 Wei Jiang
Wei Jiang Xuan-Yu Tan2
Xuan-Yu Tan2 Ming Dong
Ming Dong- 1Department of Neurology and Neuroscience Center, The First Hospital of Jilin University, Changchun, China
- 2Department of Neurosurgery, The First Hospital of Jilin University, Changchun, China
- 3Department of Emergency, The First Hospital of Jilin University, Changchun, China
- 4Department of Ophthalmology, The Second Hospital of Jilin University, Changchun, China
Neuropathic pain (NP), caused by an injury or a disease affecting the somatosensory nervous system of the central and peripheral nervous systems, has become a global health concern. Recent studies have demonstrated that epigenetic mechanisms are among those that underlie NP; thus, elucidating the molecular mechanism of DNA methylation is crucial to discovering new therapeutic methods for NP. In this review, we first briefly discuss DNA methylation, demethylation, and the associated key enzymes, such as methylases and demethylases. We then discuss the relationship between NP and DNA methylation, focusing on DNA methyltransferases including methyl-CpG-binding domain (MBD) family proteins and ten-eleven translocation (TET) enzymes. Based on experimental results of neuralgia in animal models, the mechanism of DNA methylation-related neuralgia is summarized, and useful targets for early drug intervention in NP are discussed.
Introduction
The latest and now widely accepted definition of neuropathic pain (NP) is pain caused by an injury or a disease of the somatosensory system (1). NP is a chronic disease with complex clinical symptoms, poor prognosis, and an increasing disease burden. Most importantly, treatment options are extremely limited, and some patients develop resistance to drugs of opioid analgesics (2). Peripheral nerve injury or disease can cause a series of NP symptoms including spontaneous pain, hyperalgesia, and allodynia. Allodynia refers to pain caused by stimuli that do not usually cause pain, whereas hyperalgesia refers to increased pain caused by stimuli that cause pain (3). Although distinct neuropathic syndromes induce pain, their clinical symptoms are similar. NP is divided into peripheral and central NP based on the pathogeny. Cellular and molecular changes in NP are associated with different pain pathways, among which epigenetic studies primarily focused on peripheral nerves, dorsal root ganglion (DRG), and dorsal horn (4).
The DRG is located at the junction of the peripheral and central nervous systems (5). In peripheral nerve injury, nociception is conveyed via primary sensory neurons in the DRG and back to secondary sensory neurons (6). It is well-known that neuropathic pain is related to hyperexcitation and internal firing of DRG neurons. There are two types of DRG neurons, type A and type B. Type A DRG neurons are large and are responsible for touch, vibration, and proprioception, and type B neurons are small and are responsible for nociception (7). DRG generates the fibers that convey information, including the activation of nociceptors from the skin, muscles, and joints to the spinal cord. With continuous nociceptive input, central sensitization of the spinal cord (i.e., increased reactivity of nociceptive neurons in the central nervous system to normal or subliminal afferent signals) plays an important role in pain perception (8). In response to peripheral afferent tissue injury, signals from the DRG and spinal cord activate glial cells, increase the expression of proinflammatory factors (IL-6, IL-1β, and TNF-α), increase the expression of receptors including nerve growth factor and TRPV1, alter gene expression, and decrease the expression of ion channels (i.e., sodium, voltage-gated potassium, and calcium channels; Figure 1) (9). Notably, voltage-gated potassium channels play an important role in inducing spontaneous ectopic discharge, the hyperexcitability of neurons, and neuropathic pain-like symptoms, which are described in the following sections.
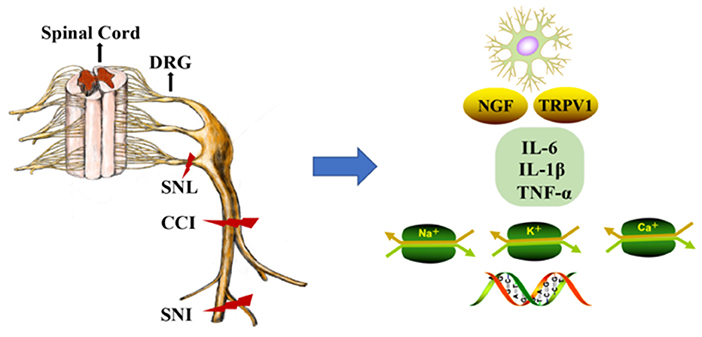
Figure 1. DNA methylation in the mechanism of neuropathic pain (NP). The dorsal root ganglion (DRG) and spinal cord produce glial cell responses, stimulating inflammatory cytokines, nerve growth factors, gene expression, and ion channels under the environment of peripheral afferent fiber injury. CCI, chronic constriction injury; SNL, spinal nerve ligation; SNI, spared nerve injury; DRG, dorsal root ganglion; NGF, nerve growth factors; Na+, Na+ channels; K+, K+ channels; Ca+, Ca+ channels.
Epigenetic processes include DNA methylation, covalent histone acetylation, and non-coding RNA expression (10). DNA is wrapped around a histone octamer consisting of dimers of histones H3, H4, H2A, and H2B. Cells regulate gene expression and the structure and function of chromatin through post-transcriptional modification of the N-terminal histone tails of nucleosomes (11, 12). Histone acetylation occurs on lysine residues and is catalyzed by the histone acetyl transferase (HAT) family, leading to transcriptional activation. Deacetylation is performed by the histone deacetylase family of enzymes (HDACs) and is involved in transcriptional inhibition (13). Methylated amino acid residues determine the inhibition or activation of gene transcription. For example, methylation of Lys9 or Lys27 of histone H3 is usually associated with gene suppression, while methylation of Lys4, Lys36, or Lys79 of H3 is usually associated with gene activation (14). MicroRNAs (miRNAs) are endogenous, non-coding functional RNAs that range from 19 to 24 nucleotides in size. They bind target mRNAs, inhibiting translation and leading to the downregulation of target proteins (15). Emerging evidence has indicated that histone acetylation and deacetylation, DNA methylation, and the regulation of miRNA are closely related to NP (16–20).
NP is involved in the activation of glial cells, the triggering of inflammatory cascades, abnormal neuronal firing, and ion channel imbalance in the central and peripheral nervous systems. Epigenetics can modulate pain responses by regulating inflammation via ion channels, receptors, and neurotransmitters. A growing number of studies have demonstrated that DNA methylation, histone acetylation, deacetylation, histone methylation, and miRNAs regulate pain through inflammatory responses (19–25). These studies suggest that harmful stimuli drive the activation of glial cells and are involved in epigenetic modification of NP. Ion-channel imbalances including sodium channels and voltage-gated calcium channels, often accompanied by spontaneous ectopic discharge and hyperexcitation, contribute to the occurrence of NP in DRG and spinal cord neurons. Furthermore, increasing evidence suggests that epigenetic modifications contribute to peripheral nerve injury by modifying ion channel status, and histone acetylation and methylation have been shown to reduce pain responses via ion-channel regulation (17, 26, 27).
