 Meng-shi Li
Meng-shi Li Yang Li
Yang Li Yang Liu
Yang Liu Xu-jie Zhou
Xu-jie Zhou Hong Zhang
Hong Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 06 May 2022
Sec. Nephrology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.905007
This article is part of the Research Topic Genetics And Epigenetics of Chronic Kidney Disease View all 17 articles
More than 200 cases of lipoprotein glomerulopathy (LPG) have been reported since it was first discovered 30 years ago. Although relatively rare, LPG is clinically an important cause of nephrotic syndrome and end-stage renal disease. Mutations in the APOE gene are the leading cause of LPG. APOE mutations are an important determinant of lipid profiles and cardiovascular health in the population and can precipitate dysbetalipoproteinemia and glomerulopathy. Apolipoprotein E-related glomerular disorders include APOE2 homozygote glomerulopathy and LPG with heterozygous APOE mutations. In recent years, there has been a rapid increase in the number of LPG case reports and some progress in research into the mechanism and animal models of LPG. We consequently need to update recent epidemiological studies and the molecular mechanisms of LPG. This endeavor may help us not only to diagnose and treat LPG in a more personized manner but also to better understand the potential relationship between lipids and the kidney.
Since it was first described in 1989 (1), lipoprotein glomerulopathy (LPG) (OMIM: 611771) has been characterized as a rare glomerular disorder leading to nephrotic syndrome and/or kidney failure (2). LPG is characterized clinically by proteinuria and elevated concentrations of triglyceride-rich lipoproteins and their remnants, and histologically characterized by lamellated lipoprotein thrombi in glomerular capillary lumina lacking foam cells. The familial occurrence of LPG has been frequently recognized. LPG is primarily associated with heterozygous APOE mutations in the low-density lipoprotein–receptor binding site or around it (3). As a “Mendelian disease” caused by a “single gene” with dominant inherited disease of incomplete penetrance, it also provides a disease model to explore pathogenic roles of APOE in some common diseases, such as Alzheimer's disease, type III hyperlipoproteinemia (HLP), and coronary artery disease (2). Trending evidence suggests that APOE gene mutations play an important role by potentially increasing the affinity of lipoproteins for the glomerular capillary wall or by enhancing the tendency of mutant apolipoproteins to form aggregates when concentrated. If left untreated, the disease usually progresses to end-stage kidney disease. Lipid-lowering medications, especially fibrates, were found to improve both clinical manifestations and histological alterations. In recent years, there has been a rapid increase in the number of LPG cases reported, and some progress in research into the mechanism and animal models of LPG. We consequently need to update recent epidemiological studies and the molecular mechanisms of LPG. Using “lipoprotein glomerulopathy” or “lipoprotein nephropathy” as key words, we retrieved data from the PubMed, Wanfang (China National Knowledge Infrastructure) and J-STAGE (online platform for Japanese academic journals) databases for literature review (Figure 1). The purpose of this review is to update the epidemiology and clinical features of lipoprotein glomerulopathy, discuss its pathogenesis, summarize current therapeutic options, and present personal perspectives for future research.
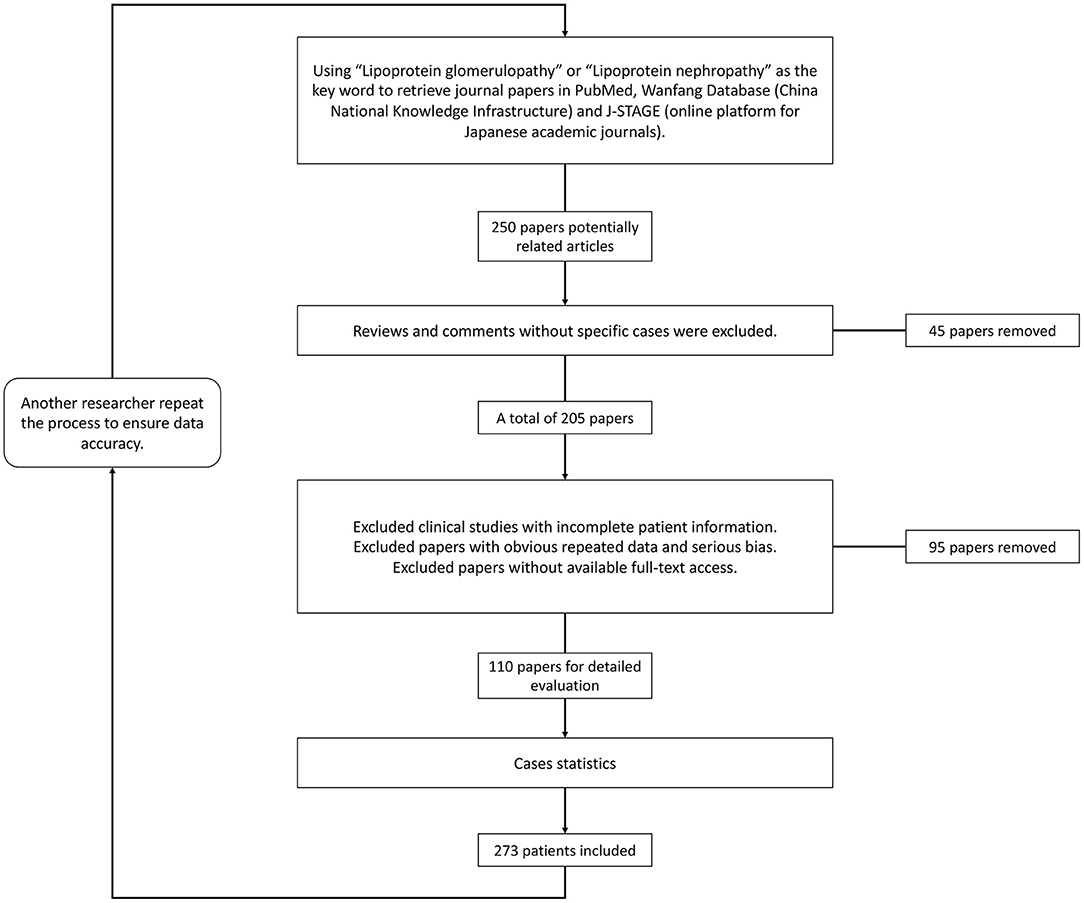
Figure 1. Flowchart of the literature review and case screening.
At the 1988 annual meeting of the Japanese Society of Nephrology, Saito et al. first reported the cases of 2 patients who had similar clinical features of renal impairment and glomerular capillary lipoprotein deposition (4) (Figure 2). In 1989, he described the case in the English literature for the first time. The absence of involvement of other organs and the characteristic morphology of the renal lesions could clearly distinguish this disease from other disorders of lipid metabolism. With the presence of lipoproteins in the glomerular deposits and abnormalities in serum lipid levels resembling the pattern observed in type III hyperlipoproteinemia (HLP), this disorder was named “lipoprotein glomerulopathy.” Type III HLP is a condition characterized by the elevation of both cholesterol and triglycerides, accumulation of incompletely catabolized triglyceride-rich lipoproteins, plamar xanthoma, and rapidly progressive atherosclerosis (5). Type III HLP has always been found in individuals who are homozygous for apoE mutations (apoE2/2) and rare in heterozygous state. In 1991, Oikawa et al. reported abnormally elevated levels of apoE in patients with LPG (6). In the report, the levels of lipoprotein components, plasma apolipoprotein profiles, and apoE isoforms were checked in 6 patients. Common features included proteinuria (1.6–10 g/d), normal lecithin-cholesterol acyltransferase (LCAT) activity, type III HLP-like lipoprotein profiles, and significantly higher levels of plasma apoE (>10 mg/dL) compared with the control patients with hyperlipidemic nephrotic syndrome without lipoprotein thrombi, or type IIb hyperlipoproteinemia without renal disease. All the patients had rare apoE isoform patterns (E2/3 in five cases and E4/4 in one case). These findings showed the first evidence that apoE hyperlipoproteinemia was associated with the apoE isoform and lipoprotein metabolic derangement. Familial occurrence of LPG was later recognized, as introduced in the below epidemiology section (7, 8). It was suggested that LPG may be an inherited disease in which abnormal lipoproteins composed of APOE mutants accumulate within the glomeruli. This finding was supported by observations of mutations named APOE Sendai (Arg145Cys) in 1997 and APOE Kyoto (Arg25Cys) in 1998. A total of 17 APOE variants associated with LPG have been identified to date, highlighting DNA analysis of the APOE gene as one of the most important tools for identifying LPG. These 17 mutations were retrieved from published case reports by manually literature searches, among which 13 have been included in the HGMD database (including APOE Kyoto, Tokyo-Maebashi, Sendai, Guangzhou, Okayama, Modena, Las Vegas, Osaka or Kurashiki, Hong Kong, Chicago, Tsukuba, E1, and Kanto, Additional File 1).
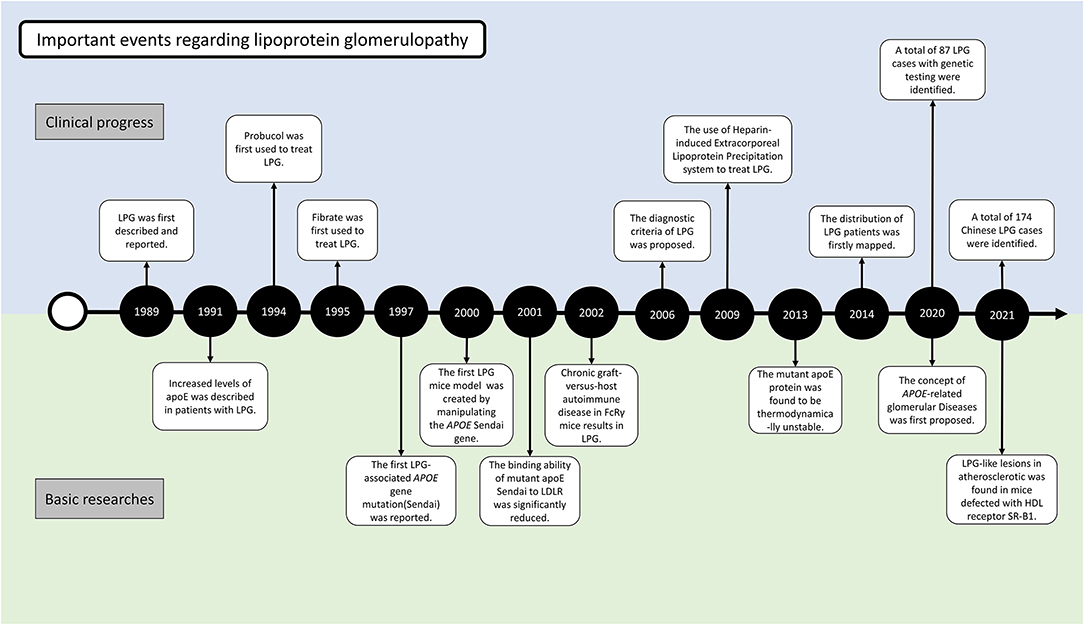
Figure 2. Milestone of LPG research in clinical and basic aspects.
Although it was initially assumed that this disease was restricted to East Asia, several cases have been reported in individuals of European ancestry. With the increased awareness of this disease, several patients with nephrotic syndrome of previously “unrecognized” cause have been grouped into LPG in recent years. The total number of reported LPG cases increased to over 200 hundred (details below).
The total number of patients was estimated to be <80 in 2011 (9). Due to increased awareness and the advance of genetic testing technology, the number of cases increased to ~150 by June 2019 (10). By an updated literature review, we estimated that the reported number would be at least 274 up to the beginning of the year 2022.
LPG showed significant regional and familial aggregation. The majority of cases were reported in Japan and China (Figures 3, 4). It is mainly distributed in southwestern and southeastern China and in central and north-central Japan (11). Some hot spots of gene mutations have been described, suggesting a founder effect of the gene mutation. APOE Sendai (Arg145Pro) was mainly observed in north-central Japan, particularly in Yamagata and Miyagi (12), whereas it has not been reported in China. APOE Kyoto (Arg25Cys) was reported as the most frequent mutation in LPG throughout the world, including in southwestern China, Japan, France and the USA. However, a report in 2014 showed that 35 patients within 31 unrelated Han families with biopsy-proven LPG resided in a small county of Sichuan Basin in southwest China, indicating regional clustering with the same genetic background. It was thus suggested that the descendants of APOE Kyoto in this area were derived from a single founder. In contrast, APOE Guangzhou and Tokyo-Maebashi were reported to be dominant in cases from the southeast area of China (7, 8, 11, 13). The association of APOE Kyoto between the Sichuan Basin and other areas is still difficult to explain. It may be possible that APOE Kyoto spread from China across multiple countries worldwide because it is historically known that the international population mobility of Chinese was more frequent than that of Japanese in ancient times (11). This theory may be similar to the idea that LPG cases may have emigrated along with tea culture to expand from China (11). Several other APOE variants have recently been reported in Western countries, including the United States, Russia, Italy, Brazil, and France. Other APOE variants associated with LPG have been detected across the world (14–20). Although many of the reported cases were reported to be sporadic, some studies reported familial clustering, supporting its genetic component with a single gene inheritance mode (21–23). But perhaps most of the family members lack of symptomatic signs or genetic testing, making them undiagnosed. No heritability studies have been documented to date. One of the largest pedigree was reported in 2008, in which 5 cases in 3 generations were observed (8). Another large pedigree with 3 cases in 2 generations was reported in 2014, together with additional 17 pedigrees with LPG (7).
