 Russell N. Low
Russell N. Low Ryan J. Low2,3
Ryan J. Low2,3 Athena Akrami
Athena Akrami- 1Retired, La Jolla, CA, United States
- 2Gatsby Computational Neuroscience Unit, University College London, London, United Kingdom
- 3Sainsbury Wellcome Centre, University College London, London, United Kingdom
The Long COVID/Post Acute Sequelae of COVID-19 (PASC) group includes patients with initial mild-to-moderate symptoms during the acute phase of the illness, in whom recovery is prolonged, or new symptoms are developed over months. Here, we propose a description of the pathophysiology of the Long COVID presentation based on inflammatory cytokine cascades and the p38 MAP kinase signaling pathways that regulate cytokine production. In this model, the SARS-CoV-2 viral infection is hypothesized to trigger a dysregulated peripheral immune system activation with subsequent cytokine release. Chronic low-grade inflammation leads to dysregulated brain microglia with an exaggerated release of central cytokines, producing neuroinflammation. Immunothrombosis linked to chronic inflammation with microclot formation leads to decreased tissue perfusion and ischemia. Intermittent fatigue, Post Exertional Malaise (PEM), CNS symptoms with “brain fog,” arthralgias, paresthesias, dysautonomia, and GI and ophthalmic problems can consequently arise as result of the elevated peripheral and central cytokines. There are abundant similarities between symptoms in Long COVID and myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). DNA polymorphisms and viral-induced epigenetic changes to cytokine gene expression may lead to chronic inflammation in Long COVID patients, predisposing some to develop autoimmunity, which may be the gateway to ME/CFS.
Highlights
- Long COVID patients include millions of people worldwide, and persistent symptoms following COVID-19 can continue for months.
- Varied and relapsing symptoms of Long COVID can be attributed to elevated peripheral and central cytokines, generated by an abnormal immune response.
- Chronic inflammation linked to immunothrombosis with microclot formation leads to decreased tissue perfusion and ischemia.
- CNS effects may be due to direct viral invasion, indirect immune response, or immunothrombosis.
- Dysregulated activation of brain microglia, due to neuroinflammation, can cause centrally mediated symptoms.
- Upregulation of the p38 MAPK pathway by SARS-Cov-2 can be a possible mechanism by which the virus increases cytokine production.
- Progression to myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and dysautonomia occurs in some patients and could involve development of autoimmunity.
1. Introduction
Long COVID, or Post Acute Sequelae of COVID-19 (PASC), is a debilitating illness with symptoms lasting more than 12 weeks following COVID-19 infection (1). Long COVID was first characterized in the COVID-19 Prolonged Symptoms Survey – Analysis Report of May 11, 2020, which described 640 patients with a protracted clinical course greater than 2 weeks (2). The report showed that, among the participants, the chance of full recovery by day 50 was less than 20%. In another study, 85% of patients who had symptoms 2 months after the initial infection continued to experience symptoms at 1 year follow up (3). The actual number of people worldwide struggling with Long COVID has not been measured systematically. However, current estimates suggest between 10% and 40% of people infected with COVID-19 have prolonged symptoms (4–7). As of January 2023, the over 670 million cases of COVID worldwide translate to at least 67 million persons with Long COVID. Among patients hospitalized for COVID-19 infection, an even larger number (up to 90%) can suffer from symptoms 8–12 weeks after admission (8–10). The incidence of Long COVID is reduced in individuals who have been vaccinated (11, 12).
Long COVID patients suffer from a plethora of symptoms, impacting their quality of life and ability to work (13). Early reports described the top 10 reported symptoms as shortness of breath, chest tightness, mild-to-moderate fatigue, chills or sweats, body aches, dry cough, fever (98.8–100°F, 37.2–37.8°C), headache, and cognitive/concentration challenges termed “brain fog” (2). Over 200 symptoms have since been described, affecting multiple organ systems (13). For example, cardiopulmonary involvement can produce chest pain, palpitations, cough and dyspnea. Gastrointestinal tract involvement (13, 14) can produce abdominal pain, and nausea while solid abdominal organ involvement of the pancreas, liver, spleen and kidney may result in end organ injury. Recognized vascular endothelial injury (15, 16) can lead to coagulopathy, pulmonary emboli and strokes. A host of neurological symptoms can result in cognitive impairment, brain fog, sleep and memory disturbances, tinnitus and vertigo (13). Involvement of the reproductive system may lead to erectile dysfunction and irregular menstruation (13). Finally, the immune system as a cause or casualty of Long COVID can result in autoimmune diseases (17). At 6 months post-infection, the most commonly reported Long COVID symptoms primarily include a combination of systemic and neurological issues such as fatigue, cognitive dysfunction, and post-exertional malaise (PEM)—wherein symptoms are markedly exacerbated by physical or mental stress/exertion, with the onset measured in hours to days following the activity (13).
Analyzing the pattern of symptoms, in terms of the underlying pathophysiology on cellular and macromolecular levels, will shed light on the Long COVID patients’ clinical plight. Questions persist about the pathophysiology of their symptoms and their eventual outcome (17). The multifactorial processes underlying Long COVID may include end organ disease, alterations in immune-inflammatory response, mitochondrial energy production, oxidative stress, and aberrant coagulation with microthrombosis. The relative contributions of each process may be different in individual patients. However, these are interrelated processes which all involve inflammatory cytokines (18). In this article, we propose a description of the pathophysiology of Long COVID symptoms based on inflammatory cytokines and a dysregulated immune-inflammatory response, and we review the possible pathways involved.
Viral infections incite a pro-inflammatory response with increased production of cytokines and chemokines (19, 20). Cytokines are small proteins involved in cell signaling. They are secreted by numerous cell types, including endothelial cells, macrophages, monocytes, and lymphocytes, and can act in an autocrine, paracrine or endocrine manner. Cytokines can be broadly categorized into different families, including interferons, interleukins, colony-stimulating factors, and tumor necrosis factors (21). Chemokines recruit inflammatory cells as part of the normal immune response (21).
Several studies have shown that both mild and severe cases of COVID-19 can result in a hyper-inflammatory response characterized by elevated levels of numerous cytokines, including IL-6, IL-8 and TNF-α (22–24). While these proinflammatory cytokines are expected to be induced upon viral infection, SARS-CoV-2 encodes several proteins that specifically evade initial type I interferon response (25, 26), resulting in a prolonged and, in certain cases, dysregulated cytokine response. A recent study shows an abnormal diffuse inflammatory cytokine profile that persists in Long COVID patients for at least 8 months, not found in asymptomatic COVID-19 survivors (27).
Studies of Long COVID patients have shown persistent deregulation of a broad range of cytokines long after infection. Specifically, it is shown that IL-1β, IL-6 and TNF-α remain elevated in Long COVID - PASC (28). This is despite the fact that peripheral blood cytokines do not need to be elevated, since they function by acting locally rather than by flowing through the blood (29). Nevertheless, persistent immune activation has been suggested to be associated with ongoing symptoms following COVID-19 (30, 31).
An abnormal immunologic response to the virus can result in an uncontrolled systemic inflammatory response with more severe infections (32). This “Cytokine Storm,” has gained widespread attention as it is associated with severely ill patients with COVID-19 pneumonia who show a progression of symptoms (23). Acute Respiratory Distress Syndrome (ARDS), requiring mechanical ventilation and ICU admission, results in a mortality rate approaching 40%–50% in COVID-19 patients (33, 34). Uncontrolled overproduction of pro-inflammatory cytokines and chemokines produces the cytokine storm, resulting in systemic inflammation, ARDS, and multiple organ failure (21, 35–37). The relationship between cytokine production and tissue damage is not necessarily causal and is clearly bidirectional. While also involving cytokines, the situation and clinical course for the Long COVID patient is quite different.
2. The cytokine release syndrome
Cytokines are small endogenous proteins that mediate communication between cells, particularly cells of the immune system. Cytokines control the growth and activity of immune cells and blood cells and are essential for directing the body’s immune-inflammatory responses and maintaining homeostasis.
While the focus on the “cytokine storm” is clinically relevant in the severely ill COVID-19 patients, the term ignores the role cytokines can play in the symptoms of less severe disease, including those of Long COVID patients.
Instead, we believe that the Cytokine Release Syndrome (CRS) description provides a more balanced presentation of the spectrum of symptoms in patients with clinical manifestations of elevated cytokine production (38, 39). CRS is a systemic inflammatory response triggered by infections, drugs, antibody-based immunotherapies, chemotherapeutic agents, and graft-vs-host disease (both acute and chronic) (38) (Figure 1).
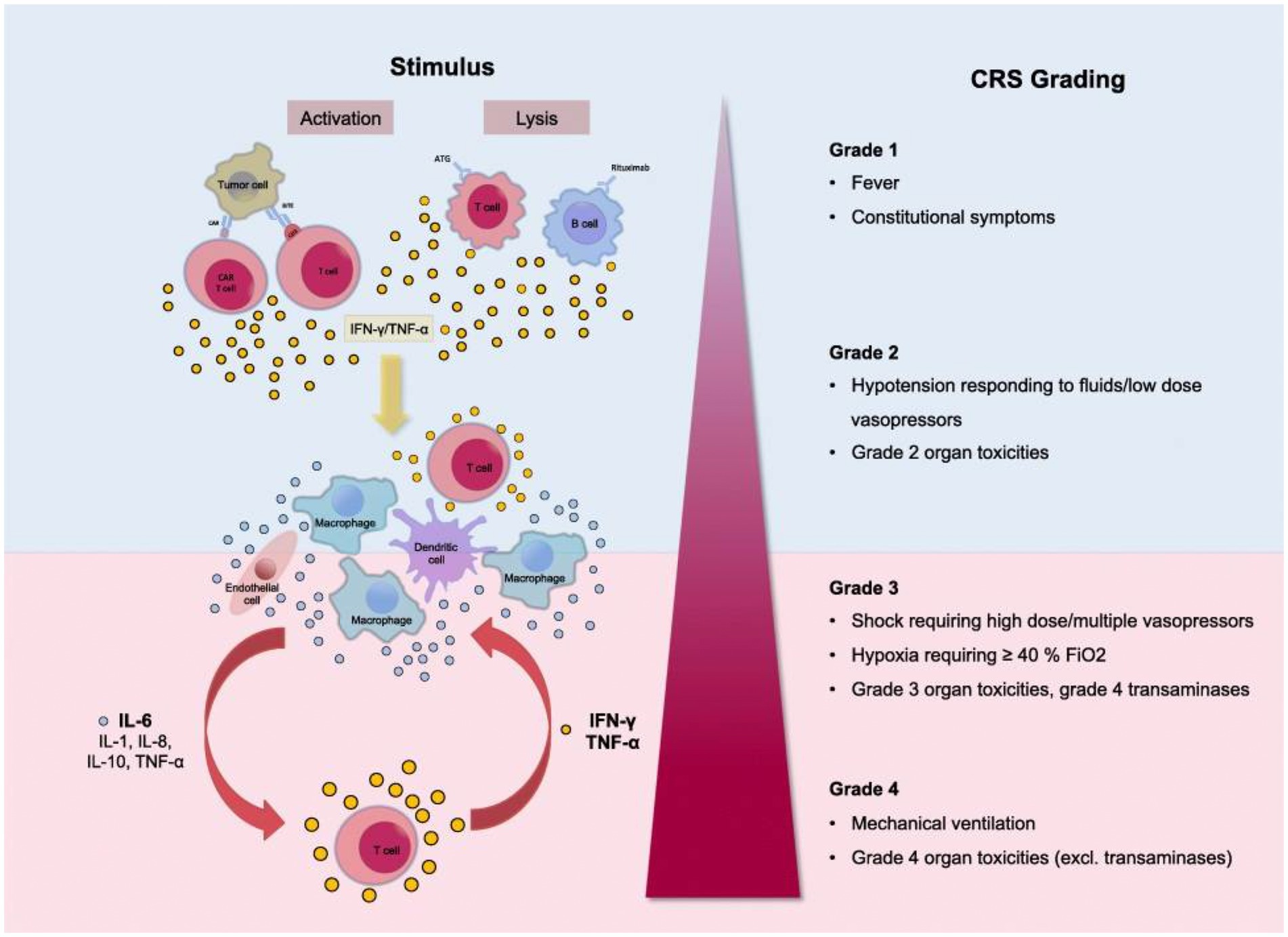
Figure 1. Grading of the Cytokine Release Syndrome. Activation of T cells induces a release of IFN-γ or TNF-α. This process activates macrophages and other immune cells, releasing proinflammatory cytokines. Large amounts of IL-6 are released, which in a positive feedback loop activates T cells and other immune cells. Reproduced from “Cytokine Release Syndrome” by AJ Shimabukuro-Vornhagen, P Gödel, M. Subklewe, HJ Stemmler, HA Schlößer, M Schlaak, et al. 2018 Immunother. Cancer 56 (http://creativecommons.org/licenses/by/4.0/).
Clinical presentation ranges from mild flu-like symptoms to life-threatening “cytokine storm.” The CRS grading system includes Grade 1 with fever and mild constitutional symptoms, Grade 2 with hypotension responding to fluids and vasopressors, Grade 3 with shock, hypoxia, and Grade 4 with respiratory failure and ARDS requiring mechanical ventilation, disseminated intravascular coagulation (38, 40). CRS is a multiorgan process affecting the lungs, CNS, spleen, gastrointestinal tract, kidneys, hepatobiliary tree, heart, musculoskeletal system, white cells, and platelets.
In general, Grade 1 CRS with fever, fatigue, headache, rash arthralgia, and myalgia correlates with the symptoms of Long COVID patients (see the section Long COVID Symptoms and Cytokine Release Syndrome Model). As with the more severe CRS grades, Grade 1 symptoms result from proinflammatory cytokine release from activated macrophages and white cells, initiating the inflammatory cascade.
These cytokines and chemokines recruit immune cells (41), which then secrete further cytokines producing a positive feedback loop. While this process occurs in the body’s normal inflammatory response, in the CRS, loss of regulation of the inflammatory cascade can result in an uncontrolled destructive process (21, 42). The cytokine cascade’s autoimmune inflammatory effect can be responsible for more damage than is produced directly by the SARS-CoV-2 virus (43).
There are critical inter-individual differences in the cytokine pathway. The COVID-19 comorbidities are characterized by low-grade inflammation and elevated cytokine levels (44). For example, the comorbidities of aging (45), obesity (46), diabetes mellitus (47, 48), and hypertension (49) are associated with increased proinflammatory cytokines and a worse COVID-19 prognosis (50).
3. Long-COVID symptoms and cytokine release syndrome model
With this knowledge of the effects of cytokines in COVID-19 patients, how can one explain the varied and relapsing symptoms of Long COVID patients? This section will review the evidence linking elevated cytokines to various symptoms commonly experienced in Long COVID patients.
3.1. Dysregulated peripheral immune response
Macrophages are the first line of innate immunity, producing various inflammatory cytokines and chemokines. Polarization between a reparative M2 and proinflammatory M1 state occurs, which can become dysregulated to favor proinflammatory cytokine release (Figure 3). This macrophage activation syndrome (MAS), present in systemic inflammatory diseases like systemic lupus erythematosus, juvenile rheumatoid arthritis and Still Disease, can result in life-threatening inflammation. Diminished function of NK cells and cytolytic CD8 T cells prolongs and amplifies the inflammatory response (51). In COVID-19, pathologic hyperinflammation due to dysregulated macrophages leads to prolonged inflammation and host damage (42, 51, 52).
3.2. A mechanism for CNS mediated symptoms
Many Long Covid symptoms may be centrally mediated by the brain, including generalized symptoms such as fatigue, malaise, fever, and dyspnea (53), as well as cognitive and neurological symptoms such as “brain fog,” concentration difficulty, memory loss, dizziness, and disequilibrium (2, 6, 54). Viral-induced CNS changes can explain the persistent and relapsing course of these symptoms in Long COVID. We describe two pathways that could produce such changes (Figure 2): (1) neuroinflammation, triggered by direct CNS infection or signals from the peripheral immune system, and (2) modulation of CNS function by a persistently activated peripheral immune system.
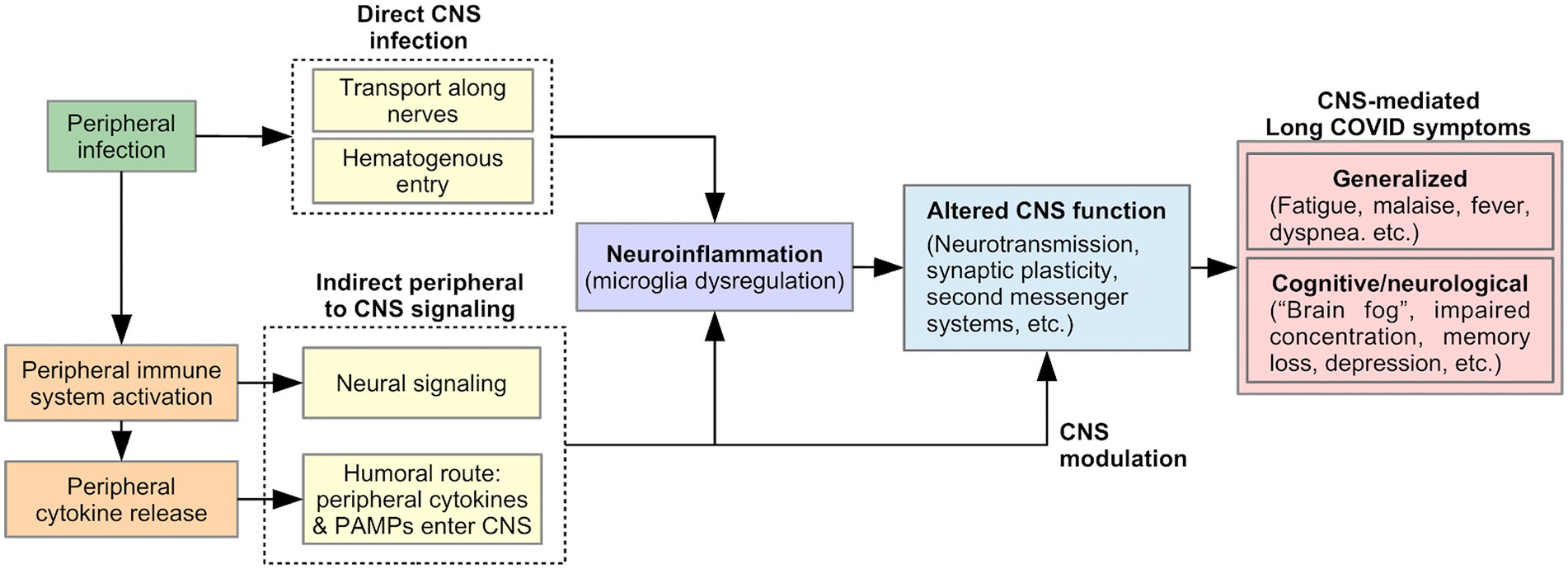
Figure 2. Proposed model of pathophysiology underlying CNS-mediated Long COVID symptoms.
