
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Med. , 29 April 2022
Sec. Gastroenterology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.891659
 Hiroyuki Suzuki1*
Hiroyuki Suzuki1* Teruko Arinaga-Hino1
Teruko Arinaga-Hino1 Tomoya Sano1Yutaro Mihara2Hironori Kusano2,3
Tomoya Sano1Yutaro Mihara2Hironori Kusano2,3 Tatsuki Mizuochi4Takao Togawa5Shogo Ito5Tatsuya Ide1Reiichiro Kuwahara1Keisuke Amano1Toshihiro Kawaguchi1Hirohisa Yano2Masayoshi Kage6
Tatsuki Mizuochi4Takao Togawa5Shogo Ito5Tatsuya Ide1Reiichiro Kuwahara1Keisuke Amano1Toshihiro Kawaguchi1Hirohisa Yano2Masayoshi Kage6 Hironori Koga1Takuji Torimura1
Hironori Koga1Takuji Torimura1
Benign recurrent intrahepatic cholestasis type 1 (BRIC1) is a rare autosomal recessive disorder that is characterized by intermittent episodes of jaundice and intense pruritus and caused by pathogenic variants of adenosine triphosphatase phospholipid transporting 8B1 (ATP8B1). The presence of genetic heterogeneity in the variants of ATP8B1 is suggested. Herein, we describe a unique clinical course in a patient with BRIC1 and a novel heterozygous pathogenic variant of ATP8B1. A 20-year-old Japanese man experienced his first cholestasis attack secondary to elevated transaminase at 17 years of age. Laboratory examinations showed no evidence of liver injury caused by viral, autoimmune, or inborn or acquired metabolic etiologies. Since the patient also had elevated transaminase and hypoalbuminemia, he was treated with ursodeoxycholic acid and prednisolone. However, these treatments did not relieve his symptoms. Histopathological assessment revealed marked cholestasis in the hepatocytes, Kupffer cells, and bile canaliculi, as well as a well-preserved intralobular bile duct arrangement and strongly expressed bile salt export pump at the canalicular membrane. Targeted next-generation sequencing detected a novel heterozygous pathogenic variant of ATP8B1 (c.1429 + 2T > G). Taken together, the patient was highly suspected of having BRIC1. Ultimately, treatment with 450 mg/day of rifampicin rapidly relieved his symptoms and shortened the symptomatic period.
Pathogenic variants in the adenosine triphosphatase phospholipid transporting 8B1 (ATP8B1) gene cause progressive familial intrahepatic cholestasis type 1 (PFIC1) and benign recurrent intrahepatic cholestasis type 1 (BRIC1) (1). The former is a progressive disease involving persistent cholestasis that eventually leads to liver cirrhosis, while the latter has a benign clinical course with recurrent cholestasis attacks and asymptomatic periods (1). ATP8B1 affects the regulation of the Farnesoid X receptor, and a deficiency of ATP8B1 protein may cause an imbalance between intestinal absorption and hepatic secretion of bile acids, leading to bile acid accumulation (2). The first cholestasis attack in patients with BRIC1 usually occurs before the second decade of life. Thereafter, several similar attacks with asymptomatic intervals ranging from several months to years may occur. During the asymptomatic intervals in these patients, biochemistry data remain normal. Each attack may last for several weeks to months without treatment, and most cases of BRIC1 do not progress to cirrhosis or end-stage liver disease. Classically, during the attacks in these patients, serum bile salt concentrations and bilirubin values are markedly elevated, whereas gamma-glutamyl transpeptidase (GGT) levels and aminotransferase activities remain within the normal range or are slightly elevated (1). To the best of our knowledge, this is the first report of a BRIC1 patient with a novel heterozygous variant of ATP8B1. Herein, we describe a unique clinical course of BRIC1 that deviates from the classical one.
