 Nicoline H. M. den Hollander
Nicoline H. M. den Hollander Bart O. Roep
Bart O. Roep- 1Department of Internal Medicine, Leiden University Medical Center, Leiden, Netherlands
- 2Graduate School, Utrecht University, Utrecht, Netherlands
Type 1 diabetes (T1D) remains a devastating disease that requires much effort to control. Life-long daily insulin injections or an insulin pump are required to avoid severe complications. With many factors contributing to disease onset, T1D is a complex disease to cure. In this review, the risk factors, pathophysiology and defect pathways are discussed. Results from (pre)clinical studies are highlighted that explore restoration of insulin production and reduction of autoimmunity. It has become clear that treatment responsiveness depends on certain pathophysiological or genetic characteristics that differ between patients. For instance, age at disease manifestation associated with efficacy of immune intervention therapies, such as depleting islet-specific effector T cells or memory B cells and increasing immune regulation. The new challenge is to determine in whom to apply which intervention strategy. Within patients with high rates of insulitis in early T1D onset, therapy depleting T cells or targeting B lymphocytes may have a benefit, whereas slow progressing T1D in adults may be better served with more sophisticated, precise and specific disease modifying therapies. Genetic barcoding and immune profiling may help determining from which new T1D endotypes patients suffer. Furthermore, progressed T1D needs replenishment of insulin production besides autoimmunity reversal, as too many beta cells are already lost or defect. Recurrent islet autoimmunity and allograft rejection or necrosis seem to be the most challenging obstacles. Since beta cells are highly immunogenic under stress, treatment might be more effective with stress reducing agents such as glucagon-like peptide 1 (GLP-1) analogs. Moreover, genetic editing by CRISPR-Cas9 allows to create hypoimmunogenic beta cells with modified human leukocyte antigen (HLA) expression that secrete immune regulating molecules. Given the differences in T1D between patients, stratification of endotypes in clinical trials seems essential for precision medicines and clinical decision making.
Introduction
Type 1 diabetes (T1D) is an auto-immune disease that causes insulin deficiency, affecting over 14 million patients worldwide. This results in blood glucose levels that are too high (hyperglycaemia) or too low (hypoglycaemia), which lead to frequent occurrence of both physical and mental difficulties: extreme thirst and hunger, muscle weakness and spasm, blurred vision, fainting, nausea, irritability, and poor concentration (1).
Patients require several insulin injections daily or an insulin pump to maintain healthy blood glucose levels. However, despite intensive monitoring and treatment most patients do not reach the guideline of a desired HbA1c for diabetics (<48 mmol/mol), let alone a healthy hemoglobin A1c (HbA1c) in general (<42 mmol/mol) (2). These guidelines are established to assure minimal risk of developing comorbidities later in life that result from poor glucose control (e.g., cardiovascular disease, damage to nerves, eyes, kidney, and feet). The mandatory effort to control blood glucose levels is very high, yet this dedication does not ascertain in-range glucose levels. Unexpected highs and lows are responsible for an average of an hour less sleep per night compared to the healthy population and an estimated 10 years shorter lifespan (2, 3). Furthermore, the overall psychological burden increases T1D patients' risk for depression. Treating insulin shortage is treating the symptoms of the disease, but not its cause; insulin is not a cure.
It has long been thought that T1D was caused by impaired antigen tolerance due to thymic dysfunction, as this is the case for several other autoimmune diseases (e.g., Myasthenia Graves, Addison, and severe combined immunodeficiency). However, recent research shows that healthy individuals also have CD8+ T cells that are specific for beta-cell proteins. Therefore, the mechanism of disease development remains elusive. Patients typically pass several physiological hallmarks before the symptoms of insulin deficiency start to show. Until today, it is still a topic of debate in which order this cascade of events takes place and how they affect disease progression. Also, many genetic and pathophysiological differences exist between patients. Altogether, it shows that T1D is a complex disease that requires more attention to ultimately cure. Most recently, the appreciation is growing that different etiopathogeneses may exist that cause disease heterogeneity, adding to the complexity and challenge to understand T1D and translate this understanding into disease intervention strategies.
T1D is complex in a sense that many factors seem to contribute to its onset. Several gene variants yield a higher risk, but environmental factors are also important. This idea arises from observational studies in which identical twins—with the same genetic background—may still have different disease outcome. Also, unrelated individuals that carry the same susceptible genes, but who live in different areas with different lifestyles, would have other risk. The incidence of T1D amongst young children increases too rapidly (2–3% annually) to be only caused by genetics (3). This suggests that certain environmental factors are needed to trigger the immune system and progress to clinical diabetes. Obesity, for example, causes chronic elevation of inflammatory cytokines, insulin resistance and metabolic and islet stress. Another hypothesis is that enteroviruses, which appear to be found in pancreatic tissue of some patients with T1D, induce HLA class 1 overexpression and CD8 recruitment (4, 5).
T1D has been proposed to undergo several stages of progression: (1) single islet autoantibody (Ab); (2) multiple islet Abs; (3) impaired glucose tolerance; (4) clinical diabetes (6). A single islet Ab may already be present before the age of 2, but this does not perse mean T1D development (stage 1). On the other hand, late-onset T1D is often accompanied by only one islet Ab. Also, the rate of progression differs between patients: some develop clinical diabetes within 1 year, some after 10 years or not at all, with rapid progression typically occurring in younger cases (7). However, if two or more antibodies are present (stage 2), further progression toward T1D is almost inevitable (84% risk), at least in the pediatric population (3).
Disease Heterogeneity
The notion that T1D appears in different flavors depending of age at onset has generated much attention lately and the concept of disease endotypes was introduced to allow the design and assignment of different therapeutic intervention strategies depending on the disease endotype (precision medicine), as now is a common practice in high-income countries or communities for some diseases, including cancer (8–11). A pathological basis for this disease heterogeneity is still relatively weak, but the most compelling evidence derives from immunohistopathological studies of the lesion in the pancreatic islets showing differences in the rate and composition of the insulitic immune cells (Figure 1) (12). Patients diagnosed below the age of 7 (an arbitrary cut-off based on the available tissues) showed higher rates of inflammation dominated by CD8 T-cells but also featuring some B lymphocytes, whereas the rate and frequency of inflammation was less in children beyond the age of 12, while B lymphocytes were rare or absent in lesions at older onset (12). Strikingly, beta-cell features differed too: in cases with younger onset, insulin and its precursor proinsulin in the same subcellular compartments of the beta-cells whereas these molecules where not overlapping, suggesting that there may be intrinsic features of beta-cells too reflecting altered cell physiology, distress and immunogenicity that may contribute to a different and possibly faster disease progression than seen amongst patients with diagnosis at older ages (12).
