 Junhui Deng
Junhui Deng Zhifen Wu
Zhifen Wu Yun He
Yun He Lirong Lin
Lirong Lin Wei Tan
Wei Tan Jurong Yang
Jurong Yang- 1The Third Affiliated Hospital of Chongqing Medical University, Chongqing, China
- 2The Fifth People's Hospital of Chongqing, Chongqing, China
A growing number of studies have confirmed that immune cells play various key roles in the pathophysiology of acute kidney injury (AKI) development. After the resident immune cells and intrinsic renal cells are damaged by ischemia and hypoxia, drugs and toxins, more immune cells will be recruited to infiltrate through the release of chemokines, while the intrinsic cells promote macrophage polarity conversion, and the immune cells will promote various programmed deaths, phenotypic conversion and cycle arrest of the intrinsic cells, ultimately leading to renal impairment and fibrosis. In the complex and dynamic immune microenvironment of AKI, the bidirectional interaction between immune cells and intrinsic renal cells affects the prognosis of the kidney and the progression of fibrosis, and determines the ultimate fate of the kidney.
Introduction
Acute kidney injury (AKI) refers to a clinical syndrome in which renal function rapidly declines in a short period of time due to various pathological factors. During the pathophysiological process of AKI, all cells in the kidney, including renal cells, resident immune cells, and infiltrating immune cells, undergo various adaptive responses mediated by complex and unclear mechanisms (1). In recent studies, single-cell sequencing of kidneys from animal models of AKI revealed spatial co-localization of renal intrinsic cells with immune cells from all stages of disease (2–5). Various studies have shown that a bidirectional interaction between renal innate cells and immune cells exists, and plays a key role in the mechanisms governing renal injury and renal repair (6, 7). In this review, we will summarize the interaction between renal intrinsic cells and immune cells in the development of AKI and provide insights into its clinical significance.
Intrinsic Cells Promote Immune Cell Infiltration
Under physiological conditions, a small number of innate immune cells reside in the normal human kidney, including macrophages, dendritic cells (DCs) and lymphocytes (8). Following injury to renal tubular epithelial cells (TECs) and endothelial cells, resident immune cells are activated and recruit circulating immune cells to infiltrate the kidneys (9–11). Renal tissue biopsies from AKI patients with sepsis were stained pathologically and displayed a large infiltration of neutrophils and macrophages within the glomeruli and tubulointerstitial regions of the kidney (12). These findings were corroborated by another study where single-cell RNA sequencing of kidney tissues from lipopolysaccharide (LPS)-induced mouse models of AKI also detected large numbers of macrophages, neutrophils, and T lymphocytes in diseased animals (13, 14). Furthermore, neutrophils, macrophages, dendritic cells, NK cells, T cells, and B cells have been reported to be recruited and activated in mouse and rat models of ischemia-reperfusion AKI (15, 16), and in cisplatin-induced AKI, with early mast cell infiltration also noted (17).
Renal intrinsic cells release various cytokines and chemokines, such as C-C motif chemokine ligand 2 (CCL2), CCL20, and C-C motif chemokine receptor 5 (CCR5) upon cell death, and release danger-associated molecular patterns (DAMPs) and activating pattern recognition receptors (PRRs), which in turn promote infiltration of renal tissue by a large number of immune cells (18–24). It has been reported that in renal injury due to ischemia/reperfusion (I/R), necrotic tubular epithelial cells and endothelial cells release DAMPs such as histones, heat shock proteins, high mobility histone (HMGB1), hyaluronic acid and biglycan, and secrete chemokines after direct interaction with Toll-like receptors 2 (TLR2) and TLR4, further driving neutrophil recruitment to the kidney (25–31). Infiltrating neutrophils interact with activated platelets to form neutrophil extracellular traps (NETs) that contribute to renal tissue injury (32). Peptidyl arginine deiminase-4 (PAD4) is a member of the peptidyl arginine deiminase family, whose function is to convert arginine in proteins to citrulline, and is an important regulator of the formation of NETs (33, 34). Rabadi et al. have repeatedly reported that in addition to being expressed in immune cells, ischemia-reperfusion induces high expression of PAD4 in TECs. This promotes nuclear translocation of PAD4 to the cytoplasm through interaction with NF-κB, and promotes neutrophil infiltration and NET formation (35, 36).
NF-κB is a key regulator of innate and adaptive immunity, and an important signaling factor for renal innate cells to promote immune cell infiltration. Highlighting its role in the inflammatory response during AKI, TEC specific NF-κB knockout mice had attenuated neutrophil and macrophage infiltration in I/R-injured AKI kidneys compared to wild-type control mice, and decreased expression vascular endothelial growth factor A(VEGFA), C-X-C motif chemokine ligand 1(CXCL1), and CXCL2 in cultured NF-κB knockout TECs in vitro (37). Moreover, Yu et al. found that NF-κB also regulated the expression of RANTES (a member of the CC chemokine family) in I/R-injured mouse renal tubular cells, promoting the release of inflammatory factors tumor necrosis factor α (TNF-α), lnterleukin-1β (IL-1β), and monocyte chemotactic protein 1 (MCP-1), thereby promoting the infiltration of T lymphocytes and macrophages into the renal interstitium (38). In addition to NF-κB, transforming growth factor β (TGF-β) signaling in TECs could also promote immune cell infiltration. By constructing mice expressing TGF-β specific receptor 1 in renal tubular epithelial cells, the researchers detected macrophages, dendritic cells, monocytes and T cells infiltration and tubular damage after stimulating the activation of TGF-β signaling pathway, confirming that renal TGF-β signaling in TECs is sufficient to induce renal tissue inflammation and lead to AKI (39).
Renal intrinsic cells can also promote infiltration by macrophages, neutrophils, and other monocytes throughIL-1.IL-33, an IL-1 family member involved in the pathogenesis of AKI, is released by endothelial cells in the kidneys of I/R-injured and cisplatin-induced AKI mice. It acts by directly targeting invariant natural killer T (iNKT) cells via binding to its receptor ST2 on the cell surface, and induces interferon (IFN) and IL-17A production, which promotes neutrophil infiltration and activation (40, 41). IL-34 is also produced and released by TECs after I/R injury in the kidney, promoting the proliferation of intrarenal and bone marrow derived macrophages, and the infiltration of circulating monocytes into the kidney through the release of chemokines, TNF-α, and MCP-1 (42). The expression and release of various cytokines and chemokines CCL2, TNF-α, and CXCL10 in TECs are regulated by the transcription factor GATA Binding Protein 2 (GATA2), a zinc finger-containing transcription factor that is specifically expressed in renal tubular cells, and maintain its cellular identity. In the study by Yu et al. (43) CCL2, TNF-α, and CXCL10 were downregulated in GATA2-deficient mice, and monocyte infiltration was significantly reduced in renal tissue. In addition to the secretion of cytokines and chemokines, TECs also communicate with immune cells via exosomes. Following injury to renal tubular epithelial cells, CD26-containing exosomes are produced and bind to the chemokine receptor C-X-C motif chemokine receptor 4 (CXCR4), leading to upregulation of downstream chemokine pathways, increased stromal-derived factor 1 (SDF1), and macrophage and neutrophil infiltration (44). TECs can also amplify the inflammatory response through the release of exosomes that contain chemokine CCL2 mRNA, which are directly transferred to macrophages, leading to enhanced macrophage migration (45).