A 20-year-old Japanese man experienced his first cholestasis attack at the age of 17 years old with the following laboratory findings; serum total bilirubin, 16.0 mg/dL; alkaline phosphatase (ALP), 682 IU/L; GGT activity, 16 IU/L; serum aspartate aminotransferase (AST), 158 IU/L; and alanine aminotransferase (ALT), 518 IU/L (Table 1). The patient had no risk factors for liver injury, such as drug use, toxin ingestion, alcohol abuse, and inborn or acquired metabolic etiologies. Parameters suggestive of autoimmune hepatitis or viral hepatitis, including antinuclear antibodies, antimitochondrial antibody, hepatitis B surface antigen, hepatitis C virus antibody, cytomegalovirus antibody, and Epstein-Barr virus, were negative (Table 1). Magnetic resonance cholangiopancreatography showed a normal intrahepatic and extrahepatic biliary tree and pancreatic ductal system. The liver tissue obtained from a percutaneous liver biopsy showed marked cholestasis in the hepatocytes, Kupffer cells, and bile canaliculi; however, no lobular inflammation, hepatic necrosis, or fibrosis were found (Figures 1A–C). He was treated with ursodeoxycholic acid (UDCA) at a dose of 600 mg/day, and its intermittent effect was obtained at the initial stage. The patient was then referred to our hospital for further examination and treatment. Serological parameters of liver fibrosis had the following values: serum type IV collagen, 237.0 ng/mL and Mac-2 binding protein glycosylation isomer, 0.23 (Table 1). Liver stiffness measured