As a more stable epigenetic modification, DNA methylation can silence or downregulate promoters or enhancers (28). Such distinct gene expression profiles could influence pain and analgesia. In recent years, some studies have demonstrated that DNA methylation is associated with the pathology of NP (29, 30). Although studies on the mechanism of DNA methylation account for an increasing proportion in NP research, the findings remain limited. In this review, we systematically describe DNA methylation and demethylation, briefly introduce the possible mechanism underlying DNA methylation-induced NP from the perspective of studies with experimental animal NP models, and aim to reveal a reliable future therapeutic target for NP.
DNA Methylation and NP
DNA Methylation, DNMTs, and Methyl-CpG-Binding Domain (MBD) Proteins
DNA methylation is important for regulating tissue-specific gene expression and may affect gene activity variably in different genomic regions. DNA methylation enables chromatin to maintain its inactive state in the following two cases: (i) silencing elements potentially harmful to DNA, such as transposons, viral DNA, and genes that should not be expressed; and (ii) avoiding the binding of transcription factors to specific sites in promoter regions or even by allowing transcription repressor binding (31). In mammals, DNA methylation is catalyzed by DNA methyltransferases (DNMTs), which predominantly add methyl groups from the S-adenosyl-l-methionine to the carbon-5 position of cytosine bases [5-methylcytosine (5mC)] mainly located at cytosine-phosphate-guanosine (CpG) islands (32, 33). Of note, the promoters of most genes, especially housekeeping genes, are located in CpG islands (34). CpG islands, especially those associated with promoters, are highly conserved between mice and humans, suggesting that these regions have important functions. Peripheral nerve injury reduces DNA methylation in the prefrontal cortex (PFC) and amygdala. The overall methylation of PFC correlates with symptom severity (35). Promoter methylation of the extracellular matrix protein gene SPARC is increased in cases of chronic lower back pain in humans and mice (36). Therefore, DNA methylation-mediated regulation of the pain-related genes in peripheral tissues and the central nervous system could be involved in the development and maintenance of NP.
The DNMT family includes DNMT1, DNMT2, DNMT3a, and DNMT3b (37–40). The main function of DNMT1 is to maintain established DNA methylation signatures in the genome and repair DNA methylation, and it is also known as maintenance methyltransferase (41). DNMT3a and DNMT3b can reversibly methylate unmethylated DNA and are, thus, classified as de novo methyltransferases (38, 39). The expression of DNMT1, DNMT3a, and DNMT3b in adult DRGs is upregulated in the NP rat model (42). DNMTs inhibit the increase in the methylation of the Mu opioid receptor (MOR) gene and prevent the decrease in MOR expression in the DRG, thereby improving morphine analgesia (43). Nerve injury significantly upregulates DNMT3a, increasing methylation of the spinal MOR gene promoter, and decreasing the expression of the MOR protein (44).
DNA methylation is mediated by three separate families of proteins—MBD, UHRF, and zinc finger proteins (45). Of these three protein families, MBD proteins are the most well-studied, especially in the context of revealing their roles in pain. In the absence of peripheral nerve injury, transcription factors (TFs) and RNA polymerase II bind to gene promoters, activating transcription. The methylation of CpG islands disrupts the binding of TFs, recruiting DNMT-mediated MBD proteins to silence gene expression (Figures 2A,B) (45, 46). The MBD family includes methyl-CpG-binding protein 2 (MeCP2) and MBD1-6 (47, 48), wherein MeCP2 primarily functions as a transcriptional repressor. MeCP2 was downregulated in a rat neuropathic pain model, with concomitant changes in the expression of HDAC1 and HDAC2 (49). Furthermore, nerve injury upregulates MeCP2 in the DRG, and the downregulation of MOR in the DRG is closely related to the increase in the expression of MeCP2. For example, MeCP2 knockout restores the expression of MOR in damaged DRGs and enhances the analgesic effect of morphine (50). Thus, DNA methylation is a complex epigenetic process, and further studies on the role of MeCP2 in NP are warranted.
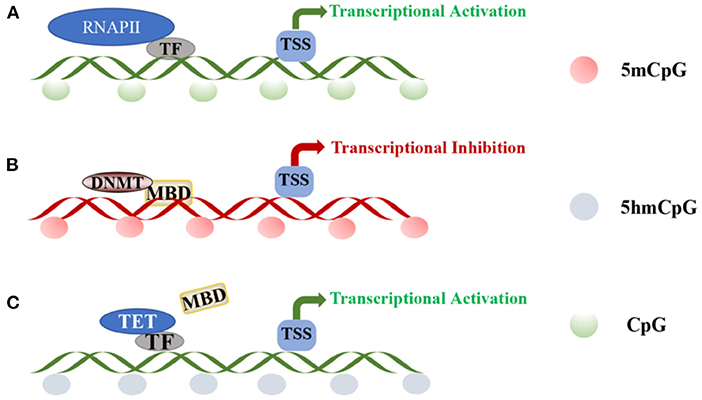
Figure 2. DNA methylation and demethylation: (A) TF and RNAPII bind to the promoter region of the gene, activating transcription at unmethylated promoters. (B) CpG island methylation is mediated by DNA methyltransferases and induces DNA methylation through MBD proteins. (C) Transcription factors recruit TET enzymes to specific sites for regulating local DNA demethylation.TF, transcription factor; RNAPII, RNA polymerase II; TSS, transcriptional start site; DNMT, DNA-methyltransferases; MBD, methyl-CpG-binding domain; TET, ten-eleven translocation.
DNMTs and NP
DNMTs, which play a key role in “reading DNA methylation,” have been studied in recent years with respect to their roles in NP. As a non-nucleoside (small molecule) DNMT inhibitor, RG108 blocks the active sites of DNMTs (51), and some studies have shown that it can relieve pain. K2p1.1 was the first K2P channel identified in mammals, and its expression is significantly downregulated after peripheral nerve injury and may lead to increased neuronal excitability (52, 53). Mao et al. (29) demonstrated that paclitaxel injection downregulated K2P1.1 in the DRG. Using a whole-cell current clamp, these authors demonstrated that neuronal excitability increased when K2P1.1 was knocked out. They also explored the underlying mechanism and found that K2p1.1 downregulation depends on the upregulation of DNMT expression. This suggested that DNA methylation may be involved in paclitaxel-induced downregulation of K2p1.1 mRNA in the DRG. The level of DNMT3a was increased in the DRG after injection of paclitaxel and, moreover, RG108 might significantly block paclitaxel-induced mechanical allodynia. Two other studies have investigated the role of RG108 as a DNMT inhibitor. Sun et al. (54) found that RG107 increased the expression of DNMT1 in the DRG and that it did so via the activation of the transcription factor cAMP response element-binding protein (CREB), causing DNA methylation of the Kcna2 (encoding Kv1.2) promoter, thereby reducing Kv1.2 expression and promoting pain. Decreased Kv1.2 expression reduced the total voltage-gated potassium current, depolarized the resting membrane potential, and induced the spontaneous ectopic discharge and hyperexcitability of neurons and neuropathic pain-like symptoms. Furthermore, RG108 administration or DNMT1 knockout reduced pain and allodynia caused by nerve damage. Specifically, DNMT1 knockout prevented neuronal hyperexcitability in the injured DRG. Another study revealed an increase in DNMT3a expression in a chronic constriction injury (CCI) model, and that MOR methylation plays an important epigenetic role in NP. DNMT3a binds to the MOR promoter, inhibits its transcription, reduces its mRNA and protein expression levels, and causes pain (44). The DNMT inhibitor RG108 was found to significantly block the increase in the methylation of the MOR promoter, consequently upregulating MOR expression to attenuate NP.