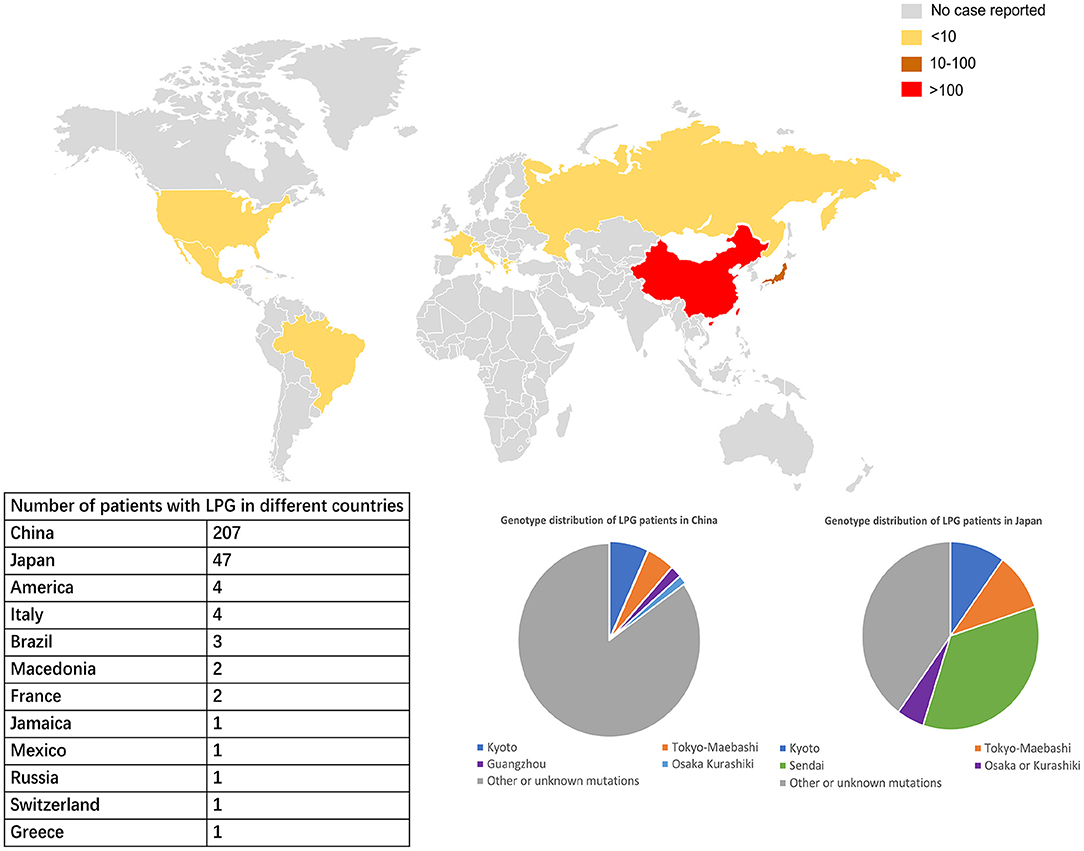
Figure 3. Worldwide distribution of LPG cases. China and Japan are the two countries with the most reported cases of LPG, but the spectra of APOE mutation in patients in these two countries are different. In China, APOE Kyoto is the major mutant, while in Japan, APOE Sendai is the most commonly one. The APOE Sendai mutation has not been reported in China thus far.
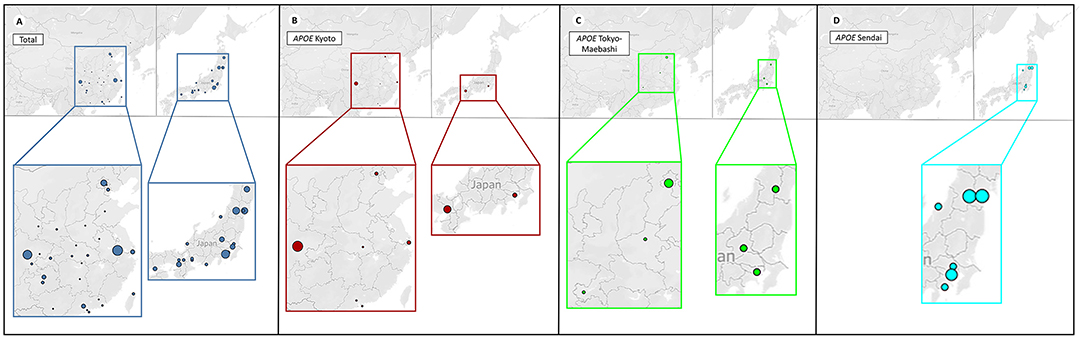
Figure 4. Distribution of patients with different APOE mutations. The four plots represent the distribution of patients with different APOE mutations in China (left) and Japan (right). For better illustration, each image is shown at the bottom enlarged. The size of each circle represents the number count of people. It can be observed that geographical clustering of LPG cases according to APOE mutation types, suggesting a founder effect. (A) Patients with LPG were mainly concentrated in the southwestern and southeastern regions in China, and the central and northeastern regions in Japan. (B) APOE Kyoto was frequently found in southwestern China, and south Japan. (C) APOE Tokyo-Maebashi was dominant in cases from Beijing in China, and the central Japan. (D) APOE Sendai was not reported in China, and distributed most in central Japan.
Apart from humans, interestingly, an observational study in 2016 reported that a captive squirrel (Sciurus vulgaris) spontaneously developed LPG-like disease. This was the first time that LPG-like disease was observed in an organism other than humans. The kidney pathology of squirrels is similar to that of human LPG, but genetic mutations have not been determined (24).
As the majority of the reports on the epidemiology of LPG were based on case reports, to systemically update this information, we conducted a literature review using the search item “lipoprotein glomerulopathy” from various databases (PubMed, China National Knowledge Infrastructure, and J-STAGE) from 1988 to January 2022. A total of 274 LPG cases were identified, and the epidemiological details can be found in Additional File 1.
According to updated statistics, China and Japan have a comparatively higher prevalence of LPG, with 207 and 47 patients, respectively. There is still lack of precise epidemiological studies in LPG. Based on the current meta data from literature review, it was estimated that the prevalence of LPG is about 3.74 per 10 million in Japan and 1.43 per 10 million in China. Surely, it may be underestimated. As mentioned above, the areas most affected by LPG were located in southwestern and southeastern China and central and northeastern Japan. A total of 20 cases have been reported in other countries and territories. Among all the cases, there were 137 males and 137 females, with an average age of diagnosis of 35 years old (from 7 to 72 years). In terms of age of onset, a total of 188 patients had documented information. There were 8, 23, 112, 42 and 3 cases with age of onset ≤ 10, 11–17, 18–45, 46–65, and >65. It suggested that about 60% of the cases were diagnosed in young adults (18–45 years old) (13).
With regard to specific mutation distribution, APOE Kyoto, APOE Tokyo-Maebashi and APOE Sendai are the three major observed forms, with 53, 13, and 14 reported patients, respectively. In China, the Sendai mutant has not yet been reported. The Kyoto mutant is mainly diagnosed in Sichuan Province in southwest China, while the Tokyo-Maebashi variant is mainly diagnosed in Beijing, suggesting that LPG patients in the same region may share a common genetic ancestor; demonstrating the “founder effect.”
The precise pathogenesis of LPG is still not well-understood. Several lines of evidence suggest that alterations in apoE structure and function play a fundamental role in the pathogenesis of LPG. It has long been hypothesized that defective lipoproteins are prone to deposit in the kidney. Indeed, all the patients reported to date were found to have heterozygous mutations in the APOE gene, along with elevated serum concentrations of apoE lipoproteins and the presence of apoE in the glomerular deposits. Disturbances in kidney structure or function may also be pivotal in the formation of lipoprotein aggregates, as the disease is kidney specific without other organs obviously affected. However, the recurrence of LPG after renal transplantation suggests that renal abnormalities may not be necessary for the development of the disease.
The APOE gene is located on chromosome 19q13.2 and comprises 4 exons with 3,603 base pairs, which are evolutionarily conserved in a variety of terrestrial and marine vertebrates (25) (Figure 5). The apoE protein is a 34 kDa circulating glycoprotein of 299 amino acids, with an additional 18 amino acids as a signal peptide. This protein can be synthesized by several cell types, in which hepatocytes account for the majority. High quantities can also be observed in brain, i.e., by astrocytes and glial cells in the cerebral cortex and by neurons in the frontal cortex and hippocampus (26).
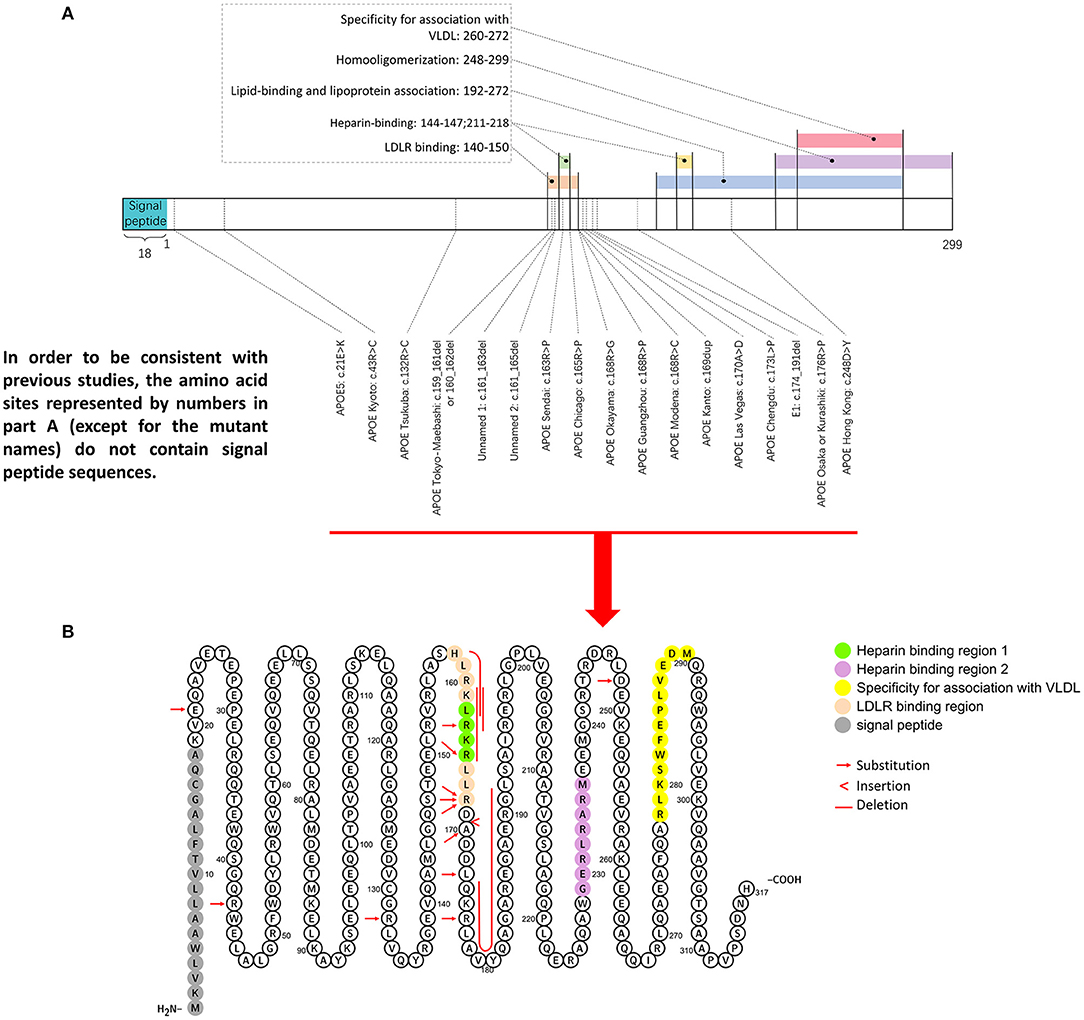
Figure 5. Representative mutation distributions of the apoE protein. (A) Seventeen mutations of the APOE gene leading to LPG have been located, most of which are concentrated at amino acid sites 140–180. This region contains various important apoE functional domains, including the LDLR binding domain and HSPG binding domain. “Hot spot” mutations suggested that the change in the binding ability of the apoE mutant to LDLR and HSPG is an important factor in LPG pathogenesis. (B) ApoE mutation sites were labeled in the amino acid sequence diagram of apoE protein. It showed that hot spot of apoE mutation is among AA 140–160.
