
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 30 August 2022
Sec. Infectious Diseases: Pathogenesis and Therapy
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.961027
 Mansoor Khaledi1
Mansoor Khaledi1 Fatemeh Sameni1
Fatemeh Sameni1 Sheida Yahyazade2
Sheida Yahyazade2 Maedeh Radandish2
Maedeh Radandish2 Parviz Owlia3*Nader Bagheri4*Hamed Afkhami5Mohamad Mahjoor6Zahra Esmaelpour7Maryam Kohansal8Farzad Aghaei4
Parviz Owlia3*Nader Bagheri4*Hamed Afkhami5Mohamad Mahjoor6Zahra Esmaelpour7Maryam Kohansal8Farzad Aghaei4Recent evidence proposed that the severity of the coronavirus disease 2019 (COVID-19) in patients is a consequence of cytokine storm, characterized by increased IL-1β, IL-6, IL-18, TNF-α, and IFN-γ. Hence, managing the cytokine storm by drugs has been suggested for the treatment of patients with severe COVID-19. Several of the proinflammatory cytokines involved in the pathogenesis of COVID-19 infection recruit a distinct intracellular signaling pathway mediated by JAKs. Consequently, JAK inhibitors, including baricitinib, pacritinib, ruxolitinib, and tofacitinib, may represent an effective therapeutic strategy for controlling the JAK to treat COVID-19. This study indicates the mechanism of cytokine storm and JAK/STAT pathway in COVID-19 as well as the medications used for JAK/STAT inhibitors.
Scientists have been familiar with the coronavirus for over half a century. The word “coronavirus,” which comes from the Latin corona (“crown”), was first used by Almeida to describe these viruses because their glycoprotein spikes resembled solar coronas (1, 2). In 1968, 229E and OC43 were found to be the first coronaviruses to infect people. However, it caused very minor infections until the severe acute respiratory syndrome (SARS-CoV-1) outbreak in 2003 and the Middle East respiratory syndrome (MERS) in 2012. Recently, the Chinese health authorities announced that there was widespread pneumonia of unknown cause in Wuhan, and the novel coronavirus genome was released and made available to the scientific community (3). The new coronavirus, tentatively called 2019-nCoV, is now called SARS-CoV-2 according to the International Committee on Classification of Viruses Research Group (4). Since, the emergence of SARS-CoV-2, over 298 million people have been infected and more than 5,469,000 deaths have occurred (5). The spike (S) glycoproteins of coronaviruses are the main targeting agents of antibodies and promote cell entry (6). cSARS-CoV-2 is spread through respiratory droplets, mainly during face-to-face contact. The mean time between exposure and onset of symptoms is 5 days, with 97.5% of them developing symptoms in 11.5 days. Fever, coughing, and shortness of breath are the most prevalent symptoms (7, 8). Asymptomatic carriers and severe clinical symptoms, which include septicemia and severe respiratory distress, are among the manifestations of COVID-19 (9). Severe symptoms requiring intensive care were experienced by about 5% of patients with COVID-19 and 20% of inpatients (10). The latest studies have shown that not only the extreme pulmonary headaches of SARS-CoV-2 but also gastrointestinal signs and symptoms and liver dysfunction were commonly detected in patients (11). Various immune sensors such as TLR7, RIG-I/MDA, and cGAS / STING contribute to the recognition of SARS-CoV-2. These sensors provoke an early response to interferon (IFN)-I, which is essential for infection control (12, 13). Neutralizing antibodies that bind to the angiotensin-converting enzyme 2 (ACE2) receptor spike (S) protein binding site may be the basis of defense, but the affinity of SARS-2 S protein's for ACE2 is much higher than that of (SARS-CoV-1), and its highly pathogenic challenges are shown in other studies (12, 14). The immune system of older people is slow, unprompted, and less effective, increasing their vulnerability to potential infections. High-risk groups have been shown to switch from congenital to adaptive inadequately, resulting in much lower antibody levels in these patients (12).
Through immune cell recruitment and cytokine production, the immune system aids in the eradication of viral infections such as influenza and coronaviruses. Immune response coordination is usually the first line of defense for the body when it comes to fighting viral infections. If the abnormal or exaggerated immune response of the host is not regulated, it might lead to severe disease (15, 16). Based on multiple studies, the most critical manifestations of COVID-19 are caused by uncontrolled immune responses and hyperinflammation (17). Feng et al. presented that SARS-CoV-2 infection triggers a hyperinflammation condition, knowns as cytokine storm, that results in acute respiratory distress syndrome (ARDS), thromboembolic diseases, encephalitis, myocardial infarction, acute kidney injury, adult vasculitis, Kawasaki-like syndrome in children, and even death (18).
The cytokine storm is an uncontrolled systemic host inflammatory reaction to many stressors such as infection, malignancy, rheumatoid disorders, and drugs (15). This situation is known as the hyperactivation of immune cells and extreme production of massive inflammatory cytokines, chemokines, and other chemical mediators (19). The term cytokine storm was first stated in the context of graft vs. host disease (GVHD) in 1993 (20). It also has been reported in several viral infections, in addition to COVID-19, like influenza H5N1, influenza H1N1, and two other coronavirus family members, SARS and MERS infections (21–24).
Acute respiratory distress syndrome is a more notable consequence in severe episodes of cytokine storm in COVID-19, involving roughly 20–40% of hospitalized patients (25, 26). Hypercytokinemia and infiltration of activated monocytes, neutrophils, B cells, and T cells to the lungs result in diffuse alveoli–capillary membrane damage, increase lung permeability, pulmonary edema, hyaline membrane formation, micro thrombosis, fibrin exudates, and fibrotic healings resulting in the end to respiratory insufficiency (27–30). Based on the fact that the presence of SARS-CoV-2 particles in bronchial and alveolar type II epithelial cells have been revealed by electron microscopy, severe lung injury in COVID-19 may be the result of both direct viral infection and cytokine storm (31, 32). Some mechanisms of cytokine storm in COVID-19 have been summarized in Figure 1.
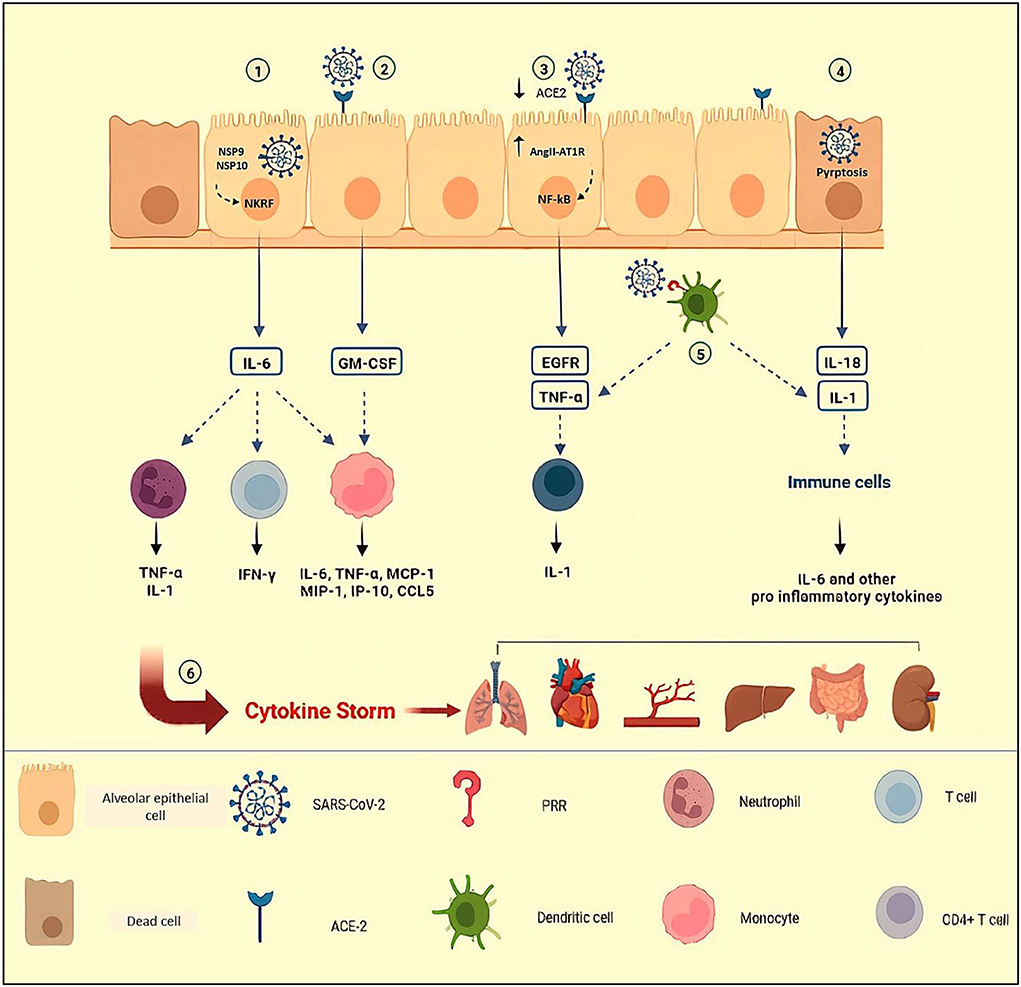
Figure 1. Some mechanisms of cytokine storm in COVID-19. (1) NSP9 and NSP 10 of SARS-CoV-2 target NKRF to promote IL-6 production. By engagement of receptor with IL-6, IFN-γ and GM-CSF are secreted by activated T cells; MCP-1, MIP-1, MIP-1, IP-1O, and CCL-5 are secreted by activated monocytes; and neutrophils release pro-inflammatory cytokines such as TNF-α and IL-1. (2) In response to SARS-CoV-2 in epithelial cells, secreted GM-CSF activates CD14+ CD16+ monocytes to produce pro-inflammatory cytokines such as IL-6 and TNF-α. (3) Attachment of SARS-CoV-2 to ACE-2, leads to a reduction in ACE-2 expression and accumulation of AngII in the host cells. The AngII and its receptor (AT1R) complex activate NF-κB, leading to the production of TNF-α and EGFR. (4) In COVID-19, increased levels of IL-1β and IL-18 indicate inflammasome activation and pyroptosis of host infected cells, triggering inflammation by recruiting immune cells and pro-inflammatory cytokine production. (5) Dendritic cells that are triggered by PRRs release TNF-α and IL-1, causing acute inflammatory responses. TNF/TNFR interactions between effector CD4+ T cells and myeloid cells have been suggested to promote IL-1β production. (6) Uncontrolled immune responses and hyperinflammation cause excessive production of pro-inflammatory cytokines, known as cytokine storms which are related to multi-organ failure and even death in COVID-19. NSP, Non-Structural Protein; NKRF, NF-κB repressor; ACE-2, Angiotensin Converting Enzyme-2; AngII, Angiotensin II; AT1R, Angiotensin II Receptor type 1; PRR, Pattern Recognition Receptors Created in Biorender.com.
Following the entry of SARS-CoV-2 into the respiratory epithelial cells, the virus stimulates an immune response with inflammatory cytokine production accompanied by a weak IFN response (33, 34). In the early phases of viral infection, the synthesis of IFN-I or IFN-α/β is the main molecule that performs an antiviral effect (35). Recently, McGonagall et al. and Ye Q et al. presented an attractive concept of the cytokine storm in COVID-19. In these articles, the cytokine storm is known to be the result of immune system failure in removing the virus. In this case, the cytokine storm is divided into two stages: the first stage is an interim immune-deficient condition and the next is a hyperactive immune situation to compensate for the failure in reduction of viral load, which manifests as a cytokine storm (36, 37). The study of Hadjadj et al. is another confirmation that a failure in initial type-I and III IFN responses to SARS-CoV-2 leads to an excessive late immune response and a severe form of COVID-19 (38). On the other hand, IFN levels rose in COVID-19 in tandem with the viral load. The postponed peak, along with a drop in lymphocyte numbers, enhanced neutrophil infiltration into the lungs, worsening the patient's condition (39, 40). Also, similar to SARS-CoV-2, delayed release of IFNs in the early stages of SARS-CoV and MERS-CoV infection hampers the immune antiviral response. Following that, the rapid production of cytokines and chemokines results in an excessive infiltration of inflammatory cells into the lungs and, as a result, lung injury (41–43).
Analysis of cytokine levels in the serum of patients with COVID-19 in a study in China showed that the levels of IL-6, IL-1β, IL-7, IL-8, IL-9, IL-10, IL-2, FGF, G-CSF, GM-CSF, IFN-γ, IP-10, MCP-1, MIP-1α, MIP-1β, RANTES, PDGF, TNF-a, and VEGF are elevated compared to healthy controls. In addition, patients hospitalized in the ICU also exhibited increased cytokine levels of IL-2, IL-7, IL-10, G-CSF, IP10, MCP-1, MIF-1α, and TNF-α than those not requiring ICU treatment (39). Based on wide data, high levels of expression of IL-5, IL-1β, IL-4, IL-10, TNF-α, IFN- γ, IP-10, and MCP-1 have been detected in the serum of patients, and the levels of IL-2R and IL-6 are positively correlated with disease severity (39, 44, 45). The cytokine storm markers observed are not unique to SARS-CoV-2; comparable findings were made in SARS-CoV-1 and MERS-CoV cohorts (46, 47).
Among other cytokines, IL-6 has been at the center of the COVID-19 pandemic (48–50). Non-structural protein (NSP) 9 and NSP10 of SARS-CoV-2 target NKRF (NF-κB repressor) to promote IL-6/IL-8 production, according to a study of peripheral blood mononuclear cells (51). IL-6 levels were a significant predictor of illness severity early in the COVID-19 pandemic (50, 52, 53). Many documents revealed that those who died because of COVID-19 had higher serum levels of interleukin IL-6 compared to survivors (53, 54).
This cytokine is produced by stromal cells, B cells, T cells, macrophages, monocytes, dendritic cells, mast cells, and non-immune cells, such as fibroblasts, endothelial cells, keratinocytes, and tumor cells (55). IL-1β and TNF-α are two pro-inflammatory cytokines involved upstream of the IL-6 signaling pathway through the activation of NF-κB and, subsequently, the virus-induced cytokine storm (56, 57). In addition to activating inflammatory cells, NF-κB stimulation causes the production of IL-6, other pro-inflammatory cytokines, and chemokines (56, 58). IL-6 binds to either membrane-bound IL-6 receptor (mIL-6R) or soluble IL-6 receptor (SIL-6R), forming a complex that acts on gp130 (cytoplasmic signaling molecules), and triggers the JAK/STAT3 signaling pathway (59). STAT3 is required for NF-κB activation to be boosted. As a result, infection with SARS-CoV-2 activates both NF-κB and STAT3 (60, 61). In this way, mbIL-6R is expressed on naive T cells, monocytes/macrophages, and neutrophils. Following the engagement of receptor with IL-6 and subsequent JAK/SATAT3 signaling, IFN-γ and GM-CSF are secreted by activated T cells; MCP-1, MIP-1, MIP-1, IP-10, and CCL-5 are secreted by activated monocytes/macrophages; and neutrophils release pro-inflammatory cytokines such as TNF-α and IL-1β (62, 63).