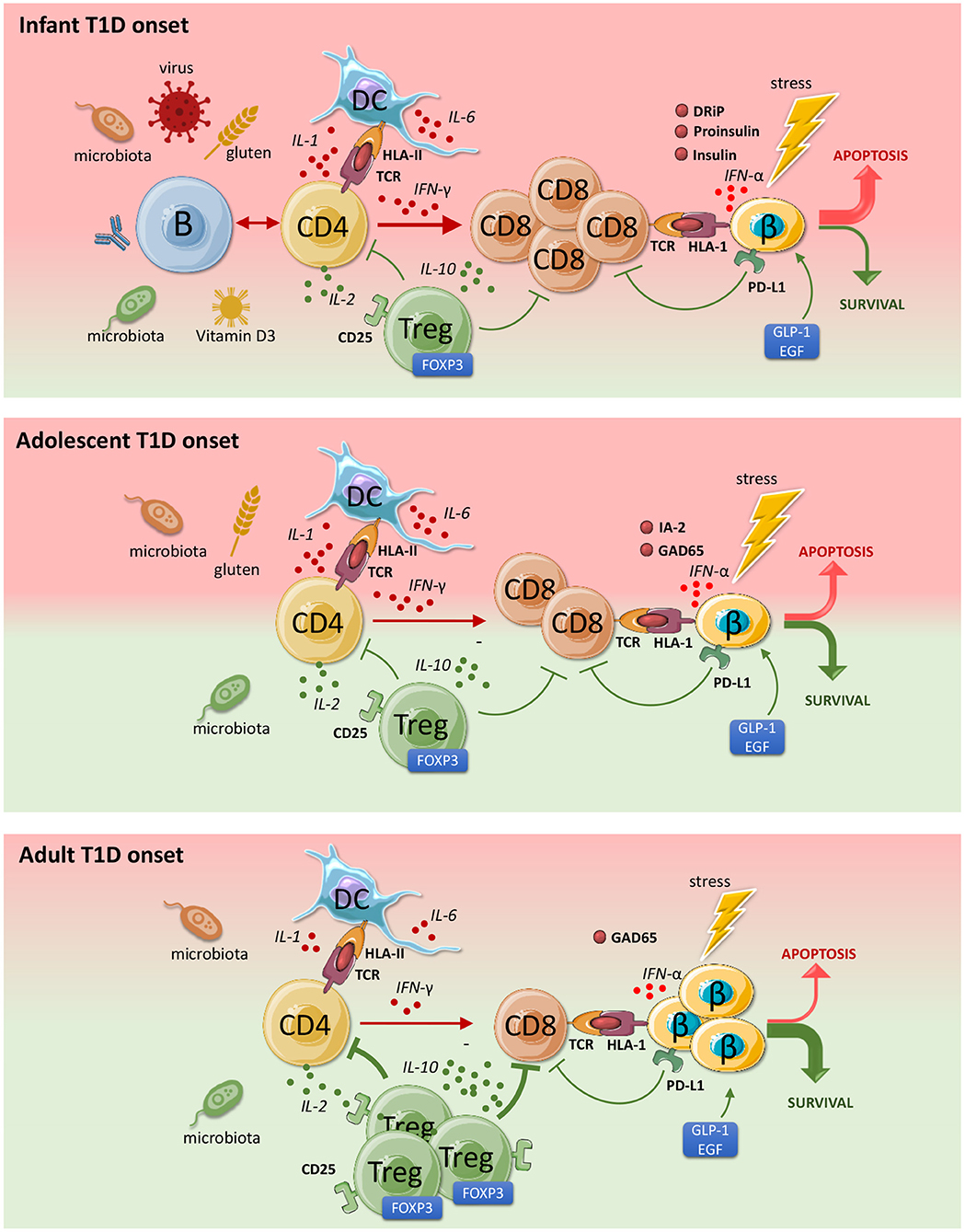
Figure 1. Pathophysiology and endotypes of type 1 diabetes (T1D). Both beta-cells and the immune system can provoke and diminish autoimmunity and beta-cell destruction. The contribution of the major cell types in disease progression differs depending on the age of disease onset. Green indicates a protective role and red points to a progressive role toward T1D development. The immune imbalance is greatest in patients developing T1D at younger ages, where the pathology is more acute and severe. With age, the degree of autoimmunity and the rate of beta-cell loss declines. CD4 T cells are activated by islet autoantigens that vary between disease endotypes. IL-2 differentially stimulates CD8+ effector T-cells as well regulatory T cells (low dose IL-2). Tregs protect against beta cell destruction. IL-6 stimulates inflammation and inhibits regulation. Activated CD8 T cells are triggered by IFN-alpha and IL-1 to attack beta cells. Due to stress, beta cells overexpress HLA Class 1 (HLA 1) and secrete IFN-alpha that provoke and attract CD8 cells. This destructive process may be inhibited by stress-reducing proteins GLP-1 and EGF. IFN-γ signaling stimulates PD-L1 expression and beta cell survival. This figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
In addition to differences in rate of disease progression and immunopathology associated with age, there are differences between patients at a given age related to, for instance, genetic background (HLA-DR3 and/or HLA-DR4; presence or absence of “protective” insulin gene variants) and ethnicity and ancestry (13). Commonly used Genetic Risk Scores designed to predict or diagnose T1D in patients of European descent did not deliver in populations of color (14).
Finally, the disease activity changes substantially with progression of disease, where insulitis appears most frequently in islets around diagnosis of T1D but is much reduced both in frequency and severity in prediabetic seropositive subjects or after diagnosis and restoration of glycaemic control with insulin therapy (12, 15, 16). This observation points out that the windows of therapeutic efficacy may depend on progression of disease, implying that some interventions specifically targeting insulitis may only be suitable or effective around the time of diagnosis, whereas other intervention strategies (for instance restoration of immune tolerance) would be better served not to be applied during the time around the medical emergency of diagnosis, insulitis and dysglycemia.
In this review we aim to find the most promising methods to cure T1D. This is investigated by sharing the latest knowledge and exploring different therapeutic approaches, covering both insulin production and auto-immunity reversal. Furthermore, the successful and less successful results from state-of-the-art studies are presented, highlighting the challenges in today's diabetes research and how to tackle these.
Key Steps to Cure T1D
To cure a patient with clinically manifested T1D, two treatment goals need to be achieved: (1) reverse or suppress autoimmunity; and (2) restore insulin production. This treatment combination ensures that the new cells can provide insulin continuously, as they are no longer destroyed by the immune system.
Suppress or Reverse Autoimmunity
Distinct cell types are associated with T1D development that may act in different patients (Figure 1). Both T cells and beta cells are involved in the pathogenesis, which likely induce disease onset in concert with dendritic cells and other antigen presenting cells (APCs), accompanied by B cells secreting islet-autoantibodies, which collectively cause beta cell destruction (8, 10, 17–21). The question remains which factor contributes to which, in whom and at which stage during disease progression. Thus, to answer this in the best possible way, researchers investigate pathophysiological changes and frequencies of immune cells in individuals that are at high-risk for T1D development (genetic or familiar susceptibility).
Over the past few years more evidence has been found regarding the imbalance between Tregs and Teff cells. The fact that non-diabetic individuals also produce islet-specific CD8 cells—although at lower levels—suggests that T1D has incorrect immune regulation by Tregs. Interestingly, genetically high-risk individuals are relatively protected when Treg levels are high (18). Furthermore, CD4+ T cells restricted by high-risk HLA class II alleles DR4-DQ8 and DR3-DQ2 present many islet-antigens such as insulin, islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP), zinc transporter 8 (ZnT8), pre-proinsulin (PPI), while also secreting cytokines, which recruits and activates increasing amounts of CD8+ cells (19). Consequently, high levels of CD8+ cells that are reactive to glutamic acid decarboxylase 65 kDa (GAD65), IA-2, PPI, ZnT8, or IGRP or other islet autoantigen have been detected in islets of T1D patients (17). Tregs of patients appear impaired in their capacity to regulate effector T-cells (22). In addition, CD4+ cells in T1D patients are thought to be relatively resistant to Tregs (23). These pathogenic T cells are more responsive to IL-6, which promotes immune responses and further suppresses Tregs (24). These findings provide more insight into the imbalanced regulatory and effector immune system and how this causes greater immune responses in T1D.
Upon stressful circumstances, beta cells—together with resident macrophages—secrete IL-1 and express the chemokine CXCL10, which attracts more T cells toward the islets (25–28). Beta cells also produce TYK2 to undergo apoptosis, while further promoting inflammation by enhancing IFN-alpha signaling, upregulating HLA class 1 molecules and attracting CD8 T cells (29). Elevated levels of proinsulin are found right after food intake, which suggests that during this stage beta cells are still intact (stage 1–3), but there is a mechanistic defect in insulin processing that is responsible for glucose intolerance (12, 30). Recent findings suggest that after prolonged stress, beta cells secrete post-translationally modified proteins that behave like autoantigens: a phenomenon that is also seen in other autoimmune diseases (e.g., multiple sclerosis and rheumatoid arthritis) (10, 31–38).
Next to initial T cell and beta cell dysregulation, B cells may also contribute to disease progression, most likely in younger patients as high levels of pancreatic B lymphocytes were found in patients aged below 7 years old (16). Less IL-10 production by regulatory T and B cells is found in children with diabetes, which cytokine is found to play a protective role against T1D development (18). Also, gut bacteria stimulate IL-10 production and maternal antibiotics intake in non-obese diabetic (NOD) mice increases the risk of diabetes development in new-borns whereas low intrauterine gluten exposure and vitamin D3 supplements in human infants decrease the risk to develop T1D (39, 40). This further supports the idea that dysbiosis contributes to early T1D development (41–44). Curiously, fecal microbiome transplantation showed promise halting disease progression in newly diagnosed T1D patients (45).
Most immunotherapy-investigating trials are aimed at preventing further progression toward clinical diabetes by immune suppression (Table 1) (46). For the antigen-specific response, a T-cell receptor/CD3 complex is required to activate the T-cells upon recognition of this specific antigen. Teplizumab and Otilixizumab, both anti-CD3 mAbs, were found effective for delaying T1D in stage 1-3 diabetes patients (47–49). A similar drug, anti-thymocyte globulin (ATG), showed some effect at stage 2-3 (50). Both treatments caused less IFN-γ and TNF secretion by the disabled CD8 memory T cells, which is desired. However, this effect sustained for only 2 years and was soon taken over by CD8 expansion and disease progression. An ultimate reset of the immune system by autologous hematopoietic stem cell therapy induced complete remission in half of the treated newly diagnosed T1D patients that sometimes lasted for more than a decade (51, 52). Efficacy was observed in a subgroup of patients characterized by low CD8 T-cell autoimmunity to islets and superior immune regulation (51).

Table 1. Treatment strategies and pitfalls.
T cells are also controlled by binding of co-stimulatory ligands and co-inhibitory ligands. These ligands are perhaps more suitable targets to restore the balance toward immune regulation instead of attack (17, 46). Several immunotherapeutic agents have been developed that target these pathways. Promising drugs are abatacept inhibiting cytotoxic T-lymphocyte-associated protein 4 (CTLA4) (53–55) and alefacept targeting CD2 (56). Monthly treatment for 2 years with abatacept significantly reduced HbA1c levels compared to placebo, even after 1 year cessation. Alefacept improved C-peptide response and beta cell function, which improved HbA1c and reduced hypoglycaemia incidence. Alefacept also impairs CD8 function, confirming that T effector cells play a big role in disrupting beta cell function. Both drugs show good results in stage 3 diabetes, yet they require prolonged administration for sustained efficacy. Of note, disease was accelerated by abatacept in patients of color, pointing to both disease heterogeneity and the need for precision medicine.
