 Cedric Happi Mbakam
Cedric Happi Mbakam Gabriel Lamothe1,2
Gabriel Lamothe1,2 Jacques P. Tremblay
Jacques P. Tremblay- 1Centre de Recherche du CHU de Québec-Université Laval, Quebec City, QC, Canada
- 2Department of Molecular Medicine, Faculty of Medicine, Laval University, Quebec City, QC, Canada
Duchenne muscular dystrophy (DMD) is an X-linked hereditary disease characterized by progressive muscle wasting due to modifications in the DMD gene (exon deletions, nonsense mutations, intra-exonic insertions or deletions, exon duplications, splice site defects, and deep intronic mutations) that result in a lack of functional dystrophin expression. Many therapeutic approaches have so far been attempted to induce dystrophin expression and improve the patient phenotype. In this manuscript, we describe the relevant updates for some therapeutic strategies for DMD aiming to restore dystrophin expression. We also present and analyze in vitro and in vivo ongoing experimental approaches to treat the disease.
Introduction
Duchenne muscular dystrophy (DMD) is a lethal X-linked recessive disease caused by mutations in the DMD gene coding for dystrophin protein (1). The DMD gene has been found to be altered by more than 4,000 mutations leading to the absence of dystrophin expression under the sarcolemma of the affected patients (2). Nowadays, many therapeutic approaches to address the DMD pathology are elaborated with the final expectation of an effective and safe treatment to cure the disease. Some of these drugs have already obtained accelerated approval as treatments for DMD. However, so far none of these approved treatments or those still in ongoing clinical trials are able to restore a permanent expression of dystrophin under the sarcolemma. In the present review, we present the major molecular approaches for therapeutics leading to dystrophin restoration.
Dystrophin Structure
Dystrophin is one of the most important proteins required for structural and functional muscle dynamism (1). It is encoded by one of the biggest known genes in humans representing about 0.1% of the entire genome (3). The DMD gene contains 79 exons representing approximately 2,200 kb. The dystrophin protein is localized under the sarcolemma, the cell membrane of the striated muscle fibers (Figure 1A). This protein is divided into four essential domains. The first domain is the actin binding amino terminal domain (ABD1), which contains two calponin homology domains (CH1 and CH2) that bind directly to actin filaments. This binding links the dystrophin to the subsarcolemmal actin network and connects it to the contractile apparatus in skeletal muscle cells (4). The second domain is the dystrophin central rod domain, which contains 24 spectrin-like repeats, each made of three alpha helices. This domain contains the second actin binding motif (ABD2), localized around the middle of the rod domain, which interacts with the ABD1, forming a strong association with actin filaments. The rod domain interacts with microtubules and membrane phospholipids, it also provides elasticity to the dystrophin and facilitates interactions with other proteins through hinges made of proline rich spacers (3, 5). The third domain is the cysteine rich domain located between the rod domain and the carboxyl terminal domain. The cysteine rich domain binds the ankyrin, a protein that plays a key role in maintaining dystrophin under the sarcolemma. This domain is required for the dystrophin function (6). The fourth and last domain of dystrophin is the carboxy terminal domain, a crucial component of dystrophin that contains two regions forming alpha helical coiled coils, a protein motif involved as hinge protein into the protein–protein interaction. This domain also provides binding sites for syntrophin and dystrobrevin (3) (Figure 1B). These four domains interact with other structures of the cell cytoskeleton at the extra cellular level with alpha-dystroglycan, at the transmembrane level with dystroglycan, sarcoglycan, and sarcospan, and at the cytoplasmic level with dystrobrevin, syntrophin, and nNOS to form what is called the dystrophin complex (3). This complex is necessary for the maintenance of the membrane stability. Any modification in the dystrophin complex structure could result in various myopathies amongst which the DMD, which is the most common myopathy.
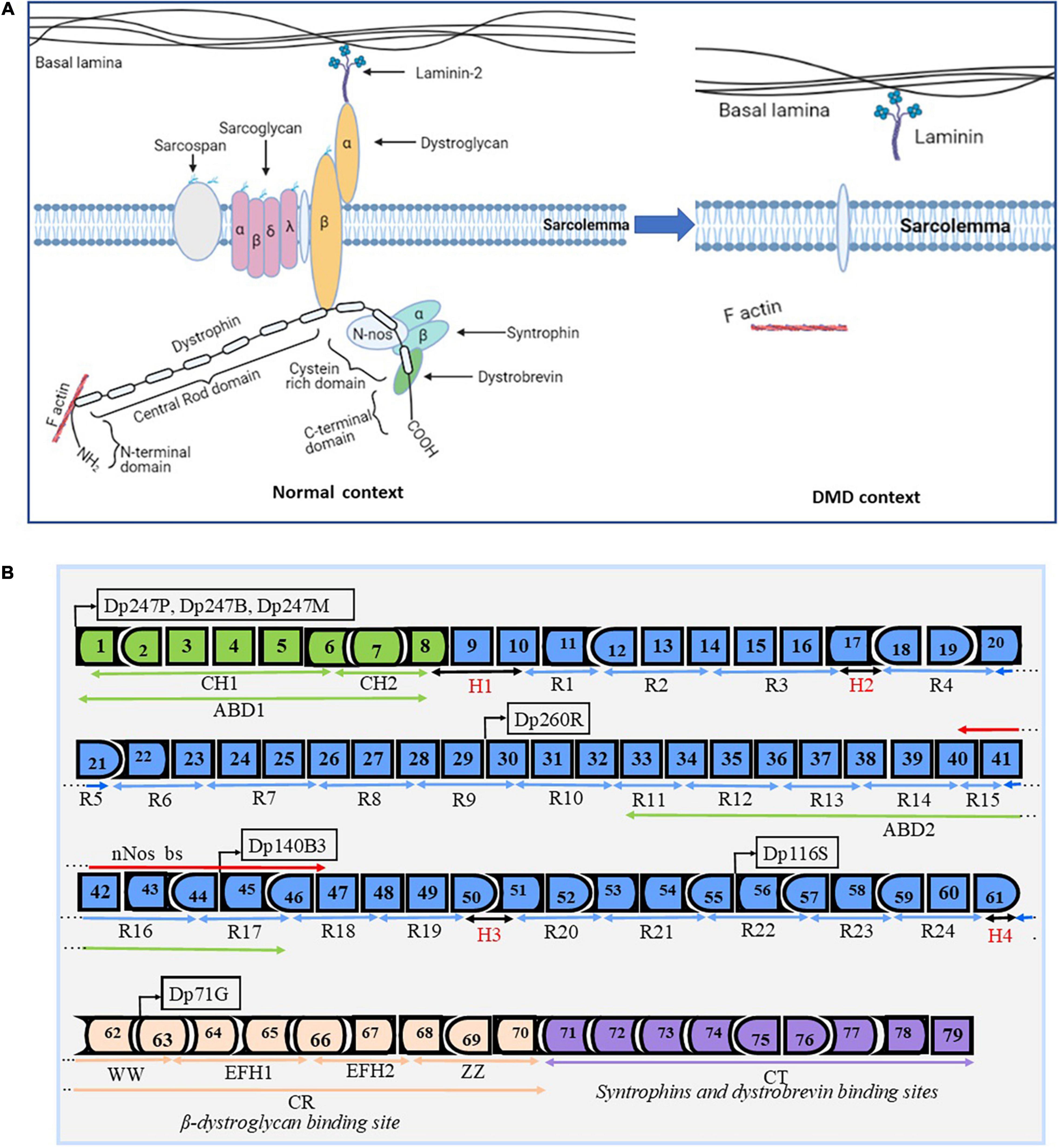
Figure 1. Dystrophin structure and protein associated domain. Design and adapted from the following references (1, 3, 147, 148). Panel (A) represents the basic structure of the dystrophin complex in the normal context and in a DMD context. In the normal context, dystrophin is present under the sarcolemma and is properly attached to other components to form the dystrophin complex. In the DMD context, there is an absence of the dystrophin complex, which results in the absence of the dystrophin complex and in muscle fiber degradation during muscle contraction. Panel (B) depicts the dystrophin gene (exons 1–79) with different functional domains: 1- the N-terminal actin binding domain (ABD1 in green) with its two calponin-homology motifs (CH1 and CH2). This domain is required for the function of the dystrophin protein. 2- The central rod domain (CR in blue color) contains 24 spectrin-like repeats (R1–R24), the normal alignment of this region is fundamental for a correctly functional dystrophin protein. This domain is interrupted and bordered by four proline rich hinge regions (H1, H2, H3, and H4). It also contains the second actin binding domain (ABD2) and the nNos binding site (nNos bs). 3- The Cysteine rich domain contains the WW domain, EF hands, and ZZ domain. This domain serves as the binding site for beta dystroglycan and it is required for a functional dystrophin. 4- The C terminal domain serves as a binding site for syntrophins and the dystrobrevin glycoprotein. This domain is also required for a functional dystrophin. The DMD gene encodes seven different protein isoforms under different tissues specific promoters (Dp427P, Dp427B, Dp427M, Dp260R, Dp140B3, Dp116S, and Dp71G, respectively for cerebellar Purkinje cells, brain, muscle, retina, and Schwann cells).
Duchenne Muscular Dystrophy Pathogenesis
Duchenne muscular dystrophy is an inherited disease characterized by progressive muscle wasting due to mutations in the DMD gene coding for the dystrophin protein (7). There are many dystrophin isoforms whose expression generally depends on tissue-specific promoters, mRNA splice events, and poly-adenylation signal sites. The muscle dystrophin protein (Dp427m), a protein of 427 kDa, is the most important and the most implicated in DMD (8). The absence of dystrophin under the sarcolemma makes the muscle fibers more vulnerable during muscle contraction. Since it is a recessive X-linked disease, males are more likely to be affected (7).
The frequency of DMD mutations may depends on geography and race. In an updated systematic review and meta-analysis study conducted by Crisafulli et al. (9), the global prevalence of DMD was estimated to be 7.1 cases per 100,000 males and 2.8 cases per 100,000 individuals in the general population. In the pediatric population, the prevalence was about 19.8 per 100,000 live male births. In another study conducted by Ryder et al. (10), the DMD prevalence was evaluated at 10.9 per 100,000 males in France, 1.9 per 100,000 males in United States, 2.2 per 100,000 males in United Kingdom and 6.1 per 100,000 males in Canada.
Single or multi-exon deletions account in average for about 60–70% of cases depending on the geographical region of the world (64% in Oceania, 66% in Europe, 70% in America, 72% in Asia, and 88% in Africa) thus representing about 2/3 of all DMD cases (11). Two regions of the DMD gene are considered hotspots for mutations (12). The first and most important is between exons 45 and 55; deletions in this zone remove the central part of the rod domain. The second hotspot concerns the region between exon 3 to exon 19 of the DMD gene. Deletions in this zone remove all or part of the actin binding amino terminal domain as well as a section of the rod domain. Some exon deletions induce a frameshift mutation that results in a premature stop codon, which alters the dystrophin protein synthesis through the translational process. In a study conducted by Bladen et al. (11), some exons were found to be the most frequently mutated in DMD patients. Exon 51 represented about 14% of total DMD mutations and 21% of exon deletions. It was followed by exon 53, which carried 10% of total DMD mutations and 15% of exon deletions. Exon 45 represented 9% of total DMD mutations and 13% of exon deletions. Exons 43 and 44 each represented 7% of total DMD mutations and 11% of exon deletions. Point mutations represented around 26% of DMD cases. These represent nonsense and missense mutations, splice site mutations, and mid intronic mutations. DMD mutations also include 10–15% exonic and intronic duplications and about 3% small indels (3, 11).
Children with DMD initially show weakness of proximal muscles, abnormal gait and calf muscle pseudo-hypertrophy by the time they are three to 5 years old. Scoliosis is generally observed by the age of 12, patients usually die before the age of 30 due to severe cardiac and respiratory complications (13, 14). De novo mutations in the DMD gene have been found to be associated with DMD pathogenesis and HLA polymorphism (15). The DMD gene is one of only a few genes that can tolerate the deletion of one or many exons and still be translated into dystrophin protein with reduced functionality. A favorable prognostic was observed in a 7 years follow-up study involving three Becker patients with in-frame deletions of exons 45–55 (16). In another case, a 61 years old man had 46% of his DMD gene deleted, however, this gene was still capable of synthesizing a functional truncated dystrophin (17). The tolerance of the DMD gene to deletions is extremely relevant in the context of certain gene therapies, which seek to remove one or many exons to restore the expression of an internally deleted dystrophin and improve the symptoms of the patients.
Molecular Biology Approaches for Duchenne Muscular Dystrophy
There are many strategies that researchers are developing to improve the phenotype of DMD patients. In this review, we discuss molecular biology approaches for the treatment of DMD: exon skipping, read through non-sense mutations, Cas9 based modification of the DMD gene, delivery of micro-dystrophin or full-length dystrophin gene, progenitor cell modulation, artificial chromosome for dystrophin transfer, gene replacement, and skeletal muscle cell transplantation.
Read Through Nonsense Mutations
This approach targets stop codon point mutations that otherwise prevent the proper translation of the dystrophin mRNA (Figure 2A). Drugs are used to disrupt translational fidelity by interacting with the ribosomal RNA to translate the full length dystrophin despite the presence of the premature stop codon (18–20). Gentamicin was the first aminoglycoside drug used for this type of approach (21). A preliminary trial carried out with two DMD and two BMD patients, both possessing nonsense mutations, and treated once a day with gentamicin at 7.5 mg/kg/day for 2 weeks did not show any dystrophin expression (22). In a subsequent trial with four DMD patients carrying point mutations, a different protocol was used (two 6-day cycles of gentamicin sulfate, at an interval of 7 weeks) and the result revealed dystrophin expression in three out of four patients with a UGA mutation (23). In a cohort study carried out over 6 months with 12 DMD patients that had point mutations in different exons, six patients saw an increase in their levels of dystrophin after the treatment with six patients reaching real increased levels (post-treatment minus pre-treatment level of dystrophin) of 10.4, 10.9, and 11% respectively for point mutations in exons 25, 33, and 35 (18). There is no updated data about the Gentamicin potential for DMD treatment perhaps due to the lack of clear evidence of clinical efficacy as well as notable side effects such as hearing loss, kidney toxicity, injection site reactions, stomach upset, and neurotoxicity (24).
