 Natalia V. Balashova1,2
Natalia V. Balashova1,2 Lev G. Zavileyskiy3
Lev G. Zavileyskiy3 Artem V. Artiukhov4,5
Artem V. Artiukhov4,5 Leonid A. Shaposhnikov6
Leonid A. Shaposhnikov6 Olga P. Sidorova7
Olga P. Sidorova7 Vladimir I. Tishkov6,8
Vladimir I. Tishkov6,8 Angela Tramonti9
Angela Tramonti9 Anastasia A. Pometun6,8
Anastasia A. Pometun6,8 Victoria I. Bunik3,4,5*
Victoria I. Bunik3,4,5*- 1Department of Clinical Laboratory Diagnostics, Faculty of Advanced Medicine, M.F. Vladimirsky Moscow Regional Research and Clinical Institute (MONIKI), Moscow, Russia
- 2Department of Dietetics and Clinical Nutritionology, Faculty of Continuing Medical Education, RUDN Medical Institute, Moscow, Russia
- 3Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, Moscow, Russia
- 4Department of Biokinetics, A. N. Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, Russia
- 5Department of Biochemistry, Sechenov University, Moscow, Russia
- 6Department of Chemical Enzymology, Faculty of Chemistry, Lomonosov Moscow State University, Moscow, Russia
- 7Department of Neurology, Faculty of Advanced Medicine, M.F. Vladimirsky Moscow Regional Research and Clinical Institute (MONIKI), Moscow, Russia
- 8Bach Institute of Biochemistry, Federal Research Centre “Fundamentals of Biotechnology” of the Russian Academy of Sciences, Moscow, Russia
- 9Institute of Molecular Biology and Pathology, Italian National Research Council, Department of Biochemical Sciences “A. Rossi Fanelli,” Sapienza University of Rome, Rome, Italy
Oxidized nicotinamide adenine dinucleotide (NAD+) is a biological molecule of systemic importance. Essential role of NAD+ in cellular metabolism relies on the substrate action in various redox reactions and cellular signaling. This work introduces an efficient enzymatic assay of NAD+ content in human blood using recombinant formate dehydrogenase (FDH, EC 1.2.1.2), and demonstrates its diagnostic potential, comparing NAD+ content in the whole blood of control subjects and patients with cardiac or neurological pathologies. In the control group (n = 22, 25–70 years old), our quantification of the blood concentration of NAD+ (18 μM, minimum 15, max 23) corresponds well to NAD+ quantifications reported in literature. In patients with demyelinating neurological diseases (n = 10, 18–55 years old), the NAD+ levels significantly (p < 0.0001) decrease (to 14 μM, min 13, max 16), compared to the control group. In cardiac patients with the heart failure of stage II and III according to the New York Heart Association (NYHA) functional classification (n = 24, 42–83 years old), the blood levels of NAD+ (13 μM, min 9, max 18) are lower than those in the control subjects (p < 0.0001) or neurological patients (p = 0.1). A better discrimination of the cardiac and neurological patients is achieved when the ratios of NAD+ to the blood creatinine levels, mean corpuscular volume or potassium ions are compared. The proposed NAD+ assay provides an easy and robust tool for clinical analyses of an important metabolic indicator in the human blood.
Introduction
Nicotinamide adenine dinucleotide (NAD, the sum of the oxidised NAD+ and reduced NADH forms) is a very important biological molecule which is involved in various metabolic and signaling pathways. Undergoing reversible reduction to NADH in many redox reactions, NAD+ is also involved in signaling of perturbed homeostasis and DNA damage response. The signaling pathways include the NAD+-degrading reactions catalyzed by protein deacylases sirtuins (1–3), poly(ADP-ribose) polymerases 1 and 2 involved in the DNA damage response (4, 5), and NAD+ hydrolyzing enzymes CD38 (6)/CD157 (7). These NAD+-dependent reactions are involved in regulation of circadian rhythms (8), aging (9–11), and immunity (12). Major portion of NAD+ resides inside the cells, where its concentration may vary from 0.01 to 1 mM, yet some studies also determine significantly lower quantities of NAD+ in the blood plasma [2–70 nM in humans (10, 13, 14) and 240–290 nM in pigs (15)].
The correlation between the concentrations of NAD+ in blood/plasma and tissues has been studied in aging (16–18). The reduction in NAD+ level with age is observed in healthy human brain (19, 20), liver (21), red blood cells (22) and macrophages (23). In plasma, the NAD+ level is shown to drop from app. 50 nM in young (20–40 years) to app. 10 nM in elderly subjects (60–87 years) (10). Human skin NAD+ content also sharply declines as people age: from 8.5 ± 1.6 ng/mg protein in newborns to 1.1 ± 0.2 in elders (>51 years) (9). A strong negative correlation is observed between NAD+ levels in skin and age in both males (r = −0.706; p = 0.001) and females (r = −0.537; p = 0.01) (9). The total pool of NAD (both NAD+ and NADH) in whole blood, however, shows a trend to a negative correlation with age in males that is not observed in females (11). In the population combined from both genders, blood NAD levels in elderly patients (75–101 years old) hospitalized for decompensated heart failure are shown to be lower (20.7 ± 3.6 μM) than those in a healthy population of 151 voluntary donors aged 19 to 68 years (23.4 ± 4.1 μM) (11).
Changed NAD levels may be associated with cancer (16, 24, 25), obesity and type 2 diabetes (16, 26, 27), various neurological disorders (28–30), intestinal inflammation (31). Many studies point to decreased NAD levels under disturbed nutrient conditions (26). In view of the wide range of pathologies potentially affecting the NAD levels and redox state, the quantitative determination of this metabolite in human blood may be of diagnostic value. Worth noting, a rapid and efficient assay of NAD+ may be extremely useful to decide on treatments of critically ill patients, as the tissue damage response is associated with the NAD+ depletion in the poly(ADP-ribose)- polymerase-catalyzed reaction (4, 5). As a basic indicator of the healthy metabolism, NAD+ level has a potential to be used as a marker of biological age or nutritional state.