Rituximab is an anti-CD20 antibody that depletes memory B cells and reduces auto-antigen presentation and attacks (57). This drug was only effective in a minority of stage 3 patients for a maximum duration of 1 year. Antibodies against insulin were less abundant, but antibodies against other beta cell derivatives remained the same. Interestingly, rituximab was more effective in younger patients, in whom pancreatic B cell infiltration is more profound. Individuals in which this treatment was less effective had increased levels of T-effector (Teff) cells, which supports the idea that treatment should target both B and T cells, at least in some cases (16, 58, 59). Efficacy of Rituximab to delay disease progression seemed limited to younger cases, whereas abatacept showed efficacy at older disease onset.
Furthermore, treatment with cytokines is being explored to enhance Treg development by increasing FOXP3 expression. Ultra-low interleukin-2 (IL-2) treatment reversed diabetes development in mice, however, in humans this effect was only temporary and led to expansion of CD8 cells. It seems that IL-2 is involved in both regulatory and Teff pathways, depending on other stimuli (60–62). Subsequently, a new treatment was developed that was more specific: stimulating CD25 and inhibiting CD122 receptor bonding (63).
Oral antigen administration effectively built tolerance and prevented development of diabetes in preclinical studies. This approach has been assessed in several clinical trials, showing some efficacy of oral insulin in a subgroup of patients but not all, and in particular in patients with higher titres of insulin autoantibodies (IAA) (17, 64, 65). Interestingly, more efficacy in delaying onset was also noted in patients with impaired glucose tolerance (IGT) than in patients without IGT who presented multiple autoantibodies. This suggests that such antigen exposure approaches are more effective in delaying further progression of stage 1 than as preventative treatment. Perhaps proinsulin would yield better outcome—especially in stage 0 patients—as this antigen is more immunogenic and its autoantibodies are present earlier during progression (66–68). Furthermore, preclinical studies suggest that low doses stimulate regulatory systems, while frequent or high doses cause depletion of effector cells (17, 69). Several other strategies to restore immune tolerance selectively without suppressing the immune system at large are currently explored that include injection with GAD65 (70, 71), proinsulin peptides in solution (72, 73) or presented by tolerogenic dendritic cells (74) and proinsulin DNA vaccination (17, 75).
Preserve or Restore Insulin Production
Current successes in reversing autoimmunity are targeting up until stage 3 diabetes, in which a certain amount of beta cells is still intact. However, in clinical diabetes (stage 4) an estimated 90% of beta cells are already lost or non-functional (senescent) (76, 77). Hence, the source of insulin production needs to be replenished.
Theoretically, there are two strategies: (1) re-activation of senescent beta cells; or (2) pancreas, islet, beta cell or stem cell transplantation. The first focuses on beta cells that are still present, but are non-functional, due to distress or other often elusive factors. The internal biosynthesis machinery of beta cells has changed to a senescent (non-insulin producing or “hibernation”) status, which is conceivably a survival response, because active beta cells would be attacked by the immune system (10). Therefore, beta cell activation is only beneficial when combined with autoimmune reversal and immunotherapy. This approach requires external factors that switch the internal system, such as transcription factors or genetic modification with vectors that alter gene expression or treatment with beta-cell tissue factors such as epidermal growth factor (EGF) and GLP-1 that revitalize and activate beta-cell functions, respectively (78, 79). The possibility to differentiate progenitor cells in pancreatic tissue is also being explored (80). The major obstacle is that the differentiation process is quite unpredictable: multiple cell fates are induced, but their cues are largely unknown. Therefore, ex vivo differentiation and transplantation seem more promising, although the first clinical trial using this method is currently ongoing (NCT04786262).
The second approach uses cells from another source, which are already producing insulin (pancreas, islets, or beta cells) or are programmed to do so (stem cells). They should respond to glucose fluctuations adequately with the right amount and pace to prevent hypo and hyperglycaemia. To function as desired, they need to be placed within an environment that contains optimal circumstances and does not reject them.
Over the past decades, several insulin-producing transplants have been studied (80). Allogeneic pancreas transplantation is the only standard therapy suitable and available for a very limited number of T1D patients that leads to durable remission in the vast majority of recipients (81). Islet transplantation is still experimental and suffers from the lack of suitable organs and organ donors and the need for better immunotherapy (to prevent recurrent islet autoimmunity and allograft rejection) and improved and prolonged islet allograft survival and function (80).
Stem cells are another source of insulin production. Embryonic stem cells are the most pluripotent, but are taken from embryos, which is ethically questionable, and they need to match the recipients HLA type to avoid rejection. Induced pluripotent stem cells (iPSCs) are a more suitable source, as they are derived from patients' own stem cells (i.e., perfect HLA-match and ethically correct). Although clonal variability exists during the differentiation process of iPSCs, this source of insulin production is mostly investigated (82–84). Stem-cell derived beta-cells appear to be hypoimmunogenic and genetic engineering of stem cell-derived beta cells could further improve their resilience, protection, survival and function (85–87). Curiously, alpha-cells turned into insulin producing cells are also resistant to immune attack by islet specific autoreactive T-cells, pointing to either more resilience or reduced immunogenicity, or a combination thereof (88).
Now that several techniques for creating insulin-producing cells have been developed, the next caveat is finding the ideal implantation site and microenvironment for these cells. Implantation into subcutaneous tissue allows monitoring (observing abnormalities), implantation and removal, while internal tissue has more vascularization. Theoretically, the liver would be a logical choice, as glycogen is stored here. Additionally, the liver is the first organ through which absorbed glucose from the intestines passes. However, as described earlier, beta cells are stressed by high glucose levels, which makes them non-functional and self-destructive. Next to glyco- and lipotoxicity, the liver has a strong immunogenic environment, which increases the risk of damaging the implant (89). Furthermore, hepatic islet infusion showed little revascularization, which led to hypoxia and central necrosis. The fact that pancreatic islets receive five times higher blood flow, stresses the necessity of vascularization (90). However, researchers found that transplantation into the pancreas often leads to unwanted (de)differentiation, because of surrounding pancreatic differentiation cues (91). They also found that, the more stem cells differentiated (into beta cells), the more damage occurred. Only pluripotent cells (i.e., non-beta cells) were able to survive longer, but did not produce insulin (i.e., insufficient glucose-sensing). Implantation in alternative locations such as bone marrow or omentum proved less successful and required more donor tissue (92, 93).
Yoshihara et al. created human islet-like organoids (HILOs) that produce insulin and are responsive to glucose (94). Several differentiation steps were needed to induce beta cell-like characteristics, including non-canonical WNT signaling (maturation) and programmed death ligand-1 (PD-L1) overexpression (immune toleration). The optimal dose and frequency were evaluated using short multiple pulse stimulation (MPS) with interferon-γ (IFN-γ), inducing overexpression of PD-L1 without dedifferentiation or beta cell death. Transplantation of these cells led to sustained (>50 days) glucose homeostasis in both immune competent and humanized diabetic mice. Although IFN-γ is known to induce cancer in humans when overexpressed in vivo, this approach would solve this problem, as PD-L1 is expressed by in vitro IFN-γ stimulation prior to transplantation (94, 95).
Despite progenitor cells being well-programmed to produce insulin, it becomes clear that interaction with the surrounding tissue needs further optimalization. Isolated islets (e.g., cell cultures) go into necrosis easily when their surrounding is not providing natural stimuli that keep them active. The vascular network diminishes, which results in less nutrients and signaling molecules. Therefore, the use of hydrogels, matrices and other biomaterial-based devices could improve tissue circumstances. A promising method is the use of membranes that are permeable for nutrients but impermeable for immune cells (80). A phase 1 trial was effective but not sustained, as hypoxia and foreign body responses occurred. Currently, one phase-2 clinical trial investigates an encapsulated version (NCT04678557), while another trial uses bigger pores to reduce hypoxia combined with immunosuppressors (NCT03163511) (80).
Transplant recipients receive immunosuppressants, but these are not sufficient to reverse pre-existent B and T cell autoimmune reactions and often harm beta-cell differentiation and function (89, 96, 97). Immunosuppressive treatment usually consists of immediate specific drugs, followed by long term broad immunotherapy that blocks graft rejection. Low-dose systemic immunosuppressants reduce graft antigen rejection and prolong beta cell functioning (98, 99). Importantly, these drugs were selected to prevent rejection but are often uncapable to reverse pre-existent immune responses, such as islet autoimmunity, and therefore unable to prevent recurrence of autoimmune mediated beta-cell destruction (100–107).
Challenges
In the twenty-first century many breakthrough solutions have been developed for a variety of diseases, however, T1D remains one of the few (severe) autoimmune diseases that cannot be cured yet by an approved therapy. Although much effort is put into different strategies, recent studies show that finding a cure for T1D is not as straightforward as earlier thought (17, 46). Therefore, it is important to map the currently faced obstacles to envision our next step.