The nucleoside DNMT inhibitor 5-azacytidine prevents the resolution of a covalent reaction intermediate, which leads to DNMT being trapped and inactivated in the form of a covalent protein–DNA adduct (55, 56). The link between 5-azacytidine and NP was subsequently further clarified, showing that nerve damage leads to an increase in DNMT3a expression, a decrease in miR-214-3p expression, and triggers colony-stimulating factor-1 (CSF1) overexpression (57). The DNMT inhibitor zebularine was also found to significantly reduce methylation of the miR-214-3p promoter, leading to an increase in miR-214-3p expression in the ipsilateral dorsal horn and a decrease in CSF-1 content, which further alleviated the pain behavior of rats after spinal nerve ligation (SNL).
Small interfering RNAs (siRNAs) and short hairpin RNAs (shRNAs) are potential epigenetic targets. Unlike DNMT inhibitors, their sequences can be designed for a specific target gene. Opioids, the gold standard for NP treatment, have unsatisfactory analgesic effects, partly due to the downregulation of opioid receptors in DRG neurons. DNMT3a reportedly inhibited expression of Oprm1 and Oprk1 and their respective proteins, namely, MOR and kappa-opioid receptor (KOR), in the DRG. Microinjection of shRNA-DNMT3a was found to increase the levels of opioid receptors and relieve pain (30). However, in another study, the expression of DNMT3b was markedly downregulated after SNL injury, leading to demethylation of the GPR151 promoter, thereby promoting the binding of the transcription factor KLF5 to the GPR151 promoter to further increase GPR151 expression. The administration of siRNA-DNMT3b further increased the GPR151 level and exacerbated pain. In contrast, DNMT3b overexpression reduced pain (58). The authors of this study showed that SNL-induced NP could decrease DNMT3b expression, and the administration of a lentivirus-carrying DNMT3b could relieve pain. They further confirmed that DNMT3b might lead to the demethylation of the CXCR3 promoter, further increase the binding of C/EBPa and CXCR3, promote transcription and expression of CXCR3, and, thereby, induce NP (59).
DNMTs and their inhibitors appear to have varying effects on NP; DNMTs do not uniformly increase or decrease NP but rather act as a methylation tool that affects NP by increasing or decreasing the expression of specific genes. DNMTs may combine with gene promoters, leading to the methylation or demethylation of genes, consequently exacerbating or relieving NP. The combination of DNA methylation or demethylation enzymes with specific gene promoters is now recognized as a mode of NP treatment in the form of methylation. Thus, understanding the methylation or demethylation of the corresponding gene promoters caused by DNMT can provide new guidance for the treatment of NP. Administering gene-specific siRNA or lentiviral therapy is a prospective treatment option in the future.
MBD and NP
Thus far, research on NP has mostly focused on MeCP2 from the MBD protein family. In the SNI and Complete Freund's Adjuvant (CFA) model, MeCP2 was found to be overexpressed in neurons and was downregulated in glial cells (49). MeCP2 was also reported to be expressed in all dorsal horn neurons of the adult spinal cord (60). Notably, intrathecal 5-azacytidine administered via spinal injection significantly inhibited the increase in global DNA methylation and MeCP2 expression in the spinal cord (61). In another study, the expression of MeCP2 in the DRG was upregulated after spared nerve injury (SNI) surgery, which mediated the upregulation of brain-derived neurotrophic factor (BDNF) expression and led to pain. Moreover, microRNAs can decrease the expression level of MeCP2, thereby inhibiting BDNF expression and reducing pain (62). Mecp2-null mice also exhibited decreased BDNF levels in the DRG and decreased pain sensitivity. Central sensitization refers to nociceptor inputs that trigger an increase in the excitability of neurons in the central pain pathway and the prolongation of synaptic potency (63). Accumulating evidence indicates that in addition to activity-dependent synaptic plasticity, changes in gene transcription contribute to the maintenance of central sensitization (37). As a sensory regulator in the nociceptive pathway, BDNF affects central sensitization (64). In CCI model rats, DNMT3a, DNMT3b, and MeCP2 expression levels increased while the MBD2 expression level decreased in the lumbar spinal cord. The authors of this study also determined that GAD-1 promoter methylation reduced the level of GAD-67 protein, which is the main inhibitor of γ-aminobutyric acid (GABA) synthetase (65). However, they did not further explain the connection between gene methylation, DNMT, and the MBD family. Therefore, the subtype changes between DNMT and MBD warrant further study.
In contrast to these conclusions, Zhang et al. (66) reported that MeCP2 can reduce pain sensitivity. These authors attenuated mechanical and thermal pain sensitivity through MeCP2 overexpression, rather than administering MeCP2 inhibitors. The mechanism was found to involve the CREB/miR-132 signaling pathway in the spinal cord. In a study of the relationship between MBD and NP, Mo et al. (67) demonstrated that MBD1 deficiency in the DRG triggered a reduction in pain hypersensitivity following peripheral nerve injury; MBD1 recruits DNMT3a to the promoters of the Oprm1 and Kcna2 genes in DRG neurons, inhibiting MOR and Kv1.2 expression and inducing NP. Furthermore, both Mbd1 knockout and intrathecal administration of siRNA-Mbd1 reduced the sensitivity of mice to pain.
DNA methylation is a highly complex process. Both methylation and demethylation regulate gene expression by inhibiting or activating genes, respectively. The differences in MeCP2 function previously discussed may be caused by its effect on different genes. Another reason for the contradictory effects of MeCP2 in NP models may be related to the temporal properties of the protein.
DNA Demethylation
DNA Demethylation and TET
A balance in DNA methylation and demethylation should be achieved in neurons. In different biological studies, DNA demethylation has been verified to be both an active and passive process. Active demethylation refers to the removal or modification of methyl groups from 5mC during certain enzymatic processes (68). Notably, along with being an intermediate form of DNA demethylation, 5-hydroxymethyl cytosine (5hmC) is an epigenetic marker enriched within promoters and gene bodies (69). The continuous reaction of ten-eleven translocation (TET) enzyme initiates DNA demethylation, and the conversion of 5mC to 5hmC is divided into two pathways—oxidation and deamination. TET proteins are large (~180–230 kDa) multidomain enzymes and include TET1, TET2, and TET3 (70), with high neuronal and low glial expression (71). Purified TET enzymes were found to modify oligonucleotide substrates containing 5mC through oxidation, and the product was authenticated as 5hmC in CpG regions (72–74). The three TET enzymes share a conserved C-terminal domain and a less-conserved N-terminal domain (75). Additionally, TET1 and TET3 may have similar functions, mainly regulating 5hmC levels at gene promoters and transcription start sites, while TET2 mainly regulates 5hmC levels in the gene body (71, 76). Therefore, although the TET family can generate 5hmC, they may regulate the expression of 5hmC at different cell sites or at different developmental stages and at different genomic sites. Several non-enzymatic proteins, such as TF, could regulate local DNA demethylation in a sequence-specific manner by recruiting TET enzymes to specific sites (Figure 2C) (77–79). Significantly high levels of 5hmC are found in adult neuronal cells (80), and 5hmC is not only an intermediate for DNA demethylation but also acts as a stable epigenetic marker, which is enriched in genomes, promoters, and transcription factor-binding sites, potentially affecting gene expression. Pan et al. (81) showed that TET1 and TET3 levels increase significantly in the spinal cord with increased 5hmC content of the whole genome. Therefore, 5hmC is a key intermediate that activates the demethylation pathway. This discovery provides a crucial clue to enrich our understanding of the mechanism of DNA demethylation.
