
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Med. , 22 July 2022
Sec. Nephrology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.937122
This article is part of the Research Topic Integrated Management of Chronic Kidney Disease Patients View all 24 articles
 Qiongxiu Zhou1,2†
Qiongxiu Zhou1,2† Qinjie Weng1†
Qinjie Weng1† Xiaoyan Zhang1
Xiaoyan Zhang1 Yunzi Liu1Jun Tong1Xu Hao1
Yunzi Liu1Jun Tong1Xu Hao1 Hao Shi1Pingyan Shen1Hong Ren1
Hao Shi1Pingyan Shen1Hong Ren1 Jingyuan Xie1*
Jingyuan Xie1* Nan Chen1*
Nan Chen1*Aim: NPHS2 is the coding gene of podocin. This study aims to investigate the association between NPHS2 p.R229Q (rs61747728), the most frequently reported missense variant of NPHS2, and focal segmental glomerular sclerosis (FSGS) or steroid-resistant nephrotic syndrome (SRNS) based on typing the variant in a Chinese FSGS/SRNS cohort and conducting a meta-analysis.
Method: We recruited patients with FSGS or SRNS and healthy individuals. To conduct a meta-analysis, all studies on p.R229Q and FSGS/SRNS were searched from public databases.
Results: In total, we enrolled 204 patients with FSGS, 61 patients with SRNS [46 with FSGS, 9 with minimal change disease (MCD), and six patients with IgA nephropathy (IgAN)], and 100 healthy controls. Unexpectedly, p.R229Q was absent in the patients from our cohort. By meta-analysis of 21 studies including 2,489 patients with FSGS/SRNS and 6,004 healthy controls, we confirmed that the A allele of p.R229Q was significantly associated with increased risk of FSGS/SRNS (allelic OR = 1.9, 95% CI = 1.44-2.52, P < 0.001). However, the subgroup analysis showed that the association between p.R229Q and FSGS/SRNS was true only in Caucasians (allelic OR = 2.14, 95%CI = 1.54-2.98, P < 0.001) and in early-onset patients (allelic OR: 2.13, 95% CI = 1.21-3.76, P = 0.009).
Conclusion: NPHS2 p.R229Q may play an important role in enhancing the susceptibility of FSGS/SRNS, especially in ethnicity of Caucasian and age of early-onset patients.
Idiopathic nephrotic syndrome (INS) is clinically characterized by edema, massive proteinuria, hypoalbuminemia, and hyperlipidemia. Although steroid is widely used for treatment of INS, about 10-20% of children (1) and 50% of adults patients (2) have steroid-resistant nephrotic syndrome (SRNS). Among different pathologic types of SRNS, focal segmental glomerulosclerosis (FSGS) is the most common cause in children (3), accounting for 60-70% (4, 5), and it also frequently occurs in adults (6).
Over the past decades, dozens of podocyte-related genes had been identified in several monogenic forms of hereditary FSGS/SRNS (7–9). Among these genes, mutations in podocin (NPHS2) had been found to play a significant role in SRNS, consisting of approximately 20 to 40% of familial and 10% of idiopathic childhood SRNS in different regions of the world (10–13). Podocin is composed of 383 amino acids localizing specifically from the insertion of the slit diaphragm (SD) to the podocyte cytoplasm (7, 14). It is required for the structural organization and regulation of the glomerular filtration barrier by interacting with other important SD molecules such as nephrin and CD2-associated protein (CD2AP) (15–18). rs61747728 (p.R229Q, G686A) is one of the most commonly reported variants in podocin. A study conducted by Tsukaguchi et al. (19) found that p.R229Q caused a decrease in the ability of podocin to bind to nephrin and was usually associated with secondary NPHS2 mutation, which enhanced the susceptibility to develop FSGS. Meanwhile, some studies reported that heterozygous p.R229Q polymorphisms were associated with SRNS when compared to a healthy population (20, 21). Furthermore, p.R229Q-podocin was found to be related with microalbuminuria in the general population (10). In contrast with these findings, p.R229Q was found not associated with SRNS or urinary albumin-creatinine ratio (ACR) in some other studies performed on White or Black individuals (10, 20, 22). These controversial findings might be due to underpowered study design or different study populations. In addition, few studies were performed on Asian population although the prevalence of SRNS was relatively higher in Asians and African Americans (3, 23, 24). The aim of this study was to investigate the association between p.R229Q and FSGS/SRNS. We first conducted a screening study on our cohort composed of adult patients who were of Chinese descent and with FSGS/SRNS. To enhance the power of this study, we performed a meta-analysis by pooling data driven from our cohort and from bodies of literature obtained from public databases.
Inclusion criteria were as follows: (1) age between 18 to 65 years old, (2) newly diagnosed nephrotic syndrome without previous usage of immunosuppressive agents within 1 month before the recruitment, and 3) renal biopsy-proved FSGS, minimal change disease (MCD), or IgA nephropathy (IgAN). We included patients with IgAN treated with full-dose prednisone as their initial therapy. SRNS was defined as less than 50%reduction in urine protein compared to baseline or urine protein higher than 3.5 g/d after treatment with a full dose of prednisone (1 mg/kg, maximum 80 mg/day) for at least 12 weeks. Healthy controls were defined as having serum creatinine bellow 100 umol/L without proteinuria or hematuria by urine routine test. All the individuals recruited in this study self-reported as having a Chinese Han origin. Informed consent was obtained from all the study participants before enrollment. The study protocol was reviewed and approved by Ruijin Hospital Human Research Ethics Committee.
Genomic DNA was extracted and purified from peripheral leukocytes in peripheral blood cells of all the included subjects. All the eight coding regions, exon-intron boundaries, 5′UTR, and 3′UTR of NPHS2 were amplified by polymerase chain reaction (PCR) (7). Primers were designed with the primier5 software or based on published information (19) (Supplementary Table 1). The PCR products were sequenced by Invitrogen. Sequence chromatograms were analyzed with the SEQUENCHER™ software (Gene Code Corp., Ann Arbor, MI, United States) by comparing with the reference sequence of NPHS2 downloaded from NCBI1.
