy in FECD has also been investigated. Since one of the known pathophysiological changes in FECD is the abnormal accumulation of collagen type VIII (COL8), investigators have been using two novel mouse models that over-express collagen 8, Col8a2L450W/L450W and Col8a2Q455K/Q455K. In both models, there is an upregulation of LC3 and autophagy proteins ATG12-5 (75). This was associated with autophagosome formation in the 40-week-old mutant endothelium. Analysis of the expression of autophagy-related genes at 40 and 80 weeks showed increased expression of the DNA-damage regulated autophagy modulator (DRAM1) and the transmembrane protein 74 (TMEM74) in the corneal endothelium of Col8a2L450W/L450W mice compared to the wild type control. The increased expression of DRAM1 and TMEM74 at 40 weeks were also observed in the corneal endothelium of Col8a2Q455K/Q455K mice, along with the phosphoinositide-3-kinase regulatory subunit 4 (PIK3R4). At 80 weeks, increased gene expression was observed in the corneal endothelium of Col8a2Q455K/Q455K mice. This included the eukaryotic translation initiation factor 2-alpha kinase 3 (EIF2AK3), the mechanistic target of rapamycin (mTOR), cathepsin S (CTSS), TMEM74, DRAM1, and tumor necrosis factor receptor superfamily member 6 (Fas), all of which were upregulated compared to the wild type control.
The solute carrying family 4 member 11 (SLC4A11) is a voltage regulated cotransporter important in mediating corneal endothelial cell function (76). A recent study evaluated the effect of SLC4A11 on mitochondria and autophagy in congenital hereditary endothelial dystrophy (CHED) (77). In that report, the authors used primary human corneal endothelial cells (pHCEnCs) with and without knockdown of SLC4A11 to compare the effect of reduced expression on the transcriptome, along with Slc4a11–/– mouse corneal endothelial cells (Slc4a11–/– MCEnCs). In both models, there was evidence of dysfunction in ion transport, metabolic alterations and mitochondria. Validation studies confirmed an increase in Ser15 phosphorylation of p53 in SLC4A11 knockdown and null cells compared to controls, indicating post-translational activation of p53 transcriptional activity (77). Similarly, there was an increase in the phosphorylation of AMPKα at Thr172, which mediates Ser15 (Ser18 for mouse) phosphorylation of p53, but no changes were detected in phosphorylation of Ser182 of AMPKβ1. Collectively, this study provided evidence to support that SLC4A11 deficiency leads to corneal endothelial dysfunction through decreased production of ATP, resulting in the activation of adenosine monophosphate-activated protein kinase (AMPK) and it downstream targets p53 and ULK (77).
Given the protective role of lithium in cells exposed to increased levels of oxidative and endoplasmic reticulum (ER) stress, Kim et al. tested the effects of lithium on corneal endothelial cell survival in vitro and in their collagen knock in mouse model, Col8a2Q455K/Q455K (78). Here, the authors show that in cells treated with either thapsigargin, an inducer of ER stress, or H2O2 to induce oxidative stress, lithium increased cell viability and autophagy. In their mouse model, the topical administration of lithium enhanced endothelial cell density (78). Electron microscopy revealed the presence of enlarged autophagosomes containing degraded mitochondria in the endothelium of lithium treated corneas. Lastly, lithium upregulated autophagy related proteins ATG5-12, p62, TMEM74, TMEM166, and TM9SF1. This suggests that lithium helped combat increased cellular stress through the induction of autophagy. Further investigations are needed to determine whether lithium or other autophagy modulating drugs will be beneficial as potential therapeutics for FECD and other endothelial dystrophies.
2.6. Antioxidant defenses
2.6.1. Oxidant-antioxidant responses in the cornea
Oxidative stress plays an essential role in the pathogenesis of many human diseases (79). Oxidative stress occurs when intracellular levels of different free radical species become elevated, leading to cellular damage. ROS is the most studied free radical and is known to play an important role in stress-related ocular conditions (80–82). In efforts to combat oxidative stress, antioxidants are produced. Antioxidants function to prevent, block and/or repair cellular and tissue damage (79). The most common antioxidant molecules in the context of intracellular activity includes superoxide dismutase, which is responsible for the conversion of superoxide anions (O2–) into H2O2 and O2, and catalase and glutathione peroxidase, which convert H2O2 into H2O and O2 (83). Thioredoxin and peroxiredoxin exhibit antioxidant activity via regulation of the dithiol/disulfide balance (84). Lastly, glutathione S-transferase can act through a Se-independent glutathione-peroxidase to reduce lipid hydroperoxides (85). In addition, non-enzymatic antioxidants also function to reduce oxidative stress. Non-enzymatic antioxidants include metal-binding proteins, ascorbic acid, vitamin E, uric acid, and glutathione (86).
The ocular surface is constantly exposed to stressors derived from the air and the environment, including pollutants, microbes, radiation, and desiccation. These stressors induce the production of free radicals, leading to increased oxidative stress-induced damage of the corneal and conjunctival epithelium. Trehalose is a disaccharide that has been shown to function as an antioxidant through the reduction of ROS (87). As we previously described in section “2.2. Dry eye disease,” trehalose is also a known inducer of autophagy (88). Indeed, evidence suggests that trehalose enhances autophagy through AMPK signaling (89, 90). Despite the ability of trehalose to mediate autophagy, its antioxidant activity appears to be autophagy independent and instead is mediated by phosphorylation of the autophagy receptor p62. Here, trehalose-mediated phosphorylation of p62 drives nuclear translocation of the nuclear factor (erythroid-derived 2)-like 2 (NRF2) protein, an integral regulator of antioxidant signaling. Similar to trehalose, melatonin has also been shown to function as both an antioxidant and an inducer of autophagy with beneficial effects on the ocular surface (50, 91–93).
NRF2 signaling and autophagy were also evaluated in murine corneal epithelial cells. In their study, Zhou et al. used both mature and progenitor corneal epithelial cells subject to H2O2-induced oxidative stress (57). They found that murine corneal epithelial progenitor cells exhibited a potent antioxidant phenotype that was mediated by NRF2 (57). This was associated with an increase in autophagic flux. In contrast to this, robust oxidative stress was induced in mature murine corneal epithelial cells and this was associated with an inhibition of autophagy. These findings suggest a potential interplay between cellular redox state and autophagy. However, much work remains to determine how the different antioxidants may interact and/or mediate autophagy in the cornea.
3. Infectious keratitis
3.1. Viral keratitis
Viral keratitis is a major cause of corneal blindness in the United States and western countries (59). The most common virus associated with keratitis and subsequent corneal scarring due to repeat infection is herpes simplex virus (HSV-1 and HSV-2) (94). Other common viruses that infect the cornea include varicella-zoster virus (VZV) or herpes zoster virus, cytomegalovirus (CMV), and Epstein-Barr virus (EBV). In rare cases, rubeola virus has also been linked with viral keratitis (95, 96). Little evidence is present in the literature on the role of the host autophagy response in the pathogenesis of viral keratitis. Past studies have highlighted the importance of autophagy in immune cells however, and a regulatory role for innate immune sensors (97–99).
Autophagy has been shown to function as a mechanism that affords virus survival inside host cells. In a Statens Seruminstitut rabbit corneal (SIRC) epithelial cell model, both HSV-1 and HSV-2 infection were shown to be capable of inducing host cell autophagy (98). This was demonstrated by confirmation of LC3 lipidation and an increase in autophagosomes. The inhibition of autophagy using the autophagy inhibitor bafilomycin decreased levels of the HSV envelope glycoprotein D, reduced viral replication, and promoted corneal epithelial cell apoptosis, suggesting that the induction of autophagy and prevention of apoptosis is important for viral replication. In contrast to this, Ames et al. showed that in the cornea, activation of the host autophagy receptor optineurin (OPTN) aids in the degradation of HSV-1 infectious particles and regulates the adaptive immune response (100). Using the cornea scarification model, they further showed that two thirds of OPTN–/– mice infected with HSV succumbed to death. Likewise, Yakoub et al. found that the induction of autophagy by starvation or pharmacological activation significantly downregulated HSV-1 replication in the corneal epithelium, retinal ganglion cells and embryonic fibroblasts (101).
Both innate and adaptive immune responses are critical to control HSV-1 infection. Of these, sustained activation of pro-inflammatory CD4+ T cells leads to the formation of a sight threatening inflammatory lesion in HSV stromal keratitis (102). Jiang et al. investigated the upstream pathways responsible for mediating CD4+ T cell activation during HSV-1 infection (99). In that study, they used mice with dendritic cells that lack ATG5, resulting in dendritic cells that are unable to undergo autophagy. They found that these mice exhibited a normal innate immune response but impaired Th cell immunity. While the absence of an adaptive immune response had no effect on overall viral load, it did decrease CD4+ T cell activation and pathological corneal inflammation.
The cytosolic DNA sensor signaling pathway, stimulator of interferon genes (STING), has also been shown to be critical in preventing HSV-1 mediated mortality during intravenous infection in mice (103). However, while STING–/– mice inoculated with HSV via the eye demonstrated worsening periocular disease and higher viral titers compared to the wild type control, they did not experience the same increase in mortality that was evident during intravenous infection. They concluded that STING is important for mediating viral loads in the cornea, but not in mortality when inoculated via the cornea. The authors further used a low virulence strain of HSV that did not antagonize host cell autophagy. In mice with an active STING response, the mutant HSV strain failed to induce death. In contrast to this, in STING–/– mice, the effect of the mutant strain on animal death was identical to that induced by a second mutant that did not antagonize host cell mitophagy. Thus, the authors concluded that activation of STING-dependent autophagy is an essential antiviral response in the central nervous system (97).
While autophagy has been shown to play a role in mediating HSV infection, it is unclear whether mitophagy is involved. In non-ocular tissue, prior work has shown that both HSV-1 and HSV-2 infection in keratinocytes promote mitochondrial dysfunction (104). Using a dermal keratinocyte cell line (HaCaT cells), changes in mitochondrial localization and morphology have been observed during the initial stages of HSV infection (104). In this work it was shown that infection triggered an increase in mitochondrial fission. This led to robust mitochondrial fragmentation and was associated with a shift in mitochondria toward a perinuclear localization. Similar to this, HSV-1 and HSV-2 infection in primary neurons in vitro also increased mitochondrial fission and decreased mitochondrial membrane potential (105). This was associated with an increase in ROS production. While an increase in mitochondrial fission and loss of mitochondrial membrane potential may promote PINK1-mediated mitophagy, no studies to date have established a link between PINK1-mediated mitophagy and HSV infection.
Like HSV, VZV is also an alpha herpesvirus that invades the nervous system. Primary infection in patients seronegative for VZV results in varicella (chickenpox). During this stage, virus particles in skin lesions undergo retrograde migration to invade either the dorsal root or trigeminal ganglia, where it remains latent for years. Reactivation of the virus, usually seen in the immune compromised or the elderly, triggers a localized mid-line respecting vesicular rash on the face known as shingles. In the United States, it is estimated that up to 20% of these cases can involve the eye and adnexa, a condition known as herpes zoster ophthalmicus (HZO) (106).