TET and NP
TET3 was suggested to be the main driving factor for the upregulation of 5hmC in the DRG after nerve injury (82). Wu et al. (83) further showed that SNL surgery could trigger methylation of Oprml1 and Kcna2, thereby increasing the amounts of 5mC with a corresponding decrease in the level of 5hmC, thus reducing the corresponding MOR and Kv1.2 protein levels and ultimately causing pain. In contrast, overexpression of DRG TET1 can block the methylation of these two genes and increase the 5hmC level, thereby reducing pain. The same authors also confirmed that overexpression of TET by DRG microinjection of the herpes simplex virus-TET1 could improve morphine analgesia and prevent morphine tolerance under NP conditions (83). However, Hsieh et al. (73) demonstrated that TET1 expression was enhanced after SNL surgery. Similar to the results of previous studies, the level of 5hmC in the dorsal horn of the ipsilateral spinal cord increased in proportion to the enhanced TET1 expression by SNL. The level of 5hmC, the excitability of dorsal horn neurons, and pain could be improved and alleviated after intrathecal siRNA-TET1 administration. Furthermore, SNL increased the binding of TET1 to the Bdnf promoter and increased the 5mC/5hmC transformation mediated by TET1 at the CpG site of the Bdnf promoter. These effects could be reversed by spinal cord-targeted injection of siRNA-TET1 (73). The same group subsequently showed that NP is related to TET1-mediated demethylation (84); melatonin was found to reverse TET expression, mGluR5 promoter demethylation, and pain hypersensitivity induced by Tet1 gene transfer. We summarize the key studies focusing on the links between DNMT, MBP, and TET proteins with NP in Table 1.
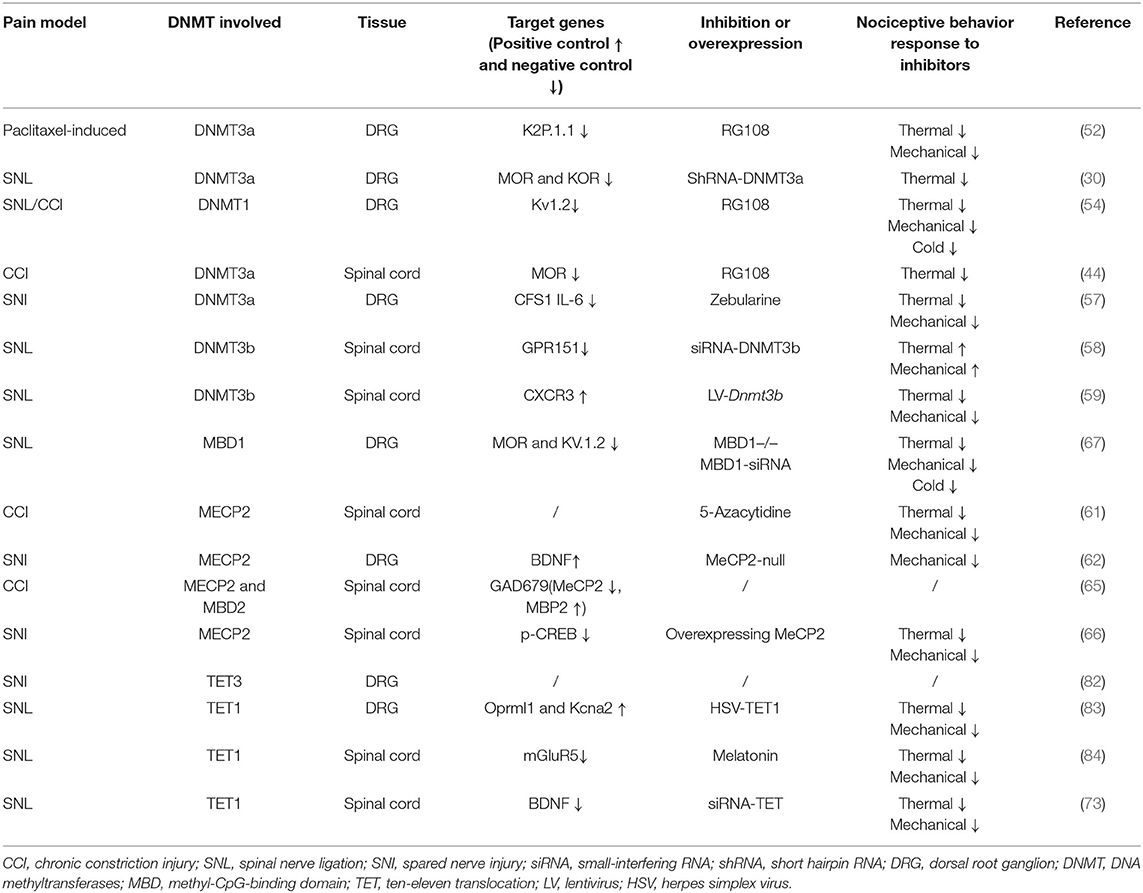
Table 1. Roles of DNMT, MBD, and TET proteins in neuropathic pain (NP) in rodent models.
Overall, these results suggest that TET-based treatment of NP remains complex because although TET1 overexpression can alleviate pain, TET reduction can also relieve pain. Moreover, similar to DNMT, the genes that TETs combine with also vary. Furthermore, the demethylation of other genes promoted by TET1 overexpression or TET inhibition under NP conditions cannot be excluded. Thus, treating NP with TET requires more in-depth research and extensive trials.
Challenges and Perspectives
NP is a complex disease with multiple pathologies. DNA methylation could act as a trigger, a downstream response mechanism, or could play a role in both processes. Epigenetic adaptations lead to chronic increases in hyperalgesia and allodynia; therefore, new treatment strategies that address NP are necessary (85). The DNMT inhibitors 5-azacitidine and decitabine are approved for treating myelodysplastic syndrome (86, 87). As a DNMT inhibitor, decitabine was approved in May 2006 for the treatment of myelodysplastic syndromes (MDS) and chronic myelogenous leukemia. Adverse reactions, such as hyperbilirubinemia, pneumonia, and constipation, appeared in phase 3 of the trial during treatment of MDS (88); however, other DNMT inhibitors have not been clinically applied. Therefore, many clinical trials are required to evaluate if they have the same side effects when applied to NP patients. Furthermore, not all drugs are tissue-specific. Thus, epigenetic treatment of NP may lead to unpredictable long-term side effects.