Relevant studies published until 1 March 2021 were searched through electronic databases of PubMed, SCOPUS, Cochrane Library, and Web of Science and using the search terms “R229Q,” “NPHS2,” “rs61747728,” “686G > A,” and “Arg229Gln.” Additional studies were also searched by reviewing the references cited in the retrieved articles.
Studies eligible for inclusion in our meta-analysis had to fulfill the following criteria: (1) studies discussing about the association between p.R229Q and SRNS (or FSGS) (e.g., SRNS group vs. control group or FSGS group vs. control group), (2) original data of genotype frequencies available, (3) SRNS was defined as patients who do not achieve complete remission within 12 weeks of glucocorticoid treatment, (4) early onset was defined as onset age ≤18 years, late onset was defined as onset age >18 years. Studies were excluded from our analysis if: (1) there was no control group, (2) genotype data were not available, (3) they were a duplicate of previous publication, and (4) they were reviews, editorials, or unpublished reports.
All the data were extracted independently by two reviewers (Qiongxiu Zhou and Xiaoyan Zhang) according to a standard protocol. The following information was extracted from the eligible studies: first author’s surname, year of publication, ethnicity of subjects, sample size, age, and distribution of p.R229Q genotype and allele frequencies in case and control groups.
In the current meta-analysis, the associations between p.R229Q and FSGS/SRNS was analyzed by calculating the pooled odds ratios (ORs) and their 95% confidence intervals (CIs) using a random-effects model. Three genetic models were used for the association study: the allelic model (A vs. G), the dominant model (GA + AA vs. GG), and the recessive model (AA vs. GG + GA) (25, 26). Subgroup analyses were conducted according to subject ethnicity and age of onset.
Hardy-Weinberg Equilibrium (HWE) was tested by chi-square test (χ2) for goodness of fit to compare the observed and expected genotype frequencies with controls using a previous meta-analysis as reference, and a P-value < 0.01 was considered as significant deviation from HWE. As deviation from HWE in subjects may bias the estimates of genetic effects in a meta-analysis, a sensitivity analysis was conducted by removing one study and recalculating the pooled OR and 95% CI to assess the stability of the results. Potential publication bias was estimated by Begg’s funnel plot. All statistical tests were performed with the STATA 11.0 software (Stata Corp., College Station, TX, United States). Two-sided P < 0.05 was considered statistically significant.
In total, there were 61 patients with SRNS and 204 patients with FSGS recruited in the screening study. Among the 61 patients with SRNS, 46 (75%) had FSGS, 9 (15%) had MCD and 6 (10%) had IgAN; 4 of the 6 patients with IgAN had FSGS changes in renal pathologic findings. The baseline characteristics of patients with SRNS and FSGS enrolled are shown in Supplementary Table 2. We also recruited 100 healthy individuals as a control group. Four synonymous single nucleotide polymorphisms (SNPs), c.187A > G (p.G34G) in exon1, c.288C > T (p.S96S) in exon2, and c.954C > T (p.A138A) and c.1038A > G (p.L346L) in exon8 of NPHS2, were identified by Sanger sequencing among all the individuals recruited in the screening study. However, p.R229Q was absent in all the patients with SRNS and FSGS.
There were 21 studies investigating the association between p.R229Q and SRNS/FSGS, including the present study. A flow chart shows the literature search for relevant studies on the association between p.R229Q and SRNS/FSGS (Supplementary Figure 1). The studies contained 2,489 cases and 6,004 controls with 12 comparisons on Caucasians, 6 on Asians, and 3 on Africans. Characteristics of these studies evaluating the association between R229Q polymorphism and SRNS/FSGS risk are shown in Table 1. The frequency distributions of genotypes were consistent with HWE.
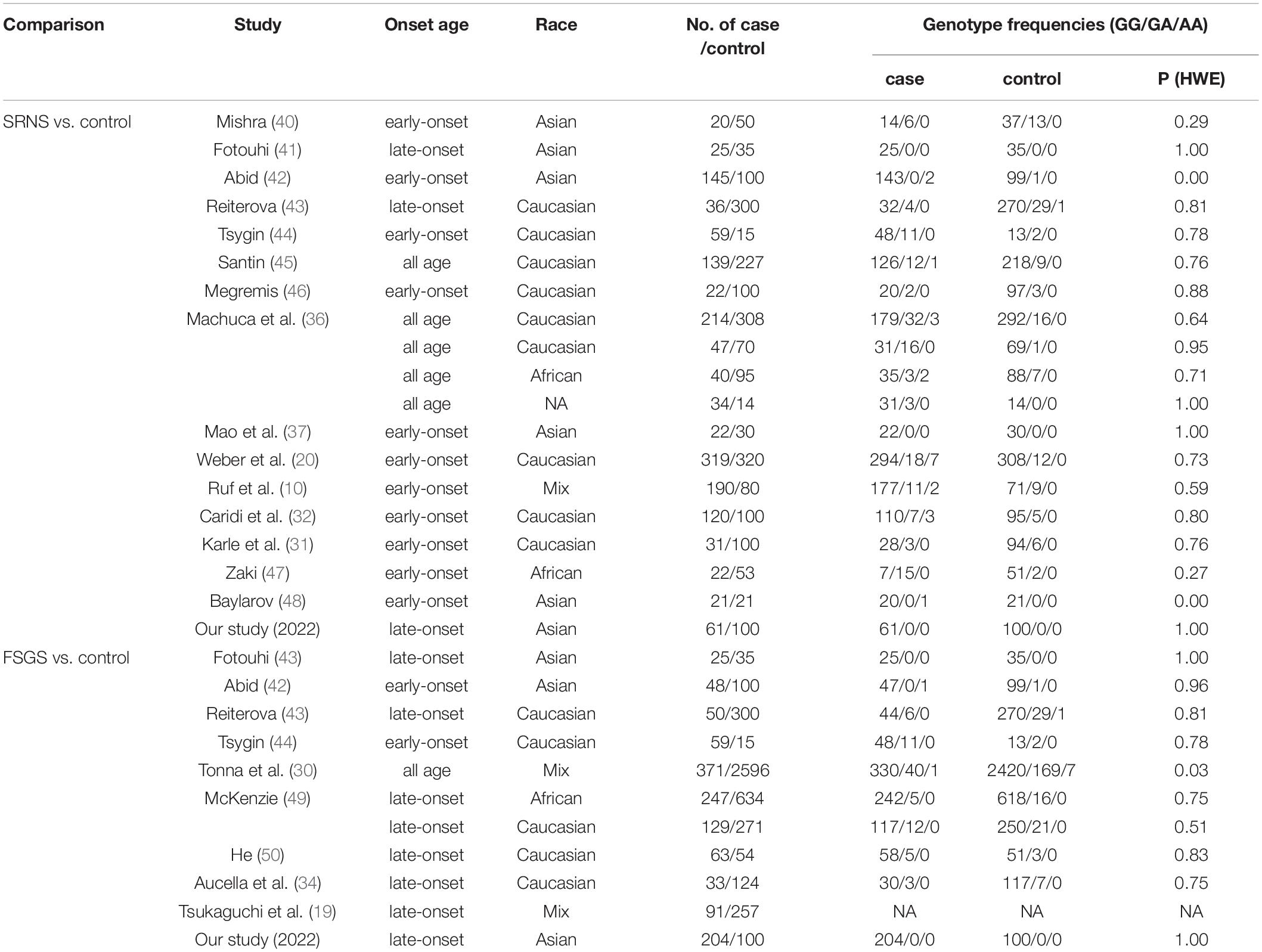
Table 1. Characteristics of studies evaluating the association between R229Q polymorphism and SRNS/FSGS risk.
