 Yue Wang
Yue Wang Yibing Fang2†
Yibing Fang2† Yifei Li
Yifei Li- 1Department of Cardiovascular Surgery, West China Hospital, Sichuan University, Chengdu, China
- 2Department of Cardiovascular Surgery, Southwest Hospital, Army Medical University, Chongqing, China
- 3Key Laboratory of Medical Electrophysiology, Ministry of Education, Medical Electrophysiological Key Laboratory of Sichuan Province, Institute of Cardiovascular Research, Southwest Medical University, Luzhou, China
- 4Key Laboratory of Birth Defects and Related Diseases of Women and Children of MOE, Department of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu, China
Background: Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a heritable heart muscle disorder that predominantly affects the right ventricle. Mutations in genes that encode components of desmosomes, the adhesive junctions that connect cardiomyocytes, are the predominant cause of ARVC. A case with novel heterozygous mutation in the LAMA2 gene is reported here. The protein encoded by LAMA2 gene is the α2 chain of laminin-211 protein, which establishes a stable relationship between the muscle fiber membrane and the extracellular matrix. We explored the potential mechanism and the relationship between the mutation and ARVC.
Case Presentation: At the age of 8, the patient developed syncope and palpitation after exercise. Dynamic electrocardiogram recorded continuous premature ventricular beats, and MRI showed the right ventricle was significantly enlarged and there were many localized distensions at the edge of the right ventricular wall. The patient was diagnosed with ARVC and received heart transplantation at the age of 14 due to severe heart dysfunction. The myocardial histological pathological staining revealed a large amount of fibrosis and adipose migration. Whole exome sequencing (WES) identified the heterozygous mutation in the LAMA2 gene [NM_000426.3: c.8842G > A (p.G2948S)]. This is the first report of these variants. Analysis was performed on genetic disorders to reveal splice site changes and damage to protein structure. LAMA2 p.G2948S predicted unstable protein structure and impaired function. Induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) were established. RNA-seq and the western blot were performed on IPSC-CMs to explore the ARVC-related signaling pathway.
Conclusion: This is the first case report to describe an ARVC phenotype in patients possessing a novel LAMA2 c.8842G > A (p.G2948S) mutation. Our results aid in understanding of the pathogenesis of ARVC. The molecular mechanism of LAMA2 leading to ARVC disease still needs further study.
Introduction
Arrhythmogenic right ventricular cardiomyopathy is an inherited cardiomyopathy characterized by the fibrofatty replacement of the myocardium predominantly in the right ventricle. Progressive loss of right ventricular myocardium and its replacement by fibrofatty tissue is the pathological hallmark of the disease (1, 2). Mutations in the genes encoding desmosome complex, which is important in regulating cell-to-cell adhesion and maintain cellular integrity, may affect protein structure of desmosome complex. The degradation of desmosome has been believed to be involved in the pathogenesis of ARVC. Until now, most of the ARVC-related genetic variants have been located in the desmosome complex. In addition, other non-desmosome genes, which participate in mediating cellular adhesion and intercellular connection, would also contribute to induce ARVC, and increasing numbers of mutations have been underlined. In detail, the reported genetic mutations of ARVC encoding proteins cover a large diverse range of biological functions, including cytoskeletal architecture formation, calcium handling, sodium transport, and cytokine signaling (2). Nevertheless, negative genetic results have been found in 35–50% of patients, suggesting that there may be unknown genes involved in the pathogenesis of ARVC (3, 4).
The LAMA2 gene encodes the α2 chain of laminin-211 protein, which establishes a stable relationship between the muscle fiber membrane and the extracellular matrix (ECM). Laminin 211 is dominantly expressed in the basement membrane of myocytes and Schwann cells. The mutations of LAMA2 have been proved to be involved in the initial of congenital muscular dystrophy type 1a (MDC1A) and limb girdle muscular dystrophy (LGMD) (5, 6). Laminin-α2 has also been proved to be expressed in other organ tissues, such as the heart (7). However, current evidence suggests that only a small proportion of patients with MDC1A is associated with cardiac-related clinical manifestations, and the correlation between LAMA2 mutation and ARVC remains unknown (8, 9). And only one record from the ClinicalVar database demonstrated LAMA2 might be related with primary dilated cardiomyopathy. Induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) have been used as a fully mimic platform to study the biological and molecular mechanism of any identified uncertain genetic variant. Several cell lines had been established to demonstrate the essential changes on ARVC-related genetic mutations. Thus, we attempt to explore the relationship between LAMA2 mutation and ARVC via iPSC-CMs.
Herein, we report a rare case with ARVC carrying a novel heterozygous mutation in the LAMA2 gene. This should be the first case report of ARVC due to a LAMA2 mutation, and we also established the first iPSC line of LAMA2 associated with ARVC, which would be differentiation into cardiomyocytes to address the potential activated ARVC-related pathogenic signaling pathways.
Case presentation
Ethics compliance
The parents of the patient provided written consent for participating in the study carried out at the West China Hospital, Sichuan University, China. This research study was approved by the Ethics Committee of West China Second University Hospital, Sichuan University, China (2014–034).
Clinical presentation and outcomes
This proband was a male who had been admitted to our hospital due to aggressive fatigue, chest tightness, and fainting for 2 years. He experienced significant chest pain and palpitations shortly before syncope, with a decrease in tolerance of daily activities. A pale face, dull heart sound, and third-degree systolic murmur in the fourth intercostal space were observed. Results of physical examination of the respiratory and nervous systems were negative. The parents of the patient declined any cardiovascular symptoms. Myocarditis has been excluded at first due to the patient, and his parents declined any no related nearby infection history. Furthermore, the patient declined any symptoms of muscular dystrophy. Then, any potential virus antibodies were negative after serum analysis. Electrocardiogram (ECG) demonstrated inversion of T waves in leads V1–V5, and epsilon waves were visible in some leads (Figure 1A). Holter presented 5,443 premature multisource ventricular beats in 24 h, with unequal pairing intervals, ventricular fusion wave, and insertion ventricular premature beats. MRI identified enlargement of the right ventricle, the thinner right ventricular wall, and localized distensions at the edge of the right ventricular wall. The myocardial contraction of the right ventricle was significantly reduced, and ejection fraction of the right ventricle was 15%. Right ventricular end-diastolic volume index = 142 ml/m2 (Figure 1B), so that, according to Task Force Criteria (10), this patient received a clinical diagnosis of ARVC based on his clinical manifestation, classic ECG presentation, and a cardiac phenomenon recorded by MRI.