The amino acid sequence of apoE can be simply divided into three parts: the N-terminal (AA 1–199), the hinge region (AA 200–215), and the C-terminal (AA 216–299). There are two important binding regions in the N-terminal sequence that have overlapping gene sequences, the low density lipoprotein receptor (LDLR) binding region (27, 28) (142–150) (29) and the heparin sulfate glycoprotein (144–147) binding region (30). Most APOE mutations associated with LPG are observed in these two regions. Other regions with functional significance included four helix regions (AA 20–160), a lipid insertion sequence (244–272) and a homo-oligomerization region (248–299) (31–34), which have been investigated less.
As the ligand for the LDL receptor family and heparan sulfate proteoglycans (HSPG), apoE associates with triglyceride-rich lipoproteins in mediating clearance of their remnants (Figure 6). When exogenous lipids enter the bloodstream from intestinal villi, new chylomicrons are formed to obtain apoC and apoE from high-density lipoprotein (HDL). Some chylomicrons are utilized by body tissues, while the remaining chylomicrons enter the liver through interactions with apoE and LDL receptor-associated proteins (LRP).
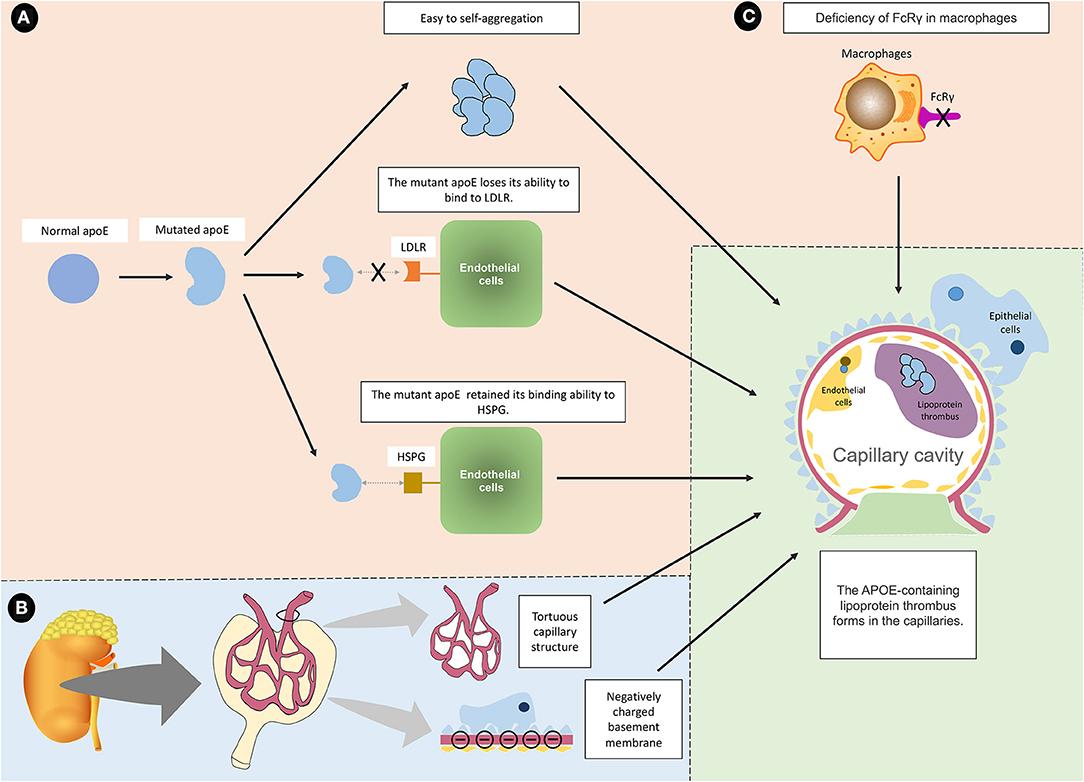
Figure 6. Summary and proposed mechanisms of LPG. (A) The mutated apoE protein is known to cause LPG through three main mechanisms. (a) The mutant protein tends to aggregate, and the aggregated macromolecules are more likely to drive the formation of lipoprotein thrombi. (b) The mutant protein loses its ability to bind to LDLR, making it difficult to eliminate. (c) The mutated protein retains the ability to bind to HSPG, which allows it to bind to endothelial cells. For the above reasons, the mutated apoE protein aggregates into macromolecules on the surface of endothelial cells without being cleared. (B) In addition to APOE mutation, it is speculated that intrinsic glomerular features may interact with apoE variants and lipoprotein abnormalities, which exacerbate the induction of LPG. For example, tortuous glomerular capillaries are conducive to the formation of thrombi. The negatively charged base membrane and the positively charged mutant protein may attract each other. (C) FcRγ deficiency may also lead to LPG because it affects the phagocytic function of macrophages.
ApoE is also important in endogenous lipid metabolism. Hepatocytes secrete VLDL containing apoE, and these VLDL are converted into IDL. The subsequent metabolism of IDL can be divided into two pathways, through interactions between apoE and LRP or metabolism into LDL (35–37). The degradation of both IDL and VLDL is partially dependent on apoE. This may explain why their concentrations are elevated in the blood of patients with LPG.
Two coding variants in the human APOE gene, rs429358 (Cys130Arg) and rs7412 (Arg176Cys), define the apoE protein as three isoforms (E2, E3, and E4) (38), among which E3 is the most common isoform, and they are different in the amino acid residues at residues 112 and 158: both cysteine—apoE2, both arginine—apoE4, and one cysteine—apoE3 (the wild type). It has been confirmed that different subtypes are associated with different predispositions to human diseases (39). The affinity between the E2 subtype and the low-density lipoprotein receptor (LDLR) is only 1% of that of the E3 subtype, resulting in lipid clearance disturbances and type III HLP. The E4 subtype is mainly associated with diseases of the central nervous system, such as Alzheimer's disease (34, 40–42). However, in LPG, the most common subtype was reported to be E3/E3 or E3/E4 (3, 9).
In 1991, abnormally elevated levels of apoE were observed in patients with LPG for the first time (6), followed by the discovery of the mutation APOE Sendai in 1997. Further APOE mutation has been suggested to be the most important etiologic factor in the pathogenesis of LPG (43, 44). Supporting evidence was obtained from animal models. In 2000, by introducing the APOE Sendai mutation into APOE-deficient mice, it was observed that both increased lipid levels and LPG-like renal pathology in the mice (45). Several other variants of APOE associated with LPG have been identified since then. A similar cause-effect of the APOE Kyoto mutation in LPG can also be observed in APOE-deficient mice, further supporting that a single gene mutation can cause LPG (46).
To date, a total of 17 APOE gene variants associated with LPG have been reported. Eight of these mutations reside in the LDLR binding region (140–150 sites), among which 5 reside in the HSPG binding region (144–147 sites). Another hot spot is located in the region spanning AA 150–180, where 5 mutations have been reported (25, 33), but few functional studies have been conducted. More recently, a case report suggesting a 28-year-old man presenting with severe proteinuria and hyperlipidemia had compound heterogeneous mutations of the APOE gene inherited from his mother (p.Arg50His) and father (APOE Kyoto). Each his parents with a heterogeneous mutation had normal kidney function without proteinuria (47). This is the first time that the combination of the 2 mutations was identified in the same case as an autosomal recessive genetic disorder. It seemed that the case showed much severe phenotype. But further precise investigation on both genetic and disease mechanisms will be needed. Since the patient carried APOE Kyoto mutation, and the new p.Arg50His variant has not been verified functionally pathogenic, therefore, this mutation is not listed in our pathogenic mutations of LPG. We assume that a dominant effect of APOE Kyoto cannot be totally rule out. Apart from this, a case was reported to have a combination of APOE Kyoto and APOE Hong Kong (Asp230Tyr) mutations (48). But as co-segregation analysis was not taken, it was difficult to confirm the significance of the respective mutation. There was also a case report of a 51-year-old Japanese woman who had 2 mutations within the same allele (a combination of APOE Chicago (Arg147Pro) and APOE5 (Glu3Lys) inherited from her mother. But her mother did not have any phenotypes (49). These data suggested LPG may be far complicated than previously speculated. Additional genetic studies may be needed, i.e., from a hypothesis-free whole genome sequencing in large cohorts.
Normal apoE protein is highly helical and has the capability to be transformed between different tertiary structures to bind lipids or proteins. In 2013, it was found that different mutated proteins [APOE Chicago (Arg147Pro), APOE Sendai (Arg145Pro), APOE Osaka or Kurashiki (Arg158Pro)] showed different protein structural stabilities (50). Of note, the common resultant characteristic of these three mutations was that arginine was replaced by a proline residue. It is universally suggested that proline residues may breakdown a transmembrane helix. Insertion of a proline residue in the middle of an α-helix is known to destabilize it by over 3 kcal/mol, effectively disrupting the helix (51, 52). These mutated apoE proteins all exhibited reduced helicity, leading to decreased structural stability evidenced as protein denaturation even at the physiological temperature of 37°C. When the hydrophobic surface is exposed, apoE may be more prone to aggregate and form large lipoprotein granules (50). Along with this idea, it was also confirmed that similar structural and functional changes can be observed in three other non-proline-substituted mutants [APOE Kyoto (Arg25Cys), APOE Tsukuba (Arg114Cys), and APOE Las Vegas (Ala152Asp)] (53). In vitro, all three mutated proteins showed decreased stability and an increased tendency to aggregate. Likewise, a study in Alzheimer's disease showed that the decreased structural stability of apoE may contribute to the formation of neurotoxic fibrils (54).
ApoE mutations may also contribute to reduced binding capacities to receptors (55, 56). It was observed that the binding ability of apoE Kyoto and apoE Sendai to LDLR was significantly reduced to just 10 and <5%, respectively, compared to the wild type (57, 58). The decrease in the apoE binding ability contributes to impaired degradation of lipoprotein and accumulation. This is consistent with the observation that hyperlipidemia is common in LPG, especially with increased VLDL and IDL. However, the binding capacity of apoE2 to LDLR was <1% of the normal value (33, 59), while apoE2-induced type III HLP only had dyslipidemia without pathological kidney changes, indicating that impaired LDLR binding abilities may not be the determining factor in kidney damage. Although the APOE Sendai mutation decreased the binding ability of apoE with LDLR to <5%, it decreased its binding ability with HSPG to 66% (57). It was suggested that the retained HSPG binding activity could enable apoE-rich lipoproteins to enter and attach to the Disse space (60, 61), which is pivotal in the initial rapid clearance step for lipoproteins. Heparan sulfate proteoglycans (HSPGs), which are also abundant in the space of Disse, may play an important role in mediating this enhanced binding. When apoE arrived and aggregated in the Disse space, they would normally enter the liver through interaction with LDLR, but the mutated apoE lost the LDLR binding ability, so they could not enter the liver to be metabolized (62, 63). Since HSPG is highly expressed in the glomerular basement membrane (64, 65), it may play a role in the renal deposition of lipoprotein. Concordant with this hypothesis, evidence has shown that the binding ability of products is enhanced in apoE Chicago to glomerular capillaries and in apoE Kyoto to human umbilical vein endothelial cells (16, 66).
As some individuals carrying APOE mutations are asymptomatic, it is speculated that some other factors may further contribute to LPG (21). Two clinical reports noted the recurrence of LPG in the transplanted kidney. Sustained inflammation induced by chronic graft-vs. host disease (GVHD) might be a predisposing factor (19, 67, 68). By injecting donor spleen cells into recipient mice for 2–4 months, it was observed that GVHD could induce LPG-like changes in Fc receptor gamma chain (FcRγ)-deficient mice, with hematuria, proteinuria, renal capillary lumen thrombosis and interstitial mononuclear cell infiltration. It was shown that macrophages from FcRγ-deficient mice had a decreased ability to clear LDL (69). Macrophages possess several different pathways in recognition and clearance of modified (oxidized) LDL, including scavenger receptors and FcRs (70, 71). Supporting this, a drastic decline of scavenger receptor CD36 was observed in LPG. It was speculated that partial reduction of modified (oxidized) LDL uptake by macrophages could result in the lipoprotein deposition in the kidney during the long course of chronic GVHD.