The minor allele frequency (MAF) (A allele) of p.R229Q was.06 in the patients with SRNS/FSGS and.03 in the healthy controls. An increased risk for p.R229Q in SRNS/FSGS was confirmed by the allelic model (OR = 1.9, 95% CI 1.44-2.52, P < 0.001, Figure 1), the recessive model (OR = 3.9, 95% CI 1.56-9.77, P = 0.004, Supplementary Figure 2) and the dominant model (OR = 1.91, 95% CI 1.37-2.73, P < 0.001, Supplementary Figure 3).
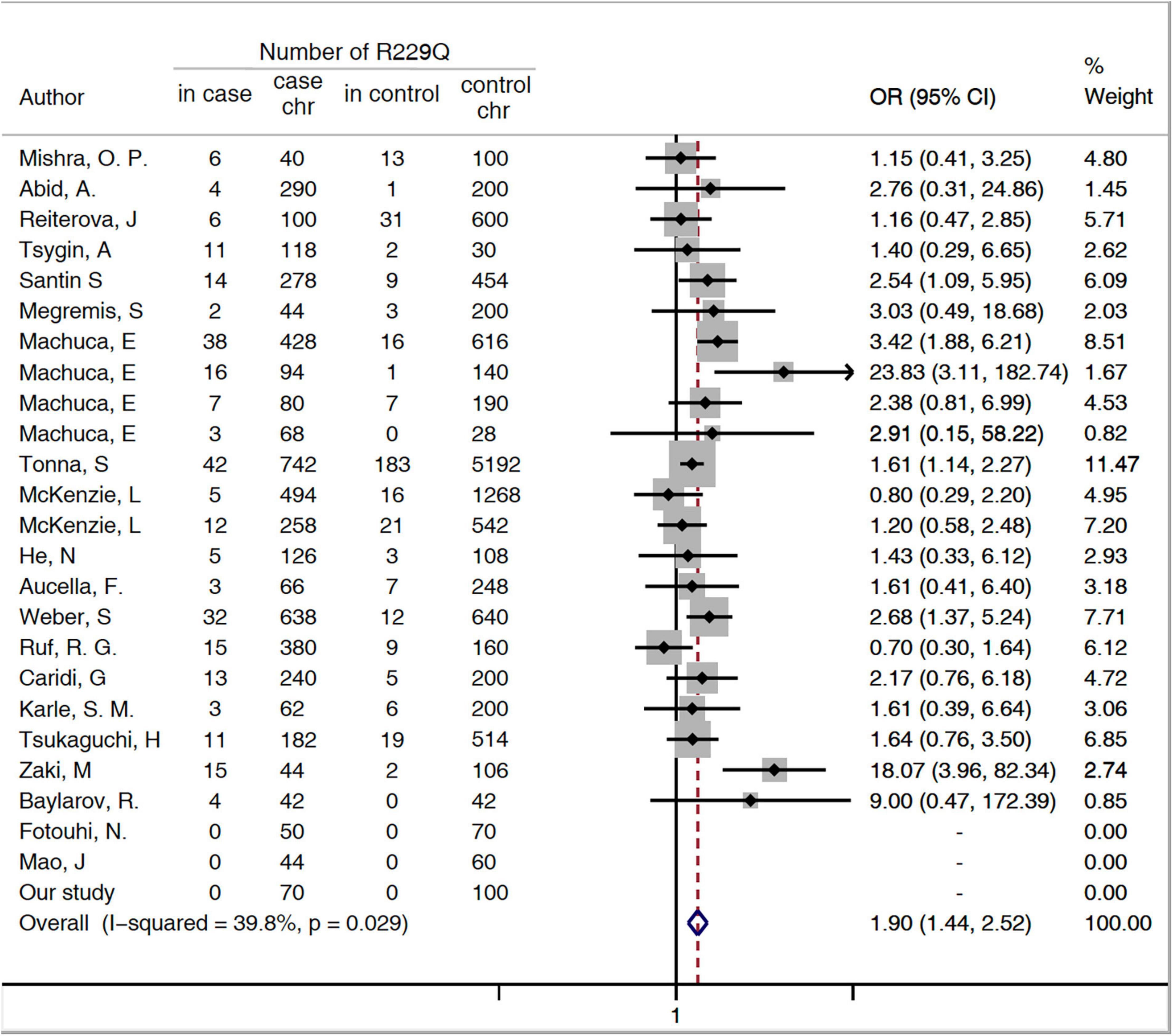
Figure 1. Forest plots of meta-analysis of association between p.R229Q and FSGS/SRNS in the allelic model. Chr, chromosome; CI, confidence interval; OR, odds risk; SRNS, steroid-resistant nephrotic syndrome; and FSGS, focal segmental glomerular sclerosis.