Several studies have demonstrated a pro-viral role for the induction of autophagy during VZV infection (107–112). This has included work showing an upregulation of autophagy during VZV infection in various models including human fibroblast cell lines, primary cells, keratinocytes, and melanocytes (108–112). In contrast to this, in a fetal lung fibroblast cell line, VZV was shown to induce autophagy, as shown by an increase in LC3 lipidation, but attenuated autophagic flux at later stages of the pathway (113). Amino acid starvation and the chemical activation of autophagy reduced viral titers, but when treated with bafilomycin, the block in autophagy increased viral titers. Taken together, this work suggests that viral induction of autophagy to induce vesicle formation without lysosomal degradation is pro-viral. In a subsequent study, it was shown that bafilomycin also functions during viral infection to block secondary envelopment due to disruption of the virus assembly complex (114). The authors speculate that these effects were due in part to the effects of bafilomycin on the Golgi complex, however, further study is necessary to confirm these findings.
In addition to autophagy, VZV infection has also been shown to alter mitochondrial morphology. Using human fetal lung fibroblasts, the authors reported on the diffusion of the immediate early p63 (IE63) protein into mitochondria during infection (115). Moreover, throughout the duration of the study, mitochondria underwent significant morphological changes, including swelling and fragmentation. Despite a report of mitochondrial abnormalities, no studies to date have evaluated the role of mitophagy in VZV infection. Moreover, all available data on the role of autophagy in VZV has been acquired using non-ocular models (116). For an in depth review on what is known on the relationship between autophagy and VZV infection, the reader is directed to the review by Heinz et al. (116).
Unlike HSV and VZV, CMV is a betaherpesvirus that causes asymptomatic infection in otherwise healthy individuals. In an immune compromised patient, dissemination of CMV occurs by invasion of CD34+ myeloid progenitor cells. Multi-organ involvement can follow as a result of viral shedding to different organs via macrophages and dendritic cells. CMV keratitis is an uncommon but known ocular manifestation causing epithelitis, stromal keratitis, and endotheliitis (59). Of relevance to this review, CMV is known to modulate autophagy in human fibroblasts and to block autophagic flux through the insulin receptor substrate (IRS1) and the tegument protein (TRS1) by degrading autophagosome cargo (117, 118).
During the initial stages of CMV infection of human skin fibroblasts and astrocytoma cells, an increase in mitochondrial biogenesis and oxidative phosphorylation has also been reported (119–121). This is due to an increased energy demand for fatty acids to facilitate formation of the viral membrane. A more recent study has shown that the viral protein pUL13 targets mitochondrial cristae architecture to promote mitochondrial respiration during infection (122). In contrast to this, the immediate early viral protein pUL37 × 1 has been shown to induce mitochondrial fragmentation in human fibroblasts (123–125). Similar to VZV, the effects of CMV on the mitochondria have been described, but the role of mitophagy is unknown. In contrast to this, studies have demonstrated that the induction of autophagy during CMV infection improves the ability of the virus to replicate (126, 127). It remains unknown whether the autophagic response during CMV infection is cell type specific and if these findings are relevant to the eye.
Epstein-Barr virus is a gammaherpesviruses that causes infectious mononucleosis (IM). The worldwide prevalence of EBV is estimated to be 90% (128). Also known as glandular fever, IM causes fever, lymphadenopathy, and pharyngitis. Chronic EBV infection is also responsible for ocular manifestations, including dry eye and infectious keratitis (129). Studies using several lymphoma and embryonic kidney cell lines have shown that EBV highjacks the autophagic degradation pathway to survive intracellularly and promote viral replication (130). This occurs following the shift from latent infection to the induction of lytic replication. This is regulated via the extracellular signal-regulated protein kinase (ERK) 1/2, protein kinase c (PKC) theta-p38 mitogen activated protein kinase (MAPK) axis or c-Jun N-terminal kinase (JNK) 1/2 pathways (131–133). EBV viral proteins including latent membrane protein-1 (LMP-1), LMP-2A and the EBV nuclear protein, EBNA-3C, are known to promote autophagy (134–136). In the absence efficient autophagosome production or the inhibition of autophagy, EBV has also been shown to recruit autophagy proteins ATG8-LC3 to its envelope in the cytosol to promote efficient envelope formation during the lytic cycle (137).
Autophagy does contribute to the anti-EBV immune response by regulating type I interferon (IFN) producing plasmacytoid dendritic cells and ROS producing monocytes (138, 139). Moreover, in lymphoma cells, autophagy is induced during the early stages of EBV infection in attempts to clear the virus, but is later attenuated by the virus during the lytic phase to promote survival (140). This was confirmed by demonstrating that the inhibition of autophagy during either the early or late phase of infection resulted in enhanced viral replication. Despite the body of work investigating the relationship between EBV and autophagy, the role of autophagy in the context of EBV keratitis is unexplored and there are no published studies on the role of mitophagy in EBV infection.
3.2. Bacterial keratitis
Bacterial invasion of the cornea can lead to sight threatening infectious keratitis. The most common risk factors for infectious keratitis include contact lens wear, trauma, and immunosuppression (141–143). In the case of contact lens-related infectious keratitis, Pseudomonas aeruginosa is the most commonly identified pathogen in culture proven cases (144). Other common bacteria include Staphylococcus aureus, Streptococcus pneumoniae, Stenotrophomonas maltophilia, Enterobacteriaceae, Klebsiella spp., Serratia spp., and Proteus spp., among others (145). Using an LC3-GFP reporter, Brothers et al. demonstrated that soluble proteins from several bacteria including S. aureus, Serratia marcescens, Escherichia coli, methicillin sensitive S. aureus (MSSA), and methicillin resistant S. aureus (MRSA) strains were capable of activating autophagy in a human corneal limbal epithelial cell line (146).
In contrast to this, Achromobacter xylosoxidans, Klebsiella pneumoniae, Acinetobacter baumannii, Enterococcus faecalis, S. maltophilia, and Moraxella spp. did not exert the same effect (146). The authors also demonstrated a greater amount of autophagy in MRSA compared to MSSA strains. Collectively, this study highlighted that a diverse array of bacterial pathogens are able to regulate host autophagy. In addition, host response varied with respect to the bacterial strain, with S. marcescens exhibiting the strongest response (146). Another study by the same group showed that S. marcescens mutants that lack function of eepR and gumB genes, both of which regulate multiple heat-stable secondary metabolites, were unable to promote autophagy in HCLE cells (147). Since targets of eepR and gumB include genes involved in the synthesis of prodigiosin (pigD), the authors tested a strain of S. marcescens with a mutation in pigD. Here they found that loss of the pigD gene abrogated autophagy.
In a different corneal epithelial cell line, Monankumar et al. reported on the ability of several different isolates of P. aeruginosa to induce autophagy (148). They found that the virulence signature played a key role. While the Type III secretion system (T3SS) promoted autophagy, T3SS mutants failed to induce autophagy. This was associated with the downregulation of Beclin 1. Treatment of cells with the autophagy inhibitors chloroquine and 3-methyl adenine increased intracellular bacterial load, while the use of Earle’s balanced salt solution to induce nutrient deprivation autophagy or rapamycin significantly reduced bacterial load in T3SS positive strains. Together, these findings demonstrate that autophagy is utilized by the host to help clear intracellular Pseudomonas from corneal epithelial cells during infection.
In non-corneal cell types, Pseudomonas’ type IV secretory system PGAP-1-like effector activates autophagy in HeLa cells via the ER stress response (149). The p21-activated kinase 1(PAK1)/Akt/mTOR pathway is involved in autophagy regulation during Pseudomonas infection in lung epithelium (150). Pseudomonas has also been shown to activate autophagy in alveolar macrophages and mast cells (151, 152). Autophagy has also been shown to prime neutrophils during bacterial sepsis (153). Neutrophils and macrophages are important players in the innate immune response during Pseudomonas keratitis (154). However, the role of immune cell autophagy in the context of ocular bacterial keratitis has not yet been explored. Further studies are required to identify the signaling pathways involved in autophagy regulation during PA keratitis.
Pseudomonas has been shown to induce mitochondrial damage in macrophages (155). Importantly, damaged mitochondrial DNA triggers activation of inflammasomes. This process is downregulated by autophagy (155). Mitophagy has been reported to be protective during PA infection by dampening the innate immune response during pathogen exposure. Pyoverdin, an iron-binding siderophore toxin from Pseudomonas alters iron and mitochondrial homeostasis to trigger mitophagy in Caenorhabditis elegans (156). Another study demonstrated the functional role of miRNA miR-302/367 in triggering mitophagy during PA infection. In this work, the upregulation of miR-302/367 promoted mitophagy in macrophages in efforts to clear the pathogen (157). PA’s type III secretion system exoenzyme effector ExoU has also been shown to induce mitochondrial dysfunction in murine macrophages.
Streptococcus pneumoniae keratitis is common among patients with other ocular diseases and following surgery or trauma to the eye (158, 159). In non-ocular systems, S. pneumoniae is known to exploit the host autophagic response for intracellular survival through choline-binding proteins (160). Studies have also identified autophagic regulation of neutrophils and macrophages during S. pneumoniae infection (161–163). At the level of the mitochondria, S. pneumoniae infection induces mitochondrial fission and mtROS production in macrophages (164). Similarly, PINK1 and Parkin-mediated mitophagy have been shown to prevent Streptococcus infection-induced lung damage by downregulating inflammasome activation and promoting apoptosis (165). The accumulation of PINK1 is known to occur in response to mitochondrial depolarization. However, studies using an in vivo mouse model have shown that despite the accumulation of PINK1 during Streptococcus infection, PINK1 was not associated with the outer mitochondrial membrane and therefore did not stimulate mitophagy. Thus, bacterial invasion was associated with frank mitochondrial damage (166).
3.3. Fungal keratitis
Fungal keratitis is a rare disease in western countries, but predominates in tropical and subtropical regions of the world. The prevalence may vary with respect to the regional climate and urbanization; however, Fusarium, Aspergillus, and Candida species are the most common fungal pathogens (167). Fungal keratitis can result in severe corneal ulceration and evisceration or enucleation of the globe. Risk factors for fungal keratitis include ocular trauma, injury, contact lens wear, corticosteroids use, and immunosuppression (168–170). Recent studies have highlighted a role for miRNAs in the regulation of autophagy in Fusarium mediated keratitis (171–173). Using a mouse model of Fusarium solani keratitis, Guo et al. demonstrated changes in the LC3II/LC3I ratio, along with loss of ATG5 compared to control and the accumulation of p62, suggesting an impairment of autophagic flux (172). This correlated with increased expression of miRNA miR-665-3P. Subsequent in vitro and in vivo blocking experiments using an antagomir targeting miRNA miR-665-3P were associated with an increase in autophagic flux. This increase corresponded to a decrease in corneal inflammation.
Work by this same group has also shown a regulatory role for miRNA miR-129-5p in negatively regulating autophagy in F. solani infected mouse corneal stromal cells (171). Here they demonstrated that subconjunctival injection of a miR-129-5p antagomir increased autophagy protein expression for LC3B and Beclin 1. They further showed that ATG14 was a direct target for miR-129-5p (171). In a third study, the same group again found impaired autophagic flux followed by upregulated miR-223-3p expression in their mouse model of F. solani infection (173). Here they found that miR-223-3p was found to negatively regulate the autophagy protein ATG16L1. Similar to their prior work, the inhibition of miR-223-3p increased autophagy while decreasing inflammation to promote healing.
