
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Med., 28 April 2022
Sec. Nephrology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.869409
 Zhen-Yu Li1,2,3,4,5
Zhen-Yu Li1,2,3,4,5 Shuang Wang1,2,3,4,5Dan-Yang Li1,2,3,4,5Dan Liu6
Shuang Wang1,2,3,4,5Dan-Yang Li1,2,3,4,5Dan Liu6 Su-Xia Wang1,2,3,4,5*
Su-Xia Wang1,2,3,4,5* Xiao-Juan Yu2,3,4,5
Xiao-Juan Yu2,3,4,5 Gang Liu2,3,4,5
Gang Liu2,3,4,5 Fu-De Zhou2,3,4,5
Fu-De Zhou2,3,4,5 Ming-Hui Zhao2,3,4,5
Ming-Hui Zhao2,3,4,5Objectives: Fibrinogen A alpha-chain amyloidosis (AFib amyloidosis) is the most common form of hereditary renal amyloidosis in the United Kingdom and Europe, but has rarely been reported in Asia. In this study, we reported two AFib amyloidosis patients in China, reviewing the literature and summarizing main characteristics of AFib amyloidosis in Asia.
Methods: Two unrelated Chinese patients were diagnosed with AFib amyloidosis by clinical presentation, renal biopsy, mass spectrometry and DNA sequencing in Peking University First Hospital of China from 2014 to 2016.
Results: Both of the patients presented with proteinuria, edema and hypertension. Renal biopsies of two patients showed extensive amyloid deposits (Congo red positive) in glomeruli, and focal tubulointerstitial amyloid deposits was also found in patient 1. Besides, hepatic involvement of amyloidosis has been detected by liver biopsy in patient 1. By electron microscopy, randomly arranged fibrils in a diameter of 8–12 nm was identified in mesangial matrix and subendothelial area of glomeruli. Immunohistochemistry demonstrated amyloid deposits were strongly positive for fibrinogen Aα in glomeruli and positive for LECT2 in the interstitium of renal medulla and the liver in Patient 1. Unevenly positive staining for both fibrinogen Aα and ApoA-I were found in Patient 2. Fibrinogen Aα was the most abundant amyloidogenic protein in both patients identified by laser microdissection and mass spectrometry-based proteomic analysis. Genetic analysis revealed the fibrinogen A a-chain gene (FGA) mutation in both patients, including a new deletion mutation [c.1639delA (p.Arg547Glyfs*21; NM_000508)] in Patient 2. Genetic analysis of the LECT2 gene in patient 1 revealed a codon change from ATC to GTC at position 172 [c.172A>G (p.Ile58Val; NM_002302)], which is a common polymorphism (SNP rs31517) in all ALECT2 amyloidosis patients.
Conclusions: We reported two AFib amyloidosis patients in China, one of them coexisted with ALECT2 amyloidosis simultaneously.
Amyloidosis is a protein misfolding disorder caused by the deposition of insoluble fibrillar amyloid in extracellular space, resulting in permanent organ damage. It can be either acquired or hereditary. Hereditary amyloidosis comprises a group of disease related to gene mutations in the coding regions of transthyretin, fibrinogen Aα chain, apolipoprotein A-I, apolipoprotein A-II, apolipoprotein C-II, apolipoprotein C-III, gelsolin, cystatin C and lysozyme. Among them, transthyretin-derived ATTR amyloidosis presents with polyneuropathy and/or cardiomyopathy, while fibrinogen A alpha-chain amyloidosis (AFib amyloidosis) presents with nephropathy typically.
AFib amyloidosis, which was first described by Benson et al. (1) in a Peruvian-Mexican family, is the most common form of hereditary renal amyloidosis in the United Kingdom and Europe (2), but has been rarely reported in Asia (3). Here we report two AFib amyloidosis patients in China, one of them coexisted with ALECT2 amyloidosis.