A subgroup analysis was performed based on disease phenotype: SRNS or FSGS. A significant increased risk of SRNS and FSGS in patients with R229Q was determined based on the allelic model (OR = 2.6, 95% CI 1.68-4.03, P = 0.024 and OR = 1.44, 1.12-1.85, 95%CI P <0.001, respectively, Figure 2). When all the patients were stratified by ethnicity, significant risks were found among Caucasians either in the allele-based analysis (OR = 2.14, 95% CI 1.54-2.98, P < 0.001, Figure 3) or in the genotype-based analysis (dominant model: OR = 2.11, 95% CI: 1.48-3, P < 0.001; recessive model: OR = 6.56, 95% CI 1.69-25.5, P = 0.007). However, no association was found in the African and Asian populations. In the subgroups stratified by onset age, the p.R229Q variant increased the risk of SRNS among the early-onset patients in the allelic (OR: 2.13, 95% CI = 1.21-3.76, P = 0.009, Figure 2), recessive (OR = 4.85, 95% CI = 1.25-18.81, P = 0.02), and dominant (OR: 2.09, 95% CI = 1.04-4.21, P = 0.04) models, while no significant association was found in all the three genetic models among the late-onset patients.
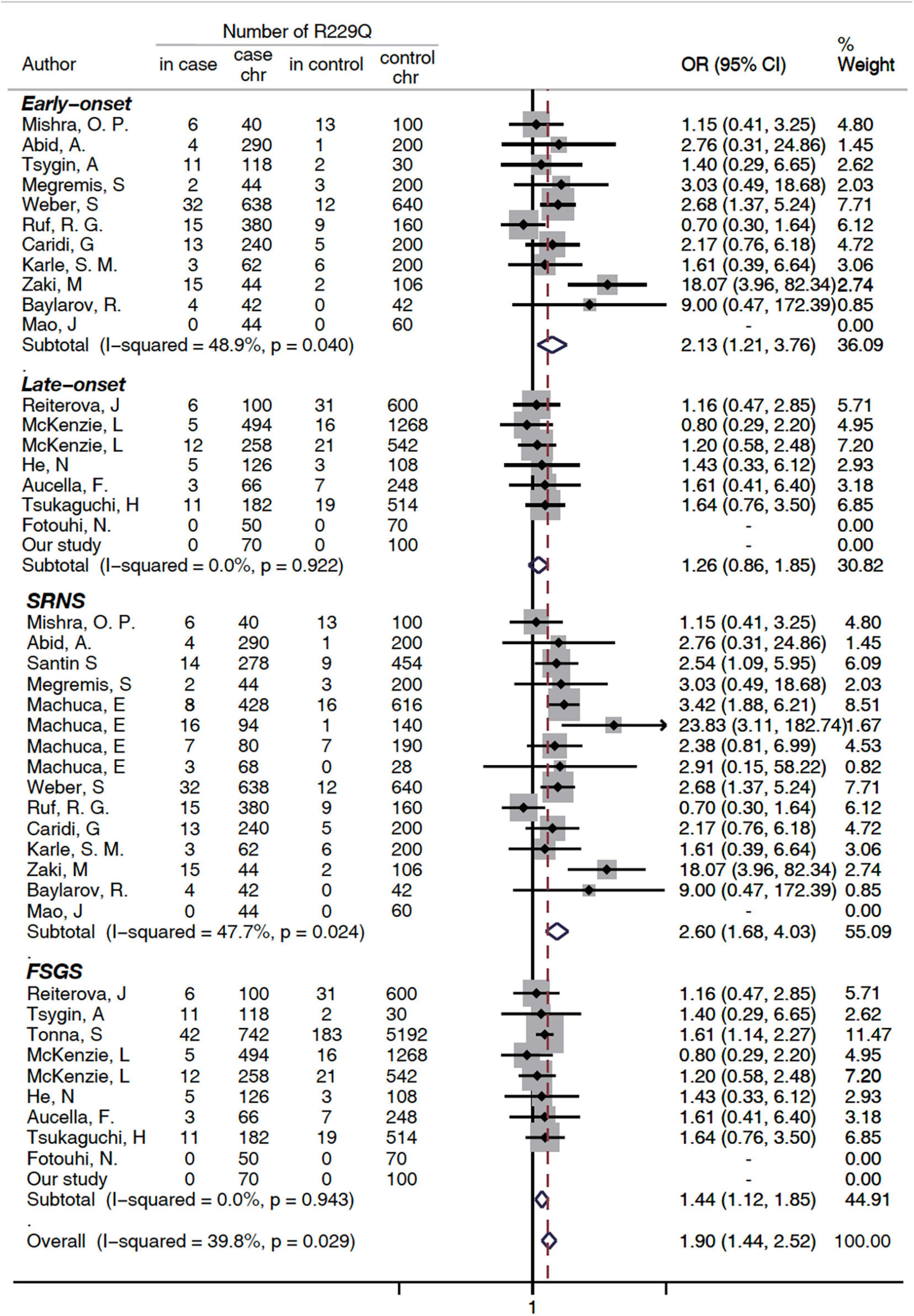
Figure 2. Subgroup analysis of association between FSGS/SRNS and the p.R229Q variant based on age of onset and disease types (allelic model). CI, confidence interval; OR, odds risk; chr, chromosome; SRNS, steroid-resistant nephrotic syndrome; and FSGS, focal segmental glomerular sclerosis. SRNS, steroid-resistant nephrotic syndrome; FSGS, focal segmental glomerular sclerosis; HWE, Hardy Weinberg Equilibrium; NA, not available (i.e., not stated).
