 Marcella Nunziato1,2†
Marcella Nunziato1,2† Federica Di Maggio1,2†
Federica Di Maggio1,2† Matilde Pensabene3
Matilde Pensabene3 Maria Valeria Esposito1,2Flavio Starnone1,2Carmine De Angelis3,4Alessandra Calabrese5Massimiliano D’Aiuto6,7
Maria Valeria Esposito1,2Flavio Starnone1,2Carmine De Angelis3,4Alessandra Calabrese5Massimiliano D’Aiuto6,7 Gerardo Botti8Sabino De Placido3,4
Gerardo Botti8Sabino De Placido3,4 Valeria D’Argenio1,9*
Valeria D’Argenio1,9* Francesco Salvatore1,2*
Francesco Salvatore1,2*- 1CEINGE–Biotecnologie Avanzate, Naples, Italy
- 2Department of Molecular Medicine and Medical Biotechnologies, University of Naples Federico II, Naples, Italy
- 3Department of Clinical Medicine and Surgery, University of Naples Federico II, Naples, Italy
- 4Department of Oncology and Hematology, Regional Reference Center for Rare Tumors, Azienda Ospedaliera Universitaria (AOU) Federico II of Naples, Naples, Italy
- 5Division of Breast Surgery, Department of Breast Disease, National Cancer Institute, Istituti di Ricovero e Cura a Carattere Scientifico (IRCCS) “Fondazione G. Pascale,” Naples, Italy
- 6Clinica Villa Fiorita, Aversa, Italy
- 7Division of Breast Oncology, National Cancer Institute, Istituti di Ricovero e Cura a Carattere Scientifico (IRCCS) “Fondazione G. Pascale,” Naples, Italy
- 8Scientific Directorate, National Cancer Institute, Istituti di Ricovero e Cura a Carattere Scientifico (IRCCS) “Fondazione G. Pascale,” Naples, Italy
- 9Department of Human Sciences and Quality of Life Promotion, San Raffaele Open University, Rome, Italy
Breast cancer is the most common neoplasia in females worldwide, about 10% being hereditary/familial and due to DNA variants in cancer-predisposing genes, such as the highly penetrant BRCA1/BRCA2 genes. However, their variants explain up to 25% of the suspected hereditary/familial cases. The availability of NGS methodologies has prompted research in this field. With the aim to improve the diagnostic sensitivity of molecular testing, a custom designed panel of 44 genes, including also non-coding regions and 5’ and 3’ UTR regions, was set up. Here, are reported the results obtained in a cohort of 64 patients, including also few males, from Southern Italy. All patients had a positive personal and/or familial history for breast and other cancers, but tested negative to routine BRCA analysis. After obtaining their written informed consent, a genomic DNA sample/patient was used to obtain an enriched DNA library, then analyzed by NGS. Sequencing data analysis allowed the identification of pathogenic variants in 12 of tested patients (19%). Interestingly, MUTYH was the most frequently altered gene, followed by RNASEL, ATM, MSH6, MRE11A, and PALB2 genes. The reported resultsreinforce the need for enlarged molecular testing beyond BRCA genes, at least in patients with a personal and familial history, strongly suggestive for a hereditary/familial form. This gives also a hint to pursue more specific precision oncology therapy.
Introduction
In 2020, breast cancer (BC) was the most common cancer in females worldwide accounting for 47.8% of total cancers and a total of 2,261,419 new cases diagnosed in the same year (24.5%). A similar trend has been observed also in Italy, where 55,133 (13.3%) new cases of BC have been registered just in 20201 (last access on January 2022) (1). Fortunately, mortality has fallen sharply over a decade due to both an even more capillary diffusion of screening programs, allowing for early diagnosis, and the availability of more efficient therapeutic strategies. Male BC is rare but very aggressive, and accounts for less than 1% of all BC cases (2).
Even if the vast majority of BCs are sporadic, about 5–10% of them are considered as hereditary and due to germline predisposing variants in cancer-related genes. The highly penetrant genes associated to hereditary BC are the well-known BRCA1 and BRCA2 (3, 4); however, it has been shown that pathogenetic variants in these 2 genes account up to 25% of all the suspected hereditary cases, thus suggesting that other genes have to be involved in this process (5).
In the last decades, the evolution of DNA sequencing technologies, through the advent and subsequent massive diffusion of next generation sequencing (NGS)-based approaches, has enhanced the study of the molecular bases of human diseases allowing clarifying the contribution of single genes to specific diseases onset (6). In particular, the parallel testing of multiple genes, through the use of multi-gene panels, has let the simultaneous and rapid analysis of cancer predisposing genes (high-, moderate- and low-penetrance genes just known in the literature, as well as new predisposing ones) in the attempt to depict a more precise picture of the molecular basis of familial BCs and correctly identify all the at-risk subjects within the affected families (7–10).
Based on these studies, the National Comprehensive Cancer Network (NCCN) guidelines for hereditary BC introduced in 2019 the possibility to additionally test 18 different genes, beyond BRCA1 and BRCA2. In particular, each of these genes has been classified as showing a very strong, strong, or just limited evidence for an increased risk of breast, ovarian, pancreatic and other cancers (11).
Moreover, recent studies have reported that a total of about 11% of all pathogenic germline variants in Caucasian patients and about 9% in Asians (affected patients) affect genes different from the traditional BRCA1/2 (12). In particular, new evidences are emerging regarding the contribution of other genes involved in DNA damage repair mechanisms, like PALB2, CHEK2, ATM, BRIP1, and others (11, 12).
It is important to underline that the early identification of germline, cancer-predisposing DNA variants plays an important role, especially in breast and ovarian cancers, both for the management of the affected patients (to drive the most proper surgical and also pharmacological approaches), as well as for the implementation of prevention programs for the at-risk family members. Thus, an enlarged molecular test may be advisable in routine diagnostic settings to increase the mutations’ carriers’ identification rate (13, 14).
In this context, a custom multi-gene panel, including 44 genes already known in the recent literature as related prevalently to breast, ovarian, colon and prostate cancers predisposition, was set up. With the aim to improve the diagnostic sensitivity of hereditary BCs molecular screening, a cohort of 64 BRCA1/2-negative, breast and ovarian cancer patients were analyzed through the above-mentioned multi-genes, custom panel enrichment method followed by NGS. Pathogenic variants in not routinely tested genes were identified. The results reported herein highlight once again the need for more enlarged molecular testing to improve the management of BC patients and of their families.
Materials and methods
Patients’ enrollment
A total of sixty-four individuals, including 57 cancer patients (54 women and 3 men), and seven unaffected women with a positive family history for breast and/or ovarian cancer, were totally enrolled in the present study among those attending to CEINGE Biotecnologie Avanzate laboratories (Napoli, Italy) to carry out the germline BRCA1/2 molecular screening from 2014 to 2018. Before the diagnostic procedure, a genetic counseling was offered to the patients in the reference clinical centers, the senology units of both “Istituto Nazionale dei Tumori–Fondazione G. Pascale” and of the Federico II University (Napoli, Italy), to assess their personal and familial cancer history and verify the presence of the criteria to be admitted to molecular testing, according to national and international guidelines (Italian Association of Medical Oncology, AIOM, NCCN). At this time, clinical data, including personal and family history, and the pedigree of each patient were collected.
The patients selected for the present study were all negative for the detection of single nucleotide substitutions and small insertion/deletions (INDELs) in BRCA1 and BRCA2 genes. In the course of the study, the BRCA1/2 molecular testing was integrated with the analysis also of the CNVs (Copy Number Variants) through the MLPA (Multiplex ligation Probe amplification) method, that was available for almost all patients, 54/64 selected for the present study. During the 5 years of observation time, a total of 655 subjects, 58 being carriers of a BRCA1/2 pathogenic variant, was analyzed. Among the remaining 597 tested negative, the patients with at least one of the following inclusion criteria were selected: (i) early onset BC (under-forty years); (ii) BC at any age but with at least one BC case in a first degree relative, or multiple BCs not in first degree relatives, or different cancer types within the family; (iii) ovarian cancer; (iv) male BC; (v) pancreatic cancer; (vi) unaffected individuals with multiple BCs or different cancer types within the family (Figure 1). All patients come from the Campania region in Southern Italy. Demographic features of the obtained study groups are summarized in Table 1, while the characteristics of each study participant are detailed in the Supplementary Table 1. Unfortunately, in this cohort of patients in our area, we have not had the possibility to analyze the relatives for being mutation carriers because of reluctance to undergo the test, since now.
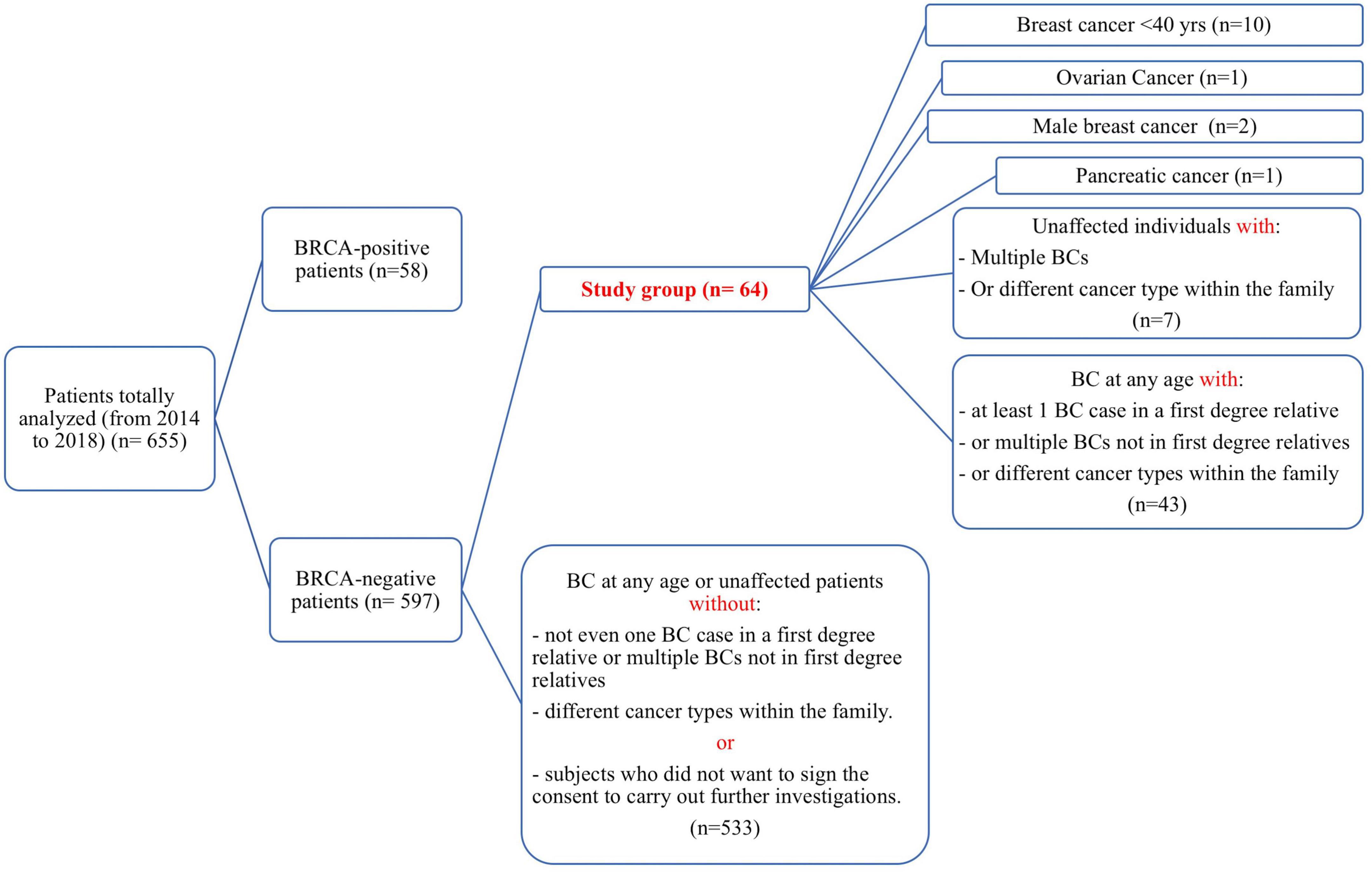
Figure 1. Strobe diagram representing the study design and the inclusion criteria over an observation time of 5 years. For each category, the corresponding number of subjects is reported in parentheses.