by transient elastography (FibroScan®) was 7.8 kPa. Further pathological assessment and immunohistochemistry studies revealed that bile salt export pump, multidrug resistance protein 3, multidrug resistance-associated protein 2, and CD10 were strongly expressed at the canalicular membrane (Figures 1D–E) and that the intralobular bile duct arrangement was well-preserved (Figure 1F). After obtaining informed consent, a genetic examination was performed to confirm the clinical diagnosis. Targeted next-generation sequencing, involving 61 genes responsible for genetic disorders of hepatic cholestasis (3), detected a heterozygous variant of ATP8B1: c.1429 + 2T > G (NM_005603.4). Thus the patient was highly suspected to have BRIC1 owing to these clinical, genetic, and pathological findings. Thereafter, similar attacks recurred with 5–6 months of asymptomatic intervals. During each attack, the patient experienced strong digestive symptoms associated with >5 kg of body weight loss and hypoalbuminemia (Figure 2). In addition, he experienced severe anorexia and frequent vomiting. There was no trace of inflammation, such as fever or an elevated C-reactive protein level, and esophagogastroduodenoscopy showed only Los Angeles grade B reflux esophagitis. Since every attack was accompanied by rapid transaminase elevation, the patient was treated with 2.5–10 mg/day of prednisolone; however, symptoms such as jaundice, pruritus, and weight loss lasted for 2–3 months. After approval by our hospital ethics committee, the