More direct evidence was obtained from humanized mice. When the APOE3 genotype or APOE Sendai mutation was introduced into FcRγ and APOE double-knockout mice, both strains of mice developed lipoprotein thrombosis, and the mice with APOE3 showed more lipoprotein thrombus in the kidney than those with the APOE Sendai genotype. This observation was absent in mice with wild-type APOE. Introduction of APOE3 in both FcRγ-deficient mice and FcRγ wild-type mice showed lipoprotein thrombosis, whereas the phenotype was much more severe in FcRγ-deficient mice (72). These results indicated macrophage impairments derived from FcRγ deficiency (73–75) were insufficient for the development of LPG, since the FcRγ deficient mice with normal murine apoE showed no lipoprotein thrombosis. However, under conditions with xenogeneic apoE, especially human apoE3, the FcRγ deficient mice may develop severe LPG.
It was also observed that a small amount of apoE can be produced by macrophages, which was considered to play a role in suppressing hyperlipidemia and arteriosclerosis. Because it was showed that expression of macrophages producing apoE Sendai in mice that received a bone marrow transplant protected against atherosclerosis while induced LPG (76–78). It was thus suggested that macrophages may play various roles in apoE related lipoprotein metabolism. Both hyperactivity or suppression can be an important factor in different types of renal lipidosis. LPG depend upon suppression of macrophages. ApoE derived from macrophages is affected by its mutation and may regulate disease activity. Functional studies are still needed in the future.
The above data is insufficient to explain why lipoprotein is specifically deposited in glomerular capillaries in LPG, which is different from other diseases such as atherosclerosis. In particular, because lesions are localized to glomeruli, intrinsic glomerular factors may interact with apoE variants and lipoprotein abnormalities to induce LPG. There might be some special factors in the glomeruli. Mutated apoE protein may present different electric charges compared to normal proteins, and therefore, they exhibit a higher affinity for negatively charged glomerular basement membranes (9). It is suggested that the presence of highly conserved acidic residues within the lipoprotein receptor (LR) modules and the positively charged region of apoE (residues 136–150) may support the hypothesis that ligand–receptor recognition is due to electrostatic interactions, so a change in electron charge might enhance the bonding (79). The tortuous structure of glomerular capillaries might also contribute to lipoprotein deposition.
It has long been believed that hyperlipidemia may play a detrimental role in kidney pathology directly or indirectly through inflammation, ROS production, endogenous electrical stress and other pathways (80, 81). Among these pathways, oxidative stress may be of special importance, as carboxymethyllysine (CML), hydroxynonenal (HNE)-protein, and malondialdehyde (MDA)-lysine were reported to be found in the kidney of a patient with LPG (68). These substances are the products of lipid peroxidation. These aldehydes may cross-link covalently with matrix tissue proteins and further alter structure and function. They may also have direct impact on parenchymal cells by cross-linking cell surface proteins to reduce intracellular responses (82–84). As the antioxidant domain of APOE overlaps with the LDLR binding region (85), mutations in the LDLR binding domain may shed unfavorable effects on its antioxidant capacity. Moreover, the roles of oxidative stress and hyperlipidemia in LPG are indirectly supported by clinical evidence that some patients experience remission after treatment with the antioxidant probucol and the lipid-lowering drug fenofibrate (10, 86).
There have been reports that no APOE mutations were found in a number of LPG patients (87). Possible explanations include changes in other unknown genes, mutations in introns or regulatory sequences, epigenetics, and environmental influences.
For example, a recent study showed that a defect in HDL receptor named scavenger receptor class B type 1 (SR-B1) was associated with LPG-like lesions in atherosclerotic mice, the severity of which can be alleviated using probucol (88).
Factors affecting CKD progression, such as fibrosis, apoptosis, local tissue injury, and infiltration of immune cells, may also be involved in LPG.
These data support a complex etiology of LPG, and additional pathogenic factors warrant further elucidation.
In 2006, a single set of diagnostic criteria for LPG was proposed by Japanese nephrologists (2). The criteria were mainly based on four items.
• different levels of proteinuria;
• dilatation of glomerular capillary lumina with pale-stained substances on light microscopy;
• fingerprint-like concentric lamellar structure in electron microscopy;
• Type III HLPs with high apoE concentrations are usually associated with a heterozygous apoE phenotype, E2/3 or E2/4, by means of isoelectric focusing electrophoresis (IEF), but sometimes with an uncommon type, e.g., E1/3 or others.
It was suggested that although genetic testing was not necessary for definitive diagnosis, it should be performed in patients with suspected LPG whenever possible to confirm the diagnosis.
Initially, LPG may cause no symptoms. Symptoms and signs are due to the buildup of waste products and fluid retention in the body (49, 89, 90). In general, nephrotic syndrome may be present in ~70% of patients (91). However, the levels of proteinuria may vary significantly among different patients. The 24 h urinary protein levels reported ranged from 0.5 to 24 g/24 h (92, 93). Without proper treatment, CKD will progress (2, 14, 94–96), and approximately half of patients will develop end-stage kidney disease over 1–27 years.
One of the most characteristic laboratory indicators of LPG is elevated serum apoE levels. It was reported that patients with LPG had a mean serum apoE concentration of 11.14–17.1 mg/dL (3.9–71.0 mg/dL), approximately twice the upper level of the normal population (2, 7, 23, 97, 98).
Another characteristic is overt dyslipidemia, with a predominance of triglycerides, mostly >6 mmol/L (15, 16, 91). In a few cases, VLDL levels were reported to be elevated (~4–6 mmol/L) (99, 100). Hyperlipidemia in LPG is similar to that in familial type III HLP. Type III HLP, first recognized in 1967, is caused by homozygous apoE2 mutations. The mutation was reported to weaken the binding force between apoE2 and LDL receptors (101, 102). Although both diseases are associated with the APOE gene, LPG and type III HLP show obvious differences. In type III HLP, atherosclerotic cardiovascular disease and xanthomatosis are common (103, 104), but they are rare in LPG.
We further checked the clinical manifestations of LPG by systematic literature review. An additional file shows this in more detail (see Additional File 2). The mean value of plasma albumin for the reported cases was 29.3 g/L (SD = 6.2 g/L, ranging from 12 to 47 g/L), and the mean 24-h urinary protein level was 4.5 g/d (SD = 3.4 g/d, ranging from 0.8 to 24 g/d). Nephrotic range proteinuria and nephrotic syndrome were key features. The mean creatinine was 129.2 μmol/L (SD = 177.5 μmol/L, and ranging from 21.2 to 1859 μmol/L), the mean urea nitrogen was 8.1 mmol/L (SD = 5.52 mmol/L, and ranging from 2.7 to 39.9 mmol/L), and the mean eGFR was 81.6 ml/min/1.73 m2 (SD = 32.5 ml/min/1.73 m2). Of these patients, 38.7% had CKD1, 35.5% had CKD2, 18.7% had CKD3, 3.2% had CKD4, and 3.9% had CKD5. In terms of blood lipids, the average triglyceride level of these patients was 3.4 mmol/L (SD = 2.0 mmol/L, ranging from 0.7 to 20.6 mmol/L) (reference range 0.6–1.7 mmol/L), which is approximately twice the normal upper limit. Total cholesterol was 6.8 mmol/L (SD = 2.4 mmol/L, and ranging from 2.8 to 22.9 mmol/L) (reference range 2.8–5.2 mmol/L), LDL was 3.6 mmol/L (SD = 1.4 mmol/L, and ranging from 1.2 to 10.6 mmol/L) (reference range 2.1–3.1 mmol/L), and HDL was 1.4 mmol/L (SD = 0.7 mmol/L, ranging from 0.5 to 8.8 mmol/L) (reference range 0.9–1.6 mmol/L). Most of the patients had significantly elevated serum apoE, with a mean of 12.1 mg/dL (SD = 6.7 mg/dL, and ranging from 3.1 to 42.3 mg/dL). Great fluctuations of standard deviations suggested great clinical heterogeneity of LPG.
We collated the clinical information of the cases with the top four APOE genotypes (Kyoto, Tokyo-Maebashi, Sendai and Osaka or Kurashiki) (Table 1). The age of onset for patients with APOE Tokyo-Maebashi seemed to be younger than that of other mutants. Patients with APOE Sendai appeared to have the lowest urinary protein and blood lipid levels. Patients with APOE Kyoto had the most severe renal manifestations. There was few clinical prognostic information available for LPG. However, according to literature review and case reports, some of the cases with LPG will progress to ESKD. These reported ESKD caused by LPG can be observed in several APOE genotypes including Kyoto, Tokyo-Maebashi, Osaka or Kurashiki, Guangzhou, and Sendai. And the reported APOE phenotypes were E2/3 and E3/3 (7, 8, 105–107). Thus, it still difficult to predict prognosis based on mutation types.
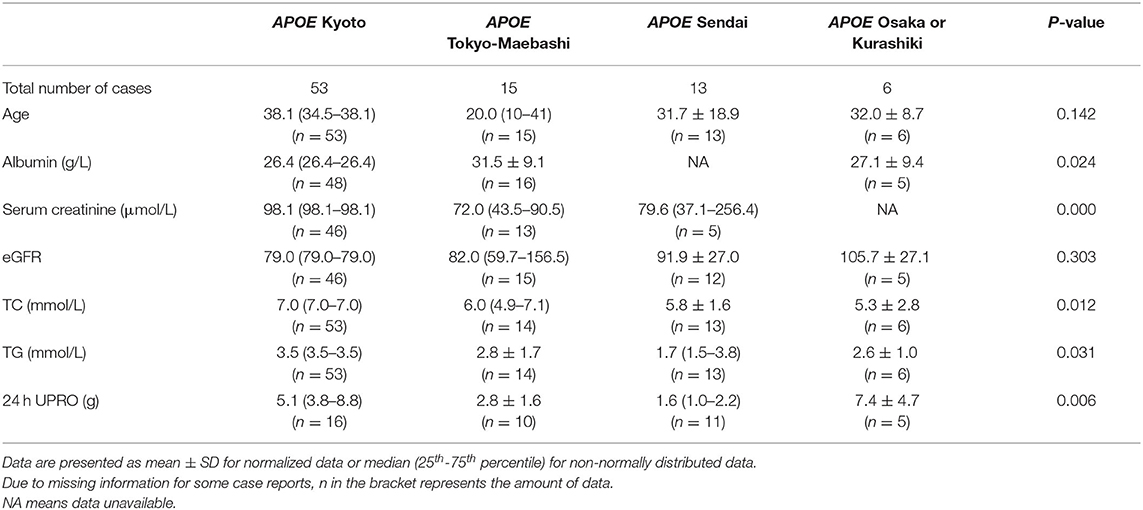
Table 1. Genotype and phenotype information based on meta data.
Other complications rarely have been reported. Known comorbidities included splenomegaly, thalassemia, psoriasis, abdominal aortic aneurysm, pleural effusion, and neurofibromatosis type I (14, 67, 99, 108, 109). However, there is no evidence of a direct link between LPG and these diseases. The only related extrarenal manifestation of LPG that has been confirmed is intravascular coagulation (110). It was reported that a 50-year-old white man had severe proteinuria with high lipid levels and a kidney pathology of LPG. He also had hypertension and coronary heart disease. The patient's heart failure was speculated to be due to cardiac amyloidosis secondary to multiple myeloma. For this reason, cardiac biopsies were requested and showed that small blood vessels in the endocardia were filled with eosinophilic substances, which were similar to those in the kidney. A more recent case reported that a 21-year-old Malaysian-Swiss male with LPG developed atypical hemolytic uremic syndrome (aHUS). Aggregation of apoE was suggested by the authors as a risk factor in initiating the occurrence of aHUS in his case (107). Regardless, the etiology was unclear and may be difficult to determine.
The most typical pathological appearance of LPG under light microscopy is the dilatation of the glomerular capillary lumen filled with eosinophilic granular and vacuolar thrombosis (Figure 7). The thrombus is positive for oil red O or Sudan red and negative for silver or PAS staining, indicating the presence of a lipid component in the thrombus (14, 22, 110–112). Using immune-electron microscopy, it has been found that lipids are surrounded by apoE and that the thromboid material is composed of lipoproteins (113). Other manifestations under light microscopy include swelling of endothelial cells and vacuolar degeneration with a small number of lipid droplets in podocytes. In the advanced stages of the disease, the mesangial cells and stroma are thickened, and there may be uneven insertion, leading to thickening of the basement membrane and the formation of the dual-track sign and eventually glomerular sclerosis. Epithelial vacuoles and granular degeneration may be observed in renal tubules in the early stages. Similar to other glomerulopathies, as the disease progresses, renal tubule atrophy, interstitial edema, monocyte infiltration and fibrosis, and thickening of the arteriole wall will be common (21, 114). A study also reported that CD68+ foam cells were present in patients' kidneys, further suggesting that macrophages may be involved in the pathogenesis of LPG.