3.2.1. Direct viral invasion of the CNS
SARS-CoV-2 is neuroinvasive, neurotropic, and neurovirulent (55). Viral proteins have been found in the brainstem and cranial nerves in autopsies (56). The route of SARS-CoV-2 entry into the CNS is currently unknown, but could include: (1) Transport along cranial and peripheral motor, sensory, or autonomic nerves (e.g., olfactory to CNS invasion) (55, 57, 58). Here, the virus invades nerve endings and is actively transported within neurons to the brain. (2) Alternatively, hematogenous spread could occur, in which the virus passes from alveolar epithelial cells or endothelial cells of other organs into the blood circulation (55). The virus penetrates the Blood Brain Barrier (BBB) through points of weakness (e.g., circumventricular organs and the choroid plexus), or by damaging the vascular endothelium of the BBB, resulting in neuroinvasion (55, 57). Neuroinvasion can trigger neuroinflammation, thereby altering CNS function and producing Long COVID symptoms, as discussed below (Figure 2).
3.2.2. Indirect immune-mediated viral effects on the CNS
While direct CNS viral infection is possible, indirect CNS involvement through a viral-mediated immune response is equally likely (56, 59). As part of the innate immune response, proinflammatory cytokines are released by activated peripheral immune cells in response to pathogen-associated molecular patterns (PAMPS) detected by pattern-recognition receptors (PRRs). Peripherally released cytokines act on the brain via two pathways of communication: (1) a neural route via primary afferent neurons, including the vagus nerve, innervating the site of the infection; (2) a humoral route, in which peripheral cytokines or PAMPS cross the BBB at the circumventricular organs and choroid plexus and stimulate the production of proinflammatory cytokines by brain immune cells (60, 61). As discussed below, these signals from the peripheral immune system to the CNS could lead to Long COVID symptoms by triggering neuroinflammation and/or by directly and persistently modulating CNS function (Figure 2).
3.3. Neuroinflammation and microglial cell dysregulation
Direct or indirect viral effects on the CNS can trigger neuroinflammation—a chronic immune response within the brain involving long-term activation of microglia, the local release of inflammatory cytokines, and resulting oxidative stress (62, 63). Neuroinflammation has been implicated in the pathogenesis of many neurodegenerative (64) and psychiatric diseases (65), and can also be triggered by viral infection (62). In a recent study, pronounced neuroinflammatory changes were found in the brainstem of post-mortem brain autopsies in COVID-19 patients (56). In these patients, evidence of SARS-CoV-2 viral proteins was found in over half of patients in the brainstem and cranial nerves. However, neuropathological changes were not associated with the presence of the virus. Instead, the CNS damage and neurological manifestations (66) were attributed to cytokine-mediated neuroimmune stimulation, also indicated by the presence of neuroinflammation in the ocular vitreous cavity of persons who recovered from COVID-19 (67).
Glial cells, such as astrocytes and microglia, play an essential role in brain neuroinflammatory insults (68). Microglia, the resident brain macrophages, acquire different functional phenotypes in response to their environment, alternating between a quiescent reparative M2 state (anti-inflammatory) and an activated proinflammatory M1 state (69, 70) (Figure 3).
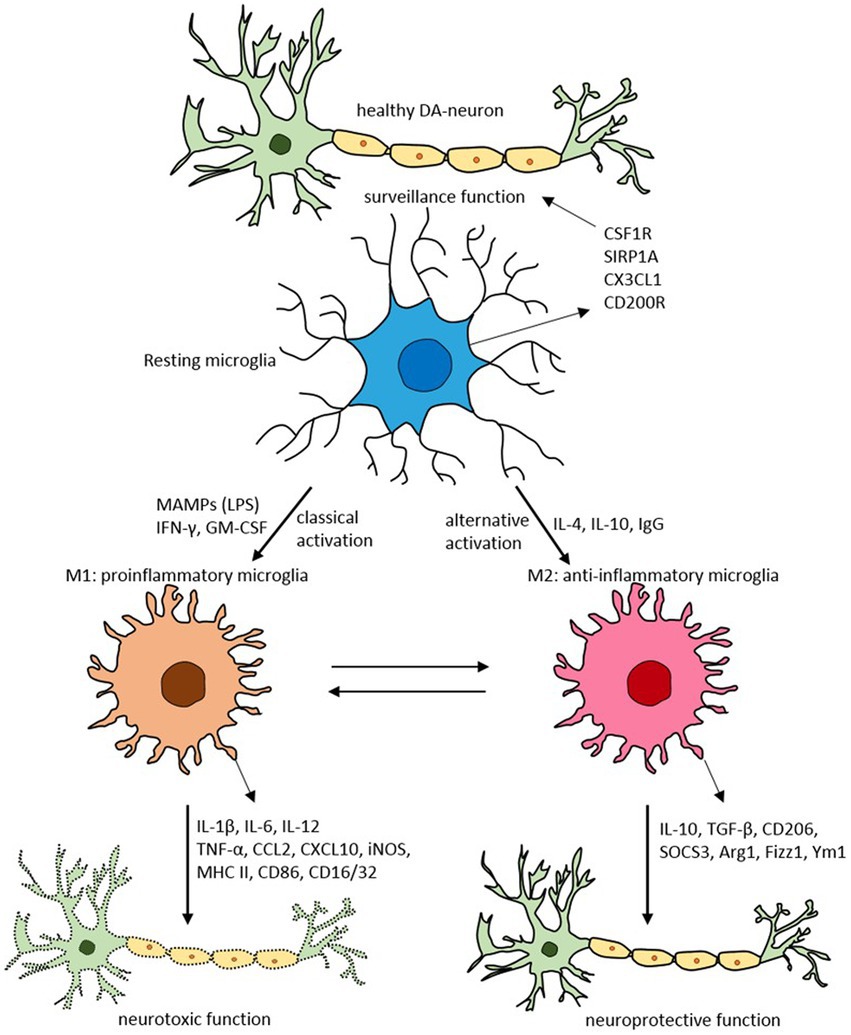
Figure 3. Schematic of microglial polarization states and functions. Reproduced from “Targeting microglial activation states as a therapeutic avenue in Parkinson’s disease” by SR Subramaniam and HJ Federoff. (http://creativecommons.org/licenses/by/4.0/).
Usually, the system is well regulated. However, with aging, chronic infection, or stress, microglia can become dysfunctional (72, 73). These dysregulated microglia are hyperreactive to signals from the peripheral immune system, producing an exaggerated and prolonged central cytokine response to an otherwise mild immune challenge (74, 75). The primed microglia then become resistant to normal regulation, failing to revert to the quiescent state after inflammation resolution (76). The dysregulation of microglia has been implicated in the pathophysiology of numerous neurodegenerative disorders (77).
3.4. Cytokine modulation of the CNS
In addition to neuroinflammation, circulating cytokines can modulate systemic functions by direct action on the brain (78, 79). This modulation is known to occur in response to viral infection. For example, even non-neurotropic viruses that do not invade the brain can cause CNS effects, triggered by a peripheral immune response (80). Human and animal studies have shown that increases in circulating cytokines can induce typical “sickness behaviors” (79, 81–83), including decreases in general activity, exploratory behavior, social and sexual interaction, food and water intake, and preference for sweets (anhedonic behavior), as well as altered sleep and impaired learning. Injection of IL-1ra (an inhibitor of pro-inflammatory IL-1) can suppress hypothalamic–pituitary–adrenal (HPA) axis activation and the associated sickness behaviors (sleep and behavioral changes) that were induced by systemic administration of an endotoxin (84).
Evidence shows that cytokines (e.g., IL-1β, IL-6, and TNF-α) play a role in complex cognitive processes via neuromodulation (85, 86), second messenger systems in neurons (87, 88), neurogenesis (86), and modification of synaptic plasticity (85). Cytokines can also induce pyrogenic responses in the brain by the synthesis of prostaglandins (89), and can additionally influence many neuroendocrine systems, the most prominent of which is the HPA-axis (89, 90). Cytokines can also influence the synthesis, secretion, and reuptake of many central neurotransmitters, including noradrenaline, dopamine, glutamate, serotonin, GABA, and acetylcholine, as well as the expression of several neuropeptides in different brain areas (89–92). The alterations in neurotransmission may lead to depression, anxiety, and mental sluggishness or “brain fog” (93, 94). Overexpression of cytokines has been associated with neuropsychiatric disorders such as depression (95–97). IL-6 has recently been implicated as a mediator in COVID-19 neuropsychiatric symptoms (98).
In summary, a peripheral inflammatory response may lead to cytokine release and mobilization of immune cells, both of which have CNS effects (94, 99). SARS-CoV-2 can affect the CNS via proinflammatory cytokines, producing neurological symptoms (99). Such “sickness behavior” usually resolves once the acute illness terminates. However, subsequent neuroinflammation and oxidative stress can produce prolonged sickness behavior, cognitive deficits, and increased sensitization to intrinsic and extrinsic stress (75). In Long COVID, dysregulation and chronic activation of cytokine signaling, as discussed later, can cause prolonged CNS effects. This model of dysregulated, activated, and primed brain cells and microglia, responding to peripheral or central cytokines, can explain the chronic and relapsing clinical course of many Long COVID symptoms.
3.5. Mild constitutional symptoms
Almost all Long COVID patients experience systemic symptoms (13). The constitutional symptoms of fever, chills, headache, arthralgia, and mild dyspnea can be attributed to cytokine release (38, 100). These symptoms of “sickness behavior” are mediated by the brain which recognizes proinflammatory cytokines as signals of sickness (see CNS Mediated Symptoms). The peripheral innate immune response to pathogen-associated molecular patterns (PAMPS) produces cytokines (IL-1, IL-6, and TNF-α) (101). Most of these proinflammatory cytokines are pyrogenic (IL-6, IL-1, IFN-γ, and TNF-α. TNF-α can have both pyrogenic and antipyretic properties) and their systemic circulation can induce a febrile response (102–104).
3.6. Neuropathic pain
Paresthesia reported in Long COVID patients (105) can be related to cytokines. In patients with diabetic neuropathy, neuropathic pain correlated with increased levels of IL-6 and IL-10 for large nerve fiber damage (106). Patients with polyneuropathies show elevated serum cytokines IL-8, TNF-α and lower expression of IL-10 (107). Disease duration positively correlates with IL-6 gene expression (107).
Post-infectious neuropathies are precipitated by an autoimmune process in which antibodies to the infectious agent cross-react with peripheral nerve myelin (108). Like other neuropathies, the process is directly related to cytokines (109), which are also a therapeutic target (110).
3.7. Extreme fatigue
Fatigue is highly prevalent among Long COVID patients (13, 111). Post-viral fatigue is well described (112). Extreme physical fatigue can have two components: actual “muscle fatigue” and “central fatigue,” related to abnormalities in deep brain circuits involved in motor planning and execution (113). For myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) patients and perhaps for Long COVID patients, the fatigue can originate centrally with decreased basal ganglia function, including psychomotor slowing (114, 115). With chronic inflammation, cytokines target the basal ganglia and dopamine function, producing persistent fatigue (116).
Cytokines cause neurotransmitter release in the brain, including serotonin, producing extreme central fatigue (117). In healthy patients, the administration of IL-6 and IL-1β produces fatigue, as well as sleep disturbances, and difficulty concentrating (118). In people with post-infection fatigue, recovering from West Nile virus, elevated levels of both proinflammatory and antiviral cytokines (IL-2, IL-6, IFN-γ, IL-10, among others) were detected (112). In patients with rheumatoid arthritis and systemic lupus erythematosus, fatigue improved with anti-IL-6 medications (119, 120). Viral-induced mitochondrial dysfunction leads to decreased ATP and increased cytokines IL-1β and IL-18 through activation of inflammasomes (121, 122). A similar mechanism can be at play in COVID19, where mitochondrial hijacking by SARS-COV-2 is suggested (123, 124).
3.8. Post-exertional malaise
Post-exertional malaise (PEM), or post-exertional symptom exacerbation (PESE), is characterized as worsening or relapse of symptoms after physical or mental activity. PEM is one of the most striking and demoralizing symptoms of Long COVID, reported by nearly 90% of patients (13). In the majority, one to two days after mild–moderate exercise, Long COVID patients experience marked worsening of symptoms with extreme fatigue, malaise, fever, shortness of breath, and neurological deficits.
Dysfunctional mitochondria can partially mediate PEM in muscles and brain microglia with less efficient glycolysis. Subsequent lactic acidosis with increasing reactive oxygen species (ROS), and elevated cytokines results in exercise-induced peripheral and central fatigue (125, 126).
For decades, exercise physiologists have known that exercise induces cytokine release (127). Suzuki (128) described a 2-15-fold increase in serum cytokines following maximal exercise. Following a marathon race, serum IL-6 levels increased 100-fold (128). Modest increases of IL-6 are expected after exercise of low-to-moderate intensity or intermittent physical activity of a shorter duration (129). Thirty minutes of acute exercise resulted in a 165% rise in IL-6 and a 32% rise in IL-8 in endurance-trained and sedentary young men (130).
Exercise-induced IL-6 responses vary with exercise intensity and duration, while exercise mode has little impact (131). Plasma IL-6 peak occurs as soon as the activity ends, returning to baseline after a few hours of recovery (127, 132). The exercise-induced cytokine release was more marked in the evening compared to the morning (133). In ME/CFS, exercise can induce significant changes in cytokine profile that are still observed 18 h following the exertion (134). Also, in ME/CFS patients experiencing PEM after moderate exercise, the severity of symptom flare is linked to cytokine activity, with increased circulating cytokines (135).
We argue that for COVID-19 patients, exercise-induced cytokines may trigger a recurrence of symptoms by reinitiating the same feedback loop pathways that caused the initial symptoms. Essentially, symptoms’ original occurrence and PEM-induced recurrence could both be due to peripheral and central cytokine cascades.
3.8.1. Psychological stress precipitates cytokine release
Intense mental concentration precipitates a recurrence or worsening of symptoms in Long COVID patients identical to physical stress. In Davis et al., 60% of participants reported relapses, triggered by stress (13). It is known that social, psychological, or mental stress is also associated with cytokine release (136). A meta-analysis of 34 studies found increases in circulating IL-6, IL-1β, IL-10, and TNF-α and stimulated IL-1β, IL-4, and interferon-γ in response to acute psychological stress (137). The peak IL-6 level was at 90 min post-stress with a > 6-fold increase over baseline.
3.8.2. The onset of symptoms post exertion
The delay in onset of symptoms following physical or mental stress is also predictable because the cytokines do not cause the symptoms; they just start the process of immune system recruitment and activation that leads to the symptoms. Cancer immunotherapy drugs produce the CRS with an onset of symptoms within a few days of the drug administration (38). This timing correlates with the delayed onset of Long COVID symptoms after exercise.
3.9. Gastrointestinal tract symptoms
More than 85% of Long COVID patients experience gastrointestinal symptoms (13). In COVID-19, recent research has shown that the infection disrupts the gut microbiome, allowing secondary pathologic bacteria to colonize the gut (138). Gut microbiome alterations have been documented in COVID-19 (139). Cytokines predispose to dysregulation of the microbiome (dysbiosis), consequently altering intestinal permeability, referred to as the “leaky gut,” allowing the bacteria to spread from the gut to the bloodstream. Entry of SARS-COV-2 into intestinal cells leads to down regulation of ACE2 receptors and subsequent decreased barrier function (140).
Ten percent (2%–50%) of COVID-19 patients report diarrhea, sometimes alternating with constipation (141). SARS-Cov-2 is present in feces in 55% of patients (142, 143). Wang et al. reported a 25-day median duration of fecal viral shedding with only 25% reporting diarrhea and 1% with nausea and vomiting. No significant differences were observed in GI symptoms between patients with positive and negative fecal viral tests (143). In patients with Long COVID, alterations in the gut microbiome have been observed (144).
The GI symptoms in Long COVID patients can be due to cytokines (145, 146) and associated cytokine-linked changes in the gut microbiome (145, 146). Diarrhea in systemic infections without intrinsic GI tract disease is well known. Pathogenesis may be related to cytokines, including IFN-γ, IL-6, and IL-10 (147). In patients with irritable bowel syndrome, pro-inflammatory cytokines, IL-6, and TNF-α, are elevated (148).
The microbiota-gut-brain axis can impact cognition and psychiatric symptoms, and lead to neurodegenerative diseases (140, 149). The interaction of gut microbiota with the brain may be through chronic inflammation, with cytokines altering maintenance of the blood brain barrier (150).
3.10. Arthralgia
Arthralgia has been described in 15% of coronavirus patients (151). More than 90% of Long COVID patients report musculoskeletal issues, including joint pain (~30% of LC patients) (13). Rheumatologists have noted that the pattern of elevated pro-inflammatory cytokines in COVID-19 are similar to those in patients with rheumatoid arthritis (151).
Possible mechanisms for the pathophysiology of joint pain/stiffness include the SARS-V2 activation of Toll-like receptors and the complement system leading to inflammation and autoantibody formation (152). Rheumatologic and autoimmune manifestations of Long COVD may be due to molecular mimicry, bystander killing, and viral persistence with polyclonal activation during longstanding presence of the virus resulting in immune mediated injury (153).
3.11. Ophthalmic problems
Ophthalmic manifestations of COVID-19 reported in hospitalized patients, include conjunctival hyperemia, and chemosis, epiphora, and increased secretions (154, 155) (156). A third of Long COVID patients report vision or other eye related issues (13). Ocular mast cells produce the inflammatory cytokines resulting in conjunctival hyperemia and chemosis (157). Visual and ocular disturbances in Long COVID patients may be similar to those reported in ME/CFS, including blurred vision, difficulty focusing, difficulty tracking lines of print, diplopia, light intolerance, and problems with peripheral vision (158). Evidence of inflammatory cells in the vitreous cavity have been found in patients recovered from COVID-19 (67).
The neuropathological mechanisms of SARS-CoV-2 infection involve multiple peripheral and cranial nerves, serving as conduits for the virus to gain access to the brainstem and olfactory cortex and to subsequently spread within the brain and to peripheral organs via cranial nerves (159). An inflammatory process in the central nervous system often manifests as optic neuritis (160). Associated neurological symptoms are seen in 25% of patients (161).
