 Judy Savige
Judy Savige Mary Huang
Mary Huang Marina Shenelli Croos Dabrera
Marina Shenelli Croos Dabrera Joel Gibson
Joel Gibson- Department of Medicine (Melbourne Health and Northern Health), Royal Melbourne Hospital, The University of Melbourne, Parkville, VIC, Australia
Alport syndrome is inherited as an X-linked (XL), autosomal recessive (AR), or autosomal dominant (AD) disease, where pathogenic COL4A3 – COL4A5 variants affect the basement membrane collagen IV α3α4α5 network. About 50% of pathogenic variants in each gene (major rearrangements and large deletions in 15%, truncating variants in 20%, splicing changes in 15%) are associated with “severe” disease with earlier onset kidney failure, and hearing loss and ocular abnormalities in males with XL inheritance and in males and females with AR disease. Severe variants are also associated with early proteinuria which is itself a risk factor for kidney failure. The other half of pathogenic variants are missense changes which are mainly Gly substitutions. These are generally associated with later onset kidney failure, hearing loss, and less often with major ocular abnormalities. Further determinants of severity for missense variants for XL disease in males, and in AD disease, include Gly versus non-Gly substitutions; increased distance from a non-collagenous interruption or terminus; and Gly substitutions with a more (Arg, Glu, Asp, Val, and Trp) or less disruptive (Ala, Ser, and Cys) residue. Understanding genotype-phenotype correlations in Alport syndrome is important because they help predict the likely age at kidney failure, and the need for early and aggressive management with renin-angiotensin system blockade and other therapies. Genotype-phenotype correlations also help standardize patients with Alport syndrome undergoing trials of clinical treatment. It is unclear whether severe variants predispose more often to kidney cysts or coincidental IgA glomerulonephritis which are recognized increasingly in COL4A3-, COL4A4 - and COL4A5-associated disease.
Introduction
Alport syndrome is the commonest inherited kidney disease and the second commonest cause of inherited kidney failure after polycystic kidney disease (1). It is characterized by persistent haematuria and a family history of haematuria or kidney failure (2). The typical clinical features are seen in males with X-linked (XL) disease due to pathogenic COL4A5 variants and in males and females with autosomal recessive (AR) disease and two pathogenic variants in COL4A3 or COL4A4. These result in haematuria, kidney failure, hearing loss and ocular abnormalities. Affected basement membranes including the glomerular membrane (GBM) are thinned, lamellated, and moth-eaten. Autosomal dominant (AD) Alport syndrome is caused by a heterozygous pathogenic variant in COL4A3 or COL4A4 and associated with isolated haematuria and a thinned rather than lamellated membrane (3, 4). With AD Alport syndrome the risk of kidney failure is small, and hearing loss and ocular abnormalities are rare (5). AD Alport syndrome is also sometimes known as thin basement membrane nephropathy and represents the carrier state for autosomal recessive Alport syndrome (3).
Structure of the Collagen IV Network
The six COL4A1–COL4A6 genes code for the collagen IV α1–α6 chains (6). These genes belong to two families, COL4A1-like (COL4A1, COL4A3, and COL4A5) and COL4A2-like (COL4A2, COL4A4, and COL4A6) which have all arisen from reduplication of COL4A1. These six genes are highly homologous and the corresponding collagen IV α chains resemble each other structurally.
Each collagen IV α chain comprises a non-collagenous amino and carboxy terminus and an intermediate collagenous sequence with Gly-Xaa-Yaa repeats (Figure 1) as well as multiple interruptions that confer flexibility (6). All collagen chains include Gly-Xaa-Yaa repeats but the collagen IV α chains differ in that they retain the non-collagenous amino and carboxy termini through which they interact to form the α1α1α2, α3α4α 5, or α5α5α6 heterotrimers and the chicken wire networks that predominate in membranes.
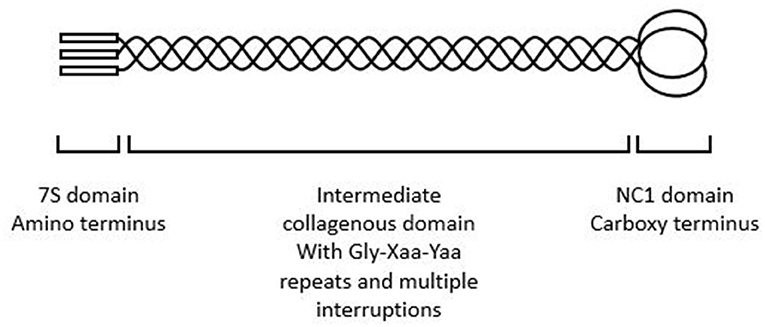
Figure 1. Collagen IV α3α4α5 heterotrimer demonstrating amino terminus, intermediate collagenous domain, and carboxy terminus. The intermediate collagenous domain comprises Gly-Xaa-Yaa repeats with multiple interruptions.
