 Jun Shimizu
Jun Shimizu Masanori A. Murayama
Masanori A. Murayama Yoshihisa Mizukami
Yoshihisa Mizukami Nagisa Arimitsu
Nagisa Arimitsu Kenji Takai
Kenji Takai Yoshishige Miyabe
Yoshishige Miyabe- 1Department of Immunology and Parasitology, St. Marianna University of School of Medicine, Kawasaki, Kanagawa, Japan
- 2Department of Animal Models for Human Diseases, Institute of Biomedical Science, Kansai Medical University, Hirakata, Osaka, Japan
Behçet disease (BD) and relapsing polychondritis (RP) are chronic multisystem disorders characterized by recurrent flare-ups of tissue inflammation. Major clinical manifestations of BD are oral aphthae, genital aphthous ulcers, skin lesions, arthritis, and uveitis. Patients with BD may develop rare but serious neural, intestinal, and vascular complications, with high relapse rates. Meanwhile, RP is characterized by the inflammation of the cartilaginous tissues of the ears, nose, peripheral joints, and tracheobronchial tree. Additionally, it affects the proteoglycan-rich structures in the eyes, inner ear, heart, blood vessels, and kidneys. The mouth and genital ulcers with inflamed cartilage (MAGIC) syndrome is a common characteristic of BD and RP. The immunopathology of these two diseases may be closely related. It is established that the genetic predisposition to BD is related to the human leukocyte antigen (HLA)-B51 gene. Skin histopathology demonstrates the overactivation of innate immunity, such as neutrophilic dermatitis/panniculitis, in patients with BD. Monocytes and neutrophils frequently infiltrate cartilaginous tissues of patients with RP. Somatic mutations in UBA1, which encodes a ubiquitylation-related enzyme, cause vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic syndrome (VEXAS) with severe systemic inflammation and activation of myeloid cells. VEXAS prompts auricular and/or nasal chondritis, with neutrophilic infiltration around the cartilage in 52–60% of patients. Thus, innate immune cells may play an important role in the initiation of inflammatory processes underlying both diseases. This review summarizes the recent advances in our understanding of the innate cell-mediated immunopathology of BD and RP, with a focus on the common and distinct features of these mechanisms.
1. Introduction
Behçet disease (BD) is an inflammatory disorder characterized by the frequent occurrence of oral ulcers, genital aphthous ulcers, and uveitis, with clinical manifestations involving the skin, cardiovascular, intestinal, and central nervous system (CNS) (1). These manifestations are important for diagnosis as there are no clinical or laboratory findings specific to BD. In 1985, an international study group developed diagnostic criteria based on the major symptoms of BD; a diagnosis is made when an individual has developed recurrent oral ulceration (at least three times over the past 12 months) with at least two of the following symptoms: persistent genital ulceration; eye lesions, such as uveitis and retinal vasculitis; skin involvement, such as erythema nodosum and thrombophlebitis; and a positive pathergy test (2). The clinical diagnostic criteria must be followed by the exclusion criteria for patients with other immune disorders presenting with common symptoms of BD. For example, chronic oral ulcerations are frequently observed in Crohn disease too (3). Additionally, Vogt-Koyanagi-Harada and Cogan syndromes should be considered during the differential diagnosis of BD in patients with uveitis (4, 5). CNS and gastrointestinal involvement are indicators of a poor prognosis in patients with neuro-BD and intestinal BD, respectively. Patients in the BD subgroup are difficult to distinguish from those with multiple sclerosis (6) and Crohn disease (7).
Relapsing polychondritis (RP) is a chronic inflammatory disorder characterized by chondritis of the auricular, nasal, joint, and tracheal cartilaginous tissues (8). In addition, it affects the proteoglycan-rich structures in the eyes, inner ear, heart, blood vessels, and kidneys. In the clinical features, respiratory involvement is associated with poor prognosis through the severe pulmonary complications such as tracheobronchomalacia and/or pulmonary infection (9). The diagnosis is usually made based on clinical symptoms using McAdam’s (10), Damiani’s (11), and/or Michet’s (12) criteria, because there are no pathognomonic clinical and laboratory features, similar to patients with BD. Approximately 20–35% of RP cases are further complicated by other immune disorders such as systemic vasculitis, rheumatoid arthritis, and systemic lupus erythematosus (10–13). In most instances, coexisting diseases precede the onset of RP (10), and in some patients, complications occur as consequent symptoms of RP (8). It is possible that the clinical course of the disease makes it difficult to obtain an early and accurate diagnosis.
Identifying new diagnostic biomarkers for human inflammatory diseases require further studies. Generally, human autoinflammatory diseases, such as familial Mediterranean fever (FMF) and tumor necrosis factor (TNF) receptor-associated periodic fever syndrome (TRAPS), are thought to be caused by abnormalities in phagocytes against pathogenic elements. In contrast, human autoimmune diseases are characterized by the overactivation of lymphocytes in response to autoantigens. In these studies, most immune disorders were suggested to be caused by a combination of autoimmune and autoinflammatory mechanisms in the disease spectrum, based on the genetic and cellular basis (14). In the immune disease spectrum, BD is associated with a mixed pattern of autoinflammatory and autoimmune diseases (Figure 1) (14).
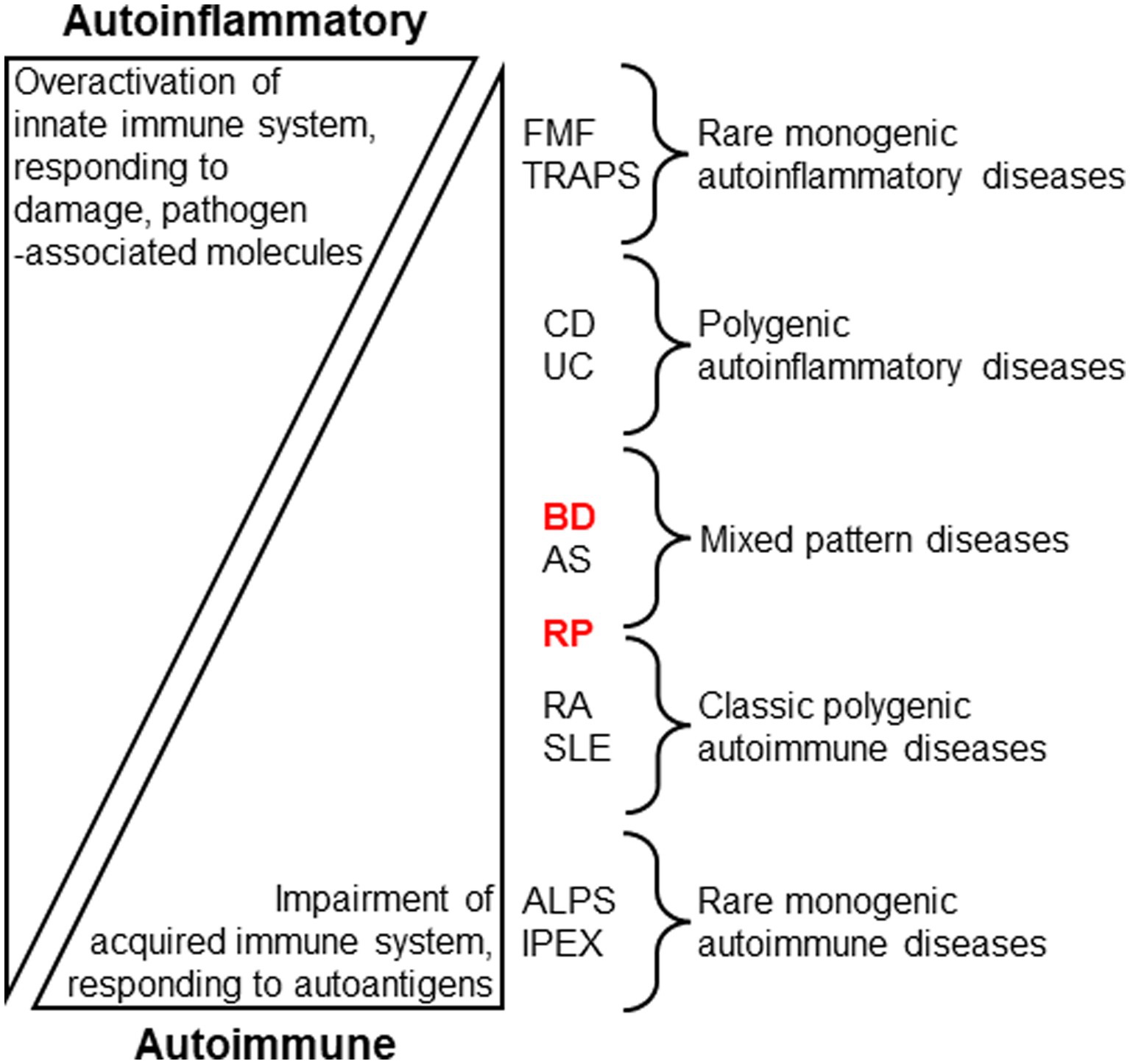
Figure 1. Stratification of human autoinflammatory and autoimmune diseases by evaluating immune conditions (14, McGonagle and McDermotte. PLoS Med. 2006; 3:e297. Modified). Innate immune overactivation via inflammatory cytokine signaling, pathogen sensing, and/or disruption of local tissue homeostasis are the proposed main causes of autoinflammatory diseases, while autoimmune diseases are associated with self-reactive lymphocytes through impaired immune tolerance. Behçet disease (BD) and relapsing polychondritis (RP) may be allocated to distinct clusters of the stratification. FMF: Familial Mediterranean fever; TRAPS: TNF receptor-associated periodic fever syndrome; CD: Crohn disease; UC: Ulcerative colitis; AS: Ankylosing spondylitis; RA: Rheumatoid arthritis; SLE: Systemic lupus erythematosus; ALPS: autoimmune lymphoproliferative syndrome; IPEX: immune dysregulation, polyendocrinopathy, enteropathy, X-linked.
In this review, we summarize current knowledge of innate cell mediated immunopathology in BD and RP to identify accurate positions of the immune disorders in the disease spectrum to facilitate the development of new therapeutic strategies.
2. Behçet disease
2.1. Epidemiology of BD
2.1.1. Environmental factors in BD
BD is prevalent along the ancient Silk Road between the Mediterranean Basin and East Asia (1). The human leukocyte antigen (HLA)-B51 gene is established as a major BD susceptibility gene, especially in the patients with ocular involvement (1). Additionally, a geological association was observed between the prevalence of BD and HLA-B51 (15). These data demonstrated that BD inflammation may be triggered by innate immunity as well as environmental factors, such as bacterial and viral agents.
Oral plaque index scores are associated with the presence of oral ulcers and BD severity (16). Dental plaque bacteria (Streptococcus sanguinis) are frequently observed in the oral cavity of patients (17). Mouthwashes containing soluble betamethasone, doxycycline, and nystatin improve oral ulcer severity scores in patients with BD (18).
Recent studies have revealed perturbation of oral and gut microbiota, especially increases in lactate-producing bacteria such as Lactobacillus and Bifidobacterium, in patients with BD compared with those in healthy individuals. Researchers have suggested pathological relationships between microbiota and immunological dysfunction in BD (Figure 2A) (19–22). In contrast to these clinical and laboratory findings, HLA-B51 transgenic mice demonstrate no obvious clinical phenotypes of BD, although stimulated neutrophils produce high levels of superoxide (23).