Intrinsic Renal Cells Promote Macrophage Polarization
Recent studies have shown that the phenotype and function of macrophages are highly plastic and can play a key role in the process of AKI and repair by altering their phenotype (46–48). Macrophages are divided into M1 (classical activation type) and M2 (selective activation type) according to their activation status. M1 macrophages differentiate in response to interferon γ (IFNγ) and/or LPS. They express high levels of nitric oxide synthase (iNOS) and produce pro-inflammatory cytokines such as TNF-α, IL-16, and IL-12 that contribute to M1 macrophage antibacterial and tumor-killing functions. In contrast, M2 macrophages are induced by IL-4/IL-13 and express high levels of insulin-like growth factor 1 (IGF-1), mannose receptor 1 (CD206), and arginase 1 (ARG-1). M2 macrophages are anti-inflammatory and have wound healing functions (49–53).
Early in AKI, TECs subjected to various injuries exert pro-inflammatory effects mainly through the release of exosomal miRNAs that transmit signals and regulate the induction of M1 macrophages. Hypoxic injury stimulates the expression of hypoxia-inducible factor-1α (HIF-1α) in renal tubular epithelial cells and promotes the release of miRNA-23a-rich exosomes to induce M1 macrophage differentiation by inhibiting the ubiquitin editor A20 (54). Interestingly, inhibition of miRNA-23a attenuated tubulointerstitial inflammation before I/R injury in mice, and lead to reduced exosome-mediated transport of miRNA-23a between TECs and macrophages.LvLL et al. found that TECs-derived exosomal miRNA-19b-3p also promoted the activation of M1 macrophages during renal injury. Specifically, TEC-derived exosomal miR-19b-3p internalized by macrophages increased NF-κB/suppressor of cytokine signaling (1SOCS-1) activation, and induced tubulointerstitial inflammation in mice that could be reversed by mIR-19b-3p inhibition (55). Clinically, high levels of miR-19b-3p positively correlated with the severity of tubulointerstitial inflammation, further confirming that exosomal miR-19b-3p mediates communication between damaged TECs and macrophages (55).
During later stages of AKI, IL-4-stimulated arginase 1(ARG-1) and CD206-positive M2 macrophages predominate and promote the transition from tubular injury to tubular repair. In vitro studies have demonstrated that renal tubular cells co-cultured with IL-4-stimulated M2 macrophages, but not IFNγ-stimulated pro-inflammatory M1 macrophages, induce tubular cell proliferation (56). Colony stimulating factors (CSF) are a group of growth factors that promote the proliferation and differentiation of myeloid progenitor cells, including macrophage colony stimulating factor (M-CSF/CSF1), granulocyte-macrophage colony stimulating factor (GM-CSF/CSF2), and granulocyte colony stimulating factor (G-CSF/CSF3) (57). In mouse models of AKI induced by I/R injury or novel diphtheria toxin (DT), CSF1 secreted by renal proximal tubular epithelial cells stimulated enhanced polarization of M2 macrophages, and mediated regenerative repair of renal tubular epithelium after AKI (58). Wang et al. demonstrated that proximal tubular CSF-1-specific knockout mice had reduced M2 macrophage polarization, delayed renal function and structural recovery, and increased tubular interstitial fibrosis in the kidney after I/R injury (59). In another study, CSF2 expression was significantly increased in HK-2 and human M1 macrophage co-cultures (60). CSF2 is secreted by HK-2 cells, but not by M1 macrophages. However, culturing macrophages with exogenous recombinant CSF2 promotes the transition from an M1 to M2 phenotype by activating phosphorylated signal transducer and activator of transcription 5(p-STAT5) in a dose- and time-dependent manner. Intraperitoneal injection of CSF2-neutralizing antibodies exacerbate renal injury and inhibit tubular proliferation, thereby decreasing survival. In contrast, administration of recombinant mouse CSF2 protein dampens septic AKI. Endogenous erythropoietin (EPO), which is synthesized and secreted by the peritubular fibroblasts, is thought to be an important protective protein in AKI that protects renal tissue by reducing apoptosis of TECs and decreasing inflammation (61, 62). It has been reported that in rhabdomyolysis-associated AKI, EPO promotes macrophage polarization toward the M2 phenotype via the Janus Kinase 2(JAK2)/STAT3/STAT6 pathway in response to IL-4 stimulation, thereby reducing M1 damage to the relevant tissues and enhancing the healing and repair mechanisms of M2 macrophages (63).
Immune Cells Promote Various Types Of Programmed Cell Deaths Of Renal Intrinsic Cells
TECs undergo various types of programmed cell death (PCD) including apoptosis, pyroptosis, necroptosis, and ferroptosis (64–67). Upon sensing local tissue injury, resident and infiltrating inflammatory cells in the kidney promote TEC PCD by producing and releasing large amounts of inflammatory cytokines and activating various pathway proteins, leading to TEC detachment, tubular occlusion, and tubular inactivation. In the early stage of AKI, M1 macrophages secrete various pro-inflammatory cytokines, including TNF-α and interleukins that binds to the corresponding receptors in TECs to promote PCD (68, 69). TNF-α has been reported to bind to the TNF receptor 1 on the surface of TECs to induce RIP3-dependent necrotizing apoptosis (70, 71). In I/R-induced AKI, TNFα has also been reported to exacerbate kidney injury through the receptor-interacting protein 3(RIP1)/RIP3 necroptosis signaling pathway (72). Type I interferon α (IFN-α) is a multifunctional active cytokine that induces apoptosis in renal TECs through activation of caspase-3, caspase-8, and caspase-9, leading to DNA fragmentation and nuclear condensation (73). Increased expression of IFN-α can be observed in renal biopsies of AKI patients after renal transplantation in clinical practice (74). Plasmacytoid DCs are a distinct DC subpopulation specialized in IFN production. Plasmacytoid DCs producing IFN-α directly induce apoptosis in TECs in vitro, suggesting that pDCs play a deleterious role in AKI via IFN-α (74). TLR9 in DCs can also be activated by mitochondrial DNA (mtDNA), by mediating IL-17A production in γδ T cells, thus promoting the development of AKI during sepsis (75). Furthermore, IL-17A has been shown to induce TEC apoptosis in AKI in sepsis. Interestingly, IL-17A knockout mice administered a cecum ligation puncture had reduced numbers of TUNEL-positive TECs, decreased cleaved caspase-3 levels, and an increased Bax/Bcl-2 expression ratio, suggesting that IL-17A plays a pathogenic role in AKI by inducing apoptosis in TECs (76). Macrophages can also regulate TEC pyroptosis by releasing exosomal miRNA. Chen et al. isolated exosomes from M1 and M2 macrophages, and miRNA sequencing revealed differential miRNA expression levels in M1 and M2 exosomes, in which miR-93-5p was involved in the regulation of mouse renal tubular epithelial cells pyroptosis. It was determined that thioredoxin-interacting protein (TXNIP) was a direct target of miR-93-5p in mouse renal tubular epithelial cells (77).
Mitogen-activated protein kinase (MAPK), an extracellular signal-regulated kinase, is another important pathway protein for macrophage function (78, 79). It is suggested that the MAPK signaling pathway plays a key role in kidney injury molecular 1(KIM-1) mediated renal injury by promoting macrophage phenotypic transformation and migration (80). Moreover, in macrophages cultured in vitro, LPS can induce inflammatory responses through activation of the MAPK pathway (81). Su et al. found that Pannexin 1 (PANX1), an ATP-releasing pathway family protein, mediates iron death in TECs via the MAPK pathway in I/R-induced AKI. Therefore, macrophages may regulate iron death in TECs by regulating MAPK (82).
