 Kai Chen1,2†
Kai Chen1,2† Yan-Yan Hu2†Lin-Lin Wang2Yun Xia2Qian Jiang2Lan Sun2Shan-Shan Qian2,3
Yan-Yan Hu2†Lin-Lin Wang2Yun Xia2Qian Jiang2Lan Sun2Shan-Shan Qian2,3 Jin-Zhao Wu2,3
Jin-Zhao Wu2,3 Liu-Qing Chen1,2*Dong-Sheng Li1,2*
Liu-Qing Chen1,2*Dong-Sheng Li1,2*- 1Hubei Key Laboratory of Infectious and Immune Skin Diseases, Wuhan No. 1 Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Department of Dermatology, Wuhan No. 1 Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3School of Medicine, Jianghan University, Wuhan, China
Port-wine stains (PWSs) are a congenital capillary malformed disorder and are caused by a number of somatic mutations that disrupt vascular development. However, the underlying genetic mutations in the pathogenesis of PWS have not yet been fully elucidated. To understand PWS genetic variations and investigate novel genetic mutations, we extracted genomic DNA from four sporadic PWS patients and then performed whole-genome sequencing (WGS). Using Sorting Intolerant from Tolerant (SIFT), PolyPhen2, Mutation Assessor, MetaSVM to identify candidate genetic mutations and whole-exome sequencing (WES) to confirm the identified variants. We found a previously reported G protein subunit alpha q (GNAQ) mutation c.548G > A, p.Arg183Gln in one case, whereas no such mutation was found in the other three samples. Moreover, six novel somatic mutations in three genes, including KCNJ12, SLC25A5, POTEE, were found in these four samples. Importantly, WES also verified the KCNJ12 (c.433G > A, p.Gly145Ser) and SLC25A5 (c.413G > A, p.Arg138His) mutations in other five sporadic PWS patients, with the frequency of 60% (3 of 5) and 40% (2 of 5), respectively. Thus, we reveal in this study two novel somatic mutations, KCNJ12 and SLC25A5, in the sporadic PWS patients for the first time. These findings highlight the genetic polymorphism of PWS and provide potential clinical prediction targets for this disease.
Introduction
Port-wine stain (PWS) is a kind of congenital capillary malformation that often occur on the face and neck, with an incidence of 3–5/1000 in newborn worldwide (1). Because of its sporadic with no heritability, a long-standing hypothesis suggests that PWS are associated with somatic mutations (2). In 2013, Shirley and colleagues performed whole-genome sequencing (WGS) of DNA from 13 patients with non-syndromic PWS, discovered a non-synonymous mutation (c.548G > A, p.Arg183Gln) in the G protein subunit alpha q (GNAQ) gene, and defined this gene as a causative mutation of PWS (3). A later finding also confirmed the presence of somatic GNAQ mutation in a sporadic PWS, as well as novel somatic mutations in several other genes (4). Similarly, next-generation sequencing data revealed the different GNAQ mutation frequencies in the structures of patients with PWS (5). Although the genetic mutation of GNAQ is important in the pathogenesis of PWS, not all PWS patients have a mutation in this gene, suggesting other genetic etiologies could also contribute to the pathologic manifestations of this disorder.
KCNJ12 (also known as IRK2, KCNJN1, and Kir2.2) is a Kir ion channel protein, which encodes potassium inwardly-rectifying channel 12 and contributes to the cardiac inward rectifier current (IK1). This protein is thought to be involved in protein homotetramerization and regulation of resting membrane potential (6). In skin development, numerous studies have shown the important roles of KCNJ12 mutations in skin cancers, such as colorectal carcinoma (CRC), head and neck squamous cell carcinoma (HNSCC), and esophageal squamosa cell carcinoma (ESCC) (7–9). Moreover, KCNJ12 mutation also was identified in ultraviolet b (UVB)-irradiated primary human keratinocytes (10). Taken together, these studies indicated the vital roles of KCNJ12 mutations in skin tumorigenesis. However, the presence of KCNJ12 mutations in PWS has not been previously reported.
Solute carrier family 25 member 5 (SLC25A5) is a member of the mitochondrial solute carrier subfamily protein and the function of this protein remains unclear, whereas SLC25A5 mutation has been reported in the samples from sporadic Alzheimer’s disease (AD) patients (11, 12). To date, no study concerning the connection between SLC25A5 mutation and PWS pathogenesis. In this study, by using WGS technology, we identified two novel genetic mutations in KCNJ12 and SLC25A5 genes in sporadic PWS patients, and the genetic variant of these two genes was validated by the whole-exome sequencing results. Identifying these somatic mutations of PWS may provide insight into the clinical prediction of this disease.
Methods
Study Approval
Each patient signed informed consent for study participation and clinical images and publication of identified information prior to the study. All the sequencing was approved by the ethics committee of Wuhan No. 1 Hospital affiliated with Tongji Medical College, Huazhong University of Science and Technology.