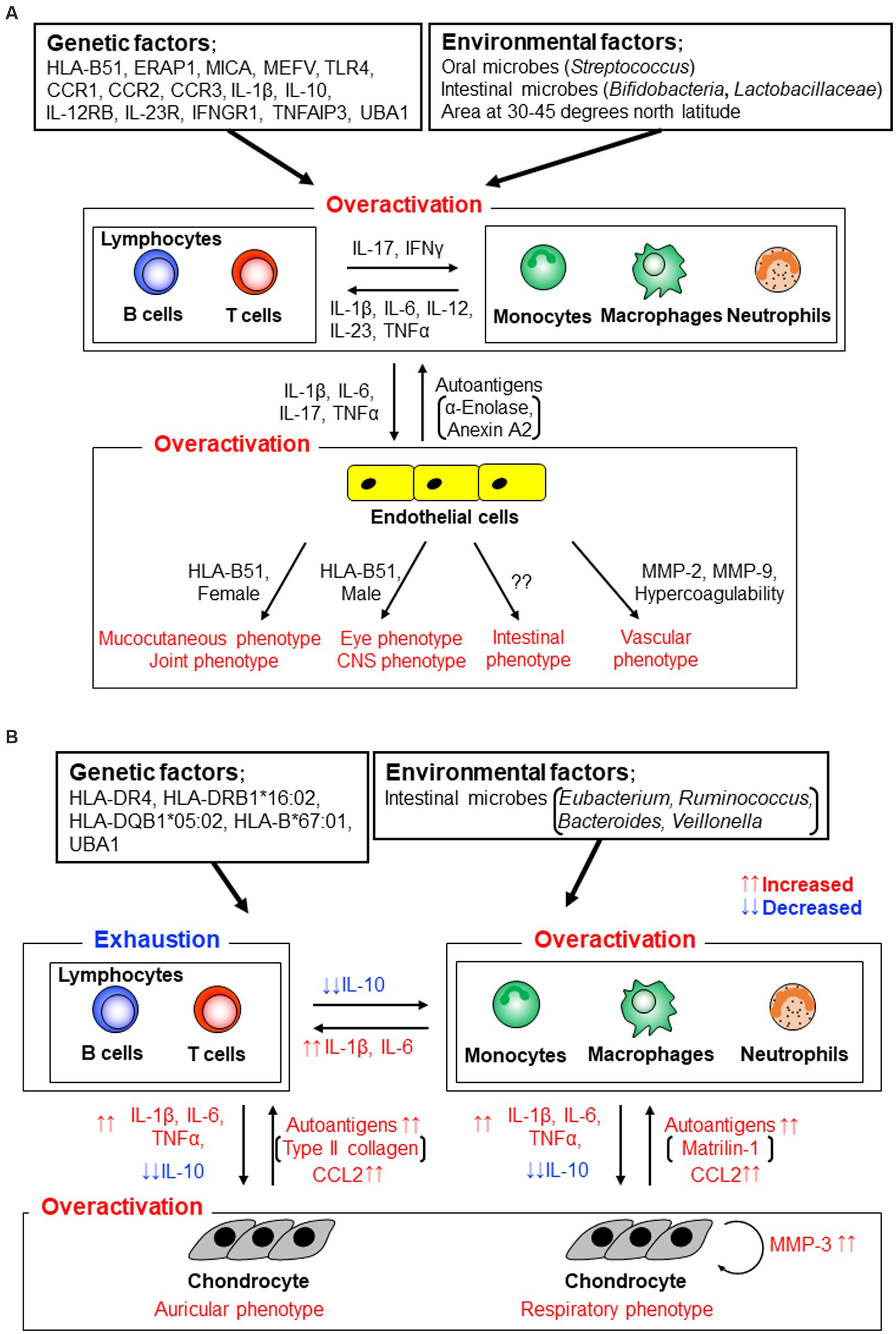
Figure 2. Stratification of human autoinflammatory and autoimmune diseases by evaluating immune conditions (14, McGonagle and McDermotte. PLoS Med. 2006; 3:e297. Modified).
2.1.2. Genetic variations in BD
Genome-wide profiling analyses revealed that, adding to HLA-B51, myeloid immune cell-related molecules, such as endoplasmic reticulum aminopeptidase-1 (ERAP1), major histocompatibility complex (MHC) class I polypeptide-related sequence-A (MICA), familial Mediterranean (MEFV) gene products, toll-like receptor-4 (TLR-4), c-c motif chemokine receptors CCR1-CCR3, interleukin (IL)-1β, IL-10, interferon (IFN)-γ receptor (IFNGR)-1, IL-23R, and IL-12RB, were risk factors of BD (24–29). These findings suggest that innate immune functions and bacterial identification systems play crucial roles in the pathogenesis of BD (Figure 2A). Lymphocytes obtained from BD patients react with human and/or mycobacterial heat shock protein peptides (30, 31). TLR-1, 2, and 4 are expressed more abundantly on neutrophils, monocytes, and lymphocytes derived from patients with BD than on those derived from healthy individuals (32).
The tumor necrosis factor-α-induced protein-3 (TNFAIP3) gene encodes A20 which regulates negatively TNFα pathway through the ubiquitin ligase activity (33). Patients with A20 haploinsufficiency develop BD phenotypes with an onset in childhood and young adulthood. Peripheral blood mononuclear cells (PBMCs) of the patients produced higher amounts of proinflammatory cytokines, such as IL-1β, TNFα, IL-17, and IL-18, in the presence of lipopolysaccharide (LPS), than those of healthy individuals.
2.1.3. Clinical phenotypes in BD
Recent clustering analyses have demonstrated that patients with BD can be divided into several subgroups according to their clinical symptoms to simplify and increase the accuracy of the clinical assessment (34, 35). For example, patients with mucocutaneous manifestations belonged to one subgroup, those with vascular manifestations belonged to another, and those with eye and/or CNS involvement belonged to another (35).
Interestingly, HLA-B51 positivity was relatively low in patients with BD with intestinal involvement, and male and female patients had eye and mucocutaneous involvement, respectively (Figure 2A) (34).
2.2. Histopathology of BD
Erythema nodosum (EN)-like lesions and papulopustular lesions are common in patients with BD (36). Histopathological examination of EN-like lesions in BD demonstrates panniculitis with vasculitis, leukocytoclastic and lymphocytic vasculitis, or phlebitis of the dermis. BD skin lesions are occasionally associated with thrombosis. The infiltrating immune cells consist mainly of neutrophils and lymphocytes (37). Similarly, in papulopustular lesions, neutrophilic infiltration was observed in the epidermis and around hair follicles. Lymphocytic infiltration has been suggested to occur as a late-stage inflammation in the neutrophilic reaction (38). Indeed, an investigation of patients with EN-like lesions revealed that neutrophilic dermatitis/panniculitis was more frequently observed in patients with BD with EN-like lesions than in patients with nodular vasculitis or EN of other immune disorders (39).
Sterile needle pricks often form inflammatory papules or pustules on the skin in patients with BD, with infiltration of neutrophils and lymphocytes as a positive pathergy test (40). Higher response rates were observed for pricks with larger gauge and/or blunt needles in patients with BD (41). A similar procedure using saliva pricks increased positive rates and was associated with disease activity (42). These data suggest that pathergy tests with needle pricks lead to the overactivation of immune cells against pathogen- and damage-associated molecular patterns in patients with BD (40).
In the pathergy test, lymphocyte and monocyte infiltrations were persistent up to 48 h after the needle prick in patients with BD compared to healthy individuals (43). Small clusters of elastase-positive neutrophils have been observed at needle prick sites in the relatively early phases of the test until 24 h after the prick (44, 45).
In the oral and genital ulcer lesions, leukocytoclastic vasculitis and lymphocyte infiltration were frequently observed in the lamina propria of the lesions (46). Intestinal ulceration is commonly found in the ileocecal region and is histologically characterized by neutrophilic and lymphocytic cell infiltration around lesions (47–49). The postmortem brain tissues of patients with neuro-BD demonstrate perivascular cuffing of macrophages and T cells in the parenchyma (50).
2.3. Peripheral blood cells in BD
2.3.1. Neutrophils in BD
Neutrophils produce reactive oxygen species (ROS) as a first-line defense against infectious pathogens (51). HLA-B51-positive neutrophils produce excessive superoxide compared to those without HLA in patients with BD and healthy individuals (23).
Neutrophil migration in patients with BD was enriched in an in vivo assay compared to that in healthy individuals, and the titers were significantly reduced in the disease remission phases (52). No significant differences were observed in superoxide production or adhesion capabilities between patients with BD and healthy individuals (52).
Neutrophil oxidative burst responses in patients with severe BD and organ involvement, such as complications of the eye, intestines, central nervous system, and cardiovascular system, were significantly higher than those in patients with mild BD (53).
2.3.2. Neutrophil extracellular traps in BD
Neutrophils activated by phorbol myristate acetate, IL-8, or LPS become flat and produce extracellular structures called NETs, which contain myeloperoxidase, neutrophil elastase, and cathepsin G (54). The nuclear enzyme protein-arginine deiminase type 4 (PAD4) citrullinates histones and promotes chromatin decondensation (55). NETs degrade virulence factors and inhibit the growth of bacteria such as Staphylococcus aureus, Salmonella typhimurium, and Shigella flexneri (54). On the other hand, damage-associated molecular patterns, such as cholesterol crystals, induced NET release of neutrophils and the NETs with cholesterol crystals promoted IL-1β production of macrophages (56). PAD4 deficient mice demonstrated reduced NET formation and a lower degree of thrombosis (57).
NETs may increase and induce thrombosis in patients with BD. Neutrophil NET release and serum levels of DNA components were significantly increased in patients with active BD compared to those in patients with inactive BD and healthy individuals (58). Neutrophil PAD4 expression level is significantly higher in patients with BD than in healthy individuals (59). Thrombin generation parameters in platelet-poor plasma obtained from patients with BD were significantly higher than those in healthy individuals, and correlated well with DNA component levels (58). NETs obtained from BD patients effectively promoted IL-8 and TNFα production of monocytes/macrophages compared with healthy individuals (60). Diffuse elastase-producing neutrophils were observed in BD skin panniculitis and vasculitis (59). NETs play a crucial role in the pathogenesis of BD.
Low-density neutrophils are immature or degranulated and recognized in human diseases (61). The frequencies of low-density neutrophils and NET production by the stimulated cells were increased in patients with BD compared to healthy individuals, but the cells exhibited decreased phagocytic capacities (62). Determining the mechanisms underlying these associations warrant further studies.
2.3.3. Monocytes/macrophages in BD
In in vitro experiments, bone marrow cells were differentiated into classically activated M1 macrophages in the presence of IFNγ and LPS and promoted production of proinflammatory cytokines, such as IL-1β, IL-6, and TNFα (63). Cells differentiate into alternatively activated M2 macrophages with IL-4 and increased IL-10 expression levels (63). M2 macrophages have been suggested to play distinct roles in lesions by reducing inflammation and promoting tissue remodeling.
Monocyte-derived macrophages treated with BD sera produced more effectively IL-12 and TNFα than those treated with sera of healthy individuals, suggesting M1 macrophage prevalence in peripheral blood of patients with BD (64). Stimulated M1 macrophages from patients with BD exhibit higher CCR1 expression levels than those from healthy individuals (65). Similarly, a gene expression profiling study demonstrated that, compared with healthy individuals, expression levels of proinflammatory monocyte-associated molecules, such as IL-1β and a CCR1 ligand CCL3, were elevated in patients with BD (66).