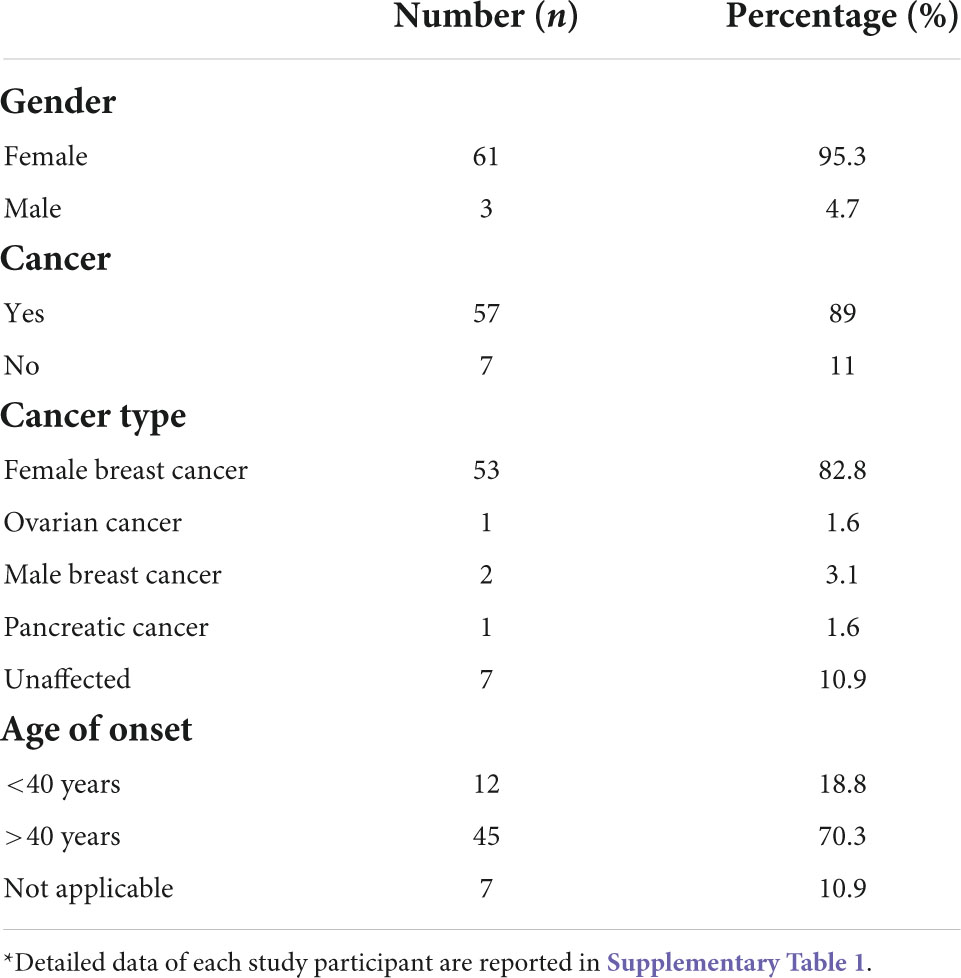
Table 1. Demographic summary of the study group.*
All patients gave their written informed consent to the study that was carried out according to the tenets of the Helsinki Declaration and approved by the Istituto Nazionale Tumori–Fondazione G. Pascale Ethics Committee (protocol number 3 of 03/25/2009).
Genomic DNA (gDNA) was extracted from peripheral blood samples using the Maxwell 16 Blood LEV DNA Purification Kit (#AS1290–Promega Corporation, WI, United States), according to manufacturer’s instructions. DNA quantity was evaluated using the QuBit 3.0 fluorimeter (#Q32850–Thermo Fisher Scientific, MA, United States), while DNA quality was assessed by 0.8% agarose gel electrophoresis.
Multi-gene panel custom design
A custom panel was chosen as methodological approach and designed with the aim to possibly highlight novel candidate genes not yet associated to hereditary BC risk. Thus, genes not included in the currently commercially available kits and for which no guidelines regarding the clinical management of the carriers have been released, were selected and analyzed in the present study. For the same reason, during the patients’ selection step the co-occurrence of different cancers within the families was carefully evaluated.
Based on these criteria, the genes to be included in the custom NGS panel were carefully selected after a deep study of the literature (9, 15, 16). Next, the custom design was realized using the web-based application HaloPlex SureDesign, we previously used for the study of other genetically heterogeneous diseases (17). The resulting custom multi-genes panel accounts for a total of 44 target genes, being equivalent to 804 target regions and 16,978 probes for a total target size of 416,043 kbp. The panel includes all the coding exons of each gene and 50 bp at exon boundaries on each side (5’ and 3’). Specifically, it comprehends genes chosen after a careful study of recent literature, in detail: 26 genes are mainly related to breast/ovarian cancers; 14 genes are related to colon cancer and four genes are related to prostate cancer (all these neoplasia are present and often mixed in the same family in the Campania population). The list of the genes included in the final panel is provided in Table 2.
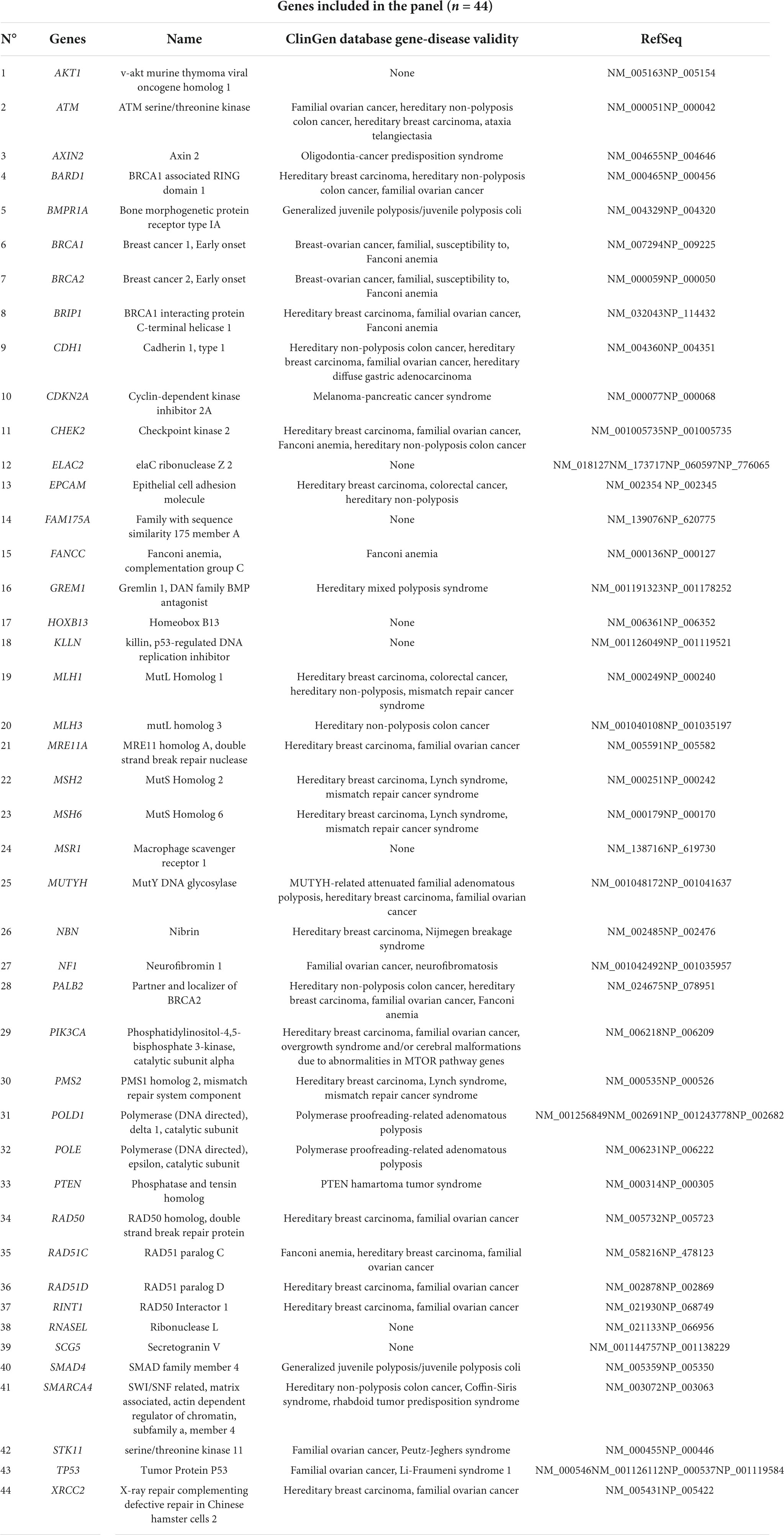
Table 2. List of the genes included in the custom panel.
Libraries preparation and sequencing
DNA libraries were obtained using the HaloPlex Target Enrichment System (#G9911C Agilent Technologies, CA, United States), following manufacturer’s instructions. The first step of the used capture methodology is a genomic DNA digestion, carried out using 16 different restriction enzymes provided by the company. Next, after the hybridization step, thousands of different targets are amplified in the same reaction using the custom specific probes, and each sample is uniquely indexed, allowing for pooling and sequencing several samples all together. More in detail, firstly, the quantity of each gDNA was assessed using the QuBit dsDNA BR Assay kit, and 225 ng of each gDNA were fragmented using eight different restriction reactions incubated 30 min at 37°C. The eight digestion reactions were, then, combined into a single hybridization mix tube (one tube/patient) that contains target-specific probes and the unique sample index.
The hybridization reaction was performed incubating the samples at 95°C for 10 min, and then at 54°C for 16 h. Next, the hybridized DNA fragments, containing biotin, were captured using streptavidin beads and a DNA ligase was added to the capture reaction to close the gaps in the circularized target DNA-probe hybrids. Finally, a PCR mix for the captured target DNA amplification step was prepared, followed by the elution with NaOH and a subsequent magnetic beads purification, using AMPure XP beads (#A63880–Beckman Coulter, Fullerton, CA, United States). The profile of each enriched library was assessed using the TapeStation 2200 using the High Sensitivity D1000 assay (#5067-5584/85), and the library quantity was measured using QuBit 3.0. Libraries were first diluted each at 10 nM and then pooled together to ensure equimolarity between samples. The final concentration of the libraries pool was 4 nM in a single tube ready to be sequenced.
Eight different sequencing runs were performed using the Illumina MiSeq platform (#MS102-2002–PE 150 × 2, San Diego, CA, United States). Eight pM of the denatured final libraries pool were combined to 25% of 8 pM PhiX, and were loaded into the MiSeq reagent cartridge, according to manufacturer’s instructions.
Bioinformatic analysis and Sanger confirmation
The sequencing platform used in this study produces raw data in FASTQ files format ready for download. FASTQ files are univocally assigned to each patient thanks to the index sequences added to gDNAs during the libraries’ preparation procedure. Data analysis was performed using the Agilent’s SureCall v4.2.0, a bioinformatic free tool that align and call all the variants found. All potentially clinical-relevant variants were confirmed using Sanger Sequencing using the same sample where NGS was performed. The method described herein is able to detect single nucleotide variants and small INDELs. This statement is based on: (i) the comparison of the variants found in BRCA1/2 by a CE_IVD BRCA1/2 test (10) and the proposed method, which showed a 100% concordance; and (ii) the confirmation by Sanger sequencing of all the potentially clinically-relevant variants identified in this study, which confirmed all of them. Since the largest INDELs identified in this study group and confirmed at Sanger comprises five nucleotides, the data presented in this paper allow to assess this as maximum number, even if we cannot exclude that it may correctly detect larger INDELs (18). Indeed, previous studies comparing different algorithms to assess INDELS-calling from NGS data report a good positive predictive value for INDELs less or equal to 10 nucleotides.