In contrast to F. solani, Aspergillus fumigatus was found to induce autophagosome formation in both human and mouse corneas (174). Treatment with autophagy inhibitors further aggravated disease severity, while the induction of autophagy decreased severity. This occurred through the regulation of polymorphonuclear cell recruitment and differentiation and balancing of the pro-inflammatory to anti-inflammatory cytokine ratio (174). A subsequent study investigating A. fumigatus also found that infection promotes autophagic flux in corneal epithelial cells in vitro and in the corneal epithelium in vivo. They further showed that the induction of autophagy is via the cGAS-STING innate immune pathway (175). In vivo studies with A. fumigatus sensitive C57BL/6 and resistant BALB/c mice have further shown that expression of the autophagy markers LAMP-1, Beclin 1, and LC3 correlated with disease severity (176). The non-canonical autophagy protein, cell cycle proliferation gene 1 (CCPG1), has also recently been implicated in the pathogenesis of A. fumigatus infection (177).
Reports on Candida albicans infection have yielded differing results arguing in support or against the role of autophagy in the host response to non-ocular infection. Using autophagy defective ATG7–/– mice, there were no differences reported in the susceptibility to fungal infection (178). Modulating autophagy had no effect on immune cell function and nucleotide polymorphisms in autophagy genes failed to correlate with cytokine production in Candidemia patients or healthy controls. However, more recent studies demonstrate the positive regulation of autophagy in the pathogenesis of C. albicans (179–182). The fungal pattern recognition receptor Dectin-1 has been shown to mediate LC3 recruitment, thereby increasing fungicidal activity in macrophages during C. albicans infection (179). In vaginal epithelium, autophagy has been shown to be essential for survival during fungal infection. This conclusion was supported by work showing that ATG5 gene knockdown increased epithelial cell death. Other studies have suggested that autophagy may function as a defense mechanism during vaginal C. albicans infection. Future work evaluating the role of C. albicans in a corneal infection model will help us to better understand the regulatory role of autophagy in host defense. To date, there have been no studies investigating the host mitophagy response during infection with fungal pathogens.
3.4. Protozoan keratitis
Protozoa that are known to cause ocular surface infections include Acanthamoeba (AC), Toxoplasma, and Leishmania (183, 184). While protozoan infections overall are rare, Acanthamoeba is the most common pathogen and is typically associated with contact lens wear. During Acanthamoeba infection, the protozoa is introduced to the eye via the contact lens. Inoculation of the lens occurs from exposure to water. Corneal trauma followed by exposure to contaminated water, soil, or agricultural work can also lead to the development of Acanthamoeba keratitis (183). One of the major barriers to treatment of Acanthamoeba keratitis is the ability of the cyst to resist killing. Autophagy has been shown to play a key role in the encystation of the amoeba, conferring protection to the pathogen (185–188). This includes autophagosomal degradation of mitochondria (189). Indeed, studies have identified several autophagy proteins that are involved in mediating encystment. These include ATG8, ATG8 isoform, ATG3, and ATG16L (185–188). Interestingly, the autophagy inhibitors 3-methyladenine (3MA) and wortmannin have been shown to be effective in reducing encystation (190). Contact lens multipurpose care solutions, which are ineffective against Acanthamoeba cysts, combined with the autophagy inhibitors 3-methyladenine or chloroquine have been shown to reduce the adhesion rate of Acanthamoeba to contact lens surfaces (191, 192). Together, these studies provide strong evidence that autophagy is a critical player in the pathogenesis of Acanthamoeba keratitis.
Toxoplasma gondii is a known etiological agent for retinitis. In some cases, T. gondii is also associated with keratitis. In contrast to Acanthamoeba, autophagy functions to clear the pathogen, making it an important component in the host defense against T. gondii ocular infection (193, 194). During infection, T. gondii forms special parasitophorous vacuoles that allow them to evade the host immune system and safely replicate intracellularly. This is associated with increased superoxide levels and robust mitochondrial dysfunction (195). The adaptive immune system has been shown to play a key role in the induction of autophagy and the subsequent degradation of these vacuoles. The CD40–CD154 ligand interaction is one mechanism whereby the infected cell upregulates Beclin 1, leading to autophagocytic removal of the pathogen (193). T. gondii however, has developed mechanisms for inhibiting autophagy and lysosomal degradation. In a mouse model of ocular Toxoplasmosis infection, the pathogen has been shown to negatively regulate autophagy by inducing prolonged epidermal growth factor (EGFR) signaling by releasing micronemal proteins with EGF domains (194). This leads to activation of PKCa/b. The use of tyrosine kinase inhibitors was further shown to block EGFR signaling, leading to Beclin 1-mediated autophagic killing. This was confirmed in a separate study that also showed that the inhibition of EGFR signaling or infection by parasites that did not express a key micronemal protein led to an increase in parasite killing through autophagosome/lysosome fusion (196).
Leishmaniasis is a tropical disease that is transmitted by sand flies after consumption of a blood meal (197). Ocular manifestations of Leishmaniasis, known as Kala-Azar, mainly affects the retina (198). In cutaneous Leishmaniasis caused by bite near the eyelid, blepharoconjunctivitis, dacryocystitis, and even keratitis can develop (199, 200). In the case of corneal disease, if not adequately treated, infection can lead to perforation. Several in vitro and in vivo studies have shown that autophagy is important in the pathogenesis of Leishmania infection (201). In chronic kidney disease, the accumulation of autophagy proteins Atg7 and LC3 has been positively correlated with Leishmania infantum disease severity suggesting that host autophagy favors pathogen survival (202). In vitro studies of Leishmania amazonensis have shown that the induction of autophagy in macrophages is associated with a high infection index in vitro (203). Similarly, the induction of autophagy in Balb/c mice has been shown to increase the number of infected macrophages and the overall parasite burden in vivo (204).
Infection of human bone marrow by the Leishmania donovani complex has also been shown to induce autophagy (205). This effect was abrogated by treatment with amphotericin B. In human monocyte-derived macrophages, the induction of autophagy using rapamycin decreased T cell proliferation, increasing parasite survival (206). In a similar study, intravenous infection of Balb/c mice with L. donovani led to the production of CD4+ and CD8+ T cells that expressed the programmed death ligand-1 (PDL-1). The inhibition of PDL-1 blocked autophagy. This enhanced T-cell protective responses and led to a reduction in the parasitic load (59). Finally, infection of bone marrow derived macrophages obtained from Balb/c mice has shown distinct RNA and protein changes during early and late stages of infection (207). This data contradicted other work by showing that autophagy induced by Leishmania promastigotes was capable of clearing the parasite. This was confirmed by knocking down expression of either Atg5 and ubiquitin, both of which blunted autophagy while increasing the overall parasitic load. A summary of the organisms and their associated autophagic pathways are detailed in Figure 5.
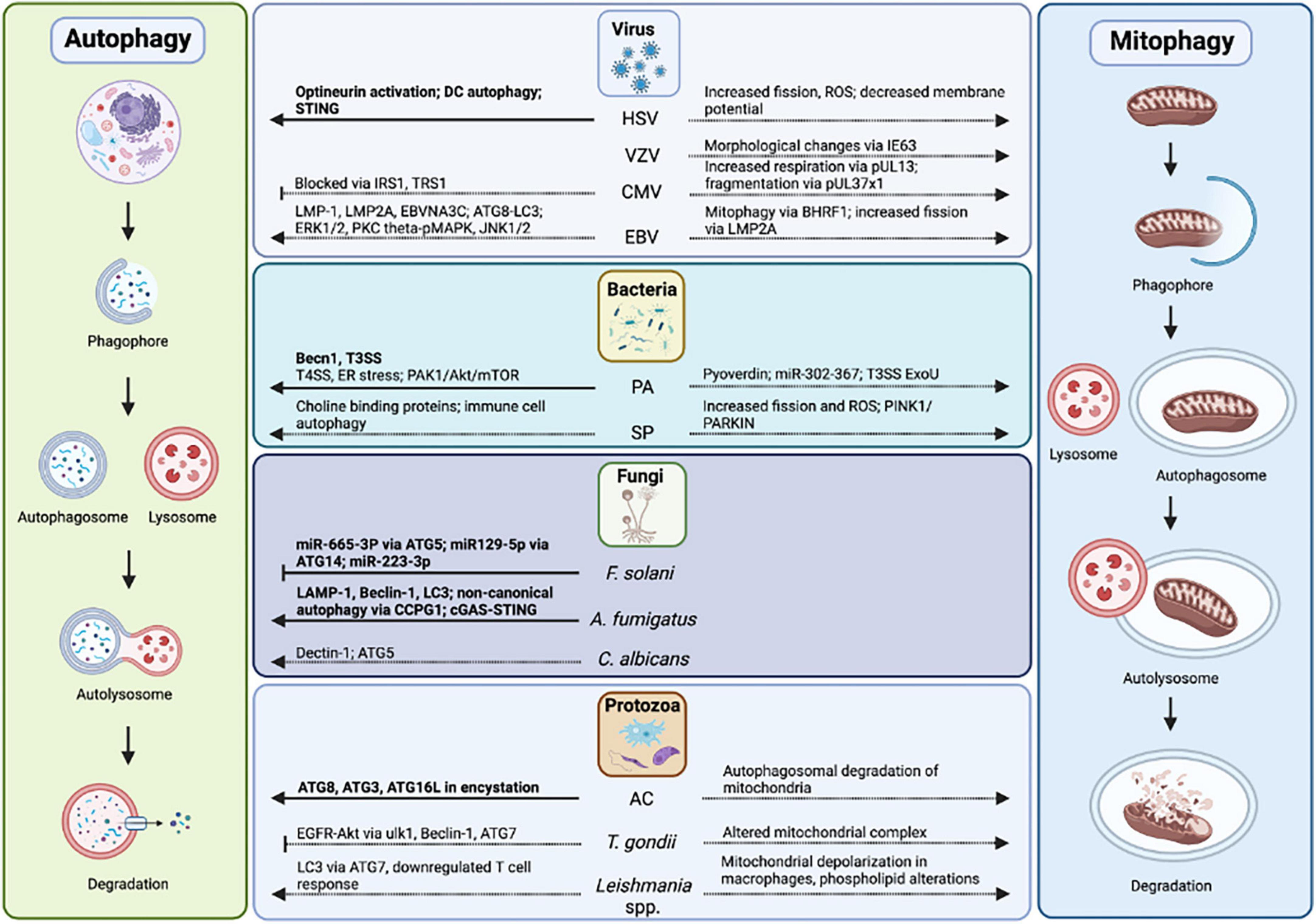
Figure 5. Autophagy and mitophagy in infectious keratitis. Autophagy has been studied in HSV, P. aeruginosa, F. solani, A. fumigatus, and Acanthamoeba-mediated keratitis. Autophagy proteins, pathways, mitophagy, and mitochondrial changes in infectious keratitis (in bold) compared to non-ocular infections.
4. Discussion
Autophagy has been investigated in the context of corneal diseases, infectious and non-infectious. In diabetes, evidence suggests there is a suppression of autophagy and mitophagy in the corneal epithelium and endothelium, in addition to trigeminal neurons. Based upon the few studies that are available, the induction of autophagy may be beneficial in resurfacing wounds in the diabetic cornea. While few studies have been performed and much work remains, the reports to date are consistent and suggestive of a modulatory role for autophagy in diabetes. The role of autophagy in dry eye disease is much less clear. The autophagic response is variable. This is likely due to differences in cell lines and animal models, types, and duration of stress. In addition, the effects of autophagy in ocular surface immune cells remains an area of heightened interest. Likewise, while Substance P, calcitonin gene related peptide, and nerve growth factor have been shown to modulate autophagy in non-ocular tissues, the role of these factors in autophagy in the corneal epithelium and corneal nerves is undefined (208–212). Moving forward, it is essential that appropriate controls are used and proper protein normalization is performed to substantiate and confirm some of these early findings.