2.3.4. Inflammasome components in BD
The inflammasome complex consists of a cytosolic nucleotide-binding domain, leucine-rich-repeat-containing (NLR) proteins, AIM2-like receptor (ALR) proteins, adaptor apoptosis-associated speck-like protein containing a CARD (ASC), and pro-caspase-1 (67). The well-studied inflammasome NLRP3 responds to and is activated by bacterial, fungal, and viral pathogen-associated molecular patterns and damage-associated molecular patterns such as ATP and uric acid crystals (68). Activated caspase-1 processes pro-IL-1β and pro-IL-18 and biologically active cytokines are secreted (68). An autoinflammatory disease, cryopyrin-associated periodic syndrome, has been suggested to be associated with NLRP3 gene mutations (69).
In the PBMCs of patients with BD, the protein and mRNA levels of NLRP3, ASC, and caspase-1 were significantly increased compared with healthy individuals (70, 71). Activated PBMCs with LPS and ATP induced significantly higher levels of IL-1β compared with cells without the stimulation (70). NLRP3 levels of cerebrospinal fluids of BD patients with CNS involvement are positively correlated with serum C-reactive protein concentrations and erythrocyte sedimentation rates (71). Patients with BD share common clinical features, at least among those with autoinflammatory diseases.
2.3.5. Eosinophils in BD
Serum immunoglobulin E (IgE) and eosinophil counts are significantly reduced in patients with BD (72). Similarly, serum eosinophil cationic protein levels are significantly lower in active patients with BD than in inactive patients (73), suggesting a role for helper type 1 (Th1)-skewed cytokine responses in the pathogenesis of BD.
2.4. Humoral mediators in BD
2.4.1. Cytokines/chemokines in BD
A literature-based meta-analysis ascertained that serum IL-1β, IL-6, and TNFα were significantly increased in patients with BD compared with healthy individuals (Figure 2A) (74). High levels of Th1 and Th17 related cytokines, such as IL-1β, IL-6, IL-12, IL-17, IL-23, IFNγ, and TNFα, were identified in an array analysis (75). BD shares skewed IL-17/IL-23 pathways and several clinical features with spondyloarthritis and Vogt-Koyanagi-Harada disease (76, 77). Serum and plasma levels of CCL2, CCL3, and CXCL10 are higher in patients with BD compared with healthy individuals (78–80). Aqueous humor CXCL16 and CX3CL1 levels are higher in patients with BD than in healthy individuals and patients with Vogt-Koyanagi-Harada disease, suggesting an enhancement of Th1 responses in BD uveitis (81).
2.4.2. Matrix metalloproteinases in BD
Serum MMP-2 and MMP-9 levels were significantly higher in patients with vasculo-BD than in healthy individuals (Figure 2A) (82), especially in patients with aneurysms, similar to patients with abdominal aortic aneurysms (83). Synovial fluid concentrations of MMP-3 are significantly lower in patients with BD than in patients with rheumatoid arthritis and are comparable to those in patients with osteoarthritis (84).
2.4.3. Autoantibodies in BD
Autoantibodies were observed in patients with BD and reacted with an endothelial cell antigen, α-enolase (a positive rate of 38% by an ELISA) (85), a ubiquitously expressed membrane protein, prohibitin (28% by an ELISA) (86), α-tropomyosin (22% by an ELISA) (87), the nuclear mitotic apparatus protein (NuMA; 28% by an ELISA) (88), a riboflavin-containing flavoprotein (41% by an ELISA) (89), a membrane protein annexin A2 (34% by an ELISA) (90), a microtubule-related protein, kinectin (23% by an immunoprecipitation assay) (91), and an actin-binding protein, cofilin-1 (13% by western blotting; Figure 2A) (92). Thus, BD demonstrates a mixed pattern of autoinflammatory and autoimmune diseases within the spectrum (14).
3. Relapsing polychondritis
3.1. Epidemiology of RP
3.1.1. Genetic variations in RP
Epidemiological studies on patients with RP have identified that the incidence rates are approximately the same in several regions of the world (93–95). HLA-DR4 appears to be a susceptibility allele for RP (96). A recent genetic study demonstrated that HLA-DRB1*16:02, HLA-DQB1*05:02, and HLA-B*67:01 are associated with RP (Figure 2B) (97). Based on our data, the authors concluded that RP may be a distinct disease from other rheumatic diseases such as rheumatoid arthritis, systemic lupus erythematosus, Takayasu arthritis, and BD.
3.1.2. Environmental factors in RP
Based on the global incidence rates mentioned above, few environmental factors have been reported to be associated with RP pathogenesis. Interestingly, similar to the data of patients with BD, a metagenomic analysis demonstrated characteristic alterations in the gut microbiota composition, such as an increase in the abundance of Eubacterium, Ruminococcus, Bacteroides, and Veillonella, in patients with RP compared with that in healthy individuals. Here, we suggest an association between gut microbes and RP immunopathogenesis (98).
3.1.3. Clinical phenotypes of RP
In the clinical manifestations, respiratory and auricular involvement, which are two key hallmark features of RP, are recognized in 40–67% and 85–90% of patients with RP, respectively, at the latest follow-up (10–13). Tracheobronchial chondritis is increasingly recognized as distinct from other pathogenic complications (Figure 2B) (99, 100). Certainly, patients with RP with respiratory involvement have progressive disease compared to those with auricular involvement (101). Similar to the ocular involvement in BD, posterior segment inflammation is associated with a weak response to treatment (102).
3.2. Histopathology of RP
In the initial stages of the disease, mononuclear cells and neutrophils infiltrate the perichondrium beside the normal cartilage tissue (103, 104). Among the inflammatory cells in granulation tissues, CD4+ Th cells and CD68+ monocytes/macrophages are prevalent (105). Damaged chondrocytes produce MMP-3 and cathepsins, and the number of proteolytic enzyme-expressing cells correlates with that of apoptotic chondrocytes. Interestingly, MMP-3 was observed in the cartilage and perichondrium, whereas MMP-8 and MMP-9 were detected only in perichondrium granulation tissues. Cartilage tissues are progressively destroyed and finally replaced by fibrous connective tissues.
Notably, 1.6–38% of patients with RP showed skin involvement (9, 10, 12, 13, 95, 106); mucosal aphthosis, nodules on the limbs, purpura, and sterile pustules were the most common dermatological manifestations (107). Skin biopsy specimens revealed leukocytoclastic vasculitis, thrombosis of the skin vessels, septal panniculitis, neutrophil infiltration, and lymphocytic vasculitis as their histological findings. About 0–12% of patients with RP develop neurological manifestations, mainly confusion, seizures, delusions, amnesia, and/or dementia (108). Histopathology of the CNS exhibited perivascular cuffs of monocytes/lymphocytes and lymphocytic infiltration in the meninges and the cerebral parenchyma of patients with RP (109–111). In contrast to BD, gastrointestinal involvement is not generally identified in patients with RP (8–13, 93, 95, 106).
3.3. Peripheral blood cells in RP
3.3.1. Neutrophils in RP
As mentioned previously, neutrophil infiltration into cartilage tissues has been recognized since the early stages of chondritis (103, 104). Leukocyte clastic vasculitis and neutrophil infiltration are frequently observed (40%) in skin biopsy specimens (107). These results suggested that neutrophil activation plays a crucial role in the initiation of chondritis in patients with RP.
3.3.2. Monocytes/macrophages in RP
Gene expression level of IL-10, a major effector cytokine of regulatory T (Treg) cells, was significantly higher in freshly isolated PBMCs from patients with RP than in those from healthy individuals (112). After the initiation of cell culture with mitogen stimulation, IL-10 gene expression level was significantly decreased in patients with RP compared to that in healthy individuals. The researchers suggested that the gene expression analysis of PBMCs revealed Treg cell exhaustion or anergy of patients with RP and the skewed T cell function associated with innate cell overactivation (Figure 2B) (113).
3.3.3. Eosinophils in RP
Similar to neutrophil infiltration in RP lesions, eosinophils have been identified in specimens from the conjunctiva (114), nasal septum (104), and skin (115), probably indicating their early involvement in the process of chondritis in patients with RP.
3.4. Humoral mediators in RP
3.4.1. Cytokines/chemokines in RP
In Th cell-related cytokines, IFNγ and IL-10 were increased in the sera of patients with RP compared with healthy individuals (116, 117). Serum levels of innate cytokines and chemokines, such as IL-8, CCL2, and CCL4, were higher in patients with RP than in healthy individuals (Figure 2B) (116, 117).
3.4.2. MMPs in RP
Serum MMP-3 levels were higher in patients with RP than in healthy individuals, likely corresponding to histopathological changes in the patients (101, 116). Lymphocytes, monocytes/macrophages, and MMP-3 positive chondrocytes were simultaneously observed in RP lesions, suggesting that these cells aggravate chondritis (105). When the concentrations were compared between RP patients with and without respiratory involvement based on the epidemiological data mentioned above, MMP-3 levels increased significantly in patients with respiratory involvement compared to those without respiratory involvement (118).
In an in vitro assay, RP PBMCs upregulated mRNA expression of inflammatory cytokines IL-1β and IL-6 against stimulation compared with those of healthy individuals (118). Expression positively correlated with serum MMP-3 only in patients with RP and respiratory involvement. These data suggested that mononuclear cells with innate cytokines play a crucial role in the inflammatory processes of RP lesions, especially in patients with RP and respiratory involvement (Figure 2B).
3.4.3. Triggering receptor expressed on myeloid cell in RP
As the molecule name, neutrophils and monocytes/macrophages express TREM-1 and promote inflammation partly through TLR-4 pathway activation (119). Soluble TREM-1 is increased in the sera of RP patients with active disease compared with those with inactive disease, suggesting its possible role as a biomarker (116).
3.4.4. Autoantibodies in RP
Several cartilage elements were identified as potential autoantigens for RP (Figure 2B). An initial report of circulating autoantibodies in patients with RP revealed that, using indirect immunofluorescence, 33% of patients had autoantibodies against type 2 collagen, and the titers increased when acute symptoms were exhibited (120). Type 2 collagen-immunized rats develop auricular chondritis in the presence of type 2 collagen-reactive antibodies (121).
Matrilin-1 is a cartilage-specific protein, and its serum concentrations were found to be significantly elevated in an RP patient with tracheal chondritis who was monitored for 2 years (122). Autoantibodies for matrilin-1 were detected using ELISA in 13% of 97 patients with RP (123). In this study, researchers ascertained that sera from RP patients with positive anti-matrilin-1 antibodies reacted with newborn mouse tracheolaryngeal cartilage, whereas sera from patients with rheumatoid arthritis did not.
Certainly, several cartilage components are associated with phenotypic differences between patients with RP with and without respiratory involvement.
4. VEXAS and RP
A cutting-edge analysis demonstrated that patients with somatic mutations in UBA1, a gene encoding the ubiquitin activating enzyme E1, developed treatment-refractory severe autoinflammatory conditions in late middle age, such as vacuole, E1 enzyme, X-linked, autoinflammatory, and somatic syndrome (VEXAS) (124). It is characterized by refractory constitutional symptoms, ear and nose chondritis, and inflammatory arthritis. Patients with VEXAS often develop hematological disorders, such as myelodysplastic syndrome (MDS) and multiple myeloma, with a poor prognosis. Hypercellular bone marrow, vacuolization of erythroid and myeloid precursors, and spontaneously activated peripheral blood myeloid cells are common laboratory findings in patients with VEXAS.