Recently, we have published the method of NAD+ quantitative determination using recombinant formate dehydrogenase (FDH) (32), whose application to the extracts of the rat brain tissue and its mitochondria has demonstrated such advantages of the assay as its high sensitivity and specificity. Compared to the “gold-standard” HPLC- and/or mass-spectrometry-based methods, our FDH assay does not require expensive consumables, neither highly professional supervision. A number of already existing biotechnological applications of FDH (33), also as fusion protein (34–36), enzyme mixture (37, 38) or in whole cell biocatalysis (39), extending from NAD(P)H regeneration to fixing atmospheric CO2 (40–43), demonstrate the enzyme robustness and utility for the environmentally friendly procedures. In this regard, development of the FDH-based clinical assays has another advantage over the currently employed assays of total NAD pool, using formazan dyes, as the most employed 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, known as MTT, is toxic for eucaryotic cells (44, 45). In the present study, we use FDH from Pseudomonas sp. that is extremely specific to NAD+, i.e., does not catalyze the reduction of NADP+, and is characterized by high catalytic efficiency and high thermal stability, compared to FDH from other sources (33, 46, 47). Employing this enzyme and developing the optimized protocol of the extraction of NAD+ from blood, we demonstrate the diagnostic potential of the assay for medical application by measuring the whole blood NAD+ in the healthy subjects and patients with neurological and cardiological pathologies.
Methods
Enrollment of Patients in the Study
The study was approved by the ethics commission of M.F. Vladimirsky Moscow Regional Research and Clinical Institute (MONIKI), decision N 17 of 10. December, 2020. All participants gave informed consent. The neurological patients with demyelinating diseases and cardiological patients with the heart failure of stage II and III according to the New York Heart Association (NYHA) functional classification, were enrolled in the study (Table 1) during 1 year. Our choice of the NYHA heart failure stages II and III was based on clinical abundance of these cardiological patients, in contrast to those of stage I, and a lower occurrence in these patients of additional deteriorations associated with the profound pathology of stage IV.

Table 1. Summary of the studied cohorts where the NAD+ content in the whole blood is quantified.
Preparation of Recombinant Formate Dehydrogenase
Recombinant FDH from Pseudomonas sp. was produced in E. coli as described in (48). The enzyme (1.1 mg/mL, app. 10 U/mg) was stored in 0.1 M sodium phosphate buffer, pH 7.0, containing 10 mM EDTA, at + 4°C.
Preparation of Methanol-Acetic Acid Extracts of Whole Blood
The blood was collected in the morning in a vacutainer tube with heparin (6 ml), aliquoted in 1 ml and frozen at −70°C. The blood levels of NAD+ did not decay upon the blood storage up to several months. Typically, the samples were accumulated and extracted within 2 months after the blood collection. A modification of the extraction protocol previously elaborated for the rat brain (49) was used. To prepare the extracts, blood samples were thawed on ice, 0.2 ml of each sample was transferred into a clean microcentrifuge tube and mixed with 1.6 ml of methanol precooled at + 4°C, using the T10 Basic ULTRA-TURRAX disperser (IKA, Staufen, Germany). 0.27 ml of 2% acetic acid was added, followed by 30 min shaking on ice in New Brunswick Excella E24R incubator (Eppendorf, Moscow, Russia) at 180 rpm. The resulting suspension was deproteinized by 20 min centrifugation at 21,500 g and 4°C, using Hitachi CT15RE centrifuge (Helicon, Moscow, Russia). The supernatant was transferred into a clean tube and stored at −70°C until analysis, usually performed the day after the extract preparations. Repeated assays of the same blood extracts before and after the storage showed that their NAD+ content was stable during several months.
NAD+ Determination Procedure
NAD+ concentration in the blood extracts was determined enzymatically as described earlier (32), using fluorescence mode of BMG ClarioSTAR Plus plate reader (Helicon, Moscow, Russia). Samples of blood extracts were shaken and 0.01, 0.015 or 0.02 ml aliquots of each sample were added into a 96-well black microplate (Greiner #655076) in duplicates. The mixture of 87% methanol/0.3% acetic acid was added to the aliquots to obtain the total volume of 0.02 ml. The blank contained 0.02 ml of the methanol-acetic acid mixture only. 0.18 ml of 0.6 M sodium formate in 0.1 M sodium phosphate buffer, pH 7.0, was added to all the wells. The background fluorescence (340/475 nm) of the samples was measured during 6 min. After registering the background levels, app. 0.03 U of FDH (app. 3 μg) was added into each well, and the fluorescence change was measured for 20 min. Usually, a plateau in the fluorescence was reached within 10 min, pointing to the completion of the reaction. NAD+ content in each well was calculated using the calibration curve employing 0.01–0.1 nmol of NAD+ and 0.03 U of FDH per well. Our comparison to the calibration with added NADH showed that the calibration employing the FDH reaction, better reproduced exact conditions of the NAD+ fluorescence assay. Simultaneously, the calibration employing the FDH reaction served as an internal control for the linearity and functionality of the enzyme assay in the selected interval of the NAD+ concentrations. The NAD+ content in the whole blood was calculated, taking into account the added extract volume and a 10.2-fold dilution of blood upon the extraction procedure.
Statistical Analysis
Comparisons between groups were made by one-way ANOVA with Tukey’s post hoc test or by Mann–Whitney’s U test in case of comparisons of two groups (*, **, ***, **** for p < 0.05, p < 0.01, p < 0.001, and p < 0.0001, respectively). Outliers were determined using the interquartile range rule (1.5 IQR criterion). Holm-Bonferroni correction was used for multiple testing adjustment. All statistical analysis and data visualization was performed using R Statistical Software (version 4.1.1; R Foundation for Statistical Computing, Vienna, Austria).
Results
In our pilot study, concentrations of NAD+ in the whole blood are compared in three different cohorts: healthy controls, cardiological patients with the heart failure of the NYHA class II and III, and neurological patients with demyelinating diseases, such as Charcot-Marie-Tooth type, Okinawa type and multiple sclerosis (Table 1). All the analyzed neuropathies are characterized by impaired myelination of nerve fibers, either in the central (multiple sclerosis) or peripheral (neuropathies of Charcot-Marie-Tooth and Okinawa types) nervous systems. Analysis of the quantified values of the whole blood NAD+ within the studied cohorts using the interquartile range rule (1.5 IQR criterion) has revealed four outliers in the group of cardiological patients, all of them determined in the patients with heart failure of the NYHA stage III. All the blood parameters inherent in the studied cohorts are shown as medians, minimum and maximum values in Supplementary Table 1, which also presents p-values characterizing significance of the group differences.
Independent of the exclusion (Figure 1) or inclusion (Supplementary Table 1) of the outliers, NAD+ concentrations in the blood of healthy subjects are significantly higher than those in the cardiological or neurological patients. No gender differences in the NAD+ levels are detected in our study. Cardiological patients with NYHA stage III show a trend to lower NAD+ levels, compared to the patients with NYHA stage II (p = 0.09, Supplementary Figure 1). Taking into account the known decreases in the blood NAD+ level with age (11) and the different median age of the studied groups (Supplementary Table 1), we have verified the finding in the cohorts using their comparison to the controls of similar age. With the studied subjects divided into the age groups of 42–68 years (cardiological patients vs. controls of the respective age) and 27–55 years (neurological patients vs. controls of respective age), significant differences in the NAD+ concentrations between the control subjects and cardiological or neurological patients are preserved (Supplementary Figure 2).