Cytokine-induced brainstem neuroinflammation could produce some of these visual problems without direct optic infection. Ocular tropism of SARS-CoV-2 was confirmed in a mouse model manifesting retinal inflammation with production of pro-inflammatory cytokines in the eyes of intranasally (IN)-infected mice (162). Finally, an autoimmune mechanism may also be at play in patients with COVID-19 optic neuritis due to anti-MOG antibodies (myelin oligodendrocyte glycoprotein) (163, 164).
3.12. Thrombotic complications of COVID-19: Relationship to cytokines
The processes of inflammation and thrombosis are intimately connected. Dysregulation can produce immunothrombosis and thrombinflammation, resulting in tissue injury from ischemia due to micro and macrovascular clotting (165).
The prothrombotic complications of SARS-CoV-2 became evident with reports of small vessel cerebral infarctions in young patients (166). Autopsies described the lungs and other organs full of micro clots (167). Myocardial infarctions, deep vein thrombosis, and systemic arterial emboli describe a systemic thrombotic process initiated by the SARS-CoV-2 virus. In a recent study, 31% of 184 COVID-19 ICU patients had thrombotic complications (166).
Inflammation-induced thrombosis is well known and, in addition, coagulation also intensifies inflammation through release of proinflammatory cytokines and growth factors, creating a vicious circle (168). The pathophysiology of thrombosis in COVID-19 may involve direct vessel endothelial injury by the SARS-CoV-2 binding to the ACE2 receptor (169). Also, pro-inflammatory cytokines increase blood viscosity (170–172), and specific cytokines, including interferon-γ, IL-6, IL-8, TNF-α, chemokine-ligand-2, IL-17A, IL-9, IL-1β, and growth-factor, have a prothrombotic effect (173, 174).
SARS-Cov-2 binds to platelet ACE2 receptors, leading to hyperactivation and platelet aggregation (175). Furthermore, spike protein promotes the release of coagulation factors and inflammatory cytokines from platelets (175). In turn, cytokines and erythropoietin potentiate platelet maturation. Platelet-producing megakaryocytes are found in unusual anatomic locations at autopsy of COVID-19 patients (176). SARS-CoV-2 induces gene expression leading to platelet hyperreactivity, contributing to COVID-19 pathophysiology (177). Critical interactions between inflammatory and thrombosis pathways, leading to SARS-CoV-2 induced vascular disease have been recently confirmed in rhesus macaques, in which disruption of endothelial cells via cytokine was observed (178).
A recently proposed microclot model suggests that small clots in the blood capillaries prevent oxygen from reaching the tissues and may cause Long COVID symptoms by limiting oxygen exchange. Plasma analysis of 70 patients with Long COVID confirmed the presence of microclots with platelet hyperactivation and resistance to fibrinolysis. Associated elevated α(2) antiplasmin and the acute inflammatory molecule Serum Amyloid A (SAA4) were observed in the supernatant and microclots (179). Twenty-four patients treated with an antiplatelet and antithrombotic regimen showed symptom improvement and reduced microclots (180).
3.13. Orthostatic tachycardia—Autonomic nervous system dysfunction
Autonomic dysfunction (dysautonomia) occurs in many Long COVID patients (181, 182). In one study, autonomic dysfunction occurred more commonly in those with neurologic symptoms (183). Dysautonomia is an autonomic nervous system (ANS) dysfunction, resulting in failure or overactivity of the ANS with alterations in blood pressure, breathing, GI tract motility, heart rate, and temperature regulation. Some Long COVID patients report orthostatic hypotension and/or postural orthostatic tachycardia (POTS) (184). POTS is a form of dysautonomia characterized by tachycardia within 10 min of standing up (>30 bpm increase from a supine position or > 120 bpm in the erect position) (185).
There is a long list of possible causes of dysautonomia. An interplay between the ANS and the immune system involves cytokines, which modulate the activity level of the sympathetic and parasympathetic nervous systems innervating multiple organs (186). POTS results from sympathetic nervous system dysfunction, and in one study was associated with increased IL-6 levels but not C-reactive protein (187).
Involvement of the brainstem and inferior frontal - olfactory regions in COVID-19 has been documented at autopsy and imaging studies (188). Many of the diverse symptoms experienced by Long COVID patients—including POTS—can be explained through the involvement of brainstem nuclei and tracts (189) which may reflect direct viral invasion, or indirect effects of neuroinflammation triggered by peripheral or central cytokine release, or effect of microclots.
An intriguing association between dysautonomia and antiphospholipid syndrome (APS, aka Hughes syndrome or “sticky blood”) has been observed (190). In addition to dysautonomia, APS is associated with a host of cardiac and neurologic manifestations. Both POTS and APS involve inflammatory cytokines (191) and can be triggered by viral infections, stress, and pregnancy. In Long COVID patients, autonomic dysfunction and some neurologic symptoms could be due to “sticky blood” and microclots resistant to fibrinolysis.
Proteomic analysis of POTS confirmed increased proteins involved in thrombogenicity and platelet activity, inflammation, cardiac contractility, skeletal muscle hypertrophy, and increased adrenergic activity (192). This proteomic footprint defines POTS, in part, as a hypercoagulable and proinflammatory state, mechanisms central to COVID-19 pathogenesis.
The occurrence of POTS after COVID-19 vaccination has been confirmed, although the incidence is five times less than that following COVID-19 infection (182, 193–195).
4. Loss of regulation of cytokine pathways in Long COVID patients
The protracted Long COVID clinical course could reflect impaired viral clearance with a reservoir of undetected viruses (196, 197). Residual viral fragments could be inciting inflammation. A recent autopsy study showed SARS-COV-2 RNA in multiple extrapulmonary anatomic sites for up to 230 days (198). Alternatively, viral reinfection could be occurring. Finally, Long COVID symptoms may reflect underlying end organ disease to the lungs, heart and other organs.
For Long COVID patients, it is more likely that the virus has been cleared and that the ongoing relapsing symptoms represent a continuing exaggerated response to cytokine release (199). Indeed, the majority of reported Long COVID symptoms can be directly attributed to cytokines. We propose that these patients exhibit an exaggerated response to the ongoing release of cytokines, which is key to understanding their disease’s pathophysiology.
Multiple checkpoint regulation sites with feedback inhibition are involved in the normal cytokine signaling pathways, allowing the cells to return to a quiescent, non-inflamed state. Autoinflammatory and autoimmune diseases can be caused by dysregulation and chronic activation of cytokine signaling (200). Dysregulation of cytokine inflammation may be occurring on a system or cellular level or a combination of both.
In patients with severe acute COVID-19, a systemic inflammatory response initiated by Acute Lung Injury (SIRS-ALI) can produce an inflammatory loop, resulting in cytokine release from numerous sources. Infection results in macrophage activation and release of inflammatory cytokines. These cytokines, IL-6, TNF, and IL-1β, promote the release of cell adhesion molecules (CAMS) and vascular endothelial growth factor in the lungs, reducing the barrier protection provided by the endothelium. Their release into the blood stimulates production and release of immature granulocytes, which can increase lung inflammation, leading to ARDS. These cytokines augment direct viral damage and persist long after the virus is cleared (201). However, since Long COVID patients do not typically present with severe pulmonary infections, this model may not directly pertain to the initiation or persistence of their inflammatory response.
4.1. System-level dysregulation of cytokine expression
Bidirectional communication between the peripheral immune system and the hypothalamic–pituitary–adrenal axis (HPA) modulates the inflammatory response. Cytokines cross the blood–brain barrier and activate the HPA axis, resulting in increased glucocorticoid secretion by the adrenal gland. Glucocorticoids, in turn, negatively feedback onto immune cells, inhibiting the expression of proinflammatory cytokines and increasing anti-inflammatory cytokines, thereby protecting the host from harmful effects of an overactive immune response. Impairments in HPA axis activity and associated hypocortisolism can result in chronic inflammatory or autoimmune diseases (202–205).
Viral-induced suppression and disruption of the host adrenal cortical response could result in transient hypoadrenalism, blocking the needed adrenal stress response (206, 207).
Notably, 39% of SARS survivors demonstrated transient hypocortisolism, developing several weeks after symptom onset and usually resolving by 1 year (208). Proposed mechanisms include hypophysitis, direct hypothalamic involvement with binding to ACE2 receptors, and mimicry with host antibodies directed against ACTH (203). Autopsy also showed adrenal gland necrosis (209) which could be secondary to direct adrenal gland infection or an autoimmune process. While intriguing, there is no correlation between SARS survivors with hypocortisolism and those who experience chronic fatigue.
In a recent study of 215 individuals, immune profiling confirmed cortisol levels approximately 50% lower among participants with Long COVID relative to matched control groups. Persistently decreased cortisol production in participants with Long COVID was observed for more than a year following acute infection (31). Lack of a rise in ACTH levels in these patients indicated a blunted compensatory response of the HPA. The same study also demonstrated exhausted T-cells, suggesting an ongoing inflammatory response.
4.2. Cellular level dysregulation of cytokine gene expression (P38 MAPK controlling pathway, polymorphisms, transcription factors, and epigenetics)
The severity and chronicity of COVID-19 symptoms may be attributed to different patterns of immunopathology (210, 211). Chronic disease was characterized by normal effector T cells pro-inflammatory response but without the normal recruitment signals to attract activated T cells (211).
On a cellular level, viruses can induce cytokine dysregulation through several mechanisms, including viral-induced epigenetic changes in gene expression, transcription factors, superimposed on underlying gene polymorphisms.
If cytokines are responsible for the myriad of chronic relapsing symptoms of the Long COVID patients, it is essential to understand the cellular controlling mechanisms that initiate or suppress cytokine release. Multiple signal transduction pathways control cytokine gene expression. The Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway (212), and the p38 mitogen-activated protein kinases (MAK) play a central role in coordinating the immune system. Through phosphorylation, these pathways respond to cytokines and other external stimuli to control cytokine gene expression.
4.2.1. p38 MAP kinase (p38 MAPK) regulation of proinflammatory cytokines
The p38 mitogen-activated protein kinases serve as essential components of an intracellular signaling pathway, essential for immune function and inflammation. P38 responds to external stresses such as infection, thermal shock, osmotic shock, and ultraviolet radiation, activating downstream targets, including several kinases, transcription factors, and cytosolic proteins. The activated transcription factor NF-kB upregulates cytokine genes, producing an inflammatory response (213). In a positive feedback loop, cytokines then activate the same p38 MAPK protein. The P38 MAPK pathway provides critical regulation over the transcription and translation of pro-inflammatory cytokine genes (214, 215) (Figure 4).
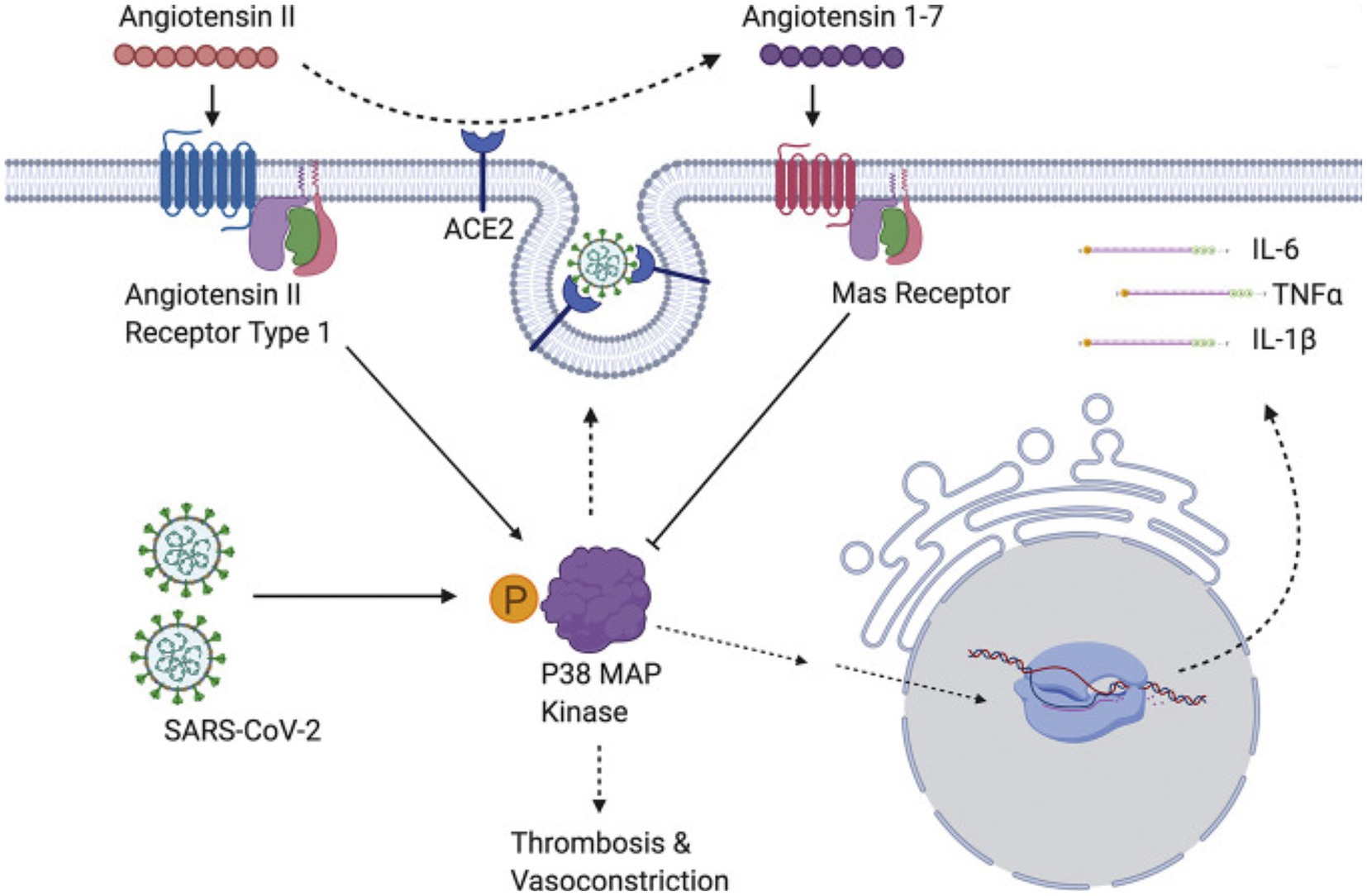
Figure 4. SARS-CoV-2 induces overwhelming inflammation by directly activating p38 and downregulating the Ang1-7 inhibitory pathway. Reproduced from “The role of antioxidants in the chemistry of oxidative stress: a review” by AM Pisoschi and A Pop. (http://creativecommons.org/licenses/by/4.0/).
Regulating the p38 MAPK pathway prevents runaway inflammation. Downregulation of p38 can occur through direct dephosphorylation (217). Downstream mitogen-activated kinases MK2 and MK3 play essential roles in regulating p38 MAPK (218). Genetically modified mice with “knockouts” of p38 MAPK or its downstream substrates MK2 and MK3 provide insights into these protein kinases’ role in disease models. In a mouse model with pemphigus vulgaris, inhibitors of p38 MAPK turned off cytokine release and prevented the blistering characteristic of the skin disease (219).
In an autoimmune encephalomyelitis model, MK2 exerts marked anti-inflammatory effects on p38 MAPK signaling. In MK2 knockout mice, in which the normal inhibitory function of the MK2 on p38 MAPK is blocked, there was a severe worsening of the clinical course (220).
The SARS-CoV virus causes p38 MAPK phosphorylation in infected cells, thereby activating physiological intracellular signaling cascades (221). Spike-mediated entry of SARS-CoV-2 triggers activation of p38 MAPK, occurring within the first 15–75 min after viral infection (222). SARS-CoV-2 directly upregulates p38 MAPK genes, rewiring host cell phosphorylations in 97 of 518 human kinases. This results in p38 MAPK activation with subsequent production of various cytokines, and the shutdown of mitotic kinases, producing cell cycle arrest (223). By potentiating the p38 pathway with gene upregulation and blocking p38 MAPK inhibitors, SARS-CoV-2 increases cytokine production, resulting in worsening inflammation and disease progression (216, 221). Conversely, inhibition of p38 signaling curtails the SARS-CoV-2 inflammatory response (222).
4.2.2. Modification of gene expression through transcription factors and epigenetics
Cytokine gene expression is controlled by a network of transcription factors, including Nuclear Factor-kB (NF-κB), which is upregulated by SARS-CoV-2 (224, 225). NF-κB induces the expression of hundreds of proinflammatory and immune genes encoding for cytokines and chemokines and is a central controlling pathway for inflammation (224). The p38 MAPK pathway regulates the transcriptional activity of NF-κB through phosphorylation (213).
Epigenetics describes the genetic and non-genetic factors that control or modify gene expression without altering the underlying DNA code. These external or environmental factors interact with the host DNA, leading to phenotypic variation. Epigenetics mechanisms include DNA methylation, histone modifications, and chromatin remodeling (226) (Figure 5).