In non-immune cells, the IL-6 amplifier (IL-6 Amp) is a hyper NF-κB activation mechanism caused by the simultaneous activation of NF-κB and STAT3. The IL-6 Amp stimulates the production of many proinflammatory cytokines, including IL-6, by hyperactivating NF-κB via STAT3. As a result, IL-6 Amp might be related to the cytokine storms in COVID-19 (61, 64).
In the case of COVID-19, IL-6 is one of the causes of lymphocytopenia via inhibiting lymphopoiesis through lymphocyte trafficking and the direct effect on progenitor cells (65, 66). The precursors for both lymphoid and myeloid fate express IL-6R. Followed by IL-6 binding, the JAK/STAT3 signaling leads to the expression of IL-6 in these cells, which suppresses lymphopoiesis (67). Chen et al. showed that SARS-CoV-2 selectively induced macrophages to produce IL-6 to directly promote lymphocyte death (68).
As a bridge between innate and adaptive immunity, IL-6 promotes TH17 and CD8+ T cells differentiation and activation, which themselves increase inflammation. In contrast, this cytokine inhibits the differentiation of regulatory CD4+ CD25+ FOXP3+ T cells (69–71).
Generally, regarding clinical symptoms, IL-6 induces cardiomyopathy, vascular leakage, activation of complement, and diffuse intravascular coagulation (72, 73).
Besides IL-6, IL-1 and TNF-α could be a predictor of cytokine storm and COVID-19 progression (74). Dendritic cells and macrophages that have been triggered by PRRs release these two cytokines, which causes acute inflammatory responses (75). TNF/TNFR interactions between effector CD4+ T cells and myeloid cells have been suggested to promote IL-1β production (76). Furthermore, in COVID-19, increased levels of IL-1β may indicate inflammasome activation and pyroptosis of host infected cells (77, 78). Microglia and IL-1 stimulation contribute to higher reactive oxygen species (ROS) and cytokine production, phagocytosis, and apoptosis inside the central nervous system (CNS) in the context of communication seen between the immune system cytokine network and the CNS cytokine network (79). Neuroinflammation, tissue damage, increased oxidative stress, and synaptic pruning dysfunction have all been reported as a result of this scenario. Inflammatory cytokines enter the circulation as well, potentially exacerbating the cytokine storm (80).
As SARS-CoV-2 enters the cells through ACE2, ACE2 expression in the cells is reduced and the serum levels of AngII increase. The AngII and its receptor (AT1R) complex activate NF-κB along with ADAM17, leading to the production of TNF-α, epidermal growth factor receptor (EGFR), and NF-κB stimulators, which is another way of TNF-α generation (81–83).
Increased expression of hyaluronic acid synthetase II in EpCAM+ lung alveolar epithelium, CD31+ lung alveolar endothelium, and fibroblasts in response to IL-1β and TNF-α leads to the accumulation of hyaluronic acid in the extracellular matrix and subsequent increase of fluid retention in airway spaces. This phenomenon creates the ground-glass appearance in the CT chest (84, 85).
Pneumonia is linked to blood levels of IL-1β, IL-7, IL-8, IL-9, and IL1-5 in COVID-19. These cytokines are produced from the injured tissue, and the same pattern was found in ICU and non-ICU patients, suggesting that they play a role in a cytokine storm. Furthermore, IL-2, IL-7, and IL-10 levels were shown to be higher in ICU and non-ICU patients (39, 86). To limit the cytokine storm, IL-10 levels are raised in the second week after symptoms start, but it is too late (87). In ICU patients, the levels of IL-4, a TH2 cytokine, and an anti-inflammatory suppressor are also raised (39).
GM-CSF is another cytokine involved in cytokine storm and is produced by macrophages, T-cells, fibroblasts, endothelial cells, epithelial cells, and tumor cells (88). In COVID-19, GM-CSF activates CD14+ CD16+ monocytes to produce pro-inflammatory cytokines such as IL-6 and TNF-α, further worsening the cytokine storm (89).
Data regarding the importance of chemokine dysregulation in COVID-19 has increasingly been reported. Transcriptome analysis of BALF samples indicated that the levels of pro-inflammatory chemokines such as CXCL1, CXCL2, CXCL6, CXCL8 (IL-8), CXCL10 (IP-10), CCL2 (MCP-1), CCL3 (MIP-1A), and CCL4 (MIP-1B) are elevated in patients with COVID-19, which correlated with disease severity. Enhanced expression of chemokine receptors such as CCR2 (CCL2 receptor) and CCR5 (CCL3 receptor) was also found, indicating that inflammatory signaling had been activated (90). High levels of macrophage chemoattractants, CXCL10 and CCL2, and neutrophil chemoattractants, CXCL8, CXCL1, and CXCL2, are consistent with these immune cells' infiltration into the lungs in SARS-CoV-2 infection leads to tissue damage (91).
The immune responses to SARS-CoV-2 infection and the role of the cytokines, chemokines, immune cells, and other mediators will be characterized with the final goal to identify targeted therapeutic strategies. Many clinical trials in the context of COVID-19 treatments confirmed this point of view, such as IL-6 blockers, IL-1 receptor antagonists, TNF-α inhibitors, JAK inhibitors, and corticosteroids (92–97).
In summary, abnormal and unregulated immune responses might be linked to the cytokine storm mechanism in COVID19, which is linked to disease severity. As a result, early cytokine storm management is critical for improving patient survival rates.
As we discussed before, in severe instances of COVID-19, immunopathologic abnormalities such as downregulation of type I and type III interferon responses, as well as elevations in proinflammatory cytokine and chemokine production, are common (38). These events are reminiscent of the JAK/STAT pathway role in SARS-CoV-2 induced cytokine storm (98). The classic type of JAK/STAT pathway activation is related to cytokine receptor signaling. The induction of JAK/STAT signaling by type I and II cytokines is shown in Figure 2.
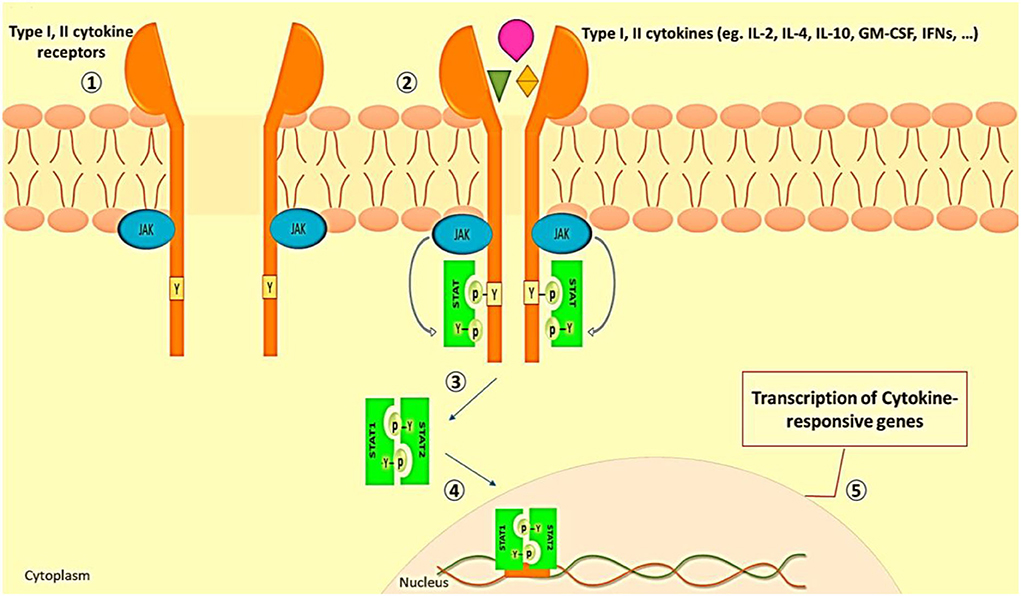
Figure 2. JAK/STAT signaling induction by type I and II cytokines. (1) In the resting state, the subunits of type I and II cytokine receptors are at a short distance from each other. (2) The engagement of ligand with receptors leads to receptor dimerization and JAK activation. (3) The activated JAKs phosphorylate the tyrosine residue of the receptor which recruits various members of STAT proteins. (4) Phosphorylated and activated STATs make themselves dimmer. (4) STATs complex translocates to the cell nucleus. (5) STATs bind to the promoter and lead to the transcription of cytokine-responsive genes (99).
According to the researches, pneumocytes, pulmonary endothelial cells, and other immune cells are recruited via the JAK/STAT pathway into the inflammation sites during COVID-19 (100–103). For instance, STAT3 is involved in the differentiation and survival of CD4+ and CD8+ T cells. Also, facilitating the stimulation of CD8+ T cells in viral infection is mediated by STAT3 (104). Followed by cytokine production in NK cells, activated JAK1 downstream of IL-15 signaling regulates the development and growth of these cells. In the same way, the engagement of IL-2 and its receptor on NK cells, JAK1, 3 and STAT1, 3, 5 leads to cytotoxicity enhancement (105). Surprisingly, when IFN-α2 and IFN-γ (type I and III IFNs) are released, the expression of STAT1 increases, causing activation of the ACE2 receptors and, as a result, boosting SARS-CoV-2 entry into the host epithelium (101). Some interactions of SARS-CoV-2 with the JAK/STAT pathway are as follows:
In severe cases of COVID-19, the production of type I and III IFNs is insufficient in the early stages of the disease (38, 106). Studies revealed that patients with severe COVID-19 sustained type I IFN and pro-inflammatory responses in the late phase of the disease as compensatory mechanisms (86, 107). In confirmation with previous findings, Pairo-Castineira et al. revealed that the low expression of IFNAR2 (type I and III IFNs receptor subunit) and high expression of TYK2 (protein of Janus kinase family) are associated with life-threatening COVID-19 (108).
During viral infection, canonical and non-canonical signaling pathways have been mentioned for IFN signaling. The IFN canonical signaling way, which is associated with JAK/STAT activation, is demonstrated in Figure 3. On the other hand, TLR-mediated or cytokines (mostly TNF) signaling pathways may redirect or synergize with IFN signaling, promoting the expression of non-canonical interferon-stimulated genes (non-ISGs) (112, 113).
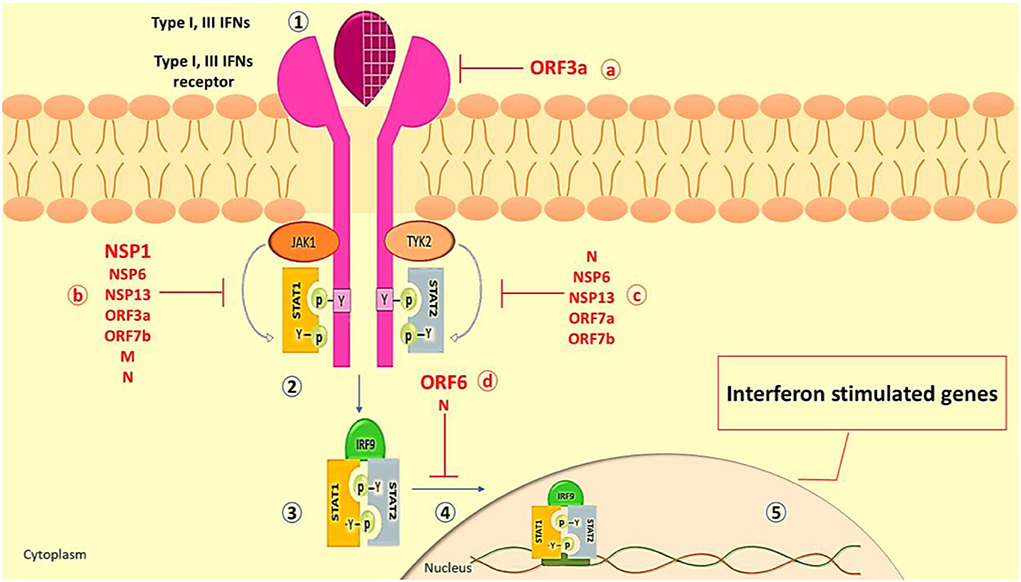
Figure 3. The Interferon type I and III canonical signaling pathway. (1) Type I and III IFNs (e.g., IFN-α, IFN-β, IFN-λ) released from virus-infected cells bind to their receptors (e.g., IFNA/LR) on neighboring cells, leading to the dimerization of receptors and activation of JAK1 and TYK2. (2) The activated JAK1 and TYK2 phosphorylate the tyrosine residue of the IFN receptor which recruits various members of the STAT family as STAT 1 and STAT2. Also, JAK1 and TYK2 add phosphorus to recruit STATs and activate them. (3) Following separation from the receptor, STAT1/STAT2 dimer interacts with IRF9. (4) The STAT1/STAT2/IRF9 complex moves to the nucleus. (5) Placement of the complex on its promoter on the gene leads to the robust expression of many classical IFN-stimulated genes (ISGs, such as ISG15, MxA, IFITM, etc.), which exert an antiviral role to restrict viral replication and spreading (109). SARS-CoV-2 inhibits IFN production and response through a variety of methods. As a result, target cells close to the original infection fail to receive essential and protective IFN signals, allowing the virus to propagate. The ORF3a can inhibit type III IFN receptors. The structural proteins of SARS-CoV-2, N, and M, and nonstructural proteins such as NSP1, NSP6, NSP13, ORF3a, and ORF7b quench IFN signaling by inhibition of STAT1 phosphorylation. (c) The phosphorylation of STAT2 in COVID-19 is repressed by N, NSP6, NSP13, ORF7a, and ORF7b proteins. (d) The translocation of the STAT1/STAT2/IRF9 into the nucleus is inhibited by N and ORF6 proteins which attenuate the transcription of the interferon-stimulated gene (13, 110, 111). IFN, Interferon; ISG, Interferon Stimulated Gene; STAT, Signal Transducer and activator of Transcription; IRF9, Interferon Regulatory Factor 9; TYK2, Tyrosine Kinase 2; JAK, Janus Kinase; MxA, Myxovirus Resistance Gene A; IFITM, Interferon Induced Transmembrane; OFR, Open Reading Frame; NSP, None Structural Protein.