The α1α1α2 network is widely distributed in basement membranes in infancy and is found in adults in the blood vessels, nerves and muscles (6). The α3α4α5 trimer is the major constituent of membranes in the glomerulus (GBM), cochlea and eye having replaced the α1α1α2 trimer at about 2 years of age. The α5α5α6 trimer is found in Bowman's capsule in the kidney and in skin.
To date, more than 5,000 pathogenic variants have been described in the COL4A3–COL4A5 genes in the two main variant databases LOVD (https://www.lovd.nl/) and Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/). These are both open access databases that accept submissions from diagnostic laboratories and from the literature. LOVD also includes clinical features for many variants which allows genotype-phenotype correlations. Many more variants have been reported for COL4A5 than for COL4A3 and COL4A4 because genetic testing has been performed more often for individuals suspected of having X-linked Alport syndrome. Although the definitions of variant types differ in different databases, in general, pathogenic variants in each of the COL4A3–COL4A5 genes include 15% major rearrangements or large deletions, 20% truncating variants, 15% splicing changes, and 45% missense variants (Table 1).

Table 1. Types of all pathogenic variants in COL4A5, COL4A3, or COL4A4.
“Severe” variants in COL4A5 are more likely to be rearrangements or deletions, truncating, or splicing changes than missense variants (7). This means that overall severe changes account for about 50% of all pathogenic variants in each of these genes, and thus that the likelihood of finding a severe variant in a person with Alport syndrome is currently also 50%.
Deletions, frameshift variants, and termination codons all result in loss of the corresponding α chain from nonsense-mediated decay and subsequently of the collagen IV α3α4α5 triple helix from affected membranes. This is replaced with the α1α1α2 network (8) which is more susceptible to proteolysis (9). Initially the GBM is thinned but repeated episodes of damage and repair result in a lamellated, moth-eaten appearance. Splicing variants have different effects depending on whether they produce a truncating change or exon skipping (10).
In contrast, the α chains resulting from a missense variant are often associated with a disrupted trimer that is retained within the podocyte ER (11, 12), increasing ER stress and podocyte loss (13, 14). Of all the missense variants, the majority (30%) are Gly substitutions in the intermediate collagenous domain. Gly is the smallest amino acid that fits within the interior of the triple helix but can be replaced by 8 different amino acids or a termination codon (7). Most Gly substitutions are pathogenic because substitution with a larger amino acid distorts triple helix formation.
Advantages of Predicting Severity of Pathogenic Variants
It is useful for the clinician to be able to predict whether a pathogenic COL4A3–COL4A5 variant is likely associated with severe or mild features in Alport syndrome. The typical severe form is seen in males with X-linked disease who have recurrent macroscopic haematuria, early onset proteinuria and kidney failure, that generally requires dialysis or a kidney transplant well before the age of 30 years (15–17). The GBM may be thinned or lamellated, with the collagen IV α5 chain absent from a kidney or skin biopsy. Lenticonus, central fleck retinopathy, and temporal retinal atrophy are common, and a maculopathy or macular hole may occur (18, 19). In contrast “mild” disease is associated with haematuria, later onset proteinuria and kidney failure often in middle age, together with a thinned, possibly partly lamellated, GBM (20). There may be a late onset hearing loss, and a peripheral retinopathy is common (21).
The severe and mild phenotypes are subtly different in other forms of inheritance. Females with a severe pathogenic COL4A5 variant have an increased risk of proteinuria, and hearing loss is common but lenticonus does not occur (22). With AR Alport syndrome, two severe variants in COL4A3 or COL4A4 have a more damaging effect than one or none (23). The relationship is less clear for severe heterozygous COL4A3 or COL4A4 variants in AD Alport syndrome since kidney failure is not common and may be due to coincidental disease.
The advantages of being able to predict severe variants include being able to institute treatment to delay kidney failure onset. Renin-angiotensin-aldosterone system blockade delays kidney failure for years in males with X-linked disease, and the delay may be sufficiently long in women that they do not require dialysis or a kidney transplant (24, 25). SGLT2 inhibitors have demonstrated promise in Alport syndrome and are currently undergoing further investigation (26, 27). Severe variants may also indicate the need for earlier, and more aggressive, or more expensive interventions as they become available (28, 29). Some treatments are specific for severe disease. Demonstrating a severe variant may also represent a persuasive argument for dissuading an affected family member from acting as a kidney donor. In addition, recognizing severe variants is important in clinical trials where standardizing the risk of kidney failure helps compare treatment outcomes.
X-Linked Alport Syndrome in Males
The location where a pathogenic variant affects the collagen α chain (amino terminus, intermediate collagenous domain, interruptions, or carboxy terminus) and the variant type (large rearrangements, truncating, splicing, or missense variants) are the major determinants of variant severity and age at kidney failure, and likelihood of hearing loss and ocular abnormalities. Because pathogenic COL4A5 variants result in kidney failure in most males the genotype-phenotype relationship is understood best in males with X-linked disease.