Two unrelated patients presenting with edema and proteinuria were found to have amyloid deposits in kidney: a 29-year-old Chinese woman (Patient 1) and a 47-year-old Chinese man (Patient 2). They underwent detailed clinical examination and laboratory tests including blood and urine biochemistry, electrocardiography, echocardiography and immunofixation electrophoresis of urine and serum.
Renal biopsies from the two patients were examined by routine light microscope (HE, PAS, Masson trichrome, Silver methenamine and Congo red), Immunofluorescence (detection for IgG, IgA, IgM, C3, C1q, FRA, Alb, κ and λ-light chain) and electron microscopy. Immunohistochemistry were performed on formalin-fixed paraffin-embedded renal tissue with antibodies against λ-light chain (1:8000; A0193, Dako), κ-light chain (1:8000; A0191, Dako), fibrinogen Aα chain (1:4000; A0080, Dako, Carpenteria, CA), lysozyme (1:200; GA009929, GTR, Shanghai, China), amyloid A (1:200; M0759, Dako, Glostrup, Denmark), transthyretin (1:2000; A0002, Dako, Carpenteria, CA), gelsolin (1:500; WH0002934M1, Sigma-Aldrich, Germany), apolipoprotein A-I (1:2000; PV9003, Calbiochem) and LECT2 (1:40, AF722, R&D Minneapolis, USA) according to standard techniques. Negative controls were included by omitting primary antibody.
Laser microdissection was performed as previously described (4). Briefly, the Congo red-positive areas of renal tissue were microdissected with laser. Proteins extracted from the microdissected areas were digested by typsin, and analyzed by liquid chromatography and electrospray tandem mass spectrometry (LC-MS/MS) to identify the component of amyloidogenic precursors.
Genomic DNA was extracted from peripheral blood leukocytes as previously described (5). Exon 5 of the fibrinogen A alpha chain gene and exon 3 of the LECT2 gene were amplified by polymerase chain reaction (PCR). Primers were designed using Primer 3.0 online. PCR products were electrophoresed through agarose gel and sequenced with ABI 3730XL (Cangso Medical Inspection, China). The sequences were analyzed using the DNASTAR software. Genomic sequences of the FGA gene (NM_000508) and the LECT2 gene (NM_002302) were used as the reference sequences. Furthermore, online tools such as HGMD Pro, PubMed and 1000Genomes were applied to evaluate the mutation genes.
Patient 1 was a 29-year-old Chinese woman. She presented with eyelid edema and lower limb edema for 6 months. She was a hepatitis B patient for 14 years and did not receive systemic therapy. Family history was negative. She had been diagnosed as AL amyloidosis in a local hospital 5 months ago and given methylprednisolone and melphalan for 6 days, then stopped due to liver injury. Her blood pressure was 130/70 mmHg when she was admitted into our hospital. The remaining physical examination was unremarkable. Investigations revealed hemoglobin concentration of 96 g/L, proteinuria of 1.74 g per 24 h, serum albumin of 28.8 g/L, serum creatinine of 182.3 μmol/L. Plasma fibrinogen (reference: 1.5–4 g/L) was normal. No evidence of a monoclonal protein was found in serum or urine. No signs of cardiac amyloidosis were found by echocardiography. Liver biopsy revealed amyloid deposition. Eight months after first admission into our hospital, her serum creatinine reached to 608 μmol/L and she started dialysis.
Patient 2 was a 47-year-old Chinese man with 10-month history of intermittent eyelid edema. His mother died of uremia. On admission, his blood pressure was 196/120 mmHg. Physical examination was unremarkable except eyelid edema. He had a nephrotic range proteinuria of 3.6 g per 24 h. Serum albumin was 33.2g/L. His renal function was normal with a serum creatinine of 93.4 μmol/L. Clotting profile was normal. Immunofixation electrophoresis of serum and urine were both negative. Mild thickened intima with a small plaque in the lower extremity artery was found by ultrasound. There was no evidence of heart involvement by echocardiography.
Clinical presentation and laboratory findings of the two patients are summarized in Table 1.