For all the variants identified, the allele frequency was verified using the gnomAD database (v.2.1.1, accessed on May 2022), the frequencies are reported in Tables 3, 4. Furthermore, different prediction tools were set up to assess pathogenicity of the variants found: namely, ClinVar (accessed on January 2022), VarSome v11.1 of December 21st 2021 (19), Alamut® software suite, Mutation Taster, ACMG (American College of Medical Genetics)/AMP (Association for Molecular Pathology) criteria and, where applicable, Human Splicing Finder v.3.1. The Deleterious Annotation of genetic variants using Neural Networks (DANN) and the Combined Annotation Dependent Depletion (CADD) scores were assessed for each variant found (Tables 3, 4); finally, BayesDel and Rare Exome Variant Ensemble Learner (REVEL) tools were used to evaluate all the variants of uncertain significance (VUSs) found (Table 4).
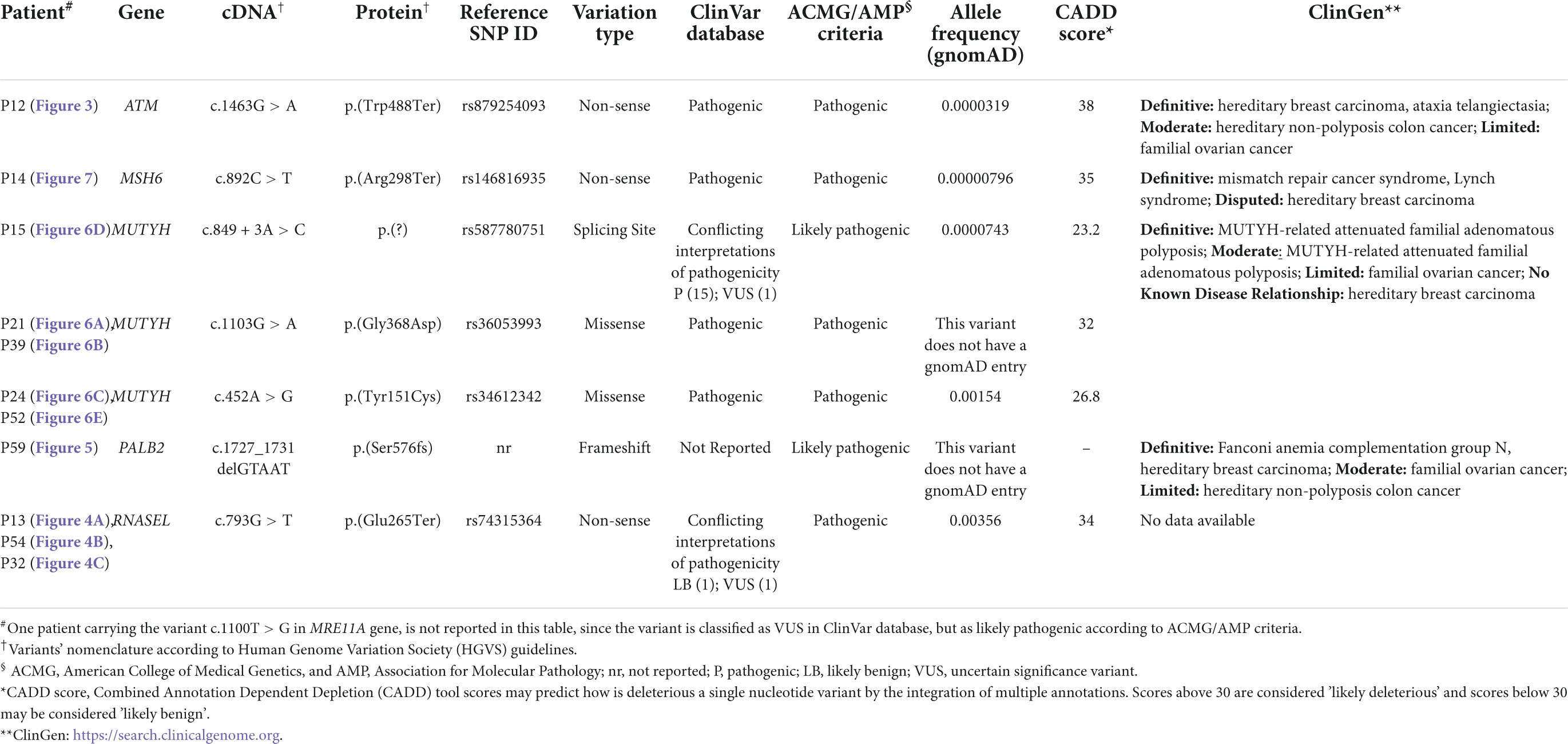
Table 3. Pathogenic variants found in genes different from the BRCAs.
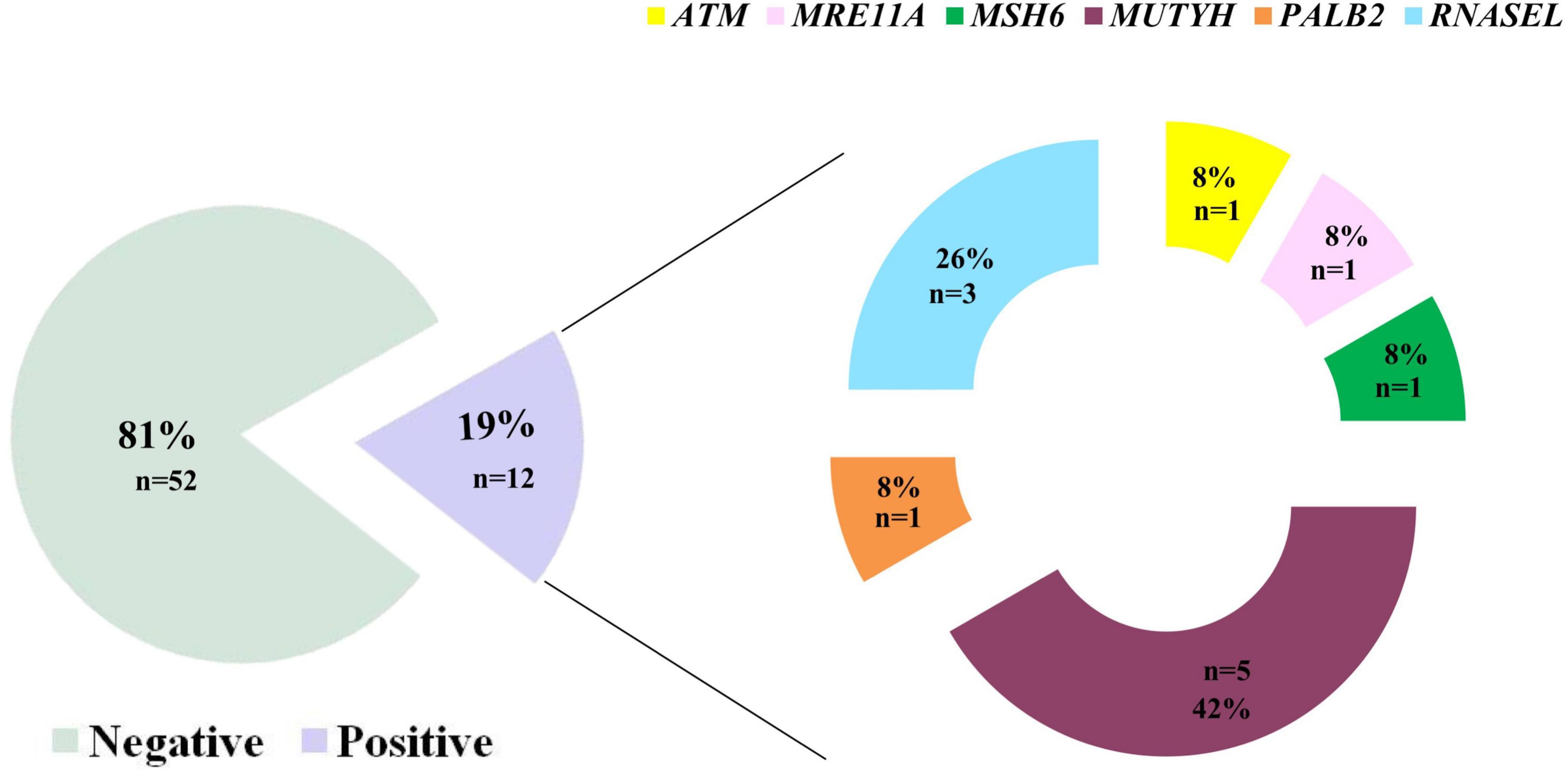
Figure 2. Results of the multi-gene panel testing based on NGS-approach in this study group (N = 63 totally analyzed patients). Eighty-three% of patients were found not to be carriers of pathogenic variants, while 17% was positive for the presence of a clinically interesting mutation. Interestingly, we highlight that MUTYH carried most of the variants identified herein (46%) followed by the RNASEL gene (27%).
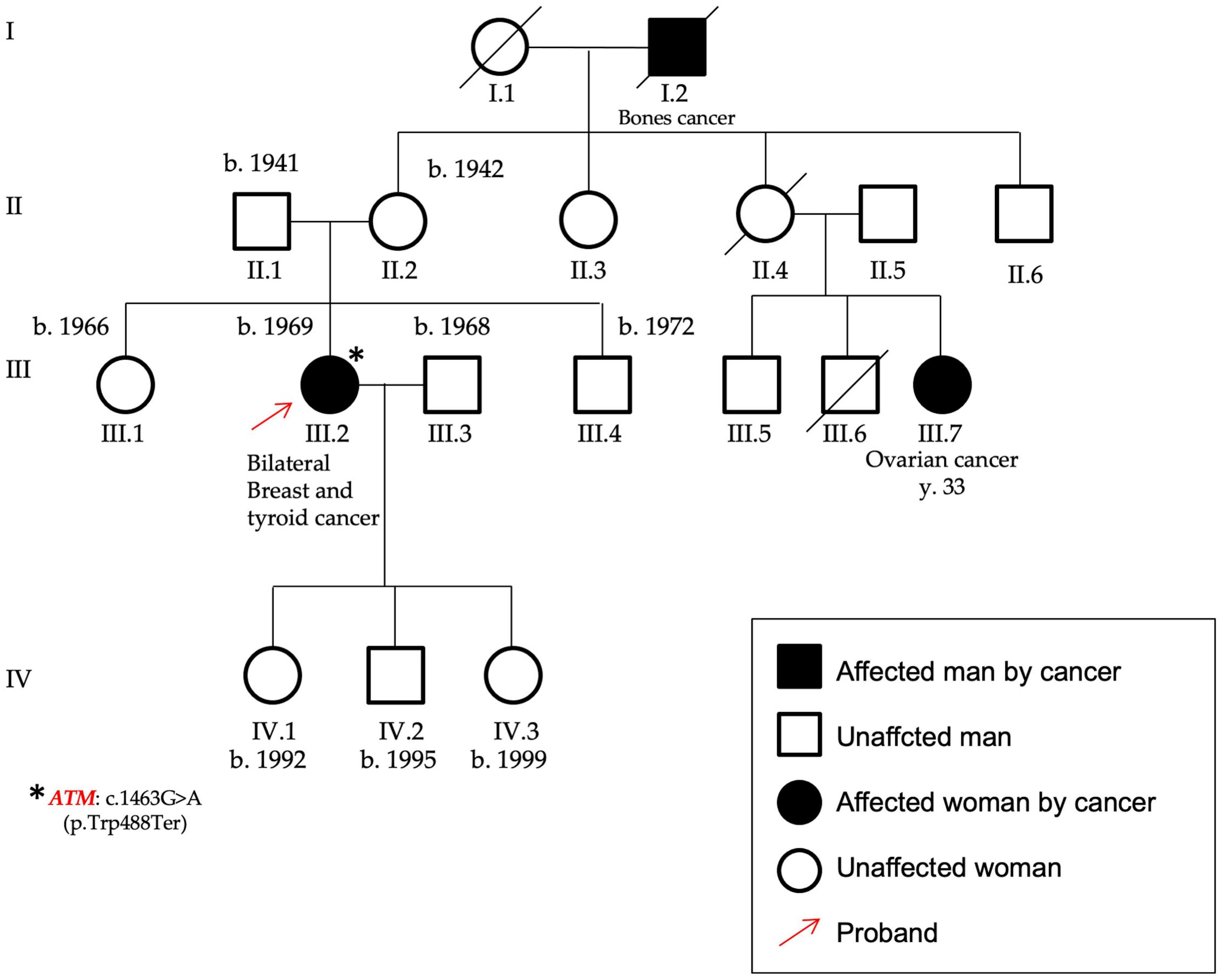
Figure 3. Pedigree of P12 carrying the variant c.1463G > A, p.(Trp488Ter), found in the ATM gene. The patient was affected by bilateral breast cancer and thyroid cancer and her maternal cousin by early onset ovarian cancer. The variant identified in this proband (highlighted by the red arrow) was a nonsense mutation in the ATM gene, as identified by the asterisk.