Many studies of COL4A5 variants in males have found that severe changes (large rearrangements, nonsense and frameshift, and splicing variants) are associated with an earlier age at kidney failure than missense changes (15–17, 30). Splice site variants often have an effect intermediate between truncating and missense variants (16).
In the COL4A5 variants in the LOVD database, the mean age at kidney failure for all males was 25.1 ± 10.6 years (n = 326). Some studies have suggested that pathogenic changes in the first 20 exons of COL4A5 and hence the amino terminus of the collagen IV α5 chain is associated with milder disease (17) and others have found more severe disease with earlier onset kidney failure (16). In LOVD, the average age at kidney failure for variants in the amino terminus was 20.3 ± 5.4 (n = 14), 25.6 ± 11.1 years for variants in the intermediate domain (n = 262) and 24.0 ± 8.2 (n = 50) for those in the carboxy terminus suggesting a more severe effect for the amino terminus compared with the intermediate domain (p = 0.08) but not the carboxy terminus (p = 0.12). However 11 of the 14 amino terminus changes were deletions which have a more severe outcome and were likely to have biased the results. Thus variant types must also be taken into account before assessing the severity associated with a variant location.
In the LOVD database, the average age at kidney failure for men with deletions was 21.1 ± 6.8 years (n = 75) which was earlier than the overall mean age (p = 0.002) and for those with nonsense variants was 20.4 ± 5.0 years (n = 33, p = 0.01). The mean age for men with canonical splice site variants was 25.2 ± 10.7 years (n = 45, p = 0.95). Donor splice site changes have been associated with earlier onset kidney failure than acceptor variants (17) but the mean age for splice site variants at −1 or −2 was 25.2 ± 11.6 years (n = 21) and 25.1 ± 10.2 (n = 24) for variants at +1 or + 2 (p = 0.98 compared with each other, and p = 0.95 compared with the overall mean). Duplications were associated with a mean age of 23.9 ± 6.1 years (n = 15, p = 0.66 compared with overall mean).
In the LOVD database, Gly substitutions in the intermediate collagenous domain [mean age at kidney failure of 28.4 ± 12.4 years (n = 52)] had a worse outcome than non-Gly substitutions [40.7 ± 17.6 years (n = 5), p = 0.03] (16).
Gly substitutions represent the commonest missense variants in COL4A5, currently accounting for most unique pathogenic variants in males (124/147, 84%).
Gly substitutions distant from a non-collagenous sequence (interruption, amino, or carboxy terminus) had a more severe phenotype with earlier onset kidney failure than Gly substitutions adjacent to a non-collagenous sequence (31). In addition substitutions with highly destabilizing residues such as Glu, Asp, Arg, Val, or Trp were associated with earlier onset kidney failure than substitutions with the less-destabilizing, Ala, Ser, or Cys (31). In LOVD, the mean age at kidney failure was 24.6 ± 8.6 for severe Gly substitutions (n = 83) and 31.1 ± 14.2 for non-severe substitutions (n = 68, p = 0.0007).
Treatment also affects the age at kidney failure for truncating variants and non-truncating variants differently. The age at kidney failure for both truncating and non-truncating variants is delayed by RAAS blockade but treatment delayed kidney failure more for non- truncating variants (30).
Severe variants are also associated with hearing loss (15–17), and ocular abnormalities, in particular, lenticonus, and fleck retinopathy (15–17). No effect of variant severity has been demonstrated on macroscopic haematuria, hypertension, or GBM thickness (15, 16).
X-Linked Alport Syndrome in Females
The correlation between COL4A5 variant location, type and phenotype has been less clear for females with X-linked Alport syndrome. Indeed an early large study found no genotype-phenotype correlation in women with X-linked disease (32). They also found no correlation between earlier age at kidney failure in men and women from the same family (32).
Part of the lack of correlation in women has been attributed to random X chromosome inactivation (33). However it is also likely to be partly due to so few women developing kidney failure and, of those who do, this occurring in later life. Women develop hearing loss and ocular abnormalities less often too. Thus recent studies have focused instead on the relationship between genotype and proteinuria since proteinuria typically precedes kidney failure (34).
Thus a recent large study of 336 females with genetically-proven X-linked Alport syndrome from 179 families where about half the pathogenic variants were severe found that 175 (73%) had proteinuria at a median age of 7 years (34). Fifty-two of the 336 (15%) had kidney failure by the age of 65 years.
Another smaller cohort found that severe variants (all those other than missense changes) were more commonly associated with proteinuria and impaired kidney function than missense variants (35). While this cohort also included occasional individuals with digenic inheritance these were unlikely to have had a significant effect.
No studies in women with pathogenic COL4A5 variants have demonstrated any genotype-phenotype correlations for hearing loss or ocular abnormalities.
In summary there may be a similar genotype-phenotype correlation in women with pathogenic COL4A5 variants and proteinuria as occurs in men with kidney failure but with a smaller effect that requires a larger cohort for its demonstration. Other factors contributing to the age at kidney failure as well as X chromosome activation, include coincidental causes including hypertension, diabetes and obesity.