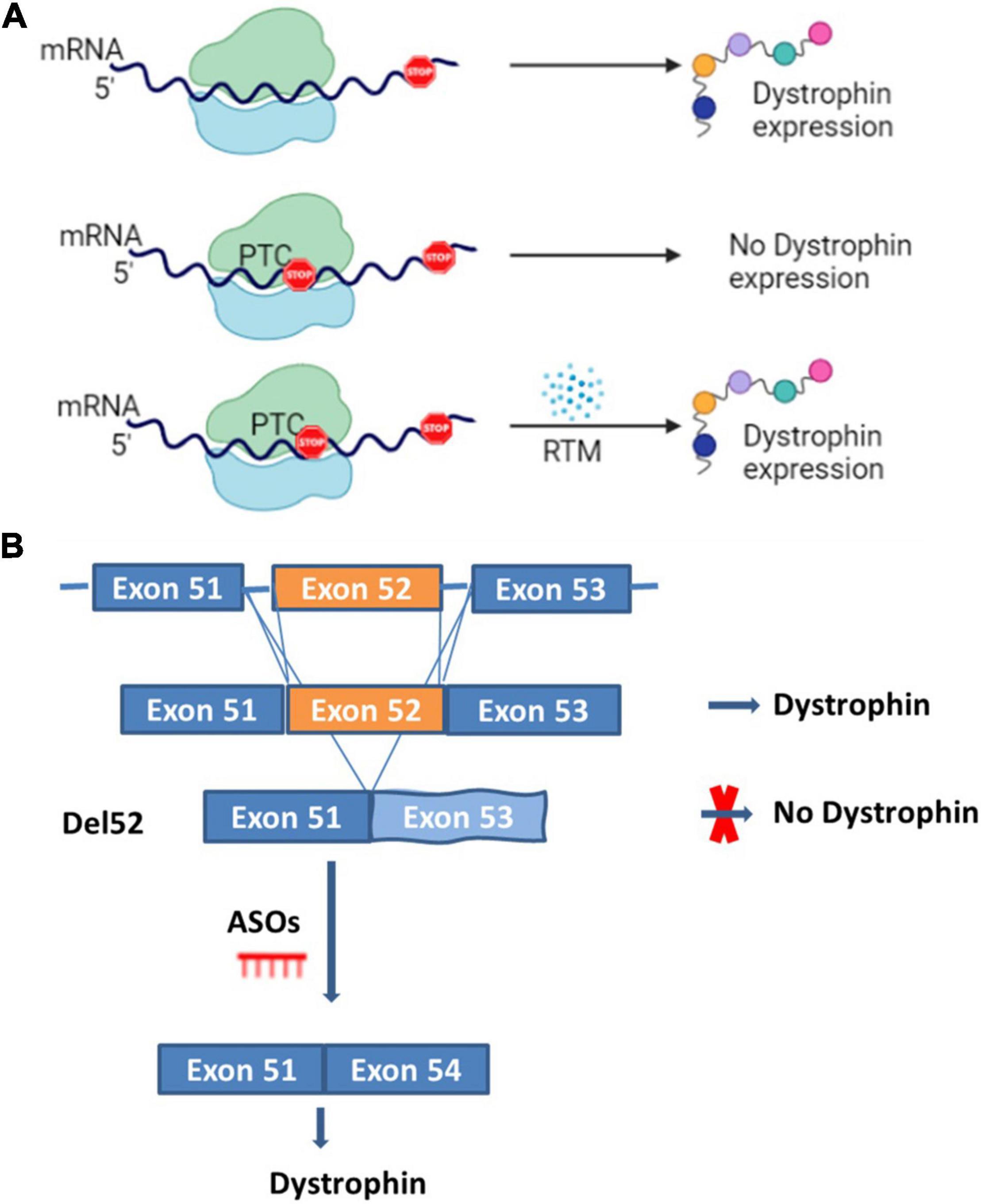
Figure 2. Read through and ASOs strategies. Panel (A) represents the normal dystrophin expression and a scenario where premature termination codon (PTC) is present and induces the absence of the dystrophin expression which can be restored by read through mutation molecules (RTM). Panel (B) represents a scenario for the DMD mutation amenable to exon 52 deletion (del52). In the presence of exon 52 there is dystrophin expression, in the case of exon 52 deletion, there is no dystrophin expression. Dystrophin expression can however be restored by inducing the skipping of exon 53 with anti-sense oligonucleotides (ASOs).
Other molecules such as Ataluren and Arbekacin sulfate (PTC Therapeutics Inc.) have also been reported to have some beneficial effects for DMD patients (20, 25). The beneficial results reported with Ataluren in a phase 2 clinical trial led to a phase 3 trial (26). Unfortunately, the phase 3 clinical trial results showed limited clinical benefits (27). In that trial carried out from 26 March 2013 to 26 August 2014, 230 participants were randomly divided into placebo and treated groups. There was no significant difference between Ataluren treated and placebo groups. Adverse effects were also reported for some patients (27). There is an ongoing phase 3 randomized, double-blind and placebo-controlled trial (NCT03179631) aiming to characterize the long-term effects of Ataluren-mediated dystrophin restoration in DMD patients. The study started in July 2017 and is expected to be completed by October 2023. In February 2021, a communication to the Duchenne community about preliminary results seemed to be encouraging (28).
Treatment Using Exon Skipping
Antisense Oligonucleotide Approach
This approach uses antisense oligonucleotides (ASOs), which are designed to bind and modulate the RNA functions through different mechanisms (Figure 2B) (29). Here, the term oligonucleotide refers to a short, unmodified, or chemically modified nucleic acid. This sequence is theoretically capable of hybridizing to a complementary nucleic acid sequence (30). ASOs used in antisense therapeutics undergo different chemical modifications to improve their metabolic stability, binding specificity and affinity (30, 31). The major modifications include the sugar modifications (2′-O-methoxyethyl, 2′-O-methyl, 2′fluoro, locked nucleic acid and constrained ethyl bicyclic nucleic acid), the phosphate group modifications (methyl phosphonate, phosphorothioate, and phosphoramidate) and both the sugar and phosphate (morpholino) (29, 30). ASOs can act by the selective cleavage of RNA through the RNase H or the Ago 2, which is active in RNA-induced silencing complex (RISC). These molecules mediate the splice modulation by targeting splice junctions or exonic/intronic splicing enhancer/silencer sites resulting in the exon skipping (29, 30). The mechanisms for the removal of the exons are not completely clear. ASOs also mediate the regulation of microRNA involved in many diseases, the mRNA polyadenylation signals and the binding between proteins and pathogenic RNA (29, 30). ASO therapeutics for DMD restore the reading frame of the mRNA by excluding some of the exons during the maturation of the messenger RNA transcript. This resulting in a truncated dystrophin protein with reduce functionality (32, 33).
Since exon deletions represent about 60–70% of the total DMD cases, there are many ongoing clinical trials aiming to restore the normal reading frame of the dystrophin mRNA by skipping certain exons (Table 1). For example, trials are ongoing to skip exon 45 (Casimersen) (NCT03532542), exon 51 (Suvodirsen, Eteplirsen) (34), and exon 53 (Golodirsen, Viltolarsen) (35, 36).
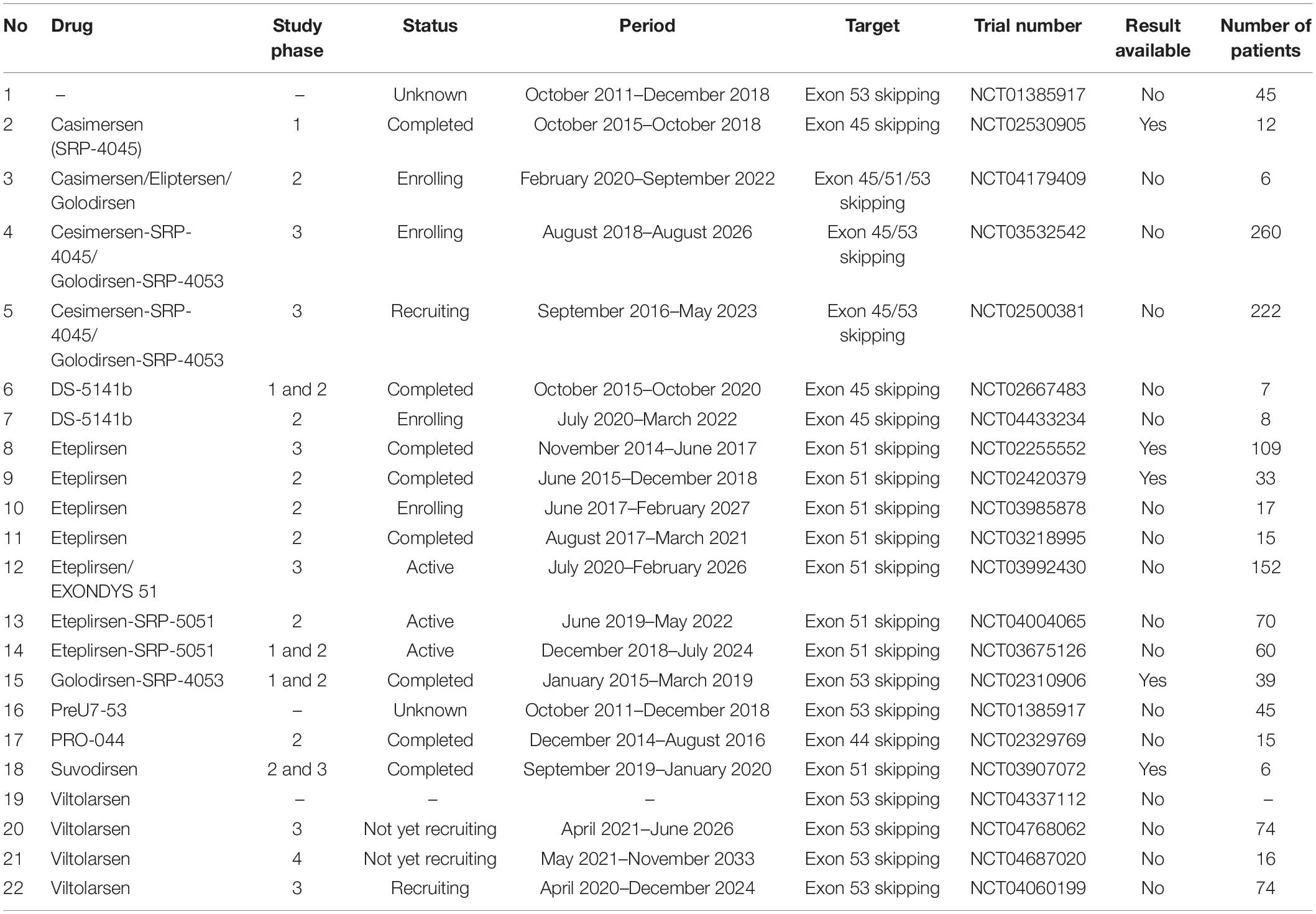
Table 1. Updated summary of exon skipping strategy for DMD.
Eteplirsen also known as Exondys 51 (Sarepta Therapeutics, Inc.) was the first drug that obtained FDA accelerated approval. In DMD patients whose mutations are amenable to exon 51 skipping, this treatment has increased dystrophin expression by 0.28% and by 0.93% after 48 and 180 weeks of treatment respectively (34, 37). The phase 2 trial (NCT03218995), which evaluated the safety, efficacy and tolerability of Eteplirsen in 15 patients aged 6–48 months ended in March 2021, and the results published in clinical trial site indicated a serious adverse event (Bronchiolitis) in 1 patient aged less than 24 months. Some adverse events (not including serious) were also indicated in many patients. A long-term phase 3 clinical trial (NCT02255552) evaluated the efficacy of Eteplirsen in 18 patients ended in July 2019. A significant difference was demonstrated in the 6 min walk test (6MWT) between the baseline and 36 months post-treatment and six patients with a mean age of 9 lost ambulation (38). There is an ongoing phase 3 randomized, double-blind, dose finding and comparison study for the safety and efficacy of high dose of Eteplirsen. This study was preceded by the first phase of an open-label dose escalation study involving 144 DMD patients that is planned to be completed by February 2026. The next generation of Eteplirsen experimented with SRP-5051 that is ongoing clinical trials phase 1 and 2 (NCT03675126 and NCT04004065) is presented to have many advantages. The most relevant is the addition of cell penetrating peptides to the phosphorodiamidate morpholino oligomer (PMO) to form peptide phosphorodiamidate morpholino oligomer (PPMO), which leads to more efficient delivery, higher level of dystrophin production and efficient dosing for patients (39). Preliminary results from phase 2 MOMENTUM study revealed that SRP-5051 for DMD patients amenable exon 51 skipping dosed monthly at 30 mg/kg lead to mean dystrophin expression of 6.55% (39).
Viltolarsen or Viltepso™ (NS Pharma, Inc.) has also been granted accelerated approval by FDA in August 2020 to treat DMD patients amenable to exon 53 skipping (35, 40). Viltolarsen has shown in a phase 1/2 trial that the intravenous infusions of 80 mg/kg/week for 24 weeks increased the mean dystrophin level by 2.8% by western blot analyses (35). The final approval of this treatment will be determined by the outcome of the ongoing phase 3 randomized, double-blind, placebo-controlled, multi-center study (NCT04060199) to assess the efficacy and safety of Viltolarsen in ambulant boys with DMD amenable to exon 53 skipping. The study is planned to be completed by December 2024. In another phase 2 trial (NCT02740972) assessing the safety and intravenous dose of NS-065/NCNP-01 in DMD boys aged 4–9 years amenable exon 53 skipping, the authors showed a proof of beneficial functional and clinical effects in the group receiving low-dose (40 mg/kg per week) or high-dose (80 mg/kg per week) of Viltolarsen (40).
Golodersen also known as Vyondys 53 (Sarepta Therapeutics, Inc.) got an accelerated approval from the FDA in December 2019. Results from the phase 1/2 trials involving 12 participants showed a dystrophin mean level of 1.019% representing an approximate 16-fold increase in truncated dystrophin. A significant increase in dystrophin positive fibers was found at week 48 compared to the baseline level (36). The phase 3 trial (NCT02500381) to evaluate the effectiveness of Golodersen is still ongoing and is expected to be completed in May 2023. Another ongoing trial (NCT03532542) aiming to evaluate the long-term safety and tolerability is also expected to be concluded by August 2026.
Casimersen also known as AMONDYS 45™ (Sarepta Therapeutics, Inc.) got an accelerated approval from the FDA in February 2021 for the treatment of DMD patients amenable to exon 45 skipping (41). This conditional approval may be associated to the preliminary results obtained after 48 weeks of treatment for the ongoing trial (NCT02500381) where the mean dystrophin protein level in the treated group was 1.736% compared to 0.925% in the placebo group. There was also an increase in the mRNA level post-treatment compared to the baseline level (42). Side effects including upper respiratory tract infections, cough, fever, headache, joint pain, and pain in the mouth and throat were observed in about 20% of the patients. The study is estimated to be completed by May 2023. In the meantime, an ongoing long-term, open-label and extension trial (NCT03532542) in which 260 participants aged 7–23 years were enrolled is aiming to evaluate the long-term effects of AMONDYS 45™. The study is estimated to be completed by August 2026 and may determine the final approval of Casimersen.