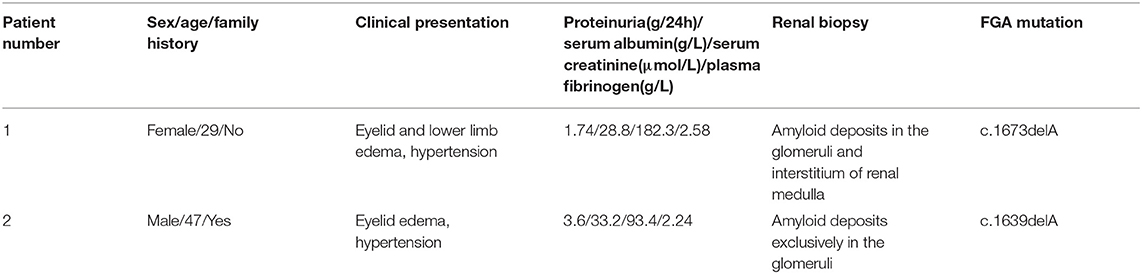
Table 1. Clinical presentation and laboratory findings of the patients.
Renal biopsies of both patients showed extensive amorphous amyloid deposits (Congo red positive) in glomeruli, resulting in striking glomerular enlargement and almost complete obliteration of the normal architecture (Figures 1A–D, Figure 2A). Congo red positive deposits produced apple green birefringence under polarized light (Figure 2C). No amyloid deposits were found in tubulointerstitium or vessels in Patient 2. But obvious amyloid deposits were found in the interstitium of renal medulla in Patient 1 (Figure 2B). Routine immunofluorescence showed negative staining for light chains (κ and λ). Electron microscope showed randomly arranged fibrils replacing mesangial matrix and subendothelial area (Figure 2D).
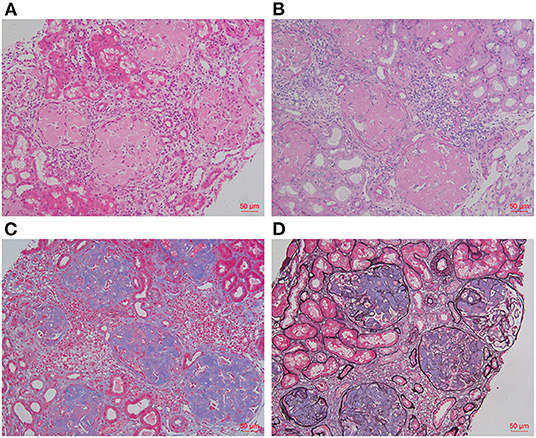
Figure 1. Histology findings of renal biopsy. HE [(A), ×200] and PAS [(B), ×200] staining showed massive amorphous eosinophilic deposits in glomeruli, leading to the obliteration of glomerular capillary loops. These amyloid deposits were pale blue by Masson trichrome staining [(C), ×200], but were not black on Silver methenamine staining [(D), ×200].
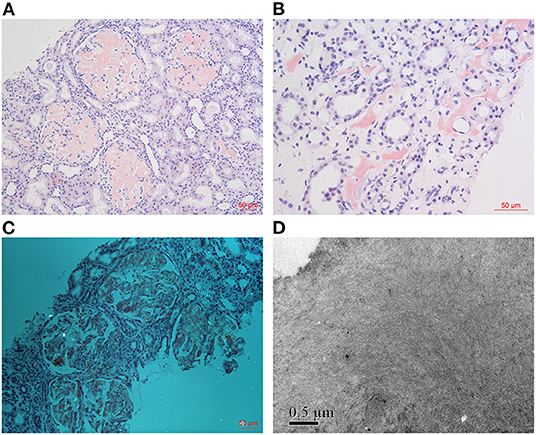
Figure 2. Histology findings (take Patient 1 as an example). Massive homogeneous and Congo red positive deposits were found in glomeruli [(A): Congo red ×200], which produce apple green birefringence under polarized light [(C): Congo red ×200]. Obvious amyloid deposits were found in the renal medulla interstitium of Patient 1 [(B): Congo red ×400]. Electron microscopy showed randomly arranged fibrils in mesangial matrix [(D): EM ×40,000].