AR Alport Syndrome
Individuals with AR Alport syndrome have two pathogenic variants in COL4A3 or COL4A4, and the COL4A3 and COL4A4 genes are affected equally often. In the LOVD database, the age at kidney failure was not different for AR and for X-linked Alport syndrome (24.4 ± 7.8 years, n = 237, p = 0.39), and in the case of AR disease, was not different for COL4A3 (23.2 ± 9.3, n = 35, p = 0.45) or COL4A4 (25.4 ± 10.3, n = 26, p = 0.55) variants (7).
Individuals with two truncating variants were more likely to develop kidney failure before 30 years of age than those with only one truncating variant, who were in turn more likely than those with no truncating variant (23). This correlation has now been confirmed in a small but unrelated cohort where both age at kidney failure and age at hearing loss were earlier with at least one truncating variant than with none (36). A further study confirmed that the presence of at least one milder (missense) variant delayed kidney failure onset (p = 0.024), and that kidney survival was further increased with two missense variants [p = 0.016; (37)]. In this study, individuals with missense variants had a later onset of kidney failure, hearing loss and ocular abnormalities (37).
AD Alport Syndrome
There are now several large studies and a meta-analysis of genotype-phenotype analyses in individuals with AD Alport syndrome (5, 31, 38, 39), but again the difficulty is that kidney failure is uncommon in these individuals, a large cohort might be needed to demonstrate a small effect, and kidney failure may result from coincidental causes. However again proteinuria may represent a surrogate marker for kidney failure (5).
A recent analysis of the UK 100K Genomes Project database demonstrated that certain variant types were associated with a higher penetrance of haematuria (31). It examined COL4A3 and COL4A4 variants that resulted in a Gly substitution and found that substitutions with a highly destabilizing residue (Arg, Glu, Asp, Val, and Trp) were associated with an increased risk of haematuria [p = 0.018; (31)], and that substitutions adjacent to a non-collagenous interruption or amino or carboxy terminus were associated less often with haematuria (p < 0.001). There was no association between haematuria and proximity to the amino terminus for missense variants that were Gly substitutions.
A further study of 240 individuals from 78 families with genetically-proven COL4A3 or COL4A4 variants, included 61 who developed kidney failure (24%) at a median of 67 years (58–73) (5). Fifty-seven percent of their variants were missense and the others were “severe.” In this study there was no difference in the age at kidney failure for truncating variants or for missense variants that were or were not Gly substitutions (p = 0.3); nor for Gly substitutions, splicing or missense variants (p = 0.90); nor Gly substitutions with Arg, Glu, or Asp or other severe non-missense variant types compared with other Gly substitutions [p = 0.5; (5)]. There was however a large intrafamilial variability in age at kidney failure.
A further study found no difference in the age at developing proteinuria or kidney failure for missense and non-missense variants (38).
However a meta-analysis of family members with 74 variants where 20 were not missense found the median age at kidney failure was 55 years for missense variants and 47 years for non-missense changes [p = 0.02; (39)].
At least two of these studies had a very wide variation in age at kidney failure (5, 39).
Hypomorphic Variants
Hypomorphic or variants associated with milder disease are recognized increasingly in the COL4A3–COL4A5 genes (Table 2). They are often clinically unrecognised and are commonly found in normal variant databases such as gnomAD (40). They may have features that are the opposite of severe variants such as occurring in women (for COL4A5 changes); and for all COL4A3–COL4A5 variants, missense variants, especially Gly substitution with Ala, Ser, or Cys; or Gly substitutions adjacent to a non-collagenous terminus or interruption; or non-Gly substitutions.
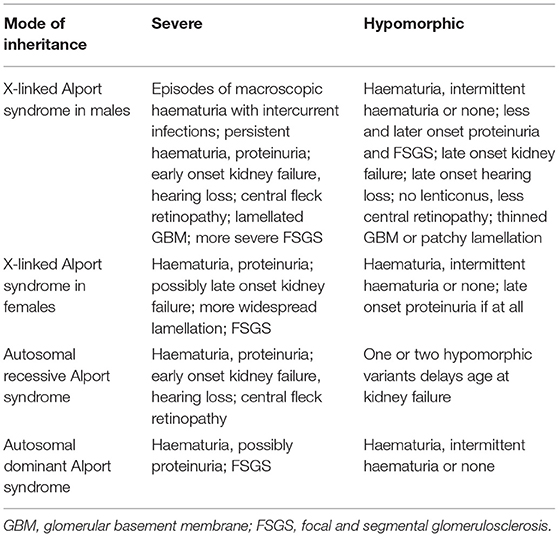
Table 2. Clinical phenotype of severe and hypomorphic variants.