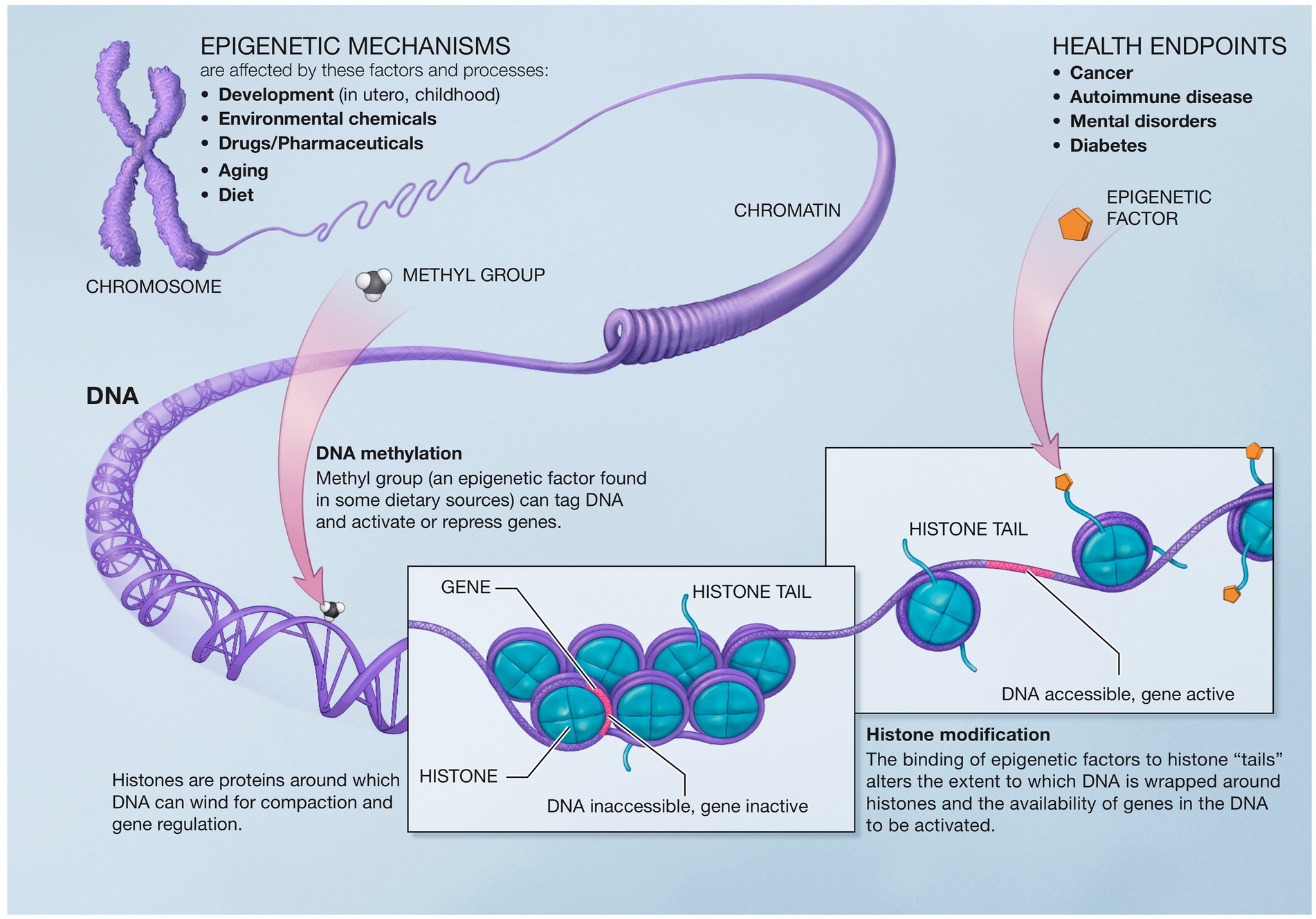
Figure 5. Epigenetic mechanisms altering gene expression. Reproduced from “A Scientific Illustration of How Epigenetic Mechanisms can Affect Health.” National Institutes of Health. (Image available for public use).
Epigenetics and transcriptional factors shape the adaptive immune response and provide a model to understand the genetic basis for the broad clinical spectrum encountered with SARS-CoV-2 infections (227, 228).
Coronavirus pathophysiology and disease severity are closely linked to epigenetic mechanisms, promoting viral entry, replication, evasion of host defenses, and immune hyperactivation (228, 229). There are remarkably 464 upregulated and 222 downregulated genes for the SARS-CoV-2 infection in the complex interaction between host and virus (230).
Transcriptional factors and viral epigenetic modification of host DNA expression result in loss of normal cytokine release regulation (226, 231). SARS-CoV-2 upregulates the p38 MAPK gene through phosphorylation, resulting in the production of a diverse spectrum of cytokines (223). These modifications can lead to increased inflammatory gene expression or loss of inhibition of the p38 MAPK signaling pathway.
Epigenomic analysis of blood hematopoietic and stem progenitor cells (HSPC) revealed sustained changes in hematopoiesis and innate immunity after COVID-19. These authors suggested that “this epigenetic reprogramming of HSPC may underlie long-term altered immune function following infection that are conveyed, through differentiation, to progeny innate immune cells.” (232).
4.2.3. Genetic polymorphisms determine differences in symptoms and clinical severity
Genetic polymorphism in the cytokine genes’ regulatory regions can explain interindividual differences in the severity and symptoms of COVID-19. Different alleles of the cytokine genes affect the level and ratio of cytokines produced, altering the autoimmune and inflammatory response (233). ACE2 polymorphism is associated with susceptibility for SARS—CoV-2 infection and possibly the severity of disease (234, 235).
COVID-19 comorbidities, hypertension, diabetes mellitus, and coronary artery disease result in a more severe clinical course with acute SARS CoV-2 infection. Polymorphisms in 11 genes associated with these comorbidities, involved in regulating immune response, cytokine activity, and viral infection, can explain variations in susceptibility and disease severity (236).
Long COVID post-viral syndrome may be related to genetic predisposition. Single nucleotide polymorphisms of cytokine genes IL-6, TNF-α, IFN-γ, and IL-10 correlate with post-viral fatigue, pain, neurocognitive difficulties, and mood disturbances, respectively. Increased fatigue post-infection is associated with the T allele of IFN-γ +874 T/A SNP (237). Overall severity and duration of post-viral symptoms are associated with polymorphisms in the IFN-γ and IL-10 genes (238).
Polymorphisms may relate to the neurologic post-viral symptoms in Long COVID. For example, a recent study (239) focused on rs2106809, a SNP of the ACE2 promoter. The authors found that allele G of rs2106809 correlated with significantly higher ACE2 expression in the brain, and hypothesized that this SNP could be involved in Long COVID symptoms such as brain fog. However, further studies specifically involving Long COVID patients would be needed to test this hypothesis, and to identify other potential genetic contributions to Long COVID. Another study (240) found that Post-COVID pain was not associated with inflammatory polymorphisms.
4.3. Other factors contributing to the cytokine release syndrome
4.3.1. Oxidative stress
An imbalance between production and accumulation of reactive oxygen species (ROS) causes oxidative stress. When oxygen-free radicals exceed antioxidant capacity, OS can trigger inflammation or result from cytokine release and inflammation (241). OS and CRS are closely related and interdependent processes. Positive feedback loops between OS and CRS result in amplifying the clinical effect and a potentially vicious cycle of OS and inflammation (242, 243). For chronic diseases, low-grade inflammation and OS are tightly linked and interdependent processes (243). The signaling link between OS and cytokines involves the p38 MAPK (244, 245). Combined antioxidant and anti-inflammatory therapy provides an intriguing two-pronged approach to treating the chronic inflammation of the Long COVID patients (246, 247).
4.3.2. Bioenergetic abnormalities
Alterations in cellular energy metabolism from aberrant mitochondrial function may result in insufficient ATP generation (248, 249). Proinflammatory cytokines and OS potentiate the mitochondrial dysfunction in ME/CFS, resulting in glucose hypometabolism and decreased ATP (125). Blockage of the TCA cycle, leading to less efficient metabolism, has been proposed (250). Mitochondria provide the metabolites essential for chromatin remodeling and DNA methylation in epigenetic DNA modifications (251). Dysfunctional mitochondria also contribute to microglial activation and neuroinflammation (252) and to central and peripheral fatigue.
Early studies suggested that SARS-CoV-2 hijacks the host cell mitochondria where it replicates, impairs mitochondrial dynamics and leads to cell death, while promoting its own survival (123, 253). Translocation of viral RNA, RNA transcripts, and proteins into the host mitochondria results in inflammasome activation and suppression of host innate and adaptive immunity (123, 253). Experimental studies in COVID-19 patients have confirmed mitochondrial dysfunction and metabolic changes with increased inefficient glycolysis (254, 255). With increasing age, a decline in ACE2 function and mitochondrial function may make older patients more susceptible to viral induced mitochondrial dysfunction and SARS CoV-2 infection (123).
Long COVID exercise limitation may be due to dysfunctional metabolism and bioenergetic abnormalities. Reduced peak oxygen consumption is observed in studies of post-COVID patients independent of cardiopulmonary status (123, 256, 257) with associated reduced oxygen extraction (257).
5. Experience from post SARS patients who experience chronic fatigue syndrome
Following other epidemics, including the 2003 SARS epidemic, a group of patients showed symptoms of chronic fatigue and sleep disturbances, very similar to those of the Long COVID patients (112, 258). Follow up of 233 survivors at 4 years showed 40.3% with ongoing chronic fatigue, with 27.1% meeting the modified 1994 CDC criteria for CFS. In personal communication with the author regarding ongoing follow up of these patients, Dr. Lam indicated that 100 patients were being followed for chronic fatigue at 4 years. Now, 18 years after the SARS outbreak, 70 patients showed interval resolution of symptoms, while 30 (13%) progressed to chronic illness with ongoing fatigue and sleep disturbance. A few patients with hypocortisolism responded to steroid replacement therapy. In the majority, there was no correlation between fatigue severity and salivary cortisol levels.
6. Progression to ME/CFS and the role of autoimmunity
Why some patients with post-viral syndrome progress to ME/CFS is a critical issue that can involve polymorphic genetic predisposition, an environmental or infectious trigger, and immune system dysregulation (259).
Autoantibodies play an important role in the COVID-19 pathophysiology (260) with dramatically increased autoantibodies compared to non-infected individuals. The wide spectrum of clinical presentations of COVID-19 may relate to altered and misdirected immune response. Whether this explains Long COVID symptoms and persistence has not been established.
However, chronic systemic inflammation leads to a host of illnesses, including autoimmune diseases (261). Since the autoimmune model for post-infectious ME/CFS is increasingly recognized (261–263), this could be a determining factor in the progression from post-viral syndrome to ME/CFS.
The viral triggering of autoimmunity is intimately associated with cytokines (195) and epigenetic alterations of gene expression (264). It is known that coronaviruses trigger autoimmunity. The SARS-CoV virus-induced autoantibodies against lung epithelial cells (265). SARS-CoV-2 infection has precipitated a host of autoimmune diseases, including multisystem inflammatory syndrome in children (MIS-C) (259, 266).
For the Long COVID patients, continuing cytokine expression can produce a low-grade chronic inflammatory state, which can further lead to activated, dysregulated microglia and neuroinflammation (267). A working hypothesis is that some patients will have a genetic polymorphism predisposing them to autoimmunity. Epigenetic changes in gene expression induced by the virus or other external factors may provide the trigger that initiates the progression to autoimmunity and ME/CFS. Recent reports confirm the development of ME/CFS in a subset of Long COVID patients with mild to moderate initial acute symptoms. Biomarkers suggest ongoing inflammation and hypoperfusion as pathomechanisms (268). In patients with unexplained dyspnea, cardiopulmonary testing confirmed circulatory impairment, abnormal ventilatory pattern and ME/CFS in Long COVID patients (269).
7. Therapeutic implications
7.1. Severe acute COVID
The use of anti-cytokine / anti-inflammatory therapies in patients with severe acute COVID-19 has been investigated (270). IL-1 blockers in COVID-19 include anakinra, canakinumab, and rilonacept. In a phase3 trial, administration of Anakinra increased full recovery and lowered mortality compared to standard care alone. On day 28, 50.4% of the anakinra group was fully recovered compared with 26.5% of the standard care group (p < 0.0.0001). Mortality was 6.9% with standard care and 3.2% with anakinra (271).
IL-6 blockers include tocilizumab, sarilumab, and siltuximab. One single institution study of tocilizumab in serve COVID-19 found no difference in mortality compared to the control group (272). In another study, tocilizumab administration decreased the likelihood of progression to mechanical ventilation or death, but did not enhance survival in non-ventilated patients (273).
Other anti-cytokine therapies in severe COVID-19 include tumor necrosis factor inhibitors. In a meta-analysis of 84 studies describing use of anti-TNF treatments it was concluded that TNF-α inhibitors are associated with a lower probability of hospitalization and severe COVID-19 when compared to any other treatment for an underlying inflammatory disease (274).
Glucocorticoids, producing a broad-spectrum immune suppression, have been widely used in severe acute COVID-19. The RECOVERY trial found a reduction in mortality amongst mechanically ventilated patients (29.3% vs. 41.4%) for those patients receiving dexamethasone (275).
7.2. Long COVID
Large national clinical trials include the RECOVER1 in the United States and STIMULATE-ICP2 in the United Kingdom. The 1.5 billion dollar RECOVER trial will test more than a dozen immunosuppressants, immune enhancing drugs, corticosteroids, and antiviral drugs to correct the immune-inflammatory dysfunction of Long COVID. The STIMULATE-ICP trial will test several medicines against Long COVID including the anti-inflammatory colchicine, two antihistamines famotidine and loratadine, and an anti-clotting drug, rivaroxaban.
For Long COVID, results of therapeutic studies are so far limited. In a recent review of pharmacologic treatment of Long COVID, Chee et al. reported on six published trials and 54 ongoing clinical trials (276). Study heterogeneity and varying definitions of “Long COVID” limit comparisons and attempts to draw conclusions.
Chronic inflammation and endothelial dysfunction was investigated in a study of Sulodexide, a novel agent that attenuates endothelial thrombosis and inflammation (277). Following a three-week course of Sulodexide, there was a significant improvement in endothelial function with alleviation of chest pain and palpitations (277). Addressing the close interplay between inflammation, endothelial dysfunction and thrombosis, ongoing studies are evaluating the use of low-dose statins, apixaban and atorvastatin, omega-3 fish oil (276, 278).
Chronic inflammation and pulmonary fibrosis was studied with therapeutic agents pirfenidone (279, 280), corticosteroids (281), and montelukast (282) for treatment of respiratory symptoms of Long COVID. In an open-label study evaluating the utility of prednisolone in patients with post COVID-19 parenchymal lung abnormalities, the administration of prednisolone 10 mg daily for 6 weeks was associated with an improvement in dyspnea and respiratory function (283). In another study, a short, four-day course of corticosteroids reduced symptoms and immunological alterations underlying Long COVID (284).
The neurocognitive symptoms of Long COVID are being studied with various pharmacologic agents including atorvastatin, vortioxetine, fampridine, RUCONEST, and intravenous gamma globulin, as well as nasal sprays containing ivermectin, and retinoic acid.
Increased oxidative stress in Long COVID patients with immune dysregulation is being evaluated with repurposing nutraceuticals and Vitamin supplements (Vitamin C and Vitamin D) as potential therapeutic strategies. Gut dysbiosis and its role in the chronic inflammation of Long COVID patients is being studied with probiotics as a treatment regimen for the treatment and management of Long COVID.
There is currently insufficient data on which to base meaningful conclusions regarding therapeutic interventions for Long COVID. There does seem to be anecdotal experience that “resetting” the immune-inflammatory system can have at least short-term benefits and relief of some symptoms. In this vein, it was observed that some patients experienced symptom relief following COVID-19 vaccination. In 11 studies evaluating the impact of vaccines on patients with Long COVID, seven found improvement in ongoing symptoms while four found no change or even worsening of symptoms (163).
There is anecdotal evidence of similar improvement in Long COVID symptoms with pregnancy, which results in major alterations in the immune-inflammatory system. This is in line with previous reports on remission of Multiple Sclerosis (285), Rheumatoid Arthritis (285, 286), and Autoimmune Hepatitis (287) during pregnancy, suggesting immunosuppressive effects of pregnancy (288). The multifaceted and complex nature of Long COVID with different subgroups and distinct pathophysiologies may require a combination of therapies, tailored to the underlying cause and individual patient symptoms. Returning the body to its homeostasis with a combination of anti-inflammatory, antihistamines, and drugs that treat blood vessel inflammation and microclots may yield results (289).
Ultimately, the prevention of Long COVID is an equally important topic to be addressed. The effect of vaccinations in decreasing Long COVID has been discussed above. Modulating and decreasing the initial immune inflammatory response in COVID-19 may be beneficial in reducing the incidence of Long COVID. A recent preprint indicated a 42% relative decrease (6.3% vs. 10.6%) in the incidence of Long COVID in participants who received early outpatient COVID-19 treatment with the drug metformin with known anti-inflammatory effects (290) compared to a matched placebo control group (291).
8. Final thoughts
The proposed explanation of the pathophysiology of the persistent Long COVID symptoms is likely incomplete. However, it provides a reasonable model to consider as part of the pathophysiology of the plight of the millions of persons who are stuck in a seemingly never-ending recovery from COVID-19. It is also important to note that cytokines do not have to be detectable in the periphery to have an effect on immune dysregulation and its possible downstream consequences (292). Moreover, methodological limitations can impact accuracy of cytokine measurements (29). Therefore, it remains a challenge to use peripheral blood cytokines measures as easily accessible biomarkers of the disease (29).
The seemingly competing mechanisms of inflammation and microthrombosis may actually be two sides of the same immunothrombotic coin. Both processes can occur and are intimately interrelated with one leading to the other. However, it is conceivable that in some Long COVID patients, one or the other processes may predominate, leading to different symptoms. This also has important implications for potential treatment options for different Long COVID patients directed at inflammation vs. clotting pathways or both.
We hope this article sheds light on how the science of cytokines and their controlling signaling pathways can explain a multitude of diverse Long COVID symptoms. Common underlying processes are likely involved in the pathophysiology of Long COVID and other chronic diseases and post-viral syndromes. Understanding this pathophysiology should help us to define and test therapeutic interventions, opening the pathway to recovery.
Summary
Dysregulated immune-inflammatory response with elevated peripheral and central cytokines can explain Long COVID symptoms. Hyperreactive brain microglia can modulate a host of CNS-mediated symptoms.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
RNL, RJL, and AA contributed to the conception and design of the study. RNL wrote the first draft of the manuscript. RNL, RJL, and AA wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
Acknowledgments
We thank M. Horan, Y. Re’em, and M. B. Barnkob for their valuable inputs on the manuscript. We thank members of the Patient-Led Research For COVID19 for their support and inspirations. We thank members of the Body Politic Online COVID19 Support Group for sharing their experiences.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
1. World Health Organization. A clinical case definition of post COVID-19 condition by a Delphi consensus, 6 October 2021. Geneva: World Health Organization (2021).
2. Assaf, G, Davis, H, McCorkell, L, Wei, H, O’Neill, B, Akrami, A, et al. What does COVID-19 recovery actually look like? An analysis of the c survey by patient-led research team [internet]. London, UK: The COVID-19 Body Politic Slack Group (2020).