As for IHC, amyloid deposits were strongly positive for fibrinogen Aα in glomeruli and also positive for LECT2 in medullary interstitium and liver in Patient 1 (Figures 3A,B), and unevenly positive for both apolipoprotein A-I (ApoA-I) and fibrinogen Aα in glomeruli of Patient 2 (Figure 3C). IHC for other amyloidogenic precursors (κ or λ light chains, Amyloid A, transthyretin, lysozyme, gesolin) were all negative.
Figure 3. IHC findings. Amyloid deposits were strongly positive for fibrinogen Aα in glomeruli [(A): IHC ×400] and positive for LECT2 in medullary interstitium [(B): IHC ×200] in Patient 1. The glomerular amyloid deposits were unevenly positive for fibrinogen Aα in Patient 2 [(C): IHC ×200].
Apolipoprotein E, serum amyloid P component and apolipoprotein A-IV, which are amyloid signature constituents, were detected in the amyloid deposits of both patients by LC-MS/MS proteomics analysis. Fibrinogen Aα was the most abundant amyloidogenic protein, with 47 spectra, 7 unique peptides for 14.31% coverage in Patient 1 and 105 spectra, 22 unique peptides for 36.02% coverage in Patient 2 (Figure 4).

Figure 4. Proteomic analysis results of Patient 1 (A) and Patient 2 (B). Yellow part represents covered amino acids.
Genetic analysis of Patient 1 showed a single nucleotide (A) deletion at position 1673 of the FGA [c.1673delA (p.Lys558Argfs*10), Figure 5A] and a codon change from ATC to GTC at position 172 of the LECT2 gene [c.172A>G (p.Ile58Val), Figure 5B). In Patient 2, genetic analysis showed a single nucleotide deletion at position 1639 of the FGA [c.1639delA (p.Arg547Glyfs*21), Figure 5C), resulting in a frame-shift mutation at codon 547.
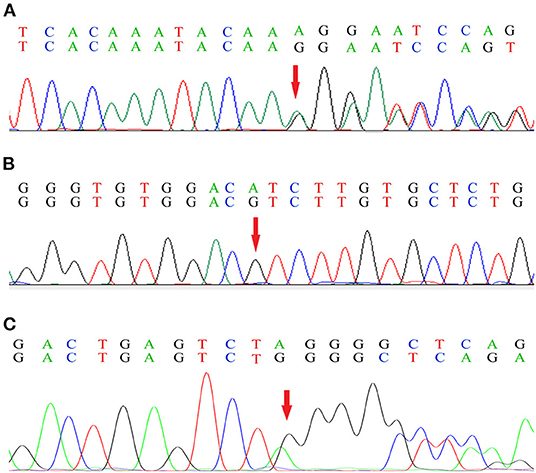
Figure 5. Genetic analysis of the FGA (A) and the LECT2 gene (B) in Patient 1, and the FGA in Patient 2 (C). The base sequence in the first line of each sub-graph is wild type, and the second line is mutant. Red arrow indicates the mutation site.
Fibrinogen is a 340-kD plasma protein produced exclusively by the liver (6) and plays an important role in the coagulation cascade. It is composed of 2 identical heterotrimers, each consisting of 3 polypeptide chains termed Aα, Bβ and γ-chain (7). These polypeptide chains are encoded by distinct genes, FGA, FGB and FGG. Mutations in these genes are responsible for dysfibrinogenemias, and heterozygous mutations in a small region of C-terminal portion of the Aα chain can cause HRA (8). Wild type fibrinogen Aα chain normally does not result in amyloid deposition (9). AFib amyloidosis is an autosomal-dominant hereditary systemic amyloidosis. To date, 17 amyloidogenic mutations have been described, including 7 missense mutations (c.1627G > A, c.1633G > A, c.1634A > T, c.1670C > A, c.1676A > T, c.1712C > A, c.1718G > T), 8 deletion mutations (c.1611delA, c.1619_1622delTTGT, c.1620delT, c.1622delT, c.1624_1627delAGTG, c.1629delG, c.1632delT, c.1673delA) and 2 insertion-deletion mutations (c.1606_1620del 1619_1620insCA, c.1720_1721delGGinsTT) (3). In our hospital, two patients were diagnosed with AFib amyloidosis, both with deletion mutations.