First of all, diversity exists between T1D patients in several areas: genetics, onset age, progression rate, autoantibodies, and beta cell number and activity (8). This inter-subject variability makes it difficult to find one clear cause, as different factors contribute to the same clinical diagnosis. Therefore, the efficacy of treatment that reverses disease progression is highly variable among patients (Table 2) (11). This also explains why treatments often do not make it further than phase II/III clinical trials: their expected effect size is not met, because treatment has no effect in certain patient groups.
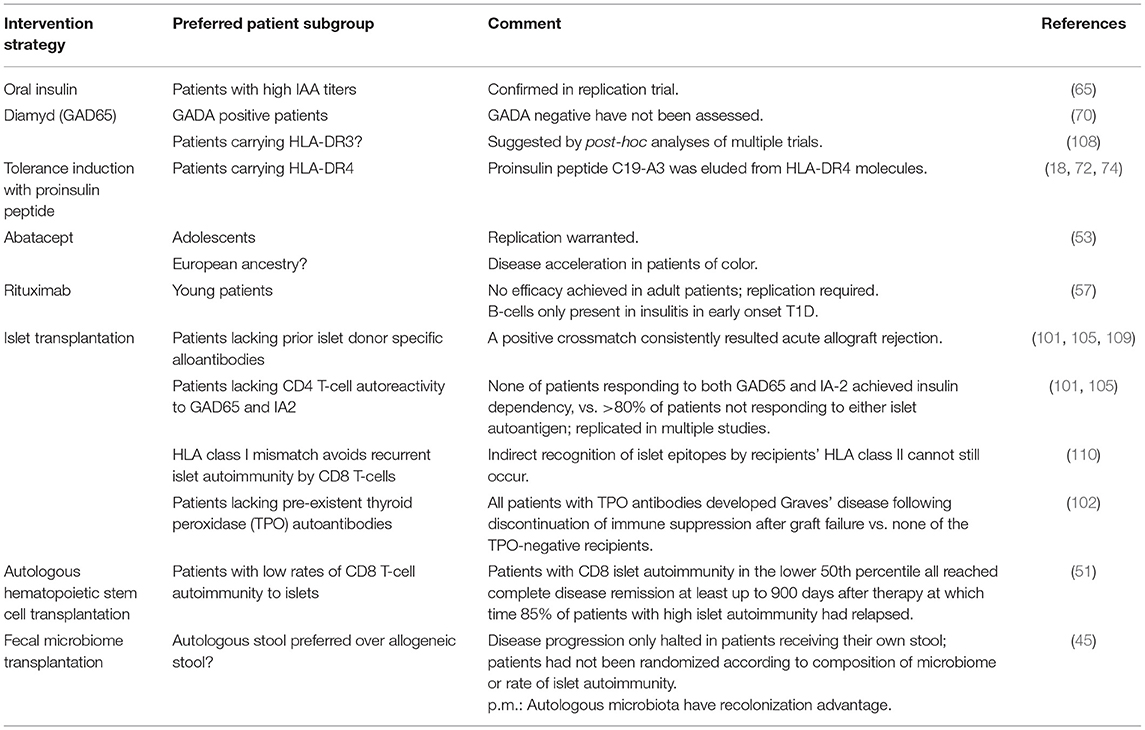
Table 2. Opportunities for precision medicine in type 1 diabetes.
Furthermore, beta cells modify their antigens under stressful circumstances. This mechanism of the immune system usually acts as surveillance against tumor cells that express neoepitopes (10). However, in this case the immune attack is targeted against beta cells, which might be an act of self-destruction; the new antigen is more immunogenic and has a higher binding affinity with cytotoxic T cells. Several studies show that post-translational modifications of beta cell epitopes are indeed strong triggers for T cell activation (10, 31–38). The neoepitope modification is thought to enhance binding affinity to high-risk HLA alleles compared to unmodified epitopes, which increases antigen presentation and recognition by T cells (111, 112). An alternative explanation for the cause of epitope modification is that the defective insulin protein is expressed under stress: an unwanted response that can be inhibited by verapamil, a drug that reduces oxidative stress and insulin-processing defects (113). Nevertheless, the formation of neoantigens complexifies the development of immunotherapy, as these antigens are highly variable between subjects. It is conceivable that immunotherapy alone will not suffice to reverse T1D durably. Combinations of beta-cell and immunotherapy may prevent continued provocations by distressed beta-cells to the immune system.
Another difficulty is translation from cell cultures to animal models to humans. Since beta cells are highly dependent on nutrients and oxygen (i.e., blood flow) and external stimuli, they function better in vivo than in vitro. On the other hand, they are more prone to re-differentiate in vivo, because of surrounding transcription factors and other signaling molecules. However, beta cells that are damaged or undergo necrosis secrete inflammatory cytokines that attract immune cells and cause damage to the well-functioning beta cells (10). In addition, a lack of good preclinical models of human T1D, the lack of access to the lesion in humans had delayed progress for decades. Since the access of diabetic pancreata through the establishment of the Network for Pancreatic Organ Donors (www.jdrfnpod.org), the insight in T1D has grown in a spectacular fashion, further underscoring the differences in autoimmune diabetes between species, but also pointing to the role of distressed islets preceding inflammation an revealing the relative paucity of inflammatory cells in human insulitis (with an striking lack of Tregs in the lesion), the low number of inflamed islets, the focal lesions, and the unexpectedly large number of insulin-positive islets seemingly unaffected at diagnosis (114).
Furthermore, transplanted cells are at risk for immune attacks and require additional immunosuppressive treatment. This becomes clear as transplanted islets slowly degrade within a few months (80, 89). Yet, broad immunosuppressants—such as cyclosporine used in the 80s—induce severe side effects such as kidney damage or infections (107, 115). Thereafter, research focused on development of more specific immunosuppressive drugs, such as IFN-γ to increase PD-L1 expression. Unfortunately, IFN-γ treatment also allows development of tumor cells as these are protected from immune surveillance as well (95).
A final hurdle is the fact that the control of T1D symptoms is relatively easy, increasing the bar for potentially risky therapeutic intervention strategies. In contrast to other autoimmune disease, where there is no alternative for immunotherapy, or cancer that would become lethal if untreated, insulin therapy deals with the hyperglycemia and diabetic complications only show years after diagnosis, masking the burning need for curative therapies. There is also a burning need to define efficacy of intervention therapy in T1D (116). At least seven types of clinical benefit were identified, varying from prevention of seroconversion in individuals at genetic risk, to prevention of epitope spreading, delay, reversal or prevention of T1D, to prevention of diabetic complication. Different immune intervention strategies may be amenable and effective in different stages of the disease and in different T1D patients (17, 46, 116).
Novel Strategies and Future Perspective
The post-translational modification of beta cell proteins (i.e., neoepitopes) explains why beta cells are suddenly not tolerated anymore, as the immune system should have been programmed to tolerate native epitopes by thymic selection. Therefore, research in antigen-based therapies should also focus on targeting neoantigens (Figure 2). Combining antigen with tolerogenic adjuvants, such as blocking T cell activation (e.g., anti-CD3) or co-stimulation (e.g., alefacept and abatacept) might limit the risk of severe side effects, which treatments were recently proven effective in mice and—to a certain extent—in humans. The major pitfall is that co-stimulation is also necessary for Tregs in humans, which might negatively affect the translation to the clinic. So, perhaps it would be beneficial to investigate a treatment regimen that depletes effector T cells, followed by Treg enhancement, as these two methods might interfere too much when administered simultaneously (17). Furthermore, as beta cell neoepitopes are a strong trigger for autoimmunity induction and are secreted under stress, agents that reduce beta cell stress (e.g., GLP-1 analogs, EGF, and Verapamil) could prevent autoimmune attacks (15, 117) (Figure 2).
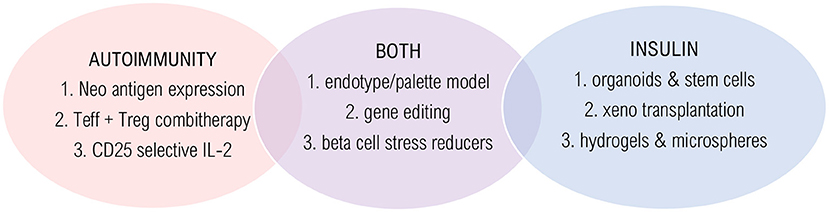
Figure 2. Innovative technologies and solutions. Promising strategies are shown that target autoimmunity (red), insulin deficiency (blue) or both (purple). Autoimmunity solutions: (1) Long-term and low-level antigen expression by AAV infection; (2) Combination therapy of effector T cell depletion followed by regulatory T cell stimulation; (3) Selective CD25-agonist/CD-122 antagonist. Insulin solutions: (1) Glucose sensing beta-cell like organoids that secrete sustained insulin by ex vivo WNT and IFN-γ signaling; (2) Humanized pancreas growth by patient-derived stem cell injection into pig blastocyst; (3) Hydrogels and TMTD-alginate microspheres to avoid rejection and necrosis. Solutions for both: (1) Trial stratification and treatment choice based on endotype; (2) Engineering hypoimmunogenic beta cells using CRISPR-Cas9; (3) Beta cell stress reducers to lower inflammation and increase beta cell function.