3. Tran, VT, Porcher, R, Pane, I, and Ravaud, P. Course of post COVID-19 disease symptoms over time in the ComPaRe long COVID prospective e-cohort. Nat Commun. (2022) 13:1812. doi: 10.1038/s41467-022-29513-z
4. Cirulli, ET, Schiabor Barrett, KM, Riffle, S, Bolze, A, Neveux, I, Dabe, S, et al. (2020). Long-term COVID-19 symptoms in a large unselected population [internet]. medRxiv [Preprint]. doi: 10.1101/2020.10.07.20208702
5. Blair, PW, Brown, DM, Jang, M, Antar, AAR, Keruly, JC, Bachu, V, et al. (2020). The clinical course of COVID-19 in the outpatient setting: a prospective cohort study [internet]. medRxiv [Preprint]. doi: 10.1101/2020.09.01.20184937
6. Ding, H, Yin, S, Cheng, Y, Cai, Y, Huang, W, and Deng, W. Neurologic manifestations of nonhospitalized patients with COVID-19 in Wuhan, China [internet]. MedComm. (2020) 1:253–6. doi: 10.1002/mco2.13
7. Tenforde, MW, Kim, SS, Lindsell, CJ, Billig Rose, E, Shapiro, NI, Files, DC, et al. Symptom duration and risk factors for delayed return to usual health among outpatients with COVID-19 in a multistate health care systems network — United States, march–June 2020. MMWR Morb Mortal Wkly Rep. (2020) 69:993–8. Available from:. doi: 10.15585/mmwr.mm6930e1
8. Carfì, A, Bernabei, R, and Landi, F, for the Gemelli Against COVID-19 Post-Acute Care Study Group. Landi F. persistent symptoms in patients after acute COVID-19. JAMA. (2020) 324:603–5. doi: 10.1001/jama.2020.12603
9. Garrigues, E, Janvier, P, Kherabi, Y, le Bot, A, Hamon, A, Gouze, H, et al. Post-discharge persistent symptoms and health-related quality of life after hospitalization for COVID-19. J Infect [Internet]. (2020) 81:e4–6. doi: 10.1016/j.jinf.2020.08.029
10. Arnold, DT, Hamilton, FW, Milne, A, Morley, A, Viner, J, Attwood, M, et al. (2020). Patient outcomes after hospitalisation with COVID-19 and implications for follow-up; results from a prospective UK cohort. medRxiv [Preprint]. doi: 10.1101/2020.08.12.20173526
11. Ayoubkhani, D, Bosworth, ML, King, S, Pouwels, KB, Glickman, M, Nafilyan, V, et al. (2022). Risk of Long Covid in people infected with SARS-CoV-2 after two doses of a COVID-19 vaccine: community-based, matched cohort study [internet]. medRxiv [Preprint]. doi: 10.1101/2022.02.23.22271388
12. Al-Aly, Z, Bowe, B, and Xie, Y. Long COVID after breakthrough SARS-CoV-2 infection. Nat Med. (2022) 28:1461–7. doi: 10.1038/s41591-022-01840-0
13. Davis, HE, Assaf, GS, McCorkell, L, Wei, H, Low, RJ, Re’em, Y, et al. Characterizing long COVID in an international cohort: 7 months of symptoms and their impact. eClinicalMedicine. (2021) 38:101019. doi: 10.1016/j.eclinm.2021.101019
14. Ghoshal, UC, Ghoshal, U, Rahman, MM, Mathur, A, Rai, S, Akhter, M, et al. Post-infection functional gastrointestinal disorders following coronavirus disease-19: a case-control study. J Gastroenterol Hepatol. (2022) 37:489–98. doi: 10.1111/jgh.15717
15. de Rooij, LPMH, Becker, LM, and Carmeliet, P. A role for the vascular endothelium in post–acute COVID-19? Circulation. (2022) 145:1503–5. doi: 10.1161/circulationaha.122.059231
16. Seitz, A, and Ong, P. Endothelial dysfunction in COVID-19: a potential predictor of long-COVID? Int J Cardiol. (2022) 349:155–6. doi: 10.1016/j.ijcard.2021.11.051
17. Davis, HE, McCorkell, L, Vogel, JM, and Topol, EJ. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol. (2023) 21:133–46. doi: 10.1038/s41579-022-00846-2
18. Proal, AD, and VanElzakker, MB. Long COVID or post-acute Sequelae of COVID-19 (PASC): an overview of biological factors that may contribute to persistent symptoms. Front Microbiol. (2021) 12:698169. doi: 10.3389/fmicb.2021.698169
19. Mogensen, TH, and Paludan, SR. Molecular pathways in virus-induced cytokine production [internet]. Microbiol Mol Biol Rev. (2001) 65:131–50. Available from:. doi: 10.1128/MMBR.65.1.131-150.2001
20. Qin, C, Zhou, L, Hu, Z, Zhang, S, Yang, S, Tao, Y, et al. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis. (2020) 71:762–8. doi: 10.1093/cid/ciaa248
21. Coperchini, F, Chiovato, L, Croce, L, Magri, F, and Rotondi, M. The cytokine storm in COVID-19: an overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. (2020) 53:25–32. doi: 10.1016/j.cytogfr.2020.05.003
22. Huang, C, Wang, Y, Li, X, Ren, L, Zhao, J, Hu, Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet [Internet]. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
23. del Valle, DM, Kim-Schulze, S, Huang, HH, Beckmann, ND, Nirenberg, S, Wang, B, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med [Internet]. (2020) 26:1636–43. doi: 10.1038/s41591-020-1051-9
24. Zheng, HY, Gao, Y, Liu, S, Sun, D, Yang, F, Zong, L, et al. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID-19 patients. Cell Mol Immunol. (2020) 17:1180–2. doi: 10.1038/s41423-020-00551-1
25. Hadjadj, J, Yatim, N, Barnabei, L, Corneau, A, Boussier, J, Smith, N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science [Internet]. (2020) 369:718–24. doi: 10.1126/science.abc6027
26. Lei, X, Dong, X, Ma, R, Wang, W, Xiao, X, Tian, Z, et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun. (2020) 11:3810. doi: 10.1038/s41467-020-17665-9
27. Phetsouphanh, C, Darley, DR, Wilson, DB, Howe, A, Munier, CML, Patel, SK, et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat Immunol. (2022) 23:210–6. doi: 10.1038/s41590-021-01113-x
28. Schultheiß, C, Willscher, E, Paschold, L, Gottschick, C, Klee, B, Glasauer, S, et al. From online data collection to identification of disease mechanisms: the IL-1ß, IL-6 and TNF-α cytokine triad is associated with post-acute sequelae of COVID-19 in a digital research cohort. SSRN Electron J [Internet]. (2021). doi: 10.2139/ssrn.3963839
29. VanElzakker, MB, Brumfield, SA, and Lara Mejia, PS. Neuroinflammation and cytokines in Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): a critical review of research methods. Front Neurol. (2018) 9:1033. doi: 10.3389/fneur.2018.01033
30. Peluso, MJ, Lu, S, Tang, AF, Durstenfeld, MS, Ho, HE, Goldberg, SA, et al. Markers of immune activation and inflammation in individuals with Postacute Sequelae of severe acute respiratory syndrome coronavirus 2 infection. J Infect Dis. (2021) 224:1839–48. doi: 10.1093/infdis/jiab490
31. Klein, J, Wood, J, Jaycox, J, Lu, P, Dhodapkar, RM, Gehlhausen, JR, et al. (2022). Distinguishing features of long COVID identified through immune profiling. medRxiv [Preprint]. doi: 10.1101/2022.08.09.22278592.
32. Cevik, M, Bamford, CGG, and Ho, A. COVID-19 pandemic—a focused review for clinicians. Clin Microbiol Infect. (2020) 26:842–7. doi: 10.1016/j.cmi.2020.04.023
33. Mehta, P, McAuley, DF, Brown, M, Sanchez, E, Tattersall, RS, Manson, JJ, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. (2020) 395:1033–4. Available from:. doi: 10.1016/S0140-6736(20)30628-0
34. Wu, C, Chen, X, Cai, Y, Xia, J’a, Zhou, X, Xu, S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. (2020) 180:934–43. doi: 10.1001/jamainternmed.2020.0994
35. Tisoncik, JR, Korth, MJ, Simmons, CP, Farrar, J, Martin, TR, and Katze, MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. (2012) 76:16–32. doi: 10.1128/MMBR.05015-11
36. Herold, T, Jurinovic, V, Arnreich, C, Lipworth, BJ, Hellmuth, JC, von Bergwelt-Baildon, M, et al. Elevated levels of IL-6 and CRP predict the need for mechanical ventilation in COVID-19 [internet]. J Allergy Clin Immunol. (2020) 146:128–36.e4. Available from:. doi: 10.1016/j.jaci.2020.05.008
37. Yang, Y, Shen, C, Li, J, Yuan, J, Yang, M, Wang, F, et al. (2020). Exuberant elevation of IP-10, MCP-3 and IL-1ra during SARS-CoV-2 infection is associated with disease severity and fatal outcome. medRxiv [Preprint]. doi: 10.1101/2020.11.26.20152520.
38. Shimabukuro-Vornhagen, A, Gödel, P, Subklewe, M, Stemmler, HJ, Schlößer, HA, Schlaak, M, et al. Cytokine release syndrome. J Immunother. Cancer. (2018) 6:56. doi: 10.1186/s40425-018-0343-9
39. Lee, DW, Gardner, R, Porter, DL, Louis, CU, Ahmed, N, Jensen, M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. (2014) 124:188–95. doi: 10.1182/blood-2014-05-552729
40. Moore, JB, and June, CH. Cytokine release syndrome in severe COVID-19. Science. (2020) 368:473–4. doi: 10.1126/science.abb8925
41. Turner, MD, Nedjai, B, Hurst, T, and Pennington, DF. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta—Mol Cell Res. (2014) 1843:2563–82. doi: 10.1016/j.bbamcr.2014.05.014
42. Merad, M, and Martin, JC. Author correction: pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. (2020) 20:448. doi: 10.1038/s41577-020-0353-y
43. Ye, Q, Wang, B, and Mao, J. The pathogenesis and treatment of the ‘cytokine storm’ in COVID-19. J Infect. (2020) 80:607–13. doi: 10.1016/j.jinf.2020.03.037
44. de Lucena, TMC, da Silva Santos, AF, de Lima, BR, de Albuquerque Borborema, ME, and de Azevêdo Silva, J. Mechanism of inflammatory response in associated comorbidities in COVID-19. Diabetes Metab Syndr. (2019) 14:597–600. doi: 10.1016/j.dsx.2020.05.025
45. Meftahi, GH, Jangravi, Z, Sahraei, H, and Bahari, Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: the contribution of “inflame-aging.”. Inflamm Res. (2020) 69:825–39. doi: 10.1007/s00011-020-01372-8
46. Rakotoarivelo, V, Lacraz, G, Mayhue, M, Brown, C, Rottembourg, D, Fradette, J, et al. Inflammatory cytokine profiles in visceral and subcutaneous adipose tissues of obese patients undergoing bariatric surgery reveal lack of correlation with obesity or diabetes [internet]. EBioMedicine. (2018) 30:237–47. doi: 10.1016/j.ebiom.2018.03.004
47. Xiao, J, Li, J, Cai, L, Chakrabarti, S, and Li, X. Cytokines and diabetes research. J Diabetes Res [Internet]. (2014) 2014:1–2. doi: 10.1155/2014/920613
48. Randeria, SN, Thomson, GJA, Nell, TA, Roberts, T, and Pretorius, E. Inflammatory cytokines in type 2 diabetes mellitus as facilitators of hypercoagulation and abnormal clot formation. Cardiovasc Diabetol. (2019) 18:72–15. doi: 10.1186/s12933-019-0870-9
49. Tanase, DM, Gosav, EM, Radu, S, Ouatu, A, Rezus, C, Ciocoiu, M, et al. Arterial hypertension and interleukins: potential therapeutic target or future diagnostic marker? Int J Hypertens. (2019) 2019:3159283:1–17. doi: 10.1155/2019/3159283
50. Palaiodimos, L, Kokkinidis, DG, Li, W, Karamanis, D, Ognibene, J, Arora, S, et al. Severe obesity, increasing age and male sex are independently associated with worse in-hospital outcomes, and higher in-hospital mortality, in a cohort of patients with COVID-19 in the Bronx. New York Metab. (2020) 108:154262. doi: 10.1016/j.metabol.2020.154262
51. Crayne, CB, Albeituni, S, Nichols, KE, and Cron, RQ. The immunology of macrophage activation syndrome [internet]. Front Immunol. (2019) 10:119. doi: 10.3389/fimmu.2019.00119
52. Kosyreva, A, Dzhalilova, D, Lokhonina, A, Vishnyakova, P, and Fatkhudinov, T. The role of macrophages in the pathogenesis of SARS-CoV-2-associated acute respiratory distress syndrome. Front Immunol. (2021) 12:682871. doi: 10.3389/fimmu.2021.682871
53. Yamato, M, and Kataoka, Y. Fatigue sensation following peripheral viral infection is triggered by neuroinflammation: who will answer these questions? Neural Regen Res. (2015) 10:203–4. doi: 10.4103/1673-5374.152369
54. Butler, M, Pollak, TA, Rooney, AG, Michael, BD, and Nicholson, TR. Neuropsychiatric complications of covid-19 [internet]. BMJ. (2020):m3871. doi: 10.1136/bmj.m3871
55. Lima, M, Siokas, V, Aloizou, AM, Liampas, I, Mentis, AA, Tsouris, Z, et al. Unraveling the possible routes of SARS-COV-2 invasion into the central nervous system. Curr Treat Options Neurol [Internet]. (2020) 22:37. doi: 10.1007/s11940-020-00647-z
56. Matschke, J, Lütgehetmann, M, Hagel, C, Sperhake, JP, Schröder, AS, Edler, C, et al. Neuropathology of patients with COVID-19 in Germany: a post-mortem case series [internet]. Lancet Neurol. (2020) 19:919–29. doi: 10.1016/S1474-4422(20)30308-2
57. Paniz-Mondolfi, A, Bryce, C, Grimes, Z, Gordon, RE, Reidy, J, Lednicky, J, et al. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) [internet]. J Med Virol. (2020) 92:699–702. doi: 10.1002/jmv.25915
58. Bohmwald, K, Gálvez, NMS, Ríos, M, and Kalergis, AM. Neurologic alterations due to respiratory virus infections. Front Cell Neurosci. (2018) 12:386. doi: 10.3389/fncel.2018.00386
59. Mohammadi, S, Moosaie, F, and Aarabi, MH. Understanding the immunologic characteristics of neurologic manifestations of SARS-CoV-2 and potential immunological mechanisms. Mol Neurobiol [Internet]. (2020). doi: 10.1007/s12035-020-02094-y
60. Dantzer, R. Neuroimmune interactions: from the brain to the immune system and vice versa. Physiol Rev. (2018) 98:477–504. doi: 10.1152/physrev.00039.2016
61. Dantzer, R, Konsman, JP, Bluthé, RM, and Kelley, KW. Neural and humoral pathways of communication from the immune system to the brain: parallel or convergent? Auton Neurosci. (2000) 85:60–5. doi: 10.1016/S1566-0702(00)00220-4
62. Streit, WJ, Mrak, RE, and Griffin, WST. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. (2004) 1:1–4. doi: 10.1186/1742-2094-1-14
63. Shabab, T, Khanabdali, R, Moghadamtousi, SZ, Kadir, HA, and Mohan, G. Neuroinflammation pathways: a general review [internet]. Int J Neurosci. (2017) 127:624–33. doi: 10.1080/00207454.2016.1212854
64. Frank-Cannon, TC, Alto, LT, McAlpine, FE, and Tansey, MG. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener. (2009) 4:1–13. doi: 10.1186/1750-1326-4-47
65. Najjar, S, Pearlman, DM, Alper, K, Najjar, A, and Devinsky, O. Neuroinflammation and psychiatric illness. J Neuroinflammation. (2013) 10:1–24. doi: 10.1186/1742-2094-10-43
66. Liotta, EM, Batra, A, Clark, JR, Shlobin, NA, Hoffman, SC, Orban, ZS, et al. Frequent neurologic manifestations and encephalopathy-associated morbidity in Covid-19 patients. Ann Clin Transl Neurol. (2020) 7:2221–30. doi: 10.1002/acn3.51210
67. Bakhoum, MF, Ritter, M, Garg, A, Chan, AX, Bakhoum, CY, and Smith, D. Subclinical ocular inflammation in persons recovered from ambulatory COVID-19 [internet]. medRxiv [Preprint]. doi: 10.1101/2020.09.22.20128140.
68. Carson, MJ, Thrash, JC, and Walter, B. The cellular response in neuroinflammation: the role of leukocytes, microglia and astrocytes in neuronal death and survival. Clin Neurosci Res. (2006) 6:237–45. doi: 10.1016/j.cnr.2006.09.004
69. Zheng, ZV, and Wong, KCG. Microglial activation and polarization after subarachnoid hemorrhage. Neuroimmunol Neuroinflamm. (2019) 2019:1. doi: 10.20517/2347-8659.2018.52
70. Kaur, C, Rathnasamy, G, and Ling, EA. Biology of microglia in the developing brain. J Neuropathol Exp Neurol. (2017) 76:736–53. doi: 10.1093/jnen/nlx056
71. Subramaniam, SR, and Federoff, HJ. Targeting microglial activation states as a therapeutic avenue in Parkinson’s disease. Front Aging Neurosci. (2017) 9:176. doi: 10.3389/fnagi.2017.00176
72. von Bernhardi, R, Eugenín-von Bernhardi, L, and Eugenín, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci. (2015) 7:124. doi: 10.3389/fnagi.2015.00124
73. Li, L, Mao, S, Wang, J, Ding, X, and Zen, JY. Viral infection and neurological disorders—potential role of extracellular nucleotides in neuroinflammation. ExRNA. (2019) 1:1–5. doi: 10.1186/s41544-019-0031-z
74. Püntener, U, Booth, SG, Perry, VH, and Teeling, JL. Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. J Neuroinflammation. (2012) 9:146. doi: 10.1186/1742-2094-9-146
75. Godbout, JP, Chen, J, Abraham, J, Richwine, AF, Berg, BM, Kelley, KW, et al. Exaggerated neuroinflammation and sickness behavior in aged mice after activation of the peripheral innate immune system [internet]. FASEB J. (2005) 19:1329–31. Available from:. doi: 10.1096/fj.05-3776fje
76. Niraula, A, Sheridan, JF, and Godbout, JP. Microglia priming with aging and stress. Neuropsychopharmacology [Internet]. (2017) 42:318–33. doi: 10.1038/npp.2016.185
77. Tay, TL, Béchade, C, D’Andrea, I, St-Pierre, MK, Henry, MS, Roumier, A, et al. Microglia gone rogue: impacts on psychiatric disorders across the lifespan. Front Mol Neurosci. (2018) 10:421. doi: 10.3389/fnmol.2017.00421
78. Rothwell, NJ, and Hopkins, SJ. Cytokines and the nervous system II: actions and mechanisms of action. Trends Neurosci. (1995) 18:130–6. doi: 10.1016/0166-2236(95)93890-A
79. Pollmacher, T, Haack, M, Schuld, A, Reichenberg, A, and Yirmiya, R. Low levels of circulating inflammatory cytokines—do they affect human brain functions? Brain Behav Immun. (2002) 16:525–32. doi: 10.1016/S0889-1591(02)00004-1
80. Barbosa-Silva, MC, Santos, LE, and Rangel, B. The impact of non-neurotropic influenza strains on the brain: a role for microglial priming? J Neurosci. (2018) 38:7758–60. doi: 10.1523/JNEUROSCI.1368-18.2018
81. Dantzer, R. Cytokine-induced sickness behavior: where do we stand? Brain Behav Immun. (2001) 15:7–24. doi: 10.1006/brbi.2000.0613
82. Yirmiya, R, Pollak, Y, Morag, M, Reichenberg, A, Barak, O, Avitsur, R, et al. Illness, cytokines, and depression [internet]. Ann N Y Acad Sci. (2000) 917:478–87. doi: 10.1111/j.1749-6632.2000.tb05412.x
83. Hou, R, Garner, M, Holmes, C, Osmond, C, Teeling, J, Lau, L, et al. Peripheral inflammatory cytokines and immune balance in generalised anxiety disorder: case-controlled study. Brain Behav Immun. (2017) 62:212–8. doi: 10.1016/j.bbi.2017.01.021
84. Rothwell, NJ, Luheshi, G, and Toulmond, S. Cytokines and their receptors in the central nervous system: physiology, pharmacology, and pathology. Pharmacol Ther. (1996) 69:85–95. doi: 10.1016/0163-7258(95)02033-0
85. Khairova, RA, Machado-Vieira, R, Du, J, and Manji, HK. A potential role for pro-inflammatory cytokines in regulating synaptic plasticity in major depressive disorder. Int J Neuropsychopharmacol. (2009) 12:561–78. doi: 10.1017/S1461145709009924
86. McAfoose, J, and Baune, BT. Evidence for a cytokine model of cognitive function. Neurosci Biobehav Rev. (2009) 33:355–66. doi: 10.1016/j.neubiorev.2008.10.005
87. Patterson, PH, and Nawa, H. Neuronal differentiation factors/cytokines and synaptic plasticity. Cells. (1993) 72:123–37. doi: 10.1016/S0092-8674(05)80032-7
88. Miller, RJ, Jung, H, Bhangoo, SK, and White, FA. Cytokine and chemokine regulation of sensory neuron function In: Sensory nerves. ed. Brendan J. Canning Berlin, Heidelberg: Springer (2009). 417–49.