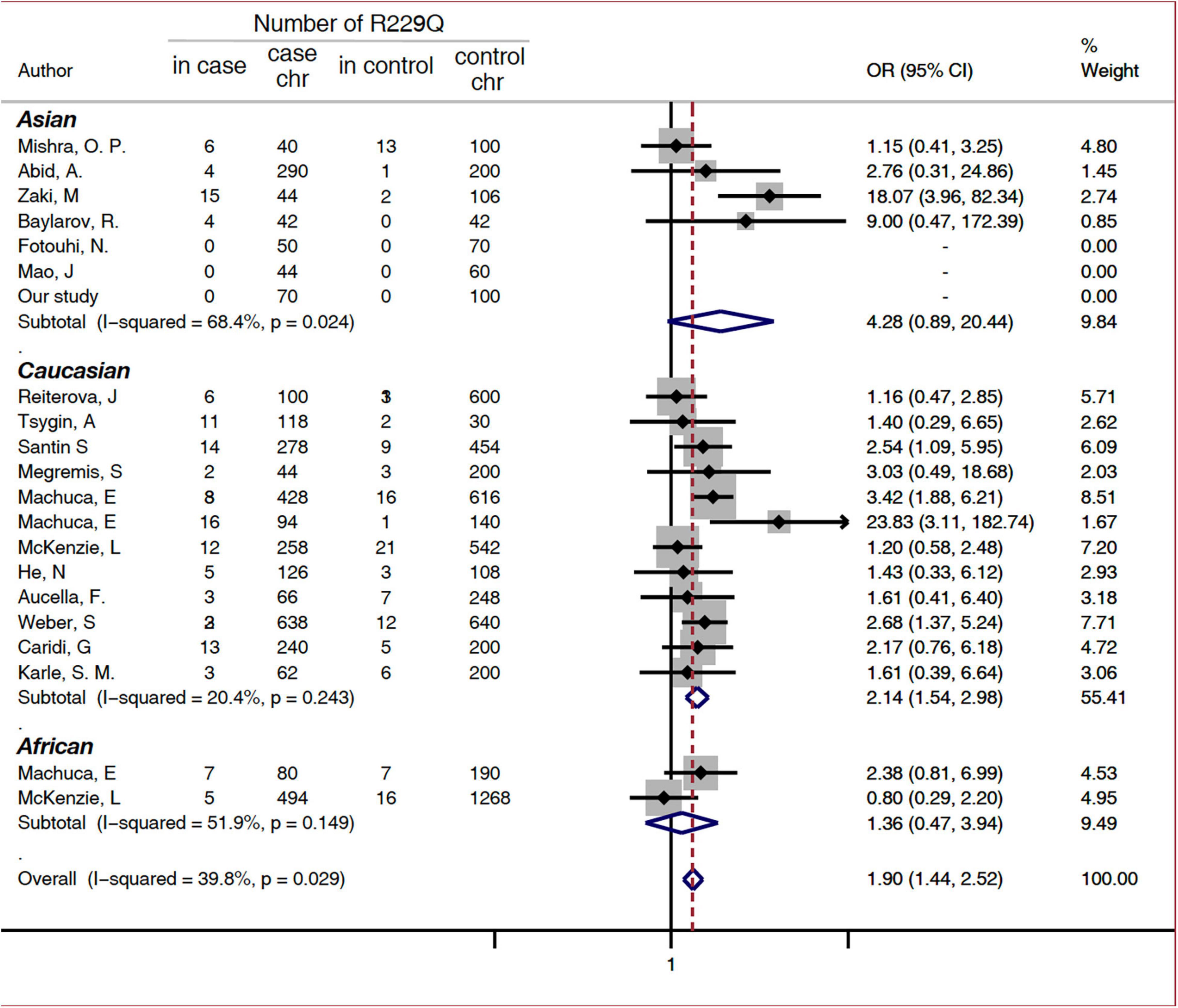
Figure 3. Subgroup analysis of association between FSGS/SRNS and the p.R229Q variant based on ethnicity (allelic model). CI, confidence interval; OR, odds risk; chr, chromosome; SRNS, steroid-resistant nephrotic syndrome; and FSGS, focal segmental glomerular sclerosis.
In this meta-analysis, significant heterogeneity (I2 = 52.1%, P = 0.003) was observed in the included dominant model, so a sensitivity analysis performed. The combined ORs were similar with one another, with a narrow range from 1.72 (95% CI: 1.32-2.26) to 2.08 (95%CI: 1.49-2.91) (Supplementary Figure 4). It indicated that our results were stable.
The publication bias of the individual studies was evaluated by Begg’s test. No significant difference was found (P = 0.15). It indicated that there was no significant publication bias in our meta-analysis (Supplementary Figure 5).
NPHS2 mutations were initially described in patients with NS from birth to 6 years of age (20, 27), while they were infrequently detected in children with non-familial SRNS (28, 29). A study on adults with FSGS/SRNS has not been fully performed. In our study, we examined the frequency of NPHS2 mutation in a cohort of adult Chinese patients with SRNS or FSGS. Four known synonymous variations (G34G, S96S, A318A, and L346L) were identified. However, the distribution of genotypes among our patients and the controls were similar, which suggested that the SNPs did not increase the risk of SRNS or FSGS. We did not find non-synonymous variations in NPHS2, and no p.R229Q was observed in patients with either SRNS or FSGS. It suggested that adult-onset NPHS2 mutation was rare in Chinese patients, and that p.R229Q had no effect on the patients, at least in our population.
In the current meta-analysis, we found that the frequency of the p.R229Q variant was significantly higher in the patients with SRNS/FSGS than in the healthy populations in the allelic (OR = 1.9, 95% CI 1.44-2.52), dominant (OR = 1.91, 95% CI 1.37-2.73), and recessive (OR = 3. 0, 95% CI 1.56-9.77) models. Furthermore, it was significant in the subgroups of either SRNS (OR 2.6, P = 0.024) or FSGS (OR 1.44, P <0.001). As FSGS was a common cause of SRNS in both children and adults, we could hypothesize that p.R229Q plays a pathogenic role in FSGS and SRNS. These findings were consistent with previous studies, suggesting that the p.R229Q allele may be a disease-causing variant, which could enhance the susceptibility of FSGS, and FSGS patients with podocin mutations were more likely to be p.R229Q in heterozygous state with one pathogenic mutation (10, 19, 20, 30–34).