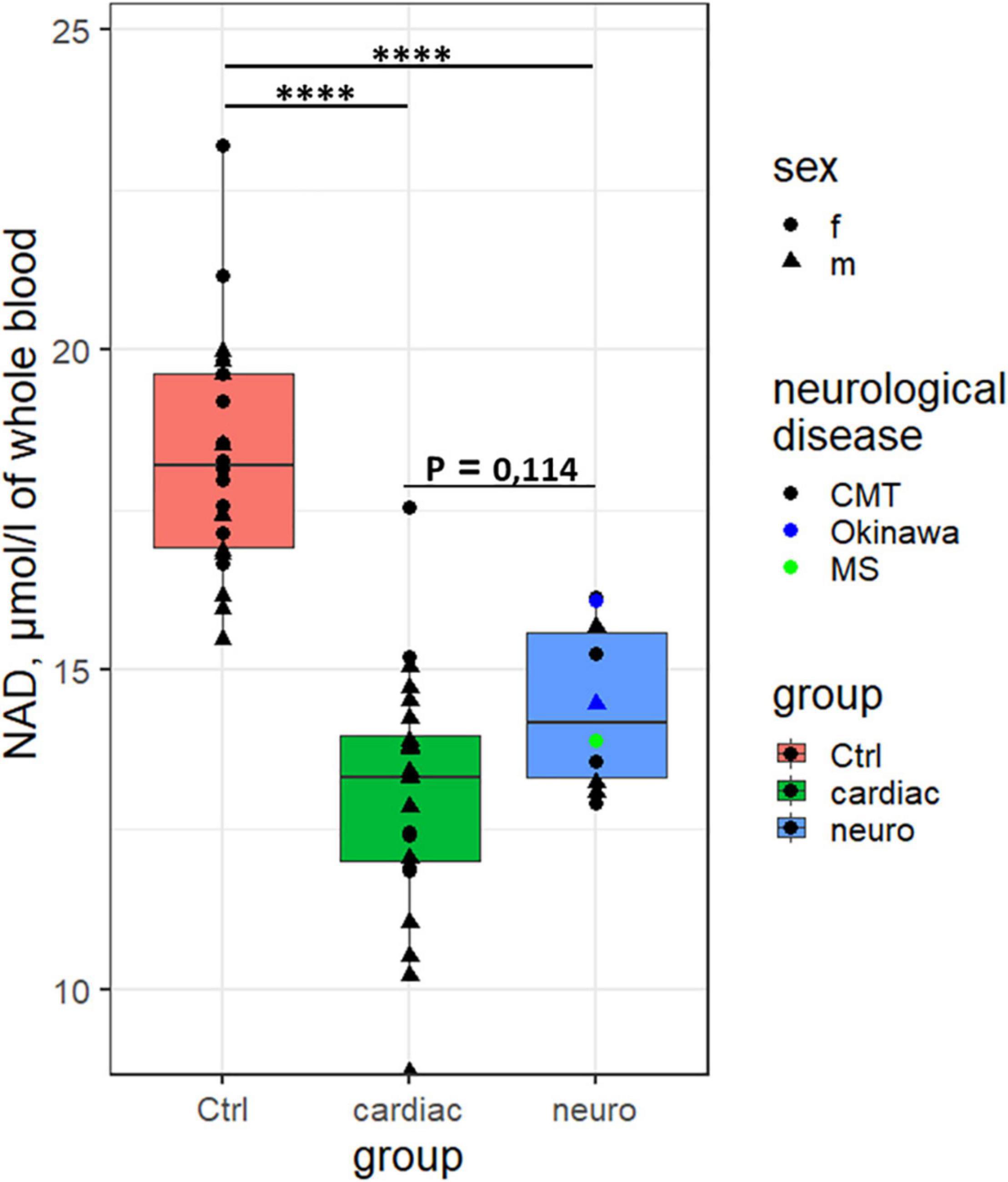
Figure 1. Differences in the concentration of NAD+ in the whole blood between the control subjects (n = 22), the cardiological (n = 24, excluding the four outliers) and neurological (n = 10) patients. ****p < 0.0001.
As the NAD+ levels alone cannot unambiguously discriminate the cardiological and neurological patients (p = 0.1, Figure 1), we have attempted to increase the discriminating power of the NAD+ assay by finding potentially different relationships between the NAD+ levels and associated blood markers in different pathologies. Correlations of the blood levels of NAD+ to the available parameters of the clinical and biochemical blood analyses of the studied cohorts indicate that the control correlations are always well separated from those in the patients, while the difference between the cardiological and neurological patients is not so strong (Supplementary Figure 3 and Supplementary Table 2). However, the NAD+ dependence on Na+ ions is opposite in the control subjects and neurological patients (Supplementary Figure 3 and Supplementary Table 2), complemented by statistically significant group differences between the median values of Na+ ions.
Analysis of the ratios of NAD+ levels to each of the available parameters, inherent in a specific blood sample, reveals that some of the ratios may be used for a better discrimination between the neurological and cardiological patients (Table 2). Figure 2 presents such ratios for the three parameters assayed in the blood along with NAD+. While one of the parameters (creatinine) is characterized by statistically significant group differences in its median values, the medians of the two other parameters (MCV, mean corpuscular volume, and K+) do not significantly differ between the groups (Supplementary Table 1). It is worth noting in this regard that the ratios presented in Figure 2 are determined in each patient. Therefore, they manifest the coupled changes better than the overall median values of the parameters. This is exemplified in Figure 2 by the statistically significant differences between the cardiological and neurological patients in their NAD+ ratios to creatinine, MCV and K+ ions. These ratios show a higher statistical significance of the differences between the neurological and cardiological patients, than NAD+ levels alone (Figures 1, 2). Thus, in addition to the blood levels of NAD+, taking into account the coupled changes in other parameters may increase the diagnostic power of the NAD+ assays.