In the limbal stem cell compartment, studies to date indicate that autophagy is protective in terms of proliferation and the DNA damage response. For much of this work, the inclusion of the Beclin 1 knockout mouse has been invaluable. In keratoconus, data in current reports is equivocal. Most reported findings are based upon epithelium harvested from the human cornea during corneal cross linking, in the case of keratoconic patients, and photorefractive keratectomy, for normal controls. There is some work that suggests that mitophagy may be downregulated in the epithelium from keratoconic eyes, but additional studies are needed. In contrast to this, the strongest data confirming an important role for mitophagy is in the corneal endothelium. This cellular monolayer plays a critical role in corneal transparency through it is ability to extrude water that is taken up by proteoglycans in the stroma. Given the high number of mitochondria that are required to maintain the pump, it is not surprising that defects in mitophagy, an essential mitochondrial quality control mechanism, have been found. Whether manipulation of these pathways may promote corneal endothelial health in patients with endothelial dystrophies remains to be proven, but is certainly an area of keen interest.
In terms of infectious disease in the cornea, huge gaps in our knowledge abound. In terms of viral infection, evidence exists to suggest that autophagy functions in a virus and cell type dependent manner. Autophagy has also been shown to affect the innate and adaptive immune systems. In the case of bacterial infection, some pathogens have been shown to induce autophagy during infection, while others have not. The strength of the response is further varied by bacteria strain and virulence. This is well exemplified by differences between methicillin resistant and methicillin sensitive S. aureus. Whether the induction of autophagy confers a gain or loss to the host remains to be determined. Mitophagy has also been shown to play a major role in the immune cell’s response. Overall, however, there is a paucity of data to define a clear role for autophagy and mitophagy in bacterial keratitis.
The same is true for fungal disease. Available data suggests that miRNAs, Beclin 1, and the cGAS-STING pathways may play key roles in mediating autophagy during fungal keratitis. To date however, not one study addresses mitophagy, despite aberrant mitochondrial damage that has been found during fungal infection in non-ocular tissues. In terms of protozoan infections, most striking and of high relevance to the cornea, is the ability of autophagy to promote Acanthamoeba encystation. As this is a particularly difficult pathogen to eradicate, the ability to block encystation and promote pathogen killing could represent a major advantageous step forward in our ability to treat these infections clinically.
Collectively, this work highlights the major gaps in knowledge that exist with respect to autophagy and mitophagy in non-infectious and infectious corneal diseases. While emerging data implicates a role for these pathways in corneal homeostasis, much work is needed to establish and refine the key players that are disturbed in corneal diseases and whether therapeutics designed to target these pathways may be beneficial.
Author contributions
RA, JS, and DR wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the NIH/NEI grants EY024546 (DR), EY029258 (DR), and EY033505 (DR), Core grant for Vision Research EY030413, and the Shirley G. and Norman Alweis Endowment Fund for Vision (DR).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Eghrari A, Riazuddin S, Gottsch J. Overview of the cornea: structure, function, and development. Prog Mol Biol Transl Sci. (2015) 134:7–23. doi: 10.1016/bs.pmbts.2015.04.001
2. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. (2019) 176:11–42. doi: 10.1016/j.cell.2018.09.048
3. Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. (2021) 40:e104705. doi: 10.15252/embj.2020104705
4. Chatterjee S, Khunti K, Davies M. Type 2 diabetes. Lancet (London, England). (2017) 389:2239–51. doi: 10.1016/S0140-6736(17)30058-2
5. Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the international diabetes federation diabetes atlas, 9(th) edition. Diabetes Res Clin Pract. (2019) 157:107843.
6. Ljubimov A. Diabetic complications in the cornea. Vis Res. (2017) 139:138–52. doi: 10.1016/j.visres.2017.03.002
7. Mansoor H, Tan H, Lin M, Mehta J, Liu Y. Diabetic corneal neuropathy. J Clin Med. (2020) 9:3956. doi: 10.3390/jcm9123956
8. Schultz R, Van Horn D, Peters M, Klewin K, Schutten W. Diabetic keratopathy. Trans Am Ophthalmol Soc. (1981) 79:180–99.
9. Chen W, Lin C, Ko P, Yeh P, Kuan Y, Hu F, et al. In vivo confocal microscopic findings of corneal wound healing after corneal epithelial debridement in diabetic vitrectomy. Ophthalmology. (2009) 116:1038–47. doi: 10.1016/j.ophtha.2009.01.002
10. Wang Y, Zhou Q, Xie L. [Diabetic keratopathy: new progresses and challenges]. Zhonghua Yan Ke Za Zhi. (2014) 50:69–72.
11. Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. (2013) 288:18077–92. doi: 10.1074/jbc.M113.474650
12. Kobayashi S, Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim Biophys Acta. (2015) 1852:252–61. doi: 10.1016/j.bbadis.2014.05.020
13. Czajka A, Ajaz S, Gnudi L, Parsade C, Jones P, Reid F, et al. Altered mitochondrial function, mitochondrial DNA and reduced metabolic flexibility in patients with diabetic nephropathy. EBioMedicine. (2015) 2:499–512. doi: 10.1016/j.ebiom.2015.04.002
14. Hu J, Kan T, Hu X. Sirt3 regulates mitophagy level to promote diabetic corneal epithelial wound healing. Exp Eye Res. (2019) 181:223–31. doi: 10.1016/j.exer.2019.02.011
15. Marcus J, Andrabi S. SIRT3 regulation under cellular stress: making sense of the ups and downs. Front Neurosci. (2018) 12:799. doi: 10.3389/fnins.2018.00799
16. Wu J, Zhang F, Yan M, Wu D, Yu Q, Zhang Y, et al. WldS enhances insulin transcription and secretion via a SIRT1-dependent pathway and improves glucose homeostasis. Diabetes. (2011) 60:3197–207. doi: 10.2337/db11-0232
17. Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. (2004) 429:771–6. doi: 10.1038/nature02583
18. Peck B, Chen C, Ho K, Di Fruscia P, Myatt S, Coombes R, et al. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther. (2010) 9:844–55. doi: 10.1158/1535-7163.MCT-09-0971
19. Zhou B, Li C, Qi W, Zhang Y, Zhang F, Wu J, et al. Downregulation of miR-181a upregulates sirtuin-1 (SIRT1) and improves hepatic insulin sensitivity. Diabetologia. (2012) 55:2032–43. doi: 10.1007/s00125-012-2539-8
20. Wang Y, Zhao X, Shi D, Chen P, Yu Y, Yang L, et al. Overexpression of SIRT1 promotes high glucose-attenuated corneal epithelial wound healing via p53 regulation of the IGFBP3/IGF-1R/AKT pathway. Invest Ophthalmol Vis Sci. (2013) 54:3806–14. doi: 10.1167/iovs.13-12091
21. Wu Y, Buckner B, Zhu M, Cavanagh H, Robertson D. Elevated IGFBP3 levels in diabetic tears: a negative regulator of IGF-1 signaling in the corneal epithelium. Ocul Surf. (2012) 10:100–7. doi: 10.1016/j.jtos.2012.01.004
22. Stuard W, Titone R, Robertson D. Tear levels of insulin-like growth factor binding protein 3 correlate with subbasal nerve plexus changes in patients with type 2 diabetes mellitus. Invest Ophthalmol Vis Sci. (2017) 58:6105–12. doi: 10.1167/iovs.17-22425
23. Stuard W, Titone R, Robertson D. Tear levels of IGFBP-3: a potential biomarker for diabetic nerve changes in the cornea. Eye Contact Lens. (2020) 46:319–25. doi: 10.1097/ICL.0000000000000700
24. Stuard W, Titone R, Robertson DM. IGFBP-3 functions as a molecular switch that mediates mitochondrial and metabolic homeostasis. FASEB J. (2022) 36:e22062. doi: 10.1096/fj.202100710RR
25. Chen C, Zhou Q, Li Z, Duan H, Liu Y, Wan L, et al. Hyperglycemia induces corneal endothelial dysfunction through attenuating mitophagy. Exp Eye Res. (2022) 215:108903. doi: 10.1016/j.exer.2021.108903
26. Titone R, Robertson D. Insulin receptor preserves mitochondrial function by binding VDAC1 in insulin insensitive mucosal epithelial cells. FASEB J. (2020) 34:754–75. doi: 10.1096/fj.201901316RR
27. Mussi N, Stuard W, Sanches J, Robertson D. Chronic hyperglycemia compromises mitochondrial function in corneal epithelial cells: implications for the diabetic cornea. Cells. (2022) 11:2567. doi: 10.3390/cells11162567
28. Cai D, Zhu M, Petroll W, Koppaka V, Robertson D. The impact of type 1 diabetes mellitus on corneal epithelial nerve morphology and the corneal epithelium. Am J Pathol. (2014) 184:2662–70. doi: 10.1016/j.ajpath.2014.06.016
29. Markoulli M, Flanagan J, Tummanapalli S, Wu J, Willcox M. The impact of diabetes on corneal nerve morphology and ocular surface integrity. Ocul Surf. (2018) 16:45–57. doi: 10.1016/j.jtos.2017.10.006
30. Petroll W, Robertson D. In vivo confocal microscopy of the cornea: new developments in image acquisition, reconstruction, and analysis using the HRT-rostock corneal module. Ocul Surf. (2015) 13:187–203. doi: 10.1016/j.jtos.2015.05.002
31. Hu J, Hu X, Kan T. MiR-34c participates in diabetic corneal neuropathy via regulation of autophagy. Invest Ophthalmol Vis Sci. (2019) 60:16–25. doi: 10.1167/iovs.18-24968
32. Hu J, Huang Y, Lin Y, Lin J. Protective effect inhibiting the expression of miR-181a on the diabetic corneal nerve in a mouse model. Exp Eye Res. (2020) 192:107925. doi: 10.1016/j.exer.2020.107925
33. Craig J, Nichols K, Akpek E, Caffery B, Dua H, Joo C, et al. TFOS DEWS II definition and classification report. Ocul Surf. (2017) 15:276–83. doi: 10.1016/j.jtos.2017.05.008