Unlike Europe, AFib amyloidosis in Asia has been rarely reported (Table 2) and has its own characteristics. The most common variant in Europe is c.1634A > T (E526V) (11), however, there is only one c.1634A > T mutation in the 8 cases of Asia, the other 7 cases are all with novel mutations, include one missense mutation, 4 deletion mutations and one insertion-deletion mutation, making diagnosis more difficult. Same as in Europe, kidney is the predominantly involved organ, which can present with proteinuria, edema, hypertension, and azotemia. But age of onset is significantly earlier (median age, 40 years old) than that in Europe [median age, 58 years old (11)], clinical symptoms seem to be more severe, some patients progressed to ESRD at a younger age. It may be related to more severe genotypes, which were resulted from frameshift mutations. Clinical presentation and gene mutation of AFib amyloidosis patients in Asia are summarized as Table 2.
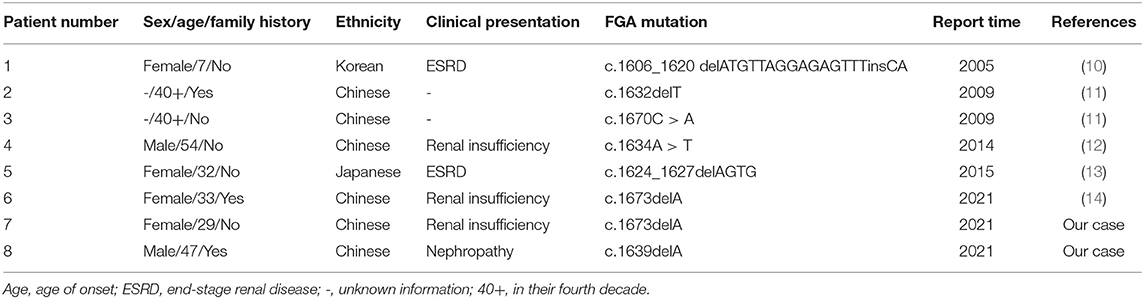
Table 2. Clinical presentation and gene mutation of AFib amyloidosis patients in Asia.
Diagnosis of AFib amyloidosis is based on clinicopathological findings, immunohistochemistry, mass spectrometry and detection of a FGA amyloidogenic genetic variant (15). On renal histology, AFib amyloidosis has its own characteristics with striking glomerular enlargement and exclusive glomerular deposition (11). It is generally recognized that there is little or no vascular or interstitial amyloid deposits in AFib amyloidosis. However, in our study, we found obvious amyloid deposits in the interstitium of renal medulla in Patient 1, which were positive for LECT2 in IHC. Genetic analysis of this patient revealed a single base transversion from A to G at position 172 of the LECT2 gene [c.172A>G (p.Ile58Val; NM_002302)], this is a common polymorphism (SNP rs31517) that are thought to be present in all ALECT2 amyloidosis patients (16). These evidences support that this patient coexisted with ALECT2 amyloidosis. Moreover, we found that the unevenly staining for amyloid precursors by IHC may lead to a diagnostic pitfall, just like Patient 2 [case 2 has been reported in ref. (13)]. Because proteomic analysis of ApoA-I and genetic analysis of the ApoA-I gene were both negative, the unevenly staining of ApoA-I by IHC was thought to be non-specific staining. This makes differential diagnosis of AFib amyloidosis more difficult. IHC is a commonly used method for the identification of amyloid type, however, it fails to identify the amyloid type in up to 30% systemic amyloidosis cases (9). As for our cases, immunohistochemistry was inconclusive in Patient 2. Mass spectrometry-based proteomic analysis is not routinely used in our country due to the high cost, but it can provide reliable information in amyloid type identification. LC-MS/MS proteomic analysis of the two patients corroborated existence of fibrinogen Aα in the amyloid deposits, leading to the diagnosis of AFib amyloidosis finally.