89. Kluger, MJ. Fever: role of pyrogens and cryogens. Physiol Rev. (1991) 71:93–127. doi: 10.1152/physrev.1991.71.1.93
90. Rivier, C, and Rivest, S (2007). Mechanisms mediating the effects of cytokines on neuroendocrine functions in the rat. Ciba Foundation Symposium 172—Corticotropin-Releasing Factor, 204–225.
91. Dunn, AJ. Systematic interleukin-1 administration stimulates hypothalamic norepinephrine metabolism parallelling the increased plasma corticosterone. Life Sci. (1988) 43:429–35. doi: 10.1016/0024-3205(88)90522-X
92. Fann, MJ, and Patterson, PH. Neuropoietic cytokines and activin a differentially regulate the phenotype of cultured sympathetic neurons. Proc Natl Acad Sci U S A. (1994) 91:43–7. doi: 10.1073/pnas.91.1.43
93. Balter, LJ, Bosch, JA, Aldred, S, Drayson, MT, Veldhuijzen van Zanten, JJCS, Higgs, S, et al. Selective effects of acute low-grade inflammation on human visual attention. Neuroimage. (2019) 202:116098. doi: 10.1016/j.neuroimage.2019.116098
94. Miller, AH, Haroon, E, Raison, CL, and Felger, JC. Cytokine targets in the brain: impact on neurotransmitters and Neurocircuits. Depress Anxiety. (2013) 30:297. doi: 10.1002/da.22084
95. Dantzer, R, O’Connor, JC, Freund, GG, Johnson, RW, and Kelley, KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. (2008) 9:46–56. doi: 10.1038/nrn2297
96. Capuron, L, and Dantzer, R. Cytokines and depression: the need for a new paradigm. Brain Behav Immun. (2003) 17 Suppl 1:S119–24. doi: 10.1016/S0889-1591(02)00078-8
97. Kronfol, Z. Cytokines and the brain: implications for clinical psychiatry [internet]. Am J Psychiatr. (2000) 157:683–94. Available from:. doi: 10.1176/appi.ajp.157.5.683
98. Kappelmann, N, Dantzer, R, and Khandaker, GM. Interleukin-6 as potential mediator of long-term neuropsychiatric symptoms of COVID-19. Psychoneuroendocrinology. (2021) 105295:105295. doi: 10.1016/j.psyneuen.2021.105295
99. Murta, V, Villarreal, A, and Ramos, AJ (2020). SARS-CoV-2 impact on the central nervous system: are astrocytes and microglia main players or merely bystanders? Available at: https://www.preprints.org/manuscript/202006.0319/v1
100. Hasselbalch, HC. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. (2013) 24:133–45. doi: 10.1016/j.cytogfr.2013.01.004
101. Dantzer, R. Cytokine, sickness behavior, and depression. Immunol Allergy Clin N Am. (2009) 29:247–64. doi: 10.1016/j.iac.2009.02.002
102. Ogoina, D. Fever, fever patterns and diseases called “fever” – a review. J Infect Public Health. (2011) 4:108–24. doi: 10.1016/j.jiph.2011.05.002
103. Conti, B, Tabarean, I, Andrei, C, and Bartfai, T. Cytokines and fever. Front Biosci. (2004) 9:1433–49. doi: 10.2741/1341
104. Netea, MG, Kullberg, BJ, and Van der Meer, JWM. Circulating cytokines as mediators of fever. Clin Infect Dis. (2000) 31:S178–84. doi: 10.1086/317513
105. Abrams, RMC, Simpson, DM, Navis, A, Jette, N, Zhou, L, and Shin, SC. Small fiber neuropathy associated with SARS-CoV-2 infection. Muscle Nerve. (2022) 65:440–3. doi: 10.1002/mus.27458
106. Magrinelli, F, Briani, C, Romano, M, Ruggero, S, Toffanin, E, Triolo, G, et al. The association between serum cytokines and damage to large and small nerve fibers in diabetic peripheral neuropathy. J Diabetes Res. (2015) 2015:1–7. doi: 10.1155/2015/547834
107. Langjahr, M, Schubert, AL, Sommer, C, and Üçeyler, N. Increased pro-inflammatory cytokine gene expression in peripheral blood mononuclear cells of patients with polyneuropathies. J Neurol. (2018) 265:618–27. doi: 10.1007/s00415-018-8748-4
108. England, J. Parainfectious neuropathies [internet]. J Neurol Sci. (2019) 405:22. Available from:. doi: 10.1016/j.jns.2019.10.060
109. Malcangio, M, Clark, AK, and Old, EA. Neuropathic pain and cytokines: current perspectives. JPR. (2013) 6:803–14. doi: 10.2147/JPR.S53660
110. Hung, AL, Lim, M, and Doshi, TL. Targeting cytokines for treatment of neuropathic pain. Scandinavian journal of pain. (2017) 17:287–93. doi: 10.1016/j.sjpain.2017.08.002
111. Ceban, F, Ling, S, Lui, LMW, Lee, Y, Gill, H, Teopiz, KM, et al. Fatigue and cognitive impairment in post-COVID-19 syndrome: a systematic review and meta-analysis. Brain Behav Immun. (2022) 101:93–135. doi: 10.1016/j.bbi.2021.12.020
112. Islam, MF, Cotler, J, and Jason, LA. Post-viral fatigue and COVID-19: lessons from past epidemics. Fatigue Biomed Health Behav. (2020) 8:61–9. doi: 10.1080/21641846.2020.1778227
113. Wan, JJ, Qin, Z, Wang, PY, Sun, Y, and Liu, X. Muscle fatigue: general understanding and treatment. Exp Mol Med. (2017) 49:e384. doi: 10.1038/emm.2017.194
114. Chaudhuri, A, and Behan, PO. Fatigue and basal ganglia. J Neurol Sci. (2000) 179:34–42. doi: 10.1016/S0022-510X(00)00411-1
115. Miller, AH, Miller, JH, Pham, DS, Beentjes, KK, Unger, ER, and Pagnoni, G. Decreased basal ganglia activation in subjects with chronic fatigue syndrome: association with symptoms of fatigue. PLoS 1. (2014) 9:e115750. doi: 10.1371/journal.pone.0115750
116. Felger, JC, and Miller, AH. Cytokine effects on the basal ganglia and dopamine function: the subcortical source of inflammatory malaise. Front Neuroendocrinol. (2012) 33:315–27. doi: 10.1016/j.yfrne.2012.09.003
117. Cordeiro, LMS, Rabelo, PCR, Moraes, MM, Teixeira-Coelho, F, Coimbra, CC, Wanner, SP, et al. Physical exercise-induced fatigue: the role of serotonergic and dopaminergic systems. Braz J Med Biol Res. (2017) 50:e6432. doi: 10.1590/1414-431x20176432
118. Späth-Schwalbe, E, Hansen, K, Schmidt, F, Schrezenmeier, H, Marshall, L, Burger, K, et al. Acute effects of recombinant human Interleukin-6 on endocrine and central nervous sleep functions in healthy men 1. J Clin Endocrinol Metab. (1998) 83:1573–9. doi: 10.1210/jcem.83.5.4795
119. Corominas, H, Alegre, C, Narváez, J, Fernández-Cid, CM, Torrente-Segarra, V, Gómez, MR, et al. Correlation of fatigue with other disease related and psychosocial factors in patients with rheumatoid arthritis treated with tocilizumab: ACT-AXIS study. Medicine [Internet]. (2019) 98:e15947. doi: 10.1097/MD.0000000000015947
120. Wendling, D, Racadot, E, and Wijdenes, J. Treatment of severe rheumatoid arthritis by anti-interleukin 6 monoclonal antibody. J Rheumatol [Internet]. (1993) 20:259–62.
121. Sarkar, S, Malovic, E, Harishchandra, DS, Ghaisas, S, Panicker, N, Charli, A, et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinsons Dis. (2017) 3:30. doi: 10.1038/s41531-017-0032-2
122. Gatti, P, Ilamathi, HS, Todkar, K, and Germain, M. Mitochondria targeted viral replication and survival strategies—prospective on SARS-CoV-2. Front Pharmacol [Internet]. (2020) 11:578599. doi: 10.3389/fphar.2020.578599
123. Singh, KK, Chaubey, G, Chen, JY, and Suravajhala, P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am J Physiol Cell Physiol. (2020) 319:C258–67. doi: 10.1152/ajpcell.00224.2020
124. Saleh, J, Peyssonnaux, C, Singh, KK, and Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion. (2020) 54:1–7. doi: 10.1016/j.mito.2020.06.008
125. Morris, G, and Maes, M. Mitochondrial dysfunctions in Myalgic encephalomyelitis / chronic fatigue syndrome explained by activated immuno-inflammatory, oxidative and nitrosative stress pathways. Metab Brain Dis. (2013) 29:19–36. doi: 10.1007/s11011-013-9435-x
126. Missiroli, S, Genovese, I, Perrone, M, Vezzani, B, Vitto, VAM, and Giorgi, C. The role of mitochondria in inflammation: from cancer to neurodegenerative disorders [internet]. J Clin Med. (2020) 9:740. Available from:. doi: 10.3390/jcm9030740
127. Keller, C, Steensberg, A, Hansen, AK, Fischer, CP, Plomgaard, P, and Pedersen, BK. Effect of exercise, training, and glycogen availability on IL-6 receptor expression in human skeletal muscle. J Appl Physiol (1985). (2005) 99:2075–9. doi: 10.1152/japplphysiol.00590.2005
128. Suzuki, K. Characterization of exercise-induced cytokine release, the impacts on the body, the mechanisms and modulations. Int J Sports Exerc Med. (2019) 5:122. doi: 10.23937/2469-5718/1510122
129. da Luz, D, and Scheffer, AL. Exercise-induced immune system response: anti-inflammatory status on peripheral and central organs. Biochim Biophys Acta Mol basis Dis. (2020) 1866:165823. doi: 10.1016/j.bbadis.2020.165823
130. Landers-Ramos, RQ, Jenkins, NT, Spangenburg, EE, Hagberg, JM, and Prior, SJ. Circulating angiogenic and inflammatory cytokine responses to acute aerobic exercise in trained and sedentary young men. Eur J Appl Physiol. (2014) 114:1377. doi: 10.1007/s00421-014-2861-6
131. Fischer, CP. Interleukin-6 in acute exercise and training: what is the biological relevance? Exerc Immunol Rev [Internet]. (2006) 12:6–33.
132. Fischer, CP. Interleukin-6 in acute exercise and training: what is the biological relevance. Exerc Immunol Rev. (2006) 12:41.
133. Kim, HK, Konishi, M, Takahashi, M, Tabata, H, Endo, N, Numao, S, et al. Effects of acute endurance exercise performed in the morning and evening on inflammatory cytokine and metabolic hormone responses. PLoS One [Internet]. (2015) 10:e0137567. doi: 10.1371/journal.pone.0137567
134. Moneghetti, KJ, Skhiri, M, Contrepois, K, Kobayashi, Y, Maecker, H, Davis, M, et al. Value of circulating cytokine profiling during submaximal exercise testing in Myalgic encephalomyelitis/chronic fatigue syndrome. Sci Rep. (2018) 8:2779. doi: 10.1038/s41598-018-20941-w
135. White, AT, Light, AR, Hughen, RW, Bateman, L, Martins, TB, Hill, HR, et al. Severity of symptom flare after moderate exercise is linked to cytokine activity in chronic fatigue syndrome. Psychophysiology. (2010) 47:615–24. doi: 10.1111/j.1469-8986.2010.00978.x
136. Finnell, JE, Lombard, CM, Melson, MN, Singh, NP, Nagarkatti, M, Nagarkatti, P, et al. The protective effects of resveratrol on social stress-induced cytokine release and depressive-like behavior. Brain Behav Immun Health. (2017) 59:147–57. doi: 10.1016/j.bbi.2016.08.019
137. Marsland, AL, Walsh, C, Lockwood, K, and John-Henderson, NA. The effects of acute psychological stress on circulating and stimulated inflammatory markers: a systematic review and meta-analysis. Brain Behav Immun. (2017) 64:208. doi: 10.1016/j.bbi.2017.01.011
138. Bernard-Raichon, L, Venzon, M, Klein, J, Axelrad, JE, Zhang, C, Sullivan, AP, et al. Gut microbiome dysbiosis in antibiotic-treated COVID-19 patients is associated with microbial translocation and bacteremia. Nat Commun. (2022) 13:5926. doi: 10.1038/s41467-022-33395-6
139. Zuo, T, Wu, X, Wen, W, and Lan, P. Gut microbiome alterations in COVID-19. Genomics Proteomics Bioinformatics. (2021) 19:679–88. doi: 10.1016/j.gpb.2021.09.004
140. Jin, B, Singh, R, Ha, SE, Zogg, H, Park, PJ, and Ro, S. Pathophysiological mechanisms underlying gastrointestinal symptoms in patients with COVID-19. World J Gastroenterol. (2021) 27:2341–52. doi: 10.3748/wjg.v27.i19.2341
141. D’Amico, F, Baumgart, DC, Danese, S, and Peyrin-Biroulet, L. Diarrhea during COVID-19 infection: pathogenesis, epidemiology, prevention, and management [internet]. Clin Gastroenterol Hepatol. (2020) 18:1663–72. Available from:. doi: 10.1016/j.cgh.2020.04.001
142. Wu, Y, Guo, C, Tang, L, Hong, Z, Zhou, J, Dong, X, et al. Prolonged presence of SARS-CoV-2 viral RNA in faecal samples. Lancet Gastroenterol Hepatol. (2020) 5:434–5. doi: 10.1016/S2468-1253(20)30083-2
143. Wang, X, Zheng, J, Guo, L, Yao, H, Wang, L, Xia, X, et al. Fecal viral shedding in COVID-19 patients: clinical significance, viral load dynamics and survival analysis. Virus Res. (2020) 289:198147. doi: 10.1016/j.virusres.2020.198147
144. Norouzi Masir, M, and Shirvaliloo, M. Symptomatology and microbiology of the gastrointestinal tract in post-COVID conditions. JGH Open. (2022) 6:667–76. doi: 10.1002/jgh3.12811
145. Gao, J. Correlation between anxiety-depression status and cytokines in diarrhea-predominant irritable bowel syndrome. Exp Ther Med. (2013) 6:93. doi: 10.3892/etm.2013.1101
146. Sartor, RB. Cytokines in intestinal inflammation: pathophysiological and clinical considerations. Gastroenterology. (1994) 106:533–9. doi: 10.1016/0016-5085(94)90614-9
147. Reisinger, EC, Fritzsche, C, Krause, R, and Krejs, GJ. Diarrhea caused by primarily non-gastrointestinal infections. Nat Clin Pract Gastroenterol Hepatol. (2005) 2:216–22. doi: 10.1038/ncpgasthep0167
148. Rana, SV, Sharma, S, Sinha, SK, Parsad, KK, Malik, A, and Singh, K. Pro-inflammatory and anti-inflammatory cytokine response in diarrhoea-predominant irritable bowel syndrome patients. Trop Gastroenterol. (2013) 33:251–6, 256. doi: 10.7869/tg.2012.66
149. Goyal, D, Ali, SA, and Singh, RK. Emerging role of gut microbiota in modulation of neuroinflammation and neurodegeneration with emphasis on Alzheimer’s disease. Prog Neuro-Psychopharmacol Biol Psychiatry. (2021) 106:110112. doi: 10.1016/j.pnpbp.2020.110112
150. Mou, Y, du, Y, Zhou, L, Yue, J, Hu, X, Liu, Y, et al. Gut microbiota interact with the brain through systemic chronic inflammation: implications on Neuroinflammation, neurodegeneration, and aging. Front Immunol. (2022) 13:796288. doi: 10.3389/fimmu.2022.796288
151. Schett, G, Manger, B, Simon, D, and Caporali, R. COVID-19 revisiting inflammatory pathways of arthritis. Nat Rev Rheumatol. (2020) 16:465–70. doi: 10.1038/s41584-020-0451-z
152. Ahmed, S, Zimba, O, and Gasparyan, AY. COVID-19 and the clinical course of rheumatic manifestations. Clin Rheumatol. (2021) 40:2611–9. doi: 10.1007/s10067-021-05691-x
153. Shah, S, Danda, D, Kavadichanda, C, Das, S, Adarsh, MB, and Negi, VS. Autoimmune and rheumatic musculoskeletal diseases as a consequence of SARS-CoV-2 infection and its treatment. Rheumatol Int. (2020) 40:1539–54. doi: 10.1007/s00296-020-04639-9
154. Wu, P, Duan, F, Luo, C, Liu, Q, Qu, X, Liang, L, et al. Characteristics of ocular findings of patients with coronavirus disease 2019 (COVID-19) in Hubei Province, China. JAMA Ophthalmol. (2020) 138:575–8. doi: 10.1001/jamaophthalmol.2020.1291
155. Pérez-Bartolomé, F, and Sánchez-Quirós, J. Ocular manifestations of SARS-CoV-2: literature review. Archivos De La Sociedad Espan~ola De Oftalmologi’a. 96:32–40. doi: 10.1016/j.oftale.2020.07.003
156. Gold, DM, and Galetta, SL. Neuro-ophthalmologic complications of coronavirus disease 2019 (COVID-19). Neurosci Lett. (2021) 742:135531. doi: 10.1016/j.neulet.2020.135531
157. Church, MK, and McGill, JI. Human ocular mast cells. Curr Opin Allergy Clin Immunol. (2002) 2:419–22. doi: 10.1097/00130832-200210000-00009
158. Badham, SP, and Hutchinson, CV. Characterising eye movement dysfunction in myalgic encephalomyelitis/chronic fatigue syndrome. German J Ophthalmol. (2013) 251:2769–76. doi: 10.1007/s00417-013-2431-3
159. Pacheco-Herrero, M, Soto-Rojas, LO, Harrington, CR, Flores-Martinez, YM, Villegas-Rojas, MM, León-Aguilar, AM, et al. Elucidating the Neuropathologic mechanisms of SARS-CoV-2 infection. Front Neurol. (2021) 12:660087. doi: 10.3389/fneur.2021.660087
161. Chen, JJ, Flanagan, EP, Jitprapaikulsan, J, López-Chiriboga, A, Fryer, JP, Leavitt, JA, et al. Myelin oligodendrocyte glycoprotein antibody–positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. (2018) 195:8–15. doi: 10.1016/j.ajo.2018.07.020
162. Jeong, GU, Kwon, HJ, Ng, WH, Liu, X, Moon, HW, Yoon, GY, et al. Ocular tropism of SARS-CoV-2 in animal models with retinal inflammation via neuronal invasion following intranasal inoculation. Nat Commun. (2022) 13:7675. doi: 10.1038/s41467-022-35225-1