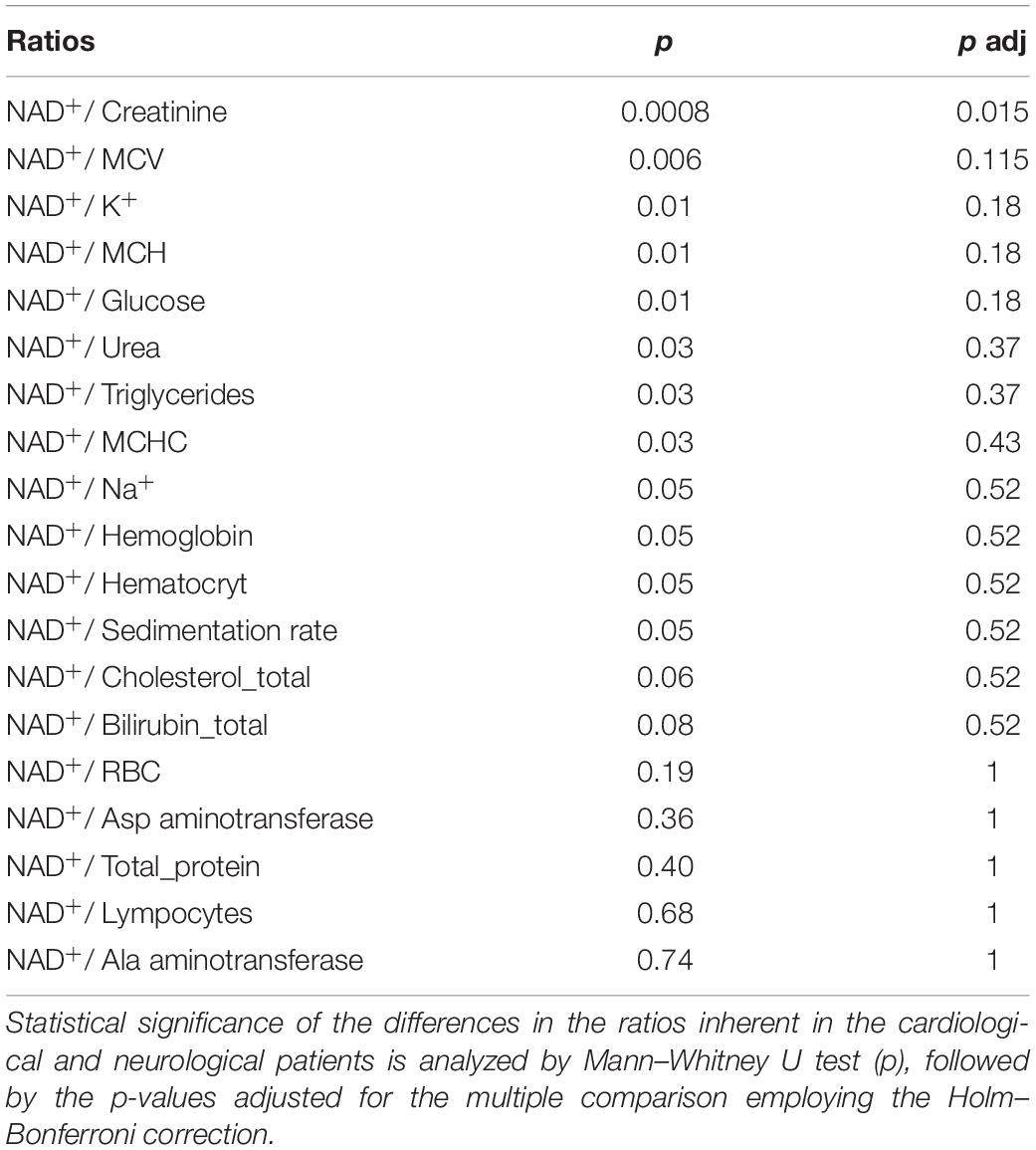
Table 2. Discriminating power of the ratios of NAD+ content to other blood parameters.
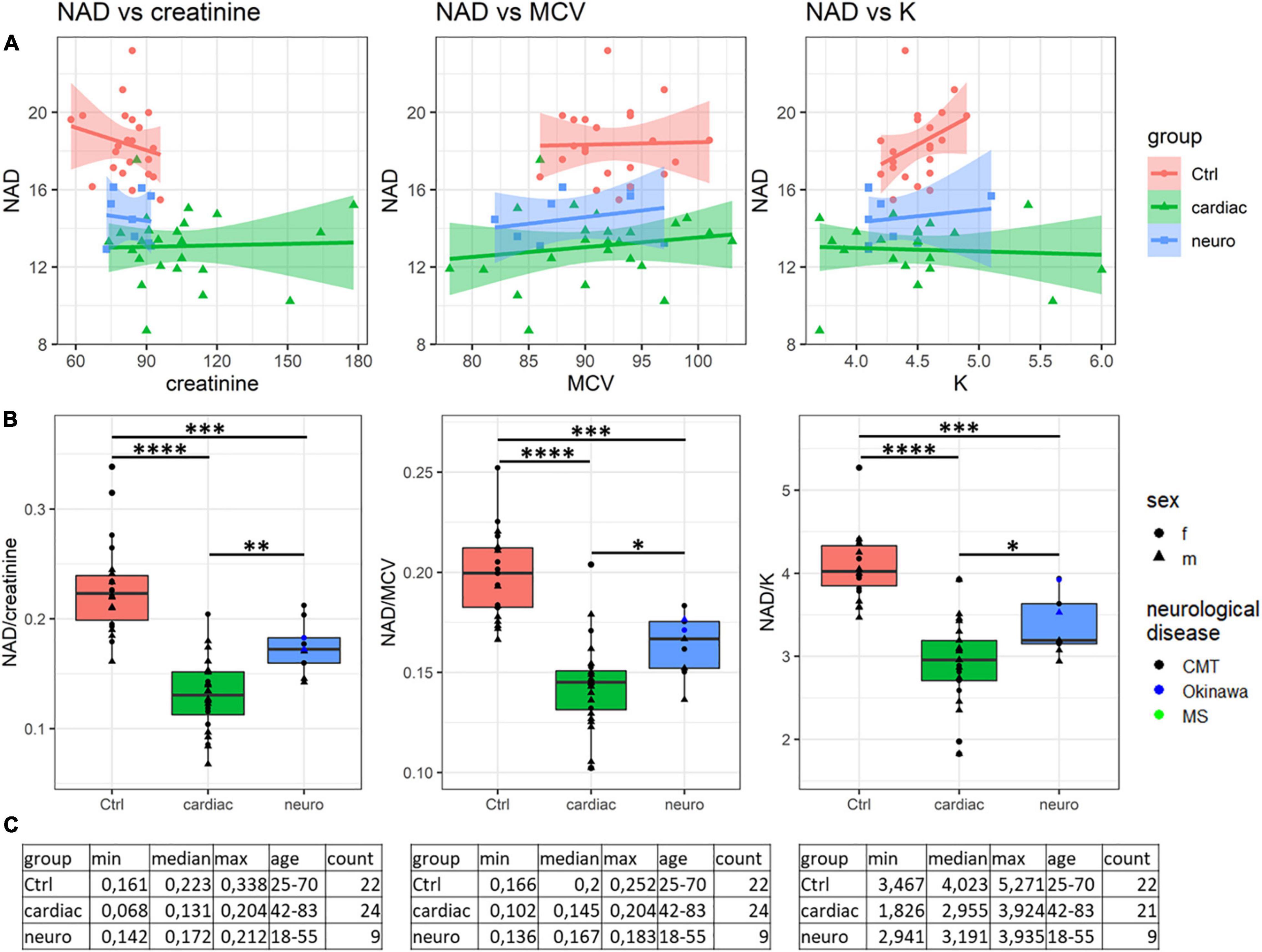
Figure 2. Analysis of discriminating potential of the coupled variations in the NAD+ concentrations and other parameters of the blood. (A) Correlations of the NAD+ concentrations with the creatinine level, mean corpuscular volume (MCV) and K+ ions. (B) Significance of the differences between the NAD+ ratios to creatinine, MCV and K+ ions, determined for each of the studied samples. (C) Tabular presentation of the ratios used in panel (B). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < <0.0001.