Different proteins in SARS-CoV-1 interact with the actions of IFNs and JAK/STAT signaling (114). Based on genetic similarity between SARS-CoV-2 and SARS-CoV-1, SARS-CoV-2 may have the same antagonist activity (115, 116). Major SARS-CoV-2 proteins involved in IFN signaling disruption are ORF3a, ORF6, and NSP1 (103, 117) (Figure 3). Generally, SARS-CoV-2 inhibits STAT1 and IFNs, resulting in the activation of the STAT3 pathway as a dominant signaling pathway, which could result in COVID-19 complications. Regarding Yang et al.'s study, inhibition of STAT1 phosphorylation by SARS-CoV-2 results in a decrease in the transcription of the ISG in dendritic cells and macrophages (103). Once STAT1 function is impaired, it is possible that signaling via STAT3 pathways will compensate (118). STAT3 inhibits IFN response through several mechanisms, such as it binds to STAT1 and prevents the formation of STAT1/STAT1, inhibits binding of STAT1/STAT2/IRF9 to the DNA, and competes with STAT1 for binding to karyopherin alpha 1 (KPNA1, a nuclear translocation factor). Following viral infection, these circumstances would result in a greater functional STAT3:STAT1 ratio (119, 120). In hospitalized patients with COVID-19, some symptoms are related to aberrant STAT3 signaling such as hyperinflammatory condition, T cell lymphopenia, coagulopathy, and fibrotic status (90, 121, 122). In the context of hyperinflammation, STAT3 participates in the cytokine storm by enhancing cytokine production by co-activating the IL-6 amplifier (123).
IL-6 is a key mediator of intracellular defense against disease and injury, acting both as an indicator of multiple distinct types of cytokine storms and a host defense agent against infection (124, 125). Hyperinflammation, such as cytokine storms, can result from excessive IL-6 production (126). IL-6 is produced and secreted by different cells, such as macrophages and monocytes, and non-immune cells, including vascular endothelial cells, mesenchymal cells, and fibroblasts (127).
The IL−6–SIL-6R complex also activates endothelial cells directly, releasing IL-6, IL-8, and MCP-1. This suggests that during a cytokine storm, IL-6 trans-signaling in the endothelium modifies the proinflammatory cytokine pathway. Therefore, IL-6 receptor-binding antibodies can be used to suppress hyperreactive immune responses (73, 128). To initiate signaling, IL-6 binds to either membrane-bound or circulating IL-6R (SIL-6R) in addition to a second glycoprotein, gp130 (59). IL-6 has pleiotropic properties related to its ubiquitous expression of gp130, whereas IL-6R is expressed solely by lymphocytes, monocytes and macrophages, and hepatocytes. Through two different signaling pathways, IL-6 promotes its effects by classic signaling and trans-signaling (129) (Figure 4). In two forms of IL-6R signaling, a hexamer complex with gp130 is produced. The JAK–STAT3 and the JAK–MAPK pathway are both used by IL-6 to send signals. The intracellular tyrosine motif in the gp130 intracellular region is responsible for biological activity. IL-6 signaling induces the production of two inhibitors, SOCS3 and SOCS1, which inhibit gp130 signaling activity (negative feedback). IL-6 attaches to a complex of mIL-6R and gp130 in traditional signaling and then activates the JAK-STAT3 pathway to relay signals. During trans-signaling, IL-6 binds to SIL-6R in the serum and tissue fluid. JAK kinases phosphorylate tyrosines in the cytoplasmic region of gp130 in response to IL-6. When JAK, which is normally attached to gp130, is phosphorylated by the stimulation of IL-6, it promotes STAT3 phosphorylation and homodimerization, allowing it to operate as a transcription factor in the nucleus, inducing the transcription of IL-6-responsive genes. JAK kinase also initiates the MAP kinase pathway by tyrosine phosphorylation of gp130 by binding SHP2. Different transcriptional actions are promoted by the JAK SHP2 MAPK pathway (130). According to that mentioned above, to reduce hyperinflammation conditions, the inhibition of the JAK/STAT and IL-6 signaling might seem to be a capable approach, but it should be used with caution in immunocompromised patients.
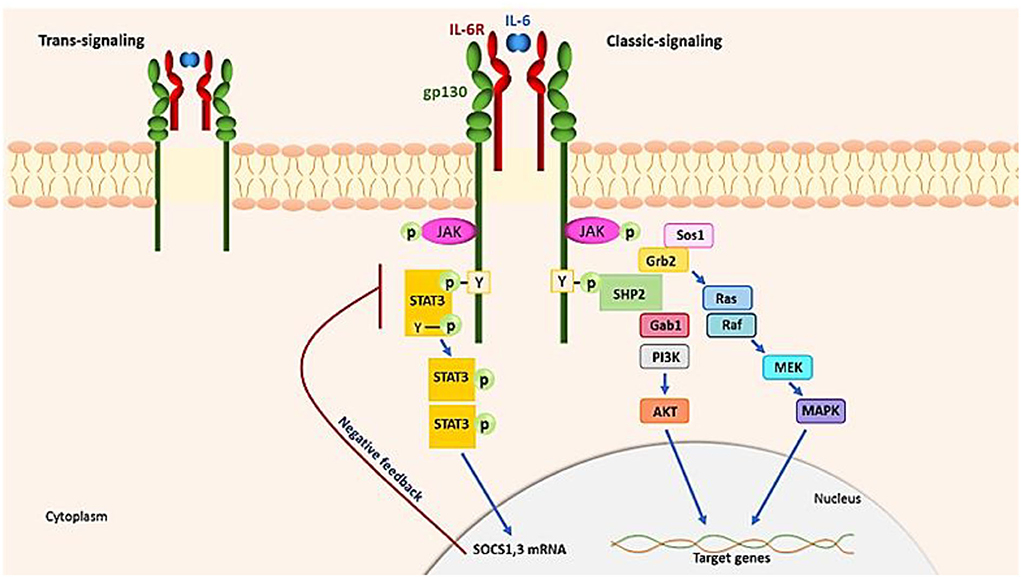
Figure 4. IL-6 signaling pathways. IL-6 requires two receptors, the IL-6R and the gp130, to begin signaling. IL-6 (classic signaling) binds to either fluid- or membrane-bound IL-6R (SIL-6R, trans-signaling). In two modalities of IL-6R signaling, a hexamer complex with gp130 is formed. The JAK–MAPK route and the JAK–STAT3 pathway are both used by IL-6 to transfer signaling. JAK kinases phosphorylate tyrosine in the cytoplasmic region of gp130, which promotes STAT3 phosphorylation and causes homodimerization, which works as a transcription factor in the nucleus.
Due to the non-selective reduction of various cytokines' action (e.g., IL-2, IL-6, IL-7, and G-CSF), JAK inhibition is hypothesized to be able to decrease cytokine storm. Non-selective immune response inhibition, on the other hand, increases the likelihood of secondary infection. Since the JAK pathway influences STAT proteins, inhibiting the JAK system may result in STAT3-dependent cytokine storm inhibition (131). Many inflammatory illnesses, such as inflammatory bowel disease (IBD), rheumatoid arthritis (RA), and psoriasis, are treated with JAK inhibitors (132). Today, various antibodies that prevent IL-6, IL-6R, or associated signaling factors are used for a wide range of inflammatory diseases. Tocilizumab, a monoclonal antibody that directly targets IL-6R, has shown to be advantageous in treating cytokine storms caused by a variety of illnesses, Castleman's illness, CAR T-cell-induced cytokine storm, and COVID-19 (133–136). Critically, patients with COVID-19 experienced pneumonia-like symptoms and substantially elevated blood IL-6 levels. At the commencement of the pandemic, Wuhan physicians used off-label tocilizumab to manage COVID-19-infected hospitalized patients (137). A retrospective cohort study by Guaraldi et al. showed that in patients with severe COVID-19 pneumonia, therapy with tocilizumab, whether given intravenously or subcutaneously, may minimize the need for invasive mechanical ventilation or mortality (138). Rosas J. et al. indicated that therapy with baricitinib and tocilizumab in patients with COVID-19 hospitalized for interstitial pneumonia did not produce significant adverse effects and might be used to treat COVID-19-related interstitial pneumonia. (139). Also, Salama et al. showed tocilizumab decreased the probability of development to the composite endpoint of mechanical ventilation or death in hospitalized patients with COVID-19 pneumonia who no longer received mechanical ventilators; however, it did now not decorate survival (NCT04372186) (140). Stone et al. determined that tocilizumab became no longer powerful for stopping intubation or dying in moderately ill hospitalized patients with COVID-19 (NCT04356937) (141). Siltuximab and Sarilumab, both IL-6 antagonists, block both traditional and trans-signaling of IL-6. Furthermore, sgp130 could bind to IL−6–SIL−6R complexes and block the trans-signalingroute (142, 143). Table 1 summarizes IL-6/JAK/STAT3 inhibitors in COVID-19 (144, 145).

Table 1. Summary of IL-6/JAK/STAT3 inhibitors in COVID-19.
Baricitinib is a JAK 1/2 inhibitor that has been shown to reduce inflammation in people with autoimmune disorders (146). Human anesthesia-related kinases are biochemically inhibited by baricitinib. The SARS-CoV-2 virus is caused by AAK1, Bike, and GAk genes (147). Baricitinib has also been shown to reduce multiple cytokines, which is suggested as a treatment for COVID-19 due to its potential anti-inflammatory and anti-viral effects (148). Baricitinib was approved by the Food and Drug Administration (FDA) in 2018 (149). It was used as an anti-rheumatic drug to treat adult patients with RA (150). It has also been demonstrated to help people with moderate to severe atopic dermatitis and reduce inflammation and itching (151). It was also used to treat a variety of autoimmune illnesses, including systemic lupus erythematosus (SLE) and atopic dermatitis (152). JAK is a tyrosine–protein kinase family and an intracellular enzyme that regulates immune cell function by modulating signals from cytokines and growth factor receptors (153). JAK proteins are classified into four groups (JAK 1, JAK 2, JAK 3, and TYK2). Various cell receptors link these proteins in different ways, resulting in heterodimers or homodimers (154). These JAK dimers phosphorylate transcription factors (STATs) and trigger the intracellular activities of inflammatory mediators, such as gene transcription, culminating in an autoimmune response (154). JAK2 and JAK1 have a greater affinity for baricitinib. It inhibits JAK proteins, prevents STATs from being phosphorylated and activated, and impacts the interferon, interleukin, and growth factor signaling pathways. Baricitinib suppresses JAK1/JAK2 expression and induces cell death in mutant cells (155).
Baricitinib plus remdesivir was better than remdesivir alone and had fewer serious side effects. It also aided in reducing recovery time and accelerating recovery, especially in patients receiving high-flow oxygen or non-invasive mechanical ventilation (156). Despite the lack of a significant decrease in disease progression, treatment with baricitinib in combination with standard therapy (including dexamethasone) may be useful. Several observational studies were performed, including small groups of hospitalized patients with COVID-19 and the elderly treated with baricitinib, and showed the first clinical evidence of improvement (133, 134). In another trial, baricitinib combined with remdesivir was found to be better than remdesivir alone in hospitalized people with COVID-19. Remdesivir produced a lot of negative side effects. The FDA granted baricitinib an emergency approval for patients with COVID-19 who needed oxygen support in the hospital (157). In the Marconi et al. study, patients received corticosteroids in the initial trial, which is now the standard of care for hospitalized patients in need of oxygen. Approximately 79 patients received systemic corticosteroids. About 19% of them received remdesivir. Eventually, 28 baricitinib and 30 (placebo) deaths occurred. However, the mortality was 28 days with baricitinib and 13 days with placebo. The benefits of baricitinib were more pronounced in patients who initially needed high-flow oxygen or ventilation. The incidence of serious side effects, including venous thromboembolism and serious infections, was similar in both groups (158). A study by Saber-Ayad et al. suggested baricitinib as a potential drug, which has a dual-mechanism drug, prevents the entry of SARS-CoV-2, and fights the cytokine storm that is the leading cause of death in COVID-19. However, subsequent studies of COVID-19 indicated that baricitinib treatment was only combined with ramedsivir. The study also showed that baricitinib-treated samples enriched baricitinib-suppressed transcripts in genes involved in the regulation of chemotaxis and the movement of immunological agents, such as neutrophils, eosinophils, monocytes, macrophages, granulocytes, natural killer cells, and lymphocytes, and also in the regulation of inflammation and humoral immune responses to viral and bacterial infections. CCL4 and family members of tumor necrosis factor (TNF, TNFSF11) were enriched. Baricitinib treatment appears to suppress tissue regeneration, ion, and membrane transduction, as well as cellular responses (159). A study by Hasan et al. showed that baricitinib could potentially suppress inflammatory cascades in severe COVID-19 pneumonia. A total of 238 patients with severe COVID-19 pneumonia were included in this prospective cohort research. For 14 days, 116 patients in the usual dose group and 122 patients in the high dose group were given 8–4 mg of baricitinib orally, and the clinical findings were compared between the groups. Baricitinib has been suggested as a viable anti-inflammatory medication for COVID-19 infection. The 8-mg daily dosage of baricitinib for 14 days led to early breathing normalization, lowered ICU and intubation needs, less 30-day mortality, and a lower 60-day readmission rate. According to this trial, fasting was superior to the standard 4-mg daily oral dosage of baricitinib in individuals with COVID-19 pneumonia (160). Baricitinib has been shown to improve the immune response to COVID-19 in several investigations. Baricitinib suppresses COVID-19-associated cytokine signaling in vitro, according to a study published by Stebbing et al. in 2020. They examined how GAK, AAK1, and BIKE were affected by a human numb-associated kinase (hNAK). Human viral infection is reduced (148). SARS-CoV-2 was mild to moderately suppressed by baricitinib and had a specific response with a substantial decrease in the number of cytokines, including IL-13, IL-1β, FGF, IL-10, IL-6, IP-10, IL-17, GM-CSF, IFN-γ, TNF-α, IL-4, IL-1ra, and MCP-1. Furthermore, baricitinib prevents viral endocytosis by inhibiting kinase signaling and inhibits cytokine release by blocking JAK1/2 signals. Baricitinib was rapidly transferred from intra-silicon studies to clinical trials (161). The study showed that healthy individuals treated with baricitinib had varying degrees of inhibition of cytokine-dependent phosphorylated STAT (pSTAT). Baricitinib inhibits signaling and also inhibits cytokines involved in COVID-19 infection, including IL-2, IL-6, IL10, IFN-c, and G-CSF. Baricitinib-induced cytokine-induced JAK/ TAT signaling resulted in a significant reduction in plasma IL-6 levels in patients with active RA who had a poor response to particular medications at week 12 (98). Baricitinib affects family members NAK AAK1, BIKE, and GAK STK16, and some facilitate the spread of coronavirus in the epithelial cells. Baricitinib showed a reduction in viral infection in infection of SARS-CoV-2 infected human liver treated with baricitinib (134). Four patients with bilateral COVID-19 pneumonia participated in this pilot study with varying degrees of illness severity; three of them were clinically unstable with moderate-to-severe illness. In March of 2020, they were all admitted from the emergency room to the ward. As would be predicted in patients with COVID-19 pneumonia, all patients showed detectable plasma IL-6 levels, as well as significantly elevated inflammatory markers (C-reactive protein [CRP]). In hospitalized patients, baricitinib therapy was associated with clinical and radiologic recovery, as well as a rapid reduction in SARS-CoV-2 viral load, inflammatory markers, and IL-6 levels. All patients had a significant detectable increase in plasma IL-6 levels (148). These findings suggest that baricitinib could be evaluated as a viable management option for patients with COVID-19 who are hospitalized and experiencing a cytokine storm and viral propagation. The discovery supports baricitinib's anti-cytokine profile and implies that it is a powerful AAK1/BIKE/GAK inhibitor that may diminish host cell infectivity. Baricitinib may reduce host cell virulence by inhibiting AAK1/BIKE/GAK in addition to confirming its anti-cytokine profile. Researchers found that baricitinib is a potent inhibitor of AAK1/BIKE/GAK and it affects host cell infectivity, confirming its anti-cytokine profile (148, 162). These data suggest that treatment with barsitinib was well tolerated in these four patients with COVID-19 bilateral pneumonia. There is evidence that treatment with baricitinib may reduce the inflammatory burden and may lead to a reduction in the severity of the disease in patients with COVID-19. This report showed that baricitinib is a cytokine inhibitor in patients with COVID-19 restricted to the intensive care unit. Baricitinib has previously been shown to inhibit IL-6 induction (148).