patient was treated with 450 mg/day of rifampicin, which was based on previous studies (4, 5). This treatment rapidly relieved his symptoms and markedly shortened the symptomatic phase compared to prednisolone (Figure 2). Rifampicin was discontinued after the biochemical data improved to normal values. With 6–9 months of asymptomatic intervals, he experienced another two similar attacks. Since each attack was also accompanied by severe anorexia and elevated aminotransferases, we added rifampicin and prednisolone on UDCA. Thereafter, the symptomatic phase was markedly shortened (Figure 2).
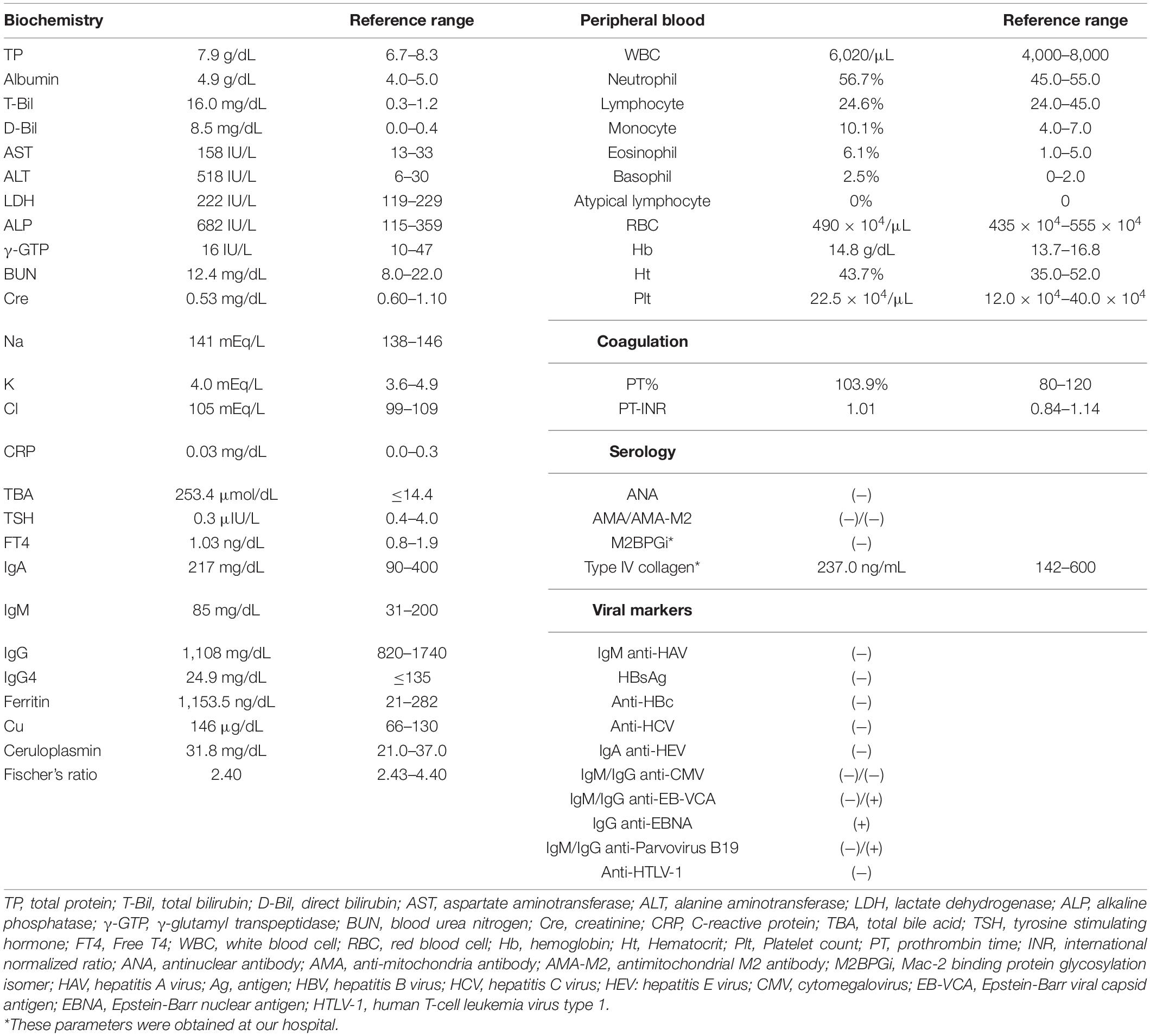
Table 1. Laboratory data on admission at previous hospital.
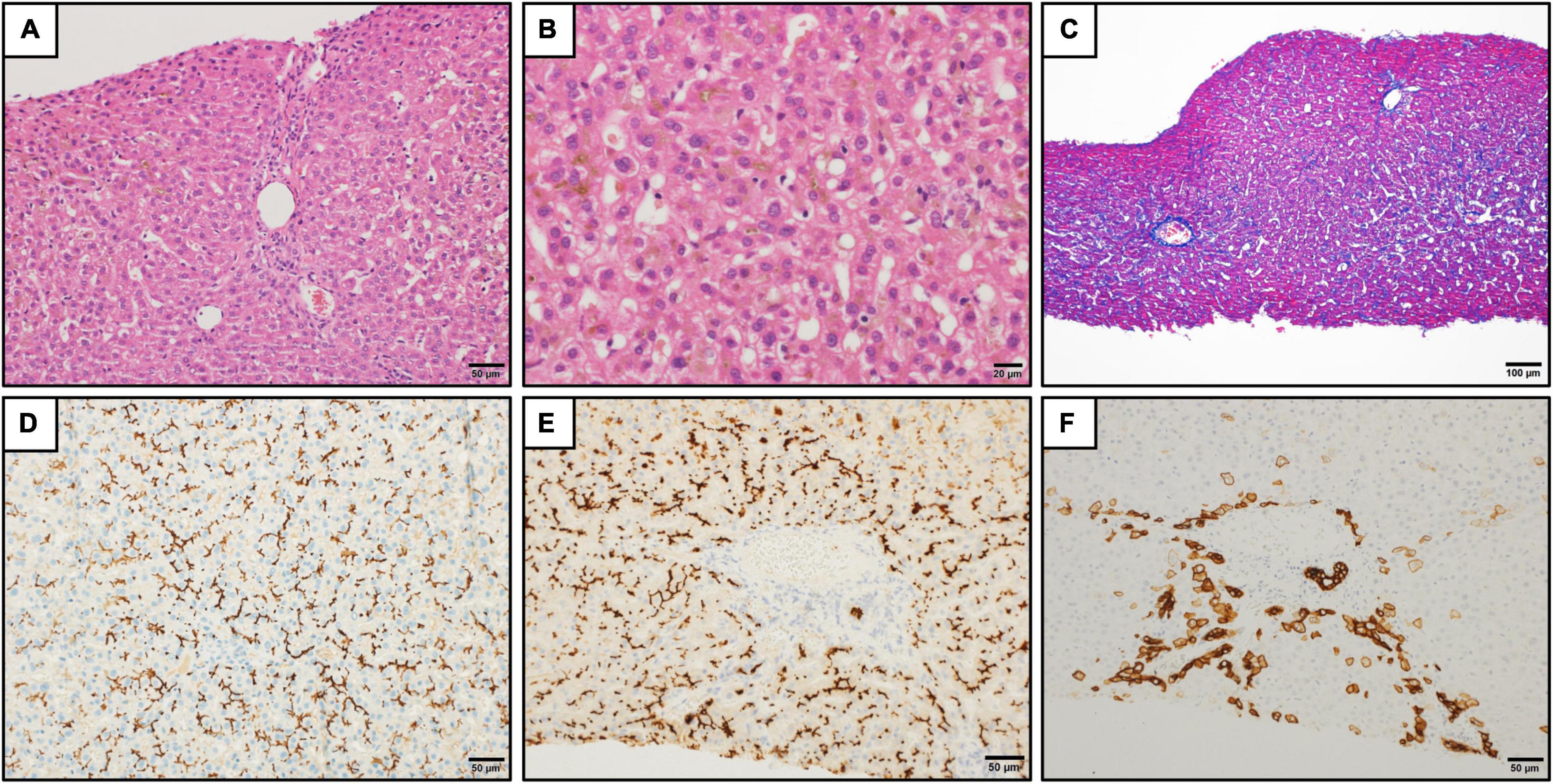
Figure 1. Histopathologic findings in the liver. (A) Liver tissue showed no significant lobular architecture (hematoxylin and eosin staining). Scale bar represents 50 μm. (B) Cholestasis was observed in the hepatocytes, Kupffer cells, and bile canaliculi at zones 2 and 3 (hematoxylin and eosin staining). Scale bar represents 20 μm. (C) There was no significant fibrosis (Azan staining). Scale bar represents 100 μm. (D–F) Immunohistochemistry for bile salt export pump (D), CD10 (E), and keratin 7 (F). The strong expression of bile salt export pump and CD10 were observed at the canalicular membrane (D,E). The keratin 7 expression was found in the hepatocytes in the periportal areas. The intralobular bile duct arrangement was preserved (F). Scale bars represent 50 μm.