When the symptoms were compared between RP patients with and without VEXAS, fever, ear chondritis, skin involvement (leukocytoclastic vasculitis and neutrophilic dermatosis), and periorbital edema were frequently observed in the patients with the syndrome (125). Notably, RP patients with VEXAS do not develop tracheobronchial chondritis during their clinical course. These data support the hypothesis that local interactions between inflammatory myeloid cells and chondrocytes/extracellular matrix are important for the initiation of chondritis in patients with RP.
5. Myelodysplastic syndrome in BD
A recent case report demonstrated that a 60-year-old man with somatic variants of UBA1 developed BD phenotypes with MDS and was resistant to aggressive treatments (126). This report demonstrates the possibility that the clinical spectrum of VEXAS can expand to BD manifestations.
In epidemiological studies, 10–20% of patients with MDS developed autoimmune manifestations (127, 128); conversely, autoimmune manifestations proceeded with the onset of MDS in 30% of patients (129). The prevalent autoimmune manifestations in a retrospective cohort study were neutrophilic dermatoses, such as Sweet syndrome, pyoderma gangrenosum, and BD (127). In this study, the deletion of 5q and trisomy 8 were associated with neutrophilic dermatosis and BD, respectively.
6. Mouth and genital ulcers with inflamed cartilage syndrome
Patients with MAGIC syndrome exhibit clinical features of both BD and RP. A prospective cohort study demonstrated good sensitivity in classifying patients according to McAdam’s or Damiani’s Criteria for RP and the International Criteria for BD (130). Interestingly, in this study, RP patients with MAGIC syndrome demonstrated higher frequency of anti-type 2 collagen autoantibodies than in those without MAGIC syndrome. This finding suggests differences in the underlying molecular mechanisms between the two respective groups of patients.
7. Innate immune responses in treatment of BD and RP
Tables 1, 2 show previously reported data on innate immune responses to therapeutic treatment in BD and RP, respectively (59, 131–149).
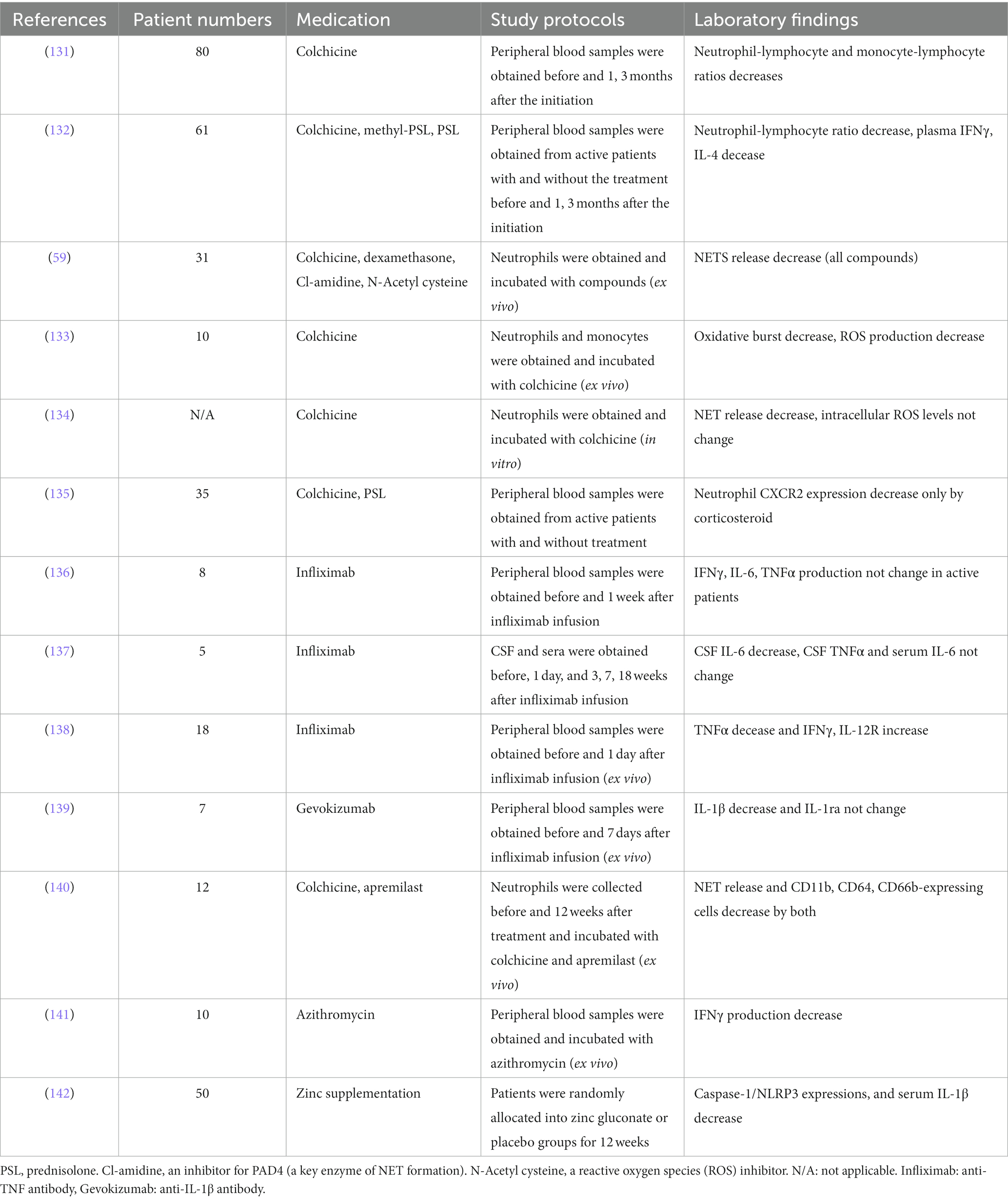
Table 1. Innate immune responses in vivo and in vitro induced by immunosuppressants in patients with Behçet disease.
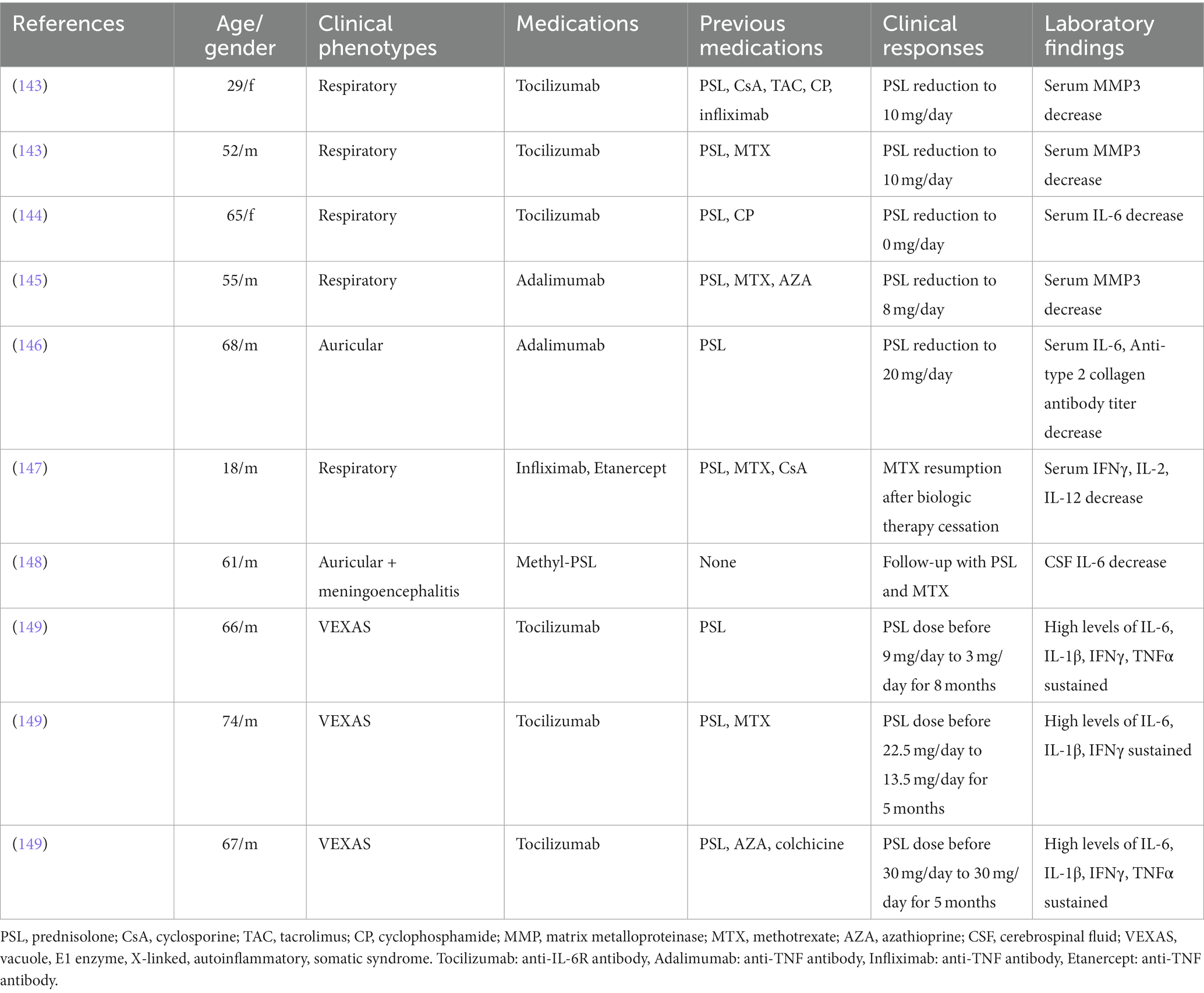
Table 2. Induction of innate immune responses by administration of immunosuppressants in patients with relapsing polychondritis.
Colchicine reduced neutrophil and monocyte infiltration into lesions by decreasing the expression levels of adhesion molecules, such as selectin P ligand (SELPLG) and platelet endothelial cell adhesion molecule-1 (PECAM-1) (150). In a study using BD neutrophils, colchicine reduced NET release to an extent similar to that of methylprednisolone, a PAD inhibitor (Cl-amidine), and an ROS inhibitor (N-acetyl cysteine) (59). Biological agents are recommended for the treatment of patients with refractory BD and RP (76, 151).
A phosphodiesterase (PDE)-4 inhibitor, apremilast, reduces degradation of cyclic adenosine monophosphate and inhibits production of proinflammatory cytokines such as IL-12, IL-23, and TNFα from PBMCs (152). Additionally, apremilast decreased the total number of oral ulcers during a 12-week placebo-controlled clinical trial (153).
Zinc plays a crucial role in innate and adaptive immune function and its depletion has led to IL-1β secretion increase of stimulated macrophages through induction of NLRP3 inflammasome (154). Zinc gluconate supplementation reduced the expression levels of serum IL-1β and white blood cell NLRP3, while decreasing the incidence of genital ulcer in patients with BD within 3 months (142).
Sustained high concentrations of inflammatory cytokines have been observed in patients with VEXAS, indicating the refractory nature of the disease, even after the initiation of an anti-IL-6 agent, tocilizumab, administration (149).