Only a few animal studies have been conducted on COVID-19, and one of those studies is Hoang et al.'s research from 2020. An 8-day treatment with baricitinib reduced macrophage production of cytokines and chemokines in the lung in a nonhuman primate animal model with SARS-CoV-2 (142). Goletti et al. (146) randomly assigned 1,033 patients to receive baricitinib and remdesivir (combined group, 515 participants) or placebo in their trial (control group, 518 patients). Remdesivir was given intravenously on day 1 at a loading dose of 200 mg, then 100 mg every day until discharge or death on day 10. For up to 14 days, baricitinib was given orally or via a nasogastric tube at a dose of 4 mg daily or 2 mg daily if renal function declined. Overall, the recovery time in the combined group was significantly shorter than the control group (mean, 7 days vs. 8 days; recovery rate ratio, 1.16; 95% confidence interval of the combined group as well as a 30% higher chance of improving the clinical condition per day). The combination group had a lower rate of significant adverse events than the control group. The combined group's recovery time was 10 days, while the control group's recovery time was 18 days. In addition, patients receiving combination therapy had the highest chance of improving their clinical condition. The highest documented efficacy of baricitinib was in patients with COVID-19 pneumonia who were receiving oxygen therapy but not invasive mechanical ventilation (146). The results of the Bronte et al. study showed that baricitinib affects the inhibition of cytokine-dependent pSTAT to varying degrees. Baricitinib inhibited cytokines involved in COVID-19 infection, including G-CSF, IFN-c, IL-6, IL10, and IL-2, (143). The study by Li et al. tested the antiviral activity of baricitinib by limiting the dose of baricitinib with a placebo. Twenty patients received baricitinib according to the study protocol. The control group was also considered. Patients were given 4 mg baricitinib twice daily for 2 days, then they received a low dose of 2 mg twice daily for 2 days, and finally 2 mg daily for patients over 75 years old, according to the inclusion criteria and pharmacokinetics of baricitinib. In patients with renal impairment, dose reduction was also investigated. The hepatotoxicity or myelotoxicity of patients in the baricitinib group was similar to that in the control group. Only one in 20 patients treated with baricitinib died after completing treatment, compared with 25 patients who died (45%) of the 56 patients in the group without baricitinib. Interestingly, patients treated with baricitinib had lower oxygen requirements compared to those from the control group (163). In the study by Pedro Abizanda et al., it was shown that patients from COVID-AGE groups who were hospitalized due to moderate to severe pneumonia were analyzed. Baricitinib treatment resulted in a significant reduction in mortality of up to 48% in patients 70 years of age or older, and an 18.5% reduction in the risk of 30-day absolute death (133). Patients who got a combination of bendamustine, lenalidomide, and dexamethasone (BRD) were enrolled in the Izumo trial. Baricitinib (14 days), dexamethasone (10 days), and remdesivir (10 days) were given to all patients. The effectiveness and side effects were assessed. In this investigation, 44 individuals with severe COVID-19 were enrolled. The 28-day mortality rate was only 2.3% (44.1 patients). For patients receiving BRD treatment, the mean hospital stay was 11 days, the recovery time was 9 days, length of stay in intensive care unit was 6 days, and duration of ventilation was they had 5 days of invasive mechanics and 5 days of supplemental oxygen therapy. Side effects occurred in 15 patients (34%). Iliopsoas hematoma, infectious endocarditis, liver dysfunction, herpes zoster, ventilator-associated pneumonia, thrombosis, and renal dysfunction, occurred in 2, 2, 11, 2, 2, 2, and 11% of patients, respectively. In severe instances of COVID-19, BRD was shown to be beneficial and had few adverse effects. The study's findings are positive. More randomized clinical trials, however, are required (164).
The aim of the study conducted by Perez-Alba et al. was to analyze the clinical outcome of dexamethasone or baricitinib and dexamethasone monotherapy in patients with severe COVID-19 pneumonia. A total of 596 patients were assessed for exclusion, with 793 being inclusion criteria. For the initial result, 197 patients were studied: 123 in the baricitinib plus dexamethasone group and 74 in the dexamethasone monotherapy group. Mechanical ventilation was used by 25.8% (197.51) and 42.9% (197.85) of patients needed ICU hospitalization. Overall, 27.9% (55/197) of people died after 30 days. The baricitinib plus dexamethasone group had a considerably reduced mortality rate than the dexamethasone monotherapy group. The two groups had the same number of nosocomial infections. Thirty-day mortality in COVID-19 pneumonia patients treated with baricitinib and dexamethasone was considerably lower than dexamethasone monotherapy. No differences in progression to invasive mechanical ventilation and nosocomial infections were observed. In hospitalized patients over 70 years of age with COVID-19 pneumonia, baricitinib is linked to an 18.5% lower risk of absolute death (130). Interestingly, a study of individuals treated with baricitinib found that ACE2 and TMPRSS2 levels were significantly reduced. In addition, baricitinib inhibits the AAK2 virus by binding to anesthetics-related kinases, and GAK-induced endocytosis, ultimately reducing the viral load (159). The ability of baricitinib to reduce the production of these cytokines, and hence immunological factors, has been demonstrated to aid patients with COVID-19 dramatically. For example, lowering serum cytokine levels, improving lymphocytes, and reducing the need for it has an oxygen flow behind it (165).
Pacritinib is an available oral activator (JAK2) and a mutant JAK2 JAK2V617F with potential anti-neoplastic activity (166). It competes for ATP binding to JAK2, which may lead to JAK2 activation, possibly the JAK-STAT output pathway, and thus become caspase-dependent apoptosis (135, 136).
According to CTI BioPharma, pacritinib's efficacy to reduce the progression of acute respiratory distress syndrome and mechanical ventilation in hospitalized patients with COVID-19 with severe coronavirus disease is being investigated in phase III clinical research. The cytokine storm, an inflammatory response that leads white blood cells to not only fight viral infections but also harm tissues, puts patients with severe COVID-19, especially those with cancer, at a significant risk for serious consequences of the condition. Pacritinib has the potential to avoid an inflammatory response to coronavirus infection and subsequent pulmonary insufficiency in the lungs, minimizing the requirement for a ventilator. Pacritinib is a kinase inhibitor for SCR1R, JAK2, and IRAK1 that is taken orally. The JAK family is required for normal blood cell growth and development, as well as the production of inflammatory cytokines and immunological responses. Blood malignancies, such as lymphoma, myeloproliferative neoplasms, and leukemia, appear to be linked to mutations in these kinases. Inflammatory and immune-mediated illnesses, such as acute graft-versus-host disease, may benefit from pacritinib treatment. Patients with COVID-19 have also been included in clinical trials to examine various cancer medications with potential therapeutic benefits for this patient population, including leronlimab (phase IIb/III), calquence (CALAVI study), and low-dose selinexor.
Researchers appear to be still exploring this medication according to various searches [198]. Pacritinib showed a satisfactory safety profile and pharmacological activity in preclinical investigations in hematologic malignancies and various cancers. Pacritinib significantly reduced the vitality and sphere-forming capacity of brain tumor-initiating cells (BTICs) in vitro, according to Jensen et al. Pacritinib was also observed to boost temozolomide activity in BTIC cells with unmethylated MGMT (O-6-methylguanine-DNA methyltransferase) promoters. Pacritinib has also been shown to be effective in treating glioblastoma multiforme (the JAK2/STAT3 inhibitor pacritinib effectively) (167). In addition, Brian and colleagues have shown that pacritinib selectively inhibits JAK2, both non-alloreactive T cells specific for nominal antigens and the development of beneficial Tregs, while limiting NK cell function in patients with COVID-19 (168). The most common side effects associated with pacritinib were thrombocytopenia and diarrhea, according to a large, randomized clinical trial (136). As of 2020, neither JAK2 inhibitor has resulted in severe infectious complications (136, 169). Furthermore, pacritinib suppresses Th1 cells that initiate cytokine release syndrome (CRS) pathogenesis through GM-CSF (pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe patients with COVID-19).
Pacritinib's effects on AAK1 are unclear. Considering that fedratinib/pacitinib spare antigen-specific T-cell activity and minimize opportunistic infections, selective JAK2 inhibitors could be initially trialed rather than broader JAK1/2 inhibitors in the treatment of COVID-19 CRS (170).
In a recent study, Kabir et al. after screening and repurposing ~300 drugs, proposed twenty-seven candidates, including pacritinib, as probable candidates for further in vitro and in vivo research for the treatment of the SARS-CoV-2 infection.
Molecular docking was used to find compounds that will inhibit the identified molecular targets, TMPRSS2-ACE2 and SARS-CoV-2, from their own comprehensive database of roughly 300 highly described existing medications with proven safety profiles (203). Inhibitors that target SARS-CoV-2 proteases, the 3CLPRO and PLPRO, which are responsible for SARS-CoV-2 duplication and spread, are evaluated in the study by Jade et al. FDA-approved medications in the Zinc database, COVID-19 chemicals in the Maybridge database and Pubchem database against 3CLPRO and PLPRO proteases, and natural compounds in the Natural Product Activity database were used in silico high-throughput screening,. Furthermore, the study results show that pacritinib, bemcentinib, ergotamine, MFCD02180753, MFCD00832476, as well as the six chemicals that fight 3CLPRO bind with strong binding free energies to their active sites. As a result, these anti-PLPRO and anti-3CLPRO drugs could be employed to treat COVID-19 infections (171).
Ruxolitinib is an EU-approved bioavailable oral inhibitor of JAK for the treatment of myelofibrosis. Some research suggests that ruxolitinib lowers gene expression. Signal transducers and activators of transcription (STAT) linked to cytokine receptors regulate the inflammatory response (66).
It disrupts the ability of blood cells to form. JAK 2 triggers STAT activation and nuclear translocation by terminating kinase activity. Ruxolitinib stops the IL-6/JAK/STAT3 pathway and can significantly reduce IL-6 levels. Excessive inflammation prevents the reduction of lung failure in patients with COVID-19.
Furthermore, in patients with COVID-19 who are experiencing a cytokine storm as a result of the host reaction, this could be advantageous (144). Furthermore, ruxolitinib's anti-inflammatory and pro-apoptotic effects on senescent cells, which are hypothesized to be employed by COVID-19 to escape and change the immune system, could play a major role in COVID-19 treatment in patients with a poor prognosis, such as elderly people (145).
Ruxolitinib has applications in the treatment of severe COVID-19 disease and its complications such as respiratory distress syndroARDS. A phase II clinical trial study using ruxolitinib prevents multiple organ failure. Various therapeutic strategies have been adopted to modulate cytokine storms, such as IL-1 and IL-6 inhibitors. It is a JAK1 and JAK2 subtype inhibitor that is both powerful and selective.
A seventy-eight-year-old woman was diagnosed with COVID-19 pneumonia by PCR on oropharyngeal and nasopharyngeal swabs, and standard treatment with ruxolitinib 5 mg daily for 2 weeks was started. In the first 3 days of taking ruxolitinib, a slight improvement in lung function was observed. Ten days after starting treatment with ruxolitinib, she showed signs of paraesophageal and Centro lobular emphysema with a decrease in bilateral vitreous opacity and symptoms associated with pulmonary fibrosis. The patient tolerated ruxolitinib treatment well, as shown by the absence of thrombocytopenia, anemia, neutropenia, and opportunistic infections, which are the main side effects. These findings show that ruxolitinib not only improves lung function reduced by SARS-CoV-2 infection, but also reduces pulmonary fibrosis, improves tissue thickness and stiffness, and makes lung function less difficult (172).
The study by Giudice et al. indicated that 17 patients with ARDS related to SARS-CoV-2 were reported. The new combination of ruxolitinib 10 mg twice a day for 14 days and Eculizumab was used to treat these individuals. This combination resulted in a considerable reduction in D-dimer levels and a significant improvement in respiratory symptoms and radiographic lung abnormalities. The results support the combined use of ruxolitinib and Eculizumab for the treatment of ARDS (173). Furthermore, in the study by Innes et al., ruxolitinib showed promise as an anti-IL6 treatment for patients with COVID-19 infection with the severe syndrome. It was resistant to anti-IL6 therapy, but inhibiting JAK/STAT with ruxolitinib showed its safety and efficacy in these conditions (174). Anti-inflammatory drugs known as Jak inhibitors can help to reduce the overactive inflammatory reaction related to COVID-19.
The clinical data of 218 patients with COVID-19 who were hospitalized with severe pneumonia and treated with ruxolitinib were investigated.