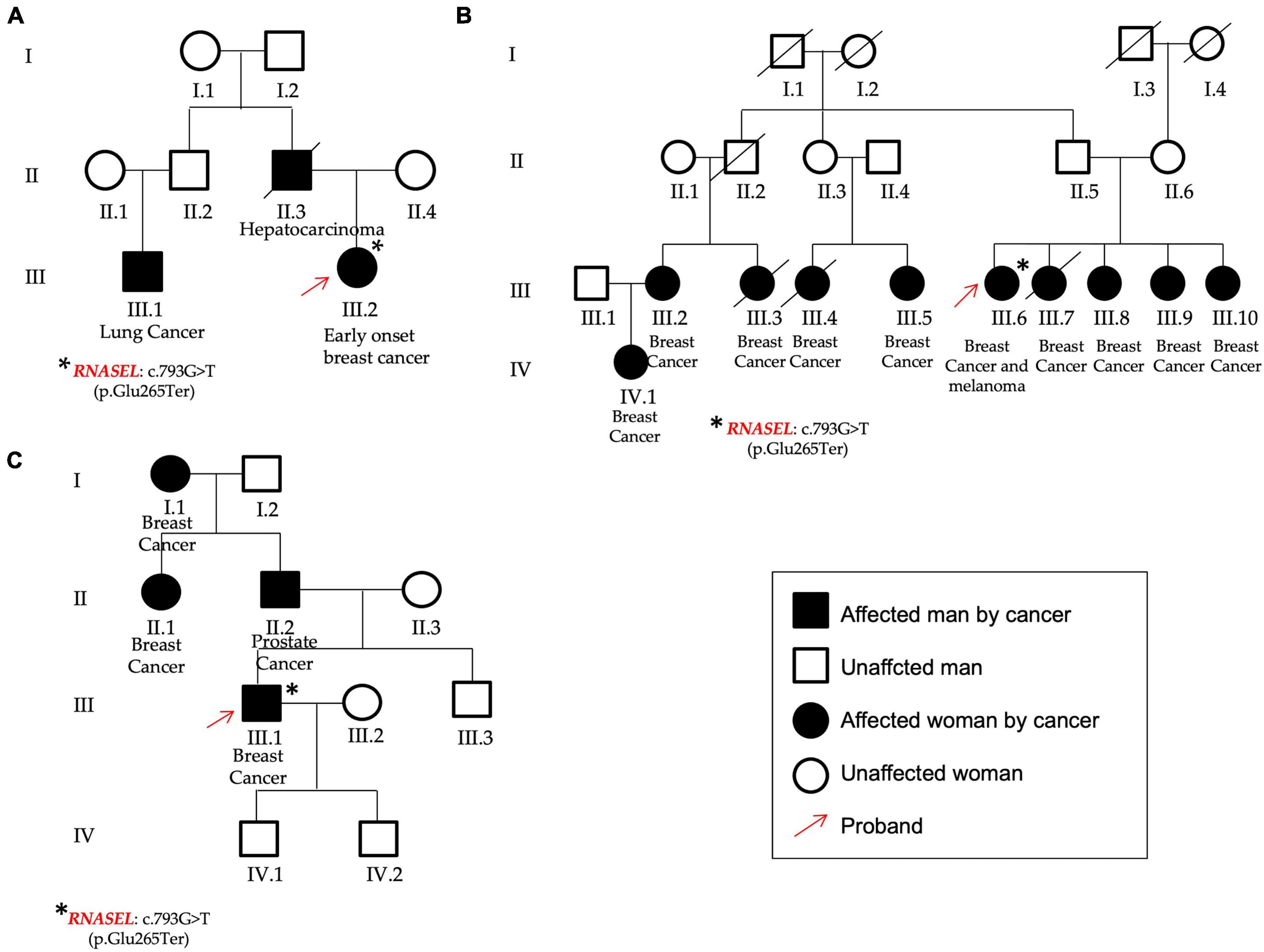
Figure 4. Pedigrees of P13 (A), P54 (B), and P32 (C) patients found to carry the same variant in the RNASEL gene. (A) The patient was affected by early onset breast cancer and showed different cases of hepatocarcinoma and lung cancer; (B) The patient affected by breast cancer and melanoma showed other cases of breast cancer from both the paternal and the maternal branches. (C) The male patient was affected by breast cancer and declared other cases both for breast and prostate cancers. Red arrows identified the three probands, asterisks the RNASEL gene variant.
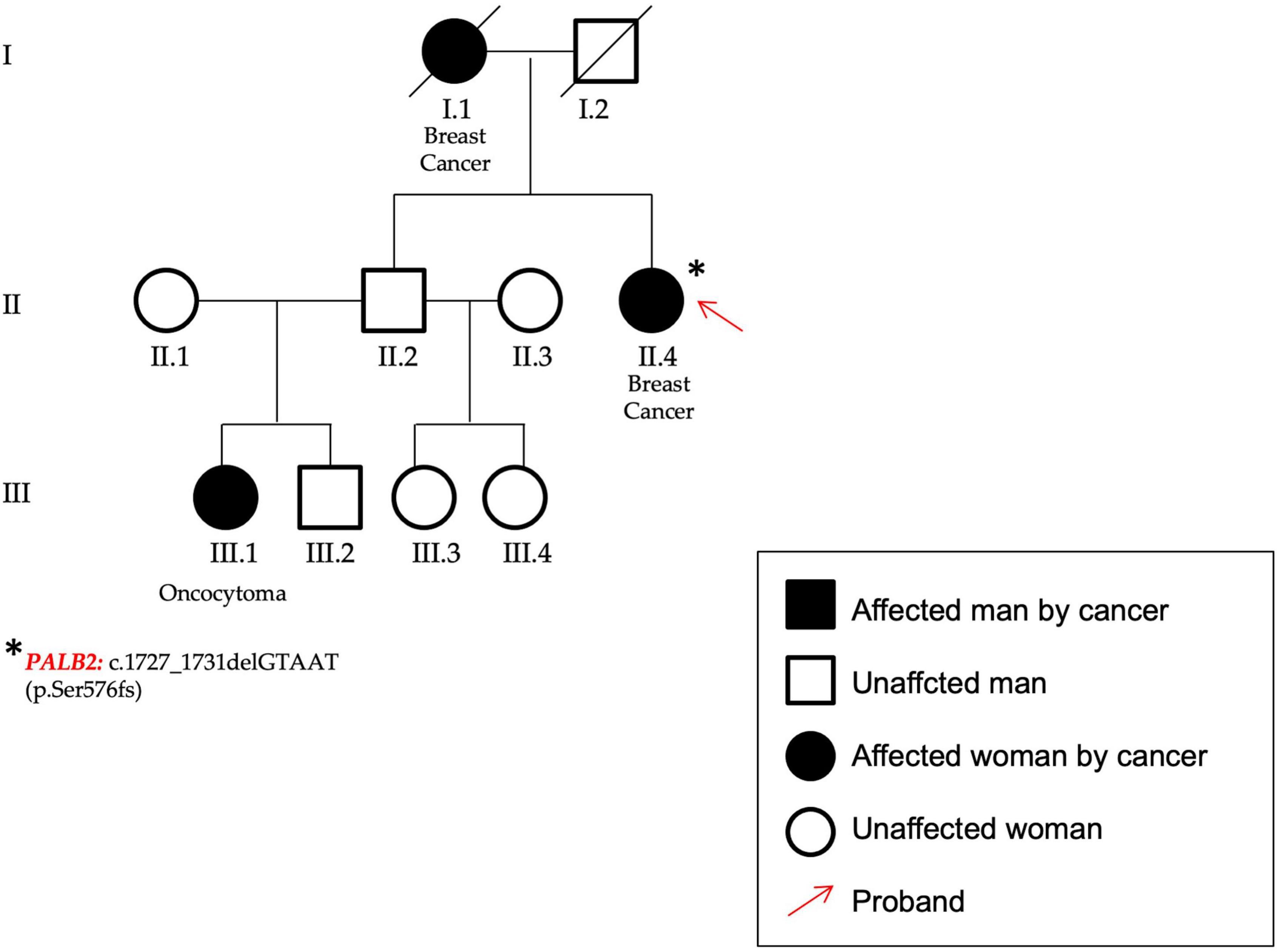
Figure 5. In figure the pedigree of P59. The patient (identified by a red arrow) affected by breast cancer showed a frameshift variant in PALB2 gene.