Images Taken
Images of the skin lesions of each patient were taken using the Canon 650D SLR camera. Dermoscopic and reflectance confocal microscopy (RCM) images were taken using the ATBM FotoFinder Bodystudio dermatoscope and VivaScope. Functional magnetic resonance imaging (MRI) was scanned using the Siemens Magnetom Verio 3.0T.
Hematoxylin-Eosin Staining
Port-wine stain lesional tissue was embedded in paraffin and cut into sections as described previously (13). The sections were stained with HE and observed under an Automated Digital Slice Scanning System (ZEISS, Axioscan 7).
DNA Extraction
Genomic DNA was extracted from PWS lesional tissue as previously described (5). DNA concentrations and quality were determined using a Multiskan Spectrum (SpectraMax 190, Thermo Scientific, Wilmington, United States).
Whole-Genome Sequencing/Whole-Exome Sequencing and Bioinformatic Analysis
Whole-genome sequencing/whole-exome sequencing was performed by Beijing Genomics Institute (BGI)-Shenzhen Biotechnology Company (Shenzhen, China). The samples were treated according to the method previously reported and pair-end 100 base reads were generated in the way of sequenced combinatorial Probe-Anchor Synthesis (cPAS) on BGISEQ-500 platform (14). The raw sequencing data were processed using the following steps: (1) Removing reads containing sequencing adapter; (2) Removing reads whose low-quality base ratio (base quality less than or equal to 5) is more than 50%; (3) Removing reads whose unknown base (‘N’ base) ratio is more than 10%. Statistical analysis of data and downstream bioinformatics analysis were performed on this filtered, high-quality data, referred to as the clean data. Clean data were aligned to the human reference genome using Burrows-Wheeler Aligner (BWA) (15). Picard was used to remove duplicated sequence reads. Realignment was performed with the Genome Analysis Toolkit (GATK) (16). Genetic variations were identified using HaplotypeCaller of GATK and annotated with SnpEff software (17). All candidate variants were filtered against public databases including the 1,000 Genomes Project, and the Single Nucleotide Polymorphism Database (dbSNP). The prediction tools, Sorting Intolerant from Tolerant (SIFT), PolyPhen2, Mutation Assessor, and MetaSVM were used to estimate the likelihood that an amino acid transition may affect the function of the protein. The mutation site that presents in at least two samples was chosen for the potential genetic variant. Variants were visualized using the Integrative Genomic Viewer (IGV).