The treatment period is given; results are given at 4, 7, 14, and 28 days. Retrospective data was gathered on clinical state, oxygen support demands, and laboratory markers. Overall, 66.5% percent of patients improved in follow-up, according to the physician's opinion. By day 7, 83.5% of them had made progress. Oxygen support status improved as well, with 21.6% of patients in ambient air on day 7 compared to 1.4% at baseline, rising to 48.2% on day 28. The reduction was significant. After 4 days an increase in CRP and lymphocyte count was observed, which appeared to be associated with a positive outcome. Approximately 87.2% of the patients were alive at the end of the observation period. There were no unexpected safety results, and patients reported minor side effects (175). In a study by Koschmieder et al. patients with COVID-19 were shown to be at risk for severe disease, especially those with comorbidities. Severe illness treatment infection with COVID-19 necessitates supportive intensive care. More specialized techniques are being considered, such as JAK inhibitors, which decrease the “cytokine storm.”
A 55-year-old patient with COVID-19 pneumonia used the JAK1/2 ruxolitinib inhibitor for primary myelofibrosis for 15 months before coronavirus infection. The patient had several serious underlying illnesses, including obesity, chronic renal disease, and arterial hypertension, and was at high risk for ARDS and COVID-19 mortality. The patient's condition remained unchanged without the requirement for mechanical ventilation because the abrupt removal of ruxolitinib could precipitate a lethal cytokine storm and ARDS. The patient tested negative for SARS-CoV-2 after 15 days and was discharged from the hospital. As a result, data suggest that ruxolitinib medication may be beneficial in preventing cytokine storms and ARDS in patients with COVID-19 pneumonia (176). COVID-19, which is caused by the novel SARS-CoV-2 virus, causes significant mortality and morbidity, and there is mounting evidence that inflammatory pathways play a role in lung injury. Severe patients with COVID-19 present an increase in inflammatory markers similar to secondary hemophagocytic lymphohistiocytosis (sHLH), which has been shown to predict mortality. There is evidence for the use of an interleukin inhibitor (IL-6) to suppress the inflammatory cytokine storm in this area. Ruxbeinib is effective in treating sHLH (147). The study by Saraceni et al. revealed that GVHD is one of the most common complications of lung damage caused by unregulated cytotoxic T cells and inflammatory cytokines. In another study, the selective JAK1/2 inhibitor ruxolitinib has shown promising results in this area. However, no clinical observations on the safety or efficacy of ruxolitinib therapy have been published in this field. In their study, Saraceni et al. described a case in which severe COVID-19 after hematopoietic stem cell transplantation in a patient with concurrent chronic GVHD, in which treatment with Ruxolitinib was well tolerated and positive (177). Ruxolitinib's immunomodulatory effect must be evaluated, and this medicine may raise the risk of opportunistic infections. In patients with an inflammatory state, disrupting the JAK signal transduction pathway could increase the risk of recognized adverse effects such as thrombocytopenia and anemia (178).
Tofacitinib is a JAK kinase inhibitor that has no effect on other kinases in the human kinome. Tofacitinib inhibits JAK3, JAK1, JAK2, and to a lesser extent TYK2 in in vitro kinase assays. Tofacitinib preferentially inhibits pairwise JAK signaling in cellular settings through cytokine receptors associated with JAK1 and/or JAK3, but selectively suppresses receptor signaling through pairs of JAK2 (179).
Tofacitinib is a tiny chemical that controls cytokines that are critical in the course of inflammatory reactions and immunological responses in people with RA.
It suppresses the synthesis of cytokines that are needed for the advancement of immunological and inflammatory responses. Tofacitinib is a tiny molecule with a novel application in the treatment of RA. COVID-19 is presently being used in several clinical trials with tofacitinib. Yale University is conducting clinical research to determine the efficacy and safety of tofacitinib in patients with moderate COVID-19 (NCT04415151). Additionally, JAK inhibitors also reduce STAT1 activity, therefore their usage in patients with COVID-19 must be done with caution (98).
Upper respiratory tract infection, nasopharyngitis, headache, and diarrhea were adverse events (AEs) associated with tofacitinib while the most common serious AEs are described as serious infections such as herpes zoster, pneumonia, urinary tract infection, and cellulitis (151). Patients on tofacitinib have had gastrointestinal perforations, lymphoma, and malignancies (except non-melanoma skin cancer) (152). A total of 289 individuals with pneumonia were given tofacitinib 10 mg twice daily in the hospital for a maximum of 14 days or until discharge in a trial by Guimares et al. Death or respiratory failure by day 28 was the primary outcome. Glucocorticoids were given to 89.3% of the patients during their stay in the hospital through day 28, and the tofacitinib group had a cumulative incidence of 18.1% mortality or respiratory failure compared to 29.0 % in the placebo group. Serious side effects appeared in 20 (14.1%) individuals in the tofacitinib group and 17 (12.0%) patients in the placebo group. Patients with COVID-19 pneumonia had a decreased risk of respiratory failure or death for 28 days after receiving tofacitinib treatment. Adverse events were reported in 26.1 % of tofacitinib patients and 22.5 % of placebo patients. Ventricular tachycardia, acute myocardial infarction, deep vein thrombosis, myocarditis, and ventricular tachycardia were all reported by individuals in the tofacitinib group as side effects. Each patient in the placebo group suffered from heart failure and hemorrhagic stroke. Serious infection occurred in 4.2% of the placebo group and 3.5% in the tofacitinib group (153). According to the findings of the study by Agrawal et al., out of 2,326 patients, 37 (1.6%) were treated with tofacitinib. Patients who got <20 mg of tofacitinib per day were 17 (45.9%) and 20 (54.1%), respectively. Tofacitinib was administered to 17 (45.9%) and 20 (54.1%) patients, respectively, in doses of 20% and <20 mg total daily. Thirty tofacitinib patients (81.1%) had ulcerative colitis compared to 946 (41.3%) patients on other IBD medications. In comparison to other medications, tofacitinib patients recovered significantly less (154). Maslennikov et al. included 32 patients in their trial who received tofacitinib and 30 individuals who did not receive any anti-cytokine medicines (control group). The tofacitinib group had a lower death rate and a lower ICU intake rate compared to the control group. The tofacitinib group had a much-reduced volume of the affected lung and a significantly higher oxygen saturation than the control group up to 10 days following delivery. CRP levels in tofacitinib patients were lower than in control patients. In the COVID-19 study, tofacitinib was proven to be a safe and effective treatment for cytokine release syndrome [221]. Alopecia areata, an autoimmune disease, damages the epithelium of the hair follicle. As a result, hair loss occurs. JAK3 has been demonstrated to have a key role in the disease's etiology. Tofacitinib is a JAK3 and JAK1 inhibitor that can suppress the production of different cytokines, such as IL-2, IL-7, and IL-6. Oral tofacitinib has been found in numerous studies to help alopecia areata sufferers regrow their hair. As a result of the recent COVID-19 outbreak, avoidance of JAK inhibitors during active infection has been suggested to prevent possible immunosuppression.
Ferreira et al. indicated that two alopecia patients with active COVID-19 infection who continued to use tofacitinib saw no deterioration of their condition (155). Moiseev et al. conducted the study to see if tofacitinib can reduce the probability of individuals with COVID-19 needing invasive mechanical dilation or dying. Patients with COVID-19 with low oxygen saturation and a prolonged fever were enrolled. The trial included 384 patients with COVID-19, 253 of whom were treated with conventional tofacitinib alone. In the tofacitinib and control groups, 12.5% (72.9) and 14.1% (185.26) of patients who needed respiratory assistance commenced mechanical ventilation or died during hospitalization, respectively (180). Hayek et al. looked at the effects of tofacitinib in combination with dexamethasone in hospitalized COVID-19-associated pneumonia patients. Tofacitinib was given universally to 138 (51.3%) of the 269 eligible patients, whereas dexamethasone was given to 131 (48.7%). A total of 44 individuals died, with 30 (68.2%) dying on dexamethasone and 14 (31.8%) dying on tofacitinib.
Tofacitinib and dexamethasone both had mortality rates of 10.1 and 22.9%, respectively. After controlling for age and clinical factors collected in the hospital, the tofacitinib group had a 70% lower chance of mortality than the dexamethasone group. With the addition of dexamethasone, COVID-19 pneumonia treatment improved quickly. This study demonstrated the potential benefits of tofacitinib in COVID-19 pneumonia. In COVID-19 pneumonia, the inclusion of tofacitinib in a therapy regimen containing dexamethasone has the potential to improve survival when compared to dexamethasone alone (181).
Nezulcitinib is a lung-selective inhibitor of the Janus kinases (JAKs), with potential anti-inflammatory and immunomodulatory activities (182). It is a panJAKi with IC50s of 10.3 nM, 10.6 nM, 10.2 nM, and 9.2 nM for JAK1, JAK2, JAK3, and TYK2, respectively, that can be administered by inhalation, and it inhibits the activity of the JAKs, thereby disrupting cytokine-induced activation of JAK-STAT signaling pathways in the airways (157). Nezulcitinib, designed for inhalation, is for the development for the treatment of acute lung injury associated with COVID-19 (182).
Upadacitinib is in a class of inhibitors medications called Janus kinase (JAK), targeting the JAK1 enzyme and decreasing the activity of the immune system (183). It is a group of four tyrosine kinases (JAK1, JAK2, JAK3, and TYK2) involved in the process of immune-mediated inflammatory diseases (IMIDs). In patients with COVID-19, IL-6 plays a role in the cytokine storm that damages the lungs. Disruption of JAK1 signaling induced by upatacitinib reduced not only the expression of T-helper 2 and 22 cytokines but also the levels of interleukin6 (IL-6), through inhibition of STAT3 phosphorylation (199).
Baricitinib is generally regarded as safe and acceptable; however, because it is an immunosuppressive medicine, it can raise the risk of serious infections (184). Urinary and upper respiratory tract infections are the most common. But herpes zoster infections are becoming more common (185). Candidiasis, pneumocystosis, tuberculosis, histoplasmosis, CMV infections, and BK virus infection are among the opportunistic illnesses that have been recorded (184).
Upper respiratory infection, nasopharyngitis, and headache are some of the side effects (186). They are prevalent among baristinib patients. The use of baricitinib for an extended period of time may increase the risk of thromboembolic illness in patients. Bacterial infections, viral infections, pneumocystis, herpes zoster, urinary tract infections, acute histoplasmosis, fungal infections (candidiasis), cryptococcosis, and pneumonia, are among the other side effects (187). Baricitinib has been linked to bone marrow suppression as well as blood abnormalities including lymphopenia, neutropenia, and anemia (186).
An increase in medium cholesterol, low-density lipoproteins (LDL), and high-density lipoproteins (HDL), without an increase in the ratio of HDL to LDL, is another side effect that usually occurs after 12 weeks of using baricitinib (188). Some people had elevated CPK levels. In clinical trials, pulmonary embolism (PE) and deep vein thrombosis (DVT) were also detected (189). Malignancies such as skin cancer and lymphoma struck a tiny fraction of the participants. Stomach discomfort, nausea, and vomiting are some of the most prevalent gastrointestinal problems (190). Severe adverse effects including gastrointestinal perforation, on the other hand, are uncommon but have been recorded in patients who had previously had diverticulitis (191). The use of baricitinib in pregnancy has not yet been extensively studied (190). In animal research, baricitinib has been found to reduce fetal weight and teratogenicity. Baricitinib was detected in rat milk throughout tests. It should be avoided by breastfeeding mothers because there is no data on its presence in human milk. Baricitinib has also been shown in animal tests to harm female fertility while having no impact on male sperm generation. Ruxolitinib has the potential to cause liver damage, hematological issues, and ambiguity. Infection care is required based on reports of viral/bacterial reactivation in patients. Hepatotoxicity of grade 3 is experienced by a patient. Two patients with anemia already had grade 3 anemia, which was treated with periodic blood samples in the ICU (191). Diarrhea, bloating, a sense of constant movement in one's body or surroundings or full feelings of a spinning sensation, skin rash, weight gain (192), and other common side effects are all common.
The cytokine storm in COVID-19 is caused by the interaction of multiple immunological components, including interleukins, chemokines, TNF-α, and IFNs. Various drugs have effective potentials in overcoming problems due to the hyperactivation of the immune system associated with the JAK/STAT signaling pathway.
This signaling pathway mediates cellular responses to proinflammatory cytokines. Therefore, inhibition of the JAK/STAT pathway may induce the inhibition of various cellular responses in COVID-19 infection.
In conclusion, JAK inhibitors including baricitinib, pacritinib, ruxolitinib, and tofacitinib have beneficial potential in the recovery and reduction of novel coronavirus disease due to anti-inflammatory and anti-viral effects.
PO, NB, MKh, and FS: design of study. PO, NB, MKh, FS, SY, and MR: acquisition of data. PO, MKh, SY, MR, MM, HA, and ZE: evaluation of data and preparation of the manuscript. PO, NB, MKh, FS, MKo, and FA: assessment of data. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Khaledi M, Yousefi Nojookambari N, Afkhami H, Sameni F, Yazdansetad S. A review on phylogenetic assessment and cytopathogenesis of filoviruses, retroviruses, and coronaviruses transmitted from bat to human. Cell Mol Res (Iranian J Biol). (2021).
3. Hamed MA. An overview on COVID-19: reality and expectation. Bull Natl Res Cent. (2020) 44:1–10. doi: 10.1186/s42269-020-00341-9
4. Ciotti M, Angeletti S, Minieri M, Giovannetti M, Benvenuto D, Pascarella S, et al. COVID-19 outbreak: an overview. Chemotherapy. (2019) 64:215–23. doi: 10.1159/000507423
5. WHO Coronavirus (COVID-19) dashboard: World Health Organization (WHO) (2022). Available online at: https://covid19.who.int/table.
6. Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. (2020) 181:281–92. e6. doi: 10.1016/j.cell.2020.02.058
7. Sameni F, Shahrjerdi S, Khorram A, Yazdani S, Hajikhani B, Nazarinejad N, et al. COVID-19 and diabetes: a narrative review. Int J Enteric Pathogens. (2021) 9:70–7. doi: 10.34172/ijep.2021.14
8. Fard NG, Khaledi M, Afkhami H, Barzan M, Farahani HE, Sameni F, et al. COVID-19 coinfection in patients with active tuberculosis: first case-report in Iran. Authorea Preprints. (2021). doi: 10.22541/au.161230839.90084658/v1
9. Dadashi M, Dadashi A, Sameni F, Sayadi S, Goudarzi M, Nasiri MJ, et al. SARS-CoV-2 and HIV co-infection; clinical features, diagnosis, and treatment strategies: a systematic review and meta-analysis. Gene Rep. (2022) 27:101624. doi: 10.1016/j.genrep.2022.101624
10. Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA. (2020) 324:782–93. doi: 10.1001/jama.2020.12839
11. Yang L, Tu L. Implications of gastrointestinal manifestations of COVID-19. Lancet Gastroenterol Hepatol. (2020) 5:629–30. doi: 10.1016/S2468-1253(20)30132-1
12. Nikolich-Zugich J, Knox KS, Rios CT, Natt B, Bhattacharya D, Fain MJ. SARS-CoV-2 and COVID-19 in older adults: what we may expect regarding pathogenesis, immune responses, and outcomes. Geroscience. (2020) 42:505–14. doi: 10.1007/s11357-020-00186-0
13. Park A, Iwasaki A. Type I and type III interferons–induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe. (2020) 27:870–8. doi: 10.1016/j.chom.2020.05.008
14. McCray Jr PB, Pewe L, Wohlford-Lenane C, Hickey M, Manzel L, Shi L, et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J Virol. (2007) 81:813–21. doi: 10.1128/JVI.02012-06
15. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. (2012) 76:16–32. doi: 10.1128/MMBR.05015-11
16. Jin YH, Cai L, Cheng ZS, Cheng H, Deng T, Fan YP, et al. (2020). A rapid advice guideline for the diagnosis and treatment of 2019 novel coronavirus (2019-nCoV) infected pneumonia (standard version). Mil Med Res. (2020) 7:4. doi: 10.1186/s40779-020-0233-6
17. Kim JS, Lee JY, Yang JW, Lee KH, Effenberger M, Szpirt W, et al. Immunopathogenesis and treatment of cytokine storm in COVID-19. Theranostics. (2021) 11:316. doi: 10.7150/thno.49713
18. Feng Y, Ling Y, Bai T, Xie Y, Huang J, Li J, et al. COVID-19 with different severities: a multicenter study of clinical features. Am J Respir Crit Care Med. (2020) 201:1380–8. doi: 10.1164/rccm.202002-0445OC
19. Teijaro JR, Walsh KB, Rice S, Rosen H, Oldstone MB. Mapping the innate signaling cascade essential for cytokine storm during influenza virus infection. Proc Nat Acad Sci. (2014) 111:3799–804. doi: 10.1073/pnas.1400593111
20. Ferrara J, Abhyankar S, Gilliland D. Cytokine storm of graftversus-host disease: A critical effector role for interleukin-1. In: Transplantation Proceedings, Vol. 25. (1993). p. 1216–7.
21. Ishikawa T. Clinical preparedness for cytokine storm induced by the highly pathogenic H5N1 influenza virus. J Pharmacogenom Pharmacoproteomics. (2012) 3:1000e131. doi: 10.4172/2153-0645.1000e131
22. Woo PC, Tung ET, Chan K-H, Lau CC, Lau SK, Yuen K-Y. Cytokine profiles induced by the novel swine-origin influenza A/H1N1 virus: implications for treatment strategies. J Infect Dis. (2010) 201:346–53. doi: 10.1086/649785
23. Lau SK, Lau CC, Chan K-H, Li CP, Chen H, Jin D-Y, et al. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: implications for pathogenesis and treatment. J Gen Virol. (2013) 94:2679–90. doi: 10.1099/vir.0.055533-0
24. Channappanavar R, Perlman S. Pathogenic human coronavirusinfections: Causes and consequences of cytokine storm and immunopathology. In: Seminars in Immunopathology, Vol. 39. Berlin; Heidelberg: Springer (2017). p. 529–39.
25. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. doi: 10.1016/S0140-6736(20)30566-3
26. Wu C, Chen X, Cai Y, Zhou X, Xu S, Huang H, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. (2020) 180:934–43. doi: 10.1001/jamainternmed.2020.0994
27. Bhatia M, Zemans RL, Jeyaseelan S. Role of chemokines in the pathogenesis of acute lung injury. Am J Respir Cell Mol Biol. (2012) 46:566–72. doi: 10.1165/rcmb.2011-0392TR
28. Marongiu F, Grandone E, Barcellona D. Pulmonary thrombosis in 2019-nCoV pneumonia? J Thromb Haemost. (2020) 18:1511–13. doi: 10.1111/jth.14818
29. Tseng C-TK, Perrone LA, Zhu H, Makino S, Peters CJ. Severe acute respiratory syndrome and the innate immune responses: modulation of effector cell function without productive infection. J Immunol. (2005) 174:7977–85. doi: 10.4049/jimmunol.174.12.7977
30. Tian S, Hu W, Niu L, Liu H, Xu H, Xiao S-Y. Pulmonary pathology of early phase SARS-CoV-2 pneumonia. J Thorac Oncol. (2020) 15:700–4. doi: 10.1016/j.jtho.2020.02.010
31. Yao X-H, He Z-C, Li T-Y, Zhang H-R, Wang Y, Mou H, et al. Pathological evidence for residual SARS-CoV-2 in pulmonary tissues of a ready-for-discharge patient. Cell Res. (2020) 30:541–3. doi: 10.1038/s41422-020-0318-5
32. Parsons PE, Eisner MD, Thompson BT, Matthay MA, Ancukiewicz M, Bernard GR, et al. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit Care Med. (2005) 33:1–6. doi: 10.1097/01.CCM.0000149854.61192.DC
33. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. (2016) 19:181–93. doi: 10.1016/j.chom.2016.01.007
34. Hussman JP. Cellular and molecular pathways of COVID-19 and potential points of therapeutic intervention. Front Pharmacol. (2020) 11:1169. doi: 10.31219/osf.io/p69g8
35. García-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. (2006) 312:879–82. doi: 10.1126/science.1125676
36. McGonagle D, Sharif K, O'Regan A, Bridgewood C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev. (2020) 19:102537. doi: 10.1016/j.autrev.2020.102537
37. Ye Q, Wang B, Mao J. The pathogenesis and treatment of theCytokine Storm'in COVID-19. J Infect. (2020) 80:607–13. doi: 10.1016/j.jinf.2020.03.037
38. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. (2020) 369:718–24. doi: 10.1126/science.abc6027
39. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
40. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. (2020) 17:533–5. doi: 10.1038/s41423-020-0402-2
41. Channappanavar R, Fehr AR, Zheng J, Wohlford-Lenane C, Abrahante JE, Mack M, et al. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest. (2019) 129:3625–39. doi: 10.1172/JCI126363
42. Mahallawi WH, Khabour OF, Zhang Q, Makhdoum HM, Suliman BA. MERS-CoV infection in humans is associated with a pro-inflammatory Th1 and Th17 cytokine profile. Cytokine. (2018) 104:8–13. doi: 10.1016/j.cyto.2018.01.025
43. Wong C, Lam C, Wu A, Ip W, Lee N, Chan I, et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol. (2004) 136:95–103. doi: 10.1111/j.1365-2249.2004.02415.x
44. Marchingo JM, Sinclair LV, Howden AJ, Cantrell DA. Quantitative analysis of how Myc controls T cell proteomes and metabolic pathways during T cell activation. Elife. (2020) 9:e53725. doi: 10.7554/eLife.53725
45. Chen L, Liu H, Liu W, Liu J, Liu K, Shang J, et al. Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia. Zhonghua Jie He He Hu Xi Za Zhi. (2020) 43:E005. doi: 10.3760/cma.j.issn.1001-0939.2020.0005
46. Zhang Y, Li J, Zhan Y, Wu L, Yu X, Zhang W, et al. Analysis of serum cytokines in patients with severe acute respiratory syndrome. Infect Immun. (2004) 72:4410–5. doi: 10.1128/IAI.72.8.4410-4415.2004
47. Min C-K, Cheon S, Ha N-Y, Sohn KM, Kim Y, Aigerim A, et al. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Sci Rep. (2016) 6:1–12. doi: 10.1038/srep25359
48. Herold T, Jurinovic V, Arnreich C, Hellmuth JC, von Bergwelt-Baildon M, Klein M, et al. Level of IL-6 predicts respiratory failure in hospitalized symptomatic COVID-19 patients. MedRxiv. (2020). doi: 10.1101/2020.04.01.20047381
49. Coomes EA, Haghbayan H. Interleukin-6 in COVID-19: a systematic review and meta-analysis. Rev Med Virol. (2020) 30:1–9. doi: 10.1002/rmv.2141
50. Ulhaq ZS, Soraya GV. Interleukin-6 as a potential biomarker of COVID-19 progression. Med Mal Infect. (2020) 50:382. doi: 10.1016/j.medmal.2020.04.002
51. Li J, Guo M, Tian X, Liu C, Wang X, Yang X, et al. Virus-host interactome and proteomic survey of PMBCs from COVID-19 patients reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. BioRxiv. (2020). doi: 10.1101/2020.03.31.019216
52. Herold T, Jurinovic V, Arnreich C, Lipworth BJ, Hellmuth JC, von Bergwelt-Baildon M, et al. Elevated levels of IL-6 and CRP predict the need for mechanical ventilation in COVID-19. J Allergy Clin Immunol. (2020) 146:128–36. e4. doi: 10.1016/j.jaci.2020.05.008
53. Gao Y, Li T, Han M, Li X, Wu D, Xu Y, et al. Diagnostic utility of clinical laboratory data determinations for patients with the severe COVID-19. J Med Virol. (2020) 92:791–6. doi: 10.1002/jmv.25770
54. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. (2020) 46:846–8. doi: 10.1007/s00134-020-05991-x
55. Scheller J, Rose-John S. Interleukin-6 and its receptor: from bench to bedside. Med Microbiol Immunol. (2006) 195:173–83. doi: 10.1007/s00430-006-0019-9
56. Rex J, Lutz A, Faletti LE, Albrecht U, Thomas M, Bode JG, et al. IL-1β and TNFα differentially influence NF-κB activity and FasL-induced apoptosis in primary murine hepatocytes during LPS-induced inflammation. Front Physiol. (2019) 10:117. doi: 10.3389/fphys.2019.00117
57. Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. (2015) 16:448–57. doi: 10.1038/ni.3153
58. Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:1–9. doi: 10.1038/sigtrans.2017.23
59. Kishimoto T, Akira S, Taga T. Interleukin-6 and its receptor: a paradigm for cytokines. Science. (1992) 258:593–7. doi: 10.1126/science.1411569
60. Wang Y, van Boxel-Dezaire AH, Cheon H, Yang J, Stark GR. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc Nat Acad Sci. (2013) 110:16975–80. doi: 10.1073/pnas.1315862110
61. Murakami M, Kamimura D, Hirano T. Pleiotropy and specificity: insights from the interleukin 6 family of cytokines. Immunity. (2019) 50:812–31. doi: 10.1016/j.immuni.2019.03.027
62. Tamura K, Kanazawa T, Tsukada S, Kobayashi T, Kawamura M, Morikawa A. Increased serum monocyte chemoattractant protein-1, macrophage inflammatory protein-1β, and interleukin-8 concentrations in hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. (2008) 51:662–8. doi: 10.1002/pbc.21660
63. Briso EM, Dienz O, Rincon M. Cutting edge: soluble IL-6R is produced by IL-6R ectodomain shedding in activated CD4 T cells. J Immunol. (2008) 180:7102–6. doi: 10.4049/jimmunol.180.11.7102
64. Lee J, Nakagiri T, Kamimura D, Harada M, Oto T, Susaki Y, et al. IL-6 amplifier activation in epithelial regions of bronchi after allogeneic lung transplantation. Int Immunol. (2013) 25:319–32. doi: 10.1093/intimm/dxs158
65. Wee JL-K, Greenwood DL, Han X, Scheerlinck J-PY. Inflammatory cytokines IL-6 and TNF-α regulate lymphocyte trafficking through the local lymph node. Vet Immunol Immunopathol. (2011) 144:95–103. doi: 10.1016/j.vetimm.2011.07.007
66. Del Valle DM, Kim-Schulze S, Hsin-Hui H, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature helps predict COVID-19 severity and death. medRxiv. (2020). doi: 10.1101/2020.05.28.20115758
67. Maeda K, Baba Y, Nagai Y, Miyazaki K, Malykhin A, Nakamura K, et al. IL-6 blocks a discrete early step in lymphopoiesis. Blood. (2005) 106:879–85. doi: 10.1182/blood-2005-02-0456
68. Feng Z, Diao B, Wang R, Wang G, Wang C, Tan Y, et al. The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) directly decimates human spleens and lymph nodes. MedRxiv. (2020). doi: 10.1101/2020.03.27.20045427
69. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
70. Yang R, Masters AR, Fortner KA, Champagne DP, Yanguas-Casás N, Silberger DJ, et al. IL-6 promotes the differentiation of a subset of naive CD8+ T cells into IL-21–producing B helper CD8+ T cells. J Exp Med. (2016) 213:2281–91. doi: 10.1084/jem.20160417
71. Dominitzki S, Fantini MC, Neufert C, Nikolaev A, Galle PR, Scheller J, et al. Cutting edge: trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+ CD25– T cells. J Immunol. (2007) 179:2041–5. doi: 10.4049/jimmunol.179.4.2041
72. Pathan N, Hemingway CA, Alizadeh AA, Stephens AC, Boldrick JC, Oragui EE, et al. Role of interleukin 6 in myocardial dysfunction of meningococcal septic shock. Lancet. (2004) 363:203–9. doi: 10.1016/S0140-6736(03)15326-3
73. Tanaka T, Narazaki M, Kishimoto T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy. (2016) 8:959–70. doi: 10.2217/imt-2016-0020
74. Del Valle DM, Kim-Schulze S, Huang H-H, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med. (2020) 26:1636–43. doi: 10.1038/s41591-020-1051-9
75. Ott LW, Resing KA, Sizemore AW, Heyen JW, Cocklin RR, Pedrick NM, et al. Tumor necrosis factor-α-and interleukin-1-induced cellular responses: coupling proteomic and genomic information. J Proteome Res. (2007) 6:2176–85. doi: 10.1021/pr060665l
76. Jain A, Irizarry-Caro RA, McDaniel MM, Chawla AS, Carroll KR, Overcast GR, et al. T cells instruct myeloid cells to produce inflammasome-independent IL-1β and cause autoimmunity. Nat Immunol. (2020) 21:65–74. doi: 10.1038/s41590-019-0559-y
77. Zhao C, Zhao W. NLRP3 inflammasome—a key player in antiviral responses. Front Immunol. (2020) 11:211. doi: 10.3389/fimmu.2020.00211
78. de Rivero Vaccari JC, Dietrich WD, Keane RW, de Rivero Vaccari JP. The inflammasome in times of COVID-19. Front Immunol. (2020) 11:2474. doi: 10.3389/fimmu.2020.583373
79. Kennedy RH, Silver R. Neuroimmune signaling: cytokines and the CNS. In: Neuroscience in the 21st Century. New York, NY: Springer New York. (2016) p. 1–41.
