 Qingdan Xu
Qingdan Xu Ping Sun
Ping Sun Chaoyi Feng
Chaoyi Feng Qian Chen
Qian Chen Xinghuai Sun
Xinghuai Sun Yuhong Chen
Yuhong Chen Guohong Tian
Guohong Tian
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Med. , 04 July 2022
Sec. Ophthalmology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.912103
This article is part of the Research Topic Eye in Systemic Diseases View all 21 articles
The T12811C mitochondrial DNA (mtDNA) mutation has been reported in Leber hereditary optic neuropathy (LHON) previously, with vision loss as the main manifestation. The involvement of other organ systems, including the central and peripheral nervous system, heart, and extraocular muscles, has not been well described. This case series report investigated four patients with T12811C mtDNA mutation, verified through a next generation sequencing. Two male patients presented with bilateral subacute visual decrease combined with involvement of multiple organ systems: leukoencephalopathy, hypertrophic cardiomyopathy, neurosensory deafness, spinal cord lesion and peripheral neuropathies. Two female patients presented with progressive ptosis and ophthalmoplegia, one of whom also manifested optic atrophy. This study found out that patients harboring T12811C mtDNA mutation manifested not only as vision loss, but also as a multi-system disorder affecting the nervous system, heart, and extraocular muscles.
Leber hereditary optic neuropathy (LHON) is a maternally inherited ocular disease leading to painless, progressive, bilateral loss of central vision in young adults, predominately affecting males (1–5). Although visual dysfunction is the most common symptom of LHON, other systemic abnormalities can also occur. These include dystonia, Parkinsonism, extra-pyramidal motor symptoms, peripheral neuropathy, cardiac conduction abnormalities, spinal cord disease, and skeletal muscle abnormalities (6–13). The maternal transmission indicates that mutations in the mitochondrial DNA (mtDNA) play key roles in the development of LHON. Three mtDNA variants are considered as primary mutations causing LHON, namely, ND4 G11778A, ND1 G3460A, and ND6 T14484C (14, 15). These three mtDNA mutations account for over 95% of LHON cases worldwide, while the remaining LHON cases may be caused by other variants (4).
The T12811C (Y159H) mutation is located in highly conserved residues in ND5 that encodes the subunit of respiratory chain complex I (16). This missense mutation results in the substitution from tyrosine to histidine. In previous investigations, Huoponen et al. reported that the ND5 T12811C mutation acted as a secondary mutation that increased the penetrance of the ND4 G11778A mutation in LHON (17). Here, we investigated the clinical phenotypes of the T12811C mutation in four Chinese patients diagnosed with LHON and other systemic diseases. The detailed clinical manifestations and genetic characteristics were evaluated. Our study reveals the genetic heterogeneity of mitochondrial diseases and expands the phenotypic spectrum of T12811C mutation.
This study was approved by the Institutional Review Board of Eye, Ear, Nose, and Throat Hospital of Fudan University (KJ2011-04). Written consent forms were obtained from all participants or their guardians. Four Han Chinese patients who had ND5 T12811C mutation were enrolled at the Neuro-Ophthalmology Division of the Eye, Ear, Nose, and Throat Hospital of Fudan University, Shanghai, China.
Detailed medical histories were recorded for all the patients, especially the family histories as well as histories of smoking and drinking. All the patients underwent complete neuro-ophthalmologic evaluations including assessment of best-corrected visual acuity (BCVA), color vision using Ishihara plates, relative afferent papillary defect (RAPD), slit-lamp biomicroscopy, fundoscopy, Goldmann/Octopus visual field perimetry, and spectral-domain optical coherence tomography (OCT). Other ancillary tests focused on neurological system, heart, and hearing were performed according to the patients’ different symptoms. Ophthalmic examinations and neurological evaluations were also conducted for all the parents.
The total DNA was isolated from peripheral blood of the four patients. For case 1, case 3, and case 4, next generation sequencing was utilized and a panel of 194 optic-atrophy associated genes were sequenced using the Illumina HiSeq 2000 (Illumina, Inc., San Diego, CA, United States) sequencing system. The panel covered the whole mtDNA genome and the nucleus genes related to mitochondrial disorders. The average coverage depth was 300 times. Ninety nine percent of the target region received 20 times coverage or more. For case 2, a small panel covering nine optic-atrophy associated nucleus genes (RTN4IP1, YME1L1, OPA1, OPA3, TMEM126A, DNM1L, ACO2, WFS1, CISD2) and the whole mtDNA genome were sequenced.
A 25-year-old man presented with painless vision loss in both eyes for 1 month. He noticed he could not see clearly in the right eye for 10 days and when first examined, both eyes’ visual acuity was abnormal. He was diagnosed with hypertrophic cardiomyopathy 7 years ago. He complained of hearing loss for years and the pure tone audiometry demonstrated moderate neurosensory hearing loss for both ears. He had no family history of similar diseases and denied smoking or drinking alcohol. The BCVA was hand motion in his right eye and 20/400 in his left eye. Color vision was 0/8 in both eyes. There was no RAPD in the right eye. Funduscopic examination showed diffuse pallor in his right eye and temporal atrophy of optic disc in his left eye, with bilateral tortuous retinal vein (Figure 1A). Goldmann visual field tests revealed central scotomas in both eyes. OCT demonstrated thinning of inferior-temporal retinal nerve fiber layer (RNFL) in the right eye and severe ganglion cell-inner plexiform layer (GCIPL) loss in both eyes (Figure 1B). T2 Flair-weighted magnetic resonance imaging (MRI) of the brain showed hyperintense signal changes involving the posterior limbs of internal capsule, occipital lobes and corpus callosum indicating leukoencephalopathy (Figure 1C). T1-weighted MRI of the orbits with gadolinium and fat suppression showed slight enhancement of the bilateral optic nerves in both axial (Figure 1D) and coronal (Figure 1E) views. The audiography demonstrated bilateral neurosensory deafness (Figure 1F). Genetic testing results showed the T12811C mutation with the heteroplasmy at the level of 99.81%. Another variant of G3946A in ND1 with the heteroplasmy of 16.45% was also detected. The patient was diagnosed with Leber plus syndrome and prescribed coenzyme Q 10 together with idebenone. His visual acuity stabilized at Snellen 20/400 for both eyes after 1 year of follow-up (Table 1).
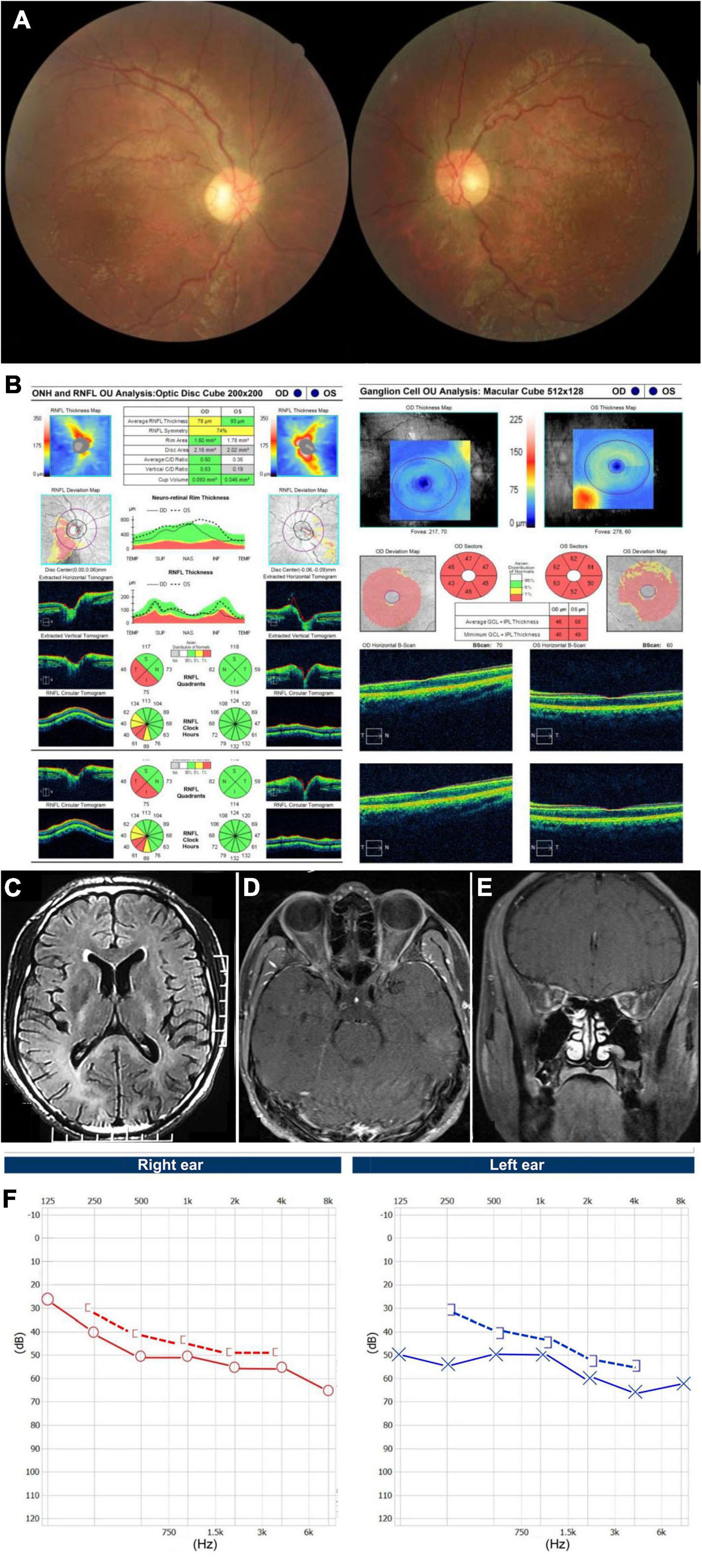
Figure 1. Accessory examinations of case 1. (A) Fundus photos showed diffuse pallor in the right eye and temporal pallor in the left eye, with tortuous retinal vein bilaterally. (B) OCT showed thinning of inferior-temporal RNFL in the right eye and severe GCIPL loss bilaterally. (C) Axial T2 Flair-weighted MRI of the brain showed hyperintense signal changes involving the posterior limbs of internal capsule, occipital lobes and corpus callosum indicating leukoencephalopathy. (D) Axial and (E) coronal T1-weighted MRI of orbits with gadolinium and fat suppression showed bilateral optic nerve slight enhancement. (F) Audiography showed bilateral neurosensory deafness.
Table 1. Clinical manifestations of the four cases.
A 58-year-old man presented with bilateral progressive visual decrease within 2 months, accompanied by weakness and numbness of his legs. He noticed both eyes blurry in the morning without periorbital pain or headache. He did not have any family history of similar diseases. He denied taking any neurotoxic medication, smoking or drinking alcohol. The BCVA was 20/200 and color vision was 0/8 in both eyes. The fundus showed temporal pallor of the bilateral optic discs (Figure 2A). The neurological examination only showed bilateral peripheral neuropathies. Muscle strength was level IV but with paresthesia of both arms and legs. Octopus visual fields showed central scotomas in both eyes (Figure 2B). The OCT showed inferior-temporal RNFL thinning in the right eye and temporal RNFL thinning in the left eye, with significant thinning of the GCIPL in both eyes (Figure 2C). The brain MRI showed scattered T2 Flair-weighted hyperintense signal and orbital MRI only showed bilateral optic nerve atrophy without enhancement in T1-weighted imaging after contrast (Figure 2D). A small pituitary tumor was reported which was irrelevant to optic neuropathy. There was no reported spinal cord lesion. Genetic testing revealed the T12811C mutation with the heteroplasmy at the level of 98.93%. The patient was diagnosed with Leber plus syndrome and prescribed cobamamide together with coenzyme Q 10 and idebenone. His visual acuity was stabilized at Snellen 20/200 for both eyes during follow-up (Table 1).
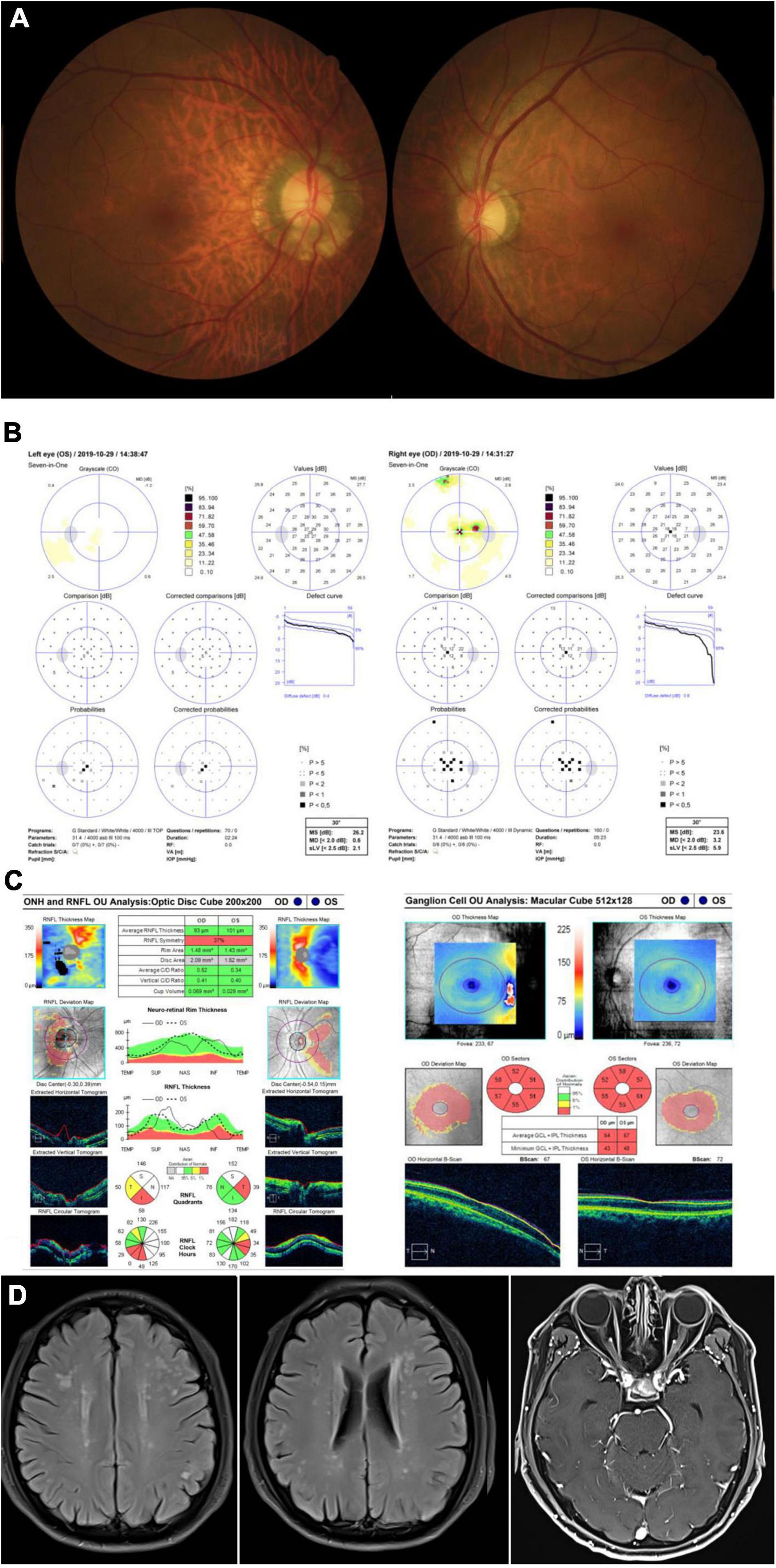
Figure 2. Accessory examinations of case 2. (A) Fundus photos showed temporal pallor of optic disc bilaterally. (B) Octopus visual fields showed central scotomas bilaterally. (C) OCT revealed inferior-temporal RNFL thinning in the right eye and temporal RNFL thinning in the left eye, with significant thinning of the GPICL bilaterally. (D) Brain MRI showed scattered T2 Flair-weighted hyperintense signal and orbital T1-weighted MRI imaging after contrast showed bilateral optic nerve atrophy without enhancement.
A 32-year-old woman complained of progressive bilateral ptosis and strabismus since she was 20 years old. She underwent strabismus surgery 10 years ago. She had no family history of similar diseases and denied smoking or drinking alcohol. Tests for ocular myasthenia gravis, including acetylcholine receptors antibody, single-fiber electromyography, repetitive nerve stimulation, and chest computed tomography scan were all negative. The BCVA was 20/60 in her right eye and 20/30 in her left eye. Color vision was 4/8 in her right eye and 6/8 in her left eye. Funduscopic examination revealed temporal pallor of the optic disc in her right eye (Figure 3A). Eye movement was impaired in both eyes, including adduction, abduction, elevation, and depression (Figure 3B). The OCT demonstrated thinning of the RNFL superiorly and inferiorly in the right eye and diffuse bilateral thinning of GCIPL (Figure 3C). The brain and orbital MRI showed thinning of extraocular muscles without other brain lesions. Genetic testing results detected the T12811C mutation only with the heteroplasmy at the level of 99.49%. No gene fragment deletion that was responsible for chronic progressive external ophthalmoplegia (CPEO) syndrome was found. She was finally diagnosed with mitochondrial disease with LHON and CPEO syndrome and prescribed coenzyme Q 10 together with idebenone (Table 1).
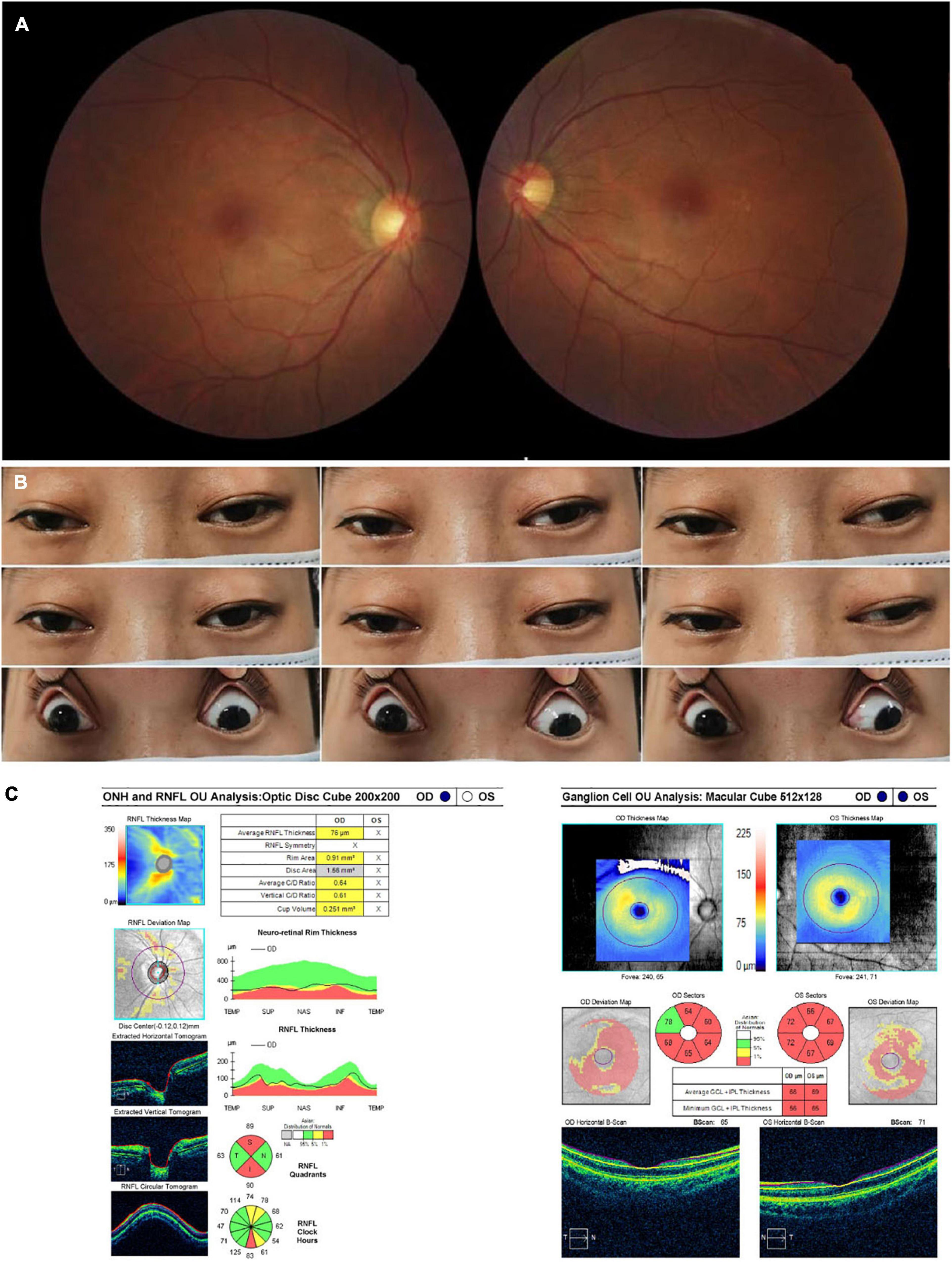
Figure 3. Accessory examinations of case 3. (A) Fundus photos showed temporal pallor in the right eye. (B) Nine cardinal eye positions showed impaired mobility in both eyes, including adduction, abduction, elevation, and depression. (C) OCT showed thinning of RNFL superiorly and inferiorly in the right eye, with diffuse thinning of GCIPL bilaterally. The RNFL thickness of the left eye was not available due to poor cooperation.
A 16-year-old woman presented with bilateral droopy eyelids and extraocular muscle weakness for about 5 years. She had no family history of similar diseases and denied smoking or drinking alcohol. The BCVA was 20/20 and color vision was 8/8 in both eyes. The fundoscopy revealed normal optic discs without retinopathy in both eyes (Figure 4A). Ophthalmic examination only revealed bilateral ptosis and exotropia. Adduction, elevation, and depression were moderately reduced in both eyes. Abduction was slightly reduced in the right eye and almost full in the left eye (Figure 4B). The OCT revealed normal retina thickness in both eyes (Figure 4C). The brain and orbital MRI showed thinning of extraocular muscles without other brain lesions. Genetic testing results showed the T12811C mutation with the heteroplasmy at the level of 99.94%. No gene fragment deletion which was usually found in CPEO patients was detected in this patient. She was finally diagnosed as ophthalmoplegia with mitochondrial disease and prescribed coenzyme Q 10 together with idebenone (Table 1).
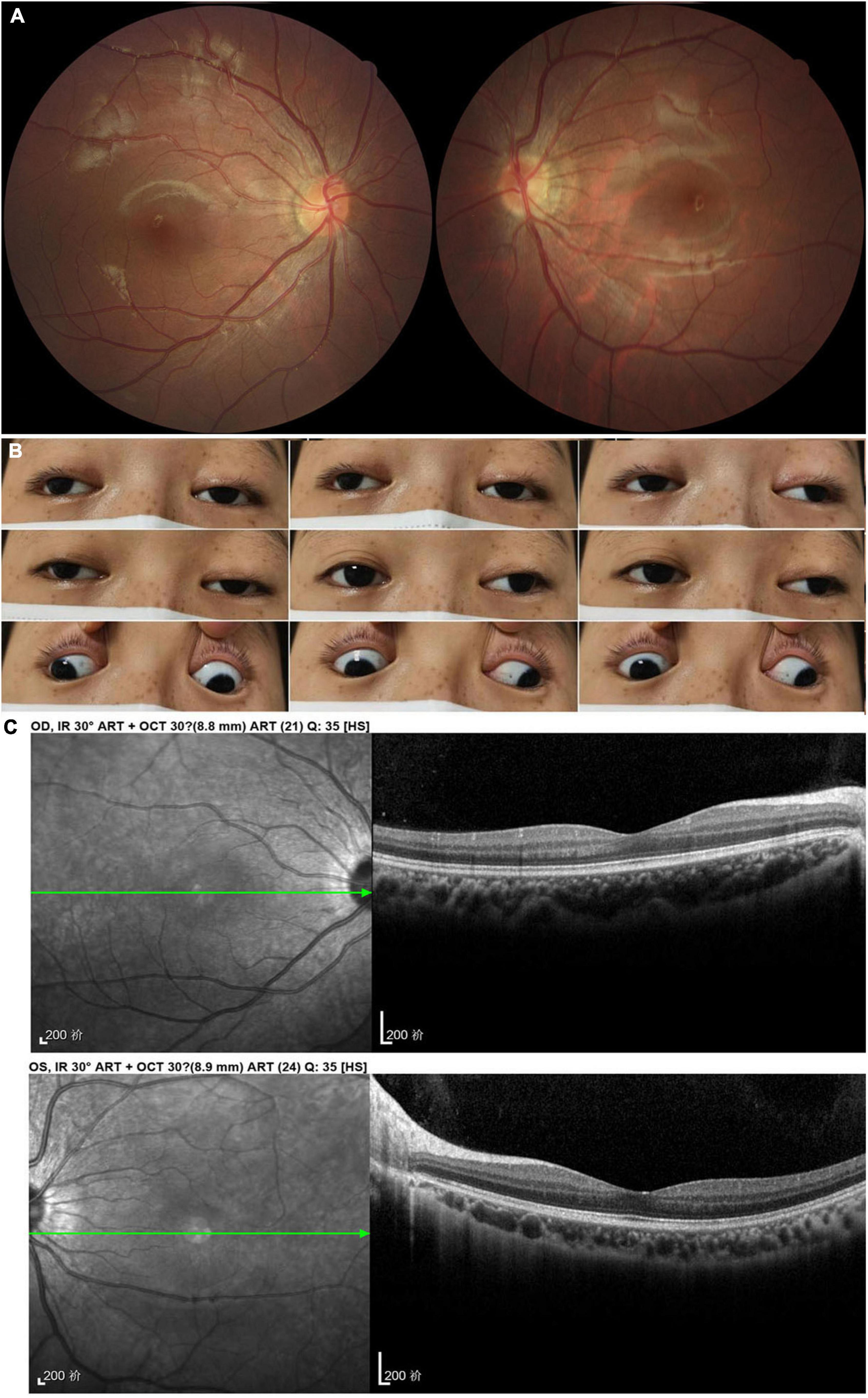
Figure 4. Accessory examinations of case 4. (A) Fundus photos showed normal optic disc and retina bilaterally. (B) Nine cardinal eye positions revealed adduction, elevation, and depression were moderately reduced bilaterally, and abduction was slightly reduced in the right eye and almost full in the left eye. (C) OCT showed normal retina thickness and macular structure bilaterally.
This case series study reported the multi-system phenotypes caused by the T12811C mutation. Our patients with an identical T12811C mutation in ND5 showed different clinical symptoms: vision loss, hypertrophic cardiomyopathy, hearing loss, paraplegia, ptosis, and extraocular muscle weakness. These clinical findings showed the broad phenotypic spectrum of the single point mutation of T12811C in mtDNA. It involved multiple systems, such as optic atrophy, cardiac abnormality, leukoencephalopathy, cochlea lesion, spinal cord lesion, peripheral neuropathies, and extraocular ophthalmoplegia.
The pathophysiology of LHON is complex. It typically manifests as an acute or subacute loss of central vision, a result of optic atrophy secondary to severe damage of retinal ganglion cells (18). The accompanying abnormalities, mainly neurological disorders, have also been reported in some patients (19). Three particular variants of G11778A, T14484C, G3460A in mtDNA are confirmed as primary mutations and explain over 95% of LHON patients worldwide (20–22). T12811C was initially published as a secondary LHON-associated mutation exacerbating the visual loss of LHON (16, 17). Recently, T12811C was reported as a primary mutation for the first time in one case, who presented as isolated LHON (23). Our group of patients manifested as multi-system disorders, thereby expanding the phenotypes in association with the T12811C mutation.
The T12811C mutation changes an evolutionarily conserved tyrosine to a histidine in the transmembrane region of ND5, the core subunit of complex I (16) (Figure 5). This mutation was predicted to alter the structure of the transmembrane region of the ND5 protein (24). ND5 plays an important role in the stability and activity of complex I (25). Researches have revealed that ND5 protein synthesis is the rate-limiting step for complex I activity and the highest rate of ND5 protein synthesis is just sufficient to maintain a normal respiratory rate (26–28). This may explain the experimental results that defects in ND5 can lead to a significant decrease in rates of mitochondrial respiration (28, 29). Hence, we consider the mutation T12811C in the ND5 gene to be pathogenic and responsible for the clinical phenotypes in our patients.
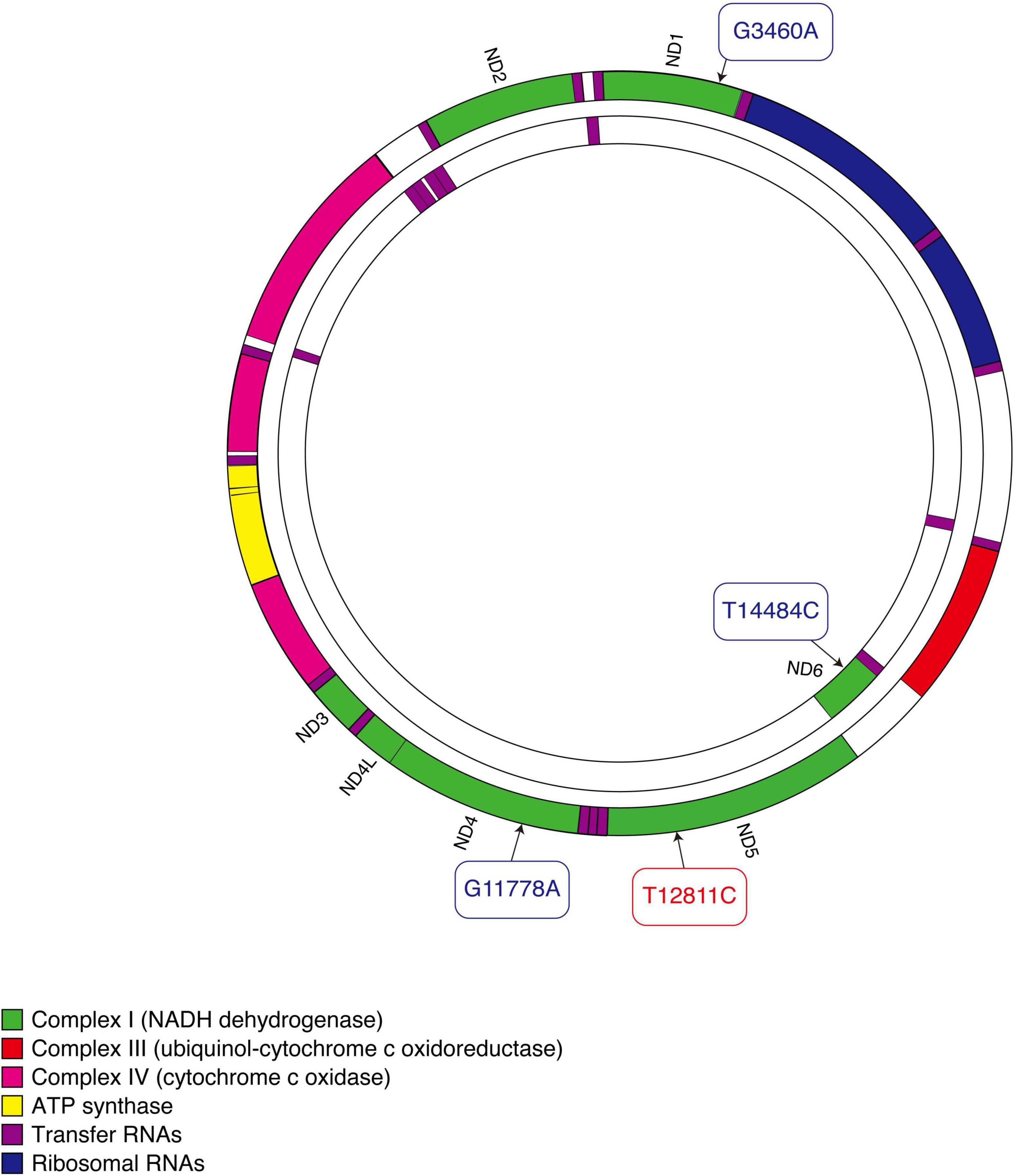
Figure 5. Schematic diagram of mitochondrial DNA. Three primary mutations causing LHON are shown in blue. The mutation that our patients carried is shown in red.
Furthermore, previous studies suggested that mutations in the ND5 gene were more likely to be associated with an extended phenotype rather than isolated visual dysfunction (30). Diseases caused by the mutations in ND5 would exhibit various degrees of clinical heterogeneity, such as Leigh syndrome (31), Idiopathic Parkinson’s disease (32), mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS) (33), myoclonic epilepsy with ragged-red fibers (MERRF) (34), and hypertrophic cardiomyopathy (35). Strikingly, neurological and muscular disorders are the main problems caused by the mutations in ND5 gene, which were also observed in our patients. Besides, deafness (36), paraplegia (37), ptosis (38), strabismus (39), and ophthalmoplegia (40) have also been occasionally seen in patients with mutations in the ND5 gene. To the best of our knowledge, our study is the first report describing ophthalmoplegia as the only symptom in such patients. Since no gene fragment deletion responsible for CPEO syndrome was found in our cases, ophthalmoplegia is most likely a phenotype of T12811C. Unfortunately, the biopsy of muscle was rejected in the two patients with opthalmoplegia according to patients’ will. The histochemistry of COX and SDH together with detected mtDNA variants in muscle sample would be a great help in the diagnosis of mitochondrial CPEO.
Some researchers have proposed hypotheses explaining how the same gene mutation can cause different phenotypes. Patients with mitochondrial diseases harbor both mutant and wild-type mtDNAs. During mitosis, the mtDNA is divided and stochastically distributed to the next generation. If the mutation load surpasses the threshold in the tissue, affected patients will present corresponding clinical manifestations (41). Since tissues with high demand for energy metabolism have lower thresholds, organs like brain, heart, skeletal muscle, optic nerve, and retina are more vulnerable to the pathogenic effects of mtDNA mutations (42). Other modifier factors including nuclear modifier genes (43), epigenetic phenomena (44), secondary mtDNA mutations (45), mitochondrial haplogroup (46), or environmental factors (47), also get involved in modifying the clinical expression and consequently cause differences in phenotypes among patients.
Similarly, additional factors may play a role in the phenotypic manifestation of the T12811C mutation as well. Here, all of our patients denied smoking and drinking alcohol, which were considered as the major environmental factors for LHON (47). However, in addition to T12811C, another variant G3946A was detected in case 1, although with relatively low heteroplasmy. The G3946A mutation can affect the assembly or turnover of complex I. It was reported to be associated with MELAS and hearing loss and the case 1 happened to have leukoencephalopathy and hearing loss as well (48). This mtDNA variant might interact with T12811C, triggering or exacerbating the expression of the phenotype. Meanwhile, all our patients harbored a single nucleotide polymorphism of A10398G, which was previously reported to increase the penetrance of primary G11778A mutation of LHON (49). However, due to its high frequency in Asian population (66%) (50), the effect of A10398G mutation on our patients remains unclear. Thus, there are still possibilities that the involved additional genetic and/or environmental factors were unrevealed in our patients.
In summary, we reported four patients with T12811C mtDNA mutation presenting with various clinical phenotypes as LHON, hypertrophic cardiomyopathy, leukoencephalopathy, cochlea lesion, spinal cord lesion, peripheral neuropathies, and ophthalmoplegia. Mitochondrial disease should be considered as one of the differential diagnoses in patients manifesting with optic atrophy or ophthalmoplegia.
The studies involving human participants were reviewed and approved by the Institutional Review Board of Eye, Ear, Nose, and Throat Hospital of Fudan University. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
YC and GT conceptualized and designed the study. GT recruited the patients and collected the data. CF and QC obtained patients’ consent, examined, and followed-up the patients. QX and PS analyzed the genetic testing results, reviewed the literature, and wrote the manuscript. XS analyzed the cases and revised the manuscript. All authors approved the final manuscript.
This work was supported by the Shanghai Committee of Science and Technology, China (grant no. 20S31905800), the National Science Foundation of China (grant no. 81870692), the Shanghai Municipal Health Commission (grant no. 20214Y0073), and the Clinical Research Plan of SHDC (grant no. SHDC2020CR6029).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to the Biobank of the Eye and ENT Hospital of Fudan University. We would also like to thank all the patients and their families.
1. Newman NJ, Wallace DC. Mitochondria and Leber’s hereditary optic neuropathy. Am J Ophthalmol. (1990) 109:726–30.
2. Yu-Wai-Man P, Griffiths PG, Hudson G, Chinnery PF. Inherited mitochondrial optic neuropathies. J Med Genet. (2009) 46:145–58. doi: 10.1136/jmg.2007.054270
3. Carelli V, La Morgia C, Valentino ML, Barboni P, Ross-Cisneros FN, Sadun AA. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim Biophys Acta. (2009) 1787:518–28. doi: 10.1016/j.bbabio.2009.02.024
4. Man PYW, Turnbull DM, Chinnery PF. Leber hereditary optic neuropathy. J Med Genet. (2002) 39: 162–9. doi: 10.1136/jmg.39.3.162
5. Sadun AA, La Morgia C, Carelli V. Leber’s hereditary optic neuropathy. Curr Treat Option Neurol. (2011) 13:109–17. doi: 10.1007/s11940-010-0100-y
6. Hanemann CO, Hefter H, Schlaug G, Seitz RJ, Freund HJ, Benecke R. Characterization of basal ganglia dysfunction in Leber ‘Plus’ disease. J Neurol. (1996) 243:297–300. doi: 10.1007/Bf00868531
7. Chu CC, Huang CC, Kao LY, Kuo HC, Yu TN, Tso DJ, et al. Clinical phenotype and the G11778A mutation of mitochondrial DNA in patients with Leber’s hereditary optic neuropathy in Taiwan. Neuro Ophthalmology. (2001) 26:207–16. doi: 10.1076/noph.26.4.207.15871
8. Bruyn GW, Vielvoye GJ, Went LN. Hereditary spastic dystonia - a new mitochondrial encephalopathy - putaminal necrosis as a diagnostic sign. J Neurol Sci. (1991) 103:195–202. doi: 10.1016/0022-510x(91)90164-3
9. Harding AE, Sweeney MG, Miller DH, Mumford CJ, Kellarwood H, Menard D, et al. Occurrence of a multiple sclerosis-like illness in women who have a Lebers hereditary optic neuropathy mitochondrial-DNA mutation. Brain. (1992) 115:979–89. doi: 10.1093/brain/115.4.979
10. Johns DR, Smith KH, Savino PJ, Miller NR. Lebers hereditary optic neuropathy - clinical manifestations of the 15257 mutation. Ophthalmology. (1993) 100:981–6.
11. Newman NJ. Lebers hereditary optic neuropathy - new genetic considerations. Arch Neurol Chicago. (1993) 50:540–8. doi: 10.1001/archneur.1993.00540050082021
12. Nikoskelainen EK, Marttila RJ, Huoponen K, Juvonen V, Lamminen T, Sonninen P, et al. Lebers plus - neurological abnormalities in patients with Lebers hereditary optic neuropathy. J Neurol Neurosurg Psychiatry. (1995) 59:160–4. doi: 10.1136/jnnp.59.2.160
13. Nikoskelainen EK, Savontaus ML, Wanne OP, Katila MJ, Nummelin KU. Leber’s hereditary optic neuroretinopathy, a maternally inherited disease. A genealogic study in four pedigrees. Arch Ophthalmol. (1987) 105:665–71. doi: 10.1001/archopht.1987.01060050083043
14. Brown MD, Torroni A, Reckord CL, Wallace DC. Phylogenetic analysis of Lebers hereditary optic neuropathy mitochondrial DNAs indicates multiple independent occurrences of the common mutations. Human Mutation. (1995) 6:311–25. doi: 10.1002/humu.1380060405
15. Mackey DA, Oostra RJ, Rosenberg T, Nikoskelainen E, BronteStewart J, Poulton J, et al. Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy. Am J Hum Genet. (1996) 59:481–5.
16. Cai W, Fu Q, Zhou X, Qu H, Tong Y, Guan MX. Mitochondrial variants may influence the phenotypic manifestation of Leber’s hereditary optic neuropathy-associated ND4 G11778A mutation. J Genet Genomics. (2008) 35:649–55. doi: 10.1016/S1673-8527(08)60086-7
17. Huoponen K, Lamminen T, Juvonen V, Aula P, Nikoskelainen E, Savontaus ML. The spectrum of mitochondrial-DNA mutations in families with Leber hereditary optic neuroretinopathy. Hum Genet. (1993) 92:379–84. doi: 10.1007/Bf01247339
18. Sundaramurthy S, SelvaKumar A, Ching J, Dharani V, Sarangapani S, Yu-Wai-Man P. Leber hereditary optic neuropathy-new insights and old challenges. Graefes Arch Clin Exp Ophthalmol. (2021) 259:2461–72. doi: 10.1007/s00417-020-04993-1
19. de Weerdt CJ, Went LN. Neurological studies in families with Leber’s optic atrophy. Acta Neurol Scand. (1971) 47:541–54. doi: 10.1111/j.1600-0404.1971.tb07507.x
20. Kim JY, Hwang JM, Chang BL, Park SS. Spectrum of the mitochondrial DNA mutations of Leber’s hereditary optic neuropathy in Koreans. J Neurol. (2003) 250:278–81. doi: 10.1007/s00415-003-0985-4
21. Mashima Y, Yamada K, Wakakura M, Kigasawa K, Kudoh J, Shimizu N, et al. Spectrum of pathogenic mitochondrial DNA mutations and clinical features in Japanese families with Leber’s hereditary optic neuropathy. Curr Eye Res. (1998) 17:403–8. doi: 10.1080/02713689808951221
22. Houshmand M, Sharifpanah F, Tabasi A, Sanati MH, Vakilian M, Lavasani S, et al. Leber’s hereditary optic neuropathy: the spectrum of mitochondrial DNA mutations in Iranian patients. Ann N Y Acad Sci. (2004) 1011:345–9. doi: 10.1196/annals.1293.035
23. Zhou HP, Ishikawa H, Yasumoto R, Sakurai K, Sawamura H. Leber hereditary optic neuropathy harboring a rare m.12811 T>C mitochondrial DNA mutation. Can J Ophthalmol. (2021) 56:e82–4. doi: 10.1016/j.jcjo.2020.12.022
24. Bi R, Zhang AM, Jia X, Zhang Q, Yao YG. Complete mitochondrial DNA genome sequence variation of Chinese families with mutation m.3635G>A and Leber hereditary optic neuropathy. Mol Vis. (2012) 18:3087–94.
25. Perales-Clemente E, Fernandez-Vizarra E, Acin-Perez R, Movilla N, Bayona-Bafaluy MP, Moreno-Loshuertos R, et al. Five entry points of the mitochondrially encoded subunits in mammalian complex I assembly. Mol Cell Biol. (2010) 30:3038–47. doi: 10.1128/Mcb.00025-10
26. Bai YD, Hu PQ, Park JS, Deng JH, Song XF, Chomyn A, et al. Genetic and functional analysis of mitochondrial DNA-encoded complex I genes. Ann N Y Acad Sci. (2004) 1011:272–83. doi: 10.1196/annals.1293.026
27. Chomyn A. Mitochondrial genetic control, of assembly and function of complex I in mammalian cells. J Bioenerg Biomembr. (2001) 33:251–7. doi: 10.1023/A:1010791204961
28. Bai YD, Shakeley RM, Attardi G. Tight control of respiration by NADH dehydrogenase ND5 subunit gene expression in mouse mitochondria. Mol Cell Biol. (2000) 20:805–15. doi: 10.1128/Mcb.20.3.805-815.2000
29. Bao HG, Zhao CJ, Li JY, Wu C. Association of MT-ND5 gene variation with mitochondrial respiratory control ratio and NADH dehydrogenase activity in tibet chicken embryos. Anim Genet. (2007) 38:514–6. doi: 10.1111/j.1365-2052.2007.01622.x
30. Kolarova H, Liskova P, Tesarova M, Kucerova Vidrova V, Forgac M, Zamecnik J, et al. Unique presentation of LHON/MELAS overlap syndrome caused by m.13046T>C in MTND5. Ophthalmic Genet. (2016) 37:419–23.
31. Petruzzella V, Di Giacinto G, Scacco S, Piemonte F, Torraco A, Carrozzo R, et al. Atypical leigh syndrome associated with the D393N mutation in the mitochondrial ND5 subunit. Neurology. (2003) 61:1017–8. doi: 10.1212/01.Wnl.0000080363.10902.E9
32. Parker WD, Parks JK. Mitochondrial ND5 mutations in idiopathic Parkinson’s disease. Biochem Bioph Res Commun. (2005) 326:667–9. doi: 10.1016/j.bbrc.2004.11.093
33. Panades-de Oliveira L, Montoya J, Emperador S, Ruiz-Pesini E, Jerico I, Arenas J, et al. A novel mutation in the mitochondrial MT-ND5 gene in a family with MELAS. The relevance of genetic analysis on targeted tissues. Mitochondrion. (2020) 50:14–8. doi: 10.1016/j.mito.2019.10.001
34. Naini AB, Lu JS, Kaufmann P, Bernstein RA, Mancuso M, Bonilla E, et al. Novel mitochondrial DNA ND5 mutation in a patient with clinical features of MELAS and MERRF. Arch Neurol Chicago. (2005) 62:473–6. doi: 10.1001/archneur.62.3.473
35. Wei YL, Yu CA, Yang P, Li AL, Wen JY, Zhao SM, et al. Novel mitochondrial DNA mutations associated with chinese familial hypertrophic cardiomyopathy. Clin Exp Pharmacol Physiol. (2009) 36:933–9. doi: 10.1111/j.1440-1681.2009.05183.x
36. Kolarova H, Liskova P, Tesarova M, Vidrova VK, Forgac M, Zamecnik J, et al. Unique presentation of LHON/MELAS overlap syndrome caused by m.13046T>C in MTND5. Ophthalmic Genet. (2016) 37:419–23. doi: 10.3109/13816810.2015.1092045
37. Wei YP, Huang Y, Yang YM, Qian M. MELAS/LS overlap syndrome associated with mitochondrial DNA mutations: clinical, genetic, and radiological studies. Front Neurol. (2021) 12:648740. doi: 10.3389/fneur.2021.648740
38. Ng YS, Lax NZ, Maddison P, Alston CL, Blakely EL, Hepplewhite PD, et al. MT-ND5 mutation exhibits highly variable neurological manifestations at low mutant load. Ebiomedicine. (2018) 30:86–93. doi: 10.1016/j.ebiom.2018.02.010
39. Han J, Lee YM, Kim SM, Han SY, Lee JB, Han SH. Ophthalmological manifestations in patients with leigh syndrome. Brit J Ophthalmol. (2015) 99:528–35. doi: 10.1136/bjophthalmol-2014-305704
40. Ayalon N, Flore LA, Christensen TG, Sam F. Mitochondrial encoded NADH dehydrogenase 5 (MT-ND5) gene point mutation presents as late onset cardiomyopathy. Int J Cardiol. (2013) 167:E143–5. doi: 10.1016/j.ijcard.2013.04.018
41. DiMauro S. A brief history of mitochondrial pathologies. Int J Mol Sci. (2019) 20:5643. doi: 10.3390/ijms20225643
42. DiMauro S, Schon EA. Mechanisms of disease: mitochondrial respiratory-chain diseases. N Engl J Med. (2003) 348:2656–68. doi: 10.1056/NEJMra022567
43. Gropman A, Chen TJ, Perng CL, Krasnewich D, Chernoff E, Tifft C, et al. Variable clinical manifestation of homoplasmic G14459A mitochondrial DNA mutation. Am J Med Genet A. (2004) 124a:377–82. doi: 10.1002/ajmg.a.20456
44. McFarland R, Clark KM, Morris AAM, Taylor RW, Macphail S, Lightowlers RN, et al. Multiple neonatal deaths due to a homoplasmic mitochondrial DNA mutation. Nat Genet. (2002) 30:145–6. doi: 10.1038/ng819
45. Jancic J, Rovcanin B, Djuric V, Pepic A, Samardzic J, Nikolic B, et al. Analysis of secondary mtDNA mutations in families with Leber’s hereditary optic neuropathy: four novel variants and their association with clinical presentation. Mitochondrion. (2020) 50:132–8. doi: 10.1016/j.mito.2019.10.011
46. Ji Y, Zhang AM, Jia X, Zhang YP, Xiao X, Li S, et al. Mitochondrial DNA haplogroups M7b1’2 and M8a affect clinical expression of Leber hereditary optic neuropathy in Chinese families with the m.11778G–>A mutation. Am J Hum Genet. (2008) 83:760–8. doi: 10.1016/j.ajhg.2008.11.002
47. Caporali L, Maresca A, Capristo M, Del Dotto V, Tagliavini F, Valentino ML, et al. Incomplete penetrance in mitochondrial optic neuropathies. Mitochondrion. (2017) 36:130–7. doi: 10.1016/j.mito.2017.07.004
48. Kirby DM, McFarland R, Ohtake A, Dunning C, Ryan MT, Wilson C, et al. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J Med Genet. (2004) 41:784–9. doi: 10.1136/jmg.2004.020537
49. Sudoyo H, Suryadi H, Lertrit P, Pramoonjago P, Lyrawati D, Marzuki S. Asian-specific mtDNA backgrounds associated with the primary G11778A mutation of Leber’s hereditary optic neuropathy. J Hum Genet. (2002) 47:594–604. doi: 10.1007/s100380200091
Keywords: Leber hereditary optic neuropathy, mitochondrial disorder, mitochondrial DNA, ophthalmoplegia, optic atrophy
Citation: Xu Q, Sun P, Feng C, Chen Q, Sun X, Chen Y and Tian G (2022) Varying Clinical Phenotypes of Mitochondrial DNA T12811C Mutation: A Case Series Report. Front. Med. 9:912103. doi: 10.3389/fmed.2022.912103
Received: 04 April 2022; Accepted: 13 June 2022;
Published: 04 July 2022.
Edited by:
Paolo Fogagnolo, University of Milan, ItalyReviewed by:
Yue-Bei Luo, Central South University, ChinaCopyright © 2022 Xu, Sun, Feng, Chen, Sun, Chen and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuhong Chen, eXVob25nY2hlbkBmdWRhbi5lZHUuY24=; Guohong Tian, dmFsZW50aWFuOTlAaG90bWFpbC5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.