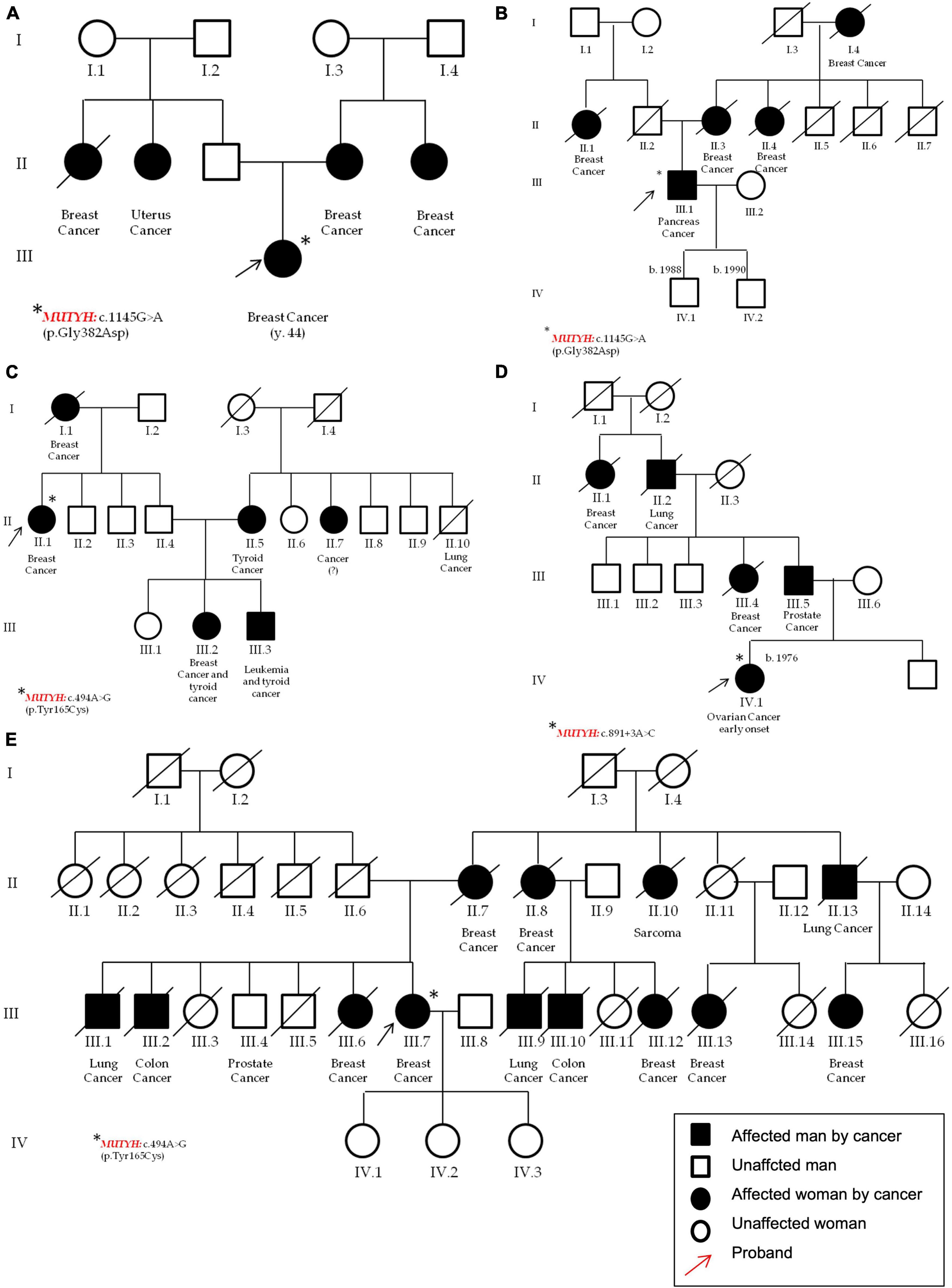
Figure 6. Pedigrees of the patients that carry pathogenic variants in the MUTYH gene. In detail, in (A) (P21) and (B) (P39) one female and one male that carry the c.1103G > A p.(Gly368Asp); in (C) (P24) a patient with c.452A > G, p.(Tyr151Cys); and in (D) (P52) a female with the c.891 + 3A > C. Pedigree (E) (P15) of the second patient carrying the MUTYH variant c.452A > G, p.(Tyr151Cys). All the probands are highlighted by red arrows and the asterisks identifies MUTYH different variants.
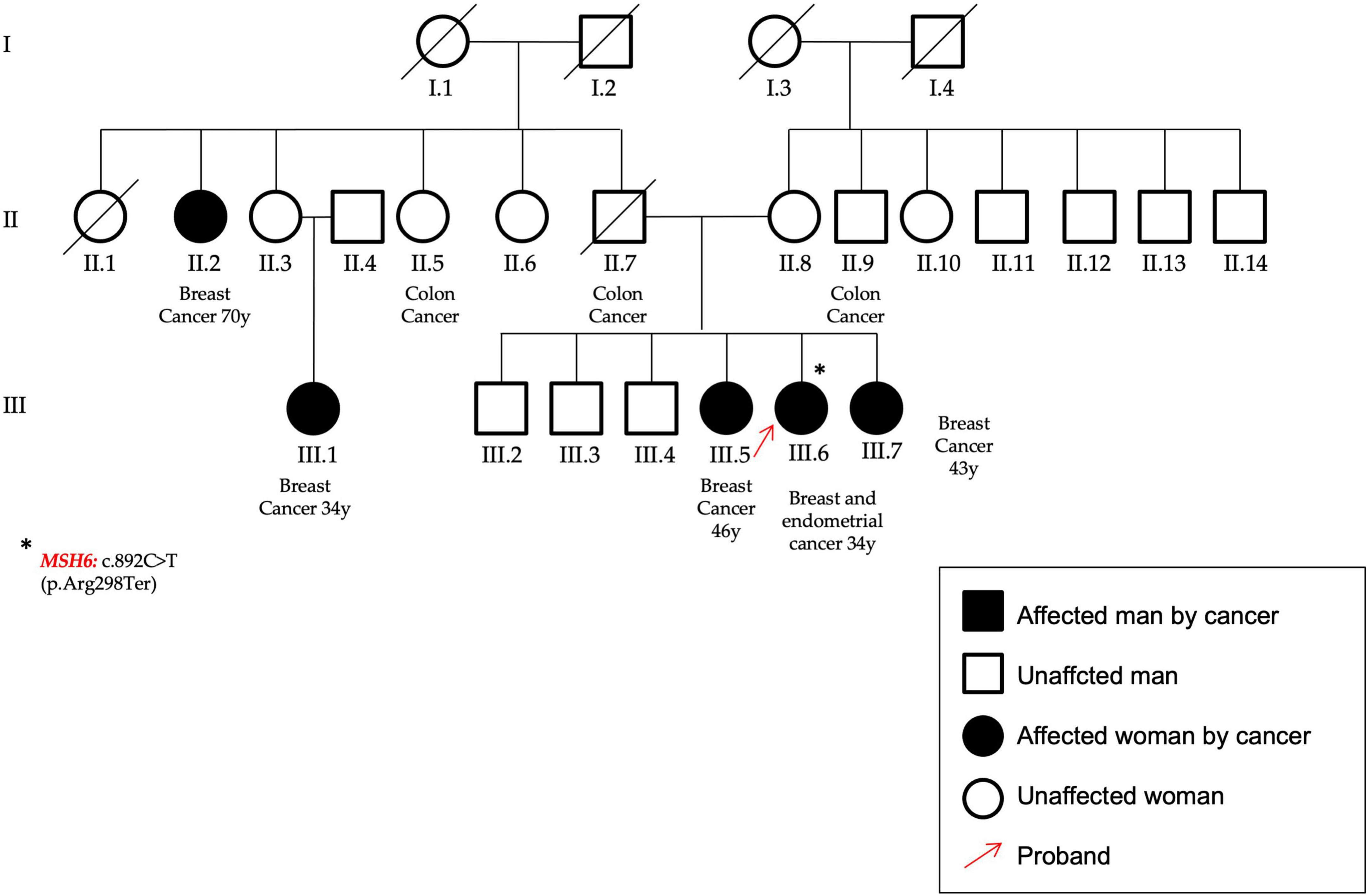
Figure 7. In the figure the pedigree of the patient (P14) that carry the variant identified in the MSH6 gene, c.892C > T–p.(Arg298Ter). She had an early onset BC and a subsequent endometrial cancer; moreover, other cases of both breast and colon cancers are reported within her family. The proband was highlighted by a red arrow and the asterisk identifies the MSH6 gene mutation.
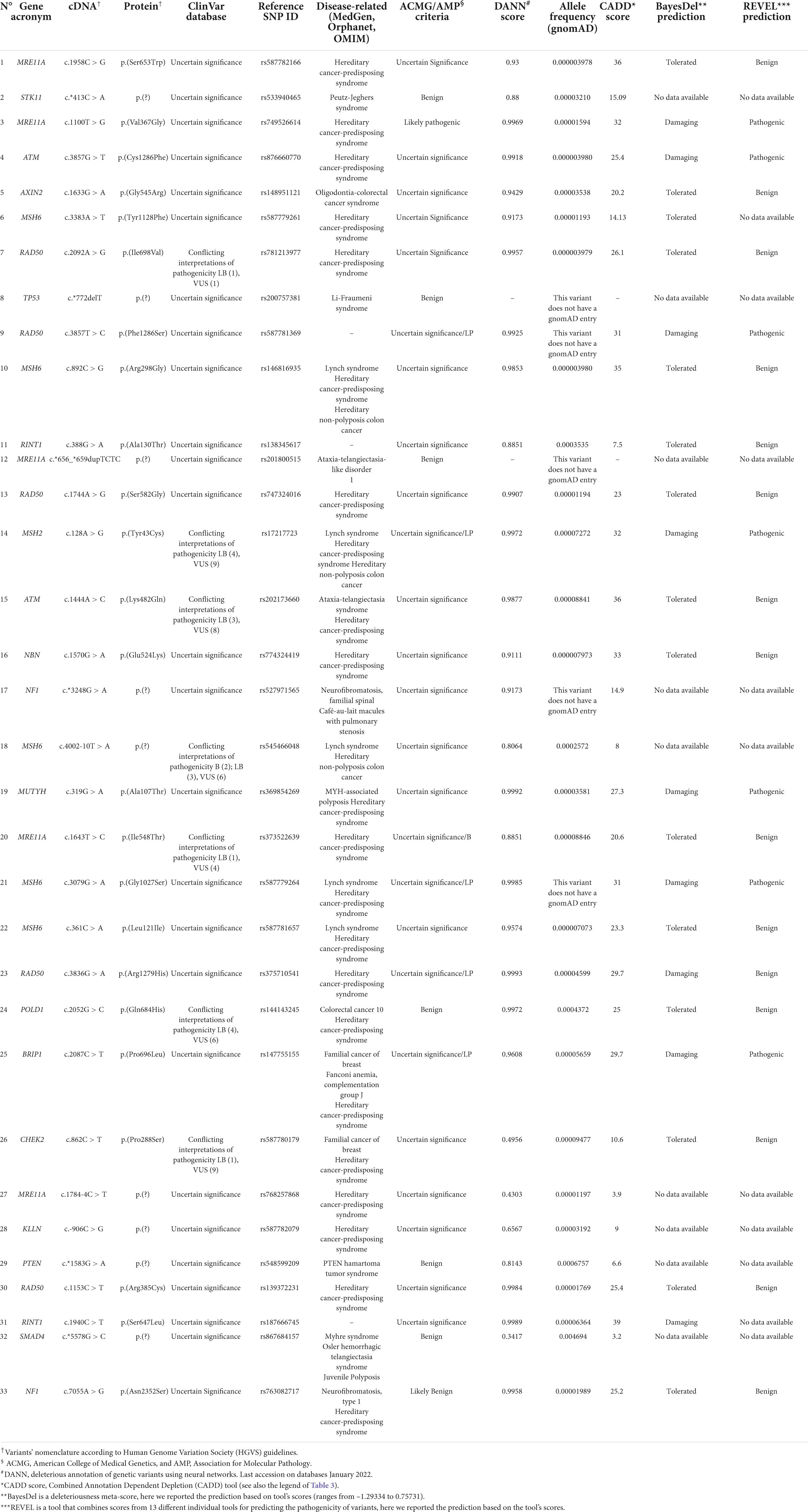
Table 4. List of the 33 variants of unknown significance (VUSs) identified in 23 patients (36%).
Results
All the 64 patients selected for this study were analyzed as described under Methods section. An average of 3,643,168 reads/sample with a mean reads’ depth in the target regions of 878X, was obtained. Sequencing data/sample are reported in the Supplementary Table 2.
The results reported herein showed that 28 patients (44%) were negative for multi-gene panel testing, 12 patients presented pathogenic/likely pathogenic variants (19%), and 23 patients (36%) showed potentially interesting variants currently classified as VUSs in ClinVar database.
Interestingly, this extended molecular screening allowed the identification of pathogenic variants in 12 different, unrelated patients (19%) resulted negative at BRCA1/2 test (concordance of mutations found in previous BRCA1/2 test was of 100%). These variants span over 6 different genes: ATM, RNASEL, PALB2, MSH6, MRE11A, and MUTYH genes, MUTYH carrying most of them (three different variants in five patients) (Figure 2 and Table 3).
In particular, as reported in Table 3, a total of eight pathogenic variants was found, of which three were nonsense variants, three were missense, one was an intronic variant affecting a splicing site, and one was a frameshift variant in the PALB2 gene, not reported in the ClinVar database, but classified as pathogenic according to the ACMG/AMP criteria (20).
In the ATM gene (NM_000051), a nonsense variant [c.1463G > A, p.(Trp488Ter)–rs879254093] in the exon 10 was found in a female patient affected by bilateral BC and also by a thyroid cancer (P12). She showed also a case of ovarian cancer (maternal cousin) (Figure 3). This variant is predicted to cause loss of normal protein function through the protein truncation or nonsense-mediated mRNA decay; loss-of-function variants in ATM gene are known in the literature to be pathogenic (21, 22).