Discussion
The NAD+ values determined in our study for the healthy volunteers are in good accordance with those of independent studies, where NAD+ in human blood is measured by mass spectrometry or NMR (Supplementary Table 3). Other enzymatic assays of NAD+ are known to employ lactate dehydrogenase or alcohol dehydrogenase. Unlike our FDH-based assay, these reactions are reversible. The reversibility makes such tests prone to conditional problems when established equilibria interfere with the completion of NAD+ transformation to NADH. In modern commercial kits, this is overcome by shifting an equilibrium through coupled reaction(s), usually including the reduction of formazan dyes, such as MTT. However, introduction of each additional coupled reaction makes the test prone to additional artifacts. Moreover, the artifacts are not possible to control when using commercial tests whose components are not disclosed. Furthermore, without additional procedures, the MTT-based tests, employing redox transformation of nicotinamide adenine dinucleotide, do not discriminate between the oxidized (NAD+) and reduced (NADH) forms of the dinucleotide. In particular, this is characteristic of the enzymatic studies mentioned in Supplementary Table 3. Although NADH and the redox ratio of NADH to NAD+ are important metabolic indicators, additional to NAD+ alone, the different chemical stability of NADH and NAD+ poses challenges for simultaneous quantification of both indicators in the same blood extract in clinical settings.
According to some estimates, the blood levels of NADH are 5–10 times lower than those of NAD+ (50), and thus should not contribute more than 20% to the total NAD pool, comprising both the oxidized and reduced dinucleotide. However, a two-fold variation in the range of the NAD pool, determined by the MTT-employing enzymatic tests (Supplementary Table 3), agrees with the notion that such tests are prone to various artifacts (32, 44, 45, 51). More importantly, comparison of the MTT-based tests relies on the arbitrary reaction time rather than the end-point titration, used in our FDH-based assay. In this case, if the sample composition differs, e.g., due to the various prescription drugs in different patients, the drugs may affect the MTT reduction, resulting in an artifactual change of the determined NAD pool. That is, the observed change may be related not to the NAD(H) content, but to the MTT reduction rate. Ascorbic acid, vitamin A, cellular sulfhydryl-containing compounds, such as reduced glutathione and coenzyme A, are known to reduce MTT to formazan (52–55). Varied content of these and other compounds in blood of different patients would thus contribute to the NAD-independent differences in the MTT-reduction. As an example, the MTT-based NADH assay in human erythrocytes shows diurnal oscillations, but they are coupled to changes in the cellular redox metabolism, including the changed expression of a highly abundant thiol-comprising redox enzyme, peroxiredoxin (56). These diurnal changes in the cellular redox metabolism raise questions regarding the nature of the MTT-detected changes, which do not necessarily reflect the NAD content. In case of the FDH reaction, the inhibitors or activators may also affect the reaction rate. However, in contrast to the MTT-based assay, the assay employing FDH is not dependent on the reaction rate or time, but based on the end-point titration of NADH itself, characterizing the complete transformation of NAD+ to NADH. Therefore, significant effects of different drugs prescribed to patients on the FDH-based NAD+ assay are not expected.
Apart from the high resistance to the artifacts discussed above, sensitivity of our fluorometric NAD+ assay corresponds to the detection limit of 5 pmoles NAD+ per 50 uL sample, or 100 nM NAD+, that is better than the detection limit of 400 nM NAD+, reported by Abcam1 or Biovision2 for the MTT-based assays.
It is worth noting that the tissue NAD+ or total NAD content may be subject to circadian changes, although the oscillation periods do not coincide in different studies (57–59). Probably, this poor coincidence is related to the fact that even in the liver tissue, where the NAD+ oscillation amplitudes are high, they are mostly within the standard errors of the NAD assays. Nevertheless, to exclude additional source of variations, the blood samples for our analyses are taken in the morning. Using the FDH-based test to detect potential circadian changes in NAD+ content in human blood may open new perspectives for chronobiological implications in medicine.
Decreased NAD+ content in demyelinating neuropathies is known from cellular and animal models of the diseases. For instance, rapid NAD+ drop upon Wallerian (injury-induced) degeneration of axons (29, 60), neurites (61) and in dissected nerves (62) suggests diminished NAD+ level in Charcot-Marie-Tooth disease and other peripheric neuropathies. Thus, our finding of decreased NAD+ levels in patients with demyelinating neuropathies is in good accordance with the model studies. Interestingly, the NAD pool is depleted in neurons exposed to toxic prion proteins, or in models of protein misfolding in Alzheimer’s and Parkinson’s diseases (63). Reduced level of total NAD in the brain tissue is observed in a mouse model of cerebellar ataxia (64), but the depletion does not occur in astrocytes or brain homogenates of scrapie-infected mice (63).
Similarly, decreased content of total NAD in myocardium is shown in mouse models of dilated cardiomyopathy (65) and failing heart (66). These model studies are in good accord with our finding of decreased levels of NAD+ in the whole human blood of cardiological patients (Figure 1). As mentioned in Introduction, the old (75–101 years) patients hospitalized for decompensated heart failure have a lower level of total NAD, compared to the younger (19–68 years) controls (11). In view of the published results on decreased NAD pool in cardiological pathologies, our finding of decreased NAD+ in the blood of cardiological patients suggests an impairment in NAD biosynthesis rather than increased NADH/NAD+ ratio in these patients. However, the abovementioned comparison of the MTT-based quantification of total NAD employs the cardiological patients and control subjects of different ages. Hence, further studies are needed to decipher the molecular mechanisms underlying decreases in NAD+ upon the cardio- and neuropathologies.