34. Seen S, Tong L. Dry eye disease and oxidative stress. Acta Ophthalmol. (2018) 96:e412–20. doi: 10.1111/aos.13526
35. Yun H, Jo Y, Kim J, Shin Y, Kim S, Choi T. Roles of autophagy in oxidative stress. Int J Mol Sci. (2020) 21:3289. doi: 10.3390/ijms21093289
36. Ma S, Yu Z, Feng S, Chen H, Chen H, Lu X. Corneal autophagy and ocular surface inflammation: a new perspective in dry eye. Exp Eye Res. (2019) 184:126–34. doi: 10.1016/j.exer.2019.04.023
37. Zhang L, Fu L, Zhang S, Zhang J, Zhao Y, Zheng Y, et al. Discovery of a small molecule targeting ULK1-modulated cell death of triple negative breast cancer in vitro and in vivo. Chem Sci. (2017) 8:2687–701. doi: 10.1039/c6sc05368h
38. Sarkar S, Davies J, Huang Z, Tunnacliffe A, Rubinsztein D. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. (2007) 282:5641–52. doi: 10.1074/jbc.M609532200
39. Panigrahi T, Shivakumar S, Shetty R, D’Souza S, Nelson E, Sethu S, et al. Trehalose augments autophagy to mitigate stress induced inflammation in human corneal cells. Ocul Surf. (2019) 17:699–713. doi: 10.1016/j.jtos.2019.08.004
40. Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema K, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. (2018) 14:1435–55. doi: 10.1080/15548627.2018.1474314
41. Shivakumar S, Panigrahi T, Shetty R, Subramani M, Ghosh A, Jeyabalan N. Chloroquine protects human corneal epithelial cells from desiccation stress induced inflammation without altering the autophagy flux. Biomed Res Int. (2018) 2018:7627329. doi: 10.1155/2018/7627329
42. Liu Z, Chen D, Chen X, Bian F, Gao N, Li J, et al. Autophagy activation protects ocular surface from inflammation in a dry eye model in vitro. Int J Mol Sci. (2020) 21:8966. doi: 10.3390/ijms21238966
43. Wang B, Peng L, Ouyang H, Wang L, He D, Zhong J, et al. Induction of DDIT4 impairs autophagy through oxidative stress in dry eye. Invest Ophthalmol Vis Sci. (2019) 60:2836–47. doi: 10.1167/iovs.19-27072
44. Qiao S, Dennis M, Song X, Vadysirisack D, Salunke D, Nash Z, et al. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat Commun. (2015) 6:7014. doi: 10.1038/ncomms8014
45. Wang N, Zimmerman K, Raab R, McKown R, Hutnik C, Talla V, et al. Lacritin rescues stressed epithelia via rapid forkhead box O3 (FOXO3)-associated autophagy that restores metabolism. J Biol Chem. (2013) 288:18146–61. doi: 10.1074/jbc.M112.436584
46. Wang W, Despanie J, Shi P, Edman-Woolcott M, Lin Y, Cui H, et al. Lacritin-mediated regeneration of the corneal epithelia by protein polymer nanoparticles. J Mater Chem B. (2014) 2:8131–41. doi: 10.1039/C4TB00979G
47. Srinivasan S, Thangavelu M, Zhang L, Green K, Nichols K. iTRAQ quantitative proteomics in the analysis of tears in dry eye patients. Invest Ophthalmol Vis Sci. (2012) 53:5052–9. doi: 10.1167/iovs.11-9022
48. Aluru S, Agarwal S, Srinivasan B, Iyer G, Rajappa S, Tatu U, et al. Lacrimal proline rich 4 (LPRR4) protein in the tear fluid is a potential biomarker of dry eye syndrome. PLoS One. (2012) 7:e51979. doi: 10.1371/journal.pone.0051979
49. McKown R, Wang N, Raab R, Karnati R, Zhang Y, Williams P, et al. Lacritin and other new proteins of the lacrimal functional unit. Exp Eye Res. (2009) 88:848–58. doi: 10.1016/j.exer.2008.09.002
50. Xu J, Chen P, Zhao G, Wei S, Li Q, Guo C, et al. Copolymer micelle-administered melatonin ameliorates hyperosmolarity-induced ocular surface damage through regulating PINK1-mediated mitophagy. Curr Eye Res. (2022) 47:688–703. doi: 10.1080/02713683.2021.2022163
51. Lavker R, Kaplan N, Wang J, Peng H. Corneal epithelial biology: lessons stemming from old to new. Exp Eye Res. (2020) 198:108094. doi: 10.1016/j.exer.2020.108094
52. Peng H, Park J, Katsnelson J, Kaplan N, Yang W, Getsios S, et al. microRNA-103/107 family regulates multiple epithelial stem cell characteristics. Stem Cells. (2015) 33:1642–56. doi: 10.1002/stem.1962
53. Boya P, Codogno P, Rodriguez-Muela N. Autophagy in stem cells: repair, remodelling and metabolic reprogramming. Development. (2018) 145:dev146506. doi: 10.1242/dev.146506
54. Robertson D, Li L, Fisher S, Pearce V, Shay J, Wright W, et al. Characterization of growth and differentiation in a telomerase-immortalized human corneal epithelial cell line. Invest Ophthalmol Vis Sci. (2005) 46:470–8. doi: 10.1167/iovs.04-0528
55. Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. (2011) 36:30–8. doi: 10.1016/j.tibs.2010.07.007
56. Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol. (2015) 35:615–21. doi: 10.1007/s10571-015-0166-x
57. Zhou J, Ge L, Jia C, Zheng X, Cui H, Zong R, et al. ROS-mediated different homeostasis of murine corneal epithelial progenitor cell line under oxidative stress. Sci Rep. (2016) 6:36481. doi: 10.1038/srep36481
58. Yildiz E, Aydemir D, Zibandeh N, Kusan E, Gumus K, Sarac OI, et al. Investigation of mitophagy biomarkers in corneal epithelium of keratoconus patients. Curr Eye Res. (2022) 47:661–9. doi: 10.1080/02713683.2022.2025846
59. Habib S, El Andaloussi A, Elmasry K, Handoussa A, Azab M, Elsawey A, et al. PDL-1 blockade prevents t cell exhaustion, inhibits autophagy, and promotes clearance of Leishmania donovani. Infect Immun. (2018) 86:e00019-18. doi: 10.1128/IAI.00019-18
60. Iqbal O, Fisher G, Vira S, Syed D, Sadeghi N, Freeman D, et al. Increased expression of secreted frizzled-related protein-1 and microtubule-associated protein light chain 3 in keratoconus. Cornea. (2013) 32:702–7. doi: 10.1097/ICO.0b013e318282987a
61. Shetty R, Sharma A, Pahuja N, Chevour P, Padmajan N, Dhamodaran K, et al. Oxidative stress induces dysregulated autophagy in corneal epithelium of keratoconus patients. PLoS One. (2017) 12:e0184628. doi: 10.1371/journal.pone.0184628
62. Zhang C, Bell W, Sundin O, De La Cruz Z, Stark W, Green W, et al. Immunohistochemistry and electron microscopy of early-onset fuchs corneal dystrophy in three cases with the same L450W COL8A2 mutation. Trans Am Ophthalmol Soc. (2006) 104:85–97.
63. Krachmer J, Purcell J Jr., Young C, Bucher K. Corneal endothelial dystrophy. a study of 64 families. Arch Ophthalmol. (1978) 96:2036–9. doi: 10.1001/archopht.1978.03910060424004
64. Baratz K, Tosakulwong N, Ryu E, Brown W, Branham K, Chen W, et al. E2-2 protein and Fuchs’s corneal dystrophy. New Eng J Med. (2010) 363:1016–24. doi: 10.1056/NEJMoa1007064
65. Jurkunas U. Fuchs endothelial corneal dystrophy through the prism of oxidative stress. Cornea. (2018) 37(Suppl. 1):S50–4. doi: 10.1097/ICO.0000000000001775
66. Jurkunas U, Bitar M, Funaki T, Azizi B. Evidence of oxidative stress in the pathogenesis of fuchs endothelial corneal dystrophy. Am J Pathol. (2010) 177:2278–89. doi: 10.2353/ajpath.2010.100279
67. Azizi B, Ziaei A, Fuchsluger T, Schmedt T, Chen Y, Jurkunas U. p53-regulated increase in oxidative-stress–induced apoptosis in fuchs endothelial corneal dystrophy: a native tissue model. Invest Ophthalmol Vis Sci. (2011) 52:9291–7. doi: 10.1167/iovs.11-8312
68. Borboli S, Colby K. Mechanisms of disease: fuchs’ endothelial dystrophy. Ophthalmol Clin North Am. (2002) 15:17–25. doi: 10.1016/s0896-1549(01)00016-5
69. Halilovic A, Schmedt T, Benischke A, Hamill C, Chen Y, Santos J, et al. Menadione-Induced DNA damage leads to mitochondrial dysfunction and fragmentation during rosette formation in fuchs endothelial corneal dystrophy. Antioxid Redox Signal. (2016) 24:1072–83. doi: 10.1089/ars.2015.6532
70. Benischke A, Vasanth S, Miyai T, Katikireddy K, White T, Chen Y, et al. Activation of mitophagy leads to decline in Mfn2 and loss of mitochondrial mass in fuchs endothelial corneal dystrophy. Sci Rep. (2017) 7:6656. doi: 10.1038/s41598-017-06523-2
71. Hamill C, Schmedt T, Jurkunas U. Fuchs endothelial cornea dystrophy: a review of the genetics behind disease development. Semin Ophthalmol. (2013) 28:281–6. doi: 10.3109/08820538.2013.825283
72. Miyai T, Vasanth S, Melangath G, Deshpande N, Kumar V, Benischke A, et al. Activation of PINK1-Parkin-Mediated mitophagy degrades mitochondrial quality control proteins in fuchs endothelial corneal dystrophy. Am J Pathol. (2019) 189:2061–76. doi: 10.1016/j.ajpath.2019.06.012
73. Bernhardt D, Müller M, Reichert A, Osiewacz H. Simultaneous impairment of mitochondrial fission and fusion reduces mitophagy and shortens replicative lifespan. Sci Rep. (2015) 5:7885. doi: 10.1038/srep07885
74. Song M, Mihara K, Chen Y, Scorrano L, Dorn G II. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. (2015) 21:273–86. doi: 10.1016/j.cmet.2014.12.011
75. Meng H, Matthaei M, Ramanan N, Grebe R, Chakravarti S, Speck C, et al. L450W and Q455K Col8a2 knock-in mouse models of Fuchs endothelial corneal dystrophy show distinct phenotypes and evidence for altered autophagy. Invest Ophthalmol Vis Sci. (2013) 54:1887–97. doi: 10.1167/iovs.12-11021
76. Jalimarada S, Ogando D, Vithana E, Bonanno J. Ion transport function of SLC4A11 in corneal endothelium. Invest Ophthalmol Vis Sci. (2013) 54:4330–40. doi: 10.1167/iovs.13-11929
77. Zhang W, Frausto R, Chung D, Griffis C, Kao L, Chen A, et al. Energy shortage in human and mouse models of SLC4A11-Associated corneal endothelial dystrophies. Invest Ophthalmol Vis Sci. (2020) 61:39. doi: 10.1167/iovs.61.8.39
78. Kim E, Meng H, Jun A. Lithium treatment increases endothelial cell survival and autophagy in a mouse model of Fuchs endothelial corneal dystrophy. Br J Ophthalmol. (2013) 97:1068–73. doi: 10.1136/bjophthalmol-2012-302881
79. Sies H, Berndt C, Jones D. Oxidative stress. Annu Rev Biochem. (2017) 86:715–48. doi: 10.1146/annurev-biochem-061516-045037
80. Saccà S, Cutolo C, Ferrari D, Corazza P, Traverso C. The eye, oxidative damage and polyunsaturated fatty acids. Nutrients. (2018) 10:668.