8. Conclusion
This review collates and summarizes the recent advances in our understanding of the innate cell mediated immunopathology of BD and RP. These studies suggest that innate immune cells are crucial in the direct initiation of local inflammation under dysregulated lymphocyte function in inflammatory diseases (Figure 1). Certain susceptibility genes characterizing BD are associated with several rare monogenic autoinflammatory diseases, such as FMF and TRAPS. Meanwhile, RP is characterized by distinct clinical phenotypes that are associated with several autoantigens in humans and mice. Adaptive immune cells and genetic/environmental factors simultaneously enhance innate immune responses in BD and RP. The identification of controllable active elements is important for the development of effective and safe treatment approaches.
Author contributions
JS and MM conceived and prepared the manuscript. YMiz, NA, KT, and YMiy prepared and edited the manuscript. JS, MM, YMiz, NA, KT, and YMiy approved the published version of the manuscript.
Funding
The work of YMiy is supported by the Japanese Society for the Promotion of Science (JSPS) KAKENHI grant number JP22K08531 and AMED under Grant Number 22jm0210069h004 and 22jm0610070h0001, Kato Memorial Bioscience Foundation, The Naito Foundation and The Uehara Memorial Foundation, MM is supported by JSPS KAKENHI (JP21K06955 and JP21H02394), SRF foundation (2022Y003), Kansai Medical University alumni association (Katano Prize) and Kansai Medical University Molecular Imaging Center of Diseases. The work of JS and MM is supported by JSPS KAKENHI (JP22K08532 and JP23K05618). The work of NA and MM is supported by JSPS KAKENHI (JP21K07379).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sakane, T, Takeno, M, Suzuki, N, and Inaba, G. Behcet's disease. N Engl J Med. (1999) 341:1284–91.
2. Criteria for diagnosis of Behcet's disease. International study group for Behcet's disease. Lancet. (1990) 335:1078–80.
3. Mays, JW, Sarmadi, M, and Moutsopoulos, NM. Oral manifestations of systemic autoimmune and inflammatory diseases: diagnosis and clinical management. J Evid Based Dent Pract. (2012) 12:265–82. doi: 10.1016/S1532-3382(12)70051-9
4. Hou, S, Li, N, Liao, X, Kijlstra, A, and Yang, P. Uveitis genetics. Exp Eye Res. (2020) 190:107853. doi: 10.1016/j.exer.2019.107853
5. Singer, O. Cogan and Behcet syndromes. Rheum Dis Clin N Am. (2015) 41:75–91.viii. doi: 10.1016/j.rdc.2014.09.007
6. Borhani-Haghighi, A, Kardeh, B, Banerjee, S, Yadollahikhales, G, Safari, A, Sahraian, MA, et al. Neuro-Behcet's disease: an update on diagnosis, differential diagnoses, and treatment. Mult Scler Relat Disord. (2019) 39:101906. doi: 10.1016/j.msard.2019.101906
7. Skef, W, Hamilton, MJ, and Arayssi, T. Gastrointestinal Behcet's disease: a review. World J Gastroenterol. (2015) 21:3801–12. doi: 10.3748/wjg.v21.i13.3801
8. Letko, E, Zafirakis, P, Baltatzis, S, Voudouri, A, Livir-Rallatos, C, and Foster, CS. Relapsing polychondritis: a clinical review. Semin Arthritis Rheum. (2002) 31:384–95. doi: 10.1053/sarh.2002.32586
9. Shimizu, J, Yamano, Y, Kawahata, K, and Suzuki, N. Elucidation of predictors of disease progression in patients with relapsing polychondritis at the onset: potential impact on patient monitoring. BMC Rheumatol. (2020) 4:41. doi: 10.1186/s41927-020-00141-8
10. McAdam, LP, O'Hanlan, MA, Bluestone, R, and Pearson, CM. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). (1976) 55:193–215.
11. Damiani, JM, and Levine, HL. Relapsing polychondritis--report of ten cases. Laryngoscope. (1979) 89:929–46.
12. Michet, CJ Jr, McKenna, CH, Luthra, HS, and O'Fallon, WM. Relapsing polychondritis. Survival and predictive role of early disease manifestations. Ann Intern Med. (1986) 104:74–8.
13. Trentham, DE, and Le, CH. Relapsing polychondritis. Ann Intern Med. (1998) 129:114–22. doi: 10.7326/0003-4819-129-2-199807150-00011
14. McGonagle, D, and McDermott, MF. A proposed classification of the immunological diseases. PLoS Med. (2006) 3:e297. doi: 10.1371/journal.pmed.0030297
15. Verity, DH, Marr, JE, Ohno, S, Wallace, GR, and Stanford, MR. Behcet's disease, the silk road and HLA-B51: historical and geographical perspectives. Tissue Antigens. (1999) 54:213–20.
16. Mumcu, G, Ergun, T, Inanc, N, Fresko, I, Atalay, T, Hayran, O, et al. Oral health is impaired in Behcet's disease and is associated with disease severity. Rheumatology (Oxford). (2004) 43:1028–33. doi: 10.1093/rheumatology/keh236
17. Yokota, K, Hayashi, S, Araki, Y, Isogai, E, Kotake, S, Yoshikawa, K, et al. Characterization of Streptococcus sanguis isolated from patients with Behçet's disease. Microbiol Immunol. (1995) 39:729–32. doi: 10.1111/j.1348-0421.1995.tb03249.x
18. Senusi, A, Kang, A, Buchanan, JAG, Adesanya, A, Aloraini, G, Stanford, M, et al. New mouthwash: an efficacious intervention for oral ulceration associated with Behçet's disease. Br J Oral Maxillofac Surg. (2020) 58:1034–9. doi: 10.1016/j.bjoms.2020.07.027
19. Seoudi, N, Bergmeier, LA, Drobniewski, F, Paster, B, and Fortune, F. The oral mucosal and salivary microbial community of Behcet's syndrome and recurrent aphthous stomatitis. J Oral Microbiol. (2015) 7:27150. doi: 10.3402/jom.v7.27150
20. Coit, P, Mumcu, G, Ture-Ozdemir, F, Unal, AU, Alpar, U, Bostanci, N, et al. Sequencing of 16S rRNA reveals a distinct salivary microbiome signature in Behcet's disease. Clin Immunol. (2016) 169:28–35. doi: 10.1016/j.clim.2016.06.002
21. Consolandi, C, Turroni, S, Emmi, G, Severgnini, M, Fiori, J, Peano, C, et al. Behçet's syndrome patients exhibit specific microbiome signature. Autoimmun Rev. (2015) 14:269–76. doi: 10.1016/j.autrev.2014.11.009
22. Shimizu, J, Kubota, T, Takada, E, Takai, K, Fujiwara, N, Arimitsu, N, et al. Bifidobacteria abundance-featured gut microbiota compositional change in patients with Behcet's disease. PLoS One. (2016) 11:e0153746. doi: 10.1371/journal.pone.0153746
23. Takeno, M, Kariyone, A, Yamashita, N, Takiguchi, M, Mizushima, Y, Kaneoka, H, et al. Excessive function of peripheral blood neutrophils from patients with Behcet's disease and from HLA-B51 transgenic mice. Arthritis Rheum. (1995) 38:426–33. doi: 10.1002/art.1780380321
24. Takeuchi, M, Mizuki, N, Meguro, A, Ombrello, MJ, Kirino, Y, Satorius, C, et al. Dense genotyping of immune-related loci implicates host responses to microbial exposure in Behçet's disease susceptibility. Nat Genet. (2017) 49:438–43. doi: 10.1038/ng.3786
25. Kirino, Y, Bertsias, G, Ishigatsubo, Y, Mizuki, N, Tugal-Tutkun, I, Seyahi, E, et al. Genome-wide association analysis identifies new susceptibility loci for Behçet's disease and epistasis between HLA-B*51 and ERAP1. Nat Genet. (2013) 45:202–7. doi: 10.1038/ng.2520
26. Kirino, Y, Zhou, Q, Ishigatsubo, Y, Mizuki, N, Tugal-Tutkun, I, Seyahi, E, et al. Targeted resequencing implicates the familial Mediterranean fever gene MEFV and the toll-like receptor 4 gene TLR4 in Behçet disease. Proc Natl Acad Sci U S A. (2013) 110:8134–9. doi: 10.1073/pnas.1306352110
27. Hughes, T, Coit, P, Adler, A, Yilmaz, V, Aksu, K, Düzgün, N, et al. Identification of multiple independent susceptibility loci in the HLA region in Behçet's disease. Nat Genet. (2013) 45:319–24. doi: 10.1038/ng.2551
28. Mizuki, N, Meguro, A, Ota, M, Ohno, S, Shiota, T, Kawagoe, T, et al. Genome-wide association studies identify IL23R-IL12RB2 and IL10 as Behçet's disease susceptibility loci. Nat Genet. (2010) 42:703–6. doi: 10.1038/ng.624
29. Remmers, EF, Cosan, F, Kirino, Y, Ombrello, MJ, Abaci, N, Satorius, C, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behçet's disease. Nat Genet. (2010) 42:698–702. doi: 10.1038/ng.625
30. Pervin, K, Childerstone, A, Shinnick, T, Mizushima, Y, van der Zee, R, Hasan, A, et al. T cell epitope expression of mycobacterial and homologous human 65-kilodalton heat shock protein peptides in short term cell lines from patients with Behçet's disease. J Immunol. (1993) 151:2273–82. doi: 10.4049/jimmunol.151.4.2273
31. Filleron, A, Tran, TA, Hubert, A, Letierce, A, Churlaud, G, Koné-Paut, I, et al. Regulatory T cell/Th17 balance in the pathogenesis of paediatric Behçet disease. Rheumatology (Oxford). (2021) 61:422–9. doi: 10.1093/rheumatology/keab253
32. van der Houwen, TB, Dik, WA, Goeijenbier, M, Hayat, M, Nagtzaam, NMA, van Hagen, M, et al. Leukocyte toll-like receptor expression in pathergy positive and negative Behçet’s disease patients. Rheumatology (Oxford). (2020) 59:3971–9. doi: 10.1093/rheumatology/keaa251
33. Zhou, Q, Wang, H, Schwartz, DM, Stoffels, M, Park, YH, Zhang, Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. (2016) 48:67–73. doi: 10.1038/ng.3459
34. Soejima, Y, Kirino, Y, Takeno, M, Kurosawa, M, Takeuchi, M, Yoshimi, R, et al. Changes in the proportion of clinical clusters contribute to the phenotypic evolution of Behçet's disease in Japan. Arthritis Res Ther. (2021) 23:49. doi: 10.1186/s13075-020-02406-6
35. Bettiol, A, Hatemi, G, Vannozzi, L, Barilaro, A, Prisco, D, and Emmi, G. Treating the different phenotypes of Behçet's syndrome. Front Immunol. (2019) 10:2830. doi: 10.3389/fimmu.2019.02830
36. Nakamura, K, Tsunemi, Y, Kaneko, F, and Alpsoy, E. Mucocutaneous manifestations of Behcet's disease. Front Med. (2020) 7:613432. doi: 10.3389/fmed.2020.613432
37. Misago, N, Tada, Y, Koarada, S, and Narisawa, Y. Erythema nodosum-like lesions in Behcet's disease: a clinicopathological study of 26 cases. Acta Derm Venereol. (2012) 92:681–6. doi: 10.2340/00015555-1349
38. Jorizzo, JL, Abernethy, JL, White, WL, Mangelsdorf, HC, Zouboulis, CC, Sarica, R, et al. Mucocutaneous criteria for the diagnosis of Behçet's disease: an analysis of clinicopathologic data from multiple international centers. J Am Acad Dermatol. (1995) 32:968–76. doi: 10.1016/0190-9622(95)91333-5
39. Demirkesen, C, Tüzüner, N, Mat, C, Senocak, M, Büyükbabani, N, Tüzün, Y, et al. Clinicopathologic evaluation of nodular cutaneous lesions of Behçet syndrome. Am J Clin Pathol. (2001) 116:341–6. doi: 10.1309/GCTH-0060-55K8-XCTT
41. Dilsen, N, Konice, M, Aral, O, Ocal, L, Inanc, M, and Gul, A. Comparative study of the skin pathergy test with blunt and sharp needles in Behcet's disease: confirmed specificity but decreased sensitivity with sharp needles. Ann Rheum Dis. (1993) 52:823–5.