Immune Cells Promote Phenotypic Conversion Of Intrinsic Renal Cells
Epithelial mesenchymal transition (EMT) of renal tubular cells is one of the most important features of tubulointerstitial fibrosis during the transition from AKI to chronic kidney disease (CKD). Increased production of pro-fibrotic cytokines such as MCP-1, complement protein 3 (C3), and TGF-β after AKI promotes EMT and leads to renal fibrosis and eventual progression to CKD (83). The inflammatory microenvironment is critical for the progression of EMT in TECs. Protein and mRNA expression of α-smooth muscle actin (α-SMA) and waveform protein increased in HK-2 cells after co-culture with M2 macrophages in Transwell chambers for 48 h, while E-cadherin significantly decreased, confirming the promotional effect of M2 macrophages on EMT (84). The investigators found that cisplatin induced only incomplete EMT in TECs alone, while conditioned medium of cisplatin-treated fibroblasts and macrophages induced complete EMT in TECs. High expression levels of α-SMA and collagen-1 in cisplatin-activated fibroblasts were detected in the triple co-culture cell model, and increased expression of ARG-1 and CD206 by macrophages were observed, indicating M2 macrophage polarization and the development of complete EMT in TECs. These data reveal a synergistic effect of TECs, fibroblasts, and M2 macrophages to promote the progression of renal fibrosis (85). Furthermore, in a mouse unilateral ureteral obstruction (UUO) model, macrophages released high levels of TGF-β1 after polarization to M2 phenotypes that promoted EMT-induced renal fibrosis. In contrast, depletion of M2 macrophages specifically inhibited EMT and subsequently reduced renal fibrosis (86, 87).
During the transition from AKI to CKD, macrophages regulate tubule cell EMT through various mechanisms. The typical epithelial cell morphology of HK-2 cells disappears after co-culture with monocytes, accompanied by decreased E-cadherin expression and increased expression of α-SMA and fibronectin, suggesting that EMT occurs in HK-2 cells by a specific mechanism of monocyte upregulation through the NF-kB signaling pathway. This phenotype was related to the upregulation of intercellular adhesion molecule 1 (ICAM-1) expression in HK-2 cells and activation of the NF-kB signaling pathway (88). HMGB-1 is a chromatin-binding protein that acts as a signal transducer by regulating IL-1 and TNF expression under inflammatory conditions (89). Human proximal tubular epithelial cells cultured using conditioned medium containing supernatants from activated peripheral blood mono-nuclear cells showed significant upregulation of HMGB-1 and induction of EMT via TGF-β, including decreased E-calmodulin expression, increased α-SMA expression, and enhanced cell migration, suggesting that HMGB-1 is a key mediator of EMT in TECs.
The complement system is another important pathogenic mediator in the pathogenesis of AKI, and its member C3 regulates renal fibrosis through the HMGB1/TGF-β1/Smad2/3 signaling pathway. In vitro experiments revealed that C3 secreted by macrophages stimulated HMGB1 translocation from the nucleus to the cytoplasm and promoted the expression of TGF-β1 in TECs and induced epithelial cell EMT (90). A study by Zheng et al. demonstrated that matrix metalloproteinase (MMP) family members secreted by macrophages in the kidney are involved in the development and progression of renal fibrosis through EMT of TECs as well as activation of resident fibroblasts, endothelial-mesenchymal transition, and pericyte-myofibroblast trans-differentiation (91). Murine renal tubular epithelial cell lines and primary renal tubular epithelial cells cultured with conditioned medium from LPS-activated macrophages significantly increased MMP-9 expression. In contrast, blockade of MMP-9 resulted in EMT inhibition in TECs (92). These results were confirmed in mouse where MMP-9 was upregulated in the early and late stages of UUO in mice, with MMP-9 cleaved bone bridge protein and macrophage infiltration significantly reduced after MMP-9 blockade. Strikingly, MMP-9 blockade significantly reduced EMT of TECs and renal significantly improved renal fibrosis (93). Pentraxin3 (PTX3), an acute-phase protein produced by resident and innate immune cells, promotes renal fibrosis by upregulating TECs EMT via a c-JunN-terminal kinase (c-JNK)-dependent mechanism. Treatment of UUO in rats and HK-2 cells with recombinant human PTX3 (Rh-PTX3) resulted in time-dependent increases in PTX3, p-JNK and waveform protein levels, as well as decreases in E-cadherin, JNK phosphorylation, and EMT markers (94). CircRNA can also act as an important mediator of macrophage promotion of tubular cell EMT, as found in the UUO-induced mouse and in vitro HK-2 cell fibrosis models. CircACTR2 in macrophages promotes macrophage inflammation and induces EMT and fibrosis in TECs via miR-561 and NLRP3 inflammasome activation (95).
Immune Cells Inhibit Tecs Cycle Arrest
The cell cycle encompasses the process of cell division and includes the G0, G1, S, G2, and M phases (96, 97). During AKI, TECs undergo cell cycle arrest in the G2/M phase and fail to differentiate, producing and secreting various pro-fibrotic factors such as TGF-β1 and connective tissue growth factor (CTGF). These factors act on peripheral cells and fibroblasts through the paracrine pathway, leading to poor renal repair and renal fibrosis (98–101). Regarding the role of immune cells and the cell cycle, it has been shown that M2 macrophages act on damaged TECs by releasing the Wnt ligand, Wnt7b, to bypass cell cycle arrest, promote cell proliferation, repair tubular basement membrane, and inhibit renal fibrosis (102).
Macrophage migration inhibitory factor (MIF), a pleiotropic cytokine in tissue injury, is produced and released by macrophages in AKI, and has been suggested to be an endogenous protective factor in the kidney (103). On MIF knockdown, TECs undergo G2/M cell cycle, expression of pro-inflammatory and pro-fibrotic mediators is increased, and renal fibrosis is exacerbated in mice with sepsis and UUO (104). It was recently found that the lipid transport protein 2 (Lcn-2) secreted by macrophages also plays a key role in TEC repair after AKI. In a co-culture system of NRK52E cells and bone marrow-derived macrophages, Lcn-2 inhibited TEC cell cycle arrest by inhibiting peroxisome proliferator-activated receptor-γ (PPAR-γ) and enhancing epithelial markers, ultimately promoting epithelial proliferation (105). Sirtuin 6 (SIRT6) is a NAD-dependent deacetylase that is highly unstable in macrophages, and can be rapidly degraded by the proteasome (106). Several studies have reported that SIRT6 expression is downregulated in multiple models of AKI, and exerts antioxidant effects and attenuates renal injury by regulating mitochondrial dynamics and inhibiting G2/M cell cycle arrest in TECs (107, 108). Trigger receptor in myeloid cells 1(TREM-1) is a regulator of TLR signaling and is highly upregulated in renal inflammatory macrophages (109). After knockdown of TREM-1 in I/R-injured renal tubular epithelial cells, the G2/M cell cycle is blocked leading to stress-induced senescence by a mechanisms associated with mitochondrial dysfunction and a decrease in cellular metabolic activity (110).