The RNASEL (NM_021133.3) nonsense variant c.793G > T, p.(Glu265Ter) (rs74315364) in the exon 2, was found in three unrelated patients: two females (P13 and P54) and one male (P32).
This gene has been already associated to hereditary prostate cancer and it has been reported that men carrying this variant show a median age at prostate cancer onset 11 years lower respect to the not-carriers (23). In this study group, one of the two females that presented the RNASEL gene variant was affected by early onset BC, showed a case of hepatocarcinoma (her father) and lung cancer too (her paternal cousin) among her family members (P13, Figure 4A). The other female was affected by BC and melanoma and presented nine other cases of breast cancer in the family (four sisters, four cousins, and one nephew) (P54, Figure 4B). Finally, the same variant was identified in a man (P32) affected by BC and showing other cases of oncological diseases not only for BC (mother and grandmother), but also for prostate cancer (his father) (Figure 4C).
In the PALB2 (NM_024675.3) gene a frameshift variant not previously reported in any database was found. The variant c.1727_1731delGTAAT p.(Ser576Lysfs*8) was classified according to ACMG/AMP criteria (20) as certainly pathogenic. The patient was affected by bilateral BC and showed other cases of breast and renal cancers within the family (P59, Figure 5).
MUTYH (NM_001048174.2) harbors most of the variants identified in this cohort. Three different variants were found in five unrelated patients; moreover, the c.1103G > A p.(Gly368Asp)—rs36053993—and the c.452A > G, p.(Tyr151Cys)—rs34612342—variants were found twice, both being very common not only in familial adenomatous polyposis, but also in the general population. In particular, the MUTYH p.(Gly368Asp) and p.(Tyr151Cys) variants are well-established pathogenic variants for MUTYH-related polyposis and are estimated to account for 50–82% of MUTYH-associated polyposis in European patients (24). In this study, the p.(Gly368Asp) was found in a female patient affected by BC and with other cases of breast and uterus cancers in her family (P21, Figure 6A), and in a man affected by pancreatic cancer with other cases of BC in his pedigree (P39, Figure 6B).
The second MUTYH variant is the c.452A > G, p.(Tyr151Cys) (rs34612342) and was identified in two women. One was affected by bilateral BC and showed different cases of breast, thyroid, and leukemia (P24, Figure 6C); the second patient was affected by BC and declared other cases of breast, lung, prostate and bones cancers (P52, Figure 6E), respectively. The third MUTYH variant was the c.849 + 3A > C (rs587780751). This variant affects a non-conserved intronic nucleotide. Mutation Taster predicts a damaging outcome for this variant, and Alamut algorithms predict the variant to alter normal splicing. In this study, the c.849 + 3A > C (rs587780751) in the MUTYH gene was found in a patient with early onset ovarian cancer and with other cases of breast, prostate and lung cancers (P15, Figure 6D).
A nonsense nucleotide substitution in the MSH6 gene (NM_000179.3) was found in a patient affected by BC (onset at 34 years of age), who developed an endometrial cancer at follow-up. She showed other cases of BC and also for colon cancer (P14, Figure 7).
Finally, one variant identified in the MRE11A gene (NM_005591.3) an interesting VUS based on ACMG/AMP criteria (20). The MRE11A variant c.1100T > G–p.(Val367Gly) (rs749526614) is currently reported as variant of uncertain significance in ClinVar database (three entries, last accession on January 2022). Indeed, it affects a highly conserved amino acidic residue but the impact of this glycine to valine substitution on protein functions is not yet established since no functional studies have been published so far to assess its consequences. On the other side, the variant has a low allelic frequency and several prediction tools suggest a deleterious effect, DANN score being 0.99, and a score of 32 obtained by using the Combined Annotation Dependent Depletion (CADD score), deleterious and pathogenic also for BayesDel and REVEL tool’s predictions, respectively (Table 4).
In addition to the above-mentioned pathogenic variants a total of 33 rare VUSs identified using ClinVar database were additionally found (see also the allele frequency obtained using gnomAD database in Table 4); 7 (about 22%) were predicted to be benign/likely benign; and the remaining 26 (about 78%) were considered VUSs, according to the ACMG/AMP criteria (20).
Discussion
Breast cancer is still the most frequent cancer in females worldwide and an important cause of cancer death (1). Early diagnosis represents the major factor able to impact BC outcome and the overall patients’ survival (25). In this context, the identification of the hereditary forms, related to germline, inherited DNA variants, is crucial to admit patients and their at-risk family members to the most proper surveillance and therapeutic programs. In particular, the availability of specific drugs effective in mutated patients has increased the request for extensive molecular testing in the presence of breast/ovarian cancers diagnosis (26). Accordingly, thanks to the diffusion of NGS-based techniques in routine diagnostic laboratories, today the molecular analysis of the BRCA1/2 genes, including single nucleotide substitutions, small INDELs, and also CNVs evaluation can be performed in a few days (27, 28). Nevertheless, though the well-established role of the BRCA1/2 genes, both of them explain up to 25% of all the suspected hereditary forms (14). So far, several studies have been carried out to fill-in this gap and identify other predisposing genes by investigating different size of genes panels (9, 29–34). Even if BRCA1/2 remain the highly penetrant genes, some evidences are accumulating, especially regarding other genes involved in DNA repair mechanisms, so that, a couple of years ago, the NCCN guidelines for hereditary cancers have been updated to include their analysis (35). Taking into consideration cohorts of patients closer to the one present in this work, positive rates of about 15% were found (about 10% in BRCA1/2 and about 5% in other genes) (31–33). Nevertheless, more recent studies show a higher fraction of non-BRCA1/2 genes mutations (11, 14) principally due to the inclusion of lower-risk patients and larger multi-gene panels tested. Based on all these data and on the observation of the frequent co-occurrence of different cancer types (not only breast and ovarian, but also colon, melanoma, pancreas and prostate cancers) within the affected families, a custom panel including 44 genes to be simultaneously analyzed through an enrichment-based protocol followed by NGS analysis was set up. Here, are reported the results obtained by the implementation of this panel to analyze a cohort of 64 subjects from Southern Italy. All these subjects were already admitted to BRCA1/2 molecular testing based on their personal and/or familial history, but tested negative. As a consequence, they were the ideal candidates for an extended molecular analysis. Interestingly, 12 additional individuals that carry pathogenic/likely pathogenic variants, being equivalent to 19% of the analyzed subjects, were identified. This result highlights the importance of enlarged molecular testing to correctly identify hereditary BCs due to DNA variants in moderate or low penetrance genes. Indeed, without this analysis, these subjects would have been considered not mutated and their cancer risk would be considered like that of the general population. In this way, instead, their inherited cancer-predisposition was identified, allowing for their better clinical management and offering the opportunity to identify the other at-risk subjects within their families.
In particular, pathogenic/likely pathogenic variants in six genes were found according to ClinVar database and ACMG/AMP classification using VarSome tool: three of which being in well-established genes associated to breast and/or ovarian cancer (ATM, PALB2 and MSH6) and 3 (MRE11A, MUTYH and RNASEL) being in good candidacy to became susceptibility genes (36).
ATM (ataxia-telangiectasia mutate) encodes a protein kinase involved in the cellular response to DNA double-strand breaks through the phosphorylation of several proteins, including BRCA1 (37). ATM gene homozygous or compound heterozygous DNA variants are associated to the onset of an autosomal recessive disease, namely ataxia telangiectasia, featured by a progressive cerebellar degeneration and oculo-cutaneous telangiectasia (37). Interestingly, ATM heterozygous variants have been associated to an increased risk of BC so that ATM is considered a moderate penetrance gene for hereditary breast and ovarian cancers (37–39). An ATM truncating variant was found in a patient with bilateral BC and a previous thyroid cancer; moreover, other cancer cases are reported within her family.
PALB2 (partner and localizer of BRCA2) encodes a protein crucial for the correct functions of both BRCA1 and BRCA2 (39, 40). While PALB2 bi-allelic variants have been associated to Fanconi’s anemia, monoallelic loss-of-function variants have been reported to be responsible for an increased risk of both breast and pancreatic cancers (41). Moreover, even if PALB2 mutation carriers’ frequency is low and varies across populations (between 1 and 2.5%), their overall BC risk has been reported to be similar to that for BRCA2 mutation carriers (42). A PALB2 gene variant, the c.1727_1731delGTAAT p.(Ser576LysfsTer8) was found in a patient affected by BC with other cases of different oncological diseases. This variant has not been previously reported; however, it has been classified as pathogenic according to ACMG/AMP guidelines and different prediction tools. Even if functional studies are required to establish its pathogenicity, since it is a deletion of five nucleotides determining at protein level a premature stop codon, its pathogenicity is plausible.
MSH6 (Mut S Homolog 6) encodes a member of the mismatch repair genes family whose pathogenic variants have been associated to Lynch syndrome (43). It has been recently assessed that MSH6 variants are associated also to an increased risk of BC, suggesting their testing in the at-risk families (39, 44). The nonsense variant was found in a female affected by BC and endometrial cancer at the age of 34 years; with three cases of colon cancer and five cases of BC in her family.
MUTYH was the most frequently mutated gene in this population, since three different pathogenic variants in five unrelated patients (two variants were found twice, see Results section for more details) were detected. Mammalian MutY homologue (MUTYH) encodes a DNA glycosylase involved in base excision repair during DNA replication and DNA damage repair (44). Inherited bi-allelic variants in this gene have been associated to the MUTYH-associated polyposis (MAP), which is a risk factor for colorectal cancer development (45). In particular, the p.(Gly368Asp) and the p.(Tyr151Cys) variants, found also in this study, are the most common in the Caucasian population, accounting for about 75% of the pathogenic variants detected in the adenomatous polyposis patients (46). A large meta-analysis defined that MUTYH bi-allelic variants are associated to a 28-fold increased risk for colorectal cancer, while monoallelic variants had a limited effect (47). This may be due to several factors, including other co-occurring risk factors, such as the age of onset (48). In addition, due to its crucial role in DNA errors correction, a MUTYH involvement in predisposing also other cancers has been proposed (48).
Currently, the role of MUTYH monoallelic variants in BC patients is quite controversial with some studies showing an association and others demonstrating that no link exists (49–54). More recently, the use of enlarged genomic testing in cancer patients has shown that MUTYH monoallelic variants are identified in BRCA1/2-negative BC patients, even if with a frequency similar to that of the expected carrier frequency (55–57). MUTYH mono-allelic variants may act as low-penetrant BC predisposing factors, thus implying that additional concomitant risk factors, like age, ethnicity, or environmental factors, are required to determine a clinical phenotype. Based on all the above, NCCN guidelines for “Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic” (version 2.2022, accessed on March 2022), includes MUTYH mono-allelic variants within the lower penetrance genes that may be added as part of multi-gene testing, but for which the association with BC is still insufficient. However, increasing knowledge about the frequency of MUTYH variants and their spectrum of associated cancers may provide in the future an additional instrument for patients’ risk stratification and for the development of novel therapies (58, 59). It has to be mentioned that Rizzolo et al. (60), carrying out a multicenter study on male BC risk in Italy, found that MUTYH pathogenic variants were associated to an increased cancer risk (60). Moreover, a recent study by Doddato et al. (36), by analyzing an Italian cohort of 200 individuals, identified MUTYH pathogenetic variants in 4 unrelated subjects (36). Even if further studies on larger groups of patients are required to confirm these data, these studies, together with the results obtained herein, suggest that MUTYH mono-allelic variants may be a currently under-estimated risk-factor for BC cancer predisposition, at least in the Southern Italian population.