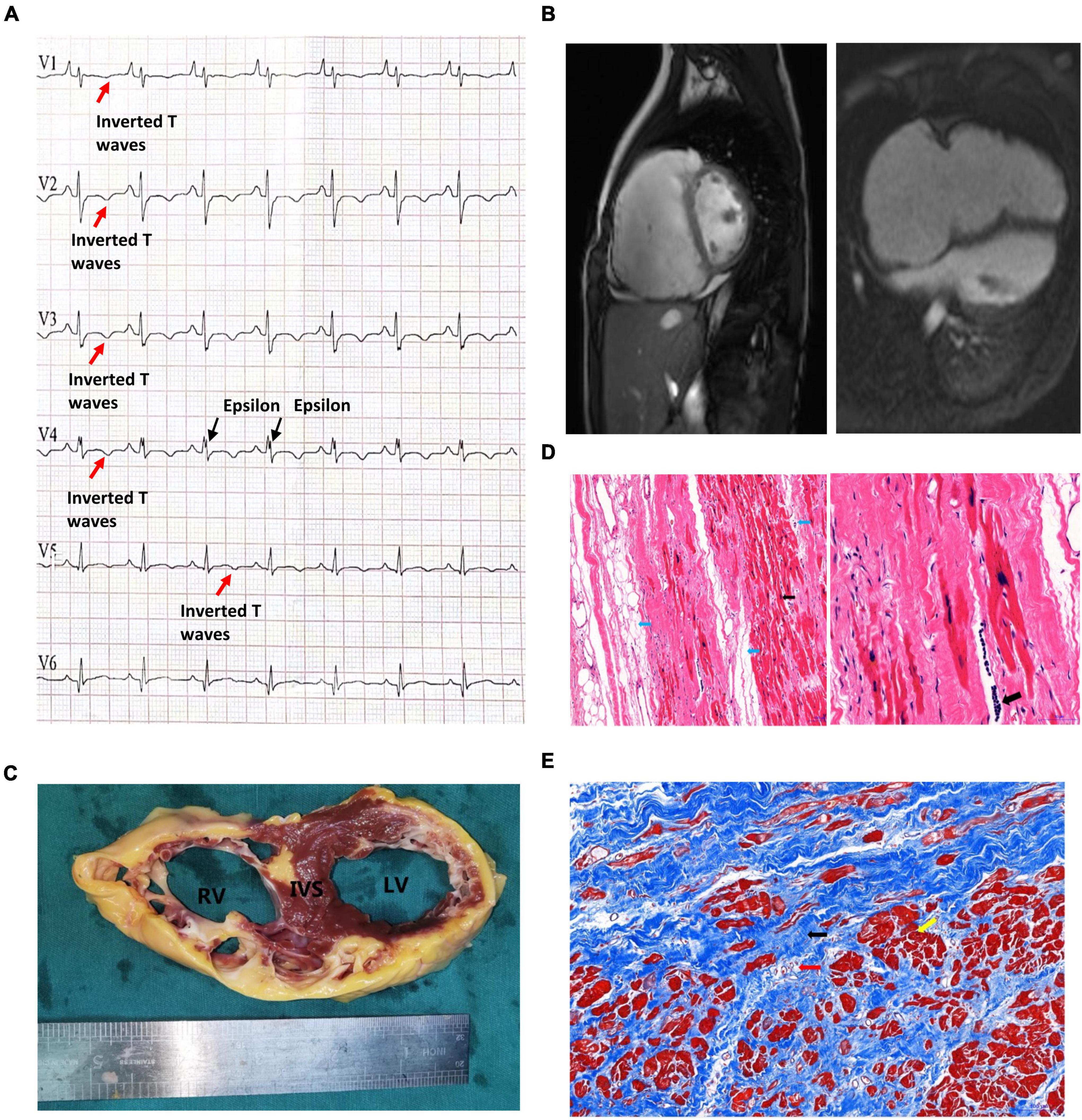
Figure 1. Clinical presentation of the proband. (A) The patient’s preoperative ECG showed inverted T waves in lead V1, V2, V3, V4, and V5, with typical Epsilon waves visible in V4. (B) Preoperative cardiac MRI of the patient indicated that the right atrium was significantly enlarged, the right ventricular wall was thinner, the right ventricular wall edge was localized in many places, and the amplitude of myocardial contraction of the right ventricle was significantly reduced. (C) The myocardium was replaced by extensive fibrous adipose tissue in the wall of the right ventricle (RV), the right ventricle was bag like and thinly dilated, part of the ventricular septum (IVS) was adipose, and part of the myocardium in the anterior and lateral wall of the left ventricle (LV) was replaced by adipose fibers. (D) HE staining of the myocardium of the right ventricle: Left: the blue arrow indicates the adipose tissue, and the myocardium of the right ventricle is surrounded by the adipose tissue and divided into cords. The black arrow shows abnormal hypertrophic cardiomyocytes with large, dark-stained nuclei and oddly shaped nuclei, broken muscle fibers in the cytoplasm of the cardiomyocytes (magnified 100×). Right: The black arrow indicates the presence of inflammatory cell infiltration between cardiomyocytes at magnified 200×. (E) Masson staining of the myocardium of the right ventricle. Note that the myocardium (the yellow arrow) is surrounded by large amount of fibrous adipocytes (the black arrow) and divided into cords or islands. The collagenous fiber bundles around small blood vessels (red arrows) are increased.
Unfortunately, due to severe and irreversible heart dysfunction, the patient received heart transplantation at the age of 14. The examination of the heart tissue revealed generally adipose infiltration in the interventricular septum and partial fiber-adipose infiltration in the myocardial tissue of the posterior wall of the left ventricle (Figure 1C). The wall of the right ventricle is extensively fiber-adipose, and only a small amount of myocardial tissue remains. The whole right ventricle is obviously dilated. And the myocardial histological pathological staining revealed a large amount of fibrosis and adipose migration (Figures 1D,E).
After the heart transplantation, the patient had a good quality of life and was followed for 3 years.