Small Nuclear RNA Approach
Another interesting approach for exon skipping is the use of small nuclear RNAs (snRNAs), which play a major role in mRNA splice modulation (43, 44). Decades ago, the snRNAs including U1snRNA and U7snRNA have been used as therapeutic approach targeting the dystrophin pre-messenger RNA to mediate skipping of exon 45 in human myoblasts (45) and exon 51 in mice (46) resulted in nearly complete restauration of dystrophin compared to wild type expression. This approach can be appropriately designed to mediate either a single or multi exon skipping (47). Recently, it has been shown that both the systemic and intramuscular injection of U7snRNA for the correction of DMD exon 2 duplication using a single complementary adeno-associated virus (scAAV) in a mouse model harboring exon 2 duplication (48), resulted in robust dystrophin expression and correction of muscle physiologic defects (49). In that study a dose-escalation experiment showed that the highest dose of scAAV U7 (7.6 × 1013 vg/kg) following a systemic injection in mouse tail vein resulted in 42–96% skipped transcripts (single or double skipping of exon 2) and 69–92% dystrophin positive fibers in skeletal muscle of 4 weeks post injection. Twelve weeks post injection permitted to achieve 85% positive fibers with progressive increase observed in absolute and specific force to finally reach complete rescue in absolute force and partial rescue of specific force. This improvement was also observed in heart muscle with up to 40% dystrophin level after immunoblotting either for 4 or 12 weeks treated mice (49).
Astellas Gene Therapies recently started a clinical trial phase 1/2 using this approach for the correction of DMD exon 2 duplication (NCT04240314) in three participants at Nationwide Children’s Hospital. The study aiming to access the systemic single dose delivery of scAAV9.U7.ACCA via peripheral vein injection is to be completed by November 2025. The scAAV9.U7.ACCA will cause the skipping of exon 2 during the splicing event to form a mRNA containing a single exon 2 or no exon 2 at all. Transcriptomic analyses indicated that scAAV9.U7.ACCA is not associated with important off target events in non-human primates (50) and in mice (51, 52). This approach is very encouraging for because of its potential safety in human. Although the preliminary clinical trial results are not yet available, the preclinical results described above seem to present this approach to be much promising that the ASOs approach where, whatever it has been granted accelerated approval, the highest editing percentage (6.55%) was shown with SRP-5051 (39).
Mechanism Based on Nuclease Activity of CRISPR Associated Protein 9 Protein
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) technology is generally considered as new, simple, and efficient approach to cut and modify a DNA strand. The CRISPR associated protein 9 (Cas9) nuclease is guided to a specific region of the DNA through a sequence of 20 nucleotides called a single guide RNA (sgRNA) (53–55). This technology was first discovered in bacteria. Many bacteria and most archaea defend themselves from invading genomic elements from bacteriophage infections and plasmid transfer using this sophisticated adaptive immune system. After being exposed, a small fragment of foreign DNA is integrated into the CRISPR array in the host genome as a new spacer sequence, which provides a memory record of this infection. In future attacks by the same invaders, the foreign DNA fragment is recognized by the CRISPR RNAs and cleaved through enzymatic processing commonly known as endonucleolytic cleavage (53–55). There are six types of nucleases divided into two major classes.
Class 1 nucleases are composed of type I, III, and IV nucleases and require a large complex of several effector proteins for the RNA guided target cleavage system. Class 2 nucleases are composed of type II, V, and VI nucleases and require only one RNA guided endonuclease to mediate the cleavage. CRISPR/Cas9 are type II endonucleases and the most commonly used types of Cas proteins for genome editing. The most commonly used Cas9 proteins for gene editing are the Streptococcus pyogenes Cas9 (SpCas9) and Staphylococcus aureus Cas9 (SaCas9). SaCas9 is about 1 kb smaller than SpCas9 and can be easily packaged with its sgRNA into a single AAV vector in what has been called an all-in-one vector. To guide the Cas9 onto a specific genomic locus, a sgRNA containing a spacer sequence of 17–24 nucleotides is required. This sgRNA also contains a constant sequence, which forms a stable complex with the Cas9 (53–55). When performing gene editing, the Cas9 nuclease that is used must be chosen based on the nucleotide sequence surrounding the target site. Each Cas9 recognizes its own sequence of two to six nucleotides called the proto spacer adjacent motif (PAM) to bind to the DNA and subsequently causes an efficient DNA break.
Various technologies were derived from the original CRISPR/Cas9 system (CRISPR-induced double strand breaks (54, 55), Cytidine Base Editing (CBE) (56), Adenine Base Editing (ABE) (56), and PRIME Editing (PE) (57). Current efforts with these technologies are ongoing to investigate the feasibility of genetically modifying the DMD gene. These approaches seek to introduce permanent modifications in the genomes of DMD patients to efficiently restore the dystrophin expression. These modifications include the correction of nonsense mutations, the insertion of one or multiple nucleotides to reframe a frameshift mutation, the modification of splicing sites to favor exon skipping and reframing, etc. (58).
Clustered Regularly Interspaced Short Palindromic Repeats-Induced Double Strand Break
Depending on the genome modification that is required, 1 or 2 sgRNAs are required with a Cas9 nuclease to induce 1 or 2 double stranded breaks at 1 or 2 specific DNA sites (Figure 3A).
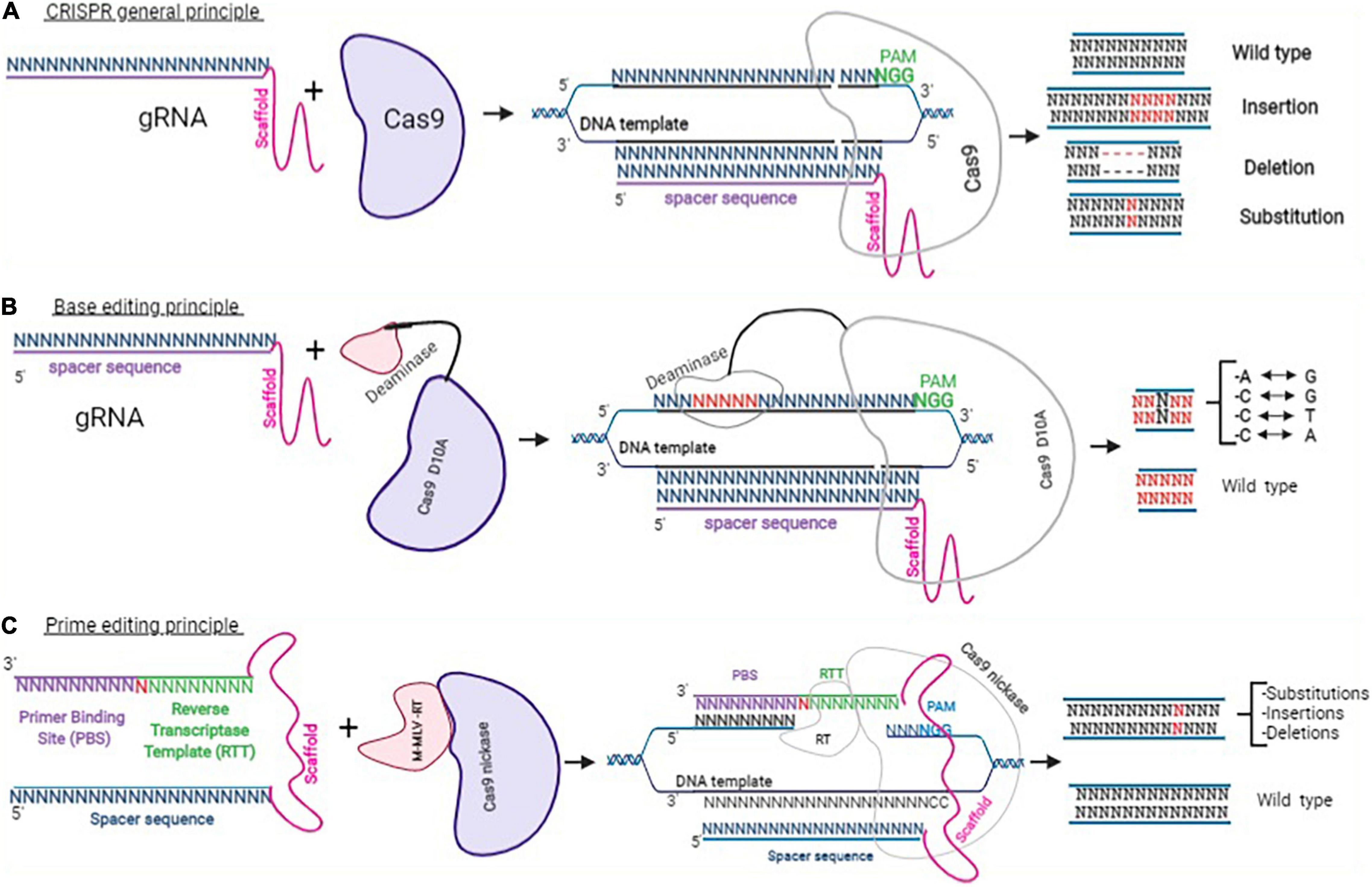
Figure 3. Clustered Regularly Interspaced Short Palindromic Repeats approaches. This figure adapted from Happi Mbakam et al. (58) represents the principle of CRISPR-Cas9 gene modification, which uses a guide RNA and a Cas9 to mediate the nuclease activity resulting in insertions, deletions, or INDELs (A). Panel (B) represents the general mechanism for base editing, which uses a sgRNA (spacer sequence + scaffold) and a Cas9 nickase fused with a cytosine or an adenine deaminase. Depending on the deaminase used, after binding with a specific DNA template, a cytosine is chemically modified into a thymine or an adenosine into a guanine. Panel (C) shows a PE2 protein (a Cas9 nickase fused with an engineered reverse transcriptase enzyme from M-MLV) and a pegRNA. The pegRNA is an extended sgRNA including the spacer sequence and the constant sequence (scaffold), a reverse transcriptase template (RTT), and a primer binding site (PBS). The binding of PE2 and pegRNA with a DNA target permits to induce insertion, deletion, or substitution of a few nucleotides.
Exon Deletion
This approach was used by our research group to target sequences in exons 50 and 54 and in exons 47 and 58 to induce deletions of part of the DMD gene using 2 sgRNAs (59, 60). This generated a hybrid exon that not only restored the normal reading frame of the DMD mRNA, but also produced a dystrophin protein with a normal spectrin-like repeat structure. This was performed in vitro in DMD patient myoblasts carrying exon deletions and in vivo in the hDMDΔ52 mouse model (59, 60). About 50% of dystrophin expression was restored in the mdx23 mice by targeting the flanking regions of exons 21 and 23 to induce an in-frame deletion using 2 sgRNAs (61).
Exon Reframing
Koo et al. (62) successfully used this approach to mediate exon reframing using 1 sgRNA. Authors showed that 8 weeks post injection of 5 × 1011 vg/kg of AAV2/9 driving the expression of CjCas9 and one sgRNA in mdx23 mouse model of DMD resulted in 39 ± 4% dystrophin positive fibers in mice with one base pair insertion (cytidine nucleotide) and 28 ± 6% of mice carrying 14 base pairs deletion. The mice showed increase in muscle-specific maximal force. Min at al. (63) also showed that the insertion of a single adenine nucleotide at 5′ boundary of exon 45 lets to the reframing of that exon and the dystrophin expression.
Exon Skipping
This approach has also been experimented in vitro for the skipping of exon 23 in DMDmdx induced multipotent progenitor cells (iMPCs). Two sgRNAs were designed to target the splice donor site of exon 23 mediating the skipping of exon 23 during the splice event. The corrected cells restored the dystrophin expression in vivo and contributed to the muscle stem cell reservoir in DMDmdx (64). Min et al. (63) used 1sgRNA to target the 5′ junction of exon 45 in a mouse carrying the deletion of exon 44 to mediate the skipping of exon 45. By skipping the exon 45, the strategy permitted to link the exon 43 to exon 46 to restore the reading frame. A systemic delivery of Cas9 and sgRNAs through peritoneal injection using two AAV9 vectors (5 × 1013 vg/kg at a ratio of 1:1) resulted in 94, 90, and 95% dystrophin expression observed by immunostaining respectively in TA, triceps, and diaphragm muscles followed by improvement in mouse muscle function. A ratio of 1 (Cas9):10 (sgRNA) was used to achieve similar results in cardiomyocytes.
However, this CRISPR double strand break technique promotes frequent non-specific insertions and deletions following off-target DNA cleavages. In the Min et al. study, up to 0.2 and 1.1% inverted terminal repeat integrations were reported respectively at the genomic and the mRNA levels. Koo et al. also reported up to 8 ± 0.7% INDELs. Happi Mbakam et al. recently presented an update of the CRISPR-Cas9 gene therapy for dystrophin replacement in DMD (58).
Base Editing
This approach uses an adenine or cytidine deaminase coupled to a Cas9 nickase (a modified Cas9, which can only cut one DNA strand) to deaminate a cytidine (C to T modification) or an adenine (A to G modification) after being guided to that specific genomic region by a sgRNA (Figure 3B) (65). This system has been used in the adult mdx4cv mouse, a dystrophic mouse model of DMD carrying a premature stop codon in exon 53 of the DMD gene, to demonstrate that base editing can restore dystrophin expression. The authors of that experiment showed that the systemic delivery of the adenine base editor (ABE) using an improved AAV vector (AAV-iNG) led to long-term and efficient restoration of up to 80% dystrophin and functional improvement of dystrophic heart (66). However, this technique is not precise enough because, other adenines or cytidines present in the editing window are susceptible to be also deaminated (65). CBEs and ABEs are also recognized to generate other off-target mutations (67).
Prime Editing
Prime editing is one of the most recent, efficient, and precise genome editing technology to be developed so far. It uses a prime editor (PE) and a prime editing guide RNA (pegRNA) (Figure 3C). The prime editor is made of a Cas9 nickase protein fused through a flexible linker to an engineered Murine Leukemia Virus Reverse transcriptase (RT). The pegRNA is a modified sgRNA, it contains the spacer sequence of the sgRNA (a sequence of 20 nucleotides designed to target a specific DNA locus), the scaffold of the sgRNA (which permits it to bind to the Cas9 protein). However, the pegRNA also contains a 3′ extension made of a primer binding site (PBS) and a reverse transcriptase template (RTT). The PBS sequence is designed to serve as primer to start the synthesis of a complementary DNA strand by the RT. The RTT sequence serves as a template for the synthesis by the RT of the intended complementary strand containing the desired modification(s) (57). The pegRNA can, in principle, change any nucleotide for any other nucleotide with precision. This technique can therefore correct point mutation, and either eliminate or insert a few nucleotides to permanently modify the genome of target cells (57, 66, 68–71). Many ongoing experiments are demonstrating the capacity of this approach to permanently correct the DMD gene as well as other genetic mutations responsible for many other hereditary diseases (69, 72).