The most frequent variant identified in this study group was the nonsense variant c.793G > T, p.(Glu265Ter) in the RNASEL gene found in three unrelated subjects. RNASEL (endoribonuclease or RNase L) encodes an enzyme involved in antiviral and anti-proliferate pathways (61). Germline variants in this gene have been associated to an increased risk of prostate cancer and it has been suggested as susceptibility gene also for other cancers (61–63). In particular, the RNASEL p.(Glu265Ter) variant was firstly described in prostate cancer patients and, since a reduction of protein activity was measured, a protein loss of function was hypothesized as pathogenetic mechanism (64). Thus, even if the role of this variant is still unclear, it was proposed as a rare founder allele in the Caucasian population (62). In the study group presented herein, three unrelated patients were found to carry the RNASEL p.(Glu265Ter) variant. No one carried other pathogenic variants in other genes and, interestingly, one of them was a male BC case whose father was affected by prostate cancer. As discussed above, the clinical significance of this variant is still controversial; moreover, as for the variants identified in MUTYH, the p.(Glu265Ter) is frequent in the Caucasian population. This finding is intriguing but, on the basis of current knowledge, it is not possible to define whether this is a casual association or whether there is a causal link.
In addition to the pathogenic variants, as usual in the case of extended molecular analysis, also several VUSs were identified using ClinVar database. One of these variants may be classified as pathogenic according to the ACMG/AMP guidelines (20), CADD score, BayesDel and REVEL predictions; it is a missense nucleotide change in the MRE11A gene involved in the repair of double-strand breaks and thus associated to breast and ovarian cancers (65). Other seven variants seem to be benign/likely benign, while the remaining 26 were classified as VUSs also in ACMG/AMP guidelines. Furthermore, the calculation of the CADD score to obtain in silico predictions, showed that nine variants could be considered harmful, but only four of these have pathogenicity predictions also for the BayesDel and REVEL tools, although are still VUSs for the ACMG/AMP criteria (66). On the one hand, these findings are currently difficult to interpret and may give inconclusive and frustrating results for patients, nevertheless their detection and annotation is crucial to increase the scientific knowledge and improve their correct classification, on the other hand those are frustrating also for patients’ relatives especially in case of family history of cancers. They are usually not tested for, but they know the existence and the possible consequences of being a mutation carrier.
Several studies have been carried out so far analyzing variable in size panels of genes and patients’ cohort (9, 29, 30). All together these studies are allowing to evaluate the prevalence of predisposing DNA variants in non-BRCA1/2 genes and to highlight some specific genes and/or variants with a higher frequency in specific geographic areas or population. Moreover, since a lot of variants with scarcely known clinical significance are emerging, accumulating genomic data is required to further classify them.
The study reported herein aims to contribute to this field by adding the data obtained by the analysis of patients from Southern Italy. In the present study, 5/64 analyzed patients carried a MUTYH monoallelic variant; this rate is so high and so unusual to be worth reporting and to admit the possibility that MUTYH variants may have a higher penetrance in the South-Italian genetic background or lifestyle/environment (67). In conclusion, the presented data support the use of enlarged multigene testing to increase the detection rate of hereditary BCs and to highlight also less frequently mutated genes and/or recurring variants in specific populations. Moreover, additional genetic variants acting as risk modifiers may be also highlighted allowing for a better BC patients stratification based on their genotype. The precision therapy goal is more and more important in these days to reach advancement also in progressively severe clinical situations.
Study limitations
A limitation of the study is that it was not able to measure weak (or even moderate) associations between relatively common variants (in MUTYH gene) and a relatively common disease (female BC) due to the small cohort. Therefore, larger studies followed by statistical data analysis are required to confirm this hypothesis. Similar issues apply also to the RNASEL p.(Glu265Ter) variant. In the present study, this variant was found in three unrelated patients allowing to speculate an intriguing association. Further studies are required to assess this hypothesis and exclude that repeated observations, of the same variant, may be due simply to random chance. Moreover, it is also possible that other variants not detectable by the used strategy (deeper intronic regions, CNVs in the non-BRCA1/2 genes, or other genes not included in the panel) may be associated to the patients’ phenotype.
Data availability statement
The original contributions presented in this study are publicly available at: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA820693/. This data can be found here: NCBI Bioproject, accession no. PRJNA820693.
Ethics statement
The studies involving human participants were reviewed and approved by Istituto Nazionale Tumori–Fondazione G. Pascale Ethics Committee (protocol number 3 of 03/25/2009). The patients/participants provided their written informed consent to participate in this study.
Author contributions
FSa and MN: conceptualization and overall supervision of methodology, data collection and discussion, project planning, and data analysis. MN, FD, VD’A, MVE, and FSt: methodology and experimental procedure and analysis of results. MP, CD, AC, MD’A, GB, and SD: patients’ enrollment, clinical data collection, and histopathology analysis. FSa and VD’A: writing and final editing of the manuscript. All authors contributed for manuscript writing also by drafting some relevant parts including tables and figures and read and agreed to the published version of the manuscript.
Funding
This research was funded by the Italian Ministry of Health (RF-2010-23183729) and Campania Region CIRO project to CEINGE-Biotecnologie Avanzate to FSa, by Campania Region SATIN 2014/2020, and by Campania Region 752/2019 and 38/2020 to FSa and VD’A.
Conflict of interest
MN, FD, MVE, FSt, VD’A, and FSa were employed or members of CEINGE-Biotecnologie avanzate.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.894358/full#supplementary-material
Footnotes
References
1. Sauer AG, Siegel RL, Jemal A, Fedewa SA. Current prevalence of major cancer risk factors and screening test use in the United States: Disparities by education and race/ethnicity. Cancer Epidemiol Biomarkers Prev. (2019) 28:629–42. doi: 10.1158/1055-9965.EPI-18-1169
2. Shah T, Shah N, Vijay DG, Patel B, Patel S. Male breast cancer: current trends—A tertiary care centre experience. Indian J Surg Oncol. (2020) 11:7–11. doi: 10.1007/s13193-019-01021-5
3. Sheikh A, Hussain SA, Ghori Q, Naeem N, Fazil A, Giri S, et al. The spectrum of genetic mutations in breast cancer. Asian Pacific J Cancer Prev. (2015) 16:2177–85. doi: 10.7314/APJCP.2015.16.6.2177
4. Hawsawi YM, Al-Numair NS, Sobahy TM, Al-Ajmi AM, Al-Harbi RM, Baghdadi MA, et al. The role of BRCA1/2 in hereditary and familial breast and ovarian cancers. Mol Genet Genomic Med. (2019) 7:e879. doi: 10.1002/mgg3.879
5. Wang YA, Jian JW, Hung CF, Peng HP, Yang CF, Cheng HCS, et al. Germline breast cancer susceptibility gene mutations and breast cancer outcomes. BMC Cancer. (2018) 18:315. doi: 10.1186/s12885-018-4229-5
6. Concolino P, Gelli G, Rizza R, Costella A, Scambia G, Capoluongo E. BRCA1 and BRCA2 testing through next generation sequencing in a small cohort of Italian breast/ovarian cancer patients: Novel pathogenic and unknown clinical significance variants. Int J Mol Sci. (2019) 20:3442. doi: 10.3390/ijms20143442
7. Fountzilas C, Kaklamani VG. Multi-gene panel testing in breast cancer management. Cancer Treat Res. (2018) 173:121–40. doi: 10.1007/978-3-319-70197-4_8
8. Catana A, Apostu AP, Antemie RG. Multi gene panel testing for hereditary breast cancer - Is it ready to be used? Med Pharm Rep. (2019) 92:220–5. doi: 10.15386/mpr-1083
9. Nunziato M, Esposito MV, Starnone F, Diroma MA, Calabrese A, del Monaco V, et al. A multi-gene panel beyond BRCA1/BRCA2 to identify new breast cancer-predisposing mutations by a picodroplet PCR followed by a next-generation sequencing strategy: A pilot study. Anal Chim Acta. (2018) 1046:154–62. doi: 10.1016/j.aca.2018.09.032
10. D’Argenio V, Esposito MV, Telese A, Precone V, Starnone F, Nunziato M, et al. The molecular analysis of BRCA1 and BRCA2: Next-generation sequencing supersedes conventional approaches. Clin Chim Acta. (2015) 446:221–5. doi: 10.1016/j.cca.2015.03.045
11. McAlarnen L, Stearns K, Uyar D. Challenges of genomic testing for hereditary breast and ovarian cancers. Appl Clin Genet. (2021) 14:1–9. doi: 10.2147/TACG.S245021
12. Da Costa E, Silva Carvalho S, Cury NM, Brotto DB, De Araujo LF, Rosa RCA, et al. Germline variants in DNA repair genes associated with hereditary breast and ovarian cancer syndrome: Analysis of a 21 gene panel in the Brazilian population. BMC Med Genomics. (2020) 13:21. doi: 10.1186/s12920-019-0652-y
13. Cock-Rada AM, Ossa CA, Garcia HI, Gomez LR. A multi-gene panel study in hereditary breast and ovarian cancer in Colombia. Fam Cancer. (2018) 17:23–30. doi: 10.1007/s10689-017-0004-z
14. Shin HC, Lee HB, Yoo TK, Lee ES, Kim RN, Park B, et al. Detection of germline mutations in breast cancer patients with clinical features of hereditary cancer syndrome using a multi-gene panel test. Cancer Res Treat. (2020) 52:697–713. doi: 10.4143/crt.2019.559
15. Judkins T, Leclair B, Bowles K, Gutin N, Trost J, McCulloch J, et al. Development and analytical validation of a 25-gene next generation sequencing panel that includes the BRCA1 and BRCA2 genes to assess hereditary cancer risk. BMC Cancer. (2015) 15:215. doi: 10.1186/s12885-015-1224-y
16. Fostira F, Saloustros E, Apostolou P, Vagena A, Kalfakakou D, Mauri D, et al. Germline deleterious mutations in genes other than BRCA2 are infrequent in male breast cancer. Breast Cancer Res Treat. (2018) 169:105–13. doi: 10.1007/s10549-018-4661-x
17. Limongelli G, Nunziato M, D’Argenio V, Esposito MV, Monda E, Mazzaccara C, et al. Yield and clinical significance of genetic screening in elite and amateur athletes. Eur J Prev Cardiol. (2020) 28:1081–90. doi: 10.1177/2047487320934265
18. Kim BY, Park JH, Jo HY, Koo SK, Park MH. Optimized detection of insertions/deletions (INDELs) in whole-exome sequencing data. PLoS One. (2017) 12:e0182272. doi: 10.1371/journal.pone.0182272
19. Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: The human genomic variant search engine. Bioinformatics. (2019) 35:1978–80. doi: 10.1093/bioinformatics/bty897
20. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24.