Molecular results
A peripheral blood sample was obtained from the patients in an EDTA anticoagulant blood sample tube stored at 4°C for less than 6 h, and DNA was extracted. Protein-coding exome enrichment was performed using the Agilent SureSelect Human All Exon V6. The whole exon sequencing (WES) of the patient and his parents were performed using the NovaSeq 6000 platform (Illumina, San Diego, CA, United States), and the raw data were processed using FastP to remove adapters and filter out low-quality reads. Paired-end reads were aligned with the ENSEMBL GRCh38/hg38 reference genome using the BWA software (version 0.7.12-r1039). Then, the SAMtools software (Version 1.3.1) was used to compare and order the results, and Sambamba (Version 0.7.1) was used to mark the repeated reads. Variant annotation was performed in accordance with database-sourced minor allele frequencies (MAFs) and practical guidelines on pathogenicity issued by the American College of Medical Genetics. The annotation of MAFs was performed based on the 1,000 Genomes, dbSNP, ESP, ExAC, Provean, Sift, Polypen2_hdiv, Polypen2_hvar, and Chigene in-house MAF databases using R software (R Foundation for Statistical Computing, Vienna, Austria). The sequencing data have been deposited in the GSA database.
Whole exon sequencing identified de novo heterozygous mutations in the LAMA2 gene at c.8842G > A, which has not been reported in any publication. This mutation was absent from the patients’ parents (Figure 2). This mutation site could be retrieved in 1000G and the ExAC database. There were 3 records of allele carriers in 1000G and 46 records of allele carriers in ExAC. Its clinical associations were found in congenital muscular dystrophy (RCV000764636.1), LAMA2-related dystrophy (RCV001086813.1), and primary dilated cardiomyopathy (RCV001293224.1). All the records were submitted by a gene sequencing company, and no available publication demonstrated the molecular mechanism and provided a detailed clinical report. Until now, no research has declaimed a clear relationship between LAMA2 c.8842G > A and ARVC. According to the American College of Medical Genetics, the variant has uncertain pathogenicity. SIFT, Polyphen_HumDiv, Polyphen_HumVar, and Mutation Taster were used for mutation locus screening. Analysis performed with MutationTaster revealed that this mutation was considered disease causing due to amino acid sequence changes, protein features affected, and splice site changes (probability = 0.999). The PolyPhen tool predicted protein structural and functional damaging for p.G2948S (Polyphen_HumDiv score = 1.000, the Polyphen_HumVar score = 0.999). The SIFT prediction on the protein batch demonstrated a damaging change as a score of 0.006. And the PROVEAN prediction on the protein batch revealed a deleterious change as a score of −4.34. All analyses were based on the P24043 FASTA sequence of the G2948S variant. However, it failed to identify any other known ARVC-related gene mutations both in exons and introns.
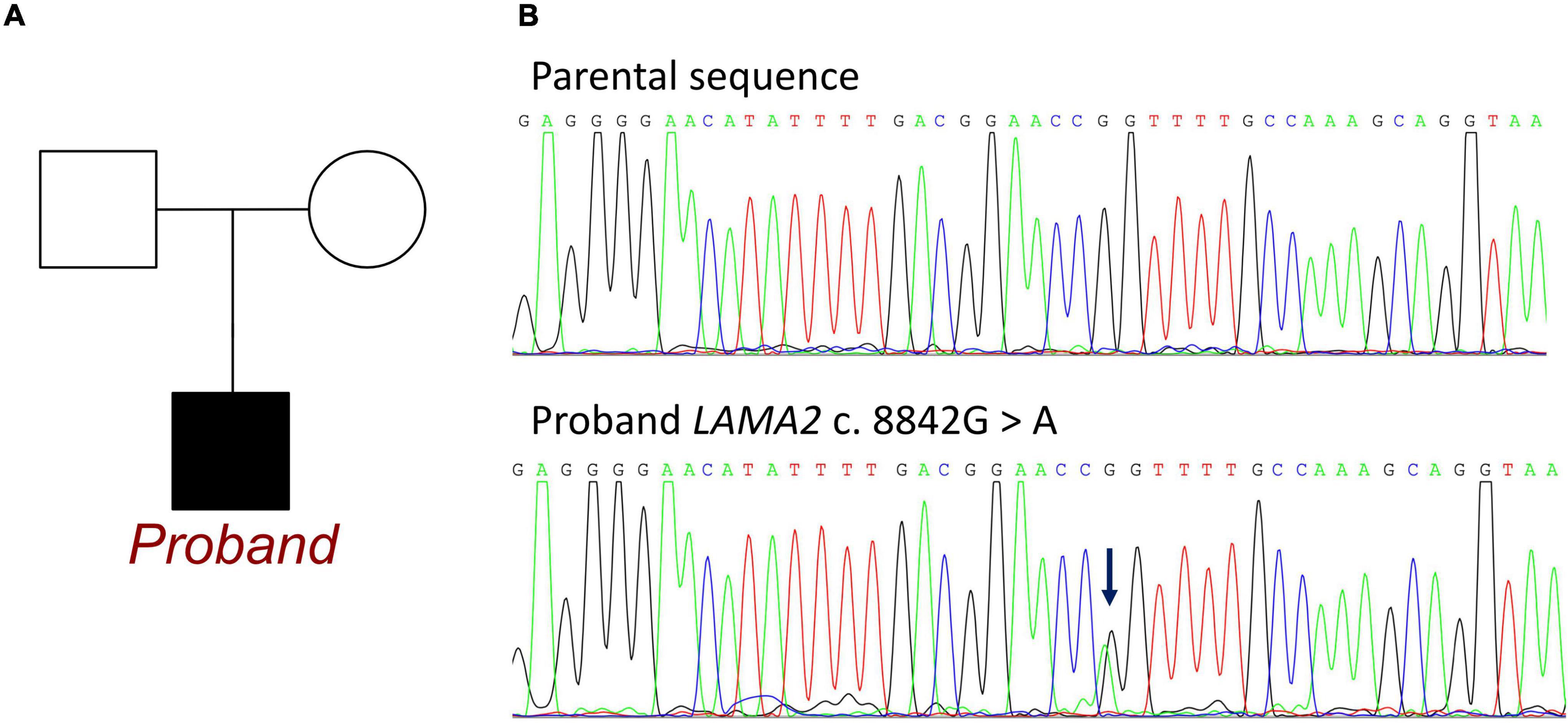
Figure 2. Information of LAMA2 c.8842G > A in the family of the proband. (A) Family map of gene mutation. (B) Sequencing of mutations. Blue arrow: The normal group showed single peak of base G, while the patient showed heterozygous state of base G peak and A peak.