Another evolving strategy is to induce Treg expansion by ectopic expression of antigen through antigen-encoding plasmid vaccines (75). In recently diagnosed patients, pro-insulin peptide injection increased IL-10 secretion and transcription factor forkhead box P3 (FOXP3) expression, and lowered beta cell-specific CD8 T cells and stress (72). Next, an intramuscular DNA vaccine to induce self-antigen expression—administered weekly for 12 weeks—induced significantly improved C-peptide levels until week 15 (75). Preclinical studies suggest that low doses of anti-CD3 combined with such antigen-delivery could build tolerance (118, 119). Given their long-term expression, adeno-associated virus (AAV) vaccines would be an interesting corrective gene delivery system for T1D, perhaps targeting multiple autoantigens, because only those CD8 T cells are reduced that are specific for the vector-expressed antigen (17, 75).
With the rapidly rising interest in the endotype concept, where subtypes of T1D could be based on pathobiological or functional mechanisms that cause T1D rather than the clinical characteristics, personalized intervention therapy comes in sight. Like asthma, treatments that target specific pathways are only effective in some patient groups, suggesting there are distinct subtypes of T1D with varying defective pathways that lead to a similar clinical result. It becomes clear that certain traits are linked, such as age, autoantibodies, and HLA-specific autoimmunity (120). For example, high-risk HLA allele DR4 is associated with early proinsulin autoantibodies. Similarly, high-risk allele DR3 is associated with GAD autoantibodies. These associations could form two distinct subtypes: proinsulin-DR4 and GAD-DR3 (8). Since drugs have very different efficacy based on low or high B cell titres, such as rituximab, B cell infiltration would also be a useful subtype.
Similarly, it would be useful to stratify clinical trials based on these variables, as described in the Palette model by McCarthy (121). He opts to cluster participants based on combinations of alleles, traits, and genes. Then, their clinical responsiveness to the investigational drug is mapped, which serves as a tool for clinical decision making: if the patient has similar characteristics to one distinct endotype, the treatment with highest benefit according to their endotype would be preferably administered. Stratification based on these endotypes would also lower variance, leading to higher power or significance, which would take precision drugs further toward phase 4 and to the market.
Lastly, whenever transplantation conditions are improved and the engrafted tissue is not rejected, there would still be a shortage of human donors, as they should be HLA-matched. Therefore, humanized pancreas growth in pigs is currently under investigation (122). This method blocks porcine pancreas growth by engineering their blastocyst and inserting patient-specific iPSCs. This technique was successful with mouse-derived stem cells, but still awaits confirmation in human trials. Another promising technique to tackle the shortage of (HLA matched) donors and graft rejection is to genetically modify beta cells using clustered regularly interspaced short palindromic repeats- associated protein 9 (CRISPR-Cas9) (70, 123). Insulin-producing beta cells were created that co-secrete IL-10, which protects them from immune attacks. Additionally, this technique could also knock-out endogenous HLA molecules and replace them with HLA-E or HLA-G to avoid attack of natural killer cells (124).
To conclude, treatment responsiveness depends on certain pathophysiological or genetic characteristics that are highly variable between patients. Therefore, future research may benefit from genome-wide association studies (GWAS) to gain insight into linked genes and characteristics, from which new T1D endotypes can be defined. Graft rejection or necrosis seem to be the most challenging obstacles, for which immune-tolerated and vascularized hydrogels and microspheres are promising strategies. Furthermore, stress reducing agents and CRISPR-Cas9 may preserve beta cell function. Since T1D remains complex and highly heterogenic, stratification of endotypes in clinical trials would help development of precision medicines and clinical decision making. Given the specular, ground-breaking and paradigm shifting new insight into the pathogenesis and diversity thereof of T1D, there is an urgent need to educate the different stakeholders (T1D patients and their loved ones, care providers, pharma, regulators and legislators) on realistic possibilities, expectation and outcomes that will differ between strategies and between patients (116). The future of T1D patients and the therapy of their disease has already changed for the better but there is still much to be gained.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Haller MJ, Atkinson MA, Schatz D. Type 1 diabetes mellitus: etiology, presentation, and management. Pediatr Clin North Am. (2005) 52:1553–78. doi: 10.1016/j.pcl.2005.07.006
2. Katsarou A, Gudbjornsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers. (2017) 3:17016. doi: 10.1038/nrdp.2017.16
3. DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet. (2018) 391:2449–62. doi: 10.1016/S0140-6736(18)31320-5
4. Dotta F, Censini S, van Halteren AG, Marselli L, Masini M, Dionisi S, et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci U S A. (2007) 104:5115–20. doi: 10.1073/pnas.0700442104
5. Richardson SJ, Rodriguez-Calvo T, Gerling IC, Mathews CE, Kaddis JS, Russell MA, et al. Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia. (2016) 59:2448–58. doi: 10.1007/s00125-016-4067-4
6. Insel RA, Dunne JL, Atkinson MA, Chiang JL, Dabelea D, Gottlieb PA, et al. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care. (2015) 38:1964–74. doi: 10.2337/dc15-1419
7. Greenbaum CJ, Speake C, Krischer J, Buckner J, Gottlieb PA, Schatz DA, et al. Strength in numbers: opportunities for enhancing the development of effective treatments for type 1 diabetes-the TrialNet experience. Diabetes. (2018) 67:1216–25. doi: 10.2337/db18-0065
8. Battaglia M, Ahmed S, Anderson MS, Atkinson MA, Becker D, Bingley PJ, et al. Introducing the endotype concept to address the challenge of disease heterogeneity in type 1 diabetes. Diabetes Care. (2020) 43:5–12. doi: 10.2337/dc19-0880
9. Claessens LA, Wesselius J, van Lummel M, Laban S, Mulder F, Mul D, et al. Clinical and genetic correlates of islet-autoimmune signatures in juvenile-onset type 1 diabetes. Diabetologia. (2020) 63:351–61. doi: 10.1007/s00125-019-05032-3
10. Roep BO, Thomaidou S, van Tienhoven R, Zaldumbide A. Type 1 diabetes mellitus as a disease of the beta-cell (do not blame the immune system?). Nat Rev Endocrinol. (2021) 17:150–61. doi: 10.1038/s41574-020-00443-4
11. Woittiez NJ, Roep BO. Impact of disease heterogeneity on treatment efficacy of immunotherapy in Type 1 diabetes: different shades of gray. Immunotherapy. (2015) 7:163–74. doi: 10.2217/imt.14.104
12. Leete P, Oram RA, McDonald TJ, Shields BM, Ziller C, TIGI Study Team, et al. Studies of insulin and proinsulin in pancreas and serum support the existence of aetiopathological endotypes of type 1 diabetes associated with age at diagnosis. Diabetologia. (2020) 63:1258–67. doi: 10.1007/s00125-020-05115-6
13. Redondo MJ, Geyer S, Steck AK, Sharp S, Wentworth JM, Weedon MN, et al. A type 1 diabetes genetic risk score predicts progression of islet autoimmunity and development of type 1 diabetes in individuals at risk. Diabetes Care. (2018) 41:1887–94. doi: 10.2337/dc18-0087
14. Kaddis JS, Perry DJ, Vu AN, Rich SS, Atkinson MA, Schatz DA, et al. Improving the prediction of type 1 diabetes across ancestries. Diabetes Care. (2022) 45:e48–50. doi: 10.2337/dc21-1254
15. von Herrath M, Bain SC, Bode B, Clausen JO, Coppieters K, Gaysina L, et al. Anti-interleukin-21 antibody and liraglutide for the preservation of beta-cell function in adults with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol. (2021) 9:212–24. doi: 10.1016/S2213-8587(21)00019-X