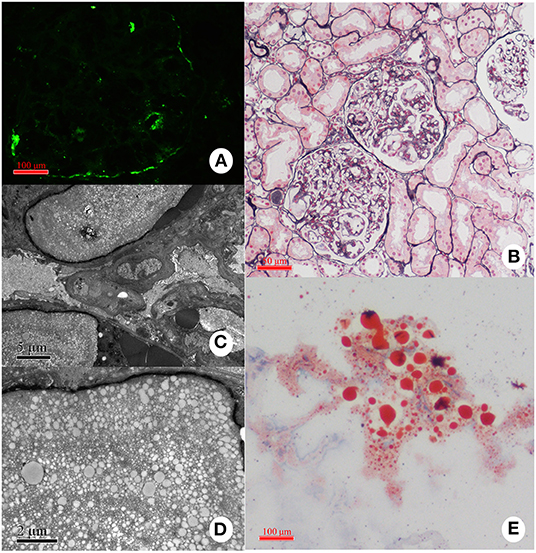
Figure 7. Representative pathological findings in a case with LPG. (A) Immunofluorescence study: the deposition of apoE is present mainly in the capillary lumina. (B) Light microscopy: Dilated capillary loops exhibiting an eosinophilic lipoprotein thrombus in the capillary lumens. (C,D) Electron microscopy: Diffuse foot-process effacement and lamellated fingerprint-like thrombi in capillary lumens, which are composed of granules and vacuoles of various sizes. (E) Oil red O staining: Numerous red droplets are seen in the thrombus-like substances in the glomerular capillaries.
The most common deposits are apoE and apoB in immunofluorescence, which might be taken as supporting evidence for the diagnosis of LPG. In the previous literature, however, immunoglobulin and complement, such as IgM, IgA, and C3 depositions, may be observed in mesangial and capillary walls in many cases, and scattered C1q deposits were also reported (16, 23, 48, 114, 115). However, most of these depositions were suggested to be non-specific, and no specific pathology pattern has been described until now.
Under electron microscopy, lipoprotein thrombosis is characterized by a fingerprint-like concentric lamellar structure, sometimes referred to as a sand stone structure (2). Some other pathological descriptions include glomerular telangiectasis, filled with protein material covered with lipid vacuoles, and lamellar vacuoles of varying sizes, with a network of vacuoles separated by high electron density bands. Lysosomes of endothelial cells and podocytes were increased, and lipid vacuoles were also found in the cytoplasm and lysosomes. Podocytes showed diffuse and complete foot process effacement with the accumulation of electron-dense material in the mesangium and glomerular basement membrane. The glomerular basement membrane demonstrated focal subcutaneous electron dense deposition (100, 110–112, 114, 116).
Because the main pathological feature of LPG is lipoprotein thrombosis, it should be differentiated from other diseases with thrombotic substances, such as macroglobulinemia nephropathy and cryoglobulinemia nephropathy.
There is still a lack of specific or targeted therapies for LPG. Most of the regimens available were based on lowering proteinuria and hyperlipidemia. In the absence of clinical trials, current data are mostly from case reports or observational studies.
Early in the history of LPG, the patient was often treated with glucocorticoids with or without immunosuppressants, similar to other nephrotic syndromes. However, it was soon proven ineffective (43, 95, 108, 117, 118). Additionally, in the twentieth century, it was reported that LPG relapsed in nearly all ESKD patients caused by LPG even with intensive immunosuppressive therapy. Therefore, kidney transplantation and immunosuppressants were not recommended because the culprit was abnormal lipoprotein components in the blood instead of the kidney. However, in some case reports, leflunomide was reported to ameliorate proteinuria in some patients. It was initially used to replace prednisone, which was found to have a complete disappearance of kidney symptoms after leflunomide was added (119). The exact mechanism of leflunomide may not be related to its immunosuppressive effect.
Regimens have achieved some success in supporting the pathologically causal role of lipoprotein and hyperlipidemia in LPG. The first drug was probucol, which reduces total TC and LDL levels and is widely used in the treatment of hypercholesterolemia (120). Probucol is also an antioxidant, and it may mitigate glomerular damage caused by oxidative stress of mutated proteins. As early as 1994, it was used in an LPG female case of 54 years old, effectively reducing hyperlipidemia, proteinuria and the complete elimination of her glomerular lipoprotein thrombosis (86). Later, different case reports confirmed the efficacy of probucol therapy (116, 121).
Fenofibrate and benzafibrate can significantly reduce blood VLDL levels by acting on peroxisomal proliferator-activated receptor (PPAR) and activating lipoprotein lipase (LPL) and thus reducing TG and LDL. A small clinical contrast study based on 35 patients supported the efficacy of fibrates in LPG. After 12 months of treatment, their lipid profiles, proteinuria, and serum albumin improved, and their serum apoE decreased significantly with stable renal function (7, 14, 93, 121, 122).
Niceritrol is a nicotinic acid derivative that has been used to lower lipoprotein levels and reduce proteinuria in patients with chronic kidney disease associated with hyperlipidemia (123, 124). It was used to relieve clinical symptoms in two patients with LPG who had failed to respond to statins (125). However, there was still a lack of more widespread attempts.
The other drugs that are still in clinical use today are statins. As a potent inhibitor of 'β-hydroxy β-methylglutaryl-CoA' ('HMG-CoA') reductase, statins can increase the expression of the LDL receptor in the hepatocytes' cell membranes. In addition, statins also have multiple effects, such as reducing TG, improving endothelial function, and anti-oxidation (126). Statins can reduce patients' blood lipid levels but can also effectively lower urinary protein levels and retain renal function. There were very few cases in which statin therapy alone was evaluated (16, 90, 94, 108, 111, 119, 125, 127).
Renin-angiotensin-aldosterone system inhibitor (RAASi) therapy shows reno-protective effects in various forms of proteinuric CKD. Therefore, although there is no evidence that ACEIs and ARBs are effective in LPG, their administration can be considered suitable both for blood pressure control and to reduce proteinuria among LPG patients. Some case reports have suggested that antilipidemic drugs combined with ACEIs and ARBs are an effective treatment for LPG (128). Recently, SGLT2 inhibitors proved to be beneficial for patients who have proteinuric CKD with or without diabetes. With evidence that SGLT2 inhibitors might be safe and useful in hereditary renal diseases such as Alport syndrome (129), whether SGLT2 inhibitors could be a potential treatment for LPG remains to be further investigated.
Adsorption therapy also showed a certain effect. LDL apheresis was used when the patient had a poor response to traditional drug therapy. However, allergic reactions to LDL-apheresis are a concern (90). In 2009, a heparin-induced extracorporeal lipoprotein precipitation (HELP) system was first used to treat a 60-year-old white woman with proteinuria, high blood pressure, and renal failure (90). The basic principle of this system is to acidify plasma pH to ~5 in vitro and then use heparin to form a polymer with LDL-cholesterol, LP (a), fibrinogen, and triglyceride to precipitate them out (130, 131). After 25 courses of treatment, the patient achieved significant clinical improvements, including decreased urinary protein, blood creatinine and lipid levels.
Protein A, immunosorbent may also be a treatment option. Since protein A has a strong affinity for the FcγR of IgG, this effect can compensate for the lack of an FcRγ. This technique has previously been shown to reduce proteinuria levels in patients with a variety of nephropathies, including diabetic nephropathy, IgA nephropathy, and amyloidosis (132). In a small pilot study involving 13 cases, immunoadsorption was administered for 10 cycles per session and 10 sessions as a course. A total of 30 L of plasma was regenerated in each course. Apart from proteinuria reduction, a repeated renal biopsy (n = 12) showed that intraglomerular lipoprotein thrombi almost disappeared. The proteinuria of six patients returned to baseline levels within 12 months. Four recurrent patients received repeat immunoadsorption treatment and showed repeated efficacy (97). The strategy is advantageous in being rapidly therapeutic and having relatively stable effects. However, disadvantages include invasive procedure complications, high cost and risk of infection.
Other lipid-lowering drugs, such as niacin (133) and apoC-III monoclonal antibodies, as well as non-pharmaceutical approaches, such as lipoprotein apheresis, may also be promising (134, 135). Other antioxidants, apart from probucol, including vitamin C, polyphenols, N-acetylcysteine, allopurinol, natural polysaccharides, pentoxifyllin, and bardoxolone methyl, may be candidates to study antioxidant efficacy, since antioxidant therapy apparently reduces CKD progression (136, 137). However, high interindividual variability and off-target effects, such as body weight reduction, need to be further investigated. The anticonvulsant topiramate, which induces weight loss and a moderate reduction in plasma lipids and glucose, has recently been reported to protect APOE-deficient mice from kidney damage. Thus, it could be investigated in drug repurposing studies for the treatment of glomerular lipidosis (138).
Gene-based therapeutics, pioneered for the treatment of monogenic inherited retinal disease, are being actively investigated as new treatments for acquired retinal disease. Gene therapy could also be tried in the future, i.e., by CRISPR/Cas9 technology, since it is widely believed that LPG is an inherited disease caused by an abnormality in the APOE gene (2, 9).
Current studies have found that several mutations of the APOE gene can lead to LPG, and most of these mutations are concentrated in some hot spots. Future functional research on LPG focusing on hot spot regions for gene mutations may be helpful to explore the specific pathogenesis of the disease and to develop target drugs. It is worth exploring whether different genotypes lead to differences in clinical phenotypes after increasing the number of patients who are enrolled and who can be stratified.
Although the symptoms of LPG are common and fixed, some cases still cannot be explained by the current theory. For example, in 2015, a patient presented with a large number of macrophage infiltrates in the glomerulus. In most cases, macrophage infiltration is not present in the glomerular capillaries of patients with LPG (91). In 2018, a new variant of the APOE gene (APOE Toyonaka) was discovered. Patients with this gene mutation presented with kidney pathology that resembled membranous nephropathy (MN) rather than LPG (139, 140). For cases such as this, it is still not clear if they belong to a new disease category or a different spectrum of LPG (27, 28, 141). APOE-related disorders were recently summarized, mainly including APOE2 homozygote glomerulopathy (HLP), MN-like APOE disease, and LPG (141). All the cases above indicate that the incidence and manifestations of LPG cannot be fully explained by existing theories. In addition to the new theory of FcRγ pathogenesis, other causes of LPG may be discovered in the future.
The current clinical diagnosis and treatment of LPG still needs to be improved. As the clinical manifestations of LPG are often non-specific, the actual incidence of LPG is underestimated. The diagnosis is beyond the scope of primary care hospitals, and conventional nephropathy treatment has proven ineffective, so the sensitivity of the diagnosis needs to be increased so that more patients can be treated properly. In terms of treatment, effective drugs are not commonly used in practice, and therapy has not been commonly used, so new drug research and development are needed. For those who have been diagnosed, indicators for clinical surveillance are still lacking. As a rare disease, an international registry may be worthwhile, as registries are essential for epidemiological data and collaborative research.
M-sL: collected data, conceived, and wrote the manuscript. YLi: collected data and wrote the manuscript. YLiu: collected data. X-jZ: revised the manuscript critically for important intellectual content and supervised the research group and has given the final approval of the version to be published. HZ: edited the manuscript and has given the final approval of the version to be published. All authors contributed to the article and approved the submitted version.
Support was provided by National Science Foundation of China (82022010, 82131430172, 81970613, 82070733, 82000680, and 82070731); Academy of Medical Sciences—Newton Advanced Fellowship (NAFR13_1033); Beijing Natural Science Foundation (Z190023); Fok Ying Tung Education Foundation (171030); and CAMS Innovation Fund for Medical Sciences (2019-I2M-5-046). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We apologize to those whose work or original publication could not be cited because of space limitations.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.905007/full#supplementary-material
LPG, Lipoprotein glomerulopathy; LCAT, Lecithin-cholesterol acyltransferase; LDLR, Low-density lipoprotein receptor; HSPG, Heparan sulfate proteoglycan; HLP, Hyperlipoproteinemia; LRP, LDL receptor-associated protein; GVHD, Graft-versus-host disease; FcRγ, Fc receptor gamma chain; CML, carboxymethyllysine; HNE, hydroxynonenal; MDA, Malondialdehyde; aHUS, Atypical hemolytic uremic syndrome; PPAR, Peroxisomal proliferator-activated receptor; LPL, Lipoprotein lipase; HMG-CoA, β-hydroxy-β-methylglutaryl-CoA; RAASi, Renin-angiotensin-aldosterone system inhibitor; HELP, Heparin-induced extracorporeal lipoprotein precipitation; MN, membranous nephropathy; IDL, Intermediate density lipoprotein; VLDL, Very low-density lipoprotein; HDL, High-density lipoprotein; TC, Total cholesterol; TG, Total triglyceride.