The results from our meta-analysis were different from Lu Lu’s study (35), which reported no association between p.R229Q and FSGS. First, we recruited more cases, with 2,489 patients with SRNS/FSGS and 6,004 controls, and there were more comparisons on different ethnicity, with 12 comparisons on Caucasians, 6 on Asians, and 3 on Africans. Secondly, Lu L’s study excluded the studies that all variant individuals are compound heterozygotes, considering that the excessive possible affecting SNPs in the NPHS2 gene related to FSGS was difficult to identify. We agree with this concern. Therefore, in our study, we constructed genetic models, the allelic (A vs. G), dominant (GA + AA vs. GG), and recessive (AA vs. GG + GA) models, to explain the allele frequency and genotype-phenotype correlation of p.R229Q in these studies. We concluded that p.R229Q might play a pathogenic role in developing SRNS/FSGS in the state of compound heterozygotes (36).
Additionally, we found that the frequency of p.R229Q was various throughout different populations. The frequency was higher in Caucasians than in Asians and Africans (36). For the Caucasians, p.R229Q increased the risk of FSGS/SRNS (allelic model: OR = 2.14, 95% CI 1.54-6.64; dominant model: OR = 2.11, 95% CI 1.48-3; recessive model: OR = 3.9, 95% CI 1.56-9.76). However, the associations with other populations were not significant. In our screening study, we did not find the p.R229Q variant in Chinese patients with adult-onset SRNS and FSGS, which was in agreement with a previous study conducted on Chinese patients with childhood-onset SRNS (37). This suggested that p.R229Q allele distribution in different races was uneven, although one explanation for this discrepancy might be that relatively small studies and low frequencies of p.R229Q in these populations could have limited the statistical power for analysis. Besides, significant results were also observed in patients with early-onset FSGS/SRNS in the allelic (OR: 2.13, 95% CI 1.21-3.76), dominant (OR: 2.11, 95% CI 1.48-3) and recessive (OR: 6.56, 95% CI 1.69-25.5) models compared with the healthy controls. We could hypothesize that p.R229Q was more likely to act as a disease modifier to increase the risk for patients with early-onset FSGS/SRNS.
Even though many studies and meta-analyses had been conducted on the genetic role of the p.R229Q variant in the previous years, most of them were performed on Caucasians. Limited studies were available on African and Asian populations. This limited our ability to reach a strong conclusion and investigate a potential function on the basis of race. Moreover, p.R229Q was recently discovered pathogenic only when trans-associated to specific mutations, and this could not be analyzed by the current meta-analysis (38, 39).
In conclusion, our study indicated that NPHS2 mutations were rare in Asian patients with sporadic adult-onset FSGS/SRNS and p.R229Q was undetectable in our cohort. NPHS2 p.R229Q may play an important role in enhancing susceptibility to FSGS/SRNS, especially in Caucasian and early-onset patients. Further studies and international multi-ethnicity approaches are needed to distinguish a pathogenic and benign p.R229Q genotype-phenotype correlation for clinical assessment.
The original contributions presented in this study are publicly available. This data can be found here: GenBank, accessions ON470453 - ON470576.
The studies involving human participants were reviewed and approved by Ruijin Hospital Human Research Ethics Committees. The patients/participants provided their written informed consent to participate in this study.
QZ and XZ completed the screening test and extracted all the data that were needed for the meta-analysis. QZ and QW drafted the original manuscript. YL, JT, and XH helped in polishing the manuscript and were responsible for the figures. JX and NC created the overall design of this study. HS, PS, and HR revised the article. All authors contributed to the article and approved the submitted version.
This study was supported by grants from the National Key Research and Development Program of China (2016YFC0904100), National Natural Science Foundation of China (Grant Nos: 81870460, 81570598, and 81370015), Science and Technology Innovation Action Plan of Shanghai Science and Technology Committee (Grant No: 17441902200), Shanghai Municipal Education Commission, Gaofeng Clinical Medicine Grant (Grant No: 20152207), Shanghai Jiao Tong University School of Medicine, Multi-Center Clinical Research Project (Grant No: DLY201510), and Shanghai Health and Family Planning Committee Hundred Talents Program (Grant No: 2018BR37).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer FL declared a shared parent affiliation with the authors JX, QW, JT, XH, HS, XZ, YL, PS, HR, and NC to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the Department of Nephrology of Ruijin Hospital for providing the clinical data for this study. We would also like to thank Jian Liu and Yuanmeng Jin for helping us get in touch with patients.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.937122/full#supplementary-material
1. Mekahli D, Liutkus A, Ranchin B, Yu A, Bessenay L, Girardin E, et al. Long-term outcome of idiopathic steroid-resistant nephrotic syndrome: a multicenter study. Pediatr Nephrol. (2009) 24:1525–32. doi: 10.1007/s00467-009-1138-5