Thus, two of the commonest hypomorphic variants are p.Gly624Asp in COL4A5 and p.Leu1474Pro in COL4A3. p.Gly624Asp in COL4A5 is found in about one in 3,000 of the European population or about one quarter of all Europeans with X-linked Alport syndrome (20, 41). This variant is located immediately adjacent to a non-collagenous interruption in the intermediate collagenous domain which contributes to its mild phenotype. It is associated with late onset kidney failure, hearing loss, but a normal ocular examination, and often a thinned rather than lamellated GBM.
The p.Leu1474Pro variant in COL4A3 occurs in up to one in 200 people. The substitution occurs in the carboxy non-collagenous domain. On its own it may be associated with a normal urinary sediment, proteinuria and FSGS (42) or in association with another COL4A3 variant, with AR Alport syndrome, and kidney failure.
Predicting Severe Variants
In summary, the determinants for variant severity appear to be the same for all the COL4A3–COL4A5 genes (Table 3). The renal physician may be assisted in deciding on the variant severity in their patient after consultation with a clinical geneticist and a multidisciplinary team. About half the pathogenic variants in each gene (major rearrangements, large deletions, truncating variants, some splicing variants) are associated with more severe disease, with earlier onset proteinuria and kidney failure, hearing loss, and ocular abnormalities in males with XL and in males and females with AR disease. For AD Alport syndrome severe changes may only increase the penetrance of haematuria. Interestingly a correlation between severe variants and proteinuria may be emerging for women with XL disease.
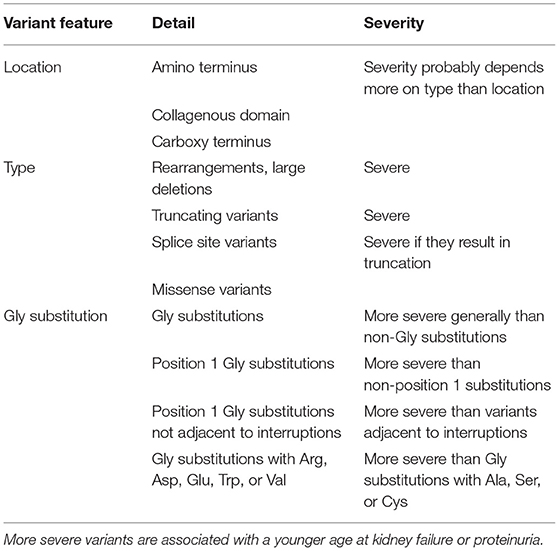
Table 3. Factors determining severity of genetic variants in the COL4A3–COL4A5 genes.
There are also two objective methods for predicting the clinical effects of COL4A3–COL4A5 variants but these are not widely available. One is immunohistochemical staining for the collagen IV α5 chain in the kidney or skin (43). Males with absent staining have a more abnormal GBM and a worse prognosis than those with positive staining (44, 45). The other method is still a research tool but uses an in vitro expression system to examine collagen IV α3α4α5 heterotrimer formation and secretion (46). Reduced formation and secretion correlate with proteinuria development and early onset kidney failure (46).
Caveats to Predicting Clinical Features From Genotype
However there are also caveats to predicting the clinical features from the genotype based on previously reported studies.
Firstly, there is a bias to reporting more severe disease. Most publications are of hospital—based series where affected individuals have the typical Alport features with kidney failure.
Secondly, sometimes the phenotype varies even in family members with the same pathogenic variant. Twenty-three families with XL Alport syndrome, each with at least 3 males with kidney failure, included 17 families (74%) where men had a consistent age at kidney failure and six with varying ages (26%) (15). Missense and splice site variants are especially associated with divergent ages. Other studies have found more consistency (15–17). Women with XL disease and males and females with AD Alport syndrome are the populations with the most variation in the age at kidney failure. This is possibly because they have a smaller genetic risk of kidney failure and their likelihood and age may be distorted by coincidental diseases such as hypertension, obesity, diabetes, or modifying variants in other filtration barrier genes (47).
Thirdly, the mode of inheritance must be considered. It has been more difficult to correlate COL4A5 variants in women with age at kidney failure because of random X chromosome inactivation. Women and girls have heterozygous COL4A5 variants twice as often as men but typically have milder disease (22). Only 20% have kidney failure by the age of 60 years but hearing loss and peripheral fleck retinopathy are common (32). Milder disease occurs because, overall with balanced X inactivation, only half the female podocytes are affected and produce defective trimers. When X inactivation is skewed, a higher or lower proportion of podocytes are affected, and the phenotype varies correspondingly. With autosomal recessive inheritance, the clinical severity depends on the consequences of both variants.
Fourthly, genetic testing techniques have changed over the past 20 years of reporting genotype-phenotype studies. Many families with typical disease underwent testing when it first became available and those tested now are often demonstrated to have milder variants (Table 4). In addition, the currently- used technique of Whole Exome Sequencing is less sensitive for detecting large deletions, and hypomorphic variants are recognized increasingly (48).