1. Watanabe Y, Yoshida F, Matsuo S, Mitarai T, Hirose S, Saito T, et al. [Lipoprotein glomerulopathy:–a new kind of glomerular disease with peculiar histological findings]. Nihon Rinsho. (1989) 47:1667–74.
2. Saito T, Matsunaga A, Oikawa S. Impact of lipoprotein glomerulopathy on the relationship between lipids and renal diseases. Am J Kidney Dis. (2006) 47:199–211. doi: 10.1053/j.ajkd.2005.10.017
3. Saito T, Matsunaga A, Ito K, Nakashima H. Topics in lipoprotein glomerulopathy: an overview. Clin Exp Nephrol. (2014) 18:214–7. doi: 10.1007/s10157-013-0887-4
4. Saito T, Sato H, Kudo K, Oikawa S, Shibata T, Hara Y, et al. Lipoprotein glomerulopathy: glomerular lipoprotein thrombi in a patient with hyperlipoproteinemia. Am J Kidney Dis. (1989) 13:148–53. doi: 10.1016/S0272-6386(89)80134-9
6. Oikawa S, Suzuki N, Sakuma E, Saito T, Namai K, Kotake H, et al. Abnormal lipoprotein and apolipoprotein pattern in lipoprotein glomerulopathy. Am J Kidney Dis. (1991) 18:553–8. doi: 10.1016/S0272-6386(12)80649-4
7. Hu Z, Huang S, Wu Y, Liu Y, Liu X, Su D, et al. Hereditary features, treatment, and prognosis of the lipoprotein glomerulopathy in patients with the APOE Kyoto mutation. Kidney Int. (2014) 85:416–24. doi: 10.1038/ki.2013.335
8. Luo B, Huang F, Liu Q, Li X, Chen W, Zhou S-F, et al. Identification of apolipoprotein E Guangzhou (arginine 150 proline), a new variant associated with lipoprotein glomerulopathy. Am J Nephrol. (2008) 28:347–53. doi: 10.1159/000111828
9. Tsimihodimos V, Elisaf M. Lipoprotein glomerulopathy. Curr Opin Lipidol. (2011) 22:262–9. doi: 10.1097/MOL.0b013e328345ebb0
10. Majeed NK, McLaughlin J, Gonzalez M. Lipoprotein glomerulopathy in a hispanic female: a case report and literature review. Can J Kidney Health Dis. (2019) 6:2054358119859576. doi: 10.1177/2054358119859576
11. Saito T, Matsunaga A. Lipoprotein glomerulopathy may provide a key to unlock the puzzles of renal lipidosis. Kidney Int. (2014) 85:243–5. doi: 10.1038/ki.2013.404
12. Toyota K, Hashimoto T, Ogino D, Matsunaga A, Ito M, Masakane I, et al. A founder haplotype of APOE-Sendai mutation associated with lipoprotein glomerulopathy. J Hum Genet. (2013) 58:254–8. doi: 10.1038/jhg.2013.8
13. Song Y, Yang C, Liu L, Wang H. Case report: a pediatric case of lipoprotein glomerulopathy in china and literature review. Front Pediatr. (2021) 9:684814. doi: 10.3389/fped.2021.684814
14. Ku M, Tao C, Zhou A-A, Cheng Y, Wan Q-J. A novel apolipoprotein E mutation (p.Arg150Cys) in a Chinese patient with lipoprotein glomerulopathy. Chin Med J. (2019) 132:237–9. doi: 10.1097/CM9.0000000000000050
15. Pêgas KL, Rohde R, Garcia CD, Bittencourt V de B, Keitel E, Poloni JAT, et al. Lipoprotein glomerulopathy: a case report of a rare disease in a Brazilian child. J Bras Nefrol. (2014) 36:93–5. doi: 10.5935/0101-2800.20140015
16. Sam R, Wu H, Yue L, Mazzone T, Schwartz MM, Arruda JAL, et al. Lipoprotein glomerulopathy: a new apolipoprotein E mutation with enhanced glomerular binding. Am J Kidney Dis. (2006) 47:539–48. doi: 10.1053/j.ajkd.2005.12.031
17. Sipovskii VG, Klemina IK, Zverkov R V, Dobronravov VA, Smirnov A V. [A case of diagnosing lipoprotein glomerulopathy in Russia]. Arkh Patol. (2016) 78:52–7. doi: 10.17116/patol201678652-57
18. Pasquariello A, Pasquariello G, Innocenti M, Minnei F, Funel N, Lorusso P, et al. Lipoprotein glomerulopathy: first report of 2 not consanguineous Italian men from the same town. J Nephrol. (2011) 24:381–5. doi: 10.5301/JN.2011.7772
19. Meyrier A, Dairou F, Callard P, Mougenot B. Lipoprotein glomerulopathy: first case in a white European. Nephrol Dial Transplant. (1995) 10:546–9. doi: 10.1093/ndt/10.4.546
20. Marinaki S, Kalaitzakis E, Kolovou K, Gakiopoulou H, Stylianou K, Papasotiriou M, et al. A case of lipoprotein glomerulopathy in a Greek Caucasian male. Int Urol Nephrol. (2022) 54:969–70. doi: 10.1007/s11255-021-02930-7
21. Yang M, Weng Q, Pan X, Hussain HMJ, Yu S, Xu J, et al. Clinical and genetic analysis of lipoprotein glomerulopathy patients caused by APOE mutations. Mol Genet Genomic Med. (2020) 8:e1281. doi: 10.1002/mgg3.1281
22. Xie W, Xie Y, Lin Z, Xu X, Zhang Y. A novel apolipoprotein E mutation caused by a five amino acid deletion in a Chinese family with lipoprotein glomerulopathy: a case report. Diagn Pathol. (2019) 14:41. doi: 10.1186/s13000-019-0820-6
23. Zhang B, Liu ZH, Zeng CH, Zheng JM, Chen HP, Li LS. Clinicopathological and genetic characteristics in Chinese patients with lipoprotein glomerulopathy. J Nephrol. (2008) 21:110–17.
24. Kobayashi R, Chambers JK, Yoshida K, Nakamura T, Yasuno K, Uchida K, et al. Spontaneous lipoprotein glomerulopathy-like nephropathy in a squirrel (Sciurus vulgaris). J zoo Wildl Med. (2016) 47:663–6. doi: 10.1638/2015-0245.1
25. Huebbe P, Rimbach G. Evolution of human apolipoprotein E (APOE) isoforms: gene structure, protein function and interaction with dietary factors. Ageing Res Rev. (2017) 37:146–61. doi: 10.1016/j.arr.2017.06.002
26. Xu PT, Gilbert JR, Qiu HL, Ervin J, Rothrock-Christian TR, Hulette C, et al. Specific regional transcription of apolipoprotein E in human brain neurons. Am J Pathol. (1999) 154:601–11. doi: 10.1016/S0002-9440(10)65305-9
27. Fukunaga M, Nagahama K, Aoki M, Shimizu A, Hara S, Matsunaga A, et al. Membranous nephropathy-like apolipoprotein E deposition disease with apolipoprotein E toyonaka (Ser197Cys) and a homozygous apolipoprotein E2/2. Case Rep Nephrol Dial. (2018) 8:45–55. doi: 10.1159/000487919
28. Sasaki M, Yasuno T, Ito K, Matsunaga A, Hisano S, Abe Y, et al. Focal segmental glomerulosclerosis with heterozygous apolipoprotein E5 (Glu3Lys). CEN Case Rep. (2018) 7:225–8. doi: 10.1007/s13730-018-0331-4
29. Guttman M, Prieto JH, Croy JE, Komives EA. Decoding of lipoprotein-receptor interactions: properties of ligand binding modules governing interactions with apolipoprotein E. Biochemistry. (2010) 49:1207–16. doi: 10.1021/bi9017208
30. Cardin AD, Hirose N, Blankenship DT, Jackson RL, Harmony JA, Sparrow DA, et al. Binding of a high reactive heparin to human apolipoprotein E: identification of two heparin-binding domains. Biochem Biophys Res Commun. (1986) 134:783–9. doi: 10.1016/S0006-291X(86)80489-2
31. Tudorache IF, Trusca VG, Gafencu AV. Apolipoprotein E - a multifunctional protein with implications in various pathologies as a result of its structural features. Comput Struct Biotechnol J. (2017) 15:359–65. doi: 10.1016/j.csbj.2017.05.003
32. Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer's diseases. Neurobiol Dis. (2014) 72 (Pt. A):3–12. doi: 10.1016/j.nbd.2014.08.025
33. Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: insights into function. Trends Biochem Sci. (2006) 31:445–54. doi: 10.1016/j.tibs.2006.06.008
34. Phillips MC. Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life. (2014) 66:616–23. doi: 10.1002/iub.1314
35. Illingworth DR. Lipoprotein metabolism. Am J Kidney Dis. (1993) 22:90–7. doi: 10.1016/S0272-6386(12)70173-7
36. Mahley RW, Innerarity TL, Rall SCJ, Weisgraber KH. Plasma lipoproteins: apolipoprotein structure and function. J Lipid Res. (1984) 25:1277–94. doi: 10.1016/S0022-2275(20)34443-6
37. Zanoni P, Velagapudi S, Yalcinkaya M, Rohrer L, von Eckardstein A. Endocytosis of lipoproteins. Atherosclerosis. (2018) 275:273–95. doi: 10.1016/j.atherosclerosis.2018.06.881
38. Palmer Allred ND, Raffield LM, Hardy JC, Hsu F-C, Divers J, Xu J, et al. APOE genotypes associate with cognitive performance but not cerebral structure: diabetes heart study mind. Diabetes Care. (2016) 39:2225–31. doi: 10.2337/dc16-0843
39. Jia L, Xu H, Chen S, Wang X, Yang J, Gong M, et al. The APOE ε4 exerts differential effects on familial and other subtypes of Alzheimer's disease. Alzheimers Dement. (2020) 16:1613–23. doi: 10.1002/alz.12153
40. Yin Y, Wang Z. ApoE and neurodegenerative diseases in aging. Adv Exp Med Biol. (2018) 1086:77–92. doi: 10.1007/978-981-13-1117-8_5
41. Muñoz SS, Garner B, Ooi L. Understanding the role of ApoE fragments in Alzheimer's disease. Neurochem Res. (2019) 44:1297–305. doi: 10.1007/s11064-018-2629-1
42. Marais AD. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology. (2019) 51:165–76. doi: 10.1016/j.pathol.2018.11.002
43. Oikawa S, Matsunaga A, Saito T, Sato H, Seki T, Hoshi K, et al. Apolipoprotein E Sendai (arginine 145–>proline): a new variant associated with lipoprotein glomerulopathy. J Am Soc Nephrol. (1997) 8:820–3. doi: 10.1681/ASN.V85820
44. Karet FE, Lifton RP. Lipoprotein glomerulopathy: a new role for apolipoprotein E? J Am Soc Nephrol. (1997) 8:840–2. doi: 10.1681/ASN.V85840
45. Ishigaki Y, Oikawa S, Suzuki T, Usui S, Magoori K, Kim DH, et al. Virus-mediated transduction of apolipoprotein E (ApoE)-sendai develops lipoprotein glomerulopathy in ApoE-deficient mice. J Biol Chem. (2000) 275:31269–73. doi: 10.1074/jbc.M005906200
46. Wu H, Yang J, Liu Y-Q, Lei S, Yang M, Yang Z, et al. Lipoprotein glomerulopathy induced by ApoE Kyoto mutation in ApoE-deficient mice. J Transl Med. (2021) 19:97. doi: 10.1186/s12967-021-02765-x
47. Li Y, Chen J, Zou Y, Wang W, Li G. Lipoprotein glomerulopathy resulting from compound heterogeneous mutations of APOE gene: a case report. Medicine. (2022) 101:e28718. doi: 10.1097/MD.0000000000028718
48. Cheung CY, Chan AOK, Chan YH, Lee KC, Chan GPT, Lau GTC, et al. A rare cause of nephrotic syndrome: lipoprotein glomerulopathy. Hong Kong Med J. (2009) 15:57–60.