Mechanism Based on Progenitor Modulation
Also considered as a stem cell disease, the DMD disease progression and severity is strongly associate to the capacity of myogenic progenitors to maintain the regeneration, growth and repair of skeletal muscle (20). Satellite cells, the prototype of muscle stem cells, have been identified as the primary quiescent progenitor for myofibers (73). The cell environment has been identified to play a key role in the maintenance of the satellite cell niche and the performance of these stem cells (74). Many other progenitors such as mesoangioblasts (75), pericytes (76), side population cells (77), muscle derived stem cells (78), PW1+ interstitial cells (79), and fibro-adipogenic progenitors (150) have also been investigated for muscle regeneration.
Histone deacetylase inhibitors (HDACi), such as valproic acid, phenylbutyrate, givinostat (ITF2357), and suberoylanilide hydroxamic acid, have been tested in DMD mouse models. Although, promising results have been obtained in mdx mice (80), the safety and tolerability of these drugs in the pediatric population remains the main concern for their use in clinical trials. However, the pharmacological effects of Givinostat, a class I and II HDACi, in pediatric patients are already known (81). By blocking the histone deacetylase enzyme, Givinostat permitted the activation of the follistatin gene leading to muscle mass increase. That effect also prevented muscle degeneration, reducing the adipogenesis process and fibrotic tissue accumulation.
Interim results from phase 1 and 2 clinical trials (NCT01761292) sponsored by Italfarmaco Inc. involving 20 DMD patients aged 7–10 years showed that Givinostat induced a significant reduction of the amount of fibrotic tissues, tissue necrosis and fatty replacement. It also significantly increased the fraction of muscle tissue in the biopsies. No improvement was observed in functional tests (82). The NCT01761292 trial has been extended to phase 2/3 clinical trial (NCT03373968) for the study of long-term safety and tolerability of Givinostat. The study is expected to be ended by December 2023. Italfarmaco Inc. communicated in February 2021 that boys from this study continued to show delayed disease progression (83). The ongoing phase 3 pivotal trial (NCT02851797) has enrolled 179 patients aged 6–17 years. The aim of this trial is to evaluate the efficacy and the safety of Givinostat and the preliminary results is expected in second quarter of 2022 (83). Givinostat has been approved by the European Medicine Agency for the treatment of DMD (84). FDA also granted in October 2020 a Rare Pediatric Disease designation to Givinostat for the treatment of DMD (83).
Gene Replacement
The gene replacement approach consists of transferring a functional copy of complete or truncated dystrophin gene into myofibers to restore the muscle strength in DMD patients. The DMD gene is one of the biggest genes measuring about 2.2 million base pairs (85). This large size makes it difficult to package. In this approach, the full-length dystrophin cDNA and mini or micro-dystrophin constructs, are investigated to develop treatments for DMD.
Artificial Chromosome Transfer of the Dystrophin Gene
Human artificial chromosome (HAC) is a kind of multipotent vector derived from native or synthetic chromosome, able to deliver a full length DMD gene in mice and eventually in humans (151). Benedetti et al. (86) developed a novel generation of HAC (DYS-HAC 2) eliminating potential immunogenic products such as the enhanced green fluorescent protein (EGFP), blasticidin (Bsd), Herpes Simplex Virus type 1-thymidine kinase (HSV1-TK), and hypoxanthine-guanine phosphoribosyl-transferase (HPRT) from DYS-HAC 1 (86). DYS-HAC 2 permits genomic integration, reversible immortalization, genetic correction, additional dystrophin expression, inducible differentiation, and cell death control.
This approach presents at least two important advantages for DMD gene therapy. The first is the ability to carry large DNA sequences that could lead to physiological expression of genes with complex transcriptional regulation such as the DMD gene. The second advantage is the episomal maintenance of a single gene copy, preventing the risk of oncogenic insertion (86). However, some limitations (low formation efficiency, the structural complexity, tissue target efficiency, and a stability problem) explain why this approach did not yet move forward to clinical trial.
Full Length Dystrophin
Meng et al. showed that DMD myoblasts transduced with a foamy virus vector expressing the full-length dystrophin transplanted intramuscularly into mdx nude mice, participated in muscle regeneration leading to the expression of full-length and functional dystrophin proteins (87).
Micro-Dystrophin
Mini or micro-dystrophin are short versions of the DMD gene made of at least N terminal domain and different parts of the DMD sequences between these N and C terminal domains (Figure 4). Several micro-dystrophins and different AAVs serotypes have been developed through decades of research on this approach (88). Nowadays, three micro-dystrophins are used in clinical trials: the five-repeat micro-dystrophin D3990 (89) (Pfizer Inc. trial for PF-06939926), the four-repeat micro-dystrophin ΔR4–23/ΔC (90) (Sarepta Inc. trial for SRP-9001) and the five-repeat micro-dystrophin mDys5R (91) (Solid GT Inc. trial for SGT-001). All the three transgenes have in common at least the N-terminal domain, the cysteine-rich domain, the spectrin-like repeats 1 and 24 (R1 and R24) and Hinges 1 and 4 (H1 and H4). All of them lack the C-terminal domain spanning from exons 71 to 79. The C-terminal domain contain the alpha1 and beta1 syntrophin biding sites and the dystrobrevin biding site, which play important role in signalization pathways and the maintenance of dystrophin complex (3). This domain has been identified to have a protective role in heart (92). Wang et al. (93) showed that compared to full-length dystrophin, micro-dystrophin (ΔR4–23/ΔC) has altered association with alpha and beta syntrophins and cavins in mdx5cv mouse hearth. The authors also indicated that the expression of micro-dystrophin in mdx5cv mice prevented the development of cardiac histopathology but did not rescue membrane localization of cavins nor did it normalize ERK signaling (93). Bourdon et al. (94) recently showed in DMDmdx rats that the ΔR4-23/ΔCT expression is sufficient to restore the interactions of most dystrophin associated protein complex (DAPC) partners in skeletal and cardiac muscles. In contrast, the addition of C terminal domain significantly increases the restoration of that interaction. The micro-dystrophin D3990 lack the nNOS binding domain and H2. The micro-dystrophin DR4–23/DC lack the nNOS binding domain and H3. The micro-dystrophin mDys5R lack H2 and H3. The H2 influence functional capacity of micro-dystrophin in mouse model (3). The nNOS is involved in important signaling transduction pathways and produces NO leading to vasodilatation which help to provide oxygen and nutrients to muscle cells (3). It is evident that each domain removed from the native dystrophin gene to build micro-dystrophin plays a role in the dynamism of the DAPC. To which extend the removal of the so qualified “unnecessary domains” in micro-dystrophin impacts the membrane dynamism and is not clear cut.
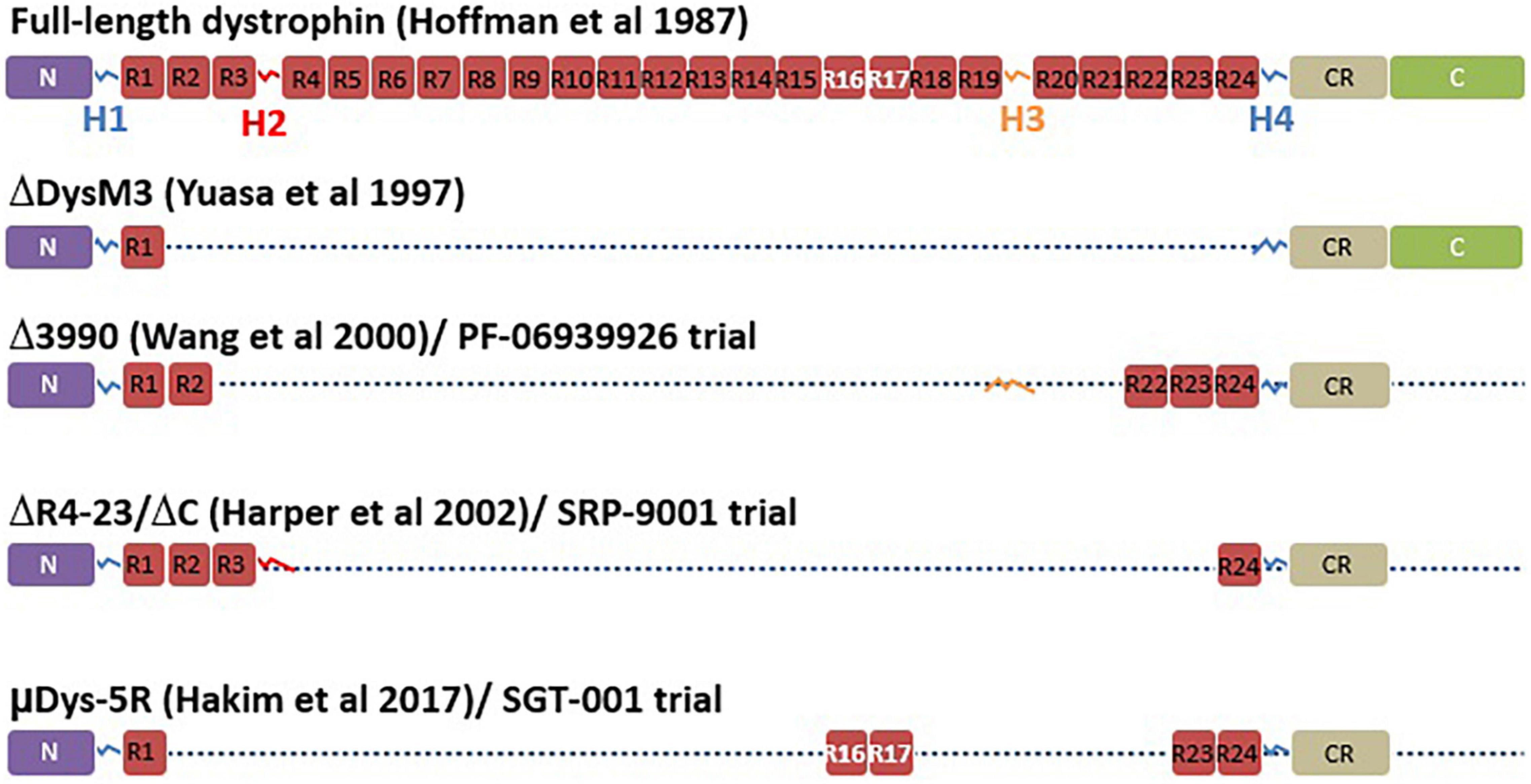
Figure 4. Micro-dystrophin variants. This figure adapted from Duan et al. (147) represents the full-length dystrophin with different domains including the N-terminal domain (N), the four hinges (H), the central rod domain (R), the cysteine rich domain (CR), and the C-terminal domain (C). The figure also illustrates the different versions of micro-dystrophin with lacking domains in use in different clinical trials including the initial micro-dystrophin constructed by Yuasa et al. (149), the Δ3990 used for the development of PF-06939926, the (ΔDysM3), the ΔR4-23/ΔC used for the development of SRP-9001, and finally the μDys-5R used for the development of SGT-001.
In 2020, Mendell et al. (95) published a preliminary result from the phase 1/2 clinical trial study (NCT03375164) sponsored by Sarepta Therapeutics Inc. that aimed to evaluate the safety and tolerability of intravenous SRP-9001 in DMD patients. This treatment involved the use of a recombinant adeno-associated virus (AAV) serotype rh74 (rAAVrh74) and codon-optimized human micro-dystrophin sequence expressed using a skeletal and cardiac muscle-specific promoter with enhanced cardiac expression (MHCK7). The micro-dystrophin sequence was made of N-terminus actin binding domain, spectrin-like repeats 1 to 3 and 24, hinges 1, 2, and 4, the cysteine-rich domain and the C-terminus domain. Four boys aged 4–7 years with exon deletions (46–50, 46–49, partial deletion exon 44) or a premature stop codon in exon 27 were enrolled. Patients received single intravenous infusion of 2 × 1014 vg/kg rAAVrh74.MHCK7 micro-dystrophin. The immunochemistry of muscle biopsy showed 81.2% of muscle fibers expressing micro-dystrophin with an intensity of 96% at the sarcolemma. Functional improvement was also observed in North Star Ambulatory Assessment (NSAA) scores. The patients were followed until March 2021 with the results yet to be published. A parallel randomized, double blind, placebo-controlled study of SRP-9001 (NCT03769116) involving 41 participants is planned to be completed by April 2026. A minimal fat infiltration was further confirmed in a case control study (96) assessing the association between treatment with rAAVrh74 and muscle quality in three patients previously involved in the Mendell et al. study (95).
There is another ongoing randomized, placebo-controlled phase 3 clinical trial sponsored by Pfizer Inc., which seeks to evaluate the efficacy and safety of PF-06939926 (NCT04281485) in 99 boys aged 4–7 years old. The mini-dystrophin gene is delivered by an AAV9 serotype, which has good affinity to skeletal muscle, nervous, heart, liver, and lung tissues (97). The study is expected to be completed by September 2028. However, in December 2021, FDA imposed a clinical hold after the death of one patient participating the non-ambulatory cohort of the phase 1b trial (NCT03362502) (98). The trial was a multicenter, open-label, single ascending dose study to evaluate the safety and tolerability of PF-06939926 in ambulatory and non-ambulatory subjects with DMD. The preliminary results presented by Pfizer Inc. in 2019 were promising and some observed adverse effects were resolved in approximately 2 weeks (99).
In September 2021, Solid Bioscience LLC reported 1.5-year data of their IGNITE trial phase 1/2 (NCT03368742), which was a controlled, open-label, single-ascending dose study to evaluate the safety, the tolerability and efficacy of SGT-001 in adolescents and children with DMD. The company indicated that SGT-001, which is a rAAV9 encoded micro-dystrophin, showed functional benefits through NSAA, 6MWT, and Forced Vital Capacity (FVC) compared to natural history data after 1.5-post treatment (100).
Some recent trials in this approach have been summarized in Table 2. The results of Mendell trial (NCT03375164), showed a great promise of this gene transfer approach although the observed mild and moderate side effects (95) and a death in Pfizer trail (NCT03362502). Specific questions about the AAVs and transgene safety and the long-term assessment in this respect need to be addressed. There is no evidence about the long-term micro-dystrophin related truncated dystrophin expression in DMD patients.