81. Nita M, Grzybowski A. The role of the reactive oxygen species and oxidative stress in the pathomechanism of the age-related ocular diseases and other pathologies of the anterior and posterior eye segments in adults. Oxid Med Cell Longev. (2016) 2016:3164734. doi: 10.1155/2016/3164734
82. Natoli R, Jiao H, Barnett N, Fernando N, Valter K, Provis J, et al. A model of progressive photo-oxidative degeneration and inflammation in the pigmented C57BL/6J mouse retina. Exp Eye Res. (2016) 147:114–27. doi: 10.1016/j.exer.2016.04.015
83. Kurutas E. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr J. (2016) 15:71. doi: 10.1186/s12937-016-0186-5
85. Singhal S, Singh S, Singhal P, Horne D, Singhal J, Awasthi S. Antioxidant role of glutathione S-transferases: 4-Hydroxynonenal, a key molecule in stress-mediated signaling. Toxicol Appl Pharmacol. (2015) 289:361–70. doi: 10.1016/j.taap.2015.10.006
86. Delamere N. Ascorbic acid and the eye. Subcell Biochem. (1996) 25:313–29. doi: 10.1007/978-1-4613-0325-1_16
87. Mizunoe Y, Kobayashi M, Sudo Y, Watanabe S, Yasukawa H, Natori D, et al. Trehalose protects against oxidative stress by regulating the Keap1-Nrf2 and autophagy pathways. Redox Biol. (2018) 15:115–24. doi: 10.1016/j.redox.2017.09.007
88. Felszeghy S, Viiri J, Paterno J, Hyttinen J, Koskela A, Chen M, et al. Loss of NRF-2 and PGC-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. (2019) 20:1–12. doi: 10.1016/j.redox.2018.09.011
89. Huynh K. Atherosclerosis: trehalose induces macrophage autophagy-lysosomal biogenesis. Nat Rev Cardiol. (2017) 14:444. doi: 10.1038/nrcardio.2017.97
90. Emanuele E. Can trehalose prevent neurodegeneration? insights from experimental studies. Curr Drug Targets. (2014) 15:551–7. doi: 10.2174/1389450115666140225104705
91. Wang B, Zuo X, Peng L, Wang X, Zeng H, Zhong J, et al. Melatonin ameliorates oxidative stress-mediated injuries through induction of HO-1 and restores autophagic flux in dry eye. Exp Eye Res. (2021) 205:108491. doi: 10.1016/j.exer.2021.108491
92. Mahalanobish S, Dutta S, Saha S, Sil P. Melatonin induced suppression of ER stress and mitochondrial dysfunction inhibited NLRP3 inflammasome activation in COPD mice. Food Chem Toxicol. (2020) 144:111588. doi: 10.1016/j.fct.2020.111588
93. Choi S, Lee E, Akuzum B, Jeong J, Maeng Y, Kim T, et al. Melatonin reduces endoplasmic reticulum stress and corneal dystrophy-associated TGFBIp through activation of endoplasmic reticulum-associated protein degradation. J Pineal Res. (2017) 63:e12426. doi: 10.1111/jpi.12426
94. Devilliers M, Ben Hadj Salah W, Barreau E, Da Cunha E, M’Garrech M, Bénichou J. [Ocular manifestations of viral diseases]. Rev Med Interne. (2021) 42:401–10. doi: 10.1016/j.revmed.2020.08.022
95. Ong A, Watson A, Subbiah S. Rubeola keratitis emergence during a recent measles outbreak in New Zealand. J Prim Health Care. (2020) 12:289–92. doi: 10.1071/HC20013
96. Pavlopoulos G, Giannakos G, Theodosiadis P, Moschos M, Iliakis E, Theodosiadis G. Rubeola keratitis: a photographic study of corneal lesions. Cornea. (2008) 27:411–6. doi: 10.1097/ICO.0b013e31816313a2
97. Parker Z, Murphy A, Leib D. Role of the DNA sensor STING in protection from lethal infection following corneal and intracerebral challenge with herpes simplex virus 1. J Virol. (2015) 89:11080–91. doi: 10.1128/JVI.00954-15
98. Petrovski G, Pasztor K, Orosz L, Albert R, Mencel E, Moe M, et al. Herpes simplex virus types 1 and 2 modulate autophagy in SIRC corneal cells. J Biosci. (2014) 39:683–92. doi: 10.1007/s12038-014-9443-y
99. Jiang Y, Yin X, Stuart P, Leib D. Dendritic cell autophagy contributes to herpes simplex virus-driven stromal keratitis and immunopathology. mBio (2015) 6:e01426–15. doi: 10.1128/mBio.01426-15
100. Ames J, Yadavalli T, Suryawanshi R, Hopkins J, Agelidis A, Patil C, et al. OPTN is a host intrinsic restriction factor against neuroinvasive HSV-1 infection. Nat Commun. (2021) 12:5401. doi: 10.1038/s41467-021-25642-z
101. Yakoub A, Shukla D. Autophagy stimulation abrogates herpes simplex virus-1 infection. Sci Rep. (2015) 5:9730. doi: 10.1038/srep09730
102. Rajasagi N, Rouse B. The role of T cells in herpes stromal keratitis. Front Immunol. (2019) 10:512. doi: 10.3389/fimmu.2019.00512
103. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. (2009) 461:788–92. doi: 10.1038/nature08476
104. Chodkowski M, Serafiñska I, Brzezicka J, Golke A, Słoñska A, Krzyżowska M, et al. Human herpesvirus type 1 and type 2 disrupt mitochondrial dynamics in human keratinocytes. Arch Virol. (2018) 163:2663–73. doi: 10.1007/s00705-018-3890-y
105. Cymerys J, Chodkowski M, Słoñska A, Krzyżowska M, Bañbura M. Disturbances of mitochondrial dynamics in cultured neurons infected with human herpesvirus type 1 and type 2. J Neurovirol. (2019) 25:765–82. doi: 10.1007/s13365-019-00762-x
106. Kalogeropoulos C, Bassukas I, Moschos M, Tabbara K. Eye and periocular skin involvement in herpes zoster infection. Med Hypothesis Discov Innov Ophthalmol. (2015) 4:142–56.
107. Buckingham E, Carpenter J, Jackson W, Grose C. Autophagy and the effects of its inhibition on varicella-zoster virus glycoprotein biosynthesis and infectivity. J Virol. (2014) 88:890–902.
108. Takahashi M, Jackson W, Laird D, Culp T, Grose C, Haynes J II, et al. Varicella-zoster virus infection induces autophagy in both cultured cells and human skin vesicles. J Virol. (2009) 83:5466–76. doi: 10.1128/JVI.02670-08
109. Carpenter J, Jackson W, Benetti L, Grose C. Autophagosome formation during varicella-zoster virus infection following endoplasmic reticulum stress and the unfolded protein response. J Virol. (2011) 85:9414–24. doi: 10.1128/JVI.00281-11
110. Eisfeld A, Turse S, Jackson S, Lerner E, Kinchington P. Phosphorylation of the varicella-zoster virus (VZV) major transcriptional regulatory protein IE62 by the VZV open reading frame 66 protein kinase. J Virol. (2006) 80:1710–23. doi: 10.1128/JVI.80.4.1710-1723.2006
111. Girsch J, Jackson W, Carpenter J, Moninger T, Jarosinski K, Grose C. Exocytosis of progeny infectious varicella-zoster virus particles via a mannose-6-phosphate receptor pathway without xenophagy following secondary envelopment. J Virol. (2020) 94:1–20. doi: 10.1128/JVI.00800-20
112. Tommasi C, Rogerson C, Depledge D, Jones M, Naeem A, Venturini C, et al. Kallikrein-Mediated cytokeratin 10 degradation is required for varicella zoster virus propagation in skin. J Invest Dermatol. (2020) 140:774–84.e11. doi: 10.1016/j.jid.2019.08.448
113. Graybill C, Morgan M, Levin M, Lee K. Varicella-zoster virus inhibits autophagosome-lysosome fusion and the degradation stage of mTOR-mediated autophagic flux. Virology. (2018) 522:220–7. doi: 10.1016/j.virol.2018.07.018
114. Girsch J, Walters K, Jackson W, Grose C. Progeny varicella-zoster virus capsids exit the nucleus but never undergo secondary envelopment during autophagic flux inhibition by bafilomycin A1. J Virol. (2019) 93:e00505–19. doi: 10.1128/JVI.00505-19
115. Keller A, Badani H, McClatchey P, Baird N, Bowlin J, Bouchard R, et al. Varicella zoster virus infection of human fetal lung cells alters mitochondrial morphology. J Neurovirol. (2016) 22:674–82. doi: 10.1007/s13365-016-0457-0
116. Heinz J, Kennedy P, Mogensen T. The role of autophagy in varicella zoster virus infection. Viruses. (2021) 13:1053. doi: 10.3390/v13061053
117. Lussignol M, Esclatine A. Cytomegalovirus and Autophagy. Amsterdam: Academic Press (2018). doi: 10.1016/B978-0-12-809819-6.00002-2
118. Lussignol M, Esclatine A. Modulation of Autophagy by Herpesvirus Proteins. Amsterdam: Academic Press (2014). doi: 10.1016/B978-0-12-801032-7.00008-3
119. Combs J, Norton E, Saifudeen Z, Bentrup K, Katakam P, Morris C, et al. Human cytomegalovirus alters host cell mitochondrial function during acute infection. J Virol. (2020) 94:e01183–19. doi: 10.1128/JVI.01183-19
120. Karniely S, Weekes M, Antrobus R, Rorbach J, van Haute L, Umrania Y, et al. Human cytomegalovirus infection upregulates the mitochondrial transcription and translation machineries. mBio (2016) 7:e00029. doi: 10.1128/mBio.00029-16
121. Kaarbø M, Ager-Wick E, Osenbroch P, Kilander A, Skinnes R, Müller F, et al. Human cytomegalovirus infection increases mitochondrial biogenesis. Mitochondrion. (2011) 11:935–45. doi: 10.1016/j.mito.2011.08.008
122. Betsinger C, Jankowski C, Hofstadter W, Federspiel J, Otter C, Jean Beltran P, et al. The human cytomegalovirus protein pUL13 targets mitochondrial cristae architecture to increase cellular respiration during infection. Proc Natl Acad Sci USA. (2021) 118:e2101675118. doi: 10.1073/pnas.2101675118
123. Bozidis P, Williamson C, Colberg-Poley A. Isolation of endoplasmic reticulum, mitochondria, and mitochondria-associated membrane fractions from transfected cells and from human cytomegalovirus-infected primary fibroblasts. Curr Protoc Cell Biol. (2007) Chapter 3:Unit 3.27.
124. Bhuvanendran S, Salka K, Rainey K, Sreetama S, Williams E, Leeker M, et al. Superresolution imaging of human cytomegalovirus vMIA localization in sub-mitochondrial compartments. Viruses. (2014) 6:1612–36. doi: 10.3390/v6041612
125. Federspiel J, Cook K, Kennedy M, Venkatesh S, Otter C, Hofstadter W, et al. Mitochondria and peroxisome remodeling across cytomegalovirus infection time viewed through the lens of inter-ViSTA. Cell Rep. (2020) 32:107943. doi: 10.1016/j.celrep.2020.107943
126. Mouna L, Hernandez E, Bonte D, Brost R, Amazit L, Delgui L, et al. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy. (2016) 12:327–42. doi: 10.1080/15548627.2015.1125071
127. Zhao J, Li Z, Wang M, Zhang Z, Ma H, Chang J, et al. Manipulation of autophagy by HCMV infection is involved in mTOR and influences the replication of virus. Acta Biochim Biophys Sin (Shanghai). (2013) 45:979–81. doi: 10.1093/abbs/gmt102
128. Sarwari N, Khoury J, Hernandez C. Chronic epstein barr virus infection leading to classical hodgkin lymphoma. BMC Hematol. (2016) 16:19. doi: 10.1186/s12878-016-0059-3
129. Matoba A. Ocular disease associated with Epstein-Barr virus infection. Surv Ophthalmol. (1990) 35:145–50. doi: 10.1016/0039-6257(90)90069-8
130. Granato M, Santarelli R, Farina A, Gonnella R, Lotti L, Faggioni A, et al. Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J Virol. (2014) 88:12715–26. doi: 10.1128/JVI.02199-14
131. Hung C, Chen L, Wang W, Chang P, Chiu Y, Hung C, et al. Regulation of autophagic activation by Rta of Epstein-Barr virus via the extracellular signal-regulated kinase pathway. J Virol. (2014) 88:12133–45. doi: 10.1128/JVI.02033-14
132. Gonnella R, Granato M, Farina A, Santarelli R, Faggioni A, Cirone M. PKC theta and p38 MAPK activate the EBV lytic cycle through autophagy induction. Biochim Biophys Acta. (2015) 1853:1586–95.