80. Liu Q. Zhou Y-h, Yang Z-q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol. (2016) 13:3–10. doi: 10.1038/cmi.2015.74
81. Xiao L, Sakagami H, Miwa N. ACE2: the key molecule for understanding the pathophysiology of severe and critical conditions of COVID-19: demon or angel? Viruses. (2020) 12:491. doi: 10.3390/v12050491
82. South AM, Diz DI, Chappell MC. COVID-19, ACE2, and the cardiovascular consequences. Am J Physiol Heart Circ Physiol. (2020). doi: 10.1152/ajpheart.00217.2020
83. Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. (2010) 2:247–57. doi: 10.1002/emmm.201000080
84. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. (2007) 7:803–15. doi: 10.1038/nri2171
85. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. (2020) 181:271–80. e8. doi: 10.1016/j.cell.2020.02.052
86. Lucas C, Wong P, Klein J, Castro TB, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. (2020) 584:463–9. doi: 10.1038/s41586-020-2588-y
87. Pedersen SF, Ho Y-C. SARS-CoV-2: a storm is raging. J Clin Invest. (2020) 130:2202–5. doi: 10.1172/JCI137647
88. Griffin JD, Cannistra SA, Demetri GD, Ernst TJ, Kanakura Y, Sullivan R. The biology of GM-CSF: regulation of production and interaction with its receptor. Int J Cell Cloning. (1990) 8:35–45. doi: 10.1002/stem.5530080705
89. Zhou Y, Fu B, Zheng X, Wang D, Zhao C, Sun R, et al. Aberrant pathogenic GM-CSF+ T cells and inflammatory CD14+ CD16+ monocytes in severe pulmonary syndrome patients of a new coronavirus. biorxiv. (2020). doi: 10.1101/2020.02.12.945576
90. Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect. (2020) 9:761–70. doi: 10.1080/22221751.2020.1747363
91. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
92. Spinelli FR, Conti F, Gadina M. HiJAKing SARS-CoV-2? The potential role of JAK inhibitors in the management of COVID-19. Science Immunol. (2020) 5:eabc5367. doi: 10.1126/sciimmunol.abc5367
93. Choy EH, De Benedetti F, Takeuchi T, Hashizume M, John MR, Kishimoto T. Translating IL-6 biology into effective treatments. Nat Rev Rheumatol. (2020) 16:335–45. doi: 10.1038/s41584-020-0419-z
94. Meng Z, Wang T, Chen L, Chen X, Li L, Qin X, et al. An experimental trial of recombinant human interferon alpha nasal drops to prevent COVID-19 in medical staff in an epidemic area. MedRxiv. (2020). doi: 10.1101/2020.04.11.20061473
95. Zhou Q, Chen V, Shannon C, Wei X, Xiang X, Wang X. Interferon-α2b treatment for COVID-19. Front Immunol. (2020) 11:1061. doi: 10.3389/fimmu.2020.01061
96. Feldmann M, Maini RN, Woody JN, Holgate ST, Winter G, Rowland M, et al. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet. (2020) 395:1407–9. doi: 10.1016/S0140-6736(20)30858-8
97. Cavalli G, De Luca G, Campochiaro C, Della-Torre E, Ripa M, Canetti D, et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: a retrospective cohort study. Lancet Rheumatol. (2020) 2:e325–e31. doi: 10.1016/S2665-9913(20)30127-2
98. Seif F, Aazami H, Khoshmirsafa M, Kamali M, Mohsenzadegan M, Pornour M, et al. JAK inhibition as a new treatment strategy for patients with COVID-19. Int Arch Allergy Immunol. (2020) 181:467–75. doi: 10.1159/000508247
99. Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK–STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. (2017) 77:521–46. doi: 10.1007/s40265-017-0701-9
100. Ziegler CG, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. (2020) 181:1016–35. e19. doi: 10.1016/j.cell.2020.04.035
101. Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. (2020) 587:657–62. doi: 10.1038/s41586-020-2601-5
102. Guillon A, Hiemstra P, Si-Tahar M. Pulmonary immune responses against SARS-CoV-2 infection: harmful or not? Intensive Care Med. (2020) 46:1897–900. doi: 10.1007/s00134-020-06170-8
103. Yang D, Chu H, Hou Y, Chai Y, Shuai H, Lee AC-Y, et al. Attenuated interferon and proinflammatory response in SARS-CoV-2–infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. J Infect Dis. (2020) 222:734–45. doi: 10.1093/infdis/jiaa356
104. Kuchipudi SV. The complex role of STAT3 in viral infections. J Immunol Res. (2015) 2015:272359. doi: 10.1155/2015/272359
105. Gotthardt D, Trifinopoulos J, Sexl V, Putz EM. JAK/STAT cytokine signaling at the crossroad of NK cell development and maturation. Front Immunol. (2019) 10:2590. doi: 10.3389/fimmu.2019.02590
106. Mommert M, Perret M, Hockin M, Viel S, Belot A, Richard JC, et al. Type-I Interferon assessment in 45 minutes using the FilmArray® PCR platform in SARS-CoV-2 and other viral infections. Eur J Immunol. (2021) 51:989–94. doi: 10.1002/eji.202048978
107. Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Science Immunol. (2020) 5:eabd1554. doi: 10.1126/sciimmunol.abd1554
108. Pairo-Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, et al. Genetic mechanisms of critical illness in Covid-19. Nature. (2021) 591:92–8. doi: 10.1038/s41586-020-03065-y
109. Lazear HM, Schoggins JW, Diamond MS. Shared and distinct functions of type I and type III interferons. Immunity. (2019) 50:907–23. doi: 10.1016/j.immuni.2019.03.025
110. Xia H, Cao Z, Xie X, Zhang X, Chen J, Wang H. Evasion of Type I interferon by SARS-CoV-2. Cell Rep. (2020) (1):108234. doi: 10.1016/j.celrep.2020.108234
111. Mu J, Fang Y, Yang Q, Shu T, Wang A, Huang M, et al. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell discovery. (2020) 6:1–4. doi: 10.1038/s41421-020-00208-3
112. Barrat FJ, Crow MK, Ivashkiv LB. Interferon target-gene expression and epigenomic signatures in health and disease. Nat Immunol. (2019) 20:1574–83. doi: 10.1038/s41590-019-0466-2
113. Sang ER, Tian Y, Miller LC, Sang Y. Epigenetic evolution of ACE2 and IL-6 genes: non-canonical interferon-stimulated genes correlate to COVID-19 susceptibility in vertebrates. Genes. (2021) 12:154. doi: 10.3390/genes12020154
114. Wathelet MG, Orr M, Frieman MB, Baric RS. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J Virol. (2007) 81:11620–33. doi: 10.1128/JVI.00702-07
115. Sardar R, Satish D, Birla S, Gupta D. Comparative analyses of SAR-CoV2 genomes from different geographical locations and other coronavirus family genomes reveals unique features potentially consequential to host-virus interaction and pathogenesis. BioRxiv. (2020). doi: 10.1101/2020.03.21.001586
116. Yuen C-K, Lam J-Y, Wong W-M, Mak L-F, Wang X, Chu H, et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg Microbes Infect. (2020) 9:1418–28. doi: 10.1080/22221751.2020.1780953
117. Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. (2020) 32:108185. doi: 10.1016/j.celrep.2020.108185
118. Shao W-H, Gamero AM, Zhen Y, Lobue MJ, Priest SO, Albandar HJ, et al. Stat1 regulates lupus-like chronic graft-versus-host disease severity via interactions with Stat3. J Immunol. (2015) 195:4136–43. doi: 10.4049/jimmunol.1501353
119. Tsai M-H, Pai L-M, Lee C-K. Fine-tuning of type I interferon response by STAT3. Front Immunol. (2019) 10:1448. doi: 10.3389/fimmu.2019.01448
120. Wang H, Yuan M, Wang S, Zhang L, Zhang R, Zou X, et al. STAT3 regulates the type I IFN-mediated antiviral response by interfering with the nuclear entry of STAT1. Int J Mol Sci. (2019) 20:4870. doi: 10.3390/ijms20194870
121. Joly BS, Siguret V, Veyradier A. Understanding pathophysiology of hemostasis disorders in critically ill patients with COVID-19. Intensive Care Med. (2020) 46:1603–6. doi: 10.1007/s00134-020-06088-1
122. Lonati C, Fumagalli J, Zanella A, Spinelli E, Mauri T. Hyaluronan in acute respiratory distress syndrome (ARDS): simply a biomarker or a deeper insight into ARDS mechanisms. J Lab Prec Med. (2018) 3:49. doi: 10.21037/jlpm.2018.05.03
123. Hojyo S, Uchida M, Tanaka K, Hasebe R, Tanaka Y, Murakami M, et al. How COVID-19 induces cytokine storm with high mortality. Inflamm Regen. (2020) 40:1–7. doi: 10.1186/s41232-020-00146-3
124. Kishimoto T. IL-6: from its discovery to clinical applications. Int Immunol. (2010) 22:347–52. doi: 10.1093/intimm/dxq030
125. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
126. Narazaki M, Kishimoto T. The two-faced cytokine IL-6 in host defense and diseases. Int J Mol Sci. (2018) 19:3528. doi: 10.3390/ijms19113528
127. Akira S, Taga T, Kishimoto T. Interleukin-6 in biology and medicine. Adv Immunol. (1993) 54:1–78. doi: 10.1016/S0065-2776(08)60532-5
128. Tanaka T, Narazaki M, Kishimoto T. Interleukin (IL-6) immunotherapy. Cold Spring Harb Perspect Biol. (2018) 10:a028456. doi: 10.1101/cshperspect.a028456
129. Kang S, Narazaki M, Metwally H, Kishimoto T. Historical overview of the interleukin-6 family cytokine. J Exp Med. (2020) 217:e20190347. doi: 10.1084/jem.20190347
130. Pérez-Alba E, Nuzzolo-Shihadeh L, Aguirre-García GM, Espinosa-Mora J, Lecona-Garcia JD, Flores-Pérez RO, et al. Baricitinib plus dexamethasone compared to dexamethasone for the treatment of severe COVID-19 pneumonia: a retrospective analysis. J Microbiol Immunol Infect. (2021) 54:787–93. doi: 10.1016/j.jmii.2021.05.009
131. Bharadwaj U, Kasembeli MM, Robinson P, Tweardy DJ. Targeting janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: rationale, progress, and caution. Pharmacol Rev. (2020) 72:486–526. doi: 10.1124/pr.119.018440
132. Schwartz DM, Bonelli M, Gadina M. O'shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol. (2016) 12:25–36. doi: 10.1038/nrrheum.2015.167
133. Abizanda P, Calbo Mayo JM, Mas Romero M, Cortés Zamora EB, Tabernero Sahuquillo MT, Romero Rizos L, et al. Baricitinib reduces 30-day mortality in older adults with moderate-to-severe COVID-19 pneumonia. J Am Geriatr Soc. (2021) 69:2752–8. doi: 10.1111/jgs.17357
134. Cantini F, Niccoli L, Matarrese D, Nicastri E, Stobbione P, Goletti D. Baricitinib therapy in COVID-19: A pilot study on safety and clinical impact. J Infect. (2020) 81:318–56. doi: 10.1016/j.jinf.2020.04.017
135. Mesa RA, Vannucchi AM, Mead A, Egyed M, Szoke A, Suvorov A, et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): an international, randomised, phase 3 trial. Lancet Haematol. (2017) 4:e225–e36. doi: 10.1016/S2352-3026(17)30027-3
136. Mascarenhas J, Hoffman R, Talpaz M, Gerds AT, Stein B, Gupta V, et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: a randomized clinical trial. JAMA Oncolo. (2018) 4:652–9. doi: 10.1001/jamaoncol.2017.5818
137. Xu X, Han M, Li T, Sun W, Wang D, Fu B, et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc Nat Acad Sci. (2020) 117:10970–5. doi: 10.1073/pnas.2005615117
138. Guaraldi G, Meschiari M, Cozzi-Lepri A, Milic J, Tonelli R, Menozzi M, et al. Tocilizumab in patients with severe COVID-19: a retrospective cohort study. Lancet Rheumatol. (2020) 2:e474–e84. doi: 10.1016/S2665-9913(20)30173-9
139. Rosas J, Liaño FP, Cantó ML, Barea JMC, Beser AR, Rabasa JTA, et al. Experience with the use of Baricitinib and tocilizumab monotherapy or combined, in patients with interstitial pneumonia secondary to coronavirus COVID19: a real-world study. Reumatol Clin. (2020). 18:150–6. doi: 10.1016/j.reuma.2020.10.009
140. Salama C, Han J, Yau L, Reiss WG, Kramer B, Neidhart JD, et al. Tocilizumab in patients hospitalized with Covid-19 pneumonia. N Engl J Med. (2021) 384:20–30. doi: 10.1056/NEJMoa2030340
141. Stone JH, Frigault MJ, Serling-Boyd NJ, Fernandes AD, Harvey L, Foulkes AS, et al. Efficacy of tocilizumab in patients hospitalized with Covid-19. N Engl J Med. (2020) 383:2333–44. doi: 10.1056/NEJMoa2028836
142. Hoang TN, Pino M, Boddapati AK, Viox EG, Starke CE, Upadhyay AA, et al. Baricitinib treatment resolves lower-airway macrophage inflammation and neutrophil recruitment in SARS-CoV-2-infected rhesus macaques. Cell. (2021) 184:460–75. e21. doi: 10.1016/j.cell.2020.11.007
143. Bronte V, Ugel S, Tinazzi E, Vella A, De Sanctis F, Canè S, et al. Baricitinib restrains the immune dysregulation in COVID-19 patients. medRxiv. (2020). doi: 10.1101/2020.06.26.20135319
144. Raman B, Cassar MP, Tunnicliffe EM, Filippini N, Griffanti L, Alfaro-Almagro F, et al. Medium-term effects of SARS-CoV-2 infection on multiple vital organs, exercise capacity, cognition, quality of life and mental health, post-hospital discharge. EClinicalMedicine. (2021) 31:100683. doi: 10.1016/j.eclinm.2020.100683
145. Cao Y, Wei J, Zou L, Jiang T, Wang G, Chen L, et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID-19): a multicenter, single-blind, randomized controlled trial. J Allergy Clin Immunol. (2020) 146:137–46. e3. doi: 10.1016/j.jaci.2020.05.019
146. Goletti D, Cantini F. Baricitinib therapy in COVID-19 pneumonia-an unmet need fulfilled. Mass Medical Soc. (2021) 384:867–9. doi: 10.1056/NEJMe2034982
147. Portsmore S, Nguyen TNT, Beacham E, Neelakantan P. Combined IL-6 and JAK-STAT inhibition therapy in COVID-19 related sHLH, potential game changer. Br J Haematol. (2020). doi: 10.1111/bjh.16966
148. Stebbing J, Krishnan V, de Bono S, Ottaviani S, Casalini G, Richardson PJ, et al. Mechanism of baricitinib supports artificial intelligence-predicted testing in COVID-19 patients. EMBO Mol Med. (2020) 12:e12697. doi: 10.15252/emmm.202012697
149. Mayence A, Vanden Eynde JJ. Baricitinib: a 2018 novel FDA-approved small molecule inhibiting janus kinases. Pharmaceuticals. (2019) 12:37. doi: 10.3390/ph12010037
150. Schmajuk G, Schneeweiss S, Katz JN, Weinblatt ME, Setoguchi S, Avorn J, et al. Treatment of older adult patients diagnosed with rheumatoid arthritis: improved but not optimal. Arthritis Care Res. (2007) 57:928–34. doi: 10.1002/art.22890
151. Xeljanz prescribing information 2014 (Available online at: http://labeling.pfizer.com/ShowLabeling.aspx?id=959.
152. Hodge JA, Kawabata TT, Krishnaswami S, Clark JD, Telliez J-B, Dowty ME, et al. The mechanism of action of tofacitinib-an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol. (2016) 34:318–28.