In the spinal cord, the barrier between blood and neuronal tissue is formed by the blood-spinal cord barrier, and in the peripheral nervous system, the endothelial blood vessels and perineurium form the blood-nerve barrier. Drugs must pass through these barriers to the peripheral or central nervous system to modulate pain. In animals, altering the route of administration, such as direct delivery to the spinal cord or brain or, more subtly, by microinjecting drugs into the DRG, could help overcome these limitations; however, this is challenging in humans. Therefore, future studies on NP should focus on the underlying epigenetic mechanisms and tissue-selective drug delivery. In addition, whole-genome methylation assays are warranted. Epigenomic editing could enable the targeting of selected modifications for the design of individualized treatments for NP patients, ideally specifically targeting the affected cells (89, 90).
The role of gender is another important consideration. Indeed, clinical studies have shown that women are at higher risk of chronic pain and exhibit greater pain sensitivity. Sex hormones, endogenous opiate functions, and genetic factors are the main reasons for these differences (91–93). For example, in preclinical trials, male and female animals respond differently to pain under the same conditions (94); female CD-1 mice required two to three times more morphine than male mice to produce the same analgesic effect (95). Furthermore, the Delta opiate receptor (DOR) helps control pain, and pain tolerance was abolished in females with DOR knockout but not in male mice (96). In the context of epigenetics and gender, gender bias in DNA methylation levels has been found in many animal and human studies (97–99). Thus, as epigenetics can regulate opiate receptors, receptors display sex differences. Currently, the available evidence does not support gender-specific DNA methylation-based treatment of NP, but this is a conceivable future outcome. Further research is now needed to elucidate the underlying epigenetic and gender-based differences in pain response.
Conclusions
Although an increasing number of studies have been conducted on NP animal models and DNA methylation, numerous promising therapeutics have failed in clinical trials (100, 101). The pain-causing gene or protein targets identified in animals may not be major contributors to pain in humans due to species differences in pain-modulation pathways. Basic cellular and molecular differences between animals and humans usually underlie these failures. Furthermore, there are also some uncertainties, such as the pathophysiological mechanism of pain in specific patients, dose selection in clinical trials, and the inability of animal models to accurately reflect the complex emotional responses humans have to pain (102, 103). In the context of difficulties in preclinical to clinical translation, improved animal models and a focus on pain circuitry are also needed to address epigenetic therapy. Given that NP remains a challenging condition to manage and the contribution of DNA methylation to this disorder is becoming increasingly recognized, the significance of DNA methylation in NP could become more apparent in coming years, and novel ways to study DNA methylation will likely be the primary focus of further studies.
Author Contributions
WJ and X-YT designed the study, reviewed the literature, and drafted the manuscript. J-ML created the summary table and helped with the writing and revision of the manuscript. PY and MD contributed significantly to the revision of the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 31872772) and the Natural Science Foundation of Jilin Province of China (Grant no. 20200201606JC) to MD.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
BDNF, brain-derived neurotrophic factor; CCI, chronic constriction injury; CFA, Complete Freund's Adjuvant; CpG, cytosine-phosphate-guanosine; CREB, cAMP response element-binding protein; CSF1, colony-stimulating factor-1; DNMTs, DNA methyltransferases; DOR, Delta opioid receptor; DRG, dorsal root ganglion; GABA, γ-aminobutyric acid; HAT, histone acetyl transferase; HDACs, histone deacetylase family of enzymes; KOR, kappa-opioid receptor; MBD, methyl-CpG-binding domain; MDS, myelodysplastic syndromes; MeCP2, methyl-CpG-binding protein 2; MOR, Mu opioid receptor; miRNAs, MicroRNAs; NP, neuropathic pain; PFC, prefrontal cortex; TET, ten-eleven translocation; TFs, transcription factors; shRNAs, short hairpin RNAs; SNL, spinal nerve ligation; SNI, spared nerve injury; siRNAs, small interfering RNAs; 5hmC, 5-hydroxymethyl cytosine; 5mC, 5-methylcytosine.
References
1. Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, et al. Neuropathic pain. Nat Rev Dis Primers. (2017) 3:17002. doi: 10.1038/nrdp.2017.2
2. Campbell JN, Meyer RA. Mechanisms of neuropathic pain. Neuron. (2006) 52:77–92. doi: 10.1016/j.neuron.2006.09.021
3. Jensen TS, Finnerup NB. Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. Lancet Neurol. (2014) 13:924–35. doi: 10.1016/S1474-4422(14)70102-4
4. Géranton SM, Tochiki KK. Regulation of gene expression and pain states by epigenetic mechanisms. Progr Mol Biol Transl Sci. (2015) 131:147–83. doi: 10.1016/bs.pmbts.2014.11.012
5. Liem L, van Dongen E, Huygen FJ, Staats P, Kramer J. The dorsal root ganglion as a therapeutic target for chronic pain. Reg Anesth Pain Med. (2016) 41:511–9. doi: 10.1097/AAP.0000000000000408
6. Yeh TY, Luo IW, Hsieh YL, Tseng TJ, Chiang H, Hsieh ST. Peripheral neuropathic pain: from experimental models to potential therapeutic targets in dorsal root ganglion neurons. Cells. (2020) 9:2725. doi: 10.3390/cells9122725
7. Kishi M, Tanabe J, Schmelzer JD, Low PA. Morphometry of dorsal root ganglion in chronic experimental diabetic neuropathy. Diabetes. (2002) 51:819–24. doi: 10.2337/diabetes.51.3.819
8. Loeser JD, Treede RD. The Kyoto protocol of IASP basic pain terminology. Pain. (2008) 137:473–7. doi: 10.1016/j.pain.2008.04.025