To understand the molecular architecture of the human LAMA2 gene, we performed comparative modeling using the SWISS-MODEL1. We estimated the change in the free energy of the model using the mutation cut-off scanning matrix (mCSM) method2, the Site Directed Mutator (SDM)3, and the DyanMut method, which can be used to analyze and visualize protein dynamics by sampling conformations and assess the impact of mutations on protein dynamics and stability4. To assess the impacts of mutations on the stability of LAMA2. We also used the DUET server5 that integrates mCSM and SDM to improve the overall prediction accuracy of the mutations under consideration. The signature vector that was ultimately generated was employed to train the predictive classification and the regression model used to calculate the change induced by mutations in terms of Gibbs folding free energy (ΔΔG).
The SWISS-MODEL tool was used to analyze stability after amino acid changes. Ramachandran plots indicated that amino acid positions were altered (Figure 3A). Rebuilding molecular structure based on a 1okq.1.A template resulted in residue changes between Gly and Alaat 2948 (Figure 3B). Five types of calculation methods all demonstrated significant destabilizing change (DynaMut −0.950 Kcal/mol; NMA-Based Predictions, 0.298 Kcal/mol; mCSM, −1.404 Kcal/mol; DUET, −1.505 Kcal/mol; SDM, −2.480 Kcal/mol; Figure 3C). Besides, change in vibrational entropy energy between wild-type and mutant Figure 3D demonstrated a decrease of molecule flexibility as −0.372 kcal/mol K. The blue-colored amino acids sequence according to the vibrational entropy change upon mutation represented a rigidification of the structure.
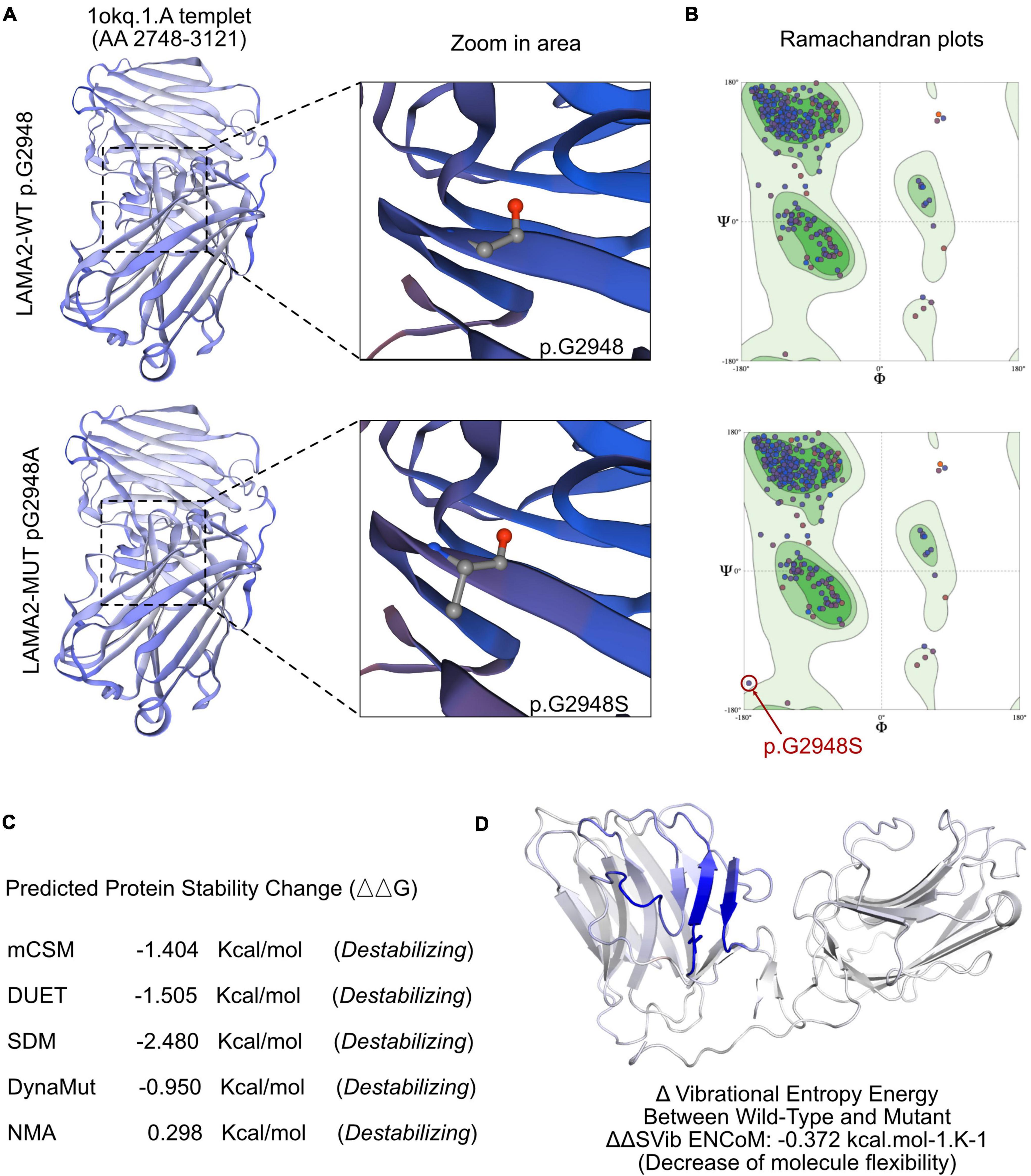
Figure 3. The effects of LAMA2 c.8842G > A mutation on the molecular structure of the protein. (A) Individual crystal structures of wild type and G2948S according to the 1okq.1.A model template. (B) Ramachandran plots of amino acid with wild type and p.G2948A. (C) The comparisons of free energy on crystal structure of the wild type sequence and the p.G2948A variant. (D) The structural changes due to the mutation of LAMA2 G2948S.