AFib amyloidosis has been rarely reported in China, Patient 1 has been misdiagnosed with AL amyloidosis in a local hospital and given inappropriate chemotherapy. Our experience suggests AFib amyloidosis should also be considered in the differential diagnosis of amyloidosis to avoid inappropriate and even harmful treatments. There is no effective therapy for AFib amyloidosis, treatment is restricted to interrupt further fibril formation, in combination with supportive care of failing organs. For those with ESRD, renal replacement therapy and kidney transplantation can be considered, but amyloid may deposit in the renal graft again. Combined liver and kidney transplantation can prevent the production of amyloidogenic fibrinogen and therefore is curative, but the procedure related mortality is high (17, 18).
Here we report two AFib amyloidosis patients in China, one of them coexisted with ALECT2 amyloidosis. Patients with AFib amyloidosis do exist in Asia, most of them had earlier onset age, with novel frameshift mutations. AFib amyloidosis may coexist with other types of amyloidosis, physicians should pay more attention to the differential diagnosis to avoid inappropriate and even harmful therapy.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Z-YL, SW, and D-YL performed the experiments of IHC, pathological studies, and gene testing. DL performed the mass spectrometry. X-JY, GL, F-DZ, and M-HZ was in charge of patient care and clinical data collection. Z-YL drafted this manuscript. S-XW designed this study and revised the manuscript critically for important intellectual content. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
This work was supported by the National Natural Science Foundation of China (No. 82170724).
1. Benson MD, Liepnieks J, Uemichi T, Wheeler G, Correa R. Hereditary renal amyloidosis associated with a mutant fibrinogen alpha-chain. Nat Genet. (1993) 3:252–5. doi: 10.1038/ng0393-252
2. Stangou AJ, Banner NR, Hendry BM, Rela M, Portmann B, Wendon J, et al. Hereditary fibrinogen A alpha-chain amyloidosis: phenotypic characterization of a systemic disease and the role of liver transplantation. Blood. (2010) 115:2998–3007. doi: 10.1182/blood-2009-06-223792
3. Yazaki M, Yoshinaga T, Sekijima Y, Kametani F, Okumura N. Hereditary fibrinogen aalpha-chain amyloidosis in asia: clinical and molecular characteristics. Int J Molec Sci. (2018) 19. doi: 10.3390/ijms19010320
4. Sethi S, Theis JD, Vrana JA, Fervenza FC, Sethi A, Qian Q, et al. Laser microdissection and proteomic analysis of amyloidosis, cryoglobulinemic GN, fibrillary GN, and immunotactoid glomerulopathy. Clin J Am Soc Nephrol. (2013) 8:915–21. doi: 10.2215/CJN.07030712
5. Talmud P, Tybjaerg-Hansen A, Bhatnagar D, Mbewu A, Miller JP, Durrington P, et al. Rapid screening for specific mutations in patients with a clinical diagnosis of familial hypercholesterolaemia. Atherosclerosis. (1991) 89:137–41. doi: 10.1016/0021-9150(91)90053-6
6. Machado JR, Silva MV, Neves PD, Oliveira FA, Correa RR, Rodrigues WV, et al. Fibrinogen A alpha-chain amyloidosis: report of the first case in Latin America. Amyloid: Int J Exper Clin Invest. (2013) 20:52–5. doi: 10.3109/13506129.2012.763029