163. Notarte, KI, Catahay, JA, Velasco, JV, Pastrana, A, ver, AT, Pangilinan, FC, et al. Impact of COVID-19 vaccination on the risk of developing long-COVID and on existing long-COVID symptoms: a systematic review. EClinicalMedicine. (2022) 53:101624. doi: 10.1016/j.eclinm.2022.101624
164. Tsouris, Z, Provatas, A, Bakirtzis, C, Aloizou, AM, Siokas, V, Tsimourtou, V, et al. Anti-MOG positive bilateral optic neuritis and brainstem encephalitis secondary to COVID-19 infection: a case report. Neurol Int. (2022) 14:991–6. doi: 10.3390/neurolint14040078
165. Stark, K, and Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat Rev Cardiol. (2021) 18:666–82. doi: 10.1038/s41569-021-00552-1
166. Klok, FA, Kruip, MJHA, van der Meer, NJM, Arbous, MS, Gommers, DAMPJ, Kant, KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. (2020) 191:145–7. doi: 10.1016/j.thromres.2020.04.013
167. Wichmann, D, Sperhake, JP, Lütgehetmann, M, Steurer, S, Edler, C, Heinemann, A, et al. Autopsy findings and venous thromboembolism in patients with COVID-19. ACP J Club. (2020) 173:1030. doi: 10.7326/L20-1206
168. Aksu, K, Donmez, A, and Keser, G. Inflammation-induced thrombosis: mechanisms, disease associations and management. Curr Pharm Des. (2012) 18:1478–93. doi: 10.2174/138161212799504731
169. Connors, JM, and Levy, JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. (2020) 135:2033–40. doi: 10.1182/blood.2020006000
170. McIntyre, TM. Inflammatory cytokines and thrombosis. Blood. (2012) 120. doi: 10.1182/blood.V120.21.SCI-44.SCI-44
171. Fibrinogen Studies Collaboration Kaptoge, S, White, IR, Thompson, SG, Wood, AM, Lewington, S, et al. Associations of plasma fibrinogen levels with established cardiovascular disease risk factors, inflammatory markers, and other characteristics: individual participant meta-analysis of 154,211 adults in 31 prospective studies. Am J Epidemiol. (2007) 166:867–79. doi: 10.1093/aje/kwm191
172. Welsh, P, Woodward, M, Rumley, A, and Lowe, G. Associations of plasma pro-inflammatory cytokines, fibrinogen, viscosity and C-reactive protein with cardiovascular risk factors and social deprivation: the fourth Glasgow MONICA study. Br J Haematol. (2008) 141:852–61. doi: 10.1111/j.1365-2141.2008.07133.x
173. Najem, MY, Couturaud, F, and Lemarié, CA. Cytokine and chemokine regulation of venous thromboembolism. J Thromb Haemost. (2020) 18:1009–19. doi: 10.1111/jth.14759
174. Branchford, BR, and Carpenter, SL. The role of inflammation in venous thromboembolism. Front Pediatr. (2018) 6:142. doi: 10.3389/fped.2018.00142
175. Zhang, S, Liu, Y, Wang, X, Yang, L, Li, H, Wang, Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. (2020) 13:120–2. doi: 10.1186/s13045-020-00954-7
176. Behrens, K, and Alexander, WS. Alexander WS. Cytokine control of megakaryopoiesis. Growth Factors. (2018) 36:89–103. doi: 10.1080/08977194.2018.1498487
177. Manne, BK, Denorme, F, Middleton, EA, Portier, I, Rowley, JW, Stubben, C, et al. Platelet gene expression and function in patients with COVID-19. Blood. (2020) 136:1317–29. doi: 10.1182/blood.2020007214
178. Aid, M, Busman-Sahay, K, Vidal, SJ, Maliga, Z, Bondoc, S, Starke, C, et al. Vascular disease and thrombosis in SARS-CoV-2-infected rhesus macaques. Cell(Cambridge,Mass). (2020) 183:1354–1366.e13. doi: 10.1016/j.cell.2020.10.005
179. Pretorius, E, Vlok, M, Venter, C, Bezuidenhout, JA, Laubscher, GJ, Steenkamp, J, et al. Persistent clotting protein pathology in Long COVID/post-acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc Diabetol. (2021) 20:172. doi: 10.1186/s12933-021-01359-7
180. Pretorius, E, Venter, C, Laubscher, GJ, Kotze, MJ, Moremi, K, Oladejo, S, et al. Prevalence of symptoms, comorbidities, fibrin amyloid microclots and platelet pathology in individuals with Long COVID/Post-Acute Sequelae of COVID-19 (PASC). Cardiovascular Diabetology. (2021) 148. doi: 10.1186/s12933-022-01579-5
181. Dani, M, Dirksen, A, Taraborrelli, P, Torocastro, M, Panagopoulos, D, Sutton, R, et al. Autonomic dysfunction in “long COVID”: rationale, physiology and management strategies [internet]. Clin Med. (2021) 21:e63–7. doi: 10.7861/clinmed.2020-0896
182. Fedorowski, A, and Sutton, R. Autonomic dysfunction and postural orthostatic tachycardia syndrome in post-acute COVID-19 syndrome. Nat Rev Cardiol. (2023) 2:1–2. doi: 10.1038/s41569-023-00842-w
183. Buoite Stella, A, Furlanis, G, Frezza, NA, Valentinotti, R, Ajcevic, M, and Manganotti, P. Autonomic dysfunction in post-COVID patients with and witfhout neurological symptoms: a prospective multidomain observational study. J Neurol. (2022) 269:587–96. doi: 10.1007/s00415-021-10735-y
184. Eshak, N, Abdelnabi, M, Ball, S, Elgwairi, E, Creed, K, Test, V, et al. Dysautonomia: an overlooked neurological manifestation in a critically ill COVID-19 patient [internet]. Am J Med Sci. (2020) 360:427–9. doi: 10.1016/j.amjms.2020.07.022
185. Astudillo, L, Laure, A, Fabry, V, Pugnet, G, Maury, P, Labrunée, M, et al. Le syndrome de tachycardie posturale (PoTS): une maladie pour l’interniste [Internet]. Rev Med Interne. (2018) 39:627–34. Available from:. doi: 10.1016/j.revmed.2018.04.017
186. Kenney, MJ, and Ganta, CK. Autonomic nervous system and immune system interactions [internet]. Comprehensive. Physiology. (2014) 4:1177–200. Available from:. doi: 10.1002/cphy.c130051
187. Okamoto, LE, Raj, SR, Gamboa, A, Shibao, CA, Arnold, AC, Garland, EM, et al. Sympathetic activation is associated with increased IL-6, but not CRP in the absence of obesity: lessons from postural tachycardia syndrome and obesity [internet]. Am J Phys Heart Circ Phys. (2015) 309:H2098–107. Available from:. doi: 10.1152/ajpheart.00409.2015
188. Guedj, E, Million, M, Dudouet, P, Tissot-Dupont, H, Bregeon, F, Cammilleri, S, et al. 18 F-FDG brain PET hypometabolism in post-SARS-CoV-2 infection: substrate for persistent/delayed disorders? Eur J Nucl Med Mol Imaging. (2021) 48:592–5. doi: 10.1007/s00259-020-04973-x
189. Yong, SJ. Persistent brainstem dysfunction in Long-COVID: a hypothesis. ACS Chem Neurosci. (2021) 12:573–80. doi: 10.1021/acschemneuro.0c00793
190. Schofield, JR, Blitshteyn, S, Shoenfeld, Y, and Hughes, GRV. Postural tachycardia syndrome (POTS) and other autonomic disorders in antiphospholipid (Hughes) syndrome (APS). Lupus. (2014) 23:697–702. doi: 10.1177/0961203314524468
191. Noureldine, HA, El Hasbani, G, Nour Eldine, M, Nour-Eldine, W, Taher, A, and Uthman, I. Postural tachycardia syndrome (POTS) and antiphospholipid syndrome (APS): what do we know so far? Rev neur, Par. (2022) 178:306–14. doi: 10.1016/j.neurol.2021.10.006
192. Johansson, M, Yan, H, Welinder, C, Végvári, Á, Hamrefors, V, Bäck, M, et al. Plasma proteomic profiling in postural orthostatic tachycardia syndrome (POTS) reveals new disease pathways. Sci Rep. (2022) 12:1–9. doi: 10.1038/s41598-022-24729-x
193. Kwan, AC, Ebinger, JE, Wei, J, le, CN, Oft, JR, Zabner, R, et al. Apparent risks of postural orthostatic tachycardia syndrome diagnoses after COVID-19 vaccination and SARS-Cov-2 infection. Nat Cardiovasc Res. (2022) 1:1187–94. doi: 10.1038/s44161-022-00177-8
194. Fedorowski, A, Li, H, Yu, X, Koelsch, KA, Harris, VM, Liles, C, et al. Antiadrenergic autoimmunity in postural tachycardia syndrome. Europace. (2017) 19:1211–9. doi: 10.1093/europace/euw154
195. Kamal, D, and Moudgil, DC. Cytokines in autoimmunity: role in induction, regulation, and treatment. J Interf Cytokine Res. (2011) 31:695–703. doi: 10.1089/jir.2011.0065
196. Gaebler, C, Wang, Z, Lorenzi, JCC, Muecksch, F, Finkin, S, Tokuyama, M, et al. Evolution of antibody immunity to SARS-CoV-2 [internet]. Cold Spring Harbor Laboratory. (2020) 591:639–644. doi: 10.1101/2020.11.03.367391
197. Avanzato, VA, Matson, MJ, Seifert, SN, Pryce, R, Williamson, BN, Anzick, SL, et al. Case study: prolonged infectious SARS-CoV-2 shedding from an asymptomatic immunocompromised cancer patient [internet]. Cell. (2020) 183:1901–1912.e9. doi: 10.1016/j.cell.2020.10.049
198. Stein, SR, Ramelli, SC, Grazioli, A, et al. SARS-CoV-2 infection and persistence in the human body and brain at autopsy. Nature. (2022) 612:758–763. doi: 10.1038/s41586-022-05542-y
199. Merad, M, and Martin, JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nature Rev Immun. (2020) 20:355–62. doi: 10.1038/s41577-020-0331-4
200. Cytokines in the balance [internet]. Nat Immunol. (2019) 20:1557–7. doi: 10.1038/s41590-019-0557-0
201. Polidoro, RB, Hagan, RS, de Santis Santiago, R, and Schmidt, NW. Overview: systemic inflammatory response derived from lung injury caused by SARS-CoV-2 infection explains severe outcomes in COVID-19. Front Immunol. (2020) 11:1626. doi: 10.3389/fimmu.2020.01626
202. Besedovsky, HO, and del Rey, A. The cytokine-HPA axis feed-back circuit. Zeitschrift für Rheumaforschung. (2000) 59:II26–30. doi: 10.1007/s003930070014
203. Somasundaram, NP, Ranathunga, I, Ratnasamy, V, Wijewickrama, PSA, Dissanayake, HA, Yogendranathan, N, et al. The impact of SARS-Cov-2 virus infection on the endocrine system. JES. (2020) 4. doi: 10.1210/jendso/bvaa082
204. Silverman, MN, Pearce, BD, Biron, CA, and Miller, AH. Immune modulation of the hypothalamic-pituitary-adrenal (HPA) Axis during viral infection. Viral Immunol. (2005) 18:41–78. doi: 10.1089/vim.2005.18.41
205. Silverman, MN, and Sternberg, EM. Glucocorticoid regulation of inflammation and its behavioral and metabolic correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci. (2012) 1261:55–63. doi: 10.1111/j.1749-6632.2012.06633.x
206. Agarwal, S, and Agarwal, SK. Endocrine changes in SARS-CoV-2 patients and lessons from SARS-CoV. Postgrad Med J. (2020) 96:412–6. doi: 10.1136/postgradmedj-2020-137934
207. Mongioì, LM, Barbagallo, F, Condorelli, RA, Cannarella, R, Aversa, A, la Vignera, S, et al. Possible long-term endocrine-metabolic complications in COVID-19: lesson from the SARS model [internet]. Endocrine. (2020) 68:467–70. doi: 10.1007/s12020-020-02349-7
208. Leow, MK, Kwek, DS, Ng, AW, Ong, KC, Kaw, GJ, and Lee, LS. Hypocortisolism in survivors of severe acute respiratory syndrome (SARS). Clin Endocr. (2005) 63:197–202. doi: 10.1111/j.1365-2265.2005.02325.x
209. Pal, R. COVID-19, hypothalamo-pituitary-adrenal axis and clinical implications. Endocrine. (2020) 68:251–2. doi: 10.1007/s12020-020-02325-1
210. Bergamaschi, L, Mescia, F, Turner, L, Hanson, A, Kotagiri, P, Dunmore, BJ, et al. (2021). Delayed bystander CD8 T cell activation, early immune pathology and persistent dysregulation characterise severe COVID-19. medRxiv [Preprint]. doi: 10.1101/2021.01.11.20248765.
211. Patterson, BK, Guevara-Coto, J, Yogendra, R, Francisco, E, Long, E, Pise, A, et al. (2020). Immune-based prediction of COVID-19 severity and chronicity decoded using machine learning [Internet]. bioRxiv [Preprint]. Available at: https://www.biorxiv.org/content/10.1101/2020.12.16.423122v1.abstract
212. Seif, F, Khoshmirsafa, M, Aazami, H, Mohsenzadegan, M, Sedighi, G, and Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun Signal [Internet]. (2017) 15:23. doi: 10.1186/s12964-017-0177-y
213. Saha, RN, Jana, M, and Pahan, K. MAPK p38 Regulates Transcriptional Activity of NF-κB in Primary Human Astrocytes via Acetylation of p65. J Immunol. (2007) 179:7101–9. doi: 10.4049/jimmunol.179.10.7101
214. Cuenda, A, and Rousseau, S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta (BBA) Mol Cell Res. (2007) 1773:1358–75. doi: 10.1016/j.bbamcr.2007.03.010
215. Bachstetter, AD, and Van Eldik, LJ. The p38 MAP kinase family as regulators of Proinflammatory cytokine production in degenerative diseases of the CNS. Aging Dis. (2010) 1:199–211. doi: 10.1101/2021.01.11.20248765
216. Grimes, JM, and Grimes, KV. p38 MAPK inhibition: a promising therapeutic approach for COVID-19. J Mol Cell Cardiol. (2020) 144:63–5. doi: 10.1016/j.yjmcc.2020.05.007
217. Huth, TK, Staines, D, and Marshall-Gradisnik, S. ERK1/2, MEK1/2 and p38 downstream signalling molecules impaired in CD56 dim CD16+ and CD56 bright CD16 dim/− natural killer cells in chronic fatigue syndrome/Myalgic encephalomyelitis patients. J Transl Med. (2016) 14:97. doi: 10.1186/s12967-016-0859-z
218. Soni, S, Anand, P, and Padwad, YS. MAPKAPK2: the master regulator of RNA-binding proteins modulates transcript stability and tumor progression. J Exp Clin Cancer Res. (2019) 38:121–18. doi: 10.1186/s13046-019-1115-1
219. Berkowitz, P, Hu, P, Warren, S, Liu, Z, Diaz, LA, and Rubenstein, DS. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci U S A. (2006) 103:12855–60. doi: 10.1073/pnas.0602973103
220. Soukup, K, Halfmann, A, le Bras, M, Sahin, E, Vittori, S, Poyer, F, et al. The MAPK-activated kinase MK2 attenuates dendritic cell–mediated Th1 differentiation and autoimmune encephalomyelitis. J Immunol. (2015) 195:541–52. doi: 10.4049/jimmunol.1401663
221. Mizutani, T, Fukushi, S, Saijo, M, Kurane, I, and Morikawa, S. Phosphorylation of p38 MAPK and its downstream targets in SARS coronavirus-infected cells. Biochem Biophys Res Commun. (2004) 319:1228–34. doi: 10.1016/j.bbrc.2004.05.107
222. Faist, A, Schloer, S, Mecate-Zambrano, A, Janowski, J, Schreiber, A, Boergeling, Y, et al. Inhibition of p38 signaling curtails the SARS-CoV-2 induced inflammatory response but retains the IFN-dependent antiviral defense of the lung epithelial barrier. Antivir Res. (2023) 209:105475. doi: 10.1016/j.antiviral.2022.105475
223. Bouhaddou, M, Memon, D, Meyer, B, White, KM, Rezelj, VV, Correa Marrero, M, et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell(Cambridge,Mass). (2020) 182:685–712.e19. doi: 10.1016/j.cell.2020.06.034
224. Liu, T, Zhang, L, Joo, D, and Sun, SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. (2017) 2. doi: 10.1038/sigtrans.2017.23
225. Hirano, T, and Murakami, M. COVID-19: a new virus, but a familiar receptor and cytokine release syndrome. Immunity. (2020) 52:731–3. doi: 10.1016/j.immuni.2020.04.003
226. Yasmin, R, Siraj, S, Hassan, A, Khan, AR, Abbasi, R, and Ahmad, N. Epigenetic regulation of inflammatory cytokines and associated genes in human malignancies. Mediat Inflamm. (2015) 2015:1–8. doi: 10.1155/2015/201703
227. Tripathi, SK, and Lahesmaa, R. Transcriptional and epigenetic regulation of T-helper lineage specification. Immunol Rev. (2014) 261:62–83. doi: 10.1111/imr.12204
228. El Baba, R, and Herbein, G. Management of epigenomic networks entailed in coronavirus infections and COVID-19. Epigenetics. (2020) 12:118. doi: 10.1186/s13148-020-00912-7
229. Pruimboom, L. Methylation pathways and SARS-CoV-2 lung infiltration and cell membrane-virus fusion are both subject to epigenetics. Front Cell Infect Microbiol. (2020) 10:290. doi: 10.3389/fcimb.2020.00290
230. Khan, MAAK, and ABMMK, Islam. (2020). SARS-CoV-2 proteins exploit host’s genetic and epigenetic mediators for the annexation of key host signaling pathways that confers its immune evasion and disease pathophysiology [internet]. bioRxiv [Preprint]. doi: 10.1101/2020.05.06.050260
231. Paschos, K, and Allday, MJ. Epigenetic reprogramming of host genes in viral and microbial pathogenesis. Trends Microbiol. (2010) 18:439–47. doi: 10.1016/j.tim.2010.07.003
232. Cheong, JG, Ravishankar, A, Sharma, S, Parkhurst, CN, Nehar-Belaid, D, Ma, S, et al. (2022). Epigenetic memory of COVID-19 in innate immune cells and their progenitors. bioRxiv [Preprint].