Summary
When the kidney is subjected to injury, renal intrinsic cells and inflammatory cells in the tissues promote and restrain each other through various mechanisms, presenting an intricate and dynamic microenvironmental state that ultimately determines cell fate decisions in the kidney (Figure 1). On the one hand, TECs, podocytes, endothelial cells, and fibroblasts promote the infiltration of immune cells in the circulation, and the polarization of macrophages, which amplifies the renal inflammatory response and promotes fibrosis after AKI. On the other hand, DCs and macrophages promote various programmed death of TECs, leading to renal tubular inactivation. Meanwhile, macrophages also balance the progression of renal fibrosis by promoting EMT and inhibiting G2/M cycle arrest of TECs.The current understanding of the interactions between renal intrinsic cells and inflammatory cells in pathology of AKI is only the tip of the iceberg, and deserves to be explored more extensively by researchers. Understanding the mechanisms mediating renal and immune cell interactions will provide new ideas and more effective targets for the prevention, intervention, and treatment of AKI.
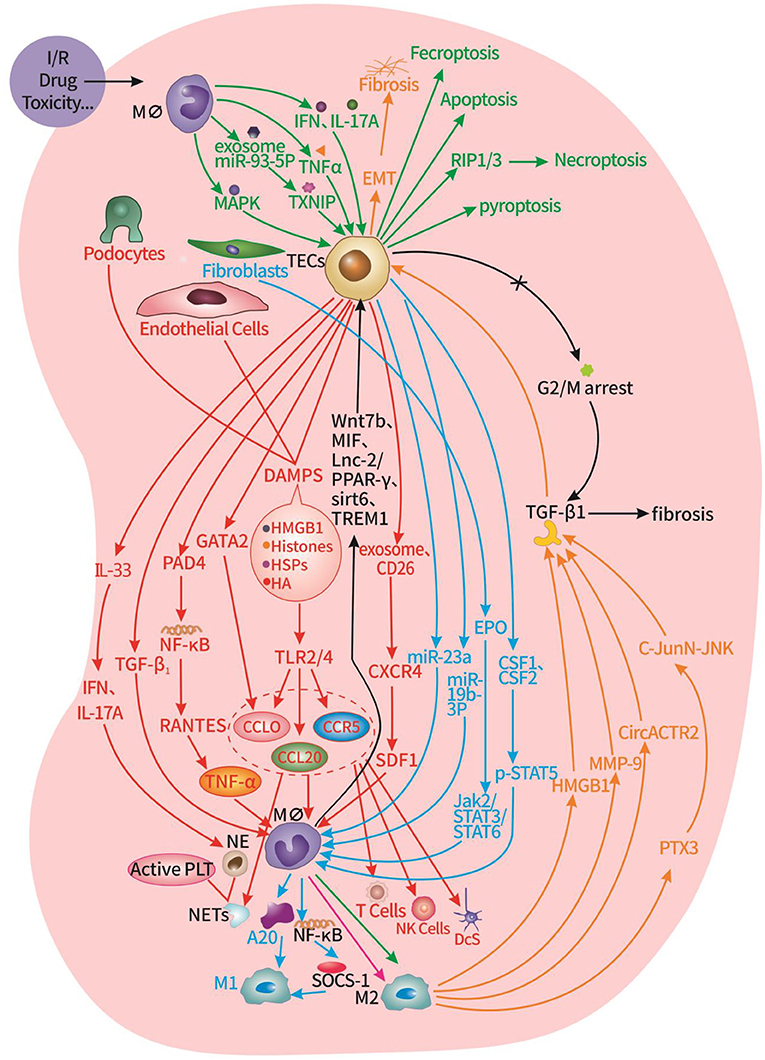
Figure 1. Interaction of innate cells and immune cells in the renal microenvironment of AKI. Red part: TECs, podocytes, endothelial cells promote the infiltration of macrophages, neutrophils, T cells, NK cells, DCs, and the formation of NETs. Blue part: TECs, fibroblasts promote the transformation of macrophages into M2 type. Green part: macrophages promote TECs apoptosis, pyroptosis, necroptosis, and ferroptosis. Orange part: macrophages promote TECs EMT. Black part: macrophages inhibit G2/M cycle arrest of TECs.
Author Contributions
JD and JY wrote the first draft. ZW, YH, LL, and WT reviewed and finalized the content of the manuscript. All authors read and approved the final version.
Funding
This research was funded by the National Natural Science Foundation of China (No. 81770682) and the Chongqing Talent Program Project (cstc2021ycjh-bgzxm0090).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Abbreviations
AKI, acute kidney injury; CKD, chronic kidney disease; CXCL1, C-X-C Motif Chemokine Ligand 1; RIP3, Receptor-Interacting Protein 3; ARG-1, arginase 1; C3, complement protein 3; CCL2, Ligand 2; CCL20, C-C Motif Chemokine Ligand 20; CCR5, C-C Motif Chemokine Receptor 5; c-JNK, c-JunN-terminal kinase; CSF, Colony stimulating factors; CTGF, connective tissue growth factor; CXCL2, C-X-C Motif Chemokine Ligand 2; CXCR4, C-X-C Motif Chemokine Receptor 4; DAMPs, danger-associated molecular patterns; DCs, dendritic cells; DT, diphtheria toxin; EMT, Epithelial mesenchymal transition; EPO, Endogenous erythropoietin; GATA2, GATA Binding Protein 2; G-CSF, granulocyte colony stimulating factor; GM-CSF, granulocyte-macrophage colony stimulating factor; HIF-1α, hypoxia-inducible factor-1α; HMGB1, high mobility histone; I/R, ischemia/reperfusion; ICAM-1, intercellular adhesion molecule 1; IFN, interferon; IGF-1, insulin-like growth factor 1; IL-1, Interleukin-1; IL-33, Interleukin-33; IL-34, Interleukin-34; iNKT cell, invariant natural killer T cell; JAK2, Janus Kinase 2; KIM-1, Kidney injury molecular 1; Lcn-2, the lipid transport protein 2; LPS, ipopolysaccharide; MAPK, Mitogen-activated protein kinase; MCP-1, monocyte chemotactic protein 1; MIF, Macrophage migration inhibitory factor; MMP, matrix metalloproteinase; NETs, neutrophil extracellular traps; PAD4, Peptidyl arginine deiminase-4; PANX1, Pannexin 1; PCD, programmed cell death; PPAR-γ, peroxisome proliferator-activated receptor-γ; PRRs, pattern recognition receptors; p-STAT-5, phosphorylated signal transducer and activator of transcription 5; PTX3, Pentraxin3; SDF1, stromal-derived factor 1; SIRT6, Sirtuin 6; SOCS-1, Suppressor Of Cytokine Signaling 1; TECs, tubular epithelial cells; TGF-β, Transforming Growth Factor β; TLR, Toll-like receptors; TNF-α, Tumor Necrosis Factor α; TREM-1, Trigger receptor in myeloid cells 1; TXNIP, thioredoxin-interacting protein; Type I interferon α, IFN-α; UUO, unilateral ureteral obstruction; VEGFA, Vascular Endothelial Growth Factor A; α-SMA, α-smooth muscle actin.