16. Leete P, Willcox A, Krogvold L, Dahl-Jorgensen K, Foulis AK, Richardson SJ, et al. Differential insulitic profiles determine the extent of beta-cell destruction and the age at onset of type 1 diabetes. Diabetes. (2016) 65:1362–9. doi: 10.2337/db15-1615
17. Roep BO, Wheeler DCS, Peakman M. Antigen-based immune modulation therapy for type 1 diabetes: the era of precision medicine. Lancet Diabetes Endocrinol. (2019) 7:65–74. doi: 10.1016/S2213-8587(18)30109-8
18. Arif S, Tree TI, Astill TP, Tremble JM, Bishop AJ, Dayan CM, et al. Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest. (2004) 113:451–63. doi: 10.1172/JCI19585
19. Arif S, Leete P, Nguyen V, Marks K, Nor NM, Estorninho M, et al. Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes. (2014) 63:3835–45. doi: 10.2337/db14-0365
20. Inshaw JRJ, Cutler AJ, Burren OS, Stefana MI, Todd JA. Approaches and advances in the genetic causes of autoimmune disease and their implications. Nat Immunol. (2018) 19:674–84. doi: 10.1038/s41590-018-0129-8
21. Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest. (2017) 127:2881–91. doi: 10.1172/JCI94549
22. Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes. (2005) 54:92–9. doi: 10.2337/diabetes.54.1.92
23. Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol. (2008) 181:7350–5. doi: 10.4049/jimmunol.181.10.7350
24. Hundhausen C, Roth A, Whalen E, Chen J, Schneider A, Long SA, et al. Enhanced T cell responses to IL-6 in type 1 diabetes are associated with early clinical disease and increased IL-6 receptor expression. Sci Transl Med. (2016) 8:356ra119. doi: 10.1126/scitranslmed.aad9943
25. Roep BO, Kleijwegt FS, van Halteren AG, Bonato V, Boggi U, Vendrame F, et al. Islet inflammation and CXCL10 in recent-onset type 1 diabetes. Clin Exp Immunol. (2010) 159:338–43. doi: 10.1111/j.1365-2249.2009.04087.x
26. Lundberg M, Krogvold L, Kuric E, Dahl-Jorgensen K, Skog O. Expression of interferon-stimulated genes in insulitic pancreatic islets of patients recently diagnosed with type 1 diabetes. Diabetes. (2016) 65:3104–10. doi: 10.2337/db16-0616
27. Arnush M, Heitmeier MR, Scarim AL, Marino MH, Manning PT, Corbett JA. IL-1 produced and released endogenously within human islets inhibits beta cell function. J Clin Invest. (1998) 102:516–26. doi: 10.1172/JCI844
28. Zirpel H, Roep BO. Islet-resident dendritic cells and macrophages in type 1 diabetes: in search of bigfoot's print. Front Endocrinol. (2021) 12:666795. doi: 10.3389/fendo.2021.666795
29. Marroqui L, Dos Santos RS, Floyel T, Grieco FA, Santin I, Op de Beeck A, et al. TYK2, a candidate gene for type 1 diabetes, modulates apoptosis and the innate immune response in human pancreatic beta-cells. Diabetes. (2015) 64:3808–17. doi: 10.2337/db15-0362
30. Sims EK, Bahnson HT, Nyalwidhe J, Haataja L, Davis AK, Speake C, et al. Proinsulin secretion is a persistent feature of type 1 diabetes. Diabetes Care. (2019) 42:258–64. doi: 10.2337/dci19-0012
31. Roep BO, Kracht MJ, van Lummel M, Zaldumbide A. A roadmap of the generation of neoantigens as targets of the immune system in type 1 diabetes. Curr Opin Immunol. (2016) 43:67–73. doi: 10.1016/j.coi.2016.09.007
32. McLaughlin RJ, Spindler MP, van Lummel M, Roep BO. Where, how, and when: positioning posttranslational modification within type 1 diabetes pathogenesis. Curr Diab Rep. (2016) 16:63. doi: 10.1007/s11892-016-0752-4
33. Kracht MJL, Zaldumbide A, Roep BO. Neoantigens and microenvironment in type 1 diabetes: lessons from antitumor immunity. Trends Endocrinol Metab. (2016) 27:353–62. doi: 10.1016/j.tem.2016.03.013
34. McLaughlin RJ, de Haan A, Zaldumbide A, de Koning EJ, de Ru AH, van Veelen PA, et al. Human islets and dendritic cells generate post-translationally modified islet autoantigens. Clin Exp Immunol. (2016) 185:133–40. doi: 10.1111/cei.12775
35. van Lummel M, Duinkerken G, van Veelen PA, de Ru A, Cordfunke R, Zaldumbide A, et al. Posttranslational modification of HLA-DQ binding islet autoantigens in type 1 diabetes. Diabetes. (2014) 63:237–47. doi: 10.2337/db12-1214
36. de Jong VM, Abreu JR, Verrijn Stuart AA, van der Slik AR, Verhaeghen K, Engelse MA, et al. Alternative splicing and differential expression of the islet autoantigen IGRP between pancreas and thymus contributes to immunogenicity of pancreatic islets but not diabetogenicity in humans. Diabetologia. (2013) 56:2651–8. doi: 10.1007/s00125-013-3034-6
37. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science. (2016) 351:711–4. doi: 10.1126/science.aad2791
38. Mannering SI, Harrison LC, Williamson NA, Morris JS, Thearle DJ, Jensen KP, et al. The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J Exp Med. (2005) 202:1191–7. doi: 10.1084/jem.20051251
39. Antvorskov JC, Halldorsson TI, Josefsen K, Svensson J, Granstrom C, Roep BO, et al. Association between maternal gluten intake and type 1 diabetes in offspring: national prospective cohort study in Denmark. BMJ. (2018) 362:k3547. doi: 10.1136/bmj.k3547
40. Hypponen E, Laara E, Reunanen A, Jarvelin MR, Virtanen SM. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet. (2001) 358:1500–3. doi: 10.1016/S0140-6736(01)06580-1
41. Erdem N, Montero E, Roep BO. Breaking and restoring immune tolerance to pancreatic beta-cells in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. (2021) 28:397–403. doi: 10.1097/MED.0000000000000646
42. Vatanen T, Kostic AD, d'Hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. (2016) 165:1551. doi: 10.1016/j.cell.2016.05.056
43. Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE. (2011) 6:e25792. doi: 10.1371/journal.pone.0025792
44. de Groot PF, Belzer C, Aydin O, Levin E, Levels JH, Aalvink S, et al. Distinct fecal and oral microbiota composition in human type 1 diabetes, an observational study. PLoS ONE. (2017) 12:e0188475. doi: 10.1371/journal.pone.0188475
45. de Groot P, Nikolic T, Pellegrini S, Sordi V, Imangaliyev S, Rampanelli E, et al. Faecal microbiota transplantation halts progression of human new-onset type 1 diabetes in a randomised controlled trial. Gut. (2021) 70:92–105. doi: 10.1136/gutjnl-2020-322630
46. Atkinson MA, Roep BO, Posgai A, Wheeler DCS, Peakman M. The challenge of modulating beta-cell autoimmunity in type 1 diabetes. Lancet Diabetes Endocrinol. (2019) 7:52–64. doi: 10.1016/S2213-8587(18)30112-8
47. Sims EK, Bundy BN, Stier K, Serti E, Lim N, Long SA, et al. Teplizumab improves and stabilizes beta cell function in antibody-positive high-risk individuals. Sci Transl Med. (2021) 13:eabc8980. doi: 10.1126/scitranslmed.abc8980
48. Keymeulen B, van Maurik A, Inman D, Oliveira J, McLaughlin R, Gittelman RM, et al. A randomised, single-blind, placebo-controlled, dose-finding safety and tolerability study of the anti-CD3 monoclonal antibody otelixizumab in new-onset type 1 diabetes. Diabetologia. (2021) 64:313–24. doi: 10.1007/s00125-020-05317-y
49. Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. (2005) 352:2598–608. doi: 10.1056/NEJMoa043980
50. Haller MJ, Schatz DA, Skyler JS, Krischer JP, Bundy BN, Miller JL, et al. Low-dose anti-thymocyte globulin (ATG) preserves beta-cell function and improves HbA1c in new-onset type 1 diabetes. Diabetes Care. (2018) 41:1917–25. doi: 10.2337/dc18-0494
51. Malmegrim KC, de Azevedo JT, Arruda LC, Abreu JR, Couri CE, de Oliveira GL, et al. Immunological balance is associated with clinical outcome after autologous hematopoietic stem cell transplantation in type 1 diabetes. Front Immunol. (2017) 8:167. doi: 10.3389/fimmu.2017.00167
52. Voltarelli JC, Couri CE, Stracieri AB, Oliveira MC, Moraes DA, Pieroni F, et al. Autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA. (2007) 297:1568–76. doi: 10.1001/jama.297.14.1568
53. Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet. (2011) 378:412–9. doi: 10.1016/S0140-6736(11)60886-6
54. Edner NM, Heuts F, Thomas N, Wang CJ, Petersone L, Kenefeck R, et al. Follicular helper T cell profiles predict response to costimulation blockade in type 1 diabetes. Nat Immunol. (2020) 21:1244–55. doi: 10.1038/s41590-020-0744-z