49. Kodera H, Mizutani Y, Sugiyama S, Miyata T, Ehara T, Matsunaga A, et al. A case of lipoprotein glomerulopathy with apoE Chicago and apoE (Glu3Lys) treated with fenofibrate. Case Rep Nephrol Dial. (2017) 7:112–20. doi: 10.1159/000478902
50. Georgiadou D, Stamatakis K, Efthimiadou EK, Kordas G, Gantz D, Chroni A, et al. Thermodynamic and structural destabilization of apoE3 by hereditary mutations associated with the development of lipoprotein glomerulopathy. J Lipid Res. (2013) 54:164–76. doi: 10.1194/jlr.M030965
51. Nilsson I, Sääf A, Whitley P, Gafvelin G, Waller C, von Heijne G. Proline-induced disruption of a transmembrane alpha-helix in its natural environment. J Mol Biol. (1998) 284:1165–75. doi: 10.1006/jmbi.1998.2217
52. Schütz AK, Rennella E, Kay LE. Exploiting conformational plasticity in the AAA+ protein VCP/p97 to modify function. Proc Natl Acad Sci U.S.A. (2017) 114:E6822–9. doi: 10.1073/pnas.1707974114
53. Katsarou M, Stratikos E, Chroni A. Thermodynamic destabilization and aggregation propensity as the mechanism behind the association of apoE3 mutants and lipoprotein glomerulopathy. J Lipid Res. (2018) 59:2339–48. doi: 10.1194/jlr.M088732
54. Hatters DM, Zhong N, Rutenber E, Weisgraber KH. Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. J Mol Biol. (2006) 361:932–44. doi: 10.1016/j.jmb.2006.06.080
55. Zhao Y, Yang Y, Xing R, Cui X, Xiao Y, Xie L, et al. Hyperlipidemia induces typical atherosclerosis development in Ldlr and Apoe deficient rats. Atherosclerosis. (2018) 271:26–35. doi: 10.1016/j.atherosclerosis.2018.02.015
56. Dybas J, Bulat K, Blat A, Mohaissen T, Wajda A, Mardyla M, et al. Age-related and atherosclerosis-related erythropathy in ApoE/LDLR(-/-) mice. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165972. doi: 10.1016/j.bbadis.2020.165972
57. Hoffmann MM, Scharnagl H, Panagiotou E, Banghard WT, Wieland H, März W. Diminished LDL receptor and high heparin binding of apolipoprotein E2 sendai associated with lipoprotein glomerulopathy. J Am Soc Nephrol. (2001) 12:524–530. doi: 10.1681/ASN.V123524
58. Matsunaga A, Sasaki J, Komatsu T, Kanatsu K, Tsuji E, Moriyama K, et al. A novel apolipoprotein E mutation, E2 (Arg25Cys), in lipoprotein glomerulopathy. Kidney Int. (1999) 56:421–7. doi: 10.1046/j.1523-1755.1999.00572.x
59. Cambruzzi E, Pêgas KL. Pathogenesis, histopathologic findings and treatment modalities of lipoprotein glomerulopathy: a review. J Bras Nefrol. (2019) 41:393–9. doi: 10.1590/2175-8239-jbn-2018-0148
60. Häussinger D, Kordes C. Space of disse: a stem cell niche in the liver. Biol Chem. (2019) 401:81–95. doi: 10.1515/hsz-2019-0283
61. Sato Y, Kinami Y, Hashiba K, Harashima H. Different kinetics for the hepatic uptake of lipid nanoparticles between the apolipoprotein E/low density lipoprotein receptor and the N-acetyl-d-galactosamine/asialoglycoprotein receptor pathway. J Control Release. (2020) 322:217–226. doi: 10.1016/j.jconrel.2020.03.006
62. Morita S. Metabolism and modification of apolipoprotein B-containing lipoproteins involved in dyslipidemia and atherosclerosis. Biol Pharm Bull. (2016) 39:1–24. doi: 10.1248/bpb.b15-00716
63. Mahley RW, Ji ZS. Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J Lipid Res. (1999) 40:1–16. doi: 10.1016/S0022-2275(20)33334-4
64. Smith RJH, Appel GB, Blom AM, Cook HT, D'Agati VD, Fakhouri F, et al. C3 glomerulopathy - understanding a rare complement-driven renal disease. Nat Rev Nephrol. (2019) 15:129–43. doi: 10.1038/s41581-018-0107-2
65. Li J-P, Kusche-Gullberg M. Heparan sulfate: biosynthesis, structure, and function. Int Rev Cell Mol Biol. (2016) 325:215–73. doi: 10.1016/bs.ircmb.2016.02.009
66. Murano T, Matsumura R, Misawa Y, Ozaki H, Miyashita Y, Yoshida S, et al. Interaction of endothelial cells and triglyceride-rich lipoproteins with apolipoprotein E (Arg25[rarr ]Cys) from a patient with lipoprotein glomerulopathy. Metabolism. (2002) 51:201–5. doi: 10.1053/meta.2002.29990
67. Andrews PA, O'Donnell PJ, Dilly SA, Snowden SA, Bewick M. Recurrence of lipoprotein glomerulopathy after renal transplantation. Nephrol Dial Transplant. (1997) 12:2442–4. doi: 10.1093/ndt/12.11.2442
68. Miyata T, Sugiyama S, Nangaku M, Suzuki D, Uragami K, Inagi R, et al. Apolipoprotein E2/E5 variants in lipoprotein glomerulopathy recurred in transplanted kidney. J Am Soc Nephrol. (1999) 10:1590–5. doi: 10.1681/ASN.V1071590
69. Kanamaru Y, Nakao A, Shirato I, Okumura K, Ogawa H, Tomino Y, et al. Chronic graft-versus-host autoimmune disease in Fc receptor gamma chain-deficient mice results in lipoprotein glomerulopathy. J Am Soc Nephrol. (2002) 13:1527–33. doi: 10.1097/01.ASN.0000015615.14428.67
70. Steinbrecher UP. Receptors for oxidized low density lipoprotein. Biochim Biophys Acta. (1999) 1436:279–98. doi: 10.1016/S0005-2760(98)00127-1
71. Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell. (1994) 76:519–29. doi: 10.1016/0092-8674(94)90115-5
72. Ito K, Nakashima H, Watanabe M, Ishimura A, Miyahara Y, Abe Y, et al. Macrophage impairment produced by Fc receptor gamma deficiency plays a principal role in the development of lipoprotein glomerulopathy in concert with apoE abnormalities. Nephrol Dial Transplant. (2012) 27:3899–907. doi: 10.1093/ndt/gfs329
73. Virella G, Muñoz JF, Galbraith GM, Gissinger C, Chassereau C, Lopes-Virella MF. Activation of human monocyte-derived macrophages by immune complexes containing low-density lipoprotein. Clin Immunol Immunopathol. (1995) 75:179–89. doi: 10.1006/clin.1995.1069
74. Stanton LW, White RT, Bryant CM, Protter AA, Endemann G. A macrophage Fc receptor for IgG is also a receptor for oxidized low density lipoprotein. J Biol Chem. (1992) 267:22446–51. doi: 10.1016/S0021-9258(18)41692-4
75. Griffith RL, Virella GT, Stevenson HC, Lopes-Virella MF. Low density lipoprotein metabolism by human macrophages activated with low density lipoprotein immune complexes. A possible mechanism of foam cell formation. J Exp Med. (1988) 168:1041–59. doi: 10.1084/jem.168.3.1041
76. Fazio S, Babaev VR, Burleigh ME, Major AS, Hasty AH, Linton MF. Physiological expression of macrophage apoE in the artery wall reduces atherosclerosis in severely hyperlipidemic mice. J Lipid Res. (2002) 43:1602–9. doi: 10.1194/jlr.M200108-JLR200
77. Dove DE, Linton MF, Fazio S. ApoE-mediated cholesterol efflux from macrophages: separation of autocrine and paracrine effects. Am J Physiol Cell Physiol. (2005) 288:C586–92. doi: 10.1152/ajpcell.00210.2004
78. Tavori H, Fan D, Giunzioni I, Zhu L, Linton MF, Fogo AB, et al. Macrophage-derived apoESendai suppresses atherosclerosis while causing lipoprotein glomerulopathy in hyperlipidemic mice. J Lipid Res. (2014) 55:2073–81. doi: 10.1194/jlr.M049874
79. Prévost M, Raussens V. Apolipoprotein E-low density lipoprotein receptor binding: study of protein-protein interaction in rationally selected docked complexes. Proteins. (2004) 55:874–84. doi: 10.1002/prot.20080
80. de Vries APJ, Ruggenenti P, Ruan XZ, Praga M, Cruzado JM, Bajema IM, et al. Fatty kidney: emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. (2014) 2:417–26. doi: 10.1016/S2213-8587(14)70065-8
81. Gai Z, Wang T, Visentin M, Kullak-Ublick GA, Fu X, Wang Z. Lipid accumulation and chronic kidney disease. Nutrients. (2019) 11:722. doi: 10.3390/nu11040722
82. Leonarduzzi G, Scavazza A, Biasi F, Chiarpotto E, Camandola S, Vogel S, et al. The lipid peroxidation end product 4-hydroxy-2,3-nonenal up-regulates transforming growth factor beta1 expression in the macrophage lineage: a link between oxidative injury and fibrosclerosis. FASEB J. (1997) 11:851–7. doi: 10.1096/fasebj.11.11.9285483
83. Akhand AA, Kato M, Suzuki H, Liu W, Du J, Hamaguchi M, et al. Carbonyl compounds cross-link cellular proteins and activate protein-tyrosine kinase p60c-Src. J Cell Biochem. (1999) 72:1–7. doi: 10.1002/(SICI)1097-4644(19990101)72:1<1::AID-JCB1>3.0.CO;2-Y
84. Miyata T, van Ypersele de Strihou C, Kurokawa K, Baynes JW. Alterations in nonenzymatic biochemistry in uremia: origin and significance of “carbonyl stress” in long-term uremic complications. Kidney Int. (1999) 55:389–99. doi: 10.1046/j.1523-1755.1999.00302.x
85. Pham T, Kodvawala A, Hui DY. The receptor binding domain of apolipoprotein E is responsible for its antioxidant activity. Biochemistry. (2005) 44:7577–82. doi: 10.1021/bi0472696
86. Amenomori M, Haneda M, Morikawa J, Nishigaki I, Maeda S, Hidaka H, et al. A case of lipoprotein glomerulopathy successfully treated with probucol. Nephron. (1994) 67:109–13. doi: 10.1159/000187897
87. Chen S, Liu Z-H, Zheng J-M, Zhang X, Li L-S. A complete genomic analysis of the apolipoprotein E gene in Chinese patients with lipoprotein glomerulopathy. J Nephrol. (2007) 20:568–75.