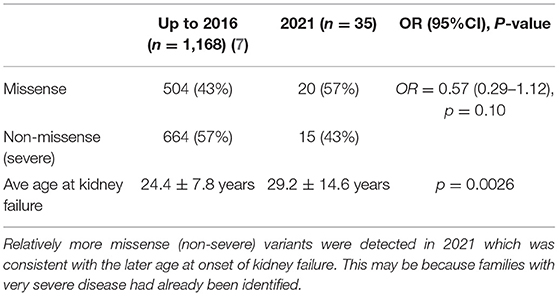
Table 4. Average age at kidney failure in males with pathogenic COL4A5 variants in LOVD in 2021 compared with 2016.
Fifthly, commonly-used treatments such as renin-angiotensin-aldosterone inhibitors modify the age at kidney failure in Alport syndrome (24, 25). Treatments may also modify the hearing loss and ocular abnormalities.
Future Studies
Future studies will focus on confirming more closely a genotype-phenotype correlation using proteinuria rather than kidney failure in women with X-linked Alport syndrome and in individuals with heterozygous COL4A3 or COL4A4 pathogenic variants. It may be possible to develop a more precise algorithm for predicting the age at kidney failure. More information will be available about hypomorphic variants, and further studies may explain whether severe COL4A3–COL4A5 variants predispose to IgA nephropathy and cystic kidney disease, or reduce the risk of diabetic nephropathy (49).
The detection of a severely-damaging genetic variant represents a powerful argument for starting treatment with ACE inhibitors, and possibly other more specific treatments much earlier in order to delay the onset of end-stage kidney failure.
Author Contributions
JS designed the project and wrote the first draft. MH, MC, and KS undertook the analysis of all the variants in the LOVD database. JG read the manuscript, provided the figure, added to the discussion, and corrected the draft. All authors reviewed and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Savige J, Gregory M, Gross O, Kashtan C, Ding J, Flinter F. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol. (2013) 24:364–75. doi: 10.1681/ASN.2012020148
3. Savige J, Rana K, Tonna S, Buzza M, Dagher H, Wang YY. Thin basement membrane nephropathy. Kidney Int. (2003) 64:1169–78. doi: 10.1046/j.1523-1755.2003.00234.x
4. Kashtan CE, Ding J, Garosi G, Heidet L, Massella L, Nakanishi K, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV alpha345: a position paper of the Alport Syndrome Classification Working Group. Kidney Int. (2018) 93:1045–51. doi: 10.1016/j.kint.2017.12.018
5. Furlano M, Martinez V, Pybus M, Arce Y, Crespi J, Venegas MDP, et al. Clinical and genetic features of autosomal dominant Alport syndrome: a cohort study. Am J Kidney Dis. (2021) 78:560–70.e1. doi: 10.1053/j.ajkd.2021.02.326
6. Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med. (2003) 348:2543–56. doi: 10.1056/NEJMra022296
7. Savige J, Storey H, Il Cheong H, Gyung Kang H, Park E, Hilbert P, et al. X-linked and autosomal recessive Alport syndrome: pathogenic variant features and further genotype-phenotype correlations. PLoS ONE. (2016) 11:e0161802. doi: 10.1371/journal.pone.0161802
8. Bateman JF, Boot-Handford RP, Lamande SR. Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat Rev Genet. (2009) 10:173–83. doi: 10.1038/nrg2520
9. Kalluri R, Shield CF, Todd P, Hudson BG, Neilson EG. Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J Clin Invest. (1997) 99:2470–78. doi: 10.1172/JCI119431
10. Horinouchi T, Nozu K, Yamamura T, Minamikawa S, Omori T, Nakanishi K, et al. Detection of splicing abnormalities and genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol. (2018) 29:2244–54. doi: 10.1681/ASN.2018030228
11. Pieri M, Stefanou C, Zaravinos A, Erguler K, Stylianou K, Lapathitis G, et al. Evidence for activation of the unfolded protein response in collagen IV nephropathies. J Am Soc Nephrol. (2014) 25:260–75. doi: 10.1681/ASN.2012121217
12. Wang D, Mohammad M, Wang Y, Tan R, Murray LS, Ricardo S, et al. The chemical chaperone, PBA, reduces ER stress and autophagy and increases collagen IV alpha5 expression in cultured fibroblasts from men with X-linked Alport syndrome and missense mutations. Kidney Int Rep. (2017) 2:739–48. doi: 10.1016/j.ekir.2017.03.004
13. Wickman L, Hodgin JB, Wang SQ, Afshinnia F, Kershaw D, Wiggins RC. Podocyte depletion in thin GBM and Alport syndrome. PLoS ONE. (2016) 11:e0155255. doi: 10.1371/journal.pone.0155255
14. Nicklason E, Mack H, Beltz J, Jacob J, Farahani M, Colville D, et al. Corneal endothelial cell abnormalities in X-linked Alport syndrome. Ophthalmic Genet. (2020) 41:13–19. doi: 10.1080/13816810.2019.1709126