55. Roep BO. New hope for immune intervention therapy in type 1 diabetes. Lancet. (2011) 378:376–8. doi: 10.1016/S0140-6736(11)60977-X
56. Rigby MR, Harris KM, Pinckney A, DiMeglio LA, Rendell MS, Felner EI, et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J Clin Invest. (2015) 125:3285–96. doi: 10.1172/JCI81722
57. Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. (2009) 361:2143–52. doi: 10.1056/NEJMoa0904452
58. Bloem SJ, Roep BO. The elusive role of B lymphocytes and islet autoantibodies in (human) type 1 diabetes. Diabetologia. (2017) 60:1185–9. doi: 10.1007/s00125-017-4284-5
59. Martin S, Wolf-Eichbaum D, Duinkerken G, Scherbaum WA, Kolb H, Noordzij JG, et al. Development of type 1 diabetes despite severe hereditary B-cell deficiency. N Engl J Med. (2001) 345:1036–40. doi: 10.1056/NEJMoa010465
60. Rosenzwajg M, Salet R, Lorenzon R, Tchitchek N, Roux A, Bernard C, et al. Low-dose IL-2 in children with recently diagnosed type 1 diabetes: a Phase I/II randomised, double-blind, placebo-controlled, dose-finding study. Diabetologia. (2020) 63:1808–21. doi: 10.1007/s00125-020-05200-w
61. Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. (2013) 1:295–305. doi: 10.1016/S2213-8587(13)70113-X
62. Waldron-Lynch F, Kareclas P, Irons K, Walker NM, Mander A, Wicker LS, et al. Rationale and study design of the Adaptive study of IL-2 dose on regulatory T cells in type 1 diabetes (DILT1D): a non-randomised, open label, adaptive dose finding trial. BMJ Open. (2014) 4:e005559. doi: 10.1136/bmjopen-2014-005559
63. Overwijk WW, Tagliaferri MA, Zalevsky J. Engineering IL-2 to give new life to T cell immunotherapy. Annu Rev Med. (2021) 72:281–311. doi: 10.1146/annurev-med-073118-011031
64. Vehik K, Cuthbertson D, Ruhlig H, Schatz DA, Peakman M, Krischer JP, et al. Long-term outcome of individuals treated with oral insulin: diabetes prevention trial-type 1 (DPT-1) oral insulin trial. Diabetes Care. (2011) 34:1585–90. doi: 10.2337/dc11-0523
65. Writing Committee for the Type 1 Diabetes TrialNet Oral Insulin Study Group, Krischer JP, Schatz DA, Bundy B, Skyler JS, Greenbaum CJ. Effect of oral insulin on prevention of diabetes in relatives of patients with type 1 diabetes: a randomized clinical trial. JAMA. (2017) 318:1891–902. doi: 10.1001/jama.2017.17070
66. Nanto-Salonen K, Kupila A, Simell S, Siljander H, Salonsaari T, Hekkala A, et al. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial. Lancet. (2008) 372:1746–55. doi: 10.1016/S0140-6736(08)61309-4
67. Achenbach P, Koczwara K, Knopff A, Naserke H, Ziegler AG, Bonifacio E. Mature high-affinity immune responses to (pro)insulin anticipate the autoimmune cascade that leads to type 1 diabetes. J Clin Invest. (2004) 114:589–97. doi: 10.1172/JCI200421307
68. Skowera A, Ellis RJ, Varela-Calvino R, Arif S, Huang GC, Van-Krinks C, et al. CTLs are targeted to kill beta cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope. J Clin Invest. (2008) 118:3390–402. doi: 10.1172/JCI35449
69. von Herrath M, Peakman M, Roep B. Progress in immune-based therapies for type 1 diabetes. Clin Exp Immunol. (2013) 172:186–202. doi: 10.1111/cei.12085
70. Ludvigsson J, Krisky D, Casas R, Battelino T, Castano L, Greening J, et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N Engl J Med. (2012) 366:433–42. doi: 10.1056/NEJMoa1107096
71. Ludvigsson J, Faresjo M, Hjorth M, Axelsson S, Cheramy M, Pihl M, et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med. (2008) 359:1909–20. doi: 10.1056/NEJMoa0804328
72. Alhadj Ali M, Liu YF, Arif S, Tatovic D, Shariff H, Gibson VB, et al. Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci Transl Med. (2017) 9:aaf779. doi: 10.1126/scitranslmed.aaf7779
73. Gibson VB, Nikolic T, Pearce VQ, Demengeot J, Roep BO, Peakman M. Proinsulin multi-peptide immunotherapy induces antigen-specific regulatory T cells and limits autoimmunity in a humanized model. Clin Exp Immunol. (2015) 182:251–60. doi: 10.1111/cei.12687
74. Nikolic T, Zwaginga JJ, Uitbeijerse BS, Woittiez NJ, de Koning EJ, Aanstoot HJ, et al. Safety and feasibility of intradermal injection with tolerogenic dendritic cells pulsed with proinsulin peptide-for type 1 diabetes. Lancet Diabetes Endocrinol. (2020) 8:470–2. doi: 10.1016/S2213-8587(20)30104-2
75. Roep BO, Solvason N, Gottlieb PA, Abreu JRF, Harrison LC, Eisenbarth GS, et al. Plasmid-encoded proinsulin preserves C-peptide while specifically reducing proinsulin-specific CD8(+) T cells in type 1 diabetes. Sci Transl Med. (2013) 5:191ra82. doi: 10.1126/scitranslmed.3006103
76. In't Veld P. Insulitis in human type 1 diabetes: a comparison between patients and animal models. Semin Immunopathol. (2014) 36:569–79. doi: 10.1007/s00281-014-0438-4
77. Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, Bhushan A. Targeted elimination of senescent beta cells prevents type 1 diabetes. Cell Metab. (2019) 29:1045–60.e10. doi: 10.1016/j.cmet.2019.01.021
78. Lam TKT, Cherney DZI. Beta cell preservation in patients with type 1 diabetes. Nat Med. (2018) 24:1089–90. doi: 10.1038/s41591-018-0144-1
79. Gromada J, Bokvist K, Ding WG, Holst JJ, Nielsen JH, Rorsman P. Glucagon-like peptide 1 (7-36) amide stimulates exocytosis in human pancreatic beta-cells by both proximal and distal regulatory steps in stimulus-secretion coupling. Diabetes. (1998) 47:57–65. doi: 10.2337/diabetes.47.1.57
80. Odorico J, Markmann J, Melton D, Greenstein J, Hwa A, Nostro C, et al. Report of the key opinion leaders meeting on stem cell-derived beta cells. Transplantation. (2018) 102:1223–9. doi: 10.1097/TP.0000000000002217
81. Smets YF, Westendorp RG, van der Pijl JW, de Charro FT, Ringers J, de Fijter JW, et al. Effect of simultaneous pancreas-kidney transplantation on mortality of patients with type-1 diabetes mellitus and end-stage renal failure. Lancet. (1999) 353:1915–9. doi: 10.1016/S0140-6736(98)07513-8
82. Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol. (2014) 32:1121–33. doi: 10.1038/nbt.3033
83. Millman JR, Xie C, Van Dervort A, Gurtler M, Pagliuca FW, Melton DA. Generation of stem cell-derived beta-cells from patients with type 1 diabetes. Nat Commun. (2016) 7:11463. doi: 10.1038/ncomms11463
84. Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, et al. Generation of functional human pancreatic beta cells in vitro. Cell. (2014) 159:428–39. doi: 10.1016/j.cell.2014.09.040
85. van der Torren CR, Zaldumbide A, Duinkerken G, Brand-Schaaf SH, Peakman M, Stange G, et al. Immunogenicity of human embryonic stem cell-derived beta cells. Diabetologia. (2017) 60:126–33. doi: 10.1007/s00125-016-4125-y
86. Zaldumbide A, Alkemade G, Carlotti F, Nikolic T, Abreu JR, Engelse MA, et al. Genetically engineered human islets protected from CD8-mediated autoimmune destruction in vivo. Mol Ther. (2013) 21:1592–601. doi: 10.1038/mt.2013.105
87. Leite NC, Pelayo GC, Melton DA. Genetic manipulation of stress pathways can protect stem-cell-derived islets from apoptosis in vitro. Stem Cell Reports. (2022) 17:766–74. doi: 10.1016/j.stemcr.2022.01.018
88. Furuyama K, Chera S, van Gurp L, Oropeza D, Ghila L, Damond N, et al. Diabetes relief in mice by glucose-sensing insulin-secreting human alpha-cells. Nature. (2019) 567:43–8. doi: 10.1038/s41586-019-0942-8
89. Harlan DM, Kenyon NS, Korsgren O, Roep BO. Immunology of Diabetes Society. Current advances and travails in islet transplantation. Diabetes. (2009) 58:2175–84. doi: 10.2337/db09-0476
90. Lammert E, Thorn P. The role of the islet niche on beta cell structure and function. J Mol Biol. (2020) 432:1407–18. doi: 10.1016/j.jmb.2019.10.032
91. Faleo G, Russ HA, Wisel S, Parent AV, Nguyen V, Nair GG, et al. Mitigating ischemic injury of stem cell-derived insulin-producing cells after transplant. Stem Cell Reports. (2017) 9:807–19. doi: 10.1016/j.stemcr.2017.07.012
92. Maffi P, Nano R, Monti P, Melzi R, Sordi V, Mercalli A, et al. Islet allotransplantation in the bone marrow of patients with type 1 diabetes: a pilot randomized trial. Transplantation. (2019) 103:839–51. doi: 10.1097/TP.0000000000002416