References
1. Radi ZA. Immunopathogenesis of acute kidney injury. Toxicol Pathol. (2018) 46:930–43. doi: 10.1177/0192623318799976
2. Rudman-Melnick V, Adam M, Potter A, Chokshi SM, Ma Q, Drake KA, et al. Single-cell profiling of aki in a murine model reveals novel transcriptional signatures, profibrotic phenotype, and epithelial-to-stromal crosstalk. J Am Soc Nephrol: JASN. (2020) 31:2793–814. doi: 10.1681/ASN.2020010052
3. Gerhardt LMS, Liu J, Koppitch K, Cippà PE, McMahon AP. Single-nuclear transcriptomics reveals diversity of proximal tubule cell states in a dynamic response to acute kidney injury. Proceed Nat Aca Sci USA. (2021) 118:18. doi: 10.1073/pnas.2026684118
4. Dixon EE, Wu H, Muto Y, Wilson PC, Humphreys BD. Spatially resolved transcriptomic analysis of acute kidney injury in a female murine model. J Am Soc Nephrol: JASN. (2022) 33:279–89. doi: 10.1681/ASN.2021081150
5. Ma Z, Hu X, Ding HF, Zhang M, Huo Y, Dong Z. Single-nucleus transcriptional profiling of chronic kidney disease after cisplatin nephrotoxicity. Am J Pathol. (2022) 192:613–28. doi: 10.1016/j.ajpath.2021.12.012
6. LaFavers K. Disruption of kidney-immune system crosstalk in sepsis with acute kidney injury: lessons learned from animal models and their application to human health. Int J Mol Sci. (2022) 23:702. doi: 10.3390/ijms23031702
7. Singbartl K, Formeck CL, Kellum JA. Kidney-immune system crosstalk in Aki. Semin Nephrol. (2019) 39:96–106. doi: 10.1016/j.semnephrol.2018.10.007
8. Becker M, Gnirck AC, Turner JE. Innate lymphoid cells in renal inflammation. Front Immunol. (2020) 11:72. doi: 10.3389/fimmu.2020.00072
9. Tittel AP, Heuser C, Ohliger C, Knolle PA, Engel DR, Kurts C. Kidney dendritic cells induce innate immunity against bacterial pyelonephritis. J Am Soc Nephrol: JASN. (2011) 22:1435–41. doi: 10.1681/ASN.2010101072
10. Park JG, Na M, Kim MG, Park SH, Lee HJ, Kim DK, et al. Immune cell composition in normal human kidneys. Sci Rep. (2020) 10:15678. doi: 10.1038/s41598-020-72821-x
11. Bonavia A, Singbartl K. A Review of the role of immune cells in acute kidney injury. Pedia Nephrol. (2018) 33:1629–39. doi: 10.1007/s00467-017-3774-5
12. Aslan A, van den Heuvel MC, Stegeman CA, Popa ER, Leliveld AM, Molema G, et al. Kidney histopathology in lethal human sepsis. Crit Care. (2018) 22:359. doi: 10.1186/s13054-018-2287-3
13. Janosevic D, Myslinski J, McCarthy TW, Zollman A, Syed F, Xuei X, et al. The orchestrated cellular and molecular responses of the kidney to endotoxin define a precise sepsis timeline. eLife. (2021) 10. doi: 10.7554/eLife.62270
14. Kurts C, Panzer U, Anders HJ, Rees AJ. The immune system and kidney disease: basic concepts and clinical implications. Nat Rev Immunol. (2013) 13:738–53. doi: 10.1038/nri3523
15. Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol. (2015) 11:88–101. doi: 10.1038/nrneph.2014.180
16. Black LM, Lever JM, Traylor AM, Chen B, Yang Z, Esman SK, et al. Divergent effects of aki to ckd models on inflammation and fibrosis. Am J Physiol Renal Physiol. (2018) 315:F1107–f18. doi: 10.1152/ajprenal.00179.2018
17. Summers SA, Chan J, Gan PY, Dewage L, Nozaki Y, Steinmetz OM, et al. Mast cells mediate acute kidney injury through the production of Tnf. J Am Soc Nephrol: JASN. (2011) 22:2226–36. doi: 10.1681/ASN.2011020182
18. Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol. (2010) 28:321–42. doi: 10.1146/annurev-immunol-030409-101311
19. Yamanishi Y, Kitaura J, Izawa K, Kaitani A, Komeno Y, Nakamura M, et al. Tim1 Is an endogenous ligand for Lmir5/Cd300b: Lmir5 deficiency ameliorates mouse kidney Ischemia/reperfusion injury. J Exp Med. (2010) 207:1501–11. doi: 10.1084/jem.20090581
20. Anders HJ. Toll-Like Receptors and danger signaling in kidney injury. J Am Soc Nephrol: JASN. (2010) 21:1270–4. doi: 10.1681/ASN.2010030233
21. Rosin DL, Okusa MD. Dangers within: damp responses to damage and cell death in kidney disease. J Am Soc Nephrol: JASN. (2011) 22:416–25. doi: 10.1681/ASN.2010040430
22. Hümmeke-Oppers F, Hemelaar P, Pickkers P. Innovative drugs to target renal inflammation in sepsis: alkaline phosphatase. Front Pharmacol. (2019) 10:919. doi: 10.3389/fphar.2019.00919
23. Kim HJ, Lee JS, Kim A, Koo S, Cha HJ, Han JA, et al. Tlr2 signaling in tubular epithelial cells regulates nk cell recruitment in kidney Ischemia-reperfusion injury. J Immunol. (2013) 191:2657–64. doi: 10.4049/jimmunol.1300358
24. González-Guerrero C, Morgado-Pascual JL, Cannata-Ortiz P, Ramos-Barron MA, Gómez-Alamillo C, Arias M, et al. Ccl20 Blockade increases the severity of nephrotoxic folic acid-induced acute kidney injury. J Pathol. (2018) 246:191–204. doi: 10.1002/path.5132
25. Sabapathy V, Venkatadri R, Dogan M, Sharma R. The Yin and Yang of alarmins in regulation of acute kidney injury. Front Med. (2020) 7:441. doi: 10.3389/fmed.2020.00441
26. Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, et al. Renal-associated Tlr2 mediates Ischemia/reperfusion injury in the kidney. J Clin Invest. (2005) 115:2894–903. doi: 10.1172/JCI22832
27. Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, et al. Tlr4 activation mediates kidney Ischemia/reperfusion injury. J Clin Invest. (2007) 117:2847–59. doi: 10.1172/JCI31008
28. Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hägele H, Lichtnekert J, et al. Histones from dying renal cells aggravate kidney injury via Tlr2 and Tlr4. J Am Soc Nephrol: JASN. (2012) 23:1375–88. doi: 10.1681/ASN.2011111077
29. Li L, Huang L, Ye H, Song SP, Bajwa A, Lee SJ, et al. Dendritic cells tolerized with adenosine A2Ar agonist attenuate acute kidney injury. J Clin Invest. (2012) 122:3931–42. doi: 10.1172/JCI63170
30. Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. (2011) 121:4210–21. doi: 10.1172/JCI45161
31. Nakazawa D, Kumar SV, Marschner J, Desai J, Holderied A, Rath L, et al. Histones and neutrophil extracellular traps enhance tubular necrosis and remote organ injury in Ischemic Aki. J Am Soc Nephrol: JASN. (2017) 28:1753–68. doi: 10.1681/ASN.2016080925
32. Jansen MP, Emal D, Teske GJ, Dessing MC, Florquin S, Roelofs JJ. Release of extracellular dna influences renal ischemia reperfusion injury by platelet activation and formation of neutrophil extracellular traps. Kidney Int. (2017) 91:352–64. doi: 10.1016/j.kint.2016.08.006
33. Jones JE, Causey CP, Knuckley B, Slack-Noyes JL, Thompson PR. Protein Arginine Deiminase 4 (Pad4): current understanding and future therapeutic potential. Curr Opin Drug Discov Devel. (2009) 12:616–27.