9. Krames ES. The dorsal root ganglion in chronic pain and as a target for neuromodulation: a review. Neuromodulation. (2015) 18:24–32; discussion doi: 10.1111/ner.12247
10. Gosden RG, Feinberg AP. Genetics and epigenetics–nature's pen-and-pencil set. N Engl J Med. (2007) 356:731–3. doi: 10.1056/NEJMe068284
11. Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med. (2017) 49:e324. doi: 10.1038/emm.2017.11
12. Li W, Nagaraja S, Delcuve GP, Hendzel MJ, Davie JR. Effects of histone acetylation, ubiquitination and variants on nucleosome stability. Biochem J. (1993) 296(Pt 3):737–44. doi: 10.1042/bj2960737
13. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. (2009) 10:32–42. doi: 10.1038/nrg2485
14. Jiang Y, Langley B, Lubin FD, Renthal W, Wood MA, Yasui DH, et al. Epigenetics in the nervous system. J Neurosci. (2008) 28:11753–9. doi: 10.1523/JNEUROSCI.3797-08.2008
15. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. (2004) 116:281–97. doi: 10.1016/S0092-8674(04)00045-5
16. Liang L, Tao YX. Expression of acetyl-histone H3 and acetyl-histone H4 in dorsal root ganglion and spinal dorsal horn in rat chronic pain models. Life Sci. (2018) 211:182–8. doi: 10.1016/j.lfs.2018.09.029
17. Matsushita Y, Araki K, Omotuyi O, Mukae T, Ueda H. HDAC inhibitors restore C-fibre sensitivity in experimental neuropathic pain model. Br J Pharmacol. (2013) 170:991–8. doi: 10.1111/bph.12366
18. Yadav R, Weng HR. EZH2 regulates spinal neuroinflammation in rats with neuropathic pain. Neuroscience. (2017) 349:106–17. doi: 10.1016/j.neuroscience.2017.02.041
19. Tan Y, Yang J, Xiang K, Tan Q, Guo Q. Suppression of microRNA-155 attenuates neuropathic pain by regulating SOCS1 signalling pathway. Neurochem Res. (2015) 40:550–60. doi: 10.1007/s11064-014-1500-2
20. Xia L, Zhang Y, Dong T. Inhibition of microRNA-221 alleviates neuropathic pain through targeting suppressor of cytokine signaling 1. J Mol Neurosci. (2016) 59:411–20. doi: 10.1007/s12031-016-0748-1
21. Cardoso AL, Guedes JR, Pereira de Almeida L, Pedroso de Lima MC. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology. (2012) 135:73–88. doi: 10.1111/j.1365-2567.2011.03514.x
22. Li L, Zhao G. Downregulation of microRNA-218 relieves neuropathic pain by regulating suppressor of cytokine signaling 3. Int J Mol Med. (2016) 37:851–8. doi: 10.3892/ijmm.2016.2455
23. Yan T, Zhang F, Sun C, Sun J, Wang Y, Xu X, et al. miR-32–5p-mediated Dusp5 downregulation contributes to neuropathic pain. Biochem Biophys Res Commun. (2018) 495:506–11. doi: 10.1016/j.bbrc.2017.11.013
24. Shi G, Shi J, Liu K, Liu N, Wang Y, Fu Z, et al. Increased miR-195 aggravates neuropathic pain by inhibiting autophagy following peripheral nerve injury. Glia. (2013) 61:504–12. doi: 10.1002/glia.22451
25. Sanna MD, Galeotti N. The HDAC1/c-JUN complex is essential in the promotion of nerve injury-induced neuropathic pain through JNK signaling. Eur J Pharmacol. (2018) 825:99–106. doi: 10.1016/j.ejphar.2018.02.034
26. Ding HH, Zhang SB, Lv YY, Ma C, Liu M, Zhang KB, et al. TNF-α/STAT3 pathway epigenetically upregulates Nav1.6 expression in DRG and contributes to neuropathic pain induced by L5-VRT. J Neuroinflamm. (2019) 16:29. doi: 10.1186/s12974-019-1421-8
27. Laumet G, Garriga J, Chen SR, Zhang Y, Li DP, Smith TM, et al. G9a is essential for epigenetic silencing of K(+) channel genes in acute-to-chronic pain transition. Nat Neurosci. (2015) 18:1746–55. doi: 10.1038/nn.4165
28. Ehrlich M. DNA hypermethylation in disease: mechanisms and clinical relevance. Epigenetics. (2019) 14:1141–63. doi: 10.1080/15592294.2019.1638701
29. Mao Q, Wu S, Gu X, Du S, Mo K, Sun L, et al. DNMT3a-triggered downregulation of K(2p) 1.1 gene in primary sensory neurons contributes to paclitaxel-induced neuropathic pain. Int J Cancer. (2019) 145:2122–34. doi: 10.1002/ijc.32155
30. Sun L, Zhao JY, Gu X, Liang L, Wu S, Mo K, et al. Nerve injury-induced epigenetic silencing of opioid receptors controlled by DNMT3a in primary afferent neurons. Pain. (2017) 158:1153–65. doi: 10.1097/j.pain.0000000000000894
31. Szyf M. Targeting DNA methylation in cancer. Ageing Res Rev. (2003) 2:299–328. doi: 10.1016/S1568-1637(03)00012-6
32. Holliday R. DNA methylation and epigenotypes. Biochemistry. (2005) 70:500–4. doi: 10.1007/s10541-005-0144-x
33. Cheng X, Roberts RJ. AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res. (2001) 29:3784–95. doi: 10.1093/nar/29.18.3784
34. Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell. (1985) 40:91–9. doi: 10.1016/0092-8674(85)90312-5
35. Tajerian M, Alvarado S, Millecamps M, Vachon P, Crosby C, Bushnell MC, et al. Peripheral nerve injury is associated with chronic, reversible changes in global DNA methylation in the mouse prefrontal cortex. PLoS ONE. (2013) 8:e55259. doi: 10.1371/journal.pone.0055259
36. Sibille KT, Witek-Janusek L, Mathews HL, Fillingim RB. Telomeres and epigenetics: potential relevance to chronic pain. Pain. (2012) 153:1789–93. doi: 10.1016/j.pain.2012.06.003
37. Liang L, Lutz BM, Bekker A, Tao YX. Epigenetic regulation of chronic pain. Epigenomics. (2015) 7:235–45. doi: 10.2217/epi.14.75
38. Jeltsch A. Molecular enzymology of mammalian DNA methyltransferases. Current Top Microbiol Immunol. (2006) 301:203–25. doi: 10.1007/3-540-31390-7_7
39. Siedlecki P, Zielenkiewicz P. Mammalian DNA methyltransferases. Acta Biochim Polon. (2006) 53:245–56. doi: 10.18388/abp.2006_3337
40. Jeltsch A, Nellen W, Lyko F. Two substrates are better than one: dual specificities for Dnmt2 methyltransferases. Trends Biochem Sci. (2006) 31:306–8. doi: 10.1016/j.tibs.2006.04.005
41. Mortusewicz O, Schermelleh L, Walter J, Cardoso MC, Leonhardt H. Recruitment of DNA methyltransferase I to DNA repair sites. Proc Natl Acad Sci USA. (2005) 102:8905–9. doi: 10.1073/pnas.0501034102
42. Pollema-Mays SL, Centeno MV, Apkarian AV, Martina M. Expression of DNA methyltransferases in adult dorsal root ganglia is cell-type specific and up regulated in a rodent model of neuropathic pain. Front Cell Neurosci. (2014) 8:217. doi: 10.3389/fncel.2014.00217