Induced pluripotent stem cell reprogramming and validation
Peripheral blood mononuclear cells (PBMCs) were isolated from 20 ml of blood obtained from the patient with LAMA2 c.8842G > A. And the wild-type control was obtained from a healthy donor, who was confirmed without any pathogenic variant, including LAMA2, by another WES. The third-generation PBMCs cells with good growth status were induced to differentiate into pluripotent stem cells. The PBMCs cells were cultured in a StemPro-34 complete medium containing cytokines IL-3, IL-6, SCF, and FLT-3 for 4 days. Sendai virus containing four reprogramming transcription factors [OCT3/4 (MOI = 3), SOX2 (MOI = 5), KLF4 (MOI = 3), and C-MYC (MOI = 5)] was added into the StemPro-34 complete culture medium (provided with a Sendai virus reprogramming kit) and cultured in a cell incubator for 24 h. For the first 3 days after transfection, the cells were maintained in the StemPro-34 complete culture medium without virus. Sendai virus-infected PBMCs were transferred to Matrigel-coated 12-well plate containing an E8 medium. iPSCs, (WCHi002-A) were manually picked at around day 20 post transfection and plated onto a Matrigel-coated 12- well plate. iPSCs differentiated to cardiomyocytes by modulating WNT signaling with CHIR99021 and IWR-1. iPSC-CMs were enriched by culture in lactate-containing media. Spontaneous beating activity can be observed in iPSC-CMs and the expression of myosin light chain 2 (MYL2) and cardiac troponin T (CTNT) can be detected by immunofluorescent staining.
Induced pluripotent stem cell-derived CMs were fixed in 4% paraformaldehyde overnight. The cells were incubated in a blocking medium (PBS containing 5% donkey serum, 0.2% Triton X-100) at 4°C overnight. For immunostaining, the sections were incubated with primary antibodies at 4°C overnight and secondary antibodies for 2 h. The primary antibodies used were as follows: sarcomeric alpha-actinin (1:200, Abcam, ab137346). After washing with a blocking buffer, the slices were incubated with optimal secondary antibodies with DAPI at room temperature for 2 h. After washing, the samples were imaged using an Olympus FV1000 confocal microscope (Figure 4A). Flow cytometry had been applied to demonstrate the purity of iPSC-CMs. Non-differentiated iPSC and differentiated iPSC were stained with cardiac troponin I (1:200, Abcam, ab56357). More than 80% of CMs had been identified (Figure 4B).
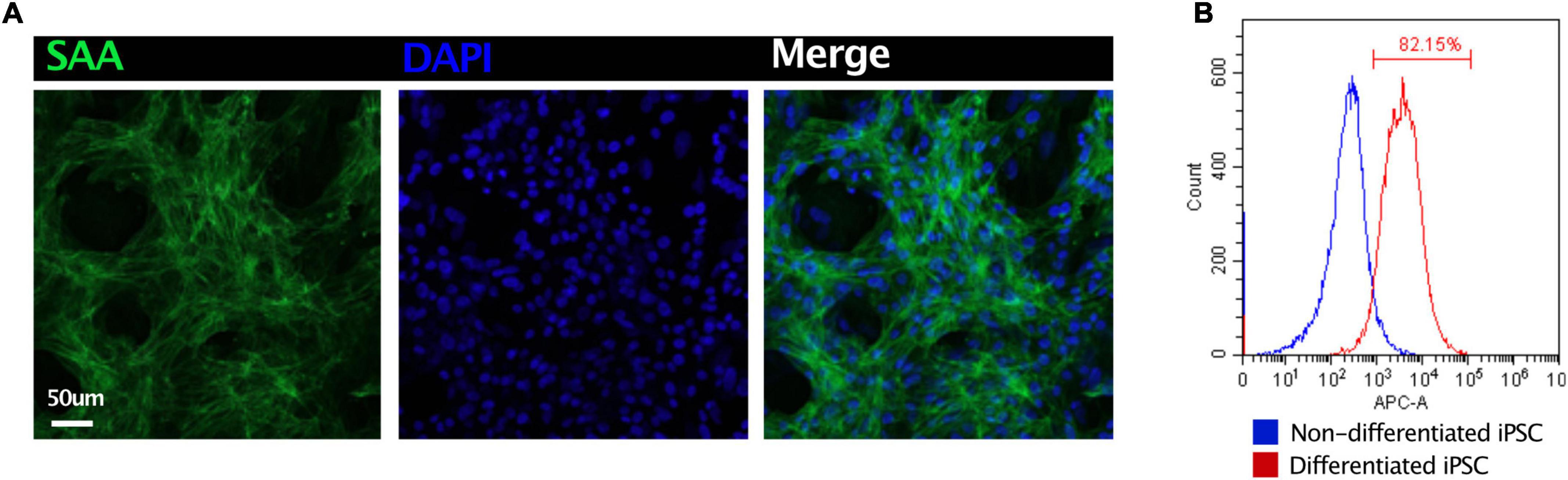
Figure 4. Quality control of iPSC-derived CMs. (A) Immunostaining of sarcomeric alpha-actinin (SAA) demonstrated the high ratio of CMs. (B) Flow cytometry revealed 82.5% iPSC had been differentiated into CMs.
The TRIzol method was used to extract total RNA from iPSC-CMs of ARVC (n = 3) and WT (n = 3) groups, and the DNase I method was used to remove DNA in the samples. An Illumina HiSeq XTEN/NovaSeq 6000 sequencing platform was used for high-throughput sequencing. Transcript abundance was determined by TopHat alignment followed by HTSeq-Count and statistical analysis by DESeq2. Enrichment analysis of differentially expressed genes (DEGs) was implemented by the clusterProfiler R package, in which gene length bias was corrected. KEGG pathways with corrected P-value less than 0.05 were considered significantly enriched by DEGs. According to the established pathophysiological characteristics of ARVC, five enrichment sets were established, including fibrosis, adipogenesis, apoptosis, intracellular calcium, and cellular connectivity (Figure 5A). The KEGG pathways of fibrosis were enriched in PI3K-Akt signaling and regulation of actin cytoskeleton (Supplementary Figure 1). The enrichment analysis for lipid metabolism revealed a general alternation of lipid metabolic dysfunction (Supplementary Figure 2). In addition, apoptosis had been recorded by KEGG enrichment (Supplementary Figure 3). Most important, ARVC was characterized as misleading calcium handling and cellular connection disorders. After KEGG analysis based on DEGs, the significant calcium signaling pathway and cGMP-PKG signaling pathway were identified (Supplementary Figure 4). Furthermore, the changes in adherens junction, cell adhesion molecules, and the hippo signaling pathway had been proved to be participated in pathological remodeling of cardiomyocyte due to LAMA2 mutation. And the enrichment of ARVC was definitely identified among all DEGs (Figure 5B).