Thus, to the best of our knowledge, declines in the blood levels of NAD+ in the neurological and cardiological patients, shown in our work (Figure 1 and Supplementary Figure 2), have not been demonstrated before, although decreased NAD levels are known in healthy aging (16–18). Our data support this finding, as we also observe a slight decline in the blood NAD+ content in healthy subjects with age (Supplementary Figure 3). For the future studies it is important to note, however, that this and other correlations may be changed in different cohorts, manifesting certain specificity of pathophysiological changes in patients (Supplementary Figure 3 and Supplementary Table 2). Owing to this, the discriminating power of NAD+ as a clinical indicator of specific pathologies or as a risk factor of their development may be increased by taking into account potential associations of the NAD+ levels with other clinically relevant parameters (Figure 2 and Table 2). Further studies in these directions may be performed using the FDH-based NAD+ assay, developed in the current work. Remarkably, the associations between perturbed levels of NAD+ and creatinine, observed in our study (Figure 2), are also known in animals with acute and chronic kidney disease (67) and in patients with pellagra (68).
Supplementation with vitamin B3 (NAD precursor) to subjects under increased risks of neurological, cardiological and other disorders is currently considered as an efficient therapeutic strategy (69). Our easy NAD+ test may be applied to reveal the best therapies to increase the NAD+ level for protection from the age-related pathologies.
As a result, the proposed FDH-based assay of NAD+ in human blood may be useful for a more precise diagnosis of different pathologies and associated risks.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Independent Ethics Committee of M.F. Vladimirsky Moscow Regional Research and Clinical Institute. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
NB and AA performed the NAD+ measurement and quantification. NB and OS provided the blood samples and medical histories analyses. LS purified the recombinant FDH. VT supervised the FDH production. LZ, AP, and VB analyzed the results. VB and AP wrote the manuscript draft. VB, AA, and AT edited the manuscript. All authors read and agreed on the submitted version of the manuscript.
Funding
This work was supported by joint grant from Russian Foundation for Basic Research (grant no. 20-54-7804) and National Research Council of Italy (grant CUP B85F20002990005).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.886485/full#supplementary-material
Footnotes
- ^ https://www.abcam.com/nadnadh-assay-kit-colorimetric-ab65348.html
- ^ https://www.biovision.com/nad-nadh-quantitation-colorimetric-kit.html
References
1. Moniot S, Weyand M, Steegborn C. Structures, substrates, and regulators of mammalian sirtuins - opportunities and challenges for drug development. Front Pharmacol. (2012) 3:16. doi: 10.3389/fphar.2012.00016
2. Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. (2014) 24:464–71. doi: 10.1016/j.tcb.2014.04.002
3. Manjula R, Anuja K, Alcain FJ. SIRT1 and SIRT2 activity control in neurodegenerative Diseases. Front Pharmacol. (2020) 11:585821. doi: 10.3389/fphar.2020.585821
4. Bai P, Canto C, Brunyanszki A, Huber A, Szanto M, Cen Y, et al. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab. (2011) 13:450–60. doi: 10.1016/j.cmet.2011.03.013
5. Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. (2011) 13:461–8. doi: 10.1016/j.cmet.2011.03.004
6. Lee HC. Structure and enzymatic functions of human CD38. Mol Med. (2006) 12:317–23. doi: 10.2119/2006â00086.Lee
7. Ortolan E, Augeri S, Fissolo G, Musso I, Funaro A. CD157: from immunoregulatory protein to potential therapeutic target. Immunol Lett. (2019) 205:59–64. doi: 10.1016/j.imlet.2018.06.007
8. Sahar S, Nin V, Barbosa MT, Chini EN, Sassone-Corsi P. Altered behavioral and metabolic circadian rhythms in mice with disrupted NAD+ oscillation. Aging. (2011) 3:794–802. doi: 10.18632/aging.100368
9. Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One. (2012) 7:e42357. doi: 10.1371/journal.pone.0042357
10. Clement J, Wong M, Poljak A, Sachdev P, Braidy N. The plasma NAD(+) metabolome is dysregulated in “Normal” aging. Rejuvenation Res. (2019) 22:121–30. doi: 10.1089/rej.2018.2077
11. Breton M, Costemale-Lacoste JF, Li Z, Lafuente-Lafuente C, Belmin J, Mericskay M. Blood NAD levels are reduced in very old patients hospitalized for heart failure. Exp Gerontol. (2020) 139:111051. doi: 10.1016/j.exger.2020.111051
12. Gasparrini M, Sorci L, Raffaelli N. Enzymology of extracellular NAD metabolism. Cell Mol Life Sci. (2021) 78:3317–31. doi: 10.1007/s00018-020-03742-1
13. Seyedsadjadi N, Berg J, Bilgin AA, Braidy N, Salonikas C, Grant R. High protein intake is associated with low plasma NAD+ levels in a healthy human cohort. PLoS One. (2018) 13:e0201968. doi: 10.1371/journal.pone.0201968
14. Grant R, Berg J, Mestayer R, Braidy N, Bennett J, Broom S, et al. A pilot study investigating changes in the human plasma and urine NAD+ metabolome during a 6 hour intravenous infusion of NAD. Front Aging Neurosci. (2019) 11:257. doi: 10.3389/fnagi.2019.00257
15. O’Reilly T, Niven DF. Levels of nicotinamide adenine dinucleotide in extracellular body fluids of pigs may be growth-limiting for Actinobacillus pleuropneumoniae and Haemophilus parasuis. Can J Vet Res. (2003) 67: 229–31.