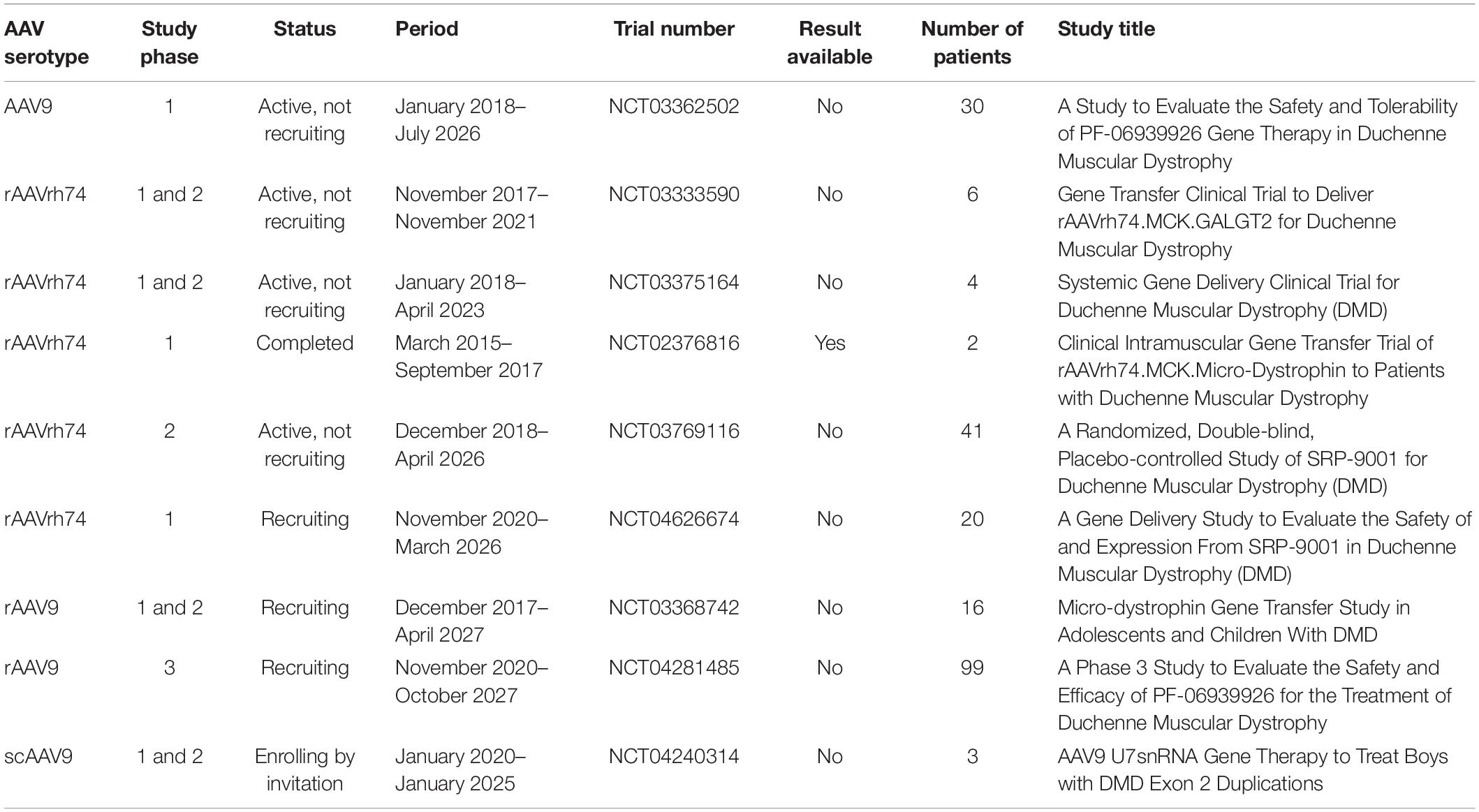
Table 2. Summary of clinical trials of gene transfer for DMD.
Mechanism Based on Skeletal Muscle Cell Transplantation
This approach is investigated for DMD and other myopathies. It is based on a specific healthy donor cell transplantation to a specific DMD patient to repair the damaged organ or to repopulate the satellite cell pool and enable the dystrophin expression (101). The engraftment of human cells in an immunodeficient mouse model requires preliminary mechanical, irradiation or chemically induced muscle damage to create a niche (102). The transplanted cells have to be able to proliferate, differentiate and integrate into the host muscle (103). The cell types usually transplanted are satellite cells, induced pluripotent stem cells, mesoangioblasts, pericytes, myoendothelial cells, and myoblasts derived from satellites cells (101). Cells can be delivered by multiple intra-muscular injections, repeated cell delivery or systemic delivery (102). In a preclinical study involving seven non-human primates, Skuk and Tremblay showed that the intra-arterial (femoral arteries) injection of myoblasts contribute to myofiber regeneration but only in muscle sites that were damaged at the time of cell injection (104). There is no recent update about the human myoblast transplantation to restore the dystrophin. Some reasons could be pointing out. The proliferation of myoblasts is labor intensive. The myoblasts injected in a muscle remain at the injection sites and thus because of large tissue surface hundreds of injection sits are required making this treatment difficult for the patients (105). Moreover, r transplanted myoblasts and finally, a high percentage of myoblasts usually died at the injection sites (106). A study of the kinetic of death of transplanted myoblasts in non-human primates over three weeks noted that transplanted myoblasts died at a significantly higher rate during the first week due to an inflammatory response that remains a critical challenge to be resolved for intra-muscular skeletal muscle cell transplantation (107).
A clinical trial conducted with nine immunosuppressed DMD patients to test a protocol including a high density of injection trajectories in skeletal muscles indicated that the transplanted cultured myoblasts from one of the patient’s parent permitted the expression of the donor dystrophin in 3.5 and up to 26% of the muscle fibers in eight patients (108). Thus, this clinical trial conducted in immunosuppressed DMD patients showed evidence that allo-transplantation under Tacrolimus immunosuppression of normal myoblasts permitted their fusion with patient myofibers leading to dystrophin expression. However, the presence of muscle fibers expressing the donor dystrophin was observed only close to the injection sites. Other investigational studies by the same authors demonstrated the pertinence and the relevance of this approach following different methodologies, which resulted in 34.5% dystrophin positive fibers in one DMD patient (105, 109).
A single blinded trial conducted in various hospitals in India with 11 participants showed the proof of principle that the transplantation of human umbilical cord mesenchymal stem cells stabilized the muscle power with no serious adverse effect (110).
Utrophin Upregulation
Utrophin is known as dystrophin homolog mostly found at the neuromuscular junction. It possess similarities with dystrophin in sequences and protein binding properties (111). Utrophin A is the predominant isoform and is found in the myotendinous junction of adult muscles and at the sarcolemma of regenerating myofibers (111). It has been shown that the activation of UTRN-A promoter mediates the upregulation of UTRN expression in mdx mice (112, 113). Some post-transcriptional effectors of utrophin upregulation have also been identified in a mouse model of DMD using an utrophin 5′/3′UTR reporter assay (114). Sengupta et al. (115) used the CRISPR-Cas9 technic to delete 500 bp IMTR containing five miRNAs binding sites within the UTRN in DMD patients derived human pluripotent stem cells (hiPSCs). The deletion of that sequence resulted in a twofold increase in utrophin level, which permitted to improve the dystrophin glycoprotein complex restoration. Some authors also successfully used this approach (112, 113, 116). The upregulation of utrophin as therapeutic approach helps in the maintenance of muscle stability and integrity thus, might functionally compensates the lack of dystrophin in DMD. Although the upregulation of utrophin has benefit in animal DMD models, there is no evidence about the impact of the persistence of that over-expression. However, this approach may be beneficial for all DMD cases since it is not mutation type-related and the UTRN is present in all DMD patients.
Clinical trials supported by Summit Therapeutics Inc. including the phase 1 placebo-controlled randomized trial (NCT02383511) in 12 healthy male volunteers, and the phase 1b placebo-controlled, randomized, double-blind study (NCT02056808) in 12 boys with DMD, demonstrated that Ezutromid (SMT C1100) was safe and well-tolerated by participants (117). The phase 2 trial (NCT02858362) involving 43 participants and consider as proof of concept study to assess activity and safety of SMT C1100 in boys with DMD was ended because of lack of efficacy (118).
Side Effects of Drugs
Each drug development approach for DMD has the main objective to develop a suitable molecule with long-term effectiveness and safety for DMD patients. At least two major reasons including the incapacity of the drug to mediate dystrophin expression and the harmful side effects of the drugs could explain the fact that some potential drug developments for DMD did not move forward. Gentamicin is one example of read through molecule that seemed to mediate interesting level of dystrophin expression but associated with important side effects such as ototoxicity, neurotoxicity, nephrotoxicity, harmful injection site reactions, nausea, stomach upset, vomiting, etc. (18, 24). ASO therapeutics mediating exon skipping also highlighted several toxicities including upper respiratory tract infections, cough, fever, headache, joint pain, pain in the mouth and throat, blood disorders, hyperglycemia, complement and coagulation cascades activation, nephrotoxicity, hypotension, etc., attributed to the chemical structure of these molecules (119). With regards to the level of dystrophin restoration that ranged from 0.2 to 6.5% editing percentages versus the side effects observed, it is questionable how easy most of these ASO therapeutics have been granted accelerated approvals for the treatment of DMD. PRO044 is an example of molecules that showed some of these side effects in DMD patients (NCT01037309). Most of times, drug developments for DMD are paused because of ineffectiveness of dystrophin expression.
The dystrophin replacement is usually associated with immune response both against dystrophin (120, 121) and AAV vector (122, 123) used to deliver the micro-gene. Mendell et al. (124) raised attention a decade ago about the consideration of cellular immunity against self and non-self dystrophin epitopes for the mini dystrophin gene transfer. As well, a 24 months study carried out in GRMD dogs for the dystrophin restauration through mini-dystrophin cMD1 gene transfer using the rAAVs2/8 indicated a transient humoral immune response against cMD1 1–2 months post injection. These reactions become undetectable after 8 months except for one dog who showed a persistent IgG response to cMD1 (121). These authors also indicated a persistent immune response against the AAV8 capsid (121). The use of immunomodulatory drugs such as rituximab and VBP6 might completely abrogate the cellular immune response against micro-dystrophin and partially the interferon gamma response (120).
The in vivo AAVs related gene transfer has been shown to be linked to adverse events including neurotoxicity, hepatotoxicity, myocarditis, oncogenicity, and thrombotic microangiopathy (125). Third death were recorded in 2020 because of hepatotoxicity during the ASPIRO clinical trial (NCT03199469) phase 1/2 for the treatment of X-Linked Myotubular Myopathy (126). The patients were treated with high dose of 3.5 × 1014 vg/kg of the AT132 which is an AAV8 vector containing a functional copy of the human MTM1 (hMTM1) gene. Despite the predicted lower dose of 1.3 × 1014 vg/kg that supposed to be safe, four other deaths were recorded in September 2021 (127). The chromosomal integration also represent a non-negligeable risk for the use of some AAV serotypes (128). This evidence highlights serious concerns about the prior validation of AAVs for the prevention of patient health.
Delivery Methods
The choice of drug delivery systems is a challenging question in drug development experiments. Although they allow for good precision and efficiency in vitro, molecules such as programable nucleases or nucleic acid are always challenging to deliver to a target site in vivo. These difficulties account for the low number of clinical studies. The choice of one method over another always depends on characteristics such as immunogenicity, packaging capacity, integration ability, long or short-term expression ability, etc. Nowadays, many approaches have been investigated such as: (1) biological delivery approaches including viral delivery systems (129) and extracellular vesicle (EV) delivery systems (130), (2) chemical delivery approaches including nanoparticles, peptide-based, and lipid particles (131), (3) physical delivery approaches including injections, electroporation, ultrasound, ballistic DNA, photoporation, magnetofection, mechanical massage, and hydrostatic pressure (131). Here we will only discuss the biological approaches explored by researchers to deliver large molecules to human or animal models.
Viral Delivery System
Many viral delivery systems are used for gene therapy including AAV, adenovirus (AdV), lentivirus, and bacterial phages (129). AAVs are frequently used in gene therapy experiments, and thus they are currently the most well characterized virus for human therapy. Their serotype diversity allows them to preferentially target a specific organ or biological system (132). Their limited size capacity, which was an important issue for gene therapy, was recently solved by the development of trans-splicing dual or triple AAV vectors but with reduced expression efficiency (133, 134). Dual AAVs coding for proteins with intein sequences have also been used because these inteins can fuse to each other and exclude themselves in the process of making the fusion protein (135).
Extracellular Vesicle Delivery System
Extracellular vesicles represent a new alternative for the delivery of nucleic acids and proteins. EVs are small lipid bilayer particles that are released by cells. They play a critical role in cell-to-cell communication by carrying and transferring their contents to other cells (130). Their contents and function strictly depend on their biogenesis. EVs can be divided into many sub-populations: micro-vesicles, exosomes, apoptotic vesicles, ectosomes, and lysosomes (130). They can be used as biomarkers of diseases by observing their contents and extrapolating the states of the cells producing them from there (130). The EV delivery method is gaining interest in science because of their natural potential to carry macromolecules such as Cas9 components. Bobis-Wozowicz et al. showed the proof of concept that programable nucleases can be packaged into EVs for genomic target editing (136). Gee et al. also demonstrated that EVs can be used as a delivery method for the CRISPR/Cas9 protein and sgRNA for exon 45 skipping in DMD induced pluripotent stem cells with 90% efficiency (137).
Perspectives
The therapeutic approach consisting of read through the mutation initially seemed to be the most effective drug development approach to handle DMD nonsense mutations allowing significant phenotypic improvement. However, success rate in this approach remains very low and usually combined adverse effects observed in treated patients. These adverse effects prevalently include ototoxicity, neurotoxicity, nephrotoxicity, harmful injection site reactions, nausea, stomach upset, etc., depending on the molecule or the strategy (18, 27). Another approach allowing exon skipping using ASOs has achieved up to 5% dystrophin expression after 24 weeks of treatment (36), which has been considerably improved to 6.5% mean dystrophin production for only three doses of a monthly administration of 30 mg/kg SRP-5051 in DMD patients with amenable exon 51 skipping in the phase 2 momentum trial of SRP-5051 (39). Many chemical improvements have been made to this drug approach to reduce adverse effects minimizing the toxicity level for improved tolerability (95). Improvements also made the drug more stable increasing the half-life to enlarge the time between two uptakes. They also permit to increase the drug affinity and specificity to the target as well as the level of dystrophin production (39, 138). One of the most recent and more promising modification made by Sarepta therapeutics for antisense therapeutics is the addition of polypeptides to PMO to form the next generation PPMO (39). Gene replacement approaches are also interesting since they would permit long-term dystrophin expression. The most advanced gene replacement approach inducing a robust dystrophin expression consists of transferring a functional copy of a micro-dystrophin gene with a recombinant AAV serotype rh74 (rAAVrh74) and codon-optimized human micro-dystrophin sequence expressed using a skeletal and cardiac muscle-specific promoter with enhanced cardiac expression (MHCK7) (95). There are some ongoing phase 3 clinical trials for this approach (NCT03769116 and NCT03362502), but results are not yet available. However, a previous trial reported some cases of advert effects mostly included nausea, vomiting and an elevated transaminase levels that should be taken into account for subsequent study designs (95).