233. Hoffmann, SC, Stanley, EM, Cox, ED, DiMercurio, BS, Koziol, DE, Harlan, DM, et al. Ethnicity greatly influences cytokine gene polymorphism distribution. Am J Transpl. (2002) 2:560–7. doi: 10.1034/j.1600-6143.2002.20611.x
234. Devaux, CA, and Camoin-Jau, L. An update on angiotensin-converting enzyme 2 structure/functions, polymorphism, and duplicitous nature in the pathophysiology of coronavirus disease 2019: implications for vascular and coagulation disease associated with severe acute respiratory syndrome coronavirus infection. Front Microbiol. (2022) 13:1042200. doi: 10.3389/fmicb.2022.1042200
235. Suh, S, Lee, S, Gym, H, Jee, SH, Yoon, S, Park, S, et al. A systematic review on papers that study on SNPs that affect SARS-CoV-2 infection & COVID-19 severity. doi: 10.21203/rs.3.rs-470358/v1
236. Feng, S, Song, F, Guo, W, Tan, J, Zhang, X, Qiao, F, et al. Potential genes associated with COVID-19 and comorbidity. Int J Med Sci. (2022) 19:402–15. doi: 10.7150/ijms.67815
237. Piraino, B, Vollmer-Conna, U, and Lloyd, AR. Genetic associations of fatigue and other symptom domains of the acute sickness response to infection. Brain Behav Immun. (2012) 26:552–8. doi: 10.1016/j.bbi.2011.12.009
238. Vollmer-Conna, U, Piraino, BF, Cameron, B, Davenport, T, Hickie, I, Wakefield, D, et al. Cytokine polymorphisms have a synergistic effect on severity of the acute sickness response to infection. Clin Infect Dis. (2008) 47:1418–25. doi: 10.1086/592967
239. Luo, YS, Luo, L, Li, W, Chen, Y, Wu, GF, Chen, F, et al. Evaluation of a Functional Single Nucleotide Polymorphism of the SARS-CoV-2 Receptor ACE2 That Is Potentially Involved in Long COVID. Front Genet. (2022) 13:931562. doi: 10.3389/fgene.2022.931562
240. Fernández-de-Las-Peñas, C, Giordano, R, Díaz-Gil, G, Gómez-Esquer, F, Ambite-Quesada, S, Palomar-Gallego, MA, et al. Post-COVID pain is not associated with inflammatory polymorphisms in people who had been hospitalized by COVID-19. JCM. (2022) 11. doi: 10.3390/jcm11195645
242. Elmarakby, AA, and Sullivan, JC. Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc Ther. (2012) 30:49–59. doi: 10.1111/j.1755-5922.2010.00218.x
243. Biswas, SK. Does the interdependence between oxidative stress and inflammation explain the antioxidant paradox? Oxid med cell Longev. (2016) 2016:1–9. doi: 10.1155/2016/5698931
244. Son, Y, Cheong, YK, Kim, NH, Chung, HT, Kang, DG, and Pae, HO. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J Signal Transduct [Internet]. (2011) 2011:1–6. doi: 10.1155/2011/792639
245. Nahirnyj, A, Livne-Bar, I, Guo, X, and Sivak, JM. ROS detoxification and Proinflammatory cytokines are linked by p38 MAPK signaling in a model of mature astrocyte activation. PLoS One. (2013) 8:e83049. doi: 10.1371/journal.pone.0083049
246. Pisoschi, AM, and Pop, A. The role of antioxidants in the chemistry of oxidative stress: a review. Eur J Med Chem. (2015) 97:55–74. doi: 10.1016/j.ejmech.2015.04.040
247. Sadarani, BN, and Majumdar, AS. Resveratrol potentiates the effect of dexamethasone in rat model of acute lung inflammation. Int Immunopharmacol. (2015) 28:773–9. doi: 10.1016/j.intimp.2015.07.038
248. Astin, R, Banerjee, A, Baker, MR, Dani, M, Ford, E, Hull, JH, et al. Long COVID: mechanisms, risk factors and recovery. Exp Physiol. (2023) 108:12–27. doi: 10.1113/EP090802
249. Wood, E, Hall, KH, and Tate, W. Role of mitochondria, oxidative stress and the response to antioxidants in myalgic encephalomyelitis/chronic fatigue syndrome: a possible approach to SARS-CoV-2 “long-haulers”? Chronic Dis Transl Med. (2021) 7:14–26. doi: 10.1016/j.cdtm.2020.11.002
250. Fluge, Ø, Mella, O, Bruland, O, Risa, K, Dyrstad, SE, Alme, K, et al. Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight. (2016) 1:e89376. doi: 10.1172/jci.insight.89376
251. Martínez-Reyes, I, and Chandel, NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. (2020) 11:102–11. doi: 10.1038/s41467-019-13668-3
252. Chiurazzi, M, Di Maro, M, Cozzolino, M, and Colantuoni, A. Mitochondrial dynamics and microglia as new targets in metabolism regulation. IJMS. (2020) 21:3450–3564. doi: 10.3390/ijms21103450
253. Ganji, R, and Reddy, PH. Impact of COVID-19 on mitochondrial-based immunity in aging and age-related diseases. Front Aging Neurosci. (2020) 12:614650. doi: 10.3389/fnagi.2020.614650
254. Ajaz, S, McPhail, MJ, Singh, KK, Mujib, S, Trovato, FM, Napoli, S, et al. Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. Am J Physiol Cell Physiol. (2021) 320:C57–65. doi: 10.1152/ajpcell.00426.2020
255. Gibellini, L, De Biasi, S, Paolini, A, Borella, R, Boraldi, F, Mattioli, M, et al. Altered bioenergetics and mitochondrial dysfunction of monocytes in patients with COVID-19 pneumonia. EMBO Mol Med. (2020) 12:e13001. doi: 10.15252/emmm.202013001
256. Baratto, C, Caravita, S, Soranna, D, Faini, A, Dewachter, C, Zambon, A, et al. Current limitations of invasive exercise hemodynamics for the diagnosis of heart failure with preserved ejection fraction [internet]. Circu Heart Fail. (2021) 14:e007555. doi: 10.1161/circheartfailure.120.007555
257. Singh, I, Joseph, P, Heerdt, PM, Cullinan, M, Lutchmansingh, DD, Gulati, M, et al. Persistent Exertional intolerance after COVID-19: insights from invasive cardiopulmonary exercise testing. Chest. (2022) 161:54–63. doi: 10.1016/j.chest.2021.08.010
258. Lam, MHB, Wing, YK, Yu, MWM, Leung, CM, Ma, RCW, Kong, APS, et al. Mental morbidities and chronic fatigue in severe acute respiratory syndrome survivors: Long-term follow-up. Arch Intern Med. (2009) 169:2142–7. doi: 10.1001/archinternmed.2009.384
259. Ehrenfeld, M, Tincani, A, Andreoli, L, Cattalini, M, Greenbaum, A, Kanduc, D, et al. Covid-19 and autoimmunity. Autoimmun Rev. (2020) 19:102597. doi: 10.1016/j.autrev.2020.102597
260. Wang, EY, Mao, T, Klein, J, Dai, Y, Huck, JD, Jaycox, JR, et al. Diverse functional autoantibodies in patients with COVID-19. Nature [Internet]. (2021) 595: 283–288. doi: 10.1038/s41586-021-03631-y
261. Furman, D, Campisi, J, Verdin, E, Carrera-Bastos, P, Targ, S, Franceschi, C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. (2019) 25:1822–32. doi: 10.1038/s41591-019-0675-0
262. Steiner, S, Becker, SC, Hartwig, J, Sotzny, F, Lorenz, S, Bauer, S, et al. Autoimmunity-related risk variants in PTPN22 and CTLA4 are associated with ME/CFS with infectious onset. Front Immunol. (2020) 11:578. doi: 10.3389/fimmu.2020.00578
263. Sotzny, F, Blanco, J, Capelli, E, Castro-Marrero, J, Steiner, S, Murovska, M, et al. Myalgic encephalomyelitis/chronic fatigue syndrome—evidence for an autoimmune disease. Autoimmun Rev. (2018) 17:601–9. doi: 10.1016/j.autrev.2018.01.009
264. Surace, AEA, and Hedrich, CM. The role of epigenetics in autoimmune/inflammatory disease. Front Immunol. (2019) 10:1525–1541. doi: 10.3389/fimmu.2019.01525
265. Lin, YS, Lin, CF, Fang, YT, Kuo, YM, Liao, PC, Yeh, TM, et al. Antibody to severe acute respiratory syndrome (SARS)-associated coronavirus spike protein domain 2 cross-reacts with lung epithelial cells and causes cytotoxicity. Clin Exp Immunol. (2005) 141:500–8. doi: 10.1111/j.1365-2249.2005.02864.x
266. Galeotti, C, and Bayry, J. Autoimmune and inflammatory diseases following COVID-19. Nat Rev Rheumatol. (2020) 16:413–4. doi: 10.1038/s41584-020-0448-7
267. Tate, W, Walker, M, Sweetman, E, Helliwell, A, Peppercorn, K, Edgar, C, et al. Molecular mechanisms of Neuroinflammation in ME/CFS and Long COVID to sustain disease and promote relapses. Front Neurol. (2022) 13:877772. doi: 10.3389/fneur.2022.877772
268. Kedor, C, Freitag, H, Meyer-Arndt, L, Wittke, K, Hanitsch, LG, Zoller, T, et al. A prospective observational study of post-COVID-19 chronic fatigue syndrome following the first pandemic wave in Germany and biomarkers associated with symptom severity. Nat Commun. (2022) 13:5104–13. doi: 10.1038/s41467-022-32507-6
269. Mancini, DM, Brunjes, DL, Lala, A, Trivieri, MG, Contreras, JP, and Natelson, BH. Use of cardiopulmonary stress testing for patients with unexplained dyspnea post-coronavirus disease. JACC Heart Fail. (2021) 9:927–37. doi: 10.1016/j.jchf.2021.10.002
270. Jamilloux, Y, Henry, T, Belot, A, Viel, S, Fauter, M, el Jammal, T, et al. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions [internet]. Autoimmun Rev. (2020) 19:102567. doi: 10.1016/j.autrev.2020.102567
271. Kyriazopoulou, E, Poulakou, G, Milionis, H, Metallidis, S, Adamis, G, Tsiakos, K, et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial [internet]. Nat Med. (2021) 27:1752–60. doi: 10.1038/s41591-021-01499-z
272. Tsai, A, Diawara, O, Nahass, RG, and Brunetti, L. Impact of tocilizumab administration on mortality in severe COVID-19. Sci Rep. (2020) 10:19131. doi: 10.1038/s41598-020-76187-y
273. Salama, C, Han, J, Yau, L, Reiss, WG, Kramer, B, Neidhart, JD, et al. Tocilizumab in patients hospitalized with Covid-19 pneumonia. N Engl J Med. (2021) 384:20–30. doi: 10.1056/NEJMoa2030340
274. Kokkotis, G, Kitsou, K, Xynogalas, I, Spoulou, V, Magiorkinis, G, Trontzas, I, et al. Systematic review with meta-analysis: COVID-19 outcomes in patients receiving anti-TNF treatments. Aliment Pharmacol Ther. (2022) 55:154–67. doi: 10.1111/apt.16717
275. The RECOVERY Collaborative Group. Dexamethasone in hospitalized patients with COVID-19. N Engl J Med. (2021) 384:693–704. doi: 10.1056/NEJMoa2021436
276. Chee, YJ, Fan, BE, Young, BE, Dalan, R, and Lye, DC. Clinical trials on the pharmacological treatment of long COVID: a systematic review. J Med Virol. (2023) 95:e28289. doi: 10.1002/jmv.28289
277. Charfeddine, S, Ibnhadjamor, H, Jdidi, J, Torjmen, S, Kraiem, S, Bahloul, A, et al. Sulodexide significantly improves endothelial dysfunction and alleviates chest pain and palpitations in patients with Long-COVID-19: insights from TUN-EndCOV study. Front Cardiovasc Med. (2022) 9:866113. doi: 10.3389/fcvm.2022.866113
278. Buchhorn, R. Therapeutic approaches to Dysautonomia in childhood, with a special focus on Long COVID [internet]. Children. (2023) 10:316. doi: 10.3390/children10020316
279. Zhan, X, Zhao, Z, and Li, T. Post-inflammatory pulmonary fibrosis in a discharged COVID-19 patient: effectively treated with Pirfenidone [internet]. Arch Pulmonol Respir Care. (2020) 6:051–3. doi: 10.17352/aprc.000053
280. Banerjee, T, Das, M, and Mitra, K. The effect of Pirfenidone on pulmonary function parameters in post recovery COVID-19 patients with pulmonary fibrosis compared to placebo in a government medical college, West Bengal [internet]. Biomedicine. (2022) 42:1005–7. doi: 10.51248/.v42i5.1996
281. Goel, N, Goyal, N, Nagaraja, R, and Kumar, R. Systemic corticosteroids for management of “long-COVID”: an evaluation after 3 months of treatment. Monaldi Arch Chest Dis [Internet]. (2021) 92:1981–1987. doi: 10.4081/monaldi.2021.1981
282. Hussein, AARM, Mohamed Hussein, AA, Ibrahim, MEA, Makhlouf, HA, Makhlouf, NA, Abd-Elaal, HK, et al. Value of montelukast as a potential treatment of post-COVID-19 persistent cough: a non-randomized controlled pilot study [internet]. Egypt J Bronchol. (2022) 16:1–5. doi: 10.1186/s43168-022-00154-6
283. Dhooria, S, Chaudhary, S, Sehgal, IS, Agarwal, R, Arora, S, Garg, M, et al. High-dose low-dose prednisolone in symptomatic patients with post-COVID-19 diffuse parenchymal lung abnormalities: an open-label, randomised trial (the COLDSTER trial). Eur Respir J [Internet]. (2022) 59:2102930. doi: 10.1183/13993003.02930-2021
284. Utrero-Rico, A, Ruiz-Ruigómez, M, Laguna-Goya, R, Arrieta-Ortubay, E, Chivite-Lacaba, M, González-Cuadrado, C, et al. A short corticosteroid course reduces symptoms and immunological alterations underlying Long-COVID [internet]. Biomedicine. (2021) 9:1540. doi: 10.3390/biomedicines9111540
285. Simone, IL, Tortorella, C, and Ghirelli, A. Influence of pregnancy in multiple sclerosis and impact of disease-modifying therapies. Front Neurol. (2021) 12:697974. doi: 10.3389/fneur.2021.697974
286. Østensen, M, and Villiger, PM. The remission of rheumatoid arthritis during pregnancy. Semin Immunopathol. (2007) 29:185–91. doi: 10.1007/s00281-007-0072-5
287. Colle, I, and Hautekeete, M. Remission of autoimmune hepatitis during pregnancy: a report of two cases. Liver. (1999) 19:55–7. doi: 10.1111/j.1478-3231.1999.tb00010.x
288. Pazos, M, Sperling, RS, Moran, TM, and Kraus, TA. The influence of pregnancy on systemic immunity. Immunol Res. (2012) 54:254–61. doi: 10.1007/s12026-012-8303-9
289. Shaffer, L. Lots of Long COVID treatment leads, but few are proven. PNAS. 119:e2213524119. doi: 10.1073/pnas.2213524119
290. Kristófi, R, and Eriksson, JW. Metformin as an anti-inflammatory agent: a short review. J Endocrinol. (2021) 251:R11–R22. doi: 10.1530/JOE-21-0194
291. Bramante, C, Buse, JB, Liebovitz, D, Nicklas, J, et al. Outpatient treatment of COVID-19 and the development of Long COVID Over 10 Months: A Multi-Center, Quadruple-Blind, Parallel Group Randomized Phase 3 Trial. (2022) Available at SSRN: https://ssrn.com/abstract=4375620 or http://dx.doi.org/10.2139/ssrn.4375620.
292. Campisi, J, Hansen, MK, O’Connor, KA, Biedenkapp, JC, Watkins, LR, Maier, SF, et al. Circulating cytokines and endotoxin are not necessary for the activation of the sickness or corticosterone response produced by peripheral E. coli challenge [internet]. J Appl Physiol. (2003) 95:1873–82. doi: 10.1152/japplphysiol.00371.2003
Keywords: COVID-19, cytokines, Long COVID, neuroinflammation, post-acute sequelae of COVID-19, Myalgic Encephalomyelitis (ME), Chronic Fatigue Syndrome, Dysautonomia
Citation: Low RN, Low RJ and Akrami A (2023) A review of cytokine-based pathophysiology of Long COVID symptoms. Front. Med. 10:1011936. doi: 10.3389/fmed.2023.1011936
Edited by:
Rodrigo L. Castillo, University of Chile, ChileReviewed by:
Jose Alegre-Martin, Vall d’Hebron University Hospital, SpainTracey A. Ignatowski, University at Buffalo, United States
Copyright © 2023 Low, Low and Akrami. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Russell N. Low, cmxvdzUyQHlhaG9vLmNvbQ==
†Retired