16. Canto C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. (2015) 22:31–53. doi: 10.1016/j.cmet.2015.05.023
17. McReynolds MR, Chellappa K, Baur JA. Age-related NAD(+) decline. Exp Gerontol. (2020) 134:110888. doi: 10.1016/j.exger.2020.110888
18. Peluso A, Damgaard MV, Mori MAS, Treebak JT. Age-dependent decline of NAD(+)-Universal truth or confounded consensus? Nutrients. (2021) 14:101. doi: 10.3390/nu14010101
19. Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci USA. (2015) 112:2876–81. doi: 10.1073/pnas.1417921112
20. Bagga P, Hariharan H, Wilson NE, Beer JC, Shinohara RT, Elliott MA, et al. Single-Voxel (1) H MR spectroscopy of cerebral nicotinamide adenine dinucleotide (NAD(+)) in humans at 7T using a 32-channel volume coil. Magn Reson Med. (2020) 83:806–14. doi: 10.1002/mrm.27971
21. Zhou CC, Yang X, Hua X, Liu J, Fan MB, Li GQ, et al. Hepatic NAD(+) deficiency as a therapeutic target for non-alcoholic fatty liver disease in ageing. Br J Pharmacol. (2016) 173:2352–68. doi: 10.1111/bph.13513
22. Chaleckis R, Murakami I, Takada J, Kondoh H, Yanagida M. Individual variability in human blood metabolites identifies age-related differences. Proc Natl Acad Sci USA. (2016) 113:4252–9. doi: 10.1073/pnas.1603023113
23. Minhas PS, Liu L, Moon PK, Joshi AU, Dove C, Mhatre S, et al. Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat Immunol. (2019) 20:50–63. doi: 10.1038/s41590-018-0255-3
24. Yaku K, Okabe K, Hikosaka K, Nakagawa T. NAD metabolism in cancer therapeutics. Front Oncol. (2018) 8:622. doi: 10.3389/fonc.2018.00622
25. Navas LE, Carnero A. NAD(+) metabolism, stemness, the immune response, and cancer. Signal Transduct Target Ther. (2021) 6:2. doi: 10.1038/s41392-020-00354-w
26. Okabe K, Yaku K, Tobe K, Nakagawa T. Implications of altered NAD metabolism in metabolic disorders. J Biomed Sci. (2019) 26:34. doi: 10.1186/s12929-019-0527-8
27. Fan L, Cacicedo JM, Ido Y. Impaired nicotinamide adenine dinucleotide (NAD(+)) metabolism in diabetes and diabetic tissues: implications for nicotinamide-related compound treatment. J Diabetes Investig. (2020) 11:1403–19. doi: 10.1111/jdi.13303
28. Yaku K, Okabe K, Nakagawa T. NAD metabolism: implications in aging and longevity. Ageing Res Rev. (2018) 47:1–17. doi: 10.1016/j.arr.2018.05.006
29. Moss KR, Hoke A. Targeting the programmed axon degeneration pathway as a potential therapeutic for charcot-marie-tooth disease. Brain Res. (2020) 1727:146539. doi: 10.1016/j.brainres.2019.146539
30. Hikosaka K, Yaku K, Okabe K, Nakagawa T. Implications of NAD metabolism in pathophysiology and therapeutics for neurodegenerative diseases. Nutr Neurosci. (2021) 24:371–83. doi: 10.1080/1028415X.2019.1637504
31. Gerner RR, Klepsch V, Macheiner S, Arnhard K, Adolph TE, Grander C, et al. NAD metabolism fuels human and mouse intestinal inflammation. Gut. (2018) 67:1813–23. doi: 10.1136/gutjnl-2017-314241
32. Artiukhov AV, Pometun AA, Zubanova SA, Tishkov VI, Bunik VI. Advantages of formate dehydrogenase reaction for efficient NAD(+) quantification in biological samples. Anal Biochem. (2020) 603:113797. doi: 10.1016/j.ab.2020.113797
33. Tishkov VI, Popov VO. Protein engineering of formate dehydrogenase. Biomol Eng. (2006) 23:89–110. doi: 10.1016/j.bioeng.2006.02.003
34. Gao X, Yang S, Zhao C, Ren Y, Wei D. Artificial multienzyme supramolecular device: highly ordered self-assembly of oligomeric enzymes in vitro and in vivo. Angew Chem Int Ed Engl. (2014) 53:14027–30. doi: 10.1002/anie.201405016
35. Jiang W, Fang BS. Construction and evaluation of a novel bifunctional phenylalanine-formate dehydrogenase fusion protein for bienzyme system with cofactor regeneration. J Ind Microbiol Biotechnol. (2016) 43:577–84. doi: 10.1007/s10295-016-1738-6
36. Kokorin A, Parshin PD, Bakkes PJ, Pometun AA, Tishkov VI, Urlacher VB. Genetic fusion of P450 BM3 and formate dehydrogenase towards self-sufficient biocatalysts with enhanced activity. Sci Rep. (2021) 11:21706. doi: 10.1038/s41598-021-00957-5
37. Seelbach K, Riebel B, Hummel W, Kula MR, Tishkov VI, Egorov AM, et al. A novel, efficient regenerating method of NADPH using a new formate dehydrogenase. Tetrahedron Lett. (1996) 37:1377–80. doi: 10.1016/0040-4039(96)00010-x
38. Jiang W, Xu CZ, Jiang SZ, Zhang TD, Wang SZ, Fang BS. Establishing a mathematical equations and improving the production of L-tert-leucine by uniform design and regression analysis. Appl Biochem Biotechnol. (2017) 181:1454–64. doi: 10.1007/s12010-016-2295-1
39. Tao R, Jiang Y, Zhu F, Yang S. A one-pot system for production of L-2-aminobutyric acid from L-threonine by L-threonine deaminase and a NADH-regeneration system based on L-leucine dehydrogenase and formate dehydrogenase. Biotechnol Lett. (2014) 36:835–41. doi: 10.1007/s10529-013-1424-y
40. Cakar MM, Ruupunen J, Mangas-Sanchez J, Birmingham WR, Yildirim D, Turunen O, et al. Engineered formate dehydrogenase from Chaetomium thermophilum, a promising enzymatic solution for biotechnical CO2 fixation. Biotechnol Lett. (2020) 42:2251–62. doi: 10.1007/s10529-020-02937-7
41. Alpdagtas S, Turunen O, Valjakka J, Binay B. The challenges of using NAD(+)-dependent formate dehydrogenases for CO2 conversion. Crit Rev Biotechnol. (2021). [Online ahead of print]. doi: 10.1080/07388551.2021.1981820
42. Choe MS, Choi S, Kim SY, Back C, Lee D, Lee HS, et al. A hybrid Ru(II)/TiO2 catalyst for steadfast photocatalytic CO2 to CO/formate conversion following a molecular catalytic route. Inorg Chem. (2021) 60:10235–48. doi: 10.1021/acs.inorgchem.1c00615
43. Unlu A, Duman-Ozdamar ZE, Caloglu B, Binay B. Enzymes for efficient CO2 conversion. Protein J. (2021) 40:489–503. doi: 10.1007/s10930-021-10007-8
44. Riss TL, Moravec RA, Niles AL, Duellman S, Benink HA, Worzella TJ, et al. Cell Viability Assays. In: Markossian S, Grossman A, Brimacombe K, Arkin M, Auld D, Austin CP editors. Assay Guidance Manual. Bethesda, MD: Eli Lilly & Company and the National Center for Advancing Translational Sciences (2004).