One of the big challenges in the gene replacement approach is the problem of transgene persistence and sustainability of the treatment. The in vivo AAVs persist in episomal forms. The therapeutic efficacy can then be significantly reduced or lost when cells proliferate (139). Since the persistence depend on the division rate, the transgene expression persistence depends on the tissue type. Nakai et al. (140) showed that up to 92% of rAAV injected in mice portal vein were loss after hepatectomy at 6–12 weeks post injection. Authors suggested that episomal rAAVs represent the major form of gene expression in the liver. Efforts are put together to develop vectors that can be less genotoxic and more persistent in episomal forms (141, 142). Such effort might be beneficial for diseases requiring long-term robust expression of transgene. The DMD patients mostly die by the age of 30 because of cardiorespiratory complications (13, 14). The DMD patients are often diagnosed late by their teen age. The capacity of micro-dystrophin to prevent such cardiorespiratory complications is currently unknown. The existing animal models for DMD are not appropriate for such a study because they die before the disease progression into cardiopathy. To address this gap, Howard et al. (143) recently developed a dystrophic dko mouse model with transgenic correction of skeletal muscles that progresses into late-stage heart failure over 12 months in a series of steps similar to human DMD cardiomyopathy. Authors showed for the first time that micro-dystrophin effectively prevents cardiac failure in mouse.
The gene modification approach using CRISPR/Cas9 technology is evolving rapidly and gaining interest for different possibilities to modify the human genome. However, it leads to frequent micro-insertions or micro-deletions (INDELs) (59). Modifications of the Cas9 gene resulted in the development of Cas9 variants with PAMless requirement, nickase and/or high-fidelity activity (144). Nowadays, there are new nuclease variants coupled with cytidine or adenine deaminase to change cytidine into thymine and adenine into guanine, respectively. This permitted the specific modification of DMD nonsense mutations with editing efficiency of 80% in the mdx4cv mice. Off-target mutations, however, are also associated with this technology, both in the conversion window and in the transcriptome of the cells. The most recent and promising gene editing technology is the CRISPR/Cas9 Prime editing technique that can change any nucleotide into any other nucleotide, as well as insert or delete nucleotides in a specific sequence (57). That technology has recently been modified for more efficiency (69, 70, 145). There are some examples, which have served as proofs of principle, that Prime editing can be used to introduce specific point mutations in many exons of the DMD gene (72). Translation to human trials for CRISPR/Cas9 derived technologies is very difficult to implement because of a set of challenges including pre-existing immune responses to Cas9, the effects of repetitive expositions, the genome stability and off target effects of the protein, the optimization for the transient expression of Cas9, etc.
Adeno-associated viruses have long been used as viral delivery method for gene therapy approaches. However, AAVs induce immune responses and thus, patients undergoing treatment using AAVs will not be able to participate in other trials using the same vector (129). This serious issue might be handled by the removal of AAVs neutralizing antibodies by plasmaphereses in AAV seropositive candidates prior to treatment (122) and by immunomodulation of anti-AAV precursor (120, 123). Another approach to solve this anti-AAV issue is the development of AAV serotypes that are less immunogenic and cannot be recognized by the host immune system (146). AAVs also have limited packaging size, a drawback which is partially circumvented using trans-splicing technics with dual or triple AAV systems to package large DNA sequences. However, the trans-splicing methods are not as efficient as the use of single AAVs (133, 134). EVs represent a potential alternative for gene therapy cargo delivery. EVs are naturally produced by the human body and can be used to transfer large molecules (136).
Conclusion
Duchenne muscular dystrophy, as a hereditary rare disease, causes serious burdens, and suffering for both patients and their families. The increased knowledge about the disease and its etiology has led to a better understanding and development of pharmacological and clinical strategies to help patients. Nowadays, interesting therapeutic approaches (Figure 5) and combined strategies for phenotypic improvement, transitive, or long-term dystrophin expression under the sarcolemma are increasing and giving hope to those afflicted with this disease. Some of these DMD treatments have already obtained pre-authorization to treat some DMD patients. Improvements are still required to increase the effectiveness and long-term safety of these treatments.
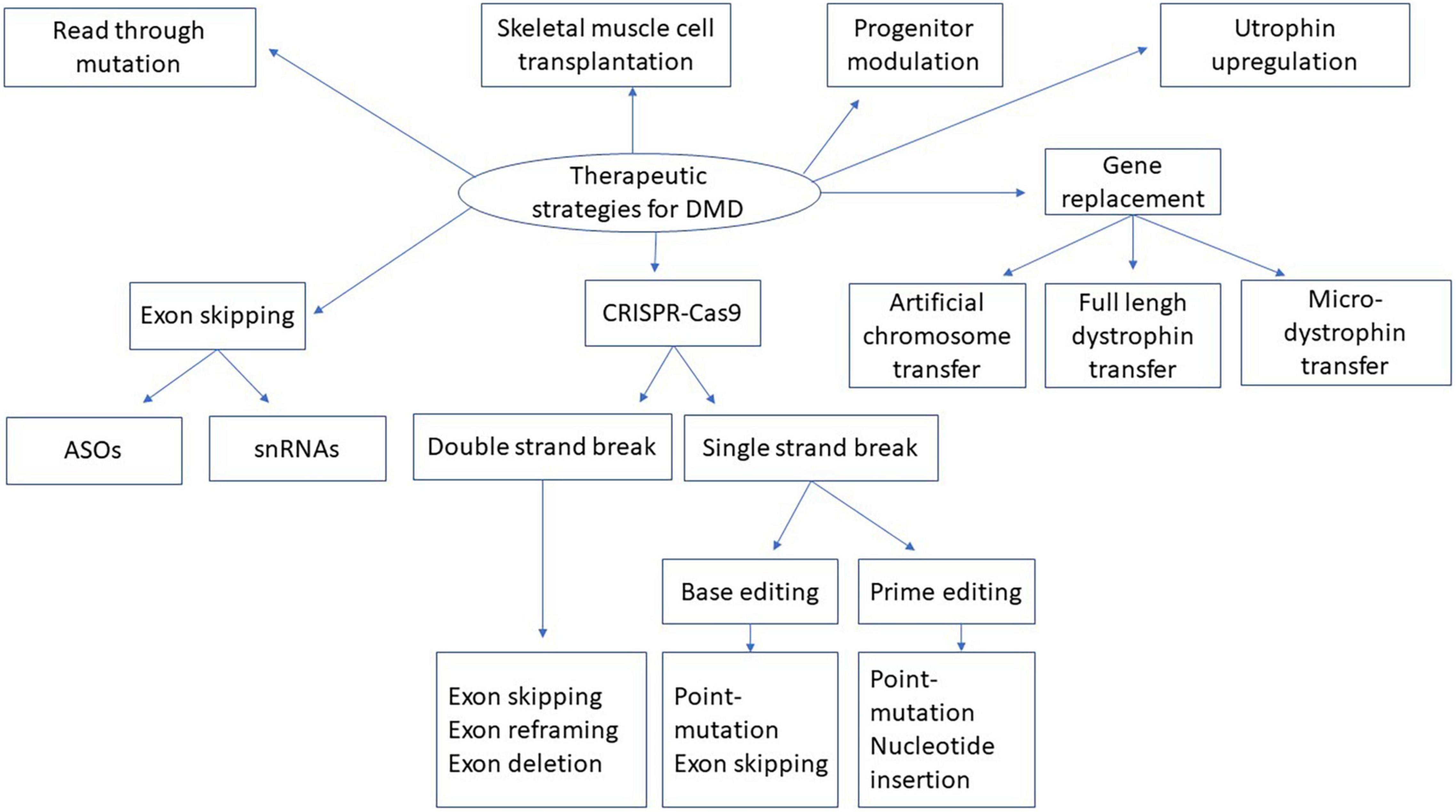
Figure 5. Therapeutic strategies for DMD. This figure represents the summary of different strategies described in the present manuscript for dystrophin replacement in DMD. snRNA means small nuclear ribonucleic acid. ASOs means antisense oligonucleotides. CRISPR-Cas9 means Clustered Regularly Interspaced Short Palindromic Repeats, CRISPR-associated protein 9.
Author Contributions
CH designed and wrote the manuscript. GL revised and improved the English spelling. JT revised and supervised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Canadian Institute of Health Research (CIHR), the Foundation for Cell and Gene Therapy (Jesse’s Journey), and the ThéCell network of the Fond de Recherche du Québec en Santé (FRQS). CH had a fellowship from the Centre Thématique de Recherche en Neurosciences (CTRN) and from the Desjardins fund from Centre de Recherche du CHU de Québec-Université Laval. GL had a CIHR and an FRQS fellowship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. (2002) 82:291–329. doi: 10.1152/physrev.00028.2001
2. Zhang Y, Li H, Min Y-L, Sanchez-Ortiz E, Huang J, Mireault AA, et al. Enhanced CRISPR-Cas9 correction of duchenne muscular dystrophy in mice by a self-complementary AAV delivery system. Sci Adv. (2020) 6:eaay6812. doi: 10.1126/sciadv.aay6812
3. Gao Q, McNally EM. The dystrophin complex: structure, function and implications for therapy. Compr Physiol. (2015) 5:1223–39. doi: 10.1002/cphy.c140048
4. Fealey ME, Horn B, Coffman C, Miller R, Lin AY, Thompson AR, et al. Dynamics of dystrophin’s actin-binding domain. Biophys J. (2018) 115:445–54. doi: 10.1016/j.bpj.2018.05.039
5. Mias-Lucquin D, Dos Santos Morais R, Chéron A, Lagarrigue M, Winder SJ, Chenuel T, et al. How the central domain of dystrophin acts to bridge F-actin to sarcolemmal lipids. J Struct Biol. (2020) 209:107411. doi: 10.1016/j.jsb.2019.107411
6. Bies RD, Caskey CT, Fenwick R. An intact cysteine-rich domain is required for dystrophin function. J Clin Invest. (1992) 90:666–72. doi: 10.1172/JCI115909
7. Nowak KJ, Davies KE. Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO Rep. (2004) 5:872–6. doi: 10.1038/sj.embor.7400221
8. Dystrophin isoforms.Dystrophin Isoforms and Their Expression [Internet]. (2006). Available online at: https://www.dmd.nl/isoforms.html (accessed May 29, 2021).
9. Crisafulli S, Sultana J, Fontana A, Salvo F, Messina S, Trifirò G. Global epidemiology of duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet J Rare Dis. (2020) 15:141. doi: 10.1186/s13023-020-01430-8
10. Ryder S, Leadley RM, Armstrong N, Westwood M, de Kock S, Butt T, et al. The burden, epidemiology, costs and treatment for duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis. (2017) 12:79. doi: 10.1186/s13023-017-0631-3
11. Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, et al. The TREAT-NMD DMD global database: analysis of more than 7,000 duchenne muscular dystrophy mutations. Hum Mutat. (2015) 36:395–402. doi: 10.1002/humu.22758
12. Liechti-Gallati S, Koenig M, Kunkel LM, Frey D, Boltshauser E, Schneider V, et al. Molecular deletion patterns in duchenne and becker type muscular dystrophy. Hum Genet. (1989) 81:343–8. doi: 10.1007/BF00283688
13. Ciafaloni E, Kumar A, Liu K, Pandya S, Westfield C, Fox DJ, et al. Age at onset of first signs or symptoms predicts age at loss of ambulation in duchenne and becker muscular dystrophy: data from the MD STARnet. J Pediatr Rehabil Med. (2016) 9:5–11. doi: 10.3233/PRM-160361
14. NIH.Duchenne Muscular Dystrophy | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program [Internet]. (2020). Available online at: https://rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy (accessed February 25, 2021).
15. Li H, Xiao L, Wang L, Lin J, Luo M, Chen M, et al. HLA polymorphism affects risk of de novo mutation of dystrophin gene and clinical severity of duchenne muscular dystrophy in a Southern Chinese population. Front Neurol. (2018) 9:970. doi: 10.3389/fneur.2018.00970
16. Nakamura A, Yoshida K, Fukushima K, Ueda H, Urasawa N, Koyama J, et al. Follow-up of three patients with a large in-frame deletion of exons 45–55 in the duchenne muscular dystrophy (DMD) gene. J Clin Neurosci. (2008) 15:757–63. doi: 10.1016/j.jocn.2006.12.012
17. England SB, Nicholson LVB, Johnson MA, Forrest SM, Love DR, Zubrzycka-Gaarn EE, et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. (1990) 343:180–2. doi: 10.1038/343180a0
18. Malik V, Rodino-Klapac LR, Viollet L, Wall C, King W, Al-Dahhak R, et al. Gentamicin-induced readthrough of stop codons in duchenne muscular dystrophy. Ann Neurol. (2010) 67:771–80. doi: 10.1002/ana.22024
19. Kaufman RJ. Correction of genetic disease by making sense from nonsense. J Clin Invest. (1999) 104:367–8. doi: 10.1172/JCI8055
20. Sun C, Shen L, Zhang Z, Xie X. Therapeutic strategies for duchenne muscular dystrophy: an update. Genes (Basel). (2020) 11:837. doi: 10.3390/genes11080837
21. Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest. (1999) 104:375–81. doi: 10.1172/JCI7866
22. Wagner KR, Hamed S, Hadley DW, Gropman AL, Burstein AH, Escolar DM, et al. Gentamicin treatment of duchenne and becker muscular dystrophy due to nonsense mutations. Ann Neurol. (2001) 49:706–11. doi: 10.1002/ana.1023
23. Politano L, Nigro G, Nigro V, Piluso G, Papparella S, Paciello O, et al. Gentamicin administration in duchenne patients with premature stop codon. preliminary results. Acta Myol. (2003) 22:15–21.
24. Hayward RS, Harding J, Molloy R, Land L, Longcroft-Neal K, Moore D, et al. Adverse effects of a single dose of gentamicin in adults: a systematic review. Br J Clin Pharmacol. (2018) 84:223–38. doi: 10.1111/bcp.13439
25. Dabrowski M, Bukowy-Bieryllo Z, Zietkiewicz E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol Med. (2018) 24:25. doi: 10.1186/s10020-018-0024-7
26. Mullard A. EMA reconsiders ‘read-through’ drug against duchenne muscular dystrophy following appeal. Nat Biotechnol. (2014) 32:706–706. doi: 10.1038/nbt0814-706
27. McDonald CM, Campbell C, Torricelli RE, Finkel RS, Flanigan KM, Goemans N, et al. Ataluren in patients with nonsense mutation duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2017) 390:1489–98. doi: 10.1016/S0140-6736(17)31611-2
28. Jesse’s Journey.PTC Therapeutics: Hosts Call to Review Results of TranslarnaTM (ataluren) | Duchenne Muscular Dystrophy | Jesse’s Journey | Canada [Internet]. Duchenne Muscular Dystrophy | Jesse’s Journey. (2021). Available online at: https://www.jessesjourney.com/ptc-therapeutics-hosts-call-to-review-results-of-translarna-ataluren/ (accessed May 30, 2021).