133. Granato M, Romeo M, Tiano M, Santarelli R, Gonnella R, Gilardini Montani M, et al. Bortezomib promotes KHSV and EBV lytic cycle by activating JNK and autophagy. Sci Rep. (2017) 7:13052. doi: 10.1038/s41598-017-13533-7
134. Lee D, Sugden B. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene. (2008) 27:2833–42. doi: 10.1038/sj.onc.1210946
135. Fotheringham J, Raab-Traub N. Epstein-Barr virus latent membrane protein 2 induces autophagy to promote abnormal acinus formation. J Virol. (2015) 89:6940–4. doi: 10.1128/JVI.03371-14
136. Bhattacharjee S, Bose P, Patel K, Roy S, Gain C, Gowda H, et al. Transcriptional and epigenetic modulation of autophagy promotes EBV oncoprotein EBNA3C induced B-cell survival. Cell Death Dis. (2018) 9:605. doi: 10.1038/s41419-018-0668-9
137. Nowag H, Guhl B, Thriene K, Romao S, Ziegler U, Dengjel J, et al. Macroautophagy proteins assist epstein barr virus production and get incorporated into the virus particles. EBioMedicine. (2014) 1:116–25. doi: 10.1016/j.ebiom.2014.11.007
138. Severa M, Giacomini E, Gafa V, Anastasiadou E, Rizzo F, Corazzari M, et al. EBV stimulates TLR- and autophagy-dependent pathways and impairs maturation in plasmacytoid dendritic cells: implications for viral immune escape. Eur J Immunol. (2013) 43:147–58. doi: 10.1002/eji.201242552
139. Gilardini Montani M, Santarelli R, Granato M, Gonnella R, Torrisi M, Faggioni A, et al. EBV reduces autophagy, intracellular ROS and mitochondria to impair monocyte survival and differentiation. Autophagy. (2019) 15:652–67. doi: 10.1080/15548627.2018.1536530
140. De Leo A, Colavita F, Ciccosanti F, Fimia G, Lieberman P, Mattia E. Inhibition of autophagy in EBV-positive Burkitt’s lymphoma cells enhances EBV lytic genes expression and replication. Cell Death Dis. (2015) 6:e1876. doi: 10.1038/cddis.2015.156
141. Lievens C, Cilimberg K, Moore A. Contact lens care tips for patients: an optometrist’s perspective. Clin Optom (Auckl). (2017) 9:113–21. doi: 10.2147/OPTO.S139651
142. Young A, Leung K, Tsim N, Hui M, Jhanji V. Risk factors, microbiological profile, and treatment outcomes of pediatric microbial keratitis in a tertiary care hospital in Hong Kong. Am J Ophthalmol. (2013) 156:1040–4.e2.
143. Keay L, Edwards K, Naduvilath T, Taylor H, Snibson G, Forde K, et al. Microbial keratitis predisposing factors and morbidity. Ophthalmology. (2006) 113:109–16.
144. Pachigolla G, Blomquist P, Cavanagh H. Microbial keratitis pathogens and antibiotic susceptibilities: a 5-year review of cases at an urban county hospital in north Texas. Eye Contact Lens. (2007) 33:45–9. doi: 10.1097/01.icl.0000234002.88643.d0
146. Brothers K, Kowalski R, Tian S, Kinchington P, Shanks R. Bacteria induce autophagy in a human ocular surface cell line. Exp Eye Res. (2018) 168:12–8. doi: 10.1016/j.exer.2017.12.010
147. Brothers K, Stella N, Shanks R. Biologically active pigment and ShlA cytolysin of Serratia marcescens induce autophagy in a human ocular surface cell line. BMC Ophthalmol. (2020) 20:120. doi: 10.1186/s12886-020-01387-z
148. Mohankumar V, Ramalingam S, Chidambaranathan G, Prajna L. Autophagy induced by type III secretion system toxins enhances clearance of Pseudomonas aeruginosa from human corneal epithelial cells. Biochem Biophys Res Commun. (2018) 503:1510–5. doi: 10.1016/j.bbrc.2018.07.071
149. Jiang F, Wang X, Wang B, Chen L, Zhao Z, Waterfield N, et al. The Pseudomonas aeruginosa type VI secretion PGAP1-like effector induces host autophagy by activating endoplasmic reticulum stress. Cell Rep. (2016) 16:1502–9. doi: 10.1016/j.celrep.2016.07.012
150. Zhu P, Bu H, Tan S, Liu J, Yuan B, Dong G, et al. A novel cochlioquinone derivative, CoB1, regulates autophagy in Pseudomonas aeruginosa infection through the PAK1/Akt1/mTOR signaling pathway. J Immunol. (2020) 205:1293–305. doi: 10.4049/jimmunol.1901346
151. Yuan K, Huang C, Fox J, Laturnus D, Carlson E, Zhang B, et al. Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J Cell Sci. (2012) 125(Pt. 2):507–15. doi: 10.1242/jcs.094573
152. Junkins R, Shen A, Rosen K, McCormick C, Lin T. Autophagy enhances bacterial clearance during P. aeruginosa lung infection. PLoS One. (2013) 8:e72263. doi: 10.1371/journal.pone.0072263
153. Park S, Shrestha S, Youn Y, Kim J, Kim S, Kim H, et al. Autophagy primes neutrophils for neutrophil extracellular trap formation during sepsis. Am J Respirat Crit Care Med. (2017) 196:577–89. doi: 10.1164/rccm.201603-0596OC
154. Hazlett L. Role of innate and adaptive immunity in the pathogenesis of keratitis. Ocul Immunol Inflamm. (2005) 13:133–8.
155. Jabir M, Hopkins L, Ritchie N, Ullah I, Bayes H, Li D, et al. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy. (2015) 11:166–82. doi: 10.4161/15548627.2014.981915
156. Kirienko N, Ausubel F, Ruvkun G. Mitophagy confers resistance to siderophore-mediated killing by Pseudomonas aeruginosa. Proc Natl Acad Sci USA. (2015) 112:1821–6. doi: 10.1073/pnas.1424954112
157. Huang T, Pu Q, Zhou C, Lin P, Gao P, Zhang X, et al. MicroRNA-302/367 cluster impacts host antimicrobial defense via regulation of mitophagic response against Pseudomonas aeruginosa infection. Front Immunol. (2020) 11:569173. doi: 10.3389/fimmu.2020.569173
158. Norina T, Raihan S, Bakiah S, Ezanee M, Liza-Sharmini A, Wan Hazzabah W. Microbial keratitis: aetiological diagnosis and clinical features in patients admitted to Hospital Universiti Sains Malaysia. Singapore Med J. (2008) 49:67–71.
159. Bharathi M, Ramakrishnan R, Vasu S, Meenakshi R, Shivkumar C, Palaniappan R. Epidemiology of bacterial keratitis in a referral centre in south India. Indian J Med Microbiol. (2003) 21:239–45.
160. Shizukuishi S, Ogawa M, Matsunaga S, Tomokiyo M, Ikebe T, Fushinobu S, et al. Streptococcus pneumoniae hijacks host autophagy by deploying CbpC as a decoy for Atg14 depletion. EMBO Rep. (2020) 21:e49232. doi: 10.15252/embr.201949232
161. Dong Y, Jin C, Ding Z, Zhu Y, He Q, Zhang X, et al. TLR4 regulates ROS and autophagy to control neutrophil extracellular traps formation against Streptococcus pneumoniae in acute otitis media. Pediatr Res. (2021) 89:785–94. doi: 10.1038/s41390-020-0964-9
162. Kim J, Paton J, Briles D, Rhee D, Pyo S. Streptococcus pneumoniae induces pyroptosis through the regulation of autophagy in murine microglia. Oncotarget. (2015) 6:44161–78. doi: 10.18632/oncotarget.6592
163. Hardy K, Tuckey A, Housley N, Andrews J, Patel M, Al-Mehdi A, et al. The Pseudomonas aeruginosa Type III secretion system exoenzyme effector ExoU induces mitochondrial damage in a murine bone marrow-derived macrophage infection model. Infect Immun. (2022) 90:e0047021. doi: 10.1128/IAI.00470-21
164. Mohasin M, Balbirnie-Cumming K, Fisk E, Prestwich E, Russell C, Marshall J, et al. Macrophages utilize mitochondrial fission to enhance mROS production during responses to Streptococcus pneumoniae. bioRxiv [preprint] (2019). doi: 10.1101/722603
165. Zhang Y, Sharma L, Losier A, Ifedigbo E, Sauler M, Bazan I, et al. Role of PINK1 mediated mitophagy during Streptococcus pneumoniae pneumonia. Am J Respir Crit Care Med. (2019) 199:A7230.