2. Korbet SM. Treatment of primary focal segmental glomerulosclerosis. Kidney Int. (2002) 62:2301–10. doi: 10.1046/j.1523-1755.2002.00674.x
3. Kim JS, Bellew CA, Silverstein DM, Aviles DH, Boineau FG, Vehaskari VM. High incidence of initial and late steroid resistance in childhood nephrotic syndrome. Kidney Int. (2005) 68:1275–81. doi: 10.1111/j.1523-1755.2005.00524.x
4. D’Agati VD. Pathobiology of focal segmental glomerulosclerosis: new developments. Curr Opin Nephrol Hypertens. (2012) 21:243–50. doi: 10.1097/MNH.0b013e32835200df
5. Chesney R. The changing face of childhood nephrotic syndrome. Kidney Int. (2004) 66:1294–302. doi: 10.1111/j.1523-1755.2004.00885.x
6. Haas M, Meehan SM, Karrison TG, Spargo BH. Changing etiologies of unexplained adult nephrotic syndrome: a comparison of renal biopsy findings from 1976-1979 and 1995-1997. Am J Kidney. (1997) 30:621–31. doi: 10.1016/s0272-6386(97)90485-6
7. Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. (2000) 24:349–54. doi: 10.1038/74166
8. Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. (2000) 24:251–6. doi: 10.1038/73456
9. Kim JM, Wu H, Green G, Winkler CA, Kopp JB, Miner JH, et al. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science. (2003) 300:1298–300. doi: 10.1126/science.1081068
10. Ruf RG, Lichtenberger A, Karle SM, Haas JP, Anacleto FE, Schultheiss M, et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J Am Soc Nephrol. (2004) 15:722–32. doi: 10.1097/01.ASN.0000113552.59155.72
11. Caridi G, Bertelli R, Carrea A, Di Duca M, Catarsi P, Artero M, et al. Prevalence, genetics, and clinical features of patients carrying podocin mutations in steroid-resistant nonfamilial focal segmental glomerulosclerosis. J Am Soc Nephrol. (2001) 12:2742–6. doi: 10.1681/ASN.V12122742
12. Pollak MR. The genetic basis of FSGS and steroid-resistant nephrosis. Semin Nephrol. (2003) 23:141–6. doi: 10.1053/snep.2003.50014
13. Niaudet P. Podocin and nephrotic syndrome: implications for the clinician. J Am Soc Nephrol. (2004) 15:832–4. doi: 10.1097/01.asn.0000118135.00519.b0
14. Roselli S, Gribouval O, Boute N, Sich M, Benessy F, Attié T, et al. Podocin localizes in the kidney to the slit diaphragm area. Am J Pathol. (2002) 160:131–9. doi: 10.1016/S0002-9440(10)64357-X
15. Huber TB, Schermer B, Muller RU, Bartram M, Calixto A, Hagmann H, et al. Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc Natl Acad Sci U.S.A. (2006) 103:17079–86. doi: 10.1073/pnas.0607465103
16. Huber TB, Schermer B, Benzing T. Podocin organizes ion channel-lipid supercomplexes: implications for mechanosensation at the slit diaphragm. Nephron Exp Nephrol. (2007) 106:e27–31. doi: 10.1159/000101789
17. Schwarz K, Simons M, Reiser J, Saleem MA, Faul C, Kriz W, et al. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest. (2001) 108:1621–9. doi: 10.1172/JCI12849
18. Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. (2005) 308:1801–4. doi: 10.1126/science.1106215
19. Tsukaguchi H, Sudhakar A, Le TC, Nguyen T, Yao J, Schwimmer JA, et al. NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele. J Clin Invest. (2002) 110:1659–66. doi: 10.1172/JCI16242
20. Weber S, Gribouval O, Esquivel EL, Morinière V, Tête MJ, Legendre C, et al. NPHS2 mutation analysis shows genetic heterogeneity of steroid-resistant nephrotic syndrome and low post-transplant recurrence. Kidney Int. (2004) 66:571–9. doi: 10.1111/j.1523-1755.2004.00776.x
21. Franceschini N, North KE, Kopp JB, McKenzie L, Winkler C. NPHS2 gene, nephrotic syndrome and focal segmental glomerulosclerosis: a HuGE review. Genet Med. (2006) 8:63–75. doi: 10.1097/01.gim.0000200947.09626.1c
22. Köttgen A, Hsu CC, Coresh J, Shuldiner AR, Berthier-Schaad Y, Gambhir TR, et al. The association of podocin R229Q polymorphism with increased albuminuria or reduced estimated GFR in a large population-based sample of US adults. Am J Kidney Dis. (2008) 52:868–75. doi: 10.1053/j.ajkd.2008.02.306
23. Sorof JM, Hawkins EP, Brewer ED, Boydstun II, Kale AS, Powell DR. Age and ethnicity affect the risk and outcome of focal segmental glomerulosclerosis. Pediatr Nephrol. (1998) 12:764–8. doi: 10.1007/s004670050542
24. Bonilla-Felix M, Parra C, Dajani T, Ferris M, Swinford RD, Portman RJ, et al. Changing patterns in the histopathology of idiopathic nephrotic syndrome in children. Kidney Int. (1999) 55:1885–90. doi: 10.1046/j.1523-1755.1999.00408.x
25. Minelli C, Thompson JR, Abrams KR, Thakkinstian A, Attia J. The choice of a genetic model in the meta-analysis of molecular association studies. Int J Epidemiol. (2005) 34:1319–28. doi: 10.1093/ije/dyi169
26. Bush WS, Moore JH. Chapter 11: genome-wide association studies. PLoS Comput Biol. (2012) 8:e1002822. doi: 10.1371/journal.pcbi.1002822
27. Hinkes B, Vlangos C, Heeringa S, Mucha B, Gbadegesin R, Liu J, et al. Specific podocin mutations correlate with age of onset in steroid-resistant nephrotic syndrome. J Am Soc Nephrol. (2008) 19:365–71. doi: 10.1681/ASN.2007040452
28. Chernin G, Heeringa SF, Gbadegesin R, Liu J, Hinkes BG, Vlangos CN, et al. Low prevalence of NPHS2 mutations in African American children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. (2008) 23:1455–60. doi: 10.1007/s00467-008-0861-7
29. Lovric S, Fang H, Vega-Warner V, Sadowski CE, Gee HY, Halbritter J, et al. Rapid detection of monogenic causes of childhood-onset steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. (2014) 9:1109–16. doi: 10.2215/CJN.09010813