42. Shenavandeh, S, Sadeghi, SMK, and Aflaki, E. Pathergy test with a 23G needle with and without self-saliva in patients with Behcet's disease, recurrent aphthous stomatitis and control group compared to the 20G test. Reumatologia. (2021) 59:302–8. doi: 10.5114/reum.2021.110567
43. Melikoglu, M, Uysal, S, Krueger, JG, Kaplan, G, Gogus, F, Yazici, H, et al. Characterization of the divergent wound-healing responses occurring in the pathergy reaction and normal healthy volunteers. J Immunol. (2006) 177:6415–21. doi: 10.4049/jimmunol.177.9.6415
44. Ergun, T, Gürbüz, O, Harvell, J, Jorizzo, J, and White, W. The histopathology of pathergy: a chronologic study of skin hyperreactivity in Behçet's disease. Int J Dermatol. (1998) 37:929–33. doi: 10.1046/j.1365-4362.1998.00474.x
45. Gül, A, Esin, S, Dilsen, N, Koniçe, M, Wigzell, H, and Biberfeld, P. Immunohistology of skin pathergy reaction in Behçet's disease. Br J Dermatol. (1995) 132:901–7. doi: 10.1111/j.1365-2133.1995.tb16946.x
46. Chun, SI, Su, WP, and Lee, S. Histopathologic study of cutaneous lesions in Behçet's syndrome. J Dermatol. (1990) 17:333–41.
47. Köklü, S, Yüksel, O, Onur, I, Ünverdi, S, Bıyıkoğlu, İ, Akbal, E, et al. Ileocolonic involvement in Behçet's disease: endoscopic and histological evaluation. Digestion. (2010) 81:214–7. doi: 10.1159/000264643
48. Hayasaki, N, Ito, M, Suzuki, T, Ina, K, Ando, T, Kusugami, K, et al. Neutrophilic phlebitis is characteristic of intestinal Behçet's disease and simple ulcer syndrome. Histopathology. (2004) 45:377–83. doi: 10.1111/j.1365-2559.2004.01954.x
49. Takada, Y, Fujita, Y, Igarashi, M, Katsumata, T, Okabe, H, Saigenji, K, et al. Intestinal Behçet's disease--pathognomonic changes in intramucosal lymphoid tissues and effect of a "rest cure" on intestinal lesions. J Gastroenterol. (1997) 32:598–604. doi: 10.1007/BF02934108
50. Hirohata, S, Kikuchi, H, Sawada, T, Nagafuchi, H, Kuwana, M, Takeno, M, et al. Clinical characteristics of neuro-Behcet's disease in Japan: a multicenter retrospective analysis. Mod Rheumatol. (2012) 22:405–13. doi: 10.3109/s10165-011-0533-5
51. Burn, GL, Foti, A, Marsman, G, Patel, DF, and Zychlinsky, A. The neutrophil. Immunity. (2021) 54:1377–91. doi: 10.1016/j.immuni.2021.06.006
52. Carletto, A, Pacor, ML, Biasi, D, Caramaschi, P, Zeminian, S, Bellavite, P, et al. Changes of neutrophil migration without modification of in vitro metabolism and adhesion in Behçet's disease. J Rheumatol. (1997) 24:1332–6.
53. Perazzio, SF, Soeiro-Pereira, PV, de Souza, AW, Condino-Neto, A, and Andrade, LE. Behçet's disease heterogeneity: cytokine production and oxidative burst of phagocytes are altered in patients with severe manifestations. Clin Exp Rheumatol. (2015) 33:S85–95.
54. Brinkmann, V, Reichard, U, Goosmann, C, Fauler, B, Uhlemann, Y, Weiss, DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
55. Wang, Y, Wysocka, J, Sayegh, J, Lee, YH, Perlin, JR, Leonelli, L, et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science. (2004) 306:279–83. doi: 10.1126/science.1101400
56. Warnatsch, A, Ioannou, M, Wang, Q, and Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. (2015) 349:316–20. doi: 10.1126/science.aaa8064
57. Martinod, K, Demers, M, Fuchs, TA, Wong, SL, Brill, A, Gallant, M, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A. (2013) 110:8674–9. doi: 10.1073/pnas.1301059110
58. le Joncour, A, Martos, R, Loyau, S, Lelay, N, Dossier, A, Cazes, A, et al. Critical role of neutrophil extracellular traps (NETs) in patients with Behcet's disease. Ann Rheum Dis. (2019) 78:1274–82. doi: 10.1136/annrheumdis-2018-214335
59. Safi, R, Kallas, R, Bardawil, T, Mehanna, CJ, Abbas, O, Hamam, R, et al. Neutrophils contribute to vasculitis by increased release of neutrophil extracellular traps in Behçet's disease. J Dermatol Sci. (2018) 92:143–50. doi: 10.1016/j.jdermsci.2018.08.010
60. Li, L, Yu, X, Liu, J, Wang, Z, Li, C, Shi, J, et al. Neutrophil extracellular traps promote aberrant macrophages activation in Behcet's disease. Front Immunol. (2020) 11:590622. doi: 10.3389/fimmu.2020.590622
61. Mistry, P, Nakabo, S, O’Neil, L, Goel, RR, Jiang, K, Carmona-Rivera, C, et al. Transcriptomic, epigenetic, and functional analyses implicate neutrophil diversity in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A. (2019) 116:25222–8. doi: 10.1073/pnas.1908576116
62. Murad, M, Low, L, Davidson, M, Murray, PI, Rauz, S, and Wallace, GR. Low density neutrophils are increased in patients with Behcet's disease but do not explain differences in neutrophil function. J Inflamm. (2022) 19:5. doi: 10.1186/s12950-022-00302-1
63. Jablonski, KA, Amici, SA, Webb, LM, Ruiz-Rosado, JD, Popovich, PG, Partida-Sanchez, S, et al. Novel markers to delineate murine M1 and M2 macrophages. PLoS One. (2015) 10:e0145342. doi: 10.1371/journal.pone.0145342
64. Wu, X, Wang, Z, Shi, J, Yu, X, Li, C, Liu, J, et al. Macrophage polarization toward M1 phenotype through NF-κB signaling in patients with Behçet's disease. Arthritis Res Ther. (2022) 24:249. doi: 10.1186/s13075-022-02938-z
65. Nakano, H, Kirino, Y, Takeno, M, Higashitani, K, Nagai, H, Yoshimi, R, et al. GWAS-identified CCR1 and IL10 loci contribute to M1 macrophage-predominant inflammation in Behçet's disease. Arthritis Res Ther. (2018) 20:124. doi: 10.1186/s13075-018-1613-0
66. Verrou, KM, Vlachogiannis, NI, Ampatziadis-Michailidis, G, Moulos, P, Pavlopoulos, GA, Hatzis, P, et al. Distinct transcriptional profile of blood mononuclear cells in Behçet's disease: insights into the central role of neutrophil chemotaxis. Rheumatology (Oxford). (2021) 60:4910–9. doi: 10.1093/rheumatology/keab052
67. Rathinam, VA, and Fitzgerald, KA. Inflammasome complexes: emerging mechanisms and effector functions. Cells. (2016) 165:792–800. doi: 10.1016/j.cell.2016.03.046
68. Lamkanfi, M, and Dixit, VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. (2012) 28:137–61. doi: 10.1146/annurev-cellbio-101011-155745
69. Booshehri, LM, and Hoffman, HM. CAPS and NLRP3. J Clin Immunol. (2019) 39:277–86. doi: 10.1007/s10875-019-00638-z
70. Kim, EH, Park, MJ, Park, S, and Lee, ES. Increased expression of the NLRP3 inflammasome components in patients with Behçet's disease. J Inflamm. (2015) 12:41. doi: 10.1186/s12950-015-0086-z
71. Hamzaoui, K, Borhani-Haghighi, A, Dhifallah, IB, and Hamzaoui, A. Elevated levels of IL-32 in cerebrospinal fluid of neuro-Behcet disease: correlation with NLRP3 inflammasome. J Neuroimmunol. (2022) 365:577820. doi: 10.1016/j.jneuroim.2022.577820
72. Chang, HK, Lee, SS, Kim, JW, Jee, YK, Kim, JU, Lee, YW, et al. The prevalence of atopy and atopic diseases in Behçet's disease. Clin Exp Rheumatol. (2003) 21:S31–4.
73. Tas, DA, Ozer, HT, and Erken, E. Serum eosinophil cationic protein levels in Behçet's disease and its relation to clinical activity. Asian Pac J Allergy Immunol. (2013) 31:67–72.
74. Hirahara, L, Takase-Minegishi, K, Kirino, Y, Iizuka-Iribe, Y, Soejima, Y, Yoshimi, R, et al. The roles of monocytes and macrophages in Behçet's disease with focus on M1 and M2 polarization. Front Immunol. (2022) 13:852297. doi: 10.3389/fimmu.2022.852297
75. Gholijani, N, Ataollahi, MR, Samiei, A, Aflaki, E, Shenavandeh, S, and Kamali-Sarvestani, E. An elevated pro-inflammatory cytokines profile in Behcet's disease: a multiplex analysis. Immunol Lett. (2017) 186:46–51. doi: 10.1016/j.imlet.2016.12.001
76. Yazici, H, Seyahi, E, Hatemi, G, and Yazici, Y. Behçet syndrome: a contemporary view. Nat Rev Rheumatol. (2018) 14:119. doi: 10.1038/nrrheum.2018.3
77. Zhong, Z, Su, G, Kijlstra, A, and Yang, P. Activation of the interleukin-23/interleukin-17 signalling pathway in autoinflammatory and autoimmune uveitis. Prog Retin Eye Res. (2021) 80:100866. doi: 10.1016/j.preteyeres.2020.100866
78. Kaburaki, T, Fujino, Y, Kawashima, H, Merino, G, Numaga, J, Chen, J, et al. Plasma and whole-blood chemokine levels in patients with Behcet's disease. Graefes Arch Clin Exp Ophthalmol. (2003) 241:353–8. doi: 10.1007/s00417-003-0668-y
79. Ozer, HT, Erken, E, Gunesacar, R, and Kara, O. Serum RANTES, MIP-1alpha, and MCP-1 levels in Behçet's disease. Rheumatol Int. (2005) 25:487–8. doi: 10.1007/s00296-004-0519-0
80. Lee, SJ, Kang, SE, Kang, EH, Choi, BY, Masek-Hammerman, K, Syed, J, et al. CXCL10/CXCR3 axis is associated with disease activity and the development of mucocutaneous lesions in patients with Behçet's disease. Sci Rep. (2017) 7:14720. doi: 10.1038/s41598-017-15189-9
81. el-Asrar, AMA, Berghmans, N, al-Obeidan, SA, Gikandi, PW, Opdenakker, G, van Damme, J, et al. Differential CXC and CX3C chemokine expression profiles in aqueous humor of patients with specific endogenous Uveitic entities. Invest Ophthalmol Vis Sci. (2018) 59:2222–8. doi: 10.1167/iovs.17-23225
82. Pay, S, Abbasov, T, Erdem, H, Musabak, U, Simsek, I, Pekel, A, et al. Serum MMP-2 and MMP-9 in patients with Behçet's disease: do their higher levels correlate to vasculo-Behçet's disease associated with aneurysm formation? Clin Exp Rheumatol. (2007) 25:S70–5.