45. Ghasemi M, Turnbull T, Sebastian S, Kempson I. The MTT assay: utility, limitations, pitfalls, and interpretation in bulk and single-cell analysis. Int J Mol Sci. (2021) 22:12827. doi: 10.3390/ijms222312827
46. Pometun AA, Kleymenov SY, Zarubina SA, Kargov IS, Parshin PD, Sadykhov EG, et al. Comparison of thermal stability of new formate dehydrogenases by differential scanning calorimetry. Mosc Univ Chem Bull. (2018) 73:80–4. doi: 10.3103/s002713141802013x
47. Tishkov VI, Pometun AA, Stepashkina AV, Fedorchuk VA, Zarubina SA I, Kargov S, et al. Rational design of practically important enzymes. Mosc Univ Chem Bull. (2018) 73:1–6. doi: 10.3103/s0027131418020153
48. Alekseeva AA, Fedorchuk VV, Zarubina SA, Sadykhov EG, Matorin AD, Savin SS, et al. The role of ala198 in the stability and coenzyme specificity of bacterial formate dehydrogenases. Acta Nat. (2015) 7:60–9. doi: 10.32607/20758251-2015-7-1-60-69
49. Ksenofontov AL, Boyko AI, Mkrtchyan GV, Tashlitsky VN, Timofeeva AV, Graf AV, et al. Analysis of free amino acids in mammalian brain homogenates. Biochemistry. (2017) 82:1183–92. doi: 10.1134/s000629791710011x
50. Nagana Gowda GA, Raftery D. Whole blood metabolomics by (1)H NMR spectroscopy provides a new opportunity to evaluate coenzymes and antioxidants. Anal chem. (2017) 89:4620–7. doi: 10.1021/acs.analchem.7b00171
51. Aleshin VA, Artiukhov AV, Oppermann H, Kazantsev AV, Lukashev NV, Bunik VI. Mitochondrial impairment may increase cellular NAD(P)H: resazurin oxidoreductase activity, perturbing the NAD(P)H-based viability assays. Cells. (2015) 4:427–51. doi: 10.3390/cells4030427
52. Pagliacci MC, Spinozzi F, Migliorati G, Fumi G, Smacchia M, Grignani F, et al. Genistein inhibits tumour cell growth in vitro but enhances mitochondrial reduction of tetrazolium salts: a further pitfall in the use of the MTT assay for evaluating cell growth and survival. Eur J Cancer. (1993) 29A:1573–7. doi: 10.1016/0959-8049(93)90297-s
53. Chakrabarti R, Kundu S, Kumar S, Chakrabarti R. Vitamin A as an enzyme that catalyzes the reduction of MTT to formazan by vitamin C. J Cell Biochem. (2000) 80:133–8. doi: 10.1002/1097-4644(20010101)80:1<133::aid-jcb120>3.0.co;2-t
54. Bernas T, Dobrucki J. Mitochondrial and nonmitochondrial reduction of MTT: interaction of MTT with TMRE, JC-1, and NAO mitochondrial fluorescent probes. Cytometry. (2002) 47:236–42. doi: 10.1002/cyto.10080
55. Ulukaya E, Colakogullari M, Wood EJ. Interference by anti-cancer chemotherapeutic agents in the MTT-tumor chemosensitivity assay. Chemotherapy. (2004) 50:43–50. doi: 10.1159/000077285
56. O’Neill JS, Reddy AB. Circadian clocks in human red blood cells. Nature. (2011) 469:498–503. doi: 10.1038/nature09702
57. Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. (2009) 324:651–4. doi: 10.1126/science.1171641
58. Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science. (2013) 342:1243417. doi: 10.1126/science.1243417
59. Levine DC, Hong H, Weidemann BJ, Ramsey KM, Affinati AH, Schmidt MS, et al. NAD(+) controls circadian reprogramming through PER2 nuclear translocation to counter aging. Mol Cell. (2020) 78:835–849.e7. doi: 10.1016/j.molcel.2020.04.010
60. Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, et al. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. (2005) 170:349–55. doi: 10.1083/jcb.200504028
61. Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science. (2015) 348:453–7. doi: 10.1126/science.1258366
62. Gilley J, Orsomando G, Nascimento-Ferreira I, Coleman MP. Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep. (2015) 10:1974–81. doi: 10.1016/j.celrep.2015.02.060
63. Zhou M, Ottenberg G, Sferrazza GF, Hubbs C, Fallahi M, Rumbaugh G, et al. Neuronal death induced by misfolded prion protein is due to NAD+ depletion and can be relieved in vitro and in vivo by NAD+ replenishment. Brain. (2015) 138:992–1008. doi: 10.1093/brain/awv002
64. Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H, et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. (2014) 157:882–96. doi: 10.1016/j.cell.2014.03.026
65. Diguet N, Trammell SAJ, Tannous C, Deloux R, Piquereau J, Mougenot N, et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation. (2018) 137:2256–73. doi: 10.1161/circulationaha.116.026099
66. Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activity. J Biol Chem. (2005) 280:43121–30. doi: 10.1074/jbc.M506162200
67. Faivre A, Katsyuba E, Verissimo T, Lindenmeyer M, Rajaram RD, Naesens M, et al. Differential role of nicotinamide adenine dinucleotide deficiency in acute and chronic kidney disease. Nephrol Dial Transplant. (2021) 36:60–8. doi: 10.1093/ndt/gfaa124
68. Creeke PI, Dibari F, Cheung E, van den Briel T, Kyroussis E, Seal AJ. Whole blood NAD and NADP concentrations are not depressed in subjects with clinical pellagra. J Nutr. (2007) 137:2013–7. doi: 10.1093/jn/137.9.2013
69. Airhart SE, Shireman LM, Risler LJ, Anderson GD, Nagana Gowda GA, Raftery D, et al. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS One. (2017) 12:e0186459. doi: 10.1371/journal.pone.0186459
Keywords: NAD+ in human blood, formate dehydrogenase, cardiac patient, neurological patient, metabolic markers, Charcot-Marie-Tooth disease
Citation: Balashova NV, Zavileyskiy LG, Artiukhov AV, Shaposhnikov LA, Sidorova OP, Tishkov VI, Tramonti A, Pometun AA and Bunik VI (2022) Efficient Assay and Marker Significance of NAD+ in Human Blood. Front. Med. 9:886485. doi: 10.3389/fmed.2022.886485
Received: 28 February 2022; Accepted: 07 April 2022;
Published: 19 May 2022.
Edited by:
Matteo Becatti, University of Firenze, ItalyReviewed by:
Baris Binay, Gebze Technical University, TurkeyMathias Mericskay, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Mirella L. Meyer-Ficca, Utah State University, United States
Copyright © 2022 Balashova, Zavileyskiy, Artiukhov, Shaposhnikov, Sidorova, Tishkov, Tramonti, Pometun and Bunik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victoria I. Bunik, YnVuaWtAYmVsb3plcnNreS5tc3UucnU=