43. Zhou XL, Yu LN, Wang Y, Tang LH, Peng YN, Cao JL, et al. Increased methylation of the MOR gene proximal promoter in primary sensory neurons plays a crucial role in the decreased analgesic effect of opioids in neuropathic pain. Mol Pain. (2014) 10:51. doi: 10.1186/1744-8069-10-51
44. Shao C, Gao Y, Jin D, Xu X, Tan S, Yu H, et al. DNMT3a methylation in neuropathic pain. J Pain Res. (2017) 10:2253–62. doi: 10.2147/JPR.S130654
45. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. (2013) 38:23–38. doi: 10.1038/npp.2012.112
46. Sweatt JD. Experience-dependent epigenetic modifications in the central nervous system. Biol Psychiatry. (2009) 65:191–7. doi: 10.1016/j.biopsych.2008.09.002
47. Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. (1998) 18:6538–47. doi: 10.1128/MCB.18.11.6538
48. Baymaz HI, Fournier A, Laget S, Ji Z, Jansen PW, Smits AH, et al. MBD5 and MBD6 interact with the human PR-DUB complex through their methyl-CpG-binding domain. Proteomics. (2014) 14:2179–89. doi: 10.1002/pmic.201400013
49. Tochiki KK, Cunningham J, Hunt SP, Géranton SM. The expression of spinal methyl-CpG-binding protein 2, DNA methyltransferases and histone deacetylases is modulated in persistent pain states. Mol Pain. (2012) 8:14. doi: 10.1186/1744-8069-8-14
50. Sun N, Yu L, Gao Y, Ma L, Ren J, Liu Y, et al. MeCP2 epigenetic silencing of Oprm1 gene in primary sensory neurons under neuropathic pain conditions. Front Neurosci. (2021) 15:743207. doi: 10.3389/fnins.2021.743207
51. Brueckner B, Lyko F. DNA methyltransferase inhibitors: old and new drugs for an epigenetic cancer therapy. Trends Pharmacol Sci. (2004) 25:551–4. doi: 10.1016/j.tips.2004.09.004
52. Mao Q, Yuan J, Ming X, Wu S, Chen L, Bekker A, et al. Role of dorsal root ganglion K2p1.1 in peripheral nerve injury-induced neuropathic pain. Mol Pain. (2017) 13:1744806917701135. doi: 10.1177/1744806917701135
53. Kindler CH, Yost CS. Two-pore domain potassium channels: new sites of local anesthetic action and toxicity. Reg Anesth Pain Med. (2005) 30:260–74. doi: 10.1016/j.rapm.2004.12.001
54. Sun L, Gu X, Pan Z, Guo X, Liu J, Atianjoh FE, et al. Contribution of DNMT1 to neuropathic pain genesis partially through epigenetically repressing Kcna2 in primary afferent neurons. J Neurosci. (2019) 39:6595–607. doi: 10.1523/JNEUROSCI.0695-19.2019
55. Brueckner B, Kuck D, Lyko F. DNA methyltransferase inhibitors for cancer therapy. Cancer J. (2007) 13:17–22. doi: 10.1097/PPO.0b013e31803c7245
56. Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc Natl Acad Sci USA. (1984) 81:6993–7. doi: 10.1073/pnas.81.22.6993
57. Liu L, Xu D, Wang T, Zhang Y, Yang X, Wang X, et al. Epigenetic reduction of miR-214–3p upregulates astrocytic colony-stimulating factor-1 and contributes to neuropathic pain induced by nerve injury. Pain. (2020) 161:96–108. doi: 10.1097/j.pain.0000000000001681
58. Jiang BC, Zhang WW, Yang T, Guo CY. Demethylation of G-protein-coupled receptor 151 promoter facilitates the binding of Krüppel-like factor 5 and enhances neuropathic pain after nerve injury in mice. J Neurosci. (2018) 38:10535–51. doi: 10.1523/JNEUROSCI.0702-18.2018
59. Jiang BC, He LN, Wu XB, Shi H, Zhang WW, Zhang ZJ, et al. Promoted interaction of C/EBPα with demethylated Cxcr3 gene promoter contributes to neuropathic pain in mice. J Neurosci. (2017) 37:685–700. doi: 10.1523/JNEUROSCI.2262-16.2016
60. Géranton SM, Morenilla-Palao C, Hunt SP. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J Neurosci. (2007) 27:6163–73. doi: 10.1523/JNEUROSCI.1306-07.2007
61. Wang Y, Liu C, Guo QL, Yan JQ, Zhu XY, Huang CS, et al. Intrathecal 5-azacytidine inhibits global DNA methylation and methyl- CpG-binding protein 2 expression and alleviates neuropathic pain in rats following chronic constriction injury. Brain Res. (2011) 1418:64–9. doi: 10.1016/j.brainres.2011.08.040
62. Manners MT, Tian Y, Zhou Z, Ajit SK. MicroRNAs downregulated in neuropathic pain regulate MeCP2 and BDNF related to pain sensitivity. FEBS Open Bio. (2015) 5:733–40. doi: 10.1016/j.fob.2015.08.010
63. Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. (2011) 152(3 Suppl.):S2–15. doi: 10.1016/j.pain.2010.09.030
64. Merighi A, Salio C, Ghirri A, Lossi L, Ferrini F, Betelli C, et al. BDNF as a pain modulator. Progress Neurobiol. (2008) 85:297–317. doi: 10.1016/j.pneurobio.2008.04.004
65. Wang Y, Lin ZP, Zheng HZ, Zhang S, Zhang ZL, Chen Y, et al. Abnormal DNA methylation in the lumbar spinal cord following chronic constriction injury in rats. Neurosci Lett. (2016) 610:1–5. doi: 10.1016/j.neulet.2015.10.048
66. Zhang R, Huang M, Cao Z, Qi J, Qiu Z, Chiang LY. MeCP2 plays an analgesic role in pain transmission through regulating CREB / miR-132 pathway. Mol Pain. (2015) 11:19. doi: 10.1186/s12990-015-0015-4
67. Mo K, Wu S. MBD1 Contributes to the genesis of acute pain and neuropathic pain by epigenetic silencing of Oprm1 and Kcna2 genes in primary sensory neurons. J. Neurosci. (2018) 38:9883–99. doi: 10.1523/JNEUROSCI.0880-18.2018
68. Ooi SK, Bestor TH. The colorful history of active DNA demethylation. Cell. (2008) 133:1145–8. doi: 10.1016/j.cell.2008.06.009
69. Kaas GA, Zhong C, Eason DE, Ross DL, Vachhani RV, Ming GL, et al. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron. (2013) 79:1086–93. doi: 10.1016/j.neuron.2013.08.032
70. Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. (2016) 30:733–50. doi: 10.1101/gad.276568.115
71. Antunes C, Sousa N, Pinto L, Marques CJ. TET enzymes in neurophysiology and brain function. Neurosci Biobehav Rev. (2019) 102:337–44. doi: 10.1016/j.neubiorev.2019.05.006
72. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. (2009) 324:930–5. doi: 10.1126/science.1170116
73. Hsieh MC, Lai CY, Ho YC, Wang HH, Cheng JK, Chau YP, et al. Tet1-dependent epigenetic modification of BDNF expression in dorsal horn neurons mediates neuropathic pain in rats. Sci Rep. (2016) 6:37411. doi: 10.1038/srep37411
74. Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. (2010) 466:1129–33. doi: 10.1038/nature09303
75. Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. (2013) 502:472–9. doi: 10.1038/nature12750
76. Li X, Yue X, Pastor WA, Lin L, Georges R, Chavez L, et al. Tet proteins influence the balance between neuroectodermal and mesodermal fate choice by inhibiting Wnt signaling. Proc Natl Acad Sci USA. (2016) 113:E8267–76. doi: 10.1073/pnas.1617802113
77. Fujiki K, Shinoda A, Kano F, Sato R, Shirahige K, Murata M. PPARγ-induced PARylation promotes local DNA demethylation by production of 5-hydroxymethylcytosine. Nat Commun. (2013) 4:2262. doi: 10.1038/ncomms3262
78. Ohlsson R, Renkawitz R, Lobanenkov V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. (2001) 17:520–7. doi: 10.1016/S0168-9525(01)02366-6
79. Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. (2016) 17:551–65. doi: 10.1038/nrg.2016.83
80. Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. (2009) 324:929–30. doi: 10.1126/science.1169786
81. Pan Z, Xue ZY, Li GF, Sun ML, Zhang M, Hao LY, et al. DNA Hydroxymethylation by ten-eleven translocation methylcytosine dioxygenase 1 and 3 regulates nociceptive sensitization in a chronic inflammatory pain model. Anesthesiology. (2017) 127:147–63. doi: 10.1097/ALN.0000000000001632
82. Chamessian AG, Qadri YJ, Cummins M, Hendrickson M, Berta T, Buchheit T, et al. 5-Hydroxymethylcytosine (5hmC) and Ten-eleven translocation 1–3 (TET1–3) proteins in the dorsal root ganglia of mouse: expression and dynamic regulation in neuropathic pain. Somatosens Mot Res. (2017) 34:72–9. doi: 10.1080/08990220.2017.1292237
83. Wu Q, Wei G, Ji F, Jia S, Wu S, Guo X, et al. TET1 overexpression mitigates neuropathic pain through rescuing the expression of μ-opioid receptor and Kv1.2 in the primary sensory neurons. Neurotherapeutics. (2019) 16:491–504. doi: 10.1007/s13311-018-00689-x
84. Hsieh MC, Ho YC, Lai CY, Chou D, Wang HH, Chen GD, et al. Melatonin impedes Tet1-dependent mGluR5 promoter demethylation to relieve pain. J Pineal Res. (2017) 63:e12436. doi: 10.1111/jpi.12436
85. Kuner R. Central mechanisms of pathological pain. Nat Med. (2010) 16:1258–66. doi: 10.1038/nm.2231
86. Kaminskas E, Farrell A, Abraham S, Baird A, Hsieh LS, Lee SL, et al. Approval summary: azacitidine for treatment of myelodysplastic syndrome subtypes. Clin Cancer Res. (2005) 11:3604–8. doi: 10.1158/1078-0432.CCR-04-2135
87. Gore SD, Jones C, Kirkpatrick P. Decitabine. Nat Rev Drug Discov. (2006) 5:891–2. doi: 10.1038/nrd2180
88. Jabbour E, Issa JP, Garcia-Manero G, Kantarjian H. Evolution of decitabine development: accomplishments, ongoing investigations, and future strategies. Cancer. (2008) 112:2341–51. doi: 10.1002/cncr.23463
89. Niederberger E, Resch E, Parnham MJ, Geisslinger G. Drugging the pain epigenome. Nat Rev Neurol. (2017) 13:434–47. doi: 10.1038/nrneurol.2017.68
90. Kungulovski G, Jeltsch A. Epigenome editing: state of the art, concepts, and perspectives. Trends Genet. (2016) 32:101–13. doi: 10.1016/j.tig.2015.12.001
91. Craft RM, Mogil JS, Aloisi AM. Sex differences in pain and analgesia: the role of gonadal hormones. Eur J Pain. (2004) 8:397–411. doi: 10.1016/j.ejpain.2004.01.003
92. Fillingim RB, Kaplan L, Staud R, Ness TJ, Glover TL, Campbell CM, et al. The A118G single nucleotide polymorphism of the mu-opioid receptor gene (OPRM1) is associated with pressure pain sensitivity in humans. J Pain. (2005) 6:159–67. doi: 10.1016/j.jpain.2004.11.008
93. Bartley EJ, Fillingim RB. Sex differences in pain: a brief review of clinical and experimental findings. Br J Anaesth. (2013) 111:52–8. doi: 10.1093/bja/aet127
94. Ram A, Edwards T, McCarty A, Afrose L, McDermott MV, Bobeck EN. GPR171 agonist reduces chronic neuropathic and inflammatory pain in male, but not female mice. Front Pain Res. (2021) 2:695396. doi: 10.3389/fpain.2021.695396
95. Rosen SF, Ham B, Haichin M, Walters IC, Tohyama S, Sotocinal SG, et al. Increased pain sensitivity and decreased opioid analgesia in T-cell-deficient mice and implications for sex differences. Pain. (2019) 160:358–66. doi: 10.1097/j.pain.0000000000001420
96. Reiss D, Maurin H, Audouard E, Martínez-Navarro M, Xue Y, Herault Y, et al. Delta opioid receptor in astrocytes contributes to neuropathic cold pain and analgesic tolerance in female mice. Front Cell Neurosci. (2021) 15:745178. doi: 10.3389/fncel.2021.745178
97. Mamrut S, Avidan N, Staun-Ram E, Ginzburg E, Truffault F, Berrih-Aknin S, et al. Integrative analysis of methylome and transcriptome in human blood identifies extensive sex- and immune cell-specific differentially methylated regions. Epigenetics. (2015) 10:943–57. doi: 10.1080/15592294.2015.1084462
98. van Dongen J, Nivard MG, Willemsen G, Hottenga JJ, Helmer Q, Dolan CV, et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat Commun. (2016) 7:11115. doi: 10.1038/ncomms11115
99. Nugent BM, Wright CL, Shetty AC, Hodes GE, Lenz KM, Mahurkar A, et al. Brain feminization requires active repression of masculinization via DNA methylation. Nat Neurosci. (2015) 18:690–7. doi: 10.1038/nn.3988
100. Hill R. NK1 (substance P) receptor antagonists–why are they not analgesic in humans? Trends Pharmacol Sci. (2000) 21:244–6. doi: 10.1016/S0165-6147(00)01502-9
101. Gavva NR, Treanor JJ, Garami A, Fang L, Surapaneni S, Akrami A, et al. Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain. (2008) 136:202–10. doi: 10.1016/j.pain.2008.01.024
102. Berge OG. Predictive validity of behavioural animal models for chronic pain. Br J Pharmacol. (2011) 164:1195–206. doi: 10.1111/j.1476-5381.2011.01300.x
Keywords: neuropathic pain, DNA methylation, gene suppression, epigenetic, treatment
Citation: Jiang W, Tan X-Y, Li J-M, Yu P and Dong M (2022) DNA Methylation: A Target in Neuropathic Pain. Front. Med. 9:879902. doi: 10.3389/fmed.2022.879902
Received: 20 February 2022; Accepted: 06 June 2022;
Published: 07 July 2022.
Edited by:
Jie Zhao, Tianjin University, ChinaReviewed by:
Jinming Han, Capital Medical University, ChinaCopyright © 2022 Jiang, Tan, Li, Yu and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Yu, eXBlbmdAamx1LmVkdS5jbg==; Ming Dong, ZG9uZ2dlQGpsdS5lZHUuY24=