15. Jais JP, Knebelmann B, Giatras I, Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol. (2000) 11:649–57. doi: 10.1681/ASN.V114649
16. Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA, Masoumi A, et al. Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol. (2010) 21:876–83. doi: 10.1681/ASN.2009070784
17. Gross O, Netzer KO, Lambrecht R, Seibold S, Weber M. Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: impact on clinical counselling. Nephrol Dial Transplant. (2002) 17:1218–27. doi: 10.1093/ndt/17.7.1218
18. Ahmed F, Kamae KK, Jones DJ, Deangelis MM, Hageman GS, Gregory MC, et al. Temporal macular thinning associated with X-linked Alport syndrome. JAMA Ophthalmol. (2013) 131:777–82. doi: 10.1001/jamaophthalmol.2013.1452
19. Chen Y, Colville D, Ierino F, Symons A, Savige J. Temporal retinal thinning and the diagnosis of Alport syndrome and thin basement membrane nephropathy. Ophthalmic Genet. (2018) 39:208–14. doi: 10.1080/13816810.2017.1401088
20. Zurowska AM, Bielska O, Daca-Roszak P, Jankowski M, Szczepanska M, Roszkowska-Bjanid D, et al. Mild X-linked Alport syndrome due to the COL4A5 G624D variant originating in the Middle Ages is predominant in Central/East Europe and causes kidney failure in midlife. Kidney Int. (2021) 99:1451–8. doi: 10.1016/j.kint.2020.10.040
21. Shaw EA, Colville D, Wang YY, Zhang KW, Dagher H, Fassett R, et al.: Characterization of the peripheral retinopathy in X-linked and autosomal recessive Alport syndrome. Nephrol Dial Transplant. (2007) 22:104–8. doi: 10.1093/ndt/gfl607
22. Savige J, Colville D, Rheault M, Gear S, Lennon R, Lagas S, et al. Alport syndrome in women and girls. Clin J Am Soc Nephrol. (2016) 11:1713–20. doi: 10.2215/CJN.00580116
23. Storey H, Savige J, Sivakumar V, Abbs S, Flinter FA: COL4A3/COL4A4 mutations and features in individuals with autosomal recessive Alport syndrome. J Am Soc Nephrol. (2013) 24:1945–54. doi: 10.1681/ASN.2012100985
24. Gross O, Licht C, Anders HJ, Hoppe B, Beck B, Tonshoff B, et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. (2012) 81:494–501. doi: 10.1038/ki.2011.407
25. Temme J, Peters F, Lange K, Pirson Y, Heidet L, Torra R, et al. Incidence of renal failure and nephroprotection by RAAS inhibition in heterozygous carriers of X-chromosomal and autosomal recessive Alport mutations. Kidney Int. (2012) 81:779–83. doi: 10.1038/ki.2011.452
26. Boeckhaus J, Gross O. Sodium-glucose cotransporter-2 inhibitors in patients with hereditary podocytopathies, Alport syndrome, and FSGS: a case series to better plan a large-scale study. Cells. (2021) 10:1815. doi: 10.3390/cells10071815
27. Mabillard H, Sayer JA. SGLT2 inhibitors - a potential treatment for Alport syndrome. Clin Sci. (2020) 134:379–88. doi: 10.1042/CS20191276
28. Omachi K, Miner JH. Alport syndrome therapeutics: ready for prime-time players. Trends Pharmacol Sci. (2019) 40:803–6. doi: 10.1016/j.tips.2019.07.012
29. Yamamura T, Horinouchi T, Adachi T, Terakawa M, Takaoka Y, Omachi K, et al. Development of an exon skipping therapy for X-linked Alport syndrome with truncating variants in COL4A5. Nat Commun. (2020) 11:2777. doi: 10.1038/s41467-020-16605-x
30. Yamamura T, Horinouchi T, Nagano C, Omori T, Sakakibara N, Aoto Y, et al. Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked Alport syndrome. Kidney Int. (2020) 98:1605–14. doi: 10.1016/j.kint.2020.06.038
31. Gibson JT, Huang M, Shenelli Croos Dabrera M, Shukla K, Rothe H, Hilbert P, et al. Genotype-phenotype correlations for COL4A3-COL4A5 variants that result in Gly substitutions in Alport syndrome. Sci Rep. (2022) 12:2722. doi: 10.1038/s41598-022-06525-9
32. Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study. J Am Soc Nephrol. (2003) 14:2603–10. doi: 10.1097/01.asn.0000090034.71205.74
33. Rheault MN, Kren SM, Hartich LA, Wall M, Thomas W, Mesa HA, et al. X-inactivation modifies disease severity in female carriers of murine X-linked Alport syndrome. Nephrol Dial Transplant. (2010) 25:764–69. doi: 10.1093/ndt/gfp551