30. Tonna SJ, Needham A, Polu K, Uscinski A, Appel GB, Falk RJ, et al. NPHS2 variation in focal and segmental glomerulosclerosis. BMC Nephrol. (2008) 9:13. doi: 10.1186/1471-2369-9-13
31. Karle SM, Uetz B, Ronner V, Glaeser L, Hildebrandt F, Fuchshuber A. Novel mutations in NPHS2 detected in both familial and sporadic steroid-resistant nephrotic syndrome. J Am Soc Nephrol. (2002) 13:388–93. doi: 10.1681/ASN.V132388
32. Caridi G, Bertelli R, Di Duca M, Dagnino M, Emma F, Muda A, et al. Broadening the spectrum of diseases related to podocin mutations. J Am Soc Nephrol. (2003) 14:1278–86. doi: 10.1097/01.ASN.0000060578.79050.E0
33. Schultheiss M, Ruf RG, Mucha BE, Wiggins R, Fuchshuber A, Lichtenberger A, et al. No evidence for genotype/phenotype correlation in NPHS1 and NPHS2 mutations. Pediatr Nephrol. (2004) 19:1340–8. doi: 10.1007/s00467-004-1629-3
34. Aucella F, De Bonis P, Gatta G, Muscarella LA, Vigilante M, di Giorgio G, et al. Molecular analysis of NPHS2 and ACTN4 genes in a series of 33 Italian patients affected by adult-onset nonfamilial focal segmental glomerulosclerosis. Nephron Clin Pract. (2005) 99:c31–6. doi: 10.1159/000082864
35. Lu L, Wan H, Yin Y, Feng WJ, Wang M, Zou YC, et al. The p.R229Q variant of the NPHS2 (podocin) gene in focal segmental glomerulosclerosis and steroid-resistant nephrotic syndrome: a meta-analysis. Int Urol Nephrol. (2014) 46:1383–93. doi: 10.1007/s11255-014-0676-3
36. Machuca E, Hummel A, Nevo F, Dantal J, Martinez F, Al-Sabban E, et al. Clinical and epidemiological assessment of steroid-resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney Int. (2009) 75:727–35. doi: 10.1038/ki.2008.650
37. Mao J, Zhang Y, Du L, Dai Y, Gu W, Liu A, et al. NPHS1 and NPHS2 gene mutations in Chinese children with sporadic nephrotic syndrome. Pediatr Res. (2007) 61:117–22. doi: 10.1203/01.pdr.0000250041.19306.3d
38. Miko A, Menyhárd M, Kaposi A, Antignac C, Tory K. The mutation-dependent pathogenicity of NPHS2 p.R229Q: a guide for clinical assessment. Hum Mutat. (2018) 39:1854–60. doi: 10.1002/humu.23660
39. Tory K, Menyhard DK, Woerner S, Nevo F, Gribouval O, Kerti A, et al. Mutation-dependent recessive inheritance of NPHS2-associated steroid-resistant nephrotic syndrome. Nat Genet. (2014) 46:299–304. doi: 10.1038/ng.2898
40. Mishra OP, Kakani N, Singh AK, Narayan G, Abhinay A, Prasad R, et al. NPHS2 R229Q polymorphism in steroid resistant nephrotic syndrome: is it responsive to immunosuppressive therapy? J Trop Pediatr. (2014) 60:231–7. doi: 10.1093/tropej/fmu006
41. Fotouhi N, Ardalan M, Jabbarpour Bonyadi M, Abdolmohammadi R, Kamalifar A, Nasri H, et al. R229Q polymorphism of NPHS2 gene in patients with late-onset steroid-resistance nephrotic syndrome: a preliminary study. Iran J Kidney Dis. (2013) 7:399–403.
42. Abid A, Khaliq S, Shahid S, Lanewala A, Mubarak M, Hashmi S, et al. A spectrum of novel NPHS1 and NPHS2 gene mutations in pediatric nephrotic syndrome patients from Pakistan. Gene. (2012) 502:133–7. doi: 10.1016/j.gene.2012.04.063
43. Reiterova J, Safrankova H, Obeidova L, Stekrova J, Maixnerova D, Merta M, et al. Mutational analysis of the NPHS2 gene in Czech patients with idiopathic nephrotic syndrome. Folia Biol. (2012) 58:64–8.
44. Tsygin A, Kornienko V, Vashurina T, Zrobok O, Matveeva M, Leonova L, et al. Prevalence of R229Q polymorphysm of NPHS2 gene in Russian children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. (2011) 26:1662.
45. Santin S, Tazon-Vega B, Silva I, Cobo MA, Gimenez I, Ruiz P, et al. Clinical value of NPHS2 analysis in early- and adult-onset steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. (2011) 6:344–54. doi: 10.2215/CJN.03770410
46. Megremis S, Mitsioni A, Mitsioni AG, Fylaktou I, Kitsiou-Tzelli S, Stefanidis CJ, et al. Nucleotide variations in the NPHS2 gene in Greek children with steroid-resistant nephrotic syndrome. Genet Test Mol Biomark. (2009) 13:249–56. doi: 10.1089/gtmb.2008.0083
47. Zaki M, El-Shaer S, Rady S, El-Salam MA, Abd-El-Salam R, Alkashlan IA, et al. Analysis of NPHS2 gene mutations in Egyptian children with nephrotic syndrome. Open Access Maced J Med Sci. (2019) 7:3145–8. doi: 10.3889/oamjms.2019.700
48. Baylarov R, Senol O, Atan M, Berdeli A. NPHS2 gene mutations in Azerbaijani children with steroid-resistant nephrotic syndrome. Saudi J Kidney Dis Transpl. (2020) 31:144–9. doi: 10.4103/1319-2442.279934
49. McKenzie LM, Hendrickson SL, Briggs WA, Dart RA, Korbet SM, Mokrzycki MH, et al. NPHS2 variation in sporadic focal segmental glomerulosclerosis. J Am Soc Nephrol. (2007) 18:2987–95. doi: 10.1681/ASN.2007030319
Keywords: focal segmental glomerular sclerosis, NPHS2, p.R229Q, steroid resistant nephrotic syndrome, meta-analysis
Citation: Zhou Q, Weng Q, Zhang X, Liu Y, Tong J, Hao X, Shi H, Shen P, Ren H, Xie J and Chen N (2022) Association Between NPHS2 p.R229Q and Focal Segmental Glomerular Sclerosis/Steroid-Resistant Nephrotic Syndrome. Front. Med. 9:937122. doi: 10.3389/fmed.2022.937122
Received: 05 May 2022; Accepted: 01 June 2022;
Published: 22 July 2022.
Edited by:
Xu-jie Zhou, Peking University, ChinaReviewed by:
Yuan-yuan Qi, First Affiliated Hospital of Zhengzhou University, ChinaCopyright © 2022 Zhou, Weng, Zhang, Liu, Tong, Hao, Shi, Shen, Ren, Xie and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingyuan Xie, bmVwaHJveGllQDE2My5jb20=; Nan Chen, Y25yajEwMEAxMjYuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.