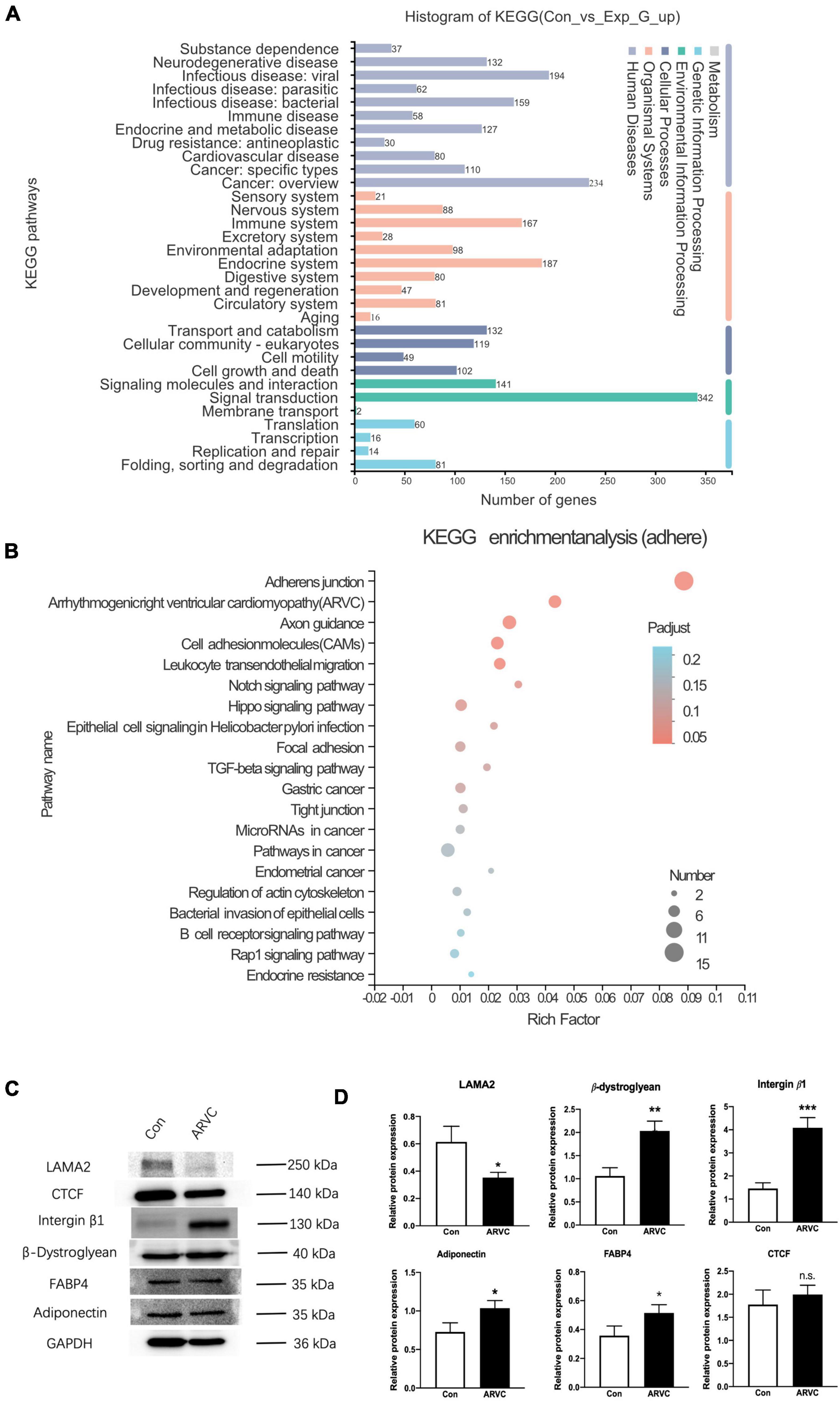
Figure 5. The analysis of iPSC-derived CMs indicated an ARVC-associated phenotype. (A) Histogram of KEGG signaling pathway classification. (B) KEGG enrichment analysis of the adherent signaling pathway. (C) Western blot results showed the protein expressions of LAMA2, CTCF, Integrin-β1, β-Dystroglycan, FABP4, and Adiponectin in myocardium cells of the control group and the ARVC group, with GAPDH as internal reference. (D) Histogram shows the protein expression differences of LAMA2, CTCF, Integrin-β1, β-Dystroglycan, FABP4, and Adiponectin between the control group and the ARVC group (N.S. The difference is meaningless; *p < 0.05; **p < 0.01; ***p < 0.001).
We queried all possible related signaling pathways of ARVC through KEGG website6, and found the LAMA2 and its related proteins integrin beta 1 (ITGB1) and β-muscle dystroglycan 1 (β-DG) are involved in the ARVC signaling pathway. The protein expressions of LAMA2, ITGB1, and β-DG were further detected by the Western blot. Normal healthy human PBMCs were successfully reprogrammed and inversely differentiated into iPSCs according to the above scheme, and were induced into cardiomyocytes as the control group. Both groups were continuously cultured in a myocardial maintenance medium for 50 days. Differentiated iPSC-CMs were lysed in RIPA lysis buffer system (Santa Cruz Biotechnology, sc-24948) with Mini Protease Inhibitor Cocktail Tablets (cOmplete, 4693124001). Total protein concentrations were normalized using BCA analysis (Life Technologies, 23227). After boiling with a 4X loading buffer for 5 min, 20-μl cell lysate of each sample was separated on a 10% gel, transferred to a PDVF membrane, and blocked by 4% BSA/TBST. Primary antibodies of LAMA2 (1:2,000, Abcam, ab140482), β-DG (1:2,000, Abcam, ab3125), ITGB1 (1:2,000, Abcam, ab52971), Adiponectin (1:2,000, Abcam, ab181281), FABP4 (1:2,000, Abcam, ab92501), CTGF (1:2,000, Abcam, ab209780), and GAPDH (1:2,000, ProteinTech, 60004-1-lg). The data were analyzed with GraphPad Prism 8.0 statistical software by t-test. LAMA2 protein expression in the ARVC group demonstrated significant decreased (p = 0.02), ITGB1 (p = 0.001), and β-DG (p = 0.004) protein expressions were significantly upregulated (Figures 5C,D).
Discussion
This study demonstrated a rare clinical case with a LAMA2 genetic heterozygous mutation (c.8842G > A, p.G2948S). The patient was initially diagnosed with ARVC based on clinical presentation and radiological images examinations according to Task Force Criteria. After several years of medication administration, the irreversible heart failure and aggressive cardiac pathological remodeling led this patient to receive a heart transplantation at the age of 14. Before the heart transplantation, WES had been applied to this boy. Interestingly, the LAMA2 c.8842G > A variant had been identified at the first time among patients with ARVC. However, the patient was absent from any other potential-related variants. So that it is critical to demonstrate the relationship between LAMA2 and ARVC. Within this research, we enrolled histological section assessment to explore the pieces of evidence on standard ARVC changes. Most important, we reprogrammed the PBMCs of this proband to iPSC. After differentiating iPSC-CMs successfully, bulk RNA-seq was involved to reveal the molecular alternations post LAMA2 mutation, which would definitely help to underline the association between LAMA2 and ARVC.