29. Sardone V, Zhou H, Muntoni F, Ferlini A, Falzarano M. Antisense oligonucleotide-based therapy for neuromuscular disease. Molecules. (2017) 22:563. doi: 10.3390/molecules22040563
30. Bennett CF, Krainer AR, Cleveland DW. Antisense oligonucleotide therapies for neurodegenerative diseases. Annu Rev Neurosci. (2019) 42:385–406. doi: 10.1146/annurev-neuro-070918-050501
31. Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. (2010) 50:259–93. doi: 10.1146/annurev.pharmtox.010909.105654
32. Fairclough RJ, Wood MJ, Davies KE. Therapy for duchenne muscular dystrophy: renewed optimism from genetic approaches. Nat Rev Genet. (2013) 14:373–8. doi: 10.1038/nrg3460
33. Guiraud S, Chen H, Burns DT, Davies KE. Advances in genetic therapeutic strategies for duchenne muscular dystrophy. Exp Physiol. (2015) 100:1458–67. doi: 10.1113/EP085308
34. Aartsma-Rus A, Krieg AMFDA. Approves eteplirsen for duchenne muscular dystrophy: the next chapter in the eteplirsen saga. Nucleic Acid Ther. (2017) 27:1–3. doi: 10.1089/nat.2016.0657
35. Komaki H, Takeshima Y, Matsumura T, Ozasa S, Funato M, Takeshita E, et al. Viltolarsen in Japanese duchenne muscular dystrophy patients: a phase 1/2 study. Ann Clin Transl Neurol. (2020) 7:2393–408. doi: 10.1002/acn3.51235
36. Frank DE, Schnell FJ, Akana C, El-Husayni SH, Desjardins CA, Morgan J, et al. Increased dystrophin production with golodirsen in patients with duchenne muscular dystrophy. Neurology. (2020) 94:e2270–82. doi: 10.1212/WNL.0000000000009233
37. FDA.FDA Grants Accelerated Approval to First Drug for Duchenne Muscular Dystrophy [Internet]. (2020). Available online at: https://www. fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug- duchenne-muscular-dystrophy (accessed June 8, 2021).
38. Brogna C, Coratti G, Pane M, Ricotti V, Messina S, D’Amico A, et al. Long-term natural history data in duchenne muscular dystrophy ambulant patients with mutations amenable to skip exons 44, 45, 51 and 53. PLoS One. (2019) 14:e0218683. doi: 10.1371/journal.pone.0218683
39. Sarepta Therapeutics.Sarepta Therapeutics Reports Positive Clinical Results from Phase 2 MOMENTUM Study of SRP-5051 in Patients with Duchenne Muscular Dystrophy Amenable to Skipping Exon 51 | Sarepta Therapeutics, Inc. [Internet]. (2021). Available online at: https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-reports- positive-clinical-results-phase-2 (accessed August 26, 2021).
40. Clemens PR, Rao VK, Connolly AM, Harper AD, Mah JK, Smith EC, et al. Safety, tolerability, and efficacy of viltolarsen in boys with duchenne muscular dystrophy amenable to exon 53 skipping: a phase 2 randomized clinical trial. JAMA Neurol. (2020) 77:982. doi: 10.1001/jamaneurol.2020.1264
41. Sarepta Therapeutics.Sarepta Therapeutics Announces FDA Approval of AMONDYS 45TM (casimersen) Injection for the Treatment of Duchenne Muscular Dystrophy (DMD) in Patients Amenable to Skipping Exon 45 | Sarepta Therapeutics, Inc. [Internet]. (2021). Available online at: https://investorrelations. sarepta.com/news-releases/news-release-details/sarepta-therapeutics- announces-fda-approval-amondys-45tm (accessed June 8, 2021).
42. Muscular Dystrophy.Amondys 45 (Casimersen) - Muscular Dystrophy News [Internet]. (2021). Available online at: https://musculardystrophynews.com/srp-4045/ (accessed June 8, 2021).
43. Kolev NG, Steitz JA. In vivo assembly of functional U7 snRNP requires RNA backbone flexibility within the Sm-binding site. Nat Struct Mol Biol. (2006) 13:347–53. doi: 10.1038/nsmb1075
44. Will CL, Lührmann R. Spliceosome structure and function. Cold Spring Harb Perspect Biol. (2011) 3:a003707. doi: 10.1101/cshperspect.a003707
45. Cazzella V, Martone J, Pinnarò C, Santini T, Twayana SS, Sthandier O, et al. Exon 45 skipping through U1-snRNA antisense molecules recovers the Dys-nNOS pathway and muscle differentiation in human DMD myoblasts. Mol Ther. (2012) 20:2134–42. doi: 10.1038/mt.2012.178
46. Goyenvalle A, Vulin A, Fougerousse F, Leturcq F, Kaplan J-C, Garcia L, et al. Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science. (2004) 306:1796–9. doi: 10.1126/science.1104297
47. Goyenvalle A, Wright J, Babbs A, Wilkins V, Garcia L, Davies KE. Engineering multiple U7snRNA constructs to induce single and multiexon-skipping for duchenne muscular dystrophy. Mol Ther. (2012) 20:1212–21. doi: 10.1038/mt.2012.26
48. Vulin A, Wein N, Simmons TR, Rutherford AM, Findlay AR, Yurkoski JA, et al. The first exon duplication mouse model of duchenne muscular dystrophy: a tool for therapeutic development. Neuromuscul Disord. (2015) 25:827–34. doi: 10.1016/j.nmd.2015.08.005
49. Simmons TR, Vetter TA, Huang N, Vulin-Chaffiol A, Wein N, Flanigan KM. Pre-clinical dose-escalation studies establish a therapeutic range for U7snRNA-mediated DMD exon 2 skipping. Mol Ther Methods Clin Dev. (2021) 21:325–40. doi: 10.1016/j.omtm.2021.03.014
50. Gushchina LV, Frair EC, Rohan N, Bradley AJ, Simmons TR, Chavan HD, et al. Lack of toxicity in nonhuman primates receiving clinically relevant doses of an AAV9.U7snRNA vector designed to induce DMD exon 2 skipping. Hum Gene Ther. (2021) 32:882–94. doi: 10.1089/hum.2020.286
51. Flanigan K, Wein N, Gushchina L, Waldrop M, Weiss RP. 140RNA-Seq shows an absence of off-target splicing effects in AAV9-U7snRNA mediated skipping of DMD exon 2. Neuromuscul Disord. (2019) 29:S89.
52. Wein N, Dunn DM, Waldrop MA, Gushchina LV, Frair EC, Weiss RB, et al. Absence of significant off-target splicing variation with a U7snRNA vector targeting DMD exon 2 duplications. Hum Gene Ther. (2021) 32:1346–59. doi: 10.1089/hum.2020.315
53. Rodríguez-Rodríguez DR, Ramírez-Solís R, Garza-Elizondo MA, De Lourdes Garza-Rodríguez M, Barrera-Saldaña HA. Genome editing: a perspective on the application of CRISPR/Cas9 to study human diseases (review). Int J Mol Med. (2019) 43:1559–74. doi: 10.3892/ijmm.2019.4112
54. Wang H, La Russa M, Qi LS. CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem. (2016) 85:227–64.
55. Jiang F, Doudna JA. CRISPR–Cas9 structures and mechanisms. Annu Rev Biophys. (2017) 46:505–29. doi: 10.1146/annurev-biophys-062215-010822
56. Porto EM, Komor AC, Slaymaker IM, Yeo GW. Base editing: advances and therapeutic opportunities. Nat Rev Drug Discov. (2020) 19:839–59. doi: 10.1038/s41573-020-0084-6
57. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. (2019) 576:149–57. doi: 10.1038/s41586-019-1711-4
58. Happi Mbakam C, Lamothe G, Tremblay G, Tremblay JP. CRISPR-Cas9 gene therapy for duchenne muscular dystrophy. Neurotherapeutics [Internet]. (2022). doi: 10.1007/s13311-022-01197-9 [Epub ahead of print].
59. Iyombe-Engembe J-P, Ouellet DL, Barbeau X, Rousseau J, Chapdelaine P, Lagüe P, et al. Efficient restoration of the dystrophin gene reading frame and protein structure in DMD myoblasts using the CinDel method. Mol Ther Nucleic Acids. (2016) 5:e283. doi: 10.1038/mtna.2015.58
60. Duchêne BL, Cherif K, Iyombe-Engembe J-P, Guyon A, Rousseau J, Ouellet DL, et al. CRISPR-induced deletion with sacas9 restores dystrophin expression in dystrophic models in vitro and in vivo. Mol Ther. (2018) 26:2604–16. doi: 10.1016/j.ymthe.2018.08.010
61. Xu L, Park KH, Zhao L, Xu J, El Refaey M, Gao Y, et al. CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol Ther. (2016) 24:564–9. doi: 10.1038/mt.2015.192
62. Koo T, Lu-Nguyen NB, Malerba A, Kim E, Kim D, Cappellari O, et al. Functional rescue of dystrophin deficiency in mice caused by frameshift mutations using campylobacter jejuni Cas9. Mol Ther. (2018) 26:1529–38. doi: 10.1016/j.ymthe.2018.03.018
63. Min Y-L, Li H, Rodriguez-Caycedo C, Mireault AA, Huang J, Shelton JM, et al. CRISPR-Cas9 corrects duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci Adv. (2019) 5:eaav4324. doi: 10.1126/sciadv.aav4324
64. Domenig SA, Bundschuh N, Lenardič A, Ghosh A, Kim I, Qabrati X, et al. CRISPR/Cas9 editing of directly reprogrammed myogenic progenitors restores dystrophin expression in a mouse model of muscular dystrophy. Stem Cell Rep. (2022) 17:321–36. doi: 10.1016/j.stemcr.2021.12.003
65. Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. (2018) 19:770–88. doi: 10.1038/s41576-018-0059-1
66. Xu L, Zhang C, Li H, Wang P, Gao Y, Mokadam NA, et al. Efficient precise in vivo base editing in adult dystrophic mice. Nat Commun. (2021) 12:3719. doi: 10.1038/s41467-021-23996-y
67. Zuo E, Sun Y, Wei W, Yuan T, Ying W, Sun H, et al. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science. (2019) 364:289–92. doi: 10.1126/science.aav9973
68. Chemello F, Chai AC, Li H, Rodriguez-Caycedo C, Sanchez-Ortiz E, Atmanli A, et al. Precise correction of duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci Adv. (2021) 7:eabg4910. doi: 10.1126/sciadv.abg4910
69. Liu P, Liang S-Q, Zheng C, Mintzer E, Zhao YG, Ponnienselvan K, et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat Commun. (2021) 12:2121. doi: 10.1038/s41467-021-22295-w
70. Chen PJ, Hussmann JA, Yan J, Knipping F, Ravisankar P, Chen P-F, et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell. (2021) 184:5635.e–52.e. doi: 10.1016/j.cell.2021.09.018
71. Tremblay G, Rousseau J, Mbakam CH, Tremblay JP. Insertion of the icelandic mutation (A673T) by prime editing: a potential preventive treatment for familial and sporadic alzheimer’s disease. CRISPR J. (2022) 5:109–22. doi: 10.1089/crispr.2021.0085
72. Rousseau J, Mbakam CH, Guyon A, Tremblay G, Begin FG, Tremblay JP. Specific mutations in genes responsible for Alzheimer and for duchenne muscular dystrophy introduced by base editing and PRIME editing. bioRxiv. (2020): doi: 10.1101/2020.07.31.230565
73. Consalvi S, Saccone V, Mozzetta C. Histone deacetylase inhibitors: a potential epigenetic treatment for c muscular dystrophy. Epigenomics. (2014) 6:547–60. doi: 10.2217/epi.14.36
74. Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev. (2013) 93:23–67. doi: 10.1152/physrev.00043.2011
75. Judson RN, Zhang R-H, Rossi FMA. Tissue-resident mesenchymal stem/progenitor cells in skeletal muscle: collaborators or saboteurs? FEBS J. (2013) 280:4100–8. doi: 10.1111/febs.12370
76. Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biology. (2007) 9:255–67. doi: 10.1038/ncb1542
77. Asakura A, Seale P, Girgis-Gabardo A, Rudnicki MA. Myogenic specification of side population cells in skeletal muscle. J Cell Biol. (2002) 159:123–34. doi: 10.1083/jcb.200202092
78. Gussoni E, Soneoka Y, Strickland CD, Buzney EA, Khan MK, Flint AF, et al. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature. (1999) 401:390–4. doi: 10.1038/43919
79. Mitchell KJ, Pannérec A, Cadot B, Parlakian A, Besson V, Gomes ER, et al. Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat Cell Biology. (2010) 12:257–66. doi: 10.1038/ncb2025
80. Minetti GC, Colussi C, Adami R, Serra C, Mozzetta C, Parente V, et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. (2006) 12:1147–50. doi: 10.1038/nm1479
81. Vojinovic J, Damjanov N, D’Urzo C, Furlan A, Susic G, Pasic S, et al. Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. (2011) 63:1452–8. doi: 10.1002/art.30238
82. Bettica P, Petrini S, D’Oria V, D’Amico A, Catteruccia M, Pane M, et al. Histological effects of givinostat in boys with duchenne muscular dystrophy. Neuromuscul Disord. (2016) 26:643–9. doi: 10.1016/j.nmd.2016.07.002
83. businesswire.Italfarmaco Provides Update on Ongoing Clinical Programs with Givinostat in Oral Presentation at XVIII International Conference on Duchenne and Becker Muscular Dystrophy [Internet]. (2021). Available online at: https://www.businesswire.com/news/home/20210222005359/en/Italfarmaco-Provides-Update-on-Ongoing-Clinical-Programs-with-Givinostat-in-Oral-Presentation-at-XVIII-International-Conference-on-Duchenne-and-Becker-Muscular-Dystrophy (accessed February 18, 2022).
84. European Medicines Agency.EU/3/12/1009: Orphan Designation for the Treatment of Duchenne Muscular Dystrophy [Internet]. (2018). Available online at: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu3121009 (accessed February 4, 2021).