166. Gao Y, Xu W, Dou X, Wang H, Zhang X, Yang S, et al. Mitochondrial DNA leakage caused by Streptococcus pneumoniae hydrogen peroxide promotes Type I IFN expression in lung cells. Front Microbiol. (2019) 10:630. doi: 10.3389/fmicb.2019.00630
167. Manikandan P, Abdel-Hadi A, Randhir Babu Singh Y, Revathi R, Anita R, Banawas S, et al. Fungal keratitis: epidemiology, rapid detection, and antifungal susceptibilities of fusarium and aspergillus isolates from corneal scrapings. Biomed Res Int. (2019) 2019:6395840. doi: 10.1155/2019/6395840
168. Tuft S, Tullo A. Fungal keratitis in the United Kingdom 2003-2005. Eye (Lond). (2009) 23:1308–13. doi: 10.1038/eye.2008.298
169. Gower E, Keay L, Oechsler R, Iovieno A, Alfonso E, Jones D, et al. Trends in fungal keratitis in the United States, 2001 to 2007. Ophthalmology. (2010) 117:2263–7. doi: 10.1016/j.ophtha.2010.03.048
171. Lin J, Lin Y, Huang Y, Hu J. Inhibiting miR-129-5p alleviates inflammation and modulates autophagy by targeting ATG14 in fungal keratitis. Exp Eye Res. (2021) 211:108731. doi: 10.1016/j.exer.2021.108731
172. Guo Q, Lin Y, Hu J. Inhibition of miR-665-3p enhances autophagy and alleviates inflammation in fusarium solani-induced keratitis. Invest Ophthalmol Vis Sci. (2021) 62:24. doi: 10.1167/iovs.62.1.24
173. Tang H, Lin Y, Huang L, Hu J. MiR-223-3p regulates autophagy and inflammation by targeting ATG16L1 in Fusarium solani-induced keratitis. Invest Ophthalmol Vis Sci. (2022) 63:41. doi: 10.1167/iovs.63.1.41
174. Li C, Li C, Lin J, Zhao G, Xu Q, Jiang N, et al. The role of autophagy in the innate immune response to fungal keratitis caused by Aspergillus fumigatus infection. Invest Ophthalmol Vis Sci. (2020) 61:25. doi: 10.1167/iovs.61.2.25
175. Han F, Guo H, Wang L, Zhang Y, Sun L, Dai C, et al. The cGAS-STING signaling pathway contributes to the inflammatory response and autophagy in Aspergillus fumigatus keratitis. Exp Eye Res. (2021) 202:108366. doi: 10.1016/j.exer.2020.108366
176. Li C, Li C, Li H, Zhao G, Lin J, Wang Q, et al. Disparate expression of autophagy in corneas of C57BL/6 mice and BALB/c mice after Aspergillus fumigatus infection. Int J Ophthalmol. (2019) 12:705–10. doi: 10.18240/ijo.2019.05.02
177. Wang L, Chen X, Yan H, Yan S, Sun X, Zhang D, et al. CCPG1 involved in corneal Aspergillus fumigatus infection. Int J Ophthalmol. (2022) 15:541–6. doi: 10.18240/ijo.2022.04.03
178. Smeekens S, Malireddi R, Plantinga T, Buffen K, Oosting M, Joosten L, et al. Autophagy is redundant for the host defense against systemic Candida albicans infections. Eur J Clin Microbiol Infect Dis. (2014) 33:711–22. doi: 10.1007/s10096-013-2002-x
179. Tam J, Mansour M, Khan N, Seward M, Puranam S, Tanne A, et al. Dectin-1-dependent LC3 recruitment to phagosomes enhances fungicidal activity in macrophages. J Infect Dis. (2014) 210:1844–54. doi: 10.1093/infdis/jiu290
180. Shroff A, Reddy K. Autophagy gene ATG5 knockdown upregulates apoptotic cell death during Candida albicans infection in human vaginal epithelial cells. Am J Reprod Immunol. (2018) 80:e13056. doi: 10.1111/aji.13056
181. Shroff A, Sequeira R, Reddy K. Human vaginal epithelial cells augment autophagy marker genes in response to Candida albicans infection. Am J Reprod Immunol. (2017) 77:e12639. doi: 10.1111/aji.12639
182. Shroff A, Sequeira R, Patel V, Reddy K. Knockout of autophagy gene, ATG5 in mice vaginal cells abrogates cytokine response and pathogen clearance during vaginal infection of Candida albicans. Cell Immunol. (2018) 324:59–73. doi: 10.1016/j.cellimm.2017.12.012
183. Padhi T, Das S, Sharma S, Rath S, Rath S, Tripathy D, et al. Ocular parasitoses: a comprehensive review. Survey Ophthalmol. (2017) 62:161–89. doi: 10.1016/j.survophthal.2016.09.005
184. Laxmanappa Hoti S, Tandon V. Ocular parasitoses and their immunology. Ocular Immunol Inflamm. (2011) 19:385–96.
185. Moon E, Chung D, Hong Y, Kong H. Autophagy protein 8 mediating autophagosome in encysting acanthamoeba. Mol Biochem Parasitol. (2009) 168:43–8. doi: 10.1016/j.molbiopara.2009.06.005
186. Moon E, Hong Y, Chung D, Kong H. Identification of atg8 isoform in encysting Acanthamoeba. Korean J Parasitol. (2013) 51:497–502. doi: 10.3347/kjp.2013.51.5.497
187. Moon E, Chung D, Hong Y, Kong H. Atg3-mediated lipidation of Atg8 is involved in encystation of Acanthamoeba. Korean J Parasitol. (2011) 49:103–8. doi: 10.3347/kjp.2011.49.2.103
188. Song S, Han B, Moon E, Lee Y, Yu H, Jha B, et al. Autophagy protein 16-mediated autophagy is required for the encystation of Acanthamoeba castellanii. Mol Biochem Parasitol. (2012) 183:158–65. doi: 10.1016/j.molbiopara.2012.02.013
189. Moon E, Hong Y, Chung D, Kong H. Cysteine protease involving in autophagosomal degradation of mitochondria during encystation of Acanthamoeba. Mol Biochem Parasitol. (2012) 185:121–6. doi: 10.1016/j.molbiopara.2012.07.008
190. Moon E, Kim S, Hong Y, Chung D, Goo Y, Kong H. Autophagy inhibitors as a potential antiamoebic treatment for Acanthamoeba keratitis. Antimicrob Agents Chemother. (2015) 59:4020–5. doi: 10.1128/AAC.05165-14
191. Lee S, Lee D, Park S, Yu H, Lee J, Lee J. Effect of multipurpose solution combined with autophagy inhibitors on adhesion of Acanthamoeba trophozoites to silicone hydrogel contact lenses. Cornea. (2017) 36:1538–43. doi: 10.1097/ICO.0000000000001340
192. Moon E, Lee S, Quan F, Kong H. Chloroquine as a possible disinfection adjunct of disinfection solutions against Acanthamoeba. Exp Parasitol. (2018) 188:102–6. doi: 10.1016/j.exppara.2018.04.005
193. Portillo J, Okenka G, Reed E, Subauste A, Van Grol J, Gentil K, et al. The CD40-autophagy pathway is needed for host protection despite IFN-Γ-dependent immunity and CD40 induces autophagy via control of P21 levels. PLoS One. (2010) 5:e14472. doi: 10.1371/journal.pone.0014472
194. Lopez Corcino Y, Gonzalez Ferrer S, Mantilla L, Trikeriotis S, Yu J, Kim S, et al. Toxoplasma gondii induces prolonged host epidermal growth factor receptor signalling to prevent parasite elimination by autophagy: perspectives for in vivo control of the parasite. Cell Microbiol. (2019) 21:e13084. doi: 10.1111/cmi.13084
195. Syn G, Anderson D, Blackwell J, Jamieson S. Toxoplasma gondii infection is associated with mitochondrial dysfunction in-vitro. Front Cell Infect Microbiol. (2017) 7:512. doi: 10.3389/fcimb.2017.00512
196. Muniz-Feliciano L, Van Grol J, Portillo J, Liew L, Liu B, Carlin C, et al. Toxoplasma gondii-induced activation of EGFR prevents autophagy protein-mediated killing of the parasite. PLoS Pathog. (2013) 9:e1003809. doi: 10.1371/journal.ppat.1003809
197. Murray H, Berman J, Davies C, Saravia N. Advances in leishmaniasis. Lancet. (2005) 366:1561–77. doi: 10.1016/S0140-6736(05)67629-5
198. Maude R, Ahmed B, Rahman A, Rahman R, Majumder M, Menezes D, et al. Retinal changes in visceral leishmaniasis by retinal photography. BMC Infect Dis. (2014) 14:527. doi: 10.1186/1471-2334-14-527
199. Durdu M, Gökçe S, Bagirova M, Yalaz M, Allahverdiyev A, Uzun S. Periocular involvement in cutaneous leishmaniasis. J Eur Acad Dermatol Venereol. (2007) 21:214–8. doi: 10.1111/j.1468-3083.2006.01903.x
200. Mignot G, Bhattacharya Y, Reddy A. Ocular leishmaniasis - a systematic review. Indian J Ophthalmol. (2021) 69:1052–60.
201. Evans R, Sundaramurthy V, Frickel E. The interplay of host autophagy and eukaryotic pathogens. Front Cell Dev Biol. (2018) 6:118. doi: 10.3389/fcell.2018.00118
202. Esch K, Schaut R, Lamb I, Clay G, Morais Lima ÁL, do Nascimento PR, et al. Activation of autophagy and nucleotide-binding domain leucine-rich repeat-containing-like receptor family, pyrin domain-containing 3 inflammasome during Leishmania infantum-associated glomerulonephritis. Am J Pathol. (2015) 185:2105–17. doi: 10.1016/j.ajpath.2015.04.017
203. Cyrino L, Araújo A, Joazeiro P, Vicente C, Giorgio S. In vivo and in vitro Leishmania amazonensis infection induces autophagy in macrophages. Tissue Cell. (2012) 44:401–8. doi: 10.1016/j.tice.2012.08.003
204. Pinheiro R, Nunes M, Pinheiro C, D’Avila H, Bozza P, Takiya C, et al. Induction of autophagy correlates with increased parasite load of Leishmania amazonensis in BALB/c but not C57BL/6 macrophages. Microbes Infect. (2009) 11:181–90. doi: 10.1016/j.micinf.2008.11.006
205. Mitroulis I, Kourtzelis I, Papadopoulos V, Mimidis K, Speletas M, Ritis K. In vivo induction of the autophagic machinery in human bone marrow cells during Leishmania donovani complex infection. Parasitol Int. (2009) 58:475–7. doi: 10.1016/j.parint.2009.07.002
206. Crauwels P, Bohn R, Thomas M, Gottwalt S, Jäckel F, Krämer S, et al. Apoptotic-like Leishmania exploit the host’s autophagy machinery to reduce T-cell-mediated parasite elimination. Autophagy. (2015) 11:285–97. doi: 10.1080/15548627.2014.998904
207. Frank B, Marcu A, de Oliveira Almeida Petersen AL, Weber H, Stigloher C, Mottram JC, et al. Autophagic digestion of Leishmania major by host macrophages is associated with differential expression of BNIP3, CTSE, and the miRNAs miR-101c, miR-129, and miR-210. Parasit Vectors. (2015) 8:404. doi: 10.1186/s13071-015-0974-3
208. Chen F, Wan Q, Li Q, Fang J, Peng L, Hu J. Substance P prevents doxorubicin-induced cardiomyocyte injury by regulating apoptosis and autophagy: in vitro and in vivo evidence. Mol Med Rep. (2022) 25:50. doi: 10.3892/mmr.2021.12566
209. Wang L, Guo L, Wang L, Zhang G, Shang J, Murao K, et al. Oxidative stress and substance P mediate psychological stress-induced autophagy and delay of hair growth in mice. Arch Dermatol Res. (2015) 307:171–81. doi: 10.1007/s00403-014-1521-3
210. Schavinski A, Machado J, Morgan H, Lautherbach N, Paula-Gomes S, Kettelhut I, et al. Calcitonin gene-related peptide exerts inhibitory effects on autophagy in the heart of mice. Peptides. (2021) 146:170677. doi: 10.1016/j.peptides.2021.170677
211. Machado J, Silveira W, Gonçalves D, Schavinski A, Khan M, Zanon N, et al. α-Calcitonin gene-related peptide inhibits autophagy and calpain systems and maintains the stability of neuromuscular junction in denervated muscles. Mol Metab. (2019) 28:91–106. doi: 10.1016/j.molmet.2019.06.024
Keywords: cornea, autophagy, mitophagy, mitochondria, epithelium, endothelium, stroma
Citation: Ayilam Ramachandran R, Sanches JM and Robertson DM (2023) The roles of autophagy and mitophagy in corneal pathology: current knowledge and future perspectives. Front. Med. 10:1064938. doi: 10.3389/fmed.2023.1064938
Received: 08 October 2022; Accepted: 16 January 2023;
Published: 21 April 2023.
Edited by:
Luigi Capasso, Azienda Sanitaria Locale Napoli 1 Centro, ItalyReviewed by:
Romina Mayra Lasagni Vitar, San Raffaele Hospital (IRCCS), ItalyHanlin Zhang, College of Letters and Science, University of California, Berkeley, United States
Copyright © 2023 Ayilam Ramachandran, Sanches and Robertson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Danielle M. Robertson, ZGFuaWVsbGUucm9iZXJ0c29uQHV0c291dGh3ZXN0ZXJuLmVkdQ==
†These authors have contributed equally to this work and share first authorship