34. Yamamura T, Nozu K, Fu XJ, Nozu Y, Ye MJ, Shono A, et al. Natural history and genotype-phenotype correlation in female X-linked Alport syndrome. Kidney Int Rep. (2017) 2:850–5. doi: 10.1016/j.ekir.2017.04.011
35. Mastrangelo A, Giani M, Groppali E, Castorina P, Solda G, Robusto M, et al. X-linked Alport syndrome in women: genotype and clinical course in 24 cases. Front Med. (2020) 7:580376. doi: 10.3389/fmed.2020.580376
36. Oka M, Nozu K, Kaito H, Fu XJ, Nakanishi K, Hashimura Y, et al. Natural history of genetically proven autosomal recessive Alport syndrome. Pediatr Nephrol. (2014) 29:1535–44. doi: 10.1007/s00467-014-2797-4
37. Lee JM, Nozu K, Choi DE, Kang HG, Ha IS, Cheong HI. Features of autosomal recessive Alport syndrome: a systematic review. J Clin Med. (2019) 8:178. doi: 10.3390/jcm8020178
38. Kamiyoshi N, Nozu K, Fu XJ, Morisada N, Nozu Y, Ye MJ, et al. Genetic, clinical, and pathologic backgrounds of patients with autosomal dominant Alport syndrome. Clin J Am Soc Nephrol. (2016) 11:1441–9. doi: 10.2215/CJN.01000116
39. Matthaiou A, Poulli T, Deltas C. Prevalence of clinical, pathological and molecular features of glomerular basement membrane nephropathy caused by COL4A3 or COL4A4 mutations: a systematic review. Clin Kidney J. (2020) 13:1025–36. doi: 10.1093/ckj/sfz176
40. Gibson J, Fieldhouse R, Chan MMY, Sadeghi-Alavijeh O, Burnett L, Izzi V, et al. Prevalence estimates of predicted pathogenic COL4A3–COL4A5 variants in a population sequencing database and their implications for Alport syndrome. J Am Soc Nephrol. (2021) 32:2273–90. doi: 10.1681/ASN.2020071065
41. Pierides A, Voskarides K, Kkolou M, Hadjigavriel M, Deltas C. X-linked, COL4A5 hypomorphic Alport mutations such as G624D and P628L may only exhibit thin basement membrane nephropathy with microhematuria and late onset kidney failure. Hippokratia. (2013) 17:207–13. Available online at: pubmed.ncbi.nlm.nih.gov/24470729/
42. Malone AF, Phelan PJ, Hall G, Cetincelik U, Homstad A, Alonso AS, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. (2014) 86:1253–59. doi: 10.1038/ki.2014.305
43. Nakanishi K, Yoshikawa N, Iijima K, Kitagawa K, Nakamura H, Ito H, et al. Immunohistochemical study of alpha 1-5 chains of type IV collagen in hereditary nephritis. Kidney Int. (1994) 46:1413–21. doi: 10.1038/ki.1994.413
44. Hashimura Y, Nozu K, Kaito H, Nakanishi K, Fu XJ, Ohtsubo H, et al. Milder clinical aspects of X-linked Alport syndrome in men positive for the collagen IV alpha5 chain. Kidney Int. (2014) 85:1208–13. doi: 10.1038/ki.2013.479
45. Said SM, Fidler ME, Valeri AM, McCann B, Fiedler W, Cornell LD, et al. Negative staining for COL4A5 correlates with worse prognosis and more severe ultrastructural alterations in males with Alport syndrome. Kidney Int Rep. (2017) 2:44–52. doi: 10.1016/j.ekir.2016.09.056
46. Kamura M, Yamamura T, Omachi K, Suico MA, Nozu K, Kaseda S, et al. Trimerization and genotype-phenotype correlation of COL4A5 mutants in Alport syndrome. Kidney Int Rep. (2020) 5:718–26. doi: 10.1016/j.ekir.2020.01.008
47. Voskarides K, Papagregoriou G, Hadjipanagi D, Petrou I, Savva I, Elia A, et al. COL4A5 and LAMA5 variants co-inherited in familial hematuria: digenic inheritance or genetic modifier effect? BMC Nephrol. (2018) 19:114. doi: 10.1186/s12882-018-0906-5
48. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
Keywords: genotype-phenotype correlation, Alport syndrome, COL4A3, COL4A4, COL4A5, XL Alport syndrome, AR Alport syndrome, AD Alport syndrome
Citation: Savige J, Huang M, Croos Dabrera MS, Shukla K and Gibson J (2022) Genotype-Phenotype Correlations for Pathogenic COL4A3–COL4A5 Variants in X-Linked, Autosomal Recessive, and Autosomal Dominant Alport Syndrome. Front. Med. 9:865034. doi: 10.3389/fmed.2022.865034
Received: 29 January 2022; Accepted: 24 March 2022;
Published: 06 May 2022.
Edited by:
Roberta Fenoglio, Ospedale San Giovanni Bosco, ItalyReviewed by:
Daw-Yang Hwang, National Health Research Institutes, TaiwanFlorian Grahammer, University Medical Center Hamburg-Eppendorf, Germany
Copyright © 2022 Savige, Huang, Croos Dabrera, Shukla and Gibson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Judy Savige, amFzYXZpZ2VAdW5pbWVsYi5lZHUuYXU=