85. Piovesan A, Caracausi M, Antonaros F, Pelleri MC, Vitale L. GeneBase 1.1: a tool to summarize data from NCBI gene datasets and its application to an update of human gene statistics. Database. (2016) 2016:baw153. doi: 10.1093/database/baw153
86. Benedetti S, Uno N, Hoshiya H, Ragazzi M, Ferrari G, Kazuki Y, et al. Reversible immortalisation enables genetic correction of human muscle progenitors and engineering of next-generation human artificial chromosomes for duchenne muscular dystrophy. EMBO Mol Med. (2018) 10:254–75. doi: 10.15252/emmm.201607284
87. Meng J, Sweeney NP, Doreste B, Muntoni F, McClure M, Morgan J. Restoration of functional full-length dystrophin after intramuscular transplantation of foamy virus-transduced myoblasts. Hum Gene Ther. (2020) 31:241–52. doi: 10.1089/hum.2019.224
88. Duan D. Systemic AAV micro-dystrophin gene therapy for duchenne muscular dystrophy. Mol Ther. (2018) 26:2337–56. doi: 10.1016/j.ymthe.2018.07.011
89. Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, et al. Modular flexibility of dystrophin: implications for gene therapy of duchenne muscular dystrophy. Nat Med. (2002) 8:253–61. doi: 10.1038/nm0302-253
90. Wang B, Li J, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci USA. (2000) 97:13714–9. doi: 10.1073/pnas.240335297
91. Hakim CH, Wasala NB, Pan X, Kodippili K, Yue Y, Zhang K, et al. A five-repeat micro-dystrophin gene ameliorated dystrophic phenotype in the severe dba/2j-mdx model of duchenne muscular dystrophy. Mol Ther Methods Clin Dev. (2017) 6:216–30. doi: 10.1016/j.omtm.2017.06.006
92. Tandon A, Jefferies JL, Villa CR, Hor KN, Wong BL, Ware SM, et al. Dystrophin genotype–cardiac phenotype correlations in duchenne and becker muscular dystrophies using cardiac magnetic resonance imaging. Am J Cardiol. (2015) 115:967–71. doi: 10.1016/j.amjcard.2015.01.030
93. Wang H, Marrosu E, Brayson D, Wasala NB, Johnson EK, Scott CS, et al. Proteomic analysis identifies key differences in the cardiac interactomes of dystrophin and micro-dystrophin. Hum Mol Genet. (2021) 30:1321–36. doi: 10.1093/hmg/ddab133
94. Bourdon A, François V, Zhang L, Lafoux A, Fraysse B, Toumaniantz G, et al. Evaluation of the dystrophin carboxy-terminal domain for micro-dystrophin gene therapy in cardiac and skeletal muscles in the DMDmdx rat model. Gene Ther. (2022) 1:1–16. doi: 10.1038/s41434-022-00317-6
95. Mendell JR, Sahenk Z, Lehman K, Nease C, Lowes LP, Miller NF, et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol. (2020) 77:1122. doi: 10.1001/jamaneurol.2020.1484
96. Willcocks RJ, Forbes SC, Walter GA, Sweeney L, Rodino-Klapac LR, Mendell JR, et al. Assessment of rAAVrh.74.MHCK7.micro-dystrophin gene therapy using magnetic resonance imaging in children with duchenne muscular dystrophy. JAMA Netw Open. (2021) 4:e2031851. doi: 10.1001/jamanetworkopen.2020.31851
97. Srivastava A. In vivo tissue-tropism of adeno-associated viral vectors. Curr Opin Virol. (2016) 21:75–80. doi: 10.1016/j.coviro.2016.08.003
98. Philippidis A. After patient death, FDA places hold on pfizer duchenne muscular dystrophy gene therapy trial. Hum Gene Ther. (2022) 33:111–5. doi: 10.1089/hum.2022.29198.bfs
99. PFIZER.Pfizer Presents Initial Clinical Data on Phase 1b Gene Therapy Study for Duchenne Muscular Dystrophy (DMD) | Pfizer [Internet]. (2019). Available online at: https://www.pfizer.com/news/press-release/pressdetail/pfizer_presents_initial_clinical_data_on_phase_1b_gene_therapy_study_for_duchenne_muscular_dystrophy_dmd (accessed February 17, 2022).
100. Solid Biosciences.Solid Biosciences Reports 1.5-Year Data from Patients in the Ongoing IGNITE DMD Phase I/II Clinical Trial of SGT-001 [Internet]. (2021). Available online at: https://www.solidbio.com/about/media/press-releases/solid-biosciences-reports-1-5-year-data-from-patients-in-the-ongoing-ignite-dmd-phase-i-ii-clinical-trial-of-sgt-001 (accessed February 18, 2022).
101. Sun C, Serra C, Lee G, Wagner K. Stem cell-based therapies for duchenne muscular dystrophy. Exp Neurol. (2020) 323:113086. doi: 10.1016/j.expneurol.2019.113086
102. Mueller AL, Bloch RJ. Skeletal muscle cell transplantation: models and methods. J Muscle Res Cell Motil. (2020) 41:297–311. doi: 10.1007/s10974-019-09550-w
103. Riederer I, Negroni E, Bencze M, Wolff A, Aamiri A, Di Santo JP, et al. Slowing down differentiation of engrafted human myoblasts into immunodeficient mice correlates with increased proliferation and migration. Mol Ther. (2012) 20:146–54. doi: 10.1038/mt.2011.193
104. Skuk D, Tremblay JP. First study of intra-arterial delivery of myogenic mononuclear cells to skeletal muscles in primates. Cell Transplant. (2014) 23(Suppl. 1):141–50. doi: 10.3727/096368914X685032
105. Skuk D, Goulet M, Roy B, Piette V, Côté CH, Chapdelaine P, et al. First test of a “high-density injection” protocol for myogenic cell transplantation throughout large volumes of muscles in a duchenne muscular dystrophy patient: eighteen months follow-up. Neuromuscul Disord. (2007) 17:38–46. doi: 10.1016/j.nmd.2006.10.003
106. Skuk D, Tremblay JP. Cell therapy in myology: dynamics of muscle precursor cell death after intramuscular administration in non-human primates. Mol Ther Methods Clin Dev. (2017) 5:232–40. doi: 10.1016/j.omtm.2017.05.002
107. Maffioletti SM, Noviello M, English K, Tedesco FS. Stem cell transplantation for muscular dystrophy: the challenge of immune response. Biomed Res Int. (2014) 2014:964010. doi: 10.1155/2014/964010
108. Skuk D, Goulet M, Roy B, Chapdelaine P, Bouchard J-P, Roy R, et al. Dystrophin expression in muscles of duchenne muscular dystrophy patients after high-density injections of normal myogenic cells. J Neuropathol Exp Neurol. (2006) 65:371–86. doi: 10.1097/01.jnen.0000218443.45782.81
109. Skuk D, Roy B, Goulet M, Chapdelaine P, Bouchard J-P, Roy R, et al. Dystrophin expression in myofibers of duchenne muscular dystrophy patients following intramuscular injections of normal myogenic cells. Mol Ther. (2004) 9:475–82. doi: 10.1016/j.ymthe.2003.11.023
110. Rajput BS, Chakrabarti SK, Dongare VS, Ramirez CM, Deb KD. Human umbilical cord mesenchymal stem cells in the treatment of duchenne muscular dystrophy: safety and feasibility study in india. J Stem Cells. (2015) 10:141–56.
111. Blake DJ, Tinsley JM, Davies KE. Utrophin: a structural and functional comparison to dystrophin. Brain Pathol. (1996) 6:37–47. doi: 10.1111/j.1750-3639.1996.tb00781.x
112. Mattei E, Corbi N, Di Certo MG, Strimpakos G, Severini C, Onori A, et al. Utrophin up-regulation by an artificial transcription factor in transgenic mice. PLoS One. (2007) 2:e774. doi: 10.1371/journal.pone.0000774
113. Pisani C, Strimpakos G, Gabanella F, Di Certo MG, Onori A, Severini C, et al. Utrophin up-regulation by artificial transcription factors induces muscle rescue and impacts the neuromuscular junction in mdx mice. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1172–82. doi: 10.1016/j.bbadis.2018.01.030
114. Loro E, Sengupta K, Bogdanovich S, Whig K, Schultz DC, Huryn DM, et al. High-throughput identification of post-transcriptional utrophin up-regulators for duchenne muscle dystrophy (DMD) therapy. Sci Rep. (2020) 10:2132.
115. Sengupta K, Mishra MK, Loro E, Spencer MJ, Pyle AD, Khurana TS. Genome editing-mediated utrophin upregulation in duchenne muscular dystrophy stem cells. Mol Ther Nucleic Acids. (2020) 22:500–9. doi: 10.1016/j.omtn.2020.08.031
116. Péladeau C, Adam N, Bronicki LM, Coriati A, Thabet M, Al-Rewashdy H, et al. Identification of therapeutics that target eEF1A2 and upregulate utrophin a translation in dystrophic muscles. Nat Commun. (2020) 11:1990. doi: 10.1038/s41467-020-15971-w
117. Muntoni F, Tejura B, Spinty S, Roper H, Hughes I, Layton G, et al. a phase 1b trial to assess the pharmacokinetics of ezutromid in pediatric duchenne muscular dystrophy patients on a balanced diet. Clin Pharmacol Drug Dev. (2019) 8:922–33. doi: 10.1002/cpdd.642
118. Muntoni F, Maresh K, Davies K, Harriman S, Layton G, Rosskamp R, et al. PhaseOut DMD: a phase 2, proof of concept, clinical study of utrophin modulation with ezutromid. Neuromuscul Disord. (2017) 27: S217.
119. Jason TLH, Koropatnick J, Berg RW. Toxicology of antisense therapeutics. Toxicol Appl Pharmacol. (2004) 201:66–83. doi: 10.1016/j.taap.2004.04.017
120. Li N, Parkes JE, Spathis R, Morales M, Mcdonald J, Kendra RM, et al. The effect of immunomodulatory treatments on anti-dystrophin immune response after aav gene therapy in dystrophin deficient mdx mice. J Neuromuscul Dis. (2021) 8:S325–40. doi: 10.3233/JND-210706
121. Le Guiner C, Servais L, Montus M, Larcher T, Fraysse B, Moullec S, et al. Long-term microdystrophin gene therapy is effective in a canine model of duchenne muscular dystrophy. Nat Commun. (2017) 8:16105. doi: 10.1038/ncomms16105
122. Chicoine LG, Montgomery CL, Bremer WG, Shontz KM, Griffin DA, Heller KN, et al. Plasmapheresis eliminates the negative impact of AAV antibodies on microdystrophin gene expression following vascular delivery. Mol Ther. (2014) 22:338–47. doi: 10.1038/mt.2013.244
123. Velazquez VM, Meadows AS, Pineda RJ, Camboni M, McCarty DM, Fu H. Effective depletion of pre-existing anti-aav antibodies requires broad immune targeting. Mol Ther Methods Clin Dev. (2017) 4:159–68. doi: 10.1016/j.omtm.2017.01.003
124. Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, et al. Dystrophin immunity in duchenne’s muscular dystrophy. N Engl J Med. (2010) 363:1429–37. doi: 10.1056/NEJMoa1000228
125. FDA.Cellular, Tissue, and Gene Therapies Advisory Committee September 2-3, 2021 Meeting Announcement - 09/02/2021 - 09/03/2021 [Internet]. (2021). Available online at: https://www.fda.gov/advisory-committees/advisory-committee-calendar/cellular-tissue-and-gene-therapies-advisory-committee-september-2-3-2021-meeting-announcement (accessed February 15, 2022).
126. Philippidis A. After third death, audentes’ AT132 remains on clinical hold. Hum Gene Ther. (2020) 31:908–10. doi: 10.1089/hum.2020.29133.bfs
127. Philippidis A. Fourth boy dies in clinical trial of astellas’ AT132. Hum Gene Ther. (2021) 32:1008–10. doi: 10.1089/hum.2021.29182.bfs
128. Dalwadi DA, Calabria A, Tiyaboonchai A, Posey J, Naugler WE, Montini E, et al. AAV integration in human hepatocytes. Mol Ther. (2021) 29:2898–909. doi: 10.1016/j.ymthe.2021.08.031
129. Xu CL, Ruan MZC, Mahajan VB, Tsang SH. Viral delivery systems for CRISPR. Viruses. (2019) 11:28. doi: 10.3390/v11010028
130. Li X, Corbett AL, Taatizadeh E, Tasnim N, Little JP, Garnis C, et al. Challenges and opportunities in exosome research—Perspectives from biology, engineering, and cancer therapy. APL Bioeng. (2019) 3:011503. doi: 10.1063/1.5087122
131. Ramamoorth M, Narvekar A. Non viral vectors in gene therapy- an overview. J Clin Diagn Res. (2015) 9:GE01–6. doi: 10.7860/JCDR/2015/10443.5394
132. Wu Z, Asokan A, Samulski RJ. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther. (2006) 14:316–27. doi: 10.1016/j.ymthe.2006.05.009
133. Koo T, Popplewell L, Athanasopoulos T, Dickson G. Triple trans-splicing adeno-associated virus vectors capable of transferring the coding sequence for full-length dystrophin protein into dystrophic mice. Hum Gene Ther. (2014) 25:98–108. doi: 10.1089/hum.2013.164
134. Reisinger E. Dual-AAV delivery of large gene sequences to the inner ear. Hear Res. (2020) 394:107857. doi: 10.1016/j.heares.2019.107857
135. Tornabene P, Trapani I, Minopoli R, Centrulo M, Lupo M, de Simone S, et al. Intein-mediated protein trans-splicing expands adeno-associated virus transfer capacity in the retina. Sci Transl Med. (2019) 11:eaav4523. doi: 10.1126/scitranslmed.aav4523
136. Bobis-Wozowicz S, Kania K, Nit K, Blazowska N, Kmiotek-Wasylewska K, Paw M, et al. Efficient in vivo genome editing mediated by stem cells-derived extracellular vesicles carrying designer nucleases. bioRxiv. (2021). doi: 10.1101/2021.02.25.432823
137. Gee P, Lung MSY, Okuzaki Y, Sasakawa N, Iguchi T, Makita Y, et al. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat Commun. (2020) 11:1334. doi: 10.1038/s41467-020-14957-y
139. Schnepp BC, Clark KR, Klemanski DL, Pacak CA, Johnson PR. Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J Virol. (2003) 77:3495–504. doi: 10.1128/jvi.77.6.3495-3504.2003
140. Nakai H, Yant SR, Storm TA, Fuess S, Meuse L, Kay MA. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J Virol. (2001) 75:6969–76. doi: 10.1128/JVI.75.15.6969-6976.2001
141. Hagedorn C, Schnödt-Fuchs M, Boehme P, Abdelrazik H, Lipps HJ, Büning HS. /MAR element facilitates episomal long-term persistence of adeno-associated virus vector genomes in proliferating cells. Hum Gene Ther. (2