Arrhythmogenic right ventricular cardiomyopathy has been characterized as a heritable disorder with palpitations, syncope, ventricular tachycardia (VT), or fibrillation. Ventricular dysfunction and heart failure can also develop can be found in some patients. This kind of disease was initially considered to be a developmental abnormality of the right ventricle. With the emerging pieces of evidence on molecular diagnosis, a novel genotype-based clinicopathology classification of arrhythmogenic cardiomyopathy (ACM) has been well established. Several specific mutations within desmosome would result in biventricular or only left ventricular phenotype, such as DSP, PLN, and CTNNA3. Besides, large unknown mutations have been claimed to be associated with left ventricular failure, leading to heart transplantation. For this patient, LAMA2 c.8842C > A could be one of the potential mutations to cause cluster 4 ACM. After the research database, only four reports have been retrieved on LAMA2 c.8842C > A. Three of them were identified among patients with muscular dystrophy, and only one record demonstrated a potential link with primary dilated cardiomyopathy. In addition, the iPSC-CMs provided a good platform to study the molecular mechanisms due to an uncertain mutation. The bulk RNA-seq results demonstrated alternations among calcium handling, impaired cell-cell interaction, reduced cellular adhesion, and dysfunction of lipid metabolism. All the changes in gene expressions and regulation fall within the pathological remodeling of ARVC. Taking the histological studies of the proband’s abandoned heart, it would draw a highly convinced association between LAMA2 c.8842C > A and ARVC.
After reviewing all the published cases of LAMA2 missense mutation, a case series of 57 particular variants was assembled and summarized in Table 1. Most of the mutations were correlated with the symptom of muscular dystrophy, and there was no association with cardiac-related clinical manifestations in theses mutations. Among them, 15 variants were located in the N-terminal domain, 18 variants were located in EGF-like domains, and 16 variants were located in LG domains. But there was no reported pathogenic missense mutation in the LG5 domain.
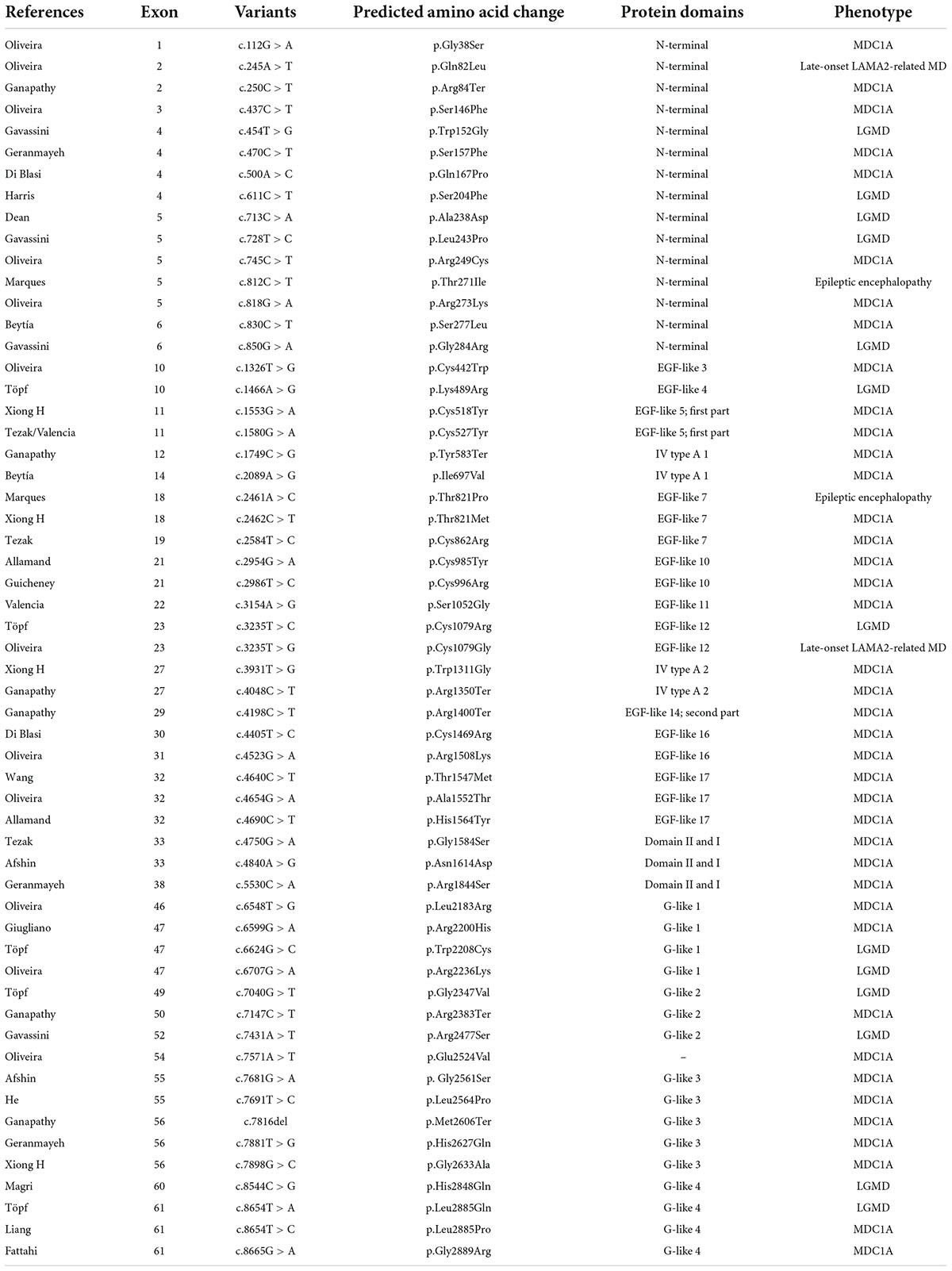
Table 1. A summary of reported cases of LAMA2 missense mutation.