Results
Clinical Characteristics
We show a representative case of a 22-year-old male patient (case 1) with a hypertrophic PWS on the left central face and lateral face (Figures 1A1–A4). The vascular morphology, including linear vessels, dots or globular vessels, and sausage-like vessels were seen in the dermoscopic picture (Figure 1B). Consistently, the RCM image indicated increased expansive blood vessels in the dermis (Figure 1C) and an MRI scan revealed obvious intracranial malformations in his brain region (Figures 1D1,D2).
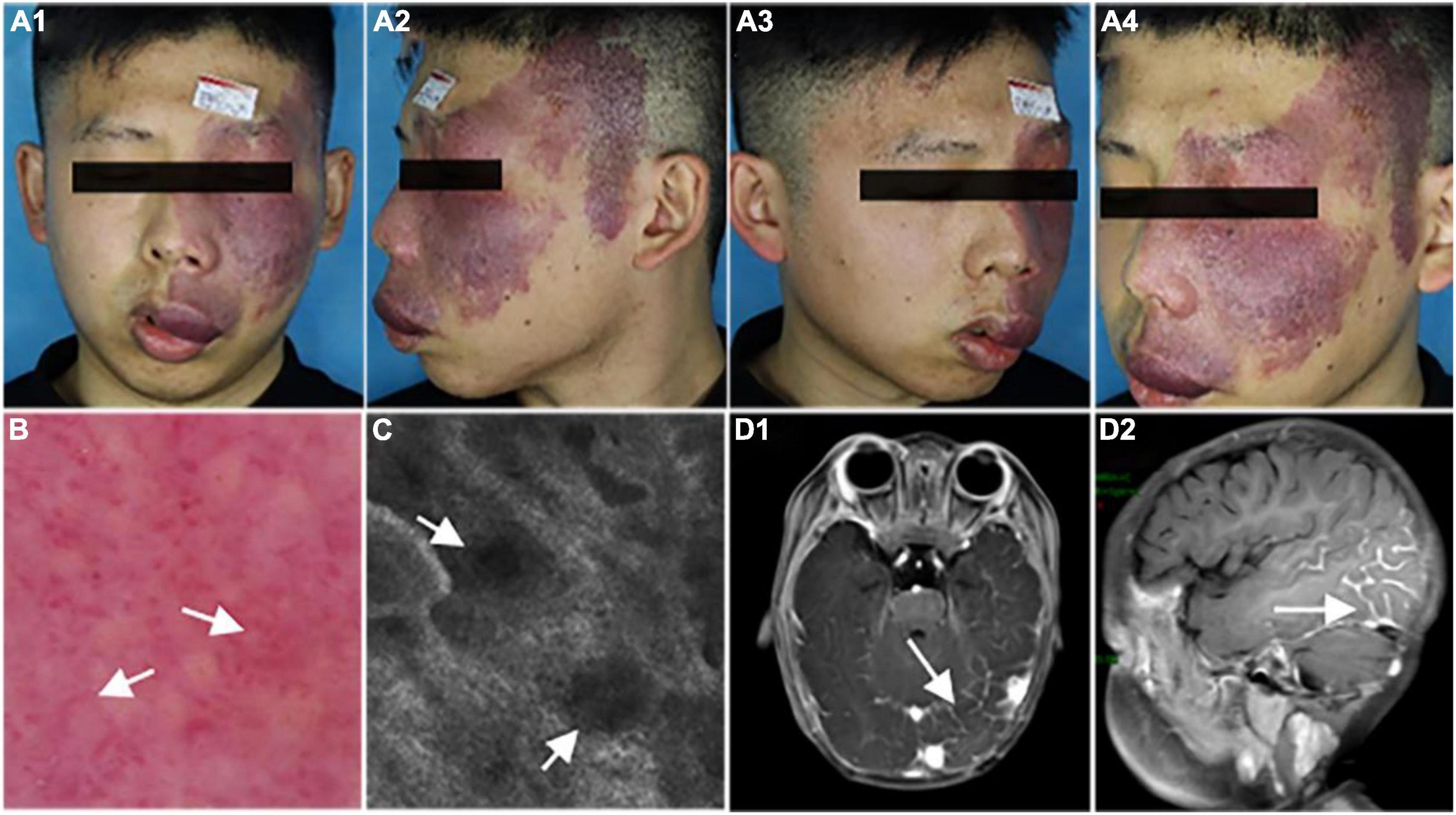
Figure 1. Representative clinical manifestations, dermoscopy finds, reflectance confocal microscopy (RCM) images, and magnetic resonance imaging (MRI) scans of the patient. (A1–A4) Port-wine stain (PWS) on the central and lateral face of the patient. (B,C) Representative dermoscopy and RCM pictures of the PWS lesions from the patient. White arrows indicate the expansive vessels. (D) Representative MRI scans of the patient. White arrows indicate an intracranial malformation in his brain tissue.
To determine the histological as well as the clinical symptoms changes, we performed a hematoxylin-eosin (HE) of the PWS lesional tissue section of this patient. Under the microscope, hyperplastic capillaries were found in the dermis (Figure 2A, HE, 5x), and ectatic vessels also were observed (Figure 2B, HE, 20x). Taken together, these results indicate that this patient is characterized by typical clinical features of PWS.
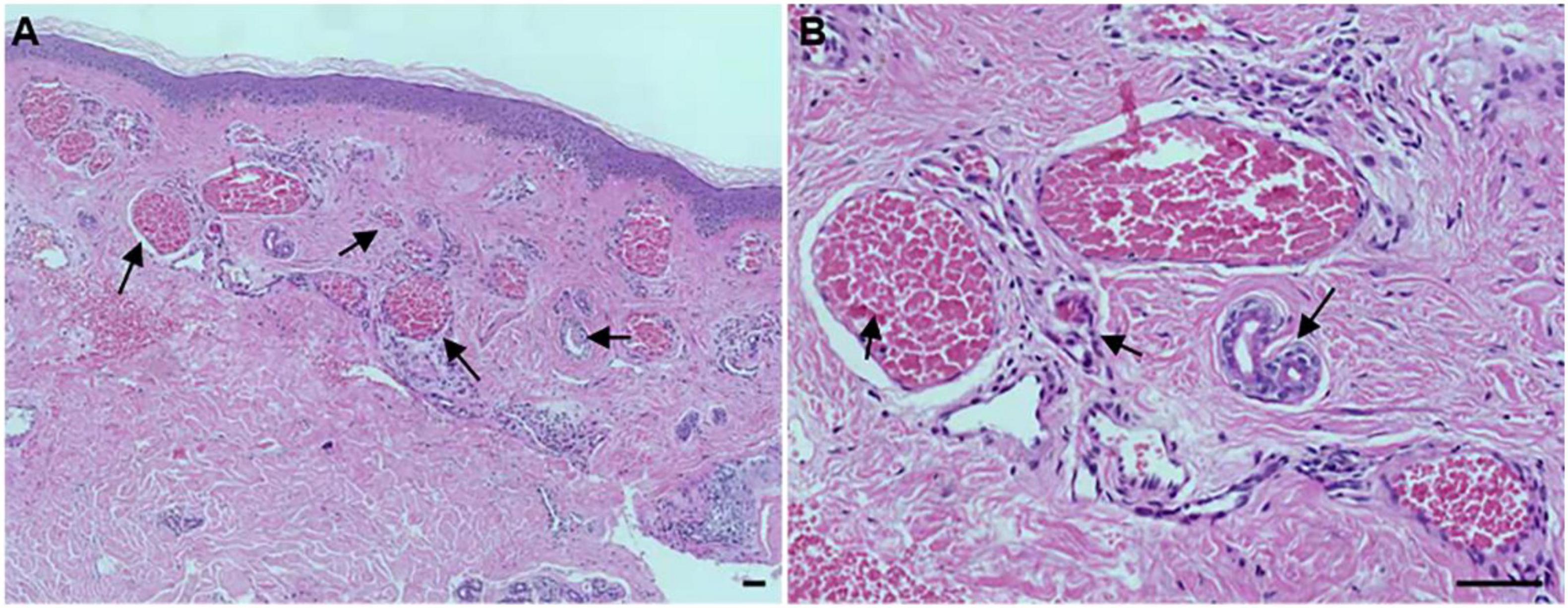
Figure 2. Histopathological examination of the port-wine stains (PWS) lesional tissue. Hematoxylin-eosin (HE)-stained PWS lesional tissue section showing ectatic capillaries. Black arrows indicate the expansive blood vessels in the dermis and filled erythrocytes in the lumen. (A): original magnification × 5. (B): original magnification × 20. Scale bars: 50 μm.