34. Raup-Konsavage WM, Wang Y, Wang WW, Feliers D, Ruan H, Reeves WB. Neutrophil Peptidyl Arginine Deiminase-4 has a pivotal role in Ischemia/reperfusion-induced acute kidney injury. Kidney Int. (2018) 93:365–74. doi: 10.1016/j.kint.2017.08.014
35. Rabadi MM, Han SJ, Kim M, D'Agati V, Lee HT. Peptidyl Arginine Deiminase-4 exacerbates Ischemic aki by finding nemo. Am J Physiol Renal Physiol. (2019) 316:F1180–f90. doi: 10.1152/ajprenal.00089.2019
36. Rabadi M, Kim M, Li H, Han SJ, Choi Y, D'Agati V, et al. Atp Induces Pad4 in renal proximal tubule cells via P2x7 receptor activation to exacerbate Ischemic Aki. Am J Physiol Renal Physiol. (2018) 314:F293–f305. doi: 10.1152/ajprenal.00364.2017
37. Markó L, Vigolo E, Hinze C, Park JK, Roël G, Balogh A, et al. Tubular epithelial Nf-Kb activity regulates Ischemic Aki. J Am Soc Nephrol: JASN. (2016) 27:2658–69. doi: 10.1681/ASN.2015070748
38. Yu TM, Palanisamy K, Sun KT, Day YJ, Shu KH, Wang IK, et al. Rantes mediates kidney Ischemia reperfusion injury through a possible role of Hif-1α and Lncrna Prins. Sci Rep. (2016) 6:18424. doi: 10.1038/srep18424
39. Gentle ME, Shi S, Daehn I, Zhang T, Qi H, Yu L, et al. Epithelial cell Tgfβ signaling induces acute tubular injury and interstitial inflammation. J Am Soc Nephrol: JASN. (2013) 24:787–99. doi: 10.1681/ASN.2012101024
40. Ferhat M, Robin A, Giraud S, Sena S, Goujon JM, Touchard G, et al. Endogenous Il-33 contributes to kidney Ischemia-reperfusion injury as an alarmin. J Am Soc Nephrol: JASN. (2018) 29:1272–88. doi: 10.1681/ASN.2017060650
41. Akcay A, Nguyen Q, He Z, Turkmen K, Won Lee D, Hernando AA, et al. Il-33 exacerbates acute kidney injury. J Am Soc Nephrol: JASN. (2011) 22:2057–67. doi: 10.1681/ASN.2010091011
42. Baek JH, Zeng R, Weinmann-Menke J, Valerius MT, Wada Y, Ajay AK, et al. Il-34 mediates acute kidney injury and worsens subsequent chronic kidney disease. J Clin Invest. (2015) 125:3198–214. doi: 10.1172/JCI81166
43. Yu L, Moriguchi T, Kaneko H, Hayashi M, Hasegawa A, Nezu M, et al. Reducing inflammatory cytokine production from renal collecting duct cells by inhibiting Gata2 ameliorates acute kidney injury. Molecular and cellular biology. (2017) 37:17. doi: 10.1128/mcb.00211-17
44. Du J, Sun Q, Wang Z, Wang F, Chen F, Wang H, et al. Tubular epithelial cells derived-exosomes containing Cd26 protects mice against renal ischemia/reperfusion injury by maintaining proliferation and dissipating inflammation. Biochem Biophys Res Commun. (2021) 553:134–40. doi: 10.1016/j.bbrc.2021.03.057
45. Lv LL, Feng Y, Wen Y, Wu WJ Ni HF, Li ZL, et al. Exosomal Ccl2 from tubular epithelial cells is critical for albumin-induced tubulointerstitial inflammation. J Am Soc Nephrol: JASN. (2018) 29:919–35. doi: 10.1681/ASN.2017050523
46. Lech M, Gröbmayr R, Ryu M, Lorenz G, Hartter I, Mulay SR, et al. Macrophage phenotype controls long-term aki outcomes–kidney regeneration versus atrophy. J Am Soc Nephrol: JASN. (2014) 25:292–304. doi: 10.1681/ASN.2013020152
47. Huen SC, Huynh L, Marlier A, Lee Y, Moeckel GW, Cantley LG. Gm-Csf promotes macrophage alternative activation after renal ischemia/reperfusion injury. J Am Soc Nephrol: JASN. (2015) 26:1334–45. doi: 10.1681/ASN.2014060612
48. Meng XM, Tang PM Li J, Lan HY. Macrophage phenotype in kidney injury and repair. Kidney Dis. (2015) 1:138–46. doi: 10.1159/000431214
49. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
50. Mosser DM, Edwards JP. Exploring the Full Spectrum of Macrophage Activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
51. Van Dyken SJ, Locksley RM. Interleukin-4- and Interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. (2013) 31:317–43. doi: 10.1146/annurev-immunol-032712-095906
52. Nakagawa M, Karim MR, Izawa T, Kuwamura M, Yamate J. Immunophenotypical Characterization of M1/M2 Macrophages and Lymphocytes in cisplatin-induced rat progressive renal fibrosis. Cells. (2021) 10:257. doi: 10.3390/cells10020257
53. Lin CC, Lu H, Wu LF, Liang Y, Chen BC, Bai YH. The role of sub-transform of macrophages in renal ischemia/reperfusion injury in rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi. (2016) 32:338–42. doi: 10.13459/j.cnki.cjap.2016.04.014
54. Li ZL, Lv LL, Tang TT, Wang B, Feng Y, Zhou LT, et al. Hif-1α inducing exosomal microrna-23a expression mediates the cross-talk between tubular epithelial cells and macrophages in tubulointerstitial inflammation. Kidney Int. (2019) 95:388–404. doi: 10.1016/j.kint.2018.09.013
55. Lv LL, Feng Y, Wu M, Wang B, Li ZL, Zhong X, et al. Exosomal Mirna-19b-3p of tubular epithelial cells promotes M1 macrophage activation in kidney injury. Cell Death Differ. (2020) 27:210–26. doi: 10.1038/s41418-019-0349-y
56. Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol: JASN. (2011) 22:317–26. doi: 10.1681/ASN.2009060615
57. Ushach I, Zlotnik A. Biological role of granulocyte macrophage colony-stimulating factor (Gm-Csf) and macrophage colony-stimulating factor (M-Csf) on cells of the myeloid lineage. J Leukoc Biol. (2016) 100:481–9. doi: 10.1189/jlb.3RU0316-144R
58. Zhang MZ, Yao B, Yang S, Jiang L, Wang S, Fan X, et al. Csf-1 signaling mediates recovery from acute kidney injury. J Clin Invest. (2012) 122:4519–32. doi: 10.1172/JCI60363
59. Wang Y, Chang J, Yao B, Niu A, Kelly E, Breeggemann MC, et al. Proximal tubule-derived colony stimulating factor-1 mediates polarization of renal macrophages and dendritic cells, and recovery in acute kidney injury. Kidney Int. (2015) 88:1274–82. doi: 10.1038/ki.2015.295
60. Li Y, Zhai P, Zheng Y, Zhang J, Kellum JA, Peng Z. Csf2 Attenuated sepsis-induced acute kidney injury by promoting alternative macrophage transition. Front Immunol. (2020) 11:1415. doi: 10.3389/fimmu.2020.01415