83. Cai, D, Sun, C, Zhang, G, Que, X, Fujise, K, Weintraub, NL, et al. A novel mechanism underlying inflammatory smooth muscle phenotype in abdominal aortic aneurysm. Circ Res. (2021) 129:e202–14. doi: 10.1161/CIRCRESAHA.121.319374
84. Pay, S, Erdem, H, Pekel, A, Simsek, I, Musabak, U, Sengul, A, et al. Synovial proinflammatory cytokines and their correlation with matrix metalloproteinase-3 expression in Behçet's disease. Does interleukin-1beta play a major role in Behçet's synovitis? Rheumatol Int. (2006) 26:608–13. doi: 10.1007/s00296-005-0040-0
85. Lee, KH, Chung, HS, Kim, HS, Oh, SH, Ha, MK, Baik, JH, et al. Human ?-enolase from endothelial cells as a target antigen of anti-endothelial cell antibody in Behçet's disease. Arthritis Rheum. (2003) 48:2025–35. doi: 10.1002/art.11074
86. Xun, Y, Chen, P, Yan, H, Yang, W, Shi, L, Chen, G, et al. Identification of prohibitin as an antigen in Behcet's disease. Biochem Biophys Res Commun. (2014) 451:389–93. doi: 10.1016/j.bbrc.2014.07.126
87. Mahesh, SP, Li, Z, Buggage, R, Mor, F, Cohen, IR, Chew, EY, et al. Alpha tropomyosin as a self-antigen in patients with Behçet's disease. Clin Exp Immunol. (2005) 140:368–75. doi: 10.1111/j.1365-2249.2005.02760.x
88. Hussain, M, Ma, F, Chen, P, Tian, Y, and Du, H. Circulation autoantibodies against C-terminus of NuMA in patients with Behcet's disease. Cent Eur J Immunol. (2020) 45:86–92. doi: 10.5114/ceji.2020.94710
89. Chen, P, Yang, W, Tian, Y, Sun, S, Chen, G, Zhang, C, et al. Electron transfer flavoprotein subunit beta is a candidate endothelial cell autoantigen in Behçet’s disease. PLoS One. (2015) 10:e0124760. doi: 10.1371/journal.pone.0124760
90. Chen, P, Yan, H, Tian, Y, Xun, Y, Shi, L, Bao, R, et al. Annexin A2 as a target endothelial cell membrane autoantigen in Behçet's disease. Sci Rep. (2015) 5:8162. doi: 10.1038/srep08162
91. Lu, Y, Ye, P, Chen, SL, Tan, EM, and Chan, EK. Identification of kinectin as a novel Behcet's disease autoantigen. Arthritis Res Ther. (2005) 7:R1133–9. doi: 10.1186/ar1798
92. Ooka, S, Nakano, H, Matsuda, T, Okamoto, K, Suematsu, N, Kurokawa, MS, et al. Proteomic surveillance of autoantigens in patients with Behcet's disease by a proteomic approach. Microbiol Immunol. (2010) 54:354–61. doi: 10.1111/j.1348-0421.2010.00215.x
93. Mathew, SD, Battafarano, DF, and Morris, MJ. Relapsing polychondritis in the Department of Defense population and review of the literature. Semin Arthritis Rheum. (2012) 42:70–83. doi: 10.1016/j.semarthrit.2011.12.007
94. Horvath, A, Pall, N, Molnar, K, Kovats, T, Surjan, G, Vicsek, T, et al. A nationwide study of the epidemiology of relapsing polychondritis. Clin Epidemiol. (2016) 8:211–30. doi: 10.2147/CLEP.S91439
95. Hazra, N, Dregan, A, Charlton, J, Gulliford, MC, and D'Cruz, DP. Incidence and mortality of relapsing polychondritis in the UK: a population-based cohort study. Rheumatology (Oxford). (2015) 54:2181–7. doi: 10.1093/rheumatology/kev240
96. Lang, B, Rothenfusser, A, Lanchbury, JS, Rauh, G, Breedveld, FC, Urlacher, A, et al. Susceptibility to relapsing polychondritis is associated with HLA-DR4. Arthritis Rheum. (1993) 36:660–4. doi: 10.1002/art.1780360513
97. Terao, C, Yoshifuji, H, Yamano, Y, Kojima, H, Yurugi, K, Miura, Y, et al. Genotyping of relapsing polychondritis identified novel susceptibility HLA alleles and distinct genetic characteristics from other rheumatic diseases. Rheumatology (Oxford). (2016) 55:1686–92. doi: 10.1093/rheumatology/kew233
98. Shimizu, J, Takai, K, Takada, E, Fujiwara, N, Arimitsu, N, Ueda, Y, et al. Possible association of proinflammatory cytokines including IL1beta and TNFalpha with enhanced Th17 cell differentiation in patients with Behcet's disease. Clin Rheumatol. (2016) 35:1857–63. doi: 10.1007/s10067-015-2966-2
99. de Montmollin, N, Dusser, D, Lorut, C, Dion, J, Costedoat-Chalumeau, N, Mouthon, L, et al. Tracheobronchial involvement of relapsing polychondritis. Autoimmun Rev. (2019) 18:102353. doi: 10.1016/j.autrev.2019.102353
100. Liu, Y, Li, X, Cheng, L, Zhan, H, Huang, Y, Li, H, et al. Progress and challenges in the use of blood biomarkers in relapsing polychondritis. Clin Exp Immunol. (2023) 212:199–211. doi: 10.1093/cei/uxad014
101. Shimizu, J, Yamano, Y, Kawahata, K, and Suzuki, N. Relapsing polychondritis patients were divided into three subgroups: patients with respiratory involvement (R subgroup), patients with auricular involvement (A subgroup), and overlapping patients with both involvements (O subgroup), and each group had distinctive clinical characteristics. Medicine (Baltimore). (2018) 97:e12837. doi: 10.1097/MD.0000000000012837
102. Yang, P, Yuan, W, du, L, Zhou, Q, Wang, C, Ye, Z, et al. Clinical features of Chinese patients with relapsing polychondritis. Br J Ophthalmol. (2019) 103:1129–32. doi: 10.1136/bjophthalmol-2018-312660
103. Kumakiri, K, Sakamoto, T, Karahashi, T, Mineta, H, and Takebayashi, S. A case of relapsing polychondritis preceded by inner ear involvement. Auris Nasus Larynx. (2005) 32:71–6. doi: 10.1016/j.anl.2004.09.012
104. Kobayashi, T, Moody, S, Komori, M, Jibatake, A, and Yaegashi, M. Early stage relapsing polychondritis diagnosed by nasal septum biopsy. Case Rep Med. (2015) 2015:307868. doi: 10.1155/2015/307868
105. Ouchi, N, Uzuki, M, Kamataki, A, Miura, Y, and Sawai, T. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. (2011) 38:730–7. doi: 10.3899/jrheum.101044
106. Zhang, L, Yun, S, Wu, T, He, Y, Guo, J, Han, L, et al. Clinical patterns and the evolution of relapsing polychondritis based on organ involvement: a Chinese retrospective cohort study. Orphanet J Rare Dis. (2021) 16:225. doi: 10.1186/s13023-021-01861-x
107. Francès, C, el Rassi, R, Laporte, JL, Rybojad, M, Papo, T, and Piette, JC. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). (2001) 80:173–9. doi: 10.1097/00005792-200105000-00003
108. Kondo, T, Fukuta, M, Takemoto, A, Takami, Y, Sato, M, Takahashi, N, et al. Limbic encephalitis associated with relapsing polychondritis responded to infliximab and maintained its condition without recurrence after discontinuation: a case report and review of the literature. Nagoya J Med Sci. (2014) 76:361–8.
109. Fujiki, F, Tsuboi, Y, Hashimoto, K, Nakajima, M, and Yamada, T. Non-herpetic limbic encephalitis associated with relapsing polychondritis. J Neurol Neurosurg Psychiatry. (2004) 75:1646–7. doi: 10.1136/jnnp.2003.035170
110. Storey, K, Matěj, R, and Rusina, R. Unusual association of seronegative, nonparaneoplastic limbic encephalitis and relapsing polychondritis in a patient with history of thymectomy for myasthenia: a case study. J Neurol. (2011) 258:159–61. doi: 10.1007/s00415-010-5691-4
111. Yan, M, Cooper, W, Harper, C, and Schwartz, R. Dementia in a patient with non-paraneoplastic limbic encephalitis associated with relapsing polychondritis. Pathology. (2006) 38:596–9. doi: 10.1080/00313020601023989
112. Shimizu, J, Kubota, T, Takada, E, Takai, K, Fujiwara, N, Arimitsu, N, et al. Propionate-producing bacteria in the intestine may associate with skewed responses of IL10-producing regulatory T cells in patients with relapsing polychondritis. PLoS One. (2018) 13:e0203657. doi: 10.1371/journal.pone.0203657
113. Shimizu, J, and Suzuki, N. Mechanical model of steady-state and inflammatory conditions in patients with relapsing polychondritis: a review. Medicine (Baltimore). (2022) 101:e28852. doi: 10.1097/MD.0000000000028852
114. Yu, EN, Jurkunas, U, Rubin, PA, Baltatzis, S, and Foster, CS. Obliterative microangiopathy presenting as chronic conjunctivitis in a patient with relapsing polychondritis. Cornea. (2006) 25:621–2. doi: 10.1097/01.ico.0000227886.26747.a9
115. Khan, JH, and Ahmed, I. A case of relapsing polychondritis involving the tragal and the Conchal bowl areas with sparing of the helix and the antihelix. J Am Acad Dermatol. (1999) 41:299–302.
116. Sato, T, Yamano, Y, Tomaru, U, Shimizu, Y, Ando, H, Okazaki, T, et al. Serum level of soluble triggering receptor expressed on myeloid cells-1 as a biomarker of disease activity in relapsing polychondritis. Mod Rheumatol. (2014) 24:129–36. doi: 10.3109/14397595.2013.852854
117. Stabler, T, Piette, JC, Chevalier, X, Marini-Portugal, A, and Kraus, VB. Serum cytokine profiles in relapsing polychondritis suggest monocyte/macrophage activation. Arthritis Rheum. (2004) 50:3663–7. doi: 10.1002/art.20613
118. Shimizu, J, Wakisaka, S, Suzuki, T, and Suzuki, N. Serum MMP3 correlated with IL1beta messenger RNA expressions of peripheral blood mononuclear cells in patients with relapsing Polychondritis with respiratory involvement. ACR Open Rheumatol. (2021) 3:636–41. doi: 10.1002/acr2.11301
119. Tammaro, A, Stroo, I, Rampanelli, E, Blank, F, Butter, LM, Claessen, N, et al. Role of TREM1-DAP12 in renal inflammation during obstructive nephropathy. PLoS One. (2013) 8:e82498. doi: 10.1371/journal.pone.0082498
120. Foidart, JM, Abe, S, Martin, GR, Zizic, TM, Barnett, EV, Lawley, TJ, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. (1978) 299:1203–7. doi: 10.1056/NEJM197811302992202
121. Cremer, MA, Pitcock, JA, Stuart, JM, Kang, AH, and Townes, AS. Auricular chondritis in rats. An experimental model of relapsing polychondritis induced with type II collagen. J Exp Med. (1981) 154:535–40.