Whole-Genome Sequencing and Somatic Variants Identification
In order to investigate the genetic variations in the PWS patient, we sequenced the whole genomes of lesional DNA samples from four sporadic PWS patients (case 1 to case 4). The demographic data and PWS features of each patient are described in Table 1. We detected a large number of somatic single nucleotide variations (SNVs) in all the samples across the chromosomes. SIFT, PolyPhen2, Mutation Assessor, and MetaSVM were used to identify pathogenic variants (18). After scoring (Mutation Assessor >3.5, MetaSVM >0, PolyPhen2 > = 0.909, SIFT < = 0.05), a previously reported GNAQ mutation c.548G > A, p.Arg183Gln was identified in one sample (case 1), whereas no such mutation was found in other three samples. Additionally, a total of six somatic variations in three genes, including KCNJ12, SLC25A5, POTEE, were found in these four sporadic PWS patients. The minor allele frequency (MAF) of each variant in the 1,000 Genomes Project also was listed in Table 2.
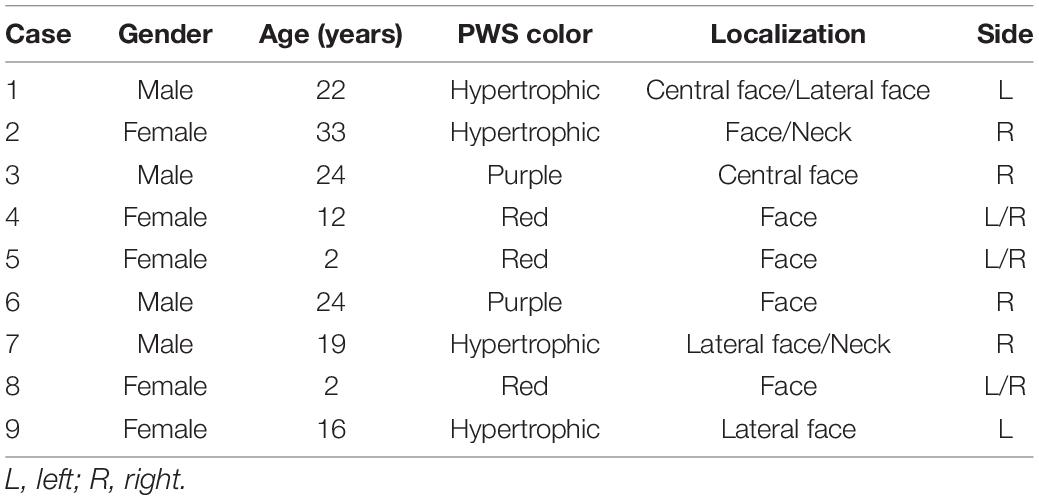
Table 1. Demographic data and port-wine stain (PWS) features of the patients in this study.