21. Yang Z, Ouyang T, Li J, Wang T, Fan Z, Fan T, et al. Prevalence and characterization of ATM germline mutations in Chinese BRCA1/2-negative breast cancer patients. Breast Cancer Res Treat. (2019) 174:639–47. doi: 10.1007/s10549-018-05124-5
22. Hall MJ, Bernhisel R, Hughes E, Larson K, Rosenthal ET, Singh NA, et al. Germline pathogenic variants in the ataxia telangiectasia mutated (ATM) gene are associated with high and moderate risks for multiple cancers. Cancer Prev Res. (2021) 14:433–40. doi: 10.1158/1940-6207.CAPR-20-0448
23. Nguyen-Dumont T, Teo ZL, Hammet F, Roberge A, Mahmoodi M, Tsimiklis H, et al. Is RNASEL: P.Glu265* a modifier of early-onset breast cancer risk for carriers of high-risk mutations? BMC Cancer. (2018) 18:165. doi: 10.1186/s12885-018-4028-z
24. Kundu S, Brinkmeyer MK, Livingston AL, David SS. Adenine removal activity and bacterial complementation with the human MutY homologue (MUTYH) and Y165C, G382D, P391L and Q324R variants associated with colorectal cancer. DNA Repair. (2009) 8:1400–10. doi: 10.1016/j.dnarep.2009.09.009.Adenine
25. Winters S, Martin C, Murphy D, Shokar NK. Breast cancer epidemiology, prevention, and screening. Prog Mol Biol Transl Sci. (2017) 151:1–32. doi: 10.1016/bs.pmbts.2017.07.002
26. Lord CJ, Ashworth A. PARP inhibitors: The first synthetic lethal targeted therapy. Science. (2017) 355:1152–8. doi: 10.1126/science.aam7344.PARP
27. Minucci A, Scambia G, Santonocito C, Concolino P, Canu G, Mignone F, et al. Clinical impact on ovarian cancer patients of massive parallel sequencing for BRCA mutation detection: The experience at Gemelli hospital and a literature review. Expert Rev Mol Diagn. (2015) 15:1383–403. doi: 10.1586/14737159.2015.1081059
28. Wallace AJ. New challenges for BRCA testing: A view from the diagnostic laboratory. Eur J Hum Genet. (2016) 24:S10–8. doi: 10.1038/ejhg.2016.94
29. Susswein LR, Marshall ML, Nusbaum R, Vogel Postula KJ, Weissman SM, Yackowski L, et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med. (2016) 18:823–32. doi: 10.1038/gim.2015.166
30. Buys SS, Sandbach JF, Gammon A, Patel G, Kidd J, Brown KL, et al. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer. (2017) 123:1721–30. doi: 10.1002/cncr.30498
31. Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, Bowles K, et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer. (2015) 121:25–33. doi: 10.1002/cncr.29010
32. Desmond A, Kurian AW, Gabree M, Mills MA, Anderson MJ, Kobayashi Y, et al. Clinical actionability of multigene panel testing for hereditary breast and ovarian cancer risk assessment. JAMA Oncol. (2015) 1:943–51. doi: 10.1001/jamaoncol.2015.2690
33. Lincoln SE, Kobayashi Y, Anderson MJ, Yang S, Desmond AJ, Mills MA, et al. A systematic comparison of traditional and multigene panel testing for hereditary breast and ovarian cancer genes in more than 1000 patients. J Mol Diagn. (2015) 17:533–44. doi: 10.1016/j.jmoldx.2015.04.009
34. Beitsch PD, Whitworth PW, Hughes K, Patel R, Rosen B, Compagnoni G, et al. Underdiagnosis of hereditary breast cancer: Are genetic testing guidelines a tool or an obstacle?. J Clin Oncol. (2019) 37:453–60. doi: 10.1200/JCO.18.01631
35. Daly MB, Pilarski R, Yurgelun MB, Berry MP, Buys SS, Dickson P, et al. Genetic/familial high-risk assessment: Breast, ovarian, and pancreatic, version 1.2020 featured updates to the NCCN guidelines. J Natl Compr Cancer Netw. (2020) 18:380–91. doi: 10.6004/jnccn.2020.0017
36. Doddato G, Valentino F, Giliberti A, Papa FT, Tita R, Bruno LP, et al. Whole exome sequencing in BRCA1-2 candidate families: The contribution of other cancer susceptibility genes. Front Oncol. (2021) 11:649435. doi: 10.3389/fonc.2021.649435
37. Marabelli M, Cheng S-C, Parmigiani G. Penetrance of ATM gene mutations in breast cancer: A meta-analysis of different measures of risk. Genet Epidemiol. (2016) 40:425–31. doi: 10.1002/gepi.21971.Penetrance
38. Hu C, Hart SN, Gnanaolivu R, Huang H, Lee KY, Na J, et al. A population-based study of genes previously implicated in breast cancer. N Engl J Med. (2021) 384:440–51. doi: 10.1056/nejmoa2005936
39. Dorling L, Carvalho S, Allen J, González-Neira A, Luccarini C, Wahlström C, et al. Breast cancer risk genes — Association analysis in more than 113,000 women. N Engl J Med. (2021) 384:428–39. doi: 10.1056/nejmoa1913948
40. Sy SMH, Huen MSY, Zhu Y, Chen J. PALB2 regulates recombinational repair through chromatin association and oligomerization. J Biol Chem. (2009) 284:18302–10. doi: 10.1074/jbc.M109.016717
41. Tischkowitz M, Xia B. PALB2/FANCN: Recombining cancer and fanconi anemia. Cancer Res. (2010) 70:7353–9. doi: 10.1158/0008-5472.CAN-10-1012
42. Thomas PS, Brown PH. Breast-cancer risk in families with mutations in PALB2. Breast Dis. (2015) 26:206–8. doi: 10.1016/j.breastdis.2015.07.017
43. Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of Lynch syndrome: 1895-2015. Nat Rev Cancer. (2015) 15:181–94. doi: 10.1038/nrc3878
44. Roberts ME, Jackson SA, Susswein LR, Zeinomar N, Ma X, Marshall ML, et al. MSH6 and PMS2 germ-line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genet Med. (2018) 20:1167–74. doi: 10.1038/gim.2017.254.MSH6
45. Nakamura T, Okabe K, Hirayama S, Chirifu M, Ikemizu S, Morioka H, et al. Structure of the mammalian adenine DNA glycosylase MUTYH: Insights into the base excision repair pathway and cancer. Nucleic Acids Res. (2021) 49:7154–63. doi: 10.1093/nar/gkab492
46. Cheadle JP, Sampson JR. MUTYH-associated polyposis-From defect in base excision repair to clinical genetic testing. DNA Repair. (2007) 6:274–9. doi: 10.1016/j.dnarep.2006.11.001
47. Theodoratou E, Campbell H, Tenesa A, Houlston R, Webb E, Lubbe S, et al. A large-scale meta-analysis to refine colorectal cancer risk estimates associated with MUTYH variants. Br J Cancer. (2010) 103:1875–84. doi: 10.1038/sj.bjc.6605966
48. Curia MC, Catalano T, Aceto GM. MUTYH: Not just polyposis. World J Clin Oncol. (2020) 11:428–49. doi: 10.5306/wjco.v11.i7.428
49. Win AK, Reece JC, Dowty JG, Buchanan DD, Clendenning M, Rosty C, et al. Risk of extracolonic cancers for people with biallelic and monoallelic mutations in MUTYH. Int J Cancer. (2016) 139:1557–63. doi: 10.1002/ijc.30197
50. Wasielewski M, Out AA, Vermeulen J, Nielsen M, Van Den Ouweland A, Tops CMJ, et al. Increased MUTYH mutation frequency among Dutch families with breast cancer and colorectal cancer. Breast Cancer Res Treat. (2010) 124:635–41. doi: 10.1007/s10549-010-0801-7
51. Zhang Y, Newcomb PA, Egan KM, Titus-Ernstoff L, Chanock S, Welch R, et al. Genetic polymorphisms in base-excision repair pathway genes and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. (2006) 15:353–8. doi: 10.1158/1055-9965.EPI-05-0653
52. Beiner ME, Zhang WW, Zhang S, Gallinger S, Sun P, Narod SA. Mutations of the MYH gene do not substantially contribute to the risk of breast cancer. Breast Cancer Res Treat. (2009) 114:575–8. doi: 10.1007/s10549-008-0042-1
53. Win AK, Cleary S, Dowty JG, Baron JA. Cancer risks for monoallelic MUTYH mutation carriers with a family history of colorectal cancer. Int J Cancer. (2011) 129:2256–62. doi: 10.1007/bf02236735
54. Out AA, Wasielewski M, Huijts PEA, Van Minderhout IJHM, Houwing-Duistermaat JJ, Tops CMJ, et al. MUTYH gene variants and breast cancer in a Dutch case-control study. Breast Cancer Res Treat. (2012) 134:219–27. doi: 10.1007/s10549-012-1965-0
55. Díaz-Gay M, Alexandrov LB. Unraveling the genomic landscape of colorectal cancer through mutational signatures. Adv Cancer Res. (2021) 151:385–424.
56. Solano AR, Mele PG, Jalil FS, Liria NC, Podesta EJ, Gutiérrez LG. Study of the genetic variants in brca1/2 and non-brca genes in a population-based cohort of 2155 breast/ovary cancer patients, including 443 triple-negative breast cancer patients, in Argentina. Cancers. (2021) 13:2711. doi: 10.3390/cancers13112711
57. Fonfria M, de Juan Jiménez I, Tena I, Chirivella I, Richart-Aznar P, Segura A, et al. Prevalence and clinicopathological characteristics of moderate and high-penetrance genes in non-BRCA1/2 breast cancer high-risk Spanish families. J Pers Med. (2021) 11:548. doi: 10.3390/jpm11060548
58. Boffetta P, Hainaut P. Module in Biomedical Sciences - Encyclopedia of Cancer. 3rd ed. London: Academic Press (2019).
59. Sharbeen G, McCarroll J, Goldstein D, Phillips PA. Exploiting base excision repair to improve therapeutic approaches for pancreatic cancer. Front Nutr. (2015) 2:10. doi: 10.3389/fnut.2015.00010
60. Rizzolo P, Silvestri V, Bucalo A, Zelli V, Valentini V, Catucci I, et al. Contribution of MUtYH variants to male breast cancer risk: Results from a multicenter study in Italy. Front Oncol. (2018) 8:583. doi: 10.3389/fonc.2018.00583
61. Rökman A, Ikonen T, Seppälä EH, Nupponen N, Autio V, Mononen N, et al. Germline alterations of the RNASEL gene, a candidate HPC1 gene at 1q25, in patients and families with prostate cancer. Am J Hum Genet. (2002) 70:1299–304. doi: 10.1086/340450
62. Bartsch DK, Fendrich V, Slater EP, Sina-Frey M, Rieder H, Greenhalf W, et al. RNASEL germline variants are associated with pancreatic cancer. Int J Cancer. (2005) 117:718–22. doi: 10.1002/ijc.21254
63. Madsen BE, Ramos EM, Boulard M, Duda K, Overgaard J, Nordsmark M, et al. Germline mutation in RNASEL predicts increased risk of head and neck, uterine cervix and breast cancer. PLoS One. (2008) 3:e2492. doi: 10.1371/journal.pone.0002492
64. Carpten J, Nupponen N, Isaacs S, Sood R, Robbins C, Xu J, et al. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat Genet. (2002) 30:181–4. doi: 10.1038/ng823
65. Damiola F, Pertesi M, Oliver J, Le Calvez-Kelm F, Voegele C, Young EL, et al. Rare key functional domain missense substitutions in MRE11A, RAD50, and NBN contribute to breast cancer susceptibility: Results from a Breast Cancer Family Registry case-control mutation-screening study. Breast Cancer Res. (2014) 16:R58. doi: 10.1186/bcr3669
66. Quang D, Chen Y, Xie XDANN. A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. (2015) 31:761–3. doi: 10.1093/bioinformatics/btu703
Keywords: breast cancer, ovarian cancer, multigene panel, DNA repair, NGS sequencing, predictive medicine, genomic predisposition to disease
Citation: Nunziato M, Di Maggio F, Pensabene M, Esposito MV, Starnone F, De Angelis C, Calabrese A, D’Aiuto M, Botti G, De Placido S, D’Argenio V and Salvatore F (2022) Multi-gene panel testing increases germline predisposing mutations’ detection in a cohort of breast/ovarian cancer patients from Southern Italy. Front. Med. 9:894358. doi: 10.3389/fmed.2022.894358
Received: 11 March 2022; Accepted: 15 July 2022;
Published: 11 August 2022.
Edited by:
Jing He, Guangzhou Medical University, ChinaReviewed by:
Serena Bonin, University of Trieste, ItalyMuhammad Usman Rashid, Shaukat Khanum Memorial Cancer Hospital and Research Center, Pakistan
Copyright © 2022 Nunziato, Di Maggio, Pensabene, Esposito, Starnone, De Angelis, Calabrese, D’Aiuto, Botti, De Placido, D’Argenio and Salvatore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valeria D’Argenio, ZGFyZ2VuaW9AY2VpbmdlLnVuaW5hLml0; Francesco Salvatore, c2FsdmF0b3JAdW5pbmEuaXQ=
†These authors have contributed equally to this work and share first authorship