93. Van Hulle F, De Groot K, Hilbrands R, Van de Velde U, Suenens K, Stange G, et al. Function and composition of pancreatic islet cell implants in omentum of type 1 diabetes patients. Am J Transplant. (2022) 22:927–36. doi: 10.1111/ajt.16884
94. Yoshihara E, O'Connor C, Gasser E, Wei Z, Oh TG, Tseng TW, et al. Immune-evasive human islet-like organoids ameliorate diabetes. Nature. (2020) 586:606–11. doi: 10.1038/s41586-020-2631-z
95. Zaidi MR, Merlino G. The two faces of interferon-gamma in cancer. Clin Cancer Res. (2011) 17:6118–24. doi: 10.1158/1078-0432.CCR-11-0482
96. Roep BO, Stobbe I, Duinkerken G, van Rood JJ, Lernmark A, Keymeulen B, et al. Auto- and alloimmune reactivity to human islet allografts transplanted into type 1 diabetic patients. Diabetes. (1999) 48:484–90. doi: 10.2337/diabetes.48.3.484
97. Moberg L, Johansson H, Lukinius A, Berne C, Foss A, Kallen R, et al. Production of tissue factor by pancreatic islet cells as a trigger of detrimental thrombotic reactions in clinical islet transplantation. Lancet. (2002) 360:2039–45. doi: 10.1016/S0140-6736(02)12020-4
98. Safley SA, Barber GF, Holdcraft RW, Gazda LS, Duncanson S, Poznansky MC, et al. Multiple clinically relevant immunotherapies prolong the function of microencapsulated porcine islet xenografts in diabetic NOD mice without the use of anti-CD154 mAb. Xenotransplantation. (2020) 27:e12577. doi: 10.1111/xen.12577
99. Stabler CL, Giraldo JA, Berman DM, Gattas-Asfura KM, Willman MA, Rabassa A, et al. Transplantation of PEGylated islets enhances therapeutic efficacy in a diabetic nonhuman primate model. Am J Transplant. (2020) 20:689–700. doi: 10.1111/ajt.15643
100. Huurman VA, van der Torren CR, Gillard P, Hilbrands R, van der Meer-Prins EP, Duinkerken G, et al. Immune responses against islet allografts during tapering of immunosuppression–a pilot study in 5 subjects. Clin Exp Immunol. (2012) 169:190–8. doi: 10.1111/j.1365-2249.2012.04605.x
101. Hilbrands R, Huurman VA, Gillard P, Velthuis JH, De Waele M, Mathieu C, et al. Differences in baseline lymphocyte counts and autoreactivity are associated with differences in outcome of islet cell transplantation in type 1 diabetic patients. Diabetes. (2009) 58:2267–76. doi: 10.2337/db09-0160
102. Gillard P, Huurman V, Van der Auwera B, Decallonne B, Poppe K, Roep BO, et al. Graves hyperthyroidism after stopping immunosuppressive therapy in type 1 diabetic Islet cell recipients with pretransplant TPO autoantibodies. Diabetes Care. (2009) 32:1817–9. doi: 10.2337/dc08-2339
103. Roelen DL, Huurman VA, Hilbrands R, Gillard P, Duinkerken G, van der Meer-Prins PW, et al. Relevance of cytotoxic alloreactivity under different immunosuppressive regimens in clinical islet cell transplantation. Clin Exp Immunol. (2009) 156:141–8. doi: 10.1111/j.1365-2249.2008.03812.x
104. Huurman VA, Velthuis JH, Hilbrands R, Tree TI, Gillard P, van der Meer-Prins PM, et al. Allograft-specific cytokine profiles associate with clinical outcome after islet cell transplantation. Am J Transplant. (2009) 9:382–8. doi: 10.1111/j.1600-6143.2008.02479.x
105. Huurman VA, Hilbrands R, Pinkse GG, Gillard P, Duinkerken G, van de Linde P, et al. Cellular islet autoimmunity associates with clinical outcome of islet cell transplantation. PLoS ONE. (2008) 3:e2435. doi: 10.1371/journal.pone.0002435
106. Huurman VA, Unger WW, Koeleman BP, Oaks MK, Chandraker AK, Terpstra OT, et al. Differential inhibition of autoreactive memory- and alloreactive naive T cell responses by soluble cytotoxic T lymphocyte antigen 4 (sCTLA4), CTLA4Ig and LEA29Y. Clin Exp Immunol. (2007) 150:487–93. doi: 10.1111/j.1365-2249.2007.03513.x
107. Huurman VA, Kalpoe JS, van de Linde P, Vaessen N, Ringers J, Kroes AC, et al. Choice of antibody immunotherapy influences cytomegalovirus viremia in simultaneous pancreas-kidney transplant recipients. Diabetes Care. (2006) 29:842–7. doi: 10.2337/diacare.29.04.06.dc05-1647
108. Hannelius U, Beam CA, Ludvigsson J. Efficacy of GAD-alum immunotherapy associated with HLA-DR3-DQ2 in recently diagnosed type 1 diabetes. Diabetologia. (2020) 63:2177–81. doi: 10.1007/s00125-020-05227-z
109. Campbell PM, Salam A, Ryan EA, Senior P, Paty BW, Bigam D, et al. Pretransplant HLA antibodies are associated with reduced graft survival after clinical islet transplantation. Am J Transplant. (2007) 7:1242–8. doi: 10.1111/j.1600-6143.2007.01777.x
110. Velthuis JH, Unger WW, Abreu JR, Duinkerken G, Franken K, Peakman M, et al. Simultaneous detection of circulating autoreactive CD8+ T-cells specific for different islet cell-associated epitopes using combinatorial MHC multimers. Diabetes. (2010) 59:1721–30. doi: 10.2337/db09-1486
111. Kracht MJ, van Lummel M, Nikolic T, Joosten AM, Laban S, van der Slik AR, et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat Med. (2017) 23:501–7. doi: 10.1038/nm.4289
112. Anderson AM, Landry LG, Alkanani AA, Pyle L, Powers AC, Atkinson MA, et al. Human islet T cells are highly reactive to preproinsulin in type 1 diabetes. Proc Natl Acad Sci U S A. (2021) 118:e2107208118. doi: 10.1073/pnas.2107208118
113. Yang L, Zhu Y, Kong D, Gong J, Yu W, Liang Y, et al. EGF suppresses the expression of miR-124a in pancreatic beta cell lines via ETS2 activation through the MEK and PI3K signaling pathways. Int J Biol Sci. (2019) 15:2561–75. doi: 10.7150/ijbs.34985
114. Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med. (2012) 209:51–60. doi: 10.1084/jem.20111187
115. Stiller CR, Dupre J, Gent M, Jenner MR, Keown PA, Laupacis A, et al. Effects of cyclosporine immunosuppression in insulin-dependent diabetes mellitus of recent onset. Science. (1984) 223:1362–7. doi: 10.1126/science.6367043
116. Roep BO, Montero E, van Tienhoven R, Atkinson MA, Schatz DA, Mathieu C. Defining a cure for type 1 diabetes: a call to action. Lancet Diabetes Endocrinol. (2021) 9:553–5. doi: 10.1016/S2213-8587(21)00181-9
117. Ovalle F, Grimes T, Xu G, Patel AJ, Grayson TB, Thielen LA, et al. Verapamil and beta cell function in adults with recent-onset type 1 diabetes. Nat Med. (2018) 24:1108–12. doi: 10.1038/s41591-018-0089-4
118. Bresson D, Togher L, Rodrigo E, Chen Y, Bluestone JA, Herold KC, et al. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J Clin Invest. (2006) 116:1371–81. doi: 10.1172/JCI27191
119. Takiishi T, Korf H, Van Belle TL, Robert S, Grieco FA, Caluwaerts S, et al. Reversal of autoimmune diabetes by restoration of antigen-specific tolerance using genetically modified Lactococcus lactis in mice. J Clin Invest. (2012) 122:1717–25. doi: 10.1172/JCI60530
120. Mathieu C, Lahesmaa R, Bonifacio E, Achenbach P, Tree T. Immunological biomarkers for the development and progression of type 1 diabetes. Diabetologia. (2018) 61:2252–8. doi: 10.1007/s00125-018-4726-8
121. McCarthy MI. Painting a new picture of personalised medicine for diabetes. Diabetologia. (2017) 60:793–9. doi: 10.1007/s00125-017-4210-x
122. Matsunari H, Watanabe M, Hasegawa K, Uchikura A, Nakano K, Umeyama K, et al. Compensation of disabled organogeneses in genetically modified pig fetuses by blastocyst complementation. Stem Cell Reports. (2020) 14:21–33. doi: 10.1016/j.stemcr.2019.11.008
123. Lim D, Sreekanth V, Cox KJ, Law BK, Wagner BK, Karp JM, et al. Engineering designer beta cells with a CRISPR-Cas9 conjugation platform. Nat Commun. (2020) 11:4043. doi: 10.1038/s41467-020-17725-0
Keywords: autoimmune disease (AD), disease heterogeneity, disease endotypes, islet autoimmunity, genetic risk score, immune intervention therapy, type 1 diabetes immunopathogenesis
Citation: Hollander NHM den and Roep BO (2022) From Disease and Patient Heterogeneity to Precision Medicine in Type 1 Diabetes. Front. Med. 9:932086. doi: 10.3389/fmed.2022.932086
Received: 29 April 2022; Accepted: 13 June 2022;
Published: 12 July 2022.
Edited by:
Ziyi Zhang, University of Toronto, CanadaReviewed by:
Joel Henrique Ellwanger, Federal University of Rio Grande do Sul, BrazilFeihan Dai, University of Toronto, Canada
Copyright © 2022 Hollander and Roep. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bart O. Roep, Ym9yb2VwQGx1bWMubmw=