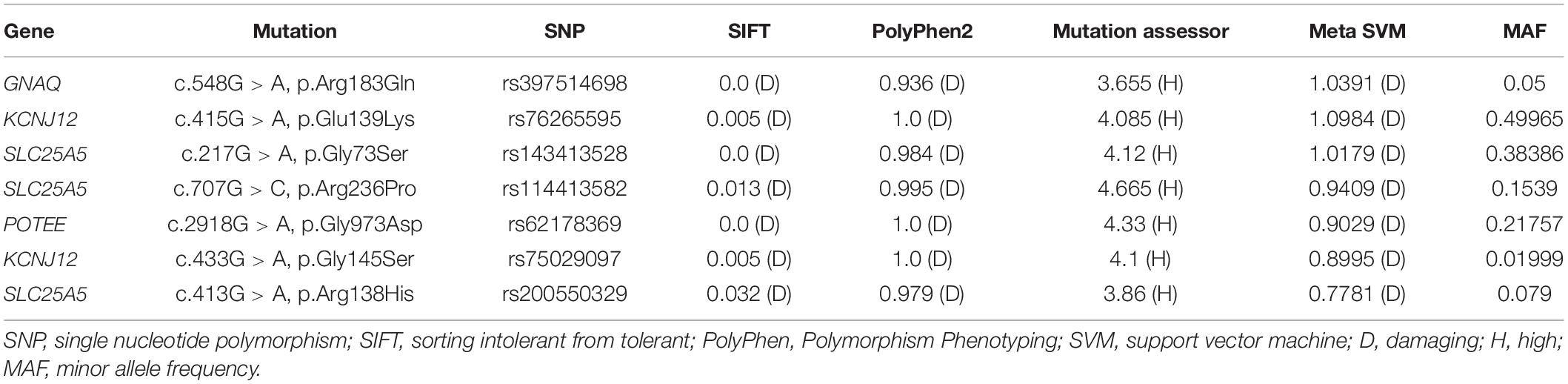
Table 2. Whole-genome sequencing identified G protein subunit alpha q (GNAQ) and other novel somatic mutations in our patients.
Whole-Exome Sequencing of KCNJ12 and SLC25A5 Variants in Other Five Port-Wine Stain Patients
We further verified the presence of these six targeted variant sites in the other five sporadic PWS patients (case 5 to case 9) via WES. The two mutations, KCNJ12 (c.433G > A, p.Gly145Ser) and SLC25A5 (c.413G > A, p.Arg138His), were validated in the patients (Figure 3), and we found the genetic variants in 60% (3 of 5) and 40% (2 of 5) of patients (Table 3), whereas no other genetic variants of those mentioned above were found in these five patients. Importantly, these two mutations were not detected in the samples from peripheral blood and matched control tissues. Therefore, KCNJ12 (c.433G > A, p.Gly145Ser) and SLC25A5 (c.413G > A, p.Arg138His) appear to be the causative somatic mutations of this disorder.
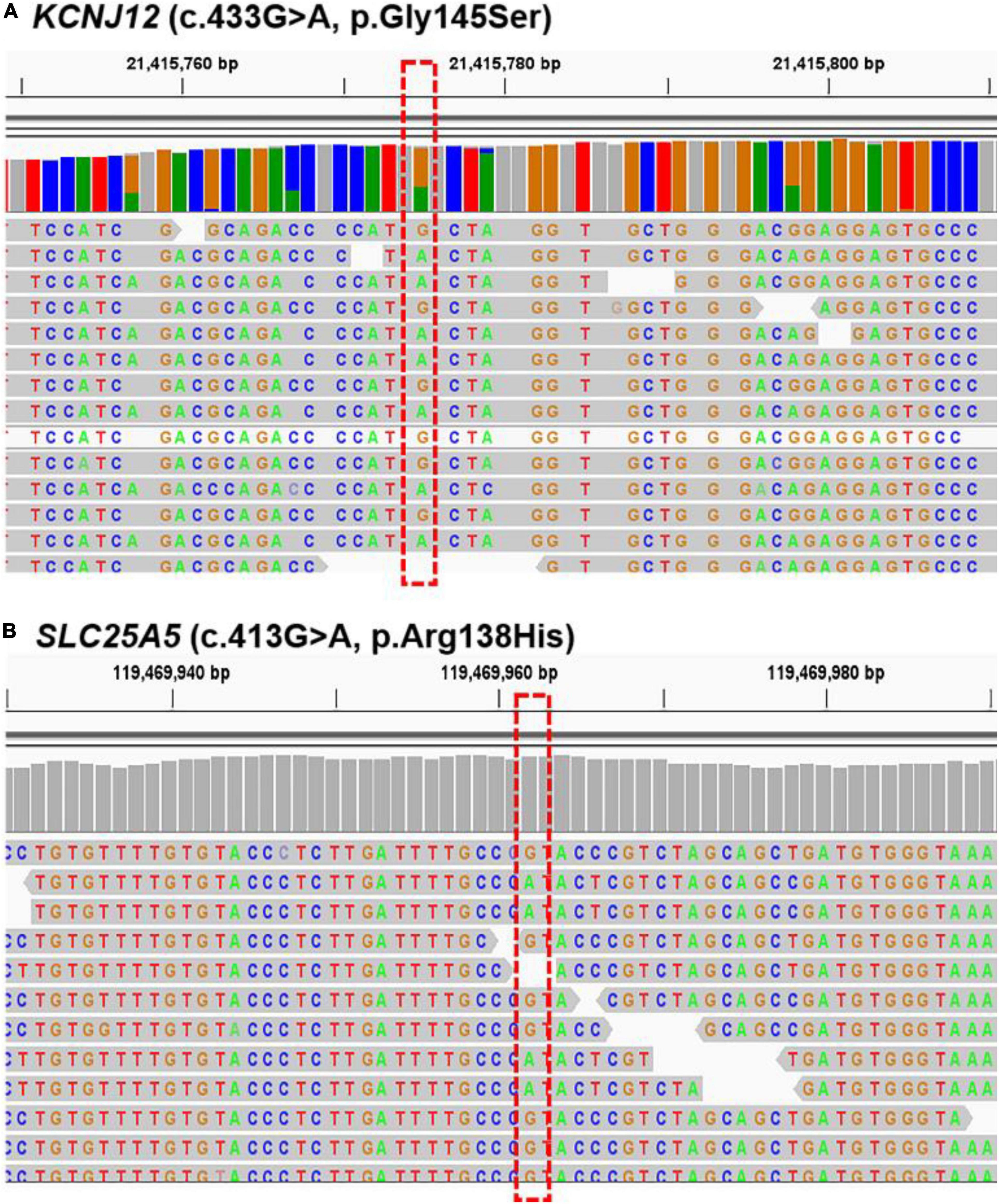
Figure 3. Sequencing results for KCNJ12 and SLC25A5 variants. Whole-exome sequencing (WES) data with the respective somatic KCNJ12 mutation (A) and SLC25A5 mutation (B) are shown from cases 8 and 9, respectively. The red dotted box indicated the mutation site.

Table 3. Whole-exome sequencing verified the KCNJ12 and SLC25A5 variants in other patients.
Discussion
In this study, we performed WGS to identify novel genetic mutations in sporadic PWS patients. Our data suggested an association between SLC25A5 mutation and increased risk of PWS. Consistently, a somatic heterozygous mutation in SLC26A4 was found in a large vestibular aqueduct syndrome (LVAS)-associated PWS patient (19). This finding strongly implies that solute carrier protein plays a pivotal role during the progression of PWS. However, the underlying mechanisms remain unknown. As reported, phosphatidylinositol 3-kinase (PI3K) and protein kinase B (AKT) were activated in blood vessels of hypertrophic PWS and participated in nodule formation (20, 21). Moreover, long non-coding RNA SLC25A5-AS1 also has been functionally implicated in controlling the PI3K/AKT pathway in gastric cancer and carcinogenesis (22, 23). Thus, our data not only presented a novel genetic variation of SLC25A5 gene in the pathogenesis of PWS but also suggested a potential regulatory signaling pathway by controlling PI3K/AKT activation.