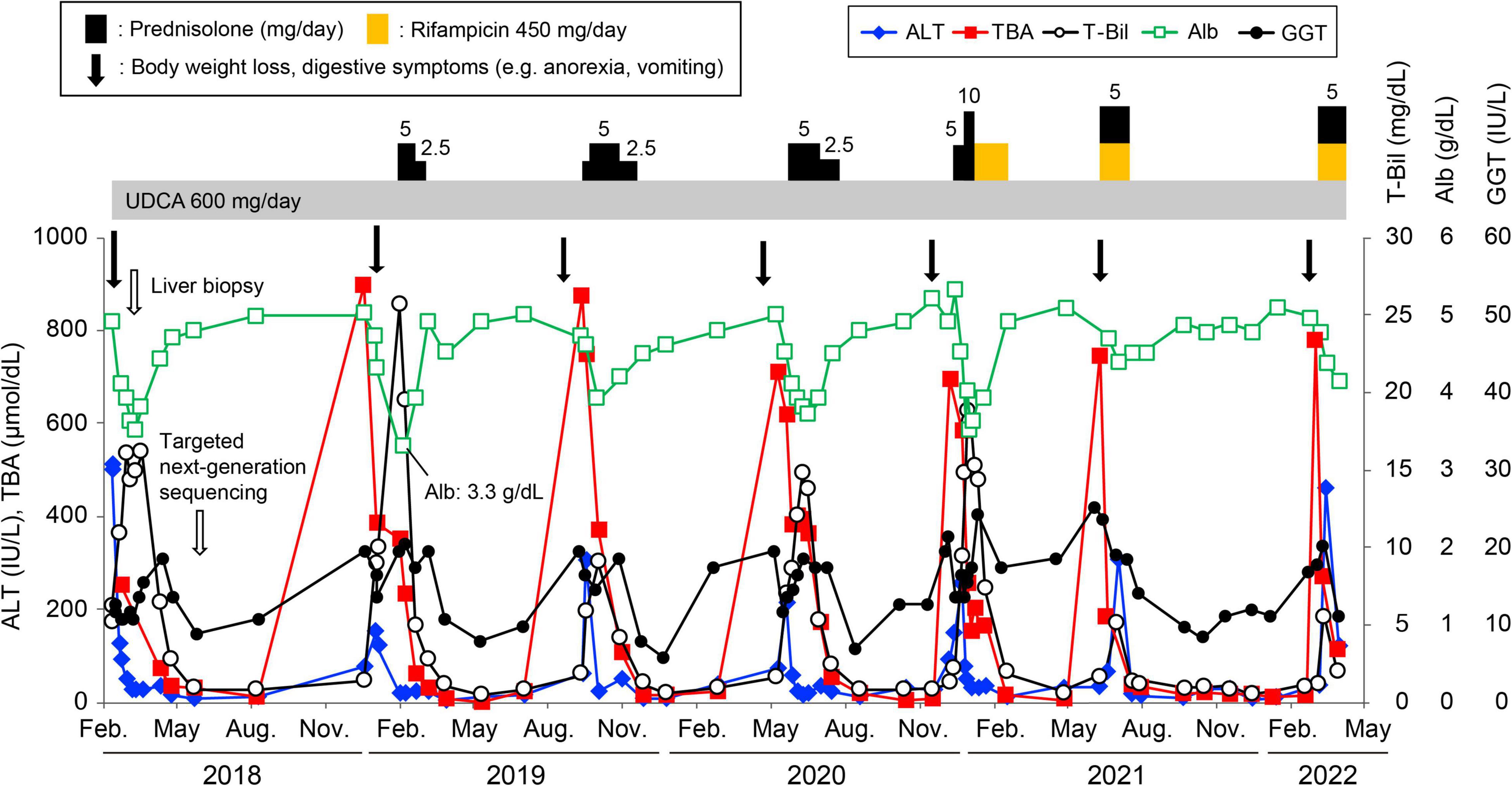
Figure 2. Clinical course of the patient. Changes in serum alanine aminotransferase (ALT, ◆), total bile acid (TBA, ■), total bilirubin (T-Bil, ○), albumin (Alb, □), and gamma-glutamyl transpeptidase (GGT, •) levels. The patient was treated with prednisolone or rifampicin with ursodeoxycholic acid during the attacks. The lowest albumin level during the follow-up period was 3.3 g/dL.
BRIC1 is a very rare autosomal recessive genetic disorder that was first described by Summerskill and Walshe in 1959. This condition is characterized by recurrent episodes of jaundice and intense pruritus, classically without the development of severe hepatic fibrosis (6). Since its discovery, many reports have been published, but the pathogenesis of BRIC1 remains unclear. Recently, molecular and genetic analyses have demonstrated that BRIC1 is caused by pathogenic variants in ATP8B1 at chromosome 18q21. In patients with ATP8B1 deficiency, the asymmetry of aminophospholipids in the hepatocanalicular membrane is disrupted, and the transport activity of the bile acid transport pump, which is an ABC transporter localized on the hepatocanalicular membrane that mediates the excretion of bile acids, is reduced, thus resulting in the development of severe intrahepatic cholestasis (7, 8). Moreover, because only a few bile acids flow into the bile canaliculus, it is considered that the cholangiocytes existing in the bile canaliculus and intrahepatic and extrahepatic biliary tree are not damaged, which could explain the normal GGT activity in these patients (7, 8). In the current case, the molecular genetic analysis revealed that the patient had a heterozygous variant of ATP8B1 with c.1429 + 2T > G. According to the guideline for determining the pathogenicity of identified variants (9), we judged the variant as a novel pathogenic variant, because this was not registered in several public databases, such as the Human Gene Mutation Database, Human Genetic Variation Database, and The Genome Aggregation Database (10) and because computational predictive programs strongly suggested its pathogenicity. In addition, we did not identify any pathogenic variant of 61 candidate genes by next-generation sequencing (3). Monoallelic pathogenic variants of ATP8B1 have been reported to be a predisposing factor for phenotypes such as drug-induced cholestasis, intrahepatic cholestasis of pregnancy type 1, and transient neonatal cholestasis (11); therefore, it is suggested that there may be undiscovered pathogenic variants of ATP8B1 in patients with BRIC1. Although the patient in this case had a variant of ATP8B1 in the heterozygous state, he was highly suspected of BRIC1 because of his clinical, genetic, and pathological findings. To the best of our knowledge, this is the first report regarding this novel pathogenic variant of ATP8B1 in a patient with BRIC1.