88. Liao J, Bai J, An X, Liu Y, Wang Y, Liu G, et al. Lipoprotein glomerulopathy-like lesions in atherosclerotic mice defected with HDL receptor SR-B1. Front Cardiovasc Med. (2021) 8:734824. doi: 10.3389/fcvm.2021.734824
89. Li W, Wang Y, Han Z, Luo C, Zhang C, Xiong J. Apolipoprotein e mutation and double filtration plasmapheresis therapy on a new Chinese patient with lipoprotein glomerulopathy. Kidney Blood Press Res. (2014) 39:330–339. doi: 10.1159/000355810
90. Russi G, Furci L, Leonelli M, Magistroni R, Romano N, Rivasi P, et al. Lipoprotein glomerulopathy treated with LDL-apheresis (heparin-induced extracorporeal lipoprotein precipitation system): a case report. J Med Case Rep. (2009) 3:1–6. doi: 10.1186/1752-1947-3-9311
91. Takasaki S, Maeda K, Joh K, Yamakage S, Fukase S, Takahashi T, et al. Macrophage infiltration into the glomeruli in lipoprotein glomerulopathy. Case Rep Nephrol Dial. (2015) 5:204–12. doi: 10.1159/000441715
92. Tokura T, Itano S, Kobayashi S, Kuwabara A, Fujimoto S, Horike H, et al. A novel mutation ApoE2 Kurashiki (R158P) in a patient with lipoprotein glomerulopathy. J Atheroscler Thromb. (2011) 18:536–41. doi: 10.5551/jat.8102
93. Arai T, Yamashita S, Yamane M, Manabe N, Matsuzaki T, Kiriyama K, et al. Disappearance of intraglomerular lipoprotein thrombi and marked improvement of nephrotic syndrome by bezafibrate treatment in a patient with lipoprotein glomerulopathy. Atherosclerosis. (2003) 169:293–9. doi: 10.1016/S0021-9150(03)00194-1
94. Wu H, Yang Y, Hu Z. The novel apolipoprotein e mutation ApoE chengdu (c.518T>C, p.L173P) in a Chinese patient with lipoprotein glomerulopathy. J Atheroscler Thromb. (2018) 25:733–40. doi: 10.5551/jat.41996
95. Wu Y, Chen X, Yang Y, Wang B, Liu X, Tao Y, et al. A case of lipoprotein glomerulopathy with thrombotic microangiopathy due to malignant hypertension. BMC Nephrol. (2013) 14:53. doi: 10.1186/1471-2369-14-53
96. Chen H, Liu Z, Gong R, Tang Z, Zeng C, Zhu M, et al. Lipoprotein glomerulopathy: clinical features and pathological characteristics in Chinese. Chin Med J. (2004) 117:1513–7. doi: 10.3760/cma.j.issn.0366-6999.2004.10.114
97. Xin Z, Zhihong L, Shijun L, Jinfeng Z, Huiping C, Caihong Z, et al. Successful treatment of patients with lipoprotein glomerulopathy by protein A immunoadsorption: a pilot study. Nephrol Dial Transplant. (2009) 24:864–9. doi: 10.1093/ndt/gfn555
98. Zhang B, Liu Z, Zeng C, Zheng J, Chen H, Zhou H, et al. Plasma level and genetic variation of apolipoprotein E in patients with lipoprotein glomerulopathy. Chin Med J. (2005) 118:555–60. doi: 10.5555/cmj.0366-6999.118.07.p555.01
99. Takasaki S, Matsunaga A, Joh K, Saito T. A case of lipoprotein glomerulopathy with a rare apolipoprotein E isoform combined with neurofibromatosis type I. CEN Case Rep. (2018) 7:127–31. doi: 10.1007/s13730-018-0309-2
100. Zou GM, Zhuo L, Tan M, Li WG. [Clinicopathologic features of lipoprotein glomerulopathy: observation of 6 cases]. Chin Med J. (2018) 98:2910–3. doi: 10.3760/cma.j.issn.0376-2491.2018.36.007
101. Blum CB. Type III hyperlipoproteinemia: still worth considering? Prog Cardiovasc Dis. (2016) 59:119–24. doi: 10.1016/j.pcad.2016.07.007
102. Sniderman AD, de Graaf J, Thanassoulis G, Tremblay AJ, Martin SS, Couture P. The spectrum of type III hyperlipoproteinemia. J Clin Lipidol. (2018) 12:1383–9. doi: 10.1016/j.jacl.2018.09.006
103. Nordestgaard BG. Triglyceride-Rich lipoproteins and atherosclerotic cardiovascular disease: new insights from epidemiology, genetics, and biology. Circ Res. (2016) 118:547–63. doi: 10.1161/CIRCRESAHA.115.306249
104. Boot CS, Luvai A, Neely RDG. The clinical and laboratory investigation of dysbetalipoproteinemia. Crit Rev Clin Lab Sci. (2020) 57:458–69. doi: 10.1080/10408363.2020.1745142
105. Koitabashi Y, Ikoma M, Miyahira T, Fujita R, Mio H, Ishida M, et al. Long-term follow-up of a paediatric case of lipoprotein glomerulopathy. Pediatr Nephrol. (1990) 4:122–8. doi: 10.1007/BF00858822
106. da Silveira-Neto JN, de Oliveira Ahn GJ, de Menezes Neves PDM, Baptista VAF, de Almeida Araújo S, Wanderley DC, et al. Lipoprotein glomerulopathy associated with the Osaka/Kurashiki APOE variant: two cases identified in Latin America. Diagn Pathol. (2021) 16:65. doi: 10.1186/s13000-021-01119-x
107. Kollbrunner L, Hirt-Minkowski P, Sanz J, Bresin E, Neuhaus TJ, Hopfer H, et al. Case report: lipoprotein glomerulopathy complicated by atypical hemolytic uremic syndrome. Front Med. (2021) 8:679048. doi: 10.3389/fmed.2021.679048
108. Chang C-F, Lin C-C, Chen J-Y, Yang A-H, Shiao M-S, Kao J-T, et al. Lipoprotein glomerulopathy associated with psoriasis vulgaris: report of 2 cases with apolipoprotein E3/3. Am J kidney Dis. (2003) 42:E18–23. doi: 10.1016/S0272-6386(03)00798-4
109. Ando M, Sasaki J, Hua H, Matsunaga A, Uchida K, Jou K, et al. A novel 18-amino acid deletion in apolipoprotein E associated with lipoprotein glomerulopathy. Kidney Int. (1999) 56:1317–23. doi: 10.1046/j.1523-1755.1999.00677.x
110. Morris CS, Bois MC, Aust CH, Thomas R, Sethi S, Maleszewski JJ. Intravascular cardiac lipoproteinosis: extrarenal manifestation of lipoprotein glomerulopathy. Cardiovasc Pathol. (2019) 42:6–9. doi: 10.1016/j.carpath.2019.04.006
111. Lui DTW, Lee ACH, Yap DYH, Chan GSW, Tan KCB. A young Chinese man with nephrotic syndrome due to lipoprotein glomerulopathy. J Clin Lipidol. (2019) 13:251–3. doi: 10.1016/j.jacl.2018.12.004
112. Bomback AS, Song H, D'Agati VD, Cohen SD, Neal A, Appel GB, et al. A new apolipoprotein E mutation, apoE Las Vegas, in a European-American with lipoprotein glomerulopathy. Nephrol Dial Transplant. (2010) 25:3442–6. doi: 10.1093/ndt/gfq389
113. Zhang P, Matalon R, Kaplan L, Kumar A, Gallo G. Lipoprotein glomerulopathy: first report in a Chinese male. Am J Kidney Dis. (1994) 24:942–50. doi: 10.1016/S0272-6386(12)81066-3
114. Boumendjel R, Papari M, Gonzalez M. A rare case of lipoprotein glomerulopathy in a white man: an emerging entity in Asia, rare in the white population. Arch Pathol Lab Med. (2010) 134:279–82. doi: 10.5858/134.2.279
115. Kinomura M, Sugiyama H, Saito T, Matsunaga A, Sada K, Kanzaki M, et al. A novel variant apolipoprotein E Okayama in a patient with lipoprotein glomerulopathy. Nephrol Dial Transplant. (2008) 23:751–6. doi: 10.1093/ndt/gfm675
116. Usui R, Takahashi M, Nitta K, Koike M. Five-year follow-up of a case of lipoprotein glomerulopathy with APOE Kyoto mutation. CEN Case Rep. (2016) 5:148–53. doi: 10.1007/s13730-016-0214-5
117. Shimizu M, Ohno T, Kimoto H, Hosono S, Nozawa M. A newborn infant with lipoprotein glomerulopathy associated with congenital nephrotic syndrome. Pediatr Int. (2001) 43:78–80. doi: 10.1046/j.1442-200x.2001.01344.x
118. Konishi K, Saruta T, Kuramochi S, Oikawa S, Saito T, Han H, et al. Association of a novel 3-amino acid deletion mutation of apolipoprotein E (Apo E Tokyo) with lipoprotein glomerulopathy. Nephron. (1999) 83:214–8. doi: 10.1159/000045513
119. Han J, Pan Y, Chen Y, Li X, Xing G, Shi J, et al. Common apolipoprotein e gene mutations contribute to lipoprotein glomerulopathy in China. Nephron Clin Pract. (2010) 114:260–7. doi: 10.1159/000276578
120. Yamashita S, Masuda D, Matsuzawa Y. Did we abandon probucol too soon? Curr Opin Lipidol. (2015) 26:304–16. doi: 10.1097/MOL.0000000000000199
121. Ieiri N, Hotta O, Taguma Y. Resolution of typical lipoprotein glomerulopathy by intensive lipid-lowering therapy. Am J Kidney Dis. (2003) 41:244–9. doi: 10.1053/ajkd.2003.50016
122. Karube M, Nakabayashi K, Fujioka Y, Yoshihara K, Yamada A, Matsunaga A, et al. Lipoprotein glomerulopathy-like disease in a patient with type III hyperlipoproteinemia due to apolipoprotein E2 (Arg158 Cys)/3 heterozygosity. Clin Exp Nephrol. (2007) 11:174–9. doi: 10.1007/s10157-007-0469-4
123. Owada A, Suda S, Hata T. Antiproteinuric effect of niceritrol, a nicotinic acid derivative, in chronic renal disease with hyperlipidemia: a randomized trial. Am J Med. (2003) 114:347–53. doi: 10.1016/S0002-9343(02)01567-X
124. Yamauchi K, Tanahashi Y, Okada M, Tsuzuki J, Sato A, Abe K, et al. Long-term effects of niceritrol on serum lipoprotein(a) and lipids in patients with high levels of lipoprotein(a). Clin Ther. (1995) 17:52–9. doi: 10.1016/0149-2918(95)80006-9
125. Hamatani H, Hiromura K, Kobatake K, Yoshida H, Kobayashi S, Yoneda N, et al. Successful treatment of lipoprotein glomerulopathy in a daughter and a mother using niceritrol. Clin Exp Nephrol. (2010) 14:619–24. doi: 10.1007/s10157-010-0333-9
126. Sirtori CR. The pharmacology of statins. Pharmacol Res. (2014) 88:3–11. doi: 10.1016/j.phrs.2014.03.002
127. Liao M-T, Tsai I-J, Cheng H-T, Lin W-C, Chang Y-W, Lin Y-H, et al. A rare cause of childhood-onset nephrotic syndrome: lipoprotein glomerulopathy. Clin Nephrol. (2012) 78:237–40. doi: 10.5414/CN106876
128. Matsunaga A, Furuyama M, Hashimoto T, Toyoda K, Ogino D, Hayasaka K. Improvement of nephrotic syndrome by intensive lipid-lowering therapy in a patient with lipoprotein glomerulopathy. Clin Exp Nephrol. (2009) 13:659–62. doi: 10.1007/s10157-009-0207-1
129. Boeckhaus J, Gross O. Sodium-Glucose cotransporter-2 inhibitors in patients with hereditary podocytopathies, alport syndrome, and FSGS: a case series to better plan a large-scale study. Cells. (2021) 10:1815. doi: 10.3390/cells10071815
130. Susca M. Heparin-Induced extracorporeal low-density lipoprotein precipitation futura, a new modification of HELP apheresis: technique and first clinical results. Ther Apher. (2001) 5:387–93. doi: 10.1046/j.1526-0968.2001.00371.x
131. Waldmann E, Parhofer KG. Apheresis for severe hypercholesterolaemia and elevated lipoprotein(a). Pathology. (2019) 51:227–32. doi: 10.1016/j.pathol.2018.10.016
132. Esnault VL, Besnier D, Testa A, Coville P, Simon P, Subra JF, et al. Effect of protein A immunoadsorption in nephrotic syndrome of various etiologies. J Am Soc Nephrol. (1999) 10:2014–7. doi: 10.1681/ASN.V1092014
133. Tuteja S. Activation of HCAR2 by niacin: benefits beyond lipid lowering. Pharmacogenomics. (2019) 20:1143–50. doi: 10.2217/pgs-2019-0092
134. Thompson G, Parhofer KG. Current role of lipoprotein apheresis. Curr Atheroscler Rep. (2019) 21:26. doi: 10.1007/s11883-019-0787-5
135. Khetarpal SA, Zeng X, Millar JS, Vitali C, Somasundara AVH, Zanoni P, et al. A human APOC3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat Med. (2017) 23:1086–94. doi: 10.1038/nm.4390
136. Ahmad KA, Yuan Yuan D, Nawaz W, Ze H, Zhuo CX, Talal B, et al. Antioxidant therapy for management of oxidative stress induced hypertension. Free Radic Res. (2017) 51:428–38. doi: 10.1080/10715762.2017.1322205
137. Huang G, Mei X, Hu J. The antioxidant activities of natural polysaccharides. Curr Drug Targets. (2017) 18:1296–300. doi: 10.2174/1389450118666170123145357
138. Manzini S, Busnelli M, Parolini C, Minoli L, Ossoli A, Brambilla E, et al. Topiramate protects apoE-deficient mice from kidney damage without affecting plasma lipids. Pharmacol Res. (2019) 141:189–200. doi: 10.1016/j.phrs.2018.12.022
139. Hirashima H, Komiya T, Toriu N, Hara S, Matsunaga A, Saito T, et al. A case of nephrotic syndrome showing contemporary presence of apolipoprotein E2 homozygote glomerulopathy and membranous nephropathy-like findings modified by apolipoprotein E Toyonaka. Clin Nephrol Case Stud. (2018) 6:45–51. doi: 10.5414/CNCS109509
140. Kato T, Ushiogi Y, Yokoyama H, Hara S, Matsunaga A, Muso E, et al. A case of apolipoprotein E toyonaka and homozygous apolipoprotein E2/2 showing non-immune membranous nephropathy-like glomerular lesions with foamy changes. CEN Case Rep. (2019) 8:106–11. doi: 10.1007/s13730-019-00380-w
Keywords: lipoprotein glomerulopathy, apolipoprotein E, epidemiology, pathogenesis, meta-analysis, treatment
Citation: Li M-s, Li Y, Liu Y, Zhou X-j and Zhang H (2022) An Updated Review and Meta Analysis of Lipoprotein Glomerulopathy. Front. Med. 9:905007. doi: 10.3389/fmed.2022.905007
Received: 26 March 2022; Accepted: 15 April 2022;
Published: 06 May 2022.
Edited by:
Jia Rao, Fudan University, ChinaReviewed by:
Gengru Jiang, Shanghai Jiao Tong University, ChinaCopyright © 2022 Li, Li, Liu, Zhou and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xu-jie Zhou, emhvdXh1amllQGJqbXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.