The laminin-211 structure consists of α2, β1, and γ1 chains. The polymeric activity of laminin-211 maps to three domains of LN: nidogen binds γ 1-Leb3, agrin binds the curl γ1 subunit, integrin α7β1 binds LG1-3, α-dystroglycan (α-DG) binds to LG4-5 with sulfated glycolipids. LG interactions provide anchoring for the cell membrane and cytoskeleton. α-DG is coupled to transmembrane β-DG, β-DG binds to cytoskeletal dystrophin, and dystrophin binds to the actin-rich cytoskeleton; this critical interaction creates a link from the basement membrane to actin cytoskeleton (11). Previous studies demonstrated homozygous or compound heterozygous mutations that result in deletion of the α2 subunit or diverse aggregation of laminin-211 lead to muscular dystrophy with peripheral nerve defects like MDC1A or LGMD.
In this case, the heterozygous mutation was located in the LG5 domain. We used SFIT, PolyPhen-2, and MutationTaster to predict the impact of this mutation on protein structure and function, showing that p.G2948S is a diseasing-causing and protein-damaging mutation. The Western blot showed that the expression of LAMA2 protein decreased in the ARVC group, and the expression of LAMA2-interacting proteins (ITGB1, β-DG) increased as a compensatory regulation. Using bulk RNA-seq, it confirmed that DEGs in iPSC-CMs of the ARVC group were abundant in signaling pathways of inflammation, fibrosis, adipose, cell connection, and apoptosis. Thus, we elucidated that p.G2948S of LAMA2 induced the change of the LG5 domain and induce the compensatory expression of ITGB1 and β-DG, and led to the activation of ARVC changes. Moreover, the impaired binding function of the LG5 domain would reshape the cytoskeleton, leading cardiac failure as a major clinical concern. There are still some limitations in this study. We did not perform the isogenic iPSC line based on this patient. At the same time, the distribution of LAMA2 by immunostaining of iPSC-CM had not been investigated, which we would carry out further analysis in the following studies.
Conclusion
In summary, this is the first case report to describe an ARVC phenotype with a LAMA2 mutation of c.8842G > A (p.G2948S), which is located in the LG5 domain. In addition, the first iPSC cell line has been established based on the proband. Histological studies and bulk RNA-seq have demonstrated a convinced association between LAMA2 c.8842C > A and ARVC. Considering 35–50% of patients with a clinical diagnosis of ARVC fail to address a particular disease-causing gene mutation, this research expands the understanding of the pathogenesis of ARVC. However, the molecular mechanism of LAMA2 mutation inducing ARVC still requires further study.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of West China Second Hospital of Sichuan University (2014-034). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
SL was the patient’s physician and responsible for the revision of the manuscript for important intellectual content. YW reviewed the literature and contributed to manuscript drafting. YW and YL performed the mutation analysis. DZ and YF contributed to the iPSC-related experiments. YL and SL conceptualized and designed the study, coordinated and supervised the data collection, and critically reviewed the manuscript for important intellectual content. All authors issued final approval for the version to be submitted.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81700360) and Technology Project of Sichuan Province of China (Nos. 2020YFS0101 and 2020YFS0102).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.922347/full#supplementary-material
Supplementary Figure 1 | KEGG enrichment analysis of the fibrosis signaling pathway.
Supplementary Figure 2 | KEGG enrichment analysis of the lipidation signaling pathway.
Supplementary Figure 3 | KEGG enrichment analysis of the apoptosis signaling pathway.
Supplementary Figure 4 | KEGG enrichment analysis of the calcium signaling pathway.
Footnotes
- ^ https://swissmodel.expasy.org/
- ^ http://biosig.unimelb.edu.au/mcsm/
- ^ http://marid.bioc.cam.ac.uk/sdm2
- ^ http://biosig.unimelb.edu.au/dynamut/
- ^ http://biosig.unimelb.edu.au/duet/
- ^ https://www.genome.jp/kegg
References
1. Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. (2017) 376:61–72. doi: 10.1056/NEJMra1509267
2. Austin KM, Trembley MA, Chandler SF, Sanders SP, Saffitz JE, Abrams DJ, et al. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. (2019) 16:519–37. doi: 10.1038/s41569-019-0200-7
3. Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C, et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ Cardiovasc Genet. (2015) 8:437–46.
4. Lazzarini E, Jongbloed JD, Pilichou K, Thiene G, Basso C, Bikker H, et al. The ARVD/C genetic variants database: 2014 update. Hum Mutat. (2015) 36:403–10.
5. Yurchenco PD, Patton BL. Developmental and pathogenic mechanisms of basement membrane assembly. Curr Pharm Des. (2009) 15:1277–94. doi: 10.2174/138161209787846766
6. Yurchenco PD. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol. (2011) 3:a004911. doi: 10.1101/cshperspect.a004911
7. Sasaki T, Giltay R, Talts U, Timpl R, Talts JF. Expression and distribution of laminin alpha1 and alpha2 chains in embryonic and adult mouse tissues: an immunochemical approach. Exp Cell Res. (2002) 275:185–99. doi: 10.1006/excr.2002.5499
8. Geranmayeh F, Clement E, Feng LH, Sewry C, Pagan J, Mein R, et al. Genotype–phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromusc Disord. (2010) 20:241–50. doi: 10.1016/j.nmd.2010.02.001
9. Jones KJ, Morgan G, Johnston H, Tobias V, Ouvrier RA, Wilkinson I, et al. The expanding phenotype of laminin 2 chain (Merosin) abnormalities: case series and review. Med Genet. (2001) 38:649–57. doi: 10.1136/jmg.38.10.649
10. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. (2010) 31:806–14. doi: 10.1093/eurheartj/ehq025
Keywords: LAMA2, ARVC, iPSC, case report, literature review
Citation: Wang Y, Fang Y, Zhang D, Li Y and Luo S (2022) A rare case of arrhythmogenic right ventricular cardiomyopathy associated with LAMA2 mutation: A case report and literature review. Front. Med. 9:922347. doi: 10.3389/fmed.2022.922347
Received: 17 April 2022; Accepted: 23 June 2022;
Published: 18 July 2022.
Edited by:
Jian Gao, Shanghai Children’s Medical Center, ChinaReviewed by:
Feng Hu, Shanghai Jiao Tong University, ChinaJiajia Yang, University of South Florida, United States
Copyright © 2022 Wang, Fang, Zhang, Li and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yifei Li, bGl5Zndjc2hAc2N1LmVkdS5jbg==; Shuhua Luo, bHVvc2h1aHVhQHNjdS5lZHUuY24=
†These authors have contributed equally to this work