Although classical BRIC1 does not progress to cirrhosis or end-stage liver disease, a previous study has reported that four cases of BRIC1 which showed progression to a more severe and permanent form of PFIC (12). This indicates the possibility of clinical continuity with an intermediate phenotype between BRIC1 and PFIC1. The clinical course in the patient in the current case differed from classical BRIC1 in that it was accompanied by very strong gastrointestinal symptoms, elevated transaminases, and hypoalbuminemia, suggesting that it may be an intermediate phenotype of these diseases. Since the phenotype in this case deviates from that in classical BRIC1, we conducted general examinations, including the evaluation of Epstein-Barr virus, cytomegalovirus, and parvovirus B19, to investigate the cause of rapid transaminase elevation. Although no obvious causes were detected, the possibility of unknown causes (including another viral infections) triggering cholestasis cannot be ruled out. In addition, this difference may also be due to the rare genetic variant of ATP8B1. In a recent Japanese nationwide survey, it was shown that there are no critical methods to distinguish between PFIC1 and BRIC1 at the early phase of the disease (13). Therefore, further accumulation and evaluation of BRIC1 cases are desirable for more appropriate follow-up of these patients.
The effects of several therapeutic strategies aimed at relieving symptoms during cholestasis attacks in patients with BRIC1, such as UDCA, corticosteroids, cholestyramine, and nasobiliary drainage, are known to be case-dependent and usually ineffective (14, 15). In this report, since the patient had elevated aminotransferases, we added prednisolone to UDCA. However, these two drugs did not seem to be effective. Treatment with rifampicin, a potent human activator of pregnane X receptor, improves pruritus, shortens the symptomatic phase, and extends the asymptomatic interval (5). Consistent with previous reports, treatment with rifampicin for the patient in the current case markedly shortened the symptom duration (4, 16). In contrast, little benefit from rifampicin regarding extension of the asymptomatic interval was obtained. It has also been reported that the long-term administration of rifampicin may result in severe hepatoxicity in patients with cholestatic disorders (16). Therefore, it is also important the timing of drug discontinuation, which needs to be further evaluated.
We reported a rare case of a young Japanese patient with BRIC1 and a novel pathogenic variant of ATP8B1. We found that his phenotype was different from those of classical BRIC1 in that it was accompanied by rapid transaminase elevation. In addition, the little benefit from prednisolone and the effectiveness of rifampicin were obtained.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study.
HS and TA-H wrote the manuscript. HKu, HY, KA, HKo, MK, and TTr supervised the study. YM, TM, TI, RK, KA, TK, and TS participated in the acquisition of data and critical revision. TTg and SI performed genetic analysis and revised the manuscript. All authors discussed the results and contributed to the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the patient and his family for being involved in this case.
1. Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. (1998) 18:219–24. doi: 10.1038/ng0398-219
2. Frankenberg T, Miloh T, Chen FY, Ananthanarayanan M, Sun AQ, Balasubramaniyan N, et al. The membrane protein ATPase class I type 8B member 1 signals through protein kinase C zeta to activate the farnesoid X receptor. Hepatol. (2008) 48:1896–905. doi: 10.1002/hep.22431
3. Ito S, Togawa T, Imagawa K, Ito K, Endo T, Sugiura T, et al. Real-life progression of the use of a genetic panel in to diagnose neonatal cholestasis. JPGN Rep. (2022) 3:e196. doi: 10.1097/PG9.0000000000000196
4. Mizuochi T, Kimura A, Tanaka A, Muto A, Nittono H, Seki Y, et al. Characterization of urinary bile acids in a pediatric BRIC-1 patient: effect of rifampicin treatment. Clin Chim Acta. (2012) 413:1301–4. doi: 10.1016/j.cca.2012.04.011
5. Marschall HU, Wagner M, Zollner G, Fickert P, Diczfalusy U, Gumhold J, et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterol. (2005) 129:476–85. doi: 10.1016/j.gastro.2005.05.009
6. Summerskill WH, Walshe JM. Benign recurrent intrahepatic “obstructive” jaundice. Lancet. (1959) 2:686–90. doi: 10.1016/s0140-6736(59)92128-2
7. Byrne JA, Strautnieks SS, Mieli-Vergani G, Higgins CF, Linton KJ, Thompson RJ. The human bile salt export pump: characterization of substrate specificity and identification of inhibitors. Gastroenterol. (2002) 123:1649–58. doi: 10.1053/gast.2002.36591
8. Noe J, Stieger B, Meier PJ. Functional expression of the canalicular bile salt export pump of human liver. Gastroenterol. (2002) 123:1659–66. doi: 10.1053/gast.2002.36587
9. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
10. gnom AD. The Genome Aggregation Database (gnomAD). (2021). Available online at: https://gnomad.broadinstitute.org (accessed on July 1, 2021).
11. Gonzales E, Spraul A, Jacquemin E. Clinical utility gene card for: progressive familial intrahepatic cholestasis type 1. Eur J Hum Genet. (2014) 22:572. doi: 10.1038/ejhg.2013.186
12. van Ooteghem NA, Klomp LW, van Berge-Henegouwen GP, Houwen RH. Benign recurrent intrahepatic cholestasis progressing to progressive familial intrahepatic cholestasis: low GGT cholestasis is a clinical continuum. J Hepatol. (2002) 36:439–43. doi: 10.1016/s0168-8278(01)00299-9
13. Mizutani A, Sabu Y, Naoi S, Ito S, Nakano S, Minowa K, et al. Assessment of adenosine triphosphatase phospholipid transporting 8B1 (ATP8B1) function in patients with cholestasis with ATP8B1 deficiency by using peripheral blood monocyte-derived macrophages. Hepatol Commun. (2021) 5:52–62. doi: 10.1002/hep4.1605
14. Sticova E, Jirsa M, Pawlowska J. New insights in genetic cholestasis: from molecular mechanisms to clinical implications. Can J Gastroenterol Hepatol. (2018) 2018:2313675. doi: 10.1155/2018/2313675
15. Chen H, Wu D, Jiang W, Lei T, Lu C, Zhou T. Case Report: a novel homozygous variant identified in a Chinese patient with benign recurrent intrahepatic cholestasis-type 1. Front Med. (2021) 8:705489. doi: 10.3389/fmed.2021.705489
Keywords: benign recurrent intrahepatic cholestasis (BRIC), ATP8B1, autosomal recessive, cholestasis, progressive familial intrahepatic cholestasis (PFIC), rifampicin
Citation: Suzuki H, Arinaga-Hino T, Sano T, Mihara Y, Kusano H, Mizuochi T, Togawa T, Ito S, Ide T, Kuwahara R, Amano K, Kawaguchi T, Yano H, Kage M, Koga H and Torimura T (2022) Case Report: A Rare Case of Benign Recurrent Intrahepatic Cholestasis-Type 1 With a Novel Heterozygous Pathogenic Variant of ATP8B1. Front. Med. 9:891659. doi: 10.3389/fmed.2022.891659
Received: 08 March 2022; Accepted: 07 April 2022;
Published: 29 April 2022.
Edited by:
Xingshun Qi, General Hospital of Shenyang Military Command, ChinaReviewed by:
Patryk Lipiński, Children’s Memorial Health Institute (IPCZD), PolandCopyright © 2022 Suzuki, Arinaga-Hino, Sano, Mihara, Kusano, Mizuochi, Togawa, Ito, Ide, Kuwahara, Amano, Kawaguchi, Yano, Kage, Koga and Torimura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroyuki Suzuki, c3V6dWtpX2hpcm95dWtpQG1lZC5rdXJ1bWUtdS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.