61. Bartnicki P, Kowalczyk M, Rysz J. The influence of the pleiotropic action of erythropoietin and its derivatives on nephroprotection. Med Sci Monit Int Med J Exp Clin Res. (2013) 19:599–605. doi: 10.12659/MSM.889023
62. Sharples EJ, Patel N, Brown P, Stewart K, Mota-Philipe H, Sheaff M, et al. Erythropoietin protects the kidney against the injury and dysfunction caused by Ischemia-reperfusion. J Am Soc Nephrol: JASN. (2004) 15:2115–24. doi: 10.1097/01.ASN.0000135059.67385.5D
63. Wang S, Zhang C, Li J, Niyazi S, Zheng L, Xu M, et al. Erythropoietin protects against rhabdomyolysis-induced acute kidney injury by modulating macrophage polarization. Cell Death Dis. (2017) 8:e2725. doi: 10.1038/cddis.2017.104
64. Wu Z, Pan J, Yang J, Zhang D. Lncrna136131 Suppresses apoptosis of renal tubular epithelial cells in acute kidney injury by targeting the Mir-378a-3p/Rab10 axis. Aging. (2022) 14:3666–86. doi: 10.18632/aging.204036
65. Deng J, Tan W, Luo Q, Lin L, Zheng L, Yang J. Long non-coding Rna Meg3 promotes renal tubular epithelial cell pyroptosis by regulating the Mir-18a-3p/Gsdmd pathway in lipopolysaccharide-induced acute kidney injury. Front Physiol. (2021) 12:663216. doi: 10.3389/fphys.2021.663216
66. Anders HJ. Necroptosis in acute kidney injury. Nephron. (2018) 139:342–8. doi: 10.1159/000489940
67. Ni L, Yuan C, Wu X. Targeting ferroptosis in acute kidney injury. Cell Death Dis. (2022) 13:182. doi: 10.1038/s41419-022-04628-9
68. Han HI, Skvarca LB, Espiritu EB, Davidson AJ, Hukriede NA. The role of macrophages during acute kidney injury: destruction and repair. Pediat Nephrol. (2019) 34:561–9. doi: 10.1007/s00467-017-3883-1
69. Novak ML, Koh TJ. Macrophage phenotypes during tissue repair. J Leukoc Biol. (2013) 93:875–81. doi: 10.1189/jlb.1012512
70. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. Rip3, an energy metabolism regulator that switches Tnf-induced cell death from apoptosis to necrosis. Science. (2009) 325:332–6. doi: 10.1126/science.1172308
71. Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, et al. A Role for Tubular necroptosis in cisplatin-induced Aki. J Am Soc Nephrol: JASN. (2015) 26:2647–58. doi: 10.1681/ASN.2014080741
72. Ke J, Zhao F, Luo Y, Deng F, Wu X. Mir-124 Negatively regulated Parp1 to alleviate renal Ischemia-reperfusion injury by inhibiting Tnfα/Rip1/Rip3 pathway. Int J Biol Sci. (2021) 17:2099–111. doi: 10.7150/ijbs.58163
73. Lechner J, Malloth N, Seppi T, Beer B, Jennings P, Pfaller W. Ifn-Alpha induces barrier destabilization and apoptosis in renal proximal tubular epithelium. Am J Physiol Cell Physiol. (2008) 294:C153–60. doi: 10.1152/ajpcell.00120.2007
74. Deng B, Lin Y, Chen Y, Ma S, Cai Q, Wang W, et al. Plasmacytoid dendritic cells promote acute kidney injury by producing Interferon-A. Cell Mol Immunol. (2021) 18:219–29. doi: 10.1038/s41423-019-0343-9
75. Naito Y, Tsuji T, Nagata S, Tsuji N, Fujikura T, Ohashi N, et al. Il-17a Activated by toll-like receptor 9 contributes to the development of septic acute kidney injury. Am J Physiol Renal Physiol. (2020) 318:F238–f47. doi: 10.1152/ajprenal.00313.2019
76. Luo CJ, Luo F, Zhang L, Xu Y, Cai GY, Fu B, et al. Knockout of Interleukin-17a protects against sepsis-associated acute kidney injury. Ann Intensive Care. (2016) 6:56. doi: 10.1186/s13613-016-0157-1
77. Juan CX, Mao Y, Cao Q, Chen Y, Zhou LB Li S, et al. Exosome-mediated pyroptosis of Mir-93-Txnip-Nlrp3 leads to functional difference between M1 and M2 macrophages in sepsis-induced acute kidney injury. J Cell Mol Med. (2021) 25:4786–99. doi: 10.1111/jcmm.16449
78. Kim EK, Choi EJ. Compromised Mapk signaling in human diseases: an update. Arch Toxicol. (2015) 89:867–82. doi: 10.1007/s00204-015-1472-2
79. Liu L, Yu H, Wu H, Yang X, Pan Y, Chen Y, et al. Toxic Proteins from Croton tiglium L. Exert a proinflammatory effect by inducing release of proinflammatory cytokines and activating the P38-Mapk signaling pathway. Mol Med Reports. (2017) 16:631–8. doi: 10.3892/mmr.2017.6617
80. Tian L, Shao X, Xie Y, Wang Q, Che X, Zhang M, et al. Kidney injury molecule-1 is elevated in nephropathy and mediates macrophage activation via the mapk signalling pathway. Cell Physiol Biochem: Int J Expl Cell Physiol, Biochem, Pharmacol. (2017) 41:769–83. doi: 10.1159/000458737
81. Li B, Liu C, Tang K, Dong X, Xue L, Su G, et al. Aquaporin-1 attenuates macrophage-mediated inflammatory responses by inhibiting P38 mitogen-activated protein kinase activation in lipopolysaccharide-induced acute kidney injury. Inflamm Res: Off J Eu Hist Res Soc. (2019) 68:1035–47. doi: 10.1007/s00011-019-01285-1
82. Su L, Jiang X, Yang C, Zhang J, Chen B, Li Y, et al. Pannexin 1 mediates ferroptosis that contributes to renal ischemia/reperfusion injury. J Biol Chem. (2019) 294:19395–404. doi: 10.1074/jbc.RA119.010949
83. Rodríguez-Romo R, Berman N, Gómez A, Bobadilla NA. Epigenetic regulation in the acute kidney injury to chronic kidney disease transition. Nephrology. (2015) 20:736–43. doi: 10.1111/nep.12521
84. Chen Z, Dong F, Lu J, Wei L, Tian L, Ge H, et al. Polarized M2c Macrophages have a promoting effect on the epithelial-to-mesenchymal transition of human renal tubular epithelial cells. Immunobiology. (2018) 223:826–33. doi: 10.1016/j.imbio.2018.08.008
85. Yu CC, Chien CT, Chang TC. M2 Macrophage polarization modulates epithelial-mesenchymal transition in cisplatin-induced tubulointerstitial fibrosis. BioMedicine. (2016) 6:5. doi: 10.7603/s40681-016-0005-5
86. Pan B, Liu G, Jiang Z, Zheng D. Regulation of renal fibrosis by macrophage polarization. Cell Physiol Biochem: Int J Exp Cell Physiol, Biochem, Pharmacol. (2015) 35:1062–9. doi: 10.1159/000373932