153. Guimarães PO, Quirk D, Furtado RH, Maia LN, Saraiva JF, Antunes MO, et al. Tofacitinib in patients hospitalized with Covid-19 pneumonia. N Engl J Med. (2021) 385:406–15. doi: 10.1056/NEJMoa2101643
154. Agrawal M, Brenner EJ, Zhang X, Modesto I, Woolcott J, Ungaro RC, et al. Characteristics and outcomes of IBD patients with COVID-19 on tofacitinib therapy in the SECURE-IBD registry. Inflamm Bowel Dis. (2021). doi: 10.1093/ibd/izaa303
155. Ferreira SB, Dias MG, Ferreira RB, Neto AN, Trüeb R, Lupi O. Rapidly progressive alopecia areata totalis in a COVID-19 patient, unresponsive to tofacitinib. J Eur Acad Dermatol Venereol. (2021). 35:e411–2. doi: 10.1111/jdv.17170
156. Kalil AC, Patterson TF, Mehta AK, Tomashek KM, Wolfe CR, Ghazaryan V, et al. Baricitinib plus remdesivir for hospitalized adults with Covid-19. N Engl J Med. (2021) 384:795–807. doi: 10.1056/NEJMoa2031994
157. Levy G, Guglielmelli P, Langmuir P, Constantinescu S. JAK inhibitors and COVID-19. J ImmunoThera Cancer. (2022) 10:e002838. doi: 10.1136/jitc-2021-002838
158. Marconi VC, Ramanan AV, de Bono S, Kartman CE, Krishnan V, Liao R, et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): a randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir Med. (2021) 9:1407–18. doi: 10.1016/S2213-2600(21)00331-3
159. Saber-Ayad M, Hammoudeh S, Abu-Gharbieh E, Hamoudi R, Tarazi H, Al-Tel TH, et al. Current Status of Baricitinib as a Repurposed Therapy for COVID-19. Pharmaceuticals. (2021) 14:680. doi: 10.3390/ph14070680
160. Hasan M, Rabbani R, Anam AM, Huq SMR, Polash MMI, Nessa SST, et al. Impact of high dose of baricitinib in severe COVID-19 pneumonia: a prospective cohort study in Bangladesh. BMC Infect Dis. (2021) 21:1–9. doi: 10.1186/s12879-021-06119-2
161. López-Cortés A, Guerrero S, Ortiz-Prado E, Yumiceba V, Vera-Gupi A, Cáceres ÁL, et al. Single-nucleus lung transcriptomics and inflammatory responses in lethal COVID-19 reveal potential drugs in advanced-stage clinical trials. (2021). doi: 10.21203/rs.3.rs-808746/v1
162. Adamczyk-Popławska M, Kwiatek A. Covid-19 therapy: What have we learned in 8 months?. Postepy Mikrobiol. (2020) 59:207–25. doi: 10.21307/PM-2020.59.3.15
163. Li B, Gould J, Yang Y, Sarkizova S, Tabaka M, Ashenberg O, et al. Cumulus provides cloud-based data analysis for large-scale single-cell and single-nucleus RNA-seq. Nat Methods. (2020) 17:793–8. doi: 10.1038/s41592-020-0905-x
164. Izumo T, Kuse N, Awano N, Tone M, Sakamoto K, Takada K, et al. Clinical impact of combination therapy with baricitinib, remdesivir, and dexamethasone in patients with severe COVID-19. Respir Investig. (2021) 59:799–803. doi: 10.1016/j.resinv.2021.07.004
165. Jorgensen SC, Tse CL, Burry L, Dresser LD. Baricitinib: a review of pharmacology, safety, and emerging clinical experience in COVID-19. Pharmacotherapy. (2020) 40:843–56. doi: 10.1002/phar.2438
166. Komrokji RS, Seymour JF, Roberts AW, Wadleigh M, To LB, Scherber R, et al. Results of a phase 2 study of pacritinib (SB1518), a JAK2/JAK2 (V617F) inhibitor, in patients with myelofibrosis. Blood. (2015) 125:2649–55. doi: 10.1182/blood-2013-02-484832
167. Verstovsek S, Odenike O, Singer JW, Granston T, Al-Fayoumi S, Deeg HJ. Phase 1/2 study of pacritinib, a next generation JAK2/FLT3 inhibitor, in myelofibrosis or other myeloid malignancies. J Hematol Oncol. (2016) 9:1–12. doi: 10.1186/s13045-016-0367-x
168. Betts BC, Bastian D, Iamsawat S, Nguyen H, Heinrichs JL, Wu Y, et al. Targeting JAK2 reduces GVHD and xenograft rejection through regulation of T cell differentiation. Proc Nat Acad Sci. (2018) 115:1582–7. doi: 10.1073/pnas.1712452115
169. Harrison CN, Schaap N, Vannucchi AM, Kiladjian J-J, Tiu RV, Zachee P, et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. (2017) 4:e317–e24. doi: 10.1016/S2352-3026(17)30088-1
170. Betts BC, Young JW. Less can be more when targeting interleukin-6-mediated cytokine release syndrome in coronavirus disease 2019. Crit Care Explor. (2020) 2:e0138. doi: 10.1097/CCE.0000000000000138
171. Jade D, Ayyamperumal S, Tallapaneni V, Nanjan CMJ, Barge S, Mohan S, et al. Virtual high throughput screening: Potential inhibitors for SARS-CoV-2 PLPRO and 3CLPRO proteases. Eur J Pharmacol. (2021) 901:174082. doi: 10.1016/j.ejphar.2021.174082
172. Ucciferri C, Auricchio A, Marinari S, Vecchiet J, Falasca K. COVID-19 in a patient with SISTEMIC sclerosis: the role of ruxolitinib. Eur J Inflammation. (2021) 19:20587392211036813. doi: 10.1177/20587392211036813
173. Giudice V, Pagliano P, Vatrella A, Masullo A, Poto S, Polverino BM, et al. Combination of ruxolitinib and eculizumab for treatment of severe SARS-CoV-2-related acute respiratory distress syndrome: a controlled study. Front Pharmacol. (2020) 11:857. doi: 10.3389/fphar.2020.00857
174. Innes AJ, Cook LB, Marks S, Bataillard E, Crossette-Thambiah C, Sivasubramaniam G, et al. Ruxolitinib for tocilizumab-refractory severe COVID-19 infection. Br J Haematol. (2020). doi: 10.1111/bjh.16979
175. Vannucchi AM, Mortara A., D'alessio A, Morelli M, Tedeschi A, Festuccia MB, et al. JAK inhibition with ruxolitinib in patients with COVID-19 and severe pneumonia: multicenter clinical experience from a compassionate use program in Italy. J Clin Med. (2021) 10:3752. doi: 10.3390/jcm10163752
176. Koschmieder S, Jost E, Cornelissen C, Müller T, Schulze-Hagen M, Bickenbach J, et al. Favorable COVID-19 course despite significant comorbidities in a ruxolitinib-treated patient with primary myelofibrosis. Eur J Haematol. (2020) 105:655–8. doi: 10.1111/ejh.13480
177. Saraceni F, Scortechini I, Mancini G, Mariani M, Federici I, Gaetani M, et al. Severe COVID-19 in a patient with chronic graft-versus-host disease after hematopoietic stem cell transplant successfully treated with ruxolitinib. Trans Infect Dis. (2021) 23:e13401. doi: 10.1111/tid.13401
178. Curran SA, Shyer JA, Angelo ETS, Talbot LR, Sharma S, Chung DJ, et al. Human dendritic cells mitigate NK-cell dysfunction mediated by nonselective JAK1/2 blockade. Cancer Immunol Res. (2017) 5:52–60. doi: 10.1158/2326-6066.CIR-16-0233
179. Roskoski Jr R. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol Res. (2016) 111:784–803. doi: 10.1016/j.phrs.2016.07.038
180. Moiseev S, Bulanov N, Zykova A, Brovko M, Novikov P, Akulkina L, et al. Tofacitinib versus standard of care treatment in patients with COVID-19: a multicenter non-randomized controlled study. Authorea Preprints. (2021). doi: 10.22541/au.162560391.18060570/v1
181. Hayek ME, Mansour M, Ndetan H, Burkes Q, Corkern R, Dulli A, et al. Anti-inflammatory treatment of COVID-19 pneumonia with tofacitinib alone or in combination with dexamethasone is safe and possibly superior to dexamethasone as a single agent in a predominantly African American cohort. Mayo Clin Proc Innov Qual Outcomes. (2021) 5:605–13. doi: 10.1016/j.mayocpiqo.2021.03.007
182. Pfeifer ND, Lo A, Bourdet DL, Colley K, Singh D. Phase I study in healthy participants to evaluate safety, tolerability, and pharmacokinetics of inhaled nezulcitinib, a potential treatment for COVID-19. Clin Transl Sci. (2021) 14:2556–65. doi: 10.1111/cts.13123
183. Kameda H, Takeuchi T, Yamaoka K, Oribe M, Kawano M, Yokoyama M, et al. Efficacy and safety of upadacitinib over 84 weeks in Japanese patients with rheumatoid arthritis (SELECT-SUNRISE). Arthritis Res Ther. (2021) 23:1–10. doi: 10.1186/s13075-020-02387-6
184. Winthrop KL, Park S, Gul A, Cardiel M, Gomez-Reino J, Tanaka Y, et al. Tuberculosis and other opportunistic infections in tofacitinib-treated patients with rheumatoid arthritis. Ann Rheum Dis. (2016) 75:1133–8. doi: 10.1136/annrheumdis-2015-207319
185. Nishiura H, Kobayashi T, Miyama T, Suzuki A., Jung S-m, Hayashi K, et al. Estimation of the asymptomatic ratio of novel coronavirus infections (COVID-19). Int J Infect Dis. (2020) 94:154–5. doi: 10.1016/j.ijid.2020.03.020
186. Regás VH-L, Culla MTD, Bellfill RL. Adverse reactions of drugs specifically used for treatment of SARS-CoV-2 infection. Med Clin (Barc). (2020) 155:448–53. doi: 10.1016/j.medcle.2020.06.026
187. Kremer JM, Genovese MC, Keystone E, Taylor PC, Zuckerman SH, Ruotolo G, et al. Effects of baricitinib on lipid, apolipoprotein, and lipoprotein particle profiles in a phase IIb study of patients with active rheumatoid arthritis. Arthritis Rheumatol. (2017) 69:943–52. doi: 10.1002/art.40036
188. Beck C, Dubois J, Grignon A, Lacroix J, David M. Incidence and risk factors of catheter-related deep vein thrombosis in a pediatric intensive care unit: a prospective study. J Pediatr. (1998) 133:237–41. doi: 10.1016/S0022-3476(98)70226-4
189. Wang P, Kandemir U, Zhang B, Wang B, Li J, Zhuang Y, et al. Incidence and risk factors of deep vein thrombosis in patients with pelvic and acetabular fractures. Clin Appl Thromb Hemost. (2019) 25:1076029619845066. doi: 10.1177/1076029619845066
190. Zheng X-Q, Huang J-F, Lin J-L, Zhu Y-X, Wang M-Q, Guo M-L, et al. Controlled release of baricitinib from a thermos-responsive hydrogel system inhibits inflammation by suppressing JAK2/STAT3 pathway in acute spinal cord injury. Colloids Surf B Biointerfaces. (2021) 199:111532. doi: 10.1016/j.colsurfb.2020.111532
191. La Rosée F, Bremer H, Gehrke I, Kehr A, Hochhaus A, Birndt S, et al. The Janus kinase 1/2 inhibitor ruxolitinib in COVID-19 with severe systemic hyperinflammation. Leukemia. (2020) 34:1805–15.
192. Micromedex I. 2022 Available online at: https://www.mayoclinic.org/drugs-supplements/ruxolitinib-oral-route/side-effects/drg-20075326
Keywords: COVID-19, JAK inhibitors, baricitinib, pacritinib, ruxolitinib, tofacitinib
Citation: Khaledi M, Sameni F, Yahyazade S, Radandish M, Owlia P, Bagheri N, Afkhami H, Mahjoor M, Esmaelpour Z, Kohansal M and Aghaei F (2022) COVID-19 and the potential of Janus family kinase (JAK) pathway inhibition: A novel treatment strategy. Front. Med. 9:961027. doi: 10.3389/fmed.2022.961027
Received: 03 June 2022; Accepted: 08 August 2022;
Published: 30 August 2022.
Edited by:
Binwu Ying, Sichuan University, ChinaReviewed by:
Sabrin Albeituni, St. Jude Children's Research Hospital, United StatesCopyright © 2022 Khaledi, Sameni, Yahyazade, Radandish, Owlia, Bagheri, Afkhami, Mahjoor, Esmaelpour, Kohansal and Aghaei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Parviz Owlia, cG93bGlhQGdtYWlsLmNvbQ==; b3dsaWFAc2hhaGVkLmFjLmly; Nader Bagheri, bmFkZXJiYWdoZXJpMzNAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.