122. Saxne, T, and Heinegard, D. Involvement of nonarticular cartilage, as demonstrated by release of a cartilage-specific protein, in rheumatoid arthritis. Arthritis Rheum. (1989) 32:1080–6.
123. Hansson, AS, Heinegard, D, Piette, JC, Burkhardt, H, and Holmdahl, R. The occurrence of autoantibodies to matrilin 1 reflects a tissue-specific response to cartilage of the respiratory tract in patients with relapsing polychondritis. Arthritis Rheum. (2001) 44:2402–12. doi: 10.1002/1529-0131(200110)44:10<2402::aid-art405>3.0.co;2-l
124. Beck, DB, Ferrada, MA, Sikora, KA, Ombrello, AK, Collins, JC, Pei, W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. (2020) 383:2628–38. doi: 10.1056/NEJMoa2026834
125. Ferrada, MA, Sikora, KA, Luo, Y, Wells, KV, Patel, B, Groarke, EM, et al. Somatic mutations in UBA1 define a distinct subset of relapsing polychondritis patients with VEXAS. Arthritis Rheum. (2021) 73:1886–95. doi: 10.1002/art.41743
126. Matsumoto, H, Asano, T, Tsuchida, N, Maeda, A, Yoshida, S, Yokose, K, et al. Behçet's disease with a somatic UBA1 variant: expanding spectrum of autoinflammatory phenotypes of VEXAS syndrome. Clin Immunol. (2022) 238:108996. doi: 10.1016/j.clim.2022.108996
127. Lee, SJ, Park, JK, Lee, EY, Joo, SH, Jung, KC, Lee, EB, et al. Certain autoimmune manifestations are associated with distinctive karyotypes and outcomes in patients with myelodysplastic syndrome: a retrospective cohort study. Medicine (Baltimore). (2016) 95:e3091. doi: 10.1097/MD.0000000000003091
128. Hochman, MJ, and DeZern, AE. Myelodysplastic syndrome and autoimmune disorders: two sides of the same coin? Lancet Haematol. (2022) 9:e523–34. doi: 10.1016/S2352-3026(22)00138-7
129. Grignano, E, Jachiet, V, Fenaux, P, Ades, L, Fain, O, and Mekinian, A. Autoimmune manifestations associated with myelodysplastic syndromes. Ann Hematol. (2018) 97:2015–23. doi: 10.1007/s00277-018-3472-9
130. Luo, Y, Bolek, EC, Quinn, KA, Wells, K, Rose, E, Sikora, K, et al. A prospective observational cohort study and systematic review of 40 patients with mouth and genital ulcers with inflamed cartilage (MAGIC) syndrome. Semin Arthritis Rheum. (2022) 52:151924. doi: 10.1016/j.semarthrit.2021.10.007
131. Demirbaş, A, and Kaya İslamoğlu, ZG. Can decreased monocyte to HDL-cholesterol ratio be a marker indicating the anti-inflammatory effect of the colchicine in Behçet's disease? A preliminary study. Dermatol Ther. (2020) 33:e14013. doi: 10.1111/dth.14013
132. Djaballah-Ider, F, and Touil-Boukoffa, C. Effect of combined colchicine-corticosteroid treatment on neutrophil/lymphocyte ratio: a predictive marker in Behçet disease activity. Inflammopharmacology. (2020) 28:819–29. doi: 10.1007/s10787-020-00701-x
133. Davtyan, TK, Mkrtchyan, NR, Manukyan, HM, and Avetisyan, SA. Dexamethasone, colchicine and iodine-lithium-alpha-dextrin act differentially on the oxidative burst and endotoxin tolerance induction in vitro in patients with Behçet's disease. Int Immunopharmacol. (2006) 6:396–407. doi: 10.1016/j.intimp.2005.09.003
134. Bettiol, A, Becatti, M, Silvestri, E, Argento, FR, Fini, E, Mannucci, A, et al. Neutrophil-mediated mechanisms of damage and in-vitro protective effect of colchicine in non-vascular Behcet's syndrome. Clin Exp Immunol. (2021) 206:410–21. doi: 10.1111/cei.13664
135. Qiao, H, Sonoda, KH, Ariyama, A, Kuratomi, Y, Kawano, Y, and Ishibashi, T. CXCR2 expression on neutrophils is upregulated during the relapsing phase of ocular Behcet disease. Curr Eye Res. (2005) 30:195–203. doi: 10.1080/02713680490904331
136. Takeuchi, M, Karasawa, Y, Harimoto, K, Tanaka, A, Shibata, M, Sato, T, et al. Analysis of Th cell-related cytokine production in Behçet disease patients with uveitis before and after infliximab treatment. Ocul Immunol Inflamm. (2017) 25:52–61. doi: 10.3109/09273948.2016.1158276
137. Kikuchi, H, Aramaki, K, and Hirohata, S. Effect of infliximab in progressive neuro-Behçet's syndrome. J Neurol Sci. (2008) 272:99–105. doi: 10.1016/j.jns.2008.05.002
138. Misumi, M, Hagiwara, E, Takeno, M, Takeda, Y, Inoue, Y, Tsuji, T, et al. Cytokine production profile in patients with Behcet's disease treated with infliximab. Cytokine. (2003) 24:210–8. doi: 10.1016/j.cyto.2003.09.003
139. Gül, A, Tugal-Tutkun, I, Dinarello, CA, Reznikov, L, Esen, BA, Mirza, A, et al. Interleukin-1β-regulating antibody XOMA 052 (gevokizumab) in the treatment of acute exacerbations of resistant uveitis of Behcet's disease: an open-label pilot study. Ann Rheum Dis. (2012) 71:563–6. doi: 10.1136/annrheumdis-2011-155143
140. le Joncour, A, Régnier, P, Maciejewski-Duval, A, Charles, E, Barete, S, Fouret, P, et al. Type‐4 Phosphodiesterase (PDE4) blockade reduces neutrophil activation in Behçet's disease Arthritis Rheum (2023), doi: 10.1002/art.42486 [Epub ahead of print].
141. Mumcu, G, İnanç, N, Özdemir, FT, Tulunay, A, Ekşioğlu-Demiralp, E, Ergun, T, et al. Effects of azithromycin on intracellular cytokine responses and mucocutaneous manifestations in Behçet's disease. Int J Dermatol. (2013) 52:1561–6. doi: 10.1111/ijd.12144
142. Faghfouri, AH, Baradaran, B, Khabbazi, A, Abdoli Shadbad, M, Papi, S, Faghfuri, E, et al. Regulation of NLRP3 inflammasome by zinc supplementation in Behçet's disease patients: a double-blind, randomized placebo-controlled clinical trial. Int Immunopharmacol. (2022) 109:108825. doi: 10.1016/j.intimp.2022.108825
143. Kawai, M, Hagihara, K, Hirano, T, Shima, Y, Kuwahara, Y, Arimitsu, J, et al. Sustained response to tocilizumab, anti-interleukin-6 receptor antibody, in two patients with refractory relapsing polychondritis. Rheumatology (Oxford). (2009) 48:318–9. doi: 10.1093/rheumatology/ken468
144. Wallace, ZS, and Stone, JH. Refractory relapsing polychondritis treated with serial success with interleukin 6 receptor blockade. J Rheumatol. (2013) 40:100–1. doi: 10.3899/jrheum.120381
145. Maekawa, M, Yoshimura, M, Kadowaki, M, Nakano, M, Moriwaki, A, Ueda, H, et al. Successful treatment of relapsing polychondritis with circumferential bronchial wall thickening including the tracheomembranous area with tumor necrosis factor-α inhibitor. Mod Rheumatol Case Rep. (2023) 7:197–201. doi: 10.1093/mrcr/rxac005
146. Nakamura, H, Suzuki, T, Nagaoka, K, Yamasaki, S, Tamai, M, Hayashi, T, et al. Efficacy of adalimumab for a refractory case of relapsing polychondritis with reduction of pro-inflammatory cytokines. Mod Rheumatol. (2011) 21:665–8. doi: 10.3109/s10165-011-0453-4
147. Kraus, VB, Stabler, T, Le, ET, Saltarelli, M, and Allen, NB. Urinary type II collagen neoepitope as an outcome measure for relapsing polychondritis. Arthritis Rheum. (2003) 48:2942–8. doi: 10.1002/art.11281
148. Matsumoto, H, Tokimura, R, Fujita, Y, Matsuoka, N, Asano, T, Sato, S, et al. Meningoencephalitis in relapsing polychondritis: a case report. Medicine (Baltimore). (2021) 100:e26315. doi: 10.1097/MD.0000000000026315
149. Kirino, Y, Takase-Minegishi, K, Tsuchida, N, Hirahara, L, Kunishita, Y, Yoshimi, R, et al. Tocilizumab in VEXAS relapsing polychondritis: a single-center pilot study in Japan. Ann Rheum Dis. (2021) 80:1501–2. doi: 10.1136/annrheumdis-2021-220876
150. Meyer-Lindemann, U, Mauersberger, C, Schmidt, AC, Moggio, A, Hinterdobler, J, Li, X, et al. Colchicine impacts leukocyte trafficking in atherosclerosis and reduces vascular inflammation. Front Immunol. (2022) 13:898690. doi: 10.3389/fimmu.2022.898690
151. Petitdemange, A, Sztejkowski, C, Damian, L, Martin, T, Mouthon, L, Amoura, Z, et al. Treatment of relapsing polychondritis: a systematic review. Clin Exp Rheumatol. (2022) 40:81–5. doi: 10.55563/clinexprheumatol/h9gq1o
152. Schafer, PH, Parton, A, Gandhi, AK, Capone, L, Adams, M, Wu, L, et al. Apremilast, a cAMP phosphodiesterase-4 inhibitor, demonstrates anti-inflammatory activity in vitro and in a model of psoriasis. Br J Pharmacol. (2010) 159:842–55. doi: 10.1111/j.1476-5381.2009.00559.x
153. Hatemi, G, Mahr, A, Ishigatsubo, Y, Song, YW, Takeno, M, Kim, D, et al. Trial of apremilast for oral ulcers in Behçet's syndrome. N Engl J Med. (2019) 381:1918–28. doi: 10.1056/NEJMoa1816594
Keywords: Behçet disease, relapsing polychondritis, neutrophils, monocytes, macrophages, cytokines, autoinflammatory disease, autoimmune disease
Citation: Shimizu J, Murayama MA, Mizukami Y, Arimitsu N, Takai K and Miyabe Y (2023) Innate immune responses in Behçet disease and relapsing polychondritis. Front. Med. 10:1055753. doi: 10.3389/fmed.2023.1055753
Edited by:
Peter Korsten, University Medical Center Göttingen, GermanyReviewed by:
Peizeng Yang, First Affiliated Hospital of Chongqing Medical University, ChinaTakayuki Katsuyama, Okayama University, Japan
Tadashi Hosoya, Tokyo Medical and Dental University, Japan
Copyright © 2023 Shimizu, Murayama, Mizukami, Arimitsu, Takai and Miyabe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshishige Miyabe, eW9zaGlzaGlnZS5taXlhYmVAbWFyaWFubmEtdS5hYy5qcA==