87. Shen B, Liu X, Fan Y, Qiu J. Macrophages regulate renal fibrosis through modulating Tgfβ superfamily signaling. Inflammation. (2014) 37:2076–84. doi: 10.1007/s10753-014-9941-y
88. Li Q, Liu BC, Lv LL, Ma KL, Zhang XL, Phillips AO. Monocytes induce proximal tubular epithelial-mesenchymal transition through Nf-Kappa B dependent upregulation of Icam-1. J Cell Biochem. (2011) 112:1585–92. doi: 10.1002/jcb.23074
89. Lynch J, Nolan S, Slattery C, Feighery R, Ryan MP, McMorrow T. High-mobility group box protein 1: a novel mediator of inflammatory-induced renal epithelial-mesenchymal transition. Am J Nephrol. (2010) 32:590–602. doi: 10.1159/000320485
90. Wang K, Wei H, Zhan J, Liang X, Zhang C, Liu Y, et al. Gspe alleviates renal fibrosis by inhibiting the activation of C3/ Hmgb1/ Tgf-B1 pathway. Chem Biol Interact. (2020) 316:108926. doi: 10.1016/j.cbi.2019.108926
91. Zhao H, Dong Y, Tian X, Tan TK, Liu Z, Zhao Y, et al. Matrix metalloproteinases contribute to kidney fibrosis in chronic kidney diseases. W J Nephrol. (2013) 2:84–9. doi: 10.5527/wjn.v2.i3.84
92. Tan TK, Zheng G, Hsu TT, Wang Y, Lee VW, Tian X, et al. Macrophage matrix metalloproteinase-9 mediates epithelial-mesenchymal transition in vitro in murine renal tubular cells. Am J Pathol. (2010) 176:1256–70. doi: 10.2353/ajpath.2010.090188
93. Tan TK, Zheng G, Hsu TT, Lee SR, Zhang J, Zhao Y, et al. Matrix metalloproteinase-9 of tubular and macrophage origin contributes to the pathogenesis of renal fibrosis via macrophage recruitment through osteopontin cleavage. Lab Invest. (2013) 93:434–49. doi: 10.1038/labinvest.2013.3
94. Hung TW, Tsai JP, Lin SH, Lee CH, Hsieh YH, Chang HR. Pentraxin 3 activates jnk signaling and regulates the epithelial-to-mesenchymal transition in renal fibrosis. Cellular Physiology Biochem: Int J Exp Cell Physiol, Biochem, Pharmacol. (2016) 40:1029–38. doi: 10.1159/000453159
95. Fu H, Gu YH, Tan J, Yang YN, Wang GH. Circactr2 in macrophages promotes renal fibrosis by activating macrophage inflammation and epithelial-mesenchymal transition of renal tubular epithelial cells. Cell Mol Life Sci: CMLS. (2022) 79:253. doi: 10.1007/s00018-022-04247-9
96. Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. (1997) 13:261–91. doi: 10.1146/annurev.cellbio.13.1.261
97. Bateman RM, Sharpe MD, Jagger JE, Ellis CG, Solé-Violán J, López-Rodríguez M, et al. 36th International Symposium on Intensive Care and Emergency Medicine: Brussels, Belgium. 15-18 March 2016. Crit Care. (2016) 20(Suppl 2):94. doi: 10.1186/s13054-016-1208-6
98. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. (2010) 16:535–43. doi: 10.1038/nm.2144
99. Moonen L, D'Haese PC, Vervaet BA. Epithelial Cell Cycle Behaviour in the Injured Kidney. Int J Mol Sci. (2018) 19:2038. doi: 10.3390/ijms19072038
100. Wang WG, Sun WX, Gao BS, Lian X, Zhou HL. Cell cycle arrest as a therapeutic target of acute kidney injury. Curr Protein Pept Sci. (2017) 18:1224–31. doi: 10.2174/1389203717666160915162238
101. Kellum JA, Chawla LS. Cell-cycle arrest and acute kidney injury: the light and the dark sides. Nephrology, dialysis, transplantation: official publication of the European dialysis and transplant association. Eu Renal Assoc. (2016) 31:16–22. doi: 10.1093/ndt/gfv130
102. Lin SL Li B, Rao S, Yeo EJ, Hudson TE, Nowlin BT, et al. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc Natl Acad Sci U S A. (2010) 107:4194–9. doi: 10.1073/pnas.0912228107
103. Urbschat A, Thiemens AK, Mertens C, Rehwald C, Meier JK, Baer PC, et al. Macrophage-Secreted Lipocalin-2 promotes regeneration of injured primary murine renal tubular epithelial cells. Int J Mol Sci. (2020) 21:38. doi: 10.3390/ijms21062038
104. Djudjaj S, Martin IV, Buhl EM, Nothofer NJ, Leng L, Piecychna M, et al. Macrophage migration inhibitory factor limits renal inflammation and fibrosis by counteracting tubular cell cycle arrest. J Am Soc Nephrol: JASN. (2017) 28:3590–604. doi: 10.1681/ASN.2017020190
105. Jung M, Brüne B, von Knethen A, Guiteras R, Cruzado JM, Hotter G, et al. Lipocalin-2 abrogates epithelial cell cycle arrest by pparγ inhibition. Lab Invest. (2018) 98:1408–22. doi: 10.1038/s41374-018-0098-4
106. Bresque M, Cal K, Pérez-Torrado V, Colman L, Rodríguez-Duarte J, Vilaseca C, et al. Sirt6 stabilization and cytoplasmic localization in macrophages regulates acute and chronic inflammation in mice. J Biol Chem. (2022) 298:101711. doi: 10.1016/j.jbc.2022.101711
107. Gao Z, Chen X, Fan Y, Zhu K, Shi M, Ding G. Sirt6 attenuates hypoxia-induced tubular epithelial cell injury via targeting G2/M phase arrest. J Cell Physiol. (2020) 235:3463–73. doi: 10.1002/jcp.29235
108. Yang X, Feng J, Liang W, Zhu Z, Chen Z, Hu J, et al. Roles of Sirt6 in kidney disease: a novel therapeutic target. Cell Mol Life Sci: CMLS. (2021) 79:53. doi: 10.1007/s00018-021-04061-9
109. Lo TH, Tseng KY, Tsao WS, Yang CY, Hsieh SL, Chiu AW, et al. Trem-1 regulates macrophage polarization in ureteral obstruction. Kidney Int. (2014) 86:1174–86. doi: 10.1038/ki.2014.205
Keywords: acute kidney injury, intrinsic renal cells, immune cells, interaction, microenvironment
Citation: Deng J, Wu Z, He Y, Lin L, Tan W and Yang J (2022) Interaction Between Intrinsic Renal Cells and Immune Cells in the Progression of Acute Kidney Injury. Front. Med. 9:954574. doi: 10.3389/fmed.2022.954574
Received: 27 May 2022; Accepted: 15 June 2022;
Published: 07 July 2022.
Edited by:
Dong Zhou, University of Connecticut, United StatesReviewed by:
Lin-Li Lv, Southeast University, ChinaYuanyuan Wang, Guizhou Medical University, China
Copyright © 2022 Deng, Wu, He, Lin, Tan and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jurong Yang, NjUwMjMwQGhvc3BpdGFsLmNxbXUuZWR1LmNu; eWpyOTIzQDE2My5jb20=