Over the past years, mutations of KCNJ12 have been found mainly in cardiovascular diseases and skin cancers (7, 24). Here, we firstly reported a novel genetic variation in the KCNJ12 gene in PWS, but the causative mechanism was still elusive. It has also been reported that topical agents can suppress pulsed dye laser (PDL)-induced angiogenesis in the skin via blocking multiple signaling pathways, such as PI3K/AKT/mammalian target of rapamycin (mTOR) and extracellular regulated kinase (ERK) pathway (25). RASA1, another published mutation gene in PWS, also was found to regulate vascular development via inhibiting the AKT pathway (26). Thus, these findings suggest a critical role for the AKT pathway in the pathophysiological process of regeneration and revascularization of blood vessels in PWS. Interestingly, a recent study reported that KCNJ12 acts as a straightforward target of miR-132-3p to modulate the AKT signaling pathway in bladder cancer oncogenesis and metastasis (27). Therefore, it is possible that KCNJ12 mutation caused PWS pathology by activating the AKT pathway, targeting KCNJ12 or the AKT signaling pathway might be a candidate intervention in the treatment of this disease.
Port-wine stains are a congenital vascular malformation and approximately 15–20% of the infant with ophthalmic (V1) trigeminal PWS are at risk of Sturge-Weber syndrome (SWS), a neurocutaneous disorder characterized by seizures, headaches, stroke, developmental delay, cognitive defects, and intellectual impairments (26, 28). Happle hypothesized that SWS and PWS have the same mutations, with the clinical manifestations dependent on where and when during development the somatic mutation occurs (2). In this study, our results confirmed the presence of two novel genetic mutations in PWS patients. As a highly expressed solute carrier protein in the cortex and the hippocampus, the increased mean precursor intensity of peptide from SLC25A5 was observed in AD, and the mutation of this gene was recognized as an important risk factor for a facial dysmorphism and seizures characterized by intellectual disability (ID) (11, 29). These lines of data suggest the crucial roles of SLC25A5 mutation in regulating neuronal pathways. Importantly, another identified gene in this study, KCNJ12, also reported an increase in epileptic granule cells and regulated the dendritic excitability (30). Moreover, KCNJ12 mutation affects the Kir ion channels-mediated neurovascular communication (31). Taken together, it is likely that these two mutations and other genetic alterations, such as GNAQ, PI3K, AKT, and RASA1, together contribute to the neuropathy in the progression of PWS, this is a matter of when the mutation was acquired.