7. Weisel JW, Litvinov RI. Mechanisms of fibrin polymerization and clinical implications. Blood. (2013) 121:1712–9. doi: 10.1182/blood-2012-09-306639
8. Paraboschi EM, Duga S, Asselta R. Fibrinogen as a pleiotropic protein causing human diseases: the mutational burden of Aalpha, Bbeta, and gamma chains. Int J Molec Sci. (2017) 18. doi: 10.3390/ijms18122711
9. Taylor GW, Gilbertson JA, Sayed R, Blanco A, Rendell NB, Rowczenio D, et al. Proteomic analysis for the diagnosis of fibrinogen aalpha-chain amyloidosis. Kidney Int Rep. (2019) 4:977–86. doi: 10.1016/j.ekir.2019.04.007
10. Kang HG, Bybee A, Ha IS, Park MS, Gilbertson JA, Cheong HI, et al. Hereditary amyloidosis in early childhood associated with a novel insertion-deletion (indel) in the fibrinogen Aalpha chain gene. Kidney Int. (2005) 68:1994–8. doi: 10.1111/j.1523-1755.2005.00653.x
11. Gillmore JD, Lachmann HJ, Rowczenio D, Gilbertson JA, Zeng CH, Liu ZH, et al. Diagnosis, pathogenesis, treatment, and prognosis of hereditary fibrinogen A alpha-chain amyloidosis. J Am Soc Nephrol. (2009) 20:444–51. doi: 10.1681/ASN.2008060614
12. Yao Y, Wang SX, Zhang YK. Hereditary fibrinogen Aalpha-chain amyloidosis caused by the E526V mutation: a case report and literature review. J Peking Univ Health Sci. (2014) 46:802–4. doi: 10.3969/j.issn.1671-167X.2014.05.026
13. Yazaki M, Yoshinaga T, Sekijima Y, Nishio S, Kanizawa Y, Kametani F, et al. The first pure form of Ostertag-type amyloidosis in Japan: a sporadic case of hereditary fibrinogen Aalpha-chain amyloidosis associated with a novel frameshift variant. Amyloid: Int J Exper Clin Invest. (2015) 22:142–4. doi: 10.3109/13506129.2015.1037389
14. Jin S, Shen Z, Li J, Lin P, Xu X, Ding X, et al. Fibrinogen A alpha-chain amyloidosis associated with a novel variant in a Chinese family. Kidney Int Rep. (2021) 6:2726–30. doi: 10.1016/j.ekir.2021.07.014
15. Tavares I, Oliveira JP, Pinho A, Moreira L, Rocha L, Santos J, et al. Unrecognized fibrinogen a alpha-chain amyloidosis: results from targeted genetic testing. Am J Kidney Dis. (2017) 70:235–43. doi: 10.1053/j.ajkd.2017.01.048
16. Ha JH, Tu HC, Wilkens S, Loh SN. Loss of bound zinc facilitates amyloid fibril formation of leukocyte cell-derived chemotaxin 2 (LECT2). J Biol Chem. (2021) 296:100446. doi: 10.1016/j.jbc.2021.100446
17. Rowczenio D, Stensland M, de Souza GA, Strom EH, Gilbertson JA, Taylor G, et al. Renal amyloidosis associated with 5 novel variants in the fibrinogen a alpha chain protein. Kidney Int Rep. (2017) 2:461–9. doi: 10.1016/j.ekir.2016.11.005
Keywords: amyloidosis, fibrinogen, LECT2, kidney, gene mutation, mass spectrometry, China
Citation: Li Z-Y, Wang S, Li D-Y, Liu D, Wang S-X, Yu X-J, Liu G, Zhou F-D and Zhao M-H (2022) Fibrinogen A Alpha-Chain Amyloidosis in Two Chinese Patients. Front. Med. 9:869409. doi: 10.3389/fmed.2022.869409
Received: 04 February 2022; Accepted: 06 April 2022;
Published: 28 April 2022.
Edited by:
Sergey BRODSKY, Ohio State University Hospital, United StatesReviewed by:
Dorota Monika Rowczenio, University College London, United KingdomCopyright © 2022 Li, Wang, Li, Liu, Wang, Yu, Liu, Zhou and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Su-Xia Wang, c3V4aWF3YW5nQGJqbXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.