In conclusion, our study identified two novel genetic mutations, SLC25A5 and KCNJ12, in the sporadic PWS patients, and provided an understanding of PWS genetic determinants. The possible importance of these mutations in the pathogenesis of PWS needs to be further explored.
Data Availability Statement
The datasets presented in this article are not readily available because of ethical/privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Wuhan No. 1 Hospital affiliated to Tongji Medical College, Huazhong University of Science and Technology. Written informed consent to participate in this study was provided by the participants or their legal guardian/next of kin. Written informed consent was obtained from the individual(s) for the publication of any identifiable images or data included in this article.
Author Contributions
L-QC and D-SL initiated and designed the study, Y-YH, L-LW, YX, QJ, and LS collected samples and validated clinical data. KC, S-SQ, and J-ZW performed the experiments and analyzed the data. KC and D-SL wrote the manuscript and revised it. All authors read and approved the final manuscript.
Funding
This study was supported partially by the National Natural Science Foundation of China (81573062), the Application Foundation, and the Advanced Program of Wuhan Science and Technology Bureau (2019010701011418).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chen J, Ghasri P, Aguilar G, van Drooge A, Wolkerstorfer A, Kelly K, et al. An overview of clinical and experimental treatment modalities for port wine stains. J Am Acad Dermatol. (2012) 67:289–304. doi: 10.1016/j.jaad.2011.11.938
2. Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. (1987) 16:899–906. doi: 10.1016/S0190-9622(87)80249-9
3. Shirley M, Tang H, Gallione C, Baugher J, Frelin L, Cohen B, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. (2013) 368:1971–9. doi: 10.1056/NEJMoa1213507
4. Lian C, Sholl L, Zakka L, Tm O, Liu C, Xu S, et al. Novel genetic mutations in a sporadic port-wine stain. JAMA Dermatol. (2014) 150:1336–40. doi: 10.1001/jamadermatol.2014.1244
5. Ma G, Yu Z, Liu F, Wang L, Yu W, Zhu J, et al. Somatic GNAQ mutation in different structures of port-wine macrocheilia. Br J Dermatol. (2018) 179:1109–14. doi: 10.1111/bjd.16830
6. Dhamoon A, Jalife J. The inward rectifier current (IK1) controls cardiac excitability and is involved in arrhythmogenesis. Heart Rhythm. (2005) 2:316–24. doi: 10.1016/j.hrthm.2004.11.012
7. Khalilipour N, Baranova A, Jebelli A, Heravi-Moussavi A, Bruskin S, Abbaszadegan M. Familial Esophageal Squamous Cell Carcinoma with damaging rare/germline mutations in KCNJ12/KCNJ18 and GPRIN2 genes. Cancer Genet. (2018) 221:46–52. doi: 10.1016/j.cancergen.2017.11.011
8. Martin D, Abba M, Molinolo A, Vitale-Cross L, Wang Z, Zaida M, et al. The head and neck cancer cell oncogenome: a platform for the development of precision molecular therapies. Oncotarget. (2014) 5:8906–23. doi: 10.18632/oncotarget.2417
9. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. (2012) 487:330–7. doi: 10.1038/nature11252
10. Shen Y, Ha W, Zeng W, Queen D, Liu L. Exome sequencing identifies novel mutation signatures of UV radiation and trichostatin A in primary human keratinocytes. Scientific Rep. (2020) 10:4943. doi: 10.1038/s41598-020-61807-4
11. Vandewalle J, Bauters M, Van Esch H, Belet S, Verbeeck J, Fieremans N, et al. The mitochondrial solute carrier SLC25A5 at Xq24 is a novel candidate gene for non-syndromic intellectual disability. Human Genet. (2013) 132:1177–85. doi: 10.1007/s00439-013-1322-3
12. Gómez-Ramos A, Podlesniy P, Soriano E, Avila J. Distinct X-chromosome SNVs from some sporadic AD samples. Scientific Rep. (2015) 5:18012. doi: 10.1038/srep18012
13. Manukhina E, Tseilikman V, Tseilikman O, Komelkova M, Kondashevskaya M, Goryacheva A, et al. Intermittent hypoxia improves behavioral and adrenal gland dysfunction induced by posttraumatic stress disorder in rats. J Appl Physiol. (2018) 125:931–7. doi: 10.1152/japplphysiol.01123.2017
14. Chen K, Wallis J, McLellan M, Larson D, Kalicki J, Pohl C, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. (2009) 6:677–81. doi: 10.1038/nmeth.1363
15. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. (2010) 26:589–95. doi: 10.1093/bioinformatics/btp698
16. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
17. Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. (2012) 6:80–92. doi: 10.4161/fly.19695
18. Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Human Mol Genet. (2015) 24:2125–37. doi: 10.1093/hmg/ddu733
19. Cong T, Liu L, Zhang H, Wang L, Jiang X. Port-wine stains associated with large vestibular aqueduct syndrome caused by mutations in GNAQ and SLC26A4 genes: a case report. J Dermatol. (2020) 47:78–81. doi: 10.1111/1346-8138.15130
20. Yin R, Gao L, Tan W, Guo W, Zhao T, Nelson J, et al. Activation of PKCα and PI3K Kinases in Hypertrophic and Nodular Port Wine Stain Lesions. Am J Dermatopathol. (2017) 39:747–52. doi: 10.1097/DAD.0000000000000785
21. Tan W, Chernova M, Gao L, Sun V, Liu H, Jia W, et al. Sustained activation of c-Jun N-terminal and extracellular signal-regulated kinases in port-wine stain blood vessels. J Am Acad Dermatol. (2014) 71:964–8. doi: 10.1016/j.jaad.2014.07.025
22. Ghafouri-Fard S, Abak A, Tondro Anamag F, Shoorei H, Majidpoor J, Taheri M. The emerging role of non-coding RNAs in the regulation of PI3K/AKT pathway in the carcinogenesis process. Biomed Pharmacother. (2021) 137:111279. doi: 10.1016/j.biopha.2021.111279
23. Li X, Yan X, Wang F, Yang Q, Luo X, Kong J, et al. Down-regulated lncRNA SLC25A5-AS1 facilitates cell growth and inhibits apoptosis via miR-19a-3p/PTEN/PI3K/AKT signalling pathway in gastric cancer. J Cell Mol Med. (2019) 23:2920–32. doi: 10.1111/jcmm.14200
24. Yuan H, Yan K, Hou D, Zhang Z, Wang H, Wang X, et al. Whole exome sequencing identifies a KCNJ12 mutation as a cause of familial dilated cardiomyopathy. Medicine. (2017) 96:e7727. doi: 10.1097/MD.0000000000007727
25. Gao L, Nadora D, Phan S, Chernova M, Sun V, Preciado S, et al. Topical axitinib suppresses angiogenesis pathways induced by pulsed dye laser. Br J Dermatol. (2015) 172:669–76. doi: 10.1111/bjd.13439
26. Nguyen V, Hochman M, Mihm M, Nelson J, Tan W. The Pathogenesis of Port Wine Stain and Sturge Weber Syndrome: complex Interactions between Genetic Alterations and Aberrant MAPK and PI3K Activation. Int J Mol Sci. (2019) 20:2243. doi: 10.3390/ijms20092243
27. Li G, Guo B, Wang H, Lin G, Lan T, Ying H, et al. CircRNA hsa_circ_0014130 function as a miR-132-3p sponge for playing oncogenic roles in bladder cancer via upregulating KCNJ12 expression. Cell Biol Toxicol. (2021). doi: 10.1007/s10565-021-09668-z
28. Sabeti S, Ball K, Burkhart C, Eichenfield L, Fernandez Faith E, Frieden I, et al. Consensus statement for the management and treatment of port-wine birthmarks in sturge-weber syndrome. JAMA Dermatol. (2021) 157:98–104. doi: 10.1001/jamadermatol.2020.4226
29. Florentinus-Mefailoski A, Bowden P, Scheltens P, Killestein J, Teunissen C, Marshall J. The plasma peptides of Alzheimer’s disease. Clin Proteom. (2021) 18:17. doi: 10.1186/s12014-021-09320-2
30. Stegen M, Kirchheim F, Hanuschkin A, Staszewski O, Veh R, Wolfart J. Adaptive intrinsic plasticity in human dentate gyrus granule cells during temporal lobe epilepsy. Cereb Cortex. (2012) 22:2087–101. doi: 10.1093/cercor/bhr294
Keywords: whole genome sequencing (WGS), port-wine stains (PWS), KCNJ12, SLC25A5, mutation
Citation: Chen K, Hu Y-Y, Wang L-L, Xia Y, Jiang Q, Sun L, Qian S-S, Wu J-Z, Chen L-Q and Li D-S (2022) Whole-Genome Sequencing Identified KCNJ12 and SLC25A5 Mutations in Port-Wine Stains. Front. Med. 9:905902. doi: 10.3389/fmed.2022.905902
Received: 28 March 2022; Accepted: 06 June 2022;
Published: 20 July 2022.
Edited by:
Kyle T. Amber, Rush University, United StatesReviewed by:
Guolong Zhang, Tongji University, ChinaLin Gao, Fourth Military Medical University, China
Copyright © 2022 Chen, Hu, Wang, Xia, Jiang, Sun, Qian, Wu, Chen and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong-Sheng Li, TERTMTIwNEAxMjYuY29t; Liu-Qing Chen, Y2hscTM1QDEyNi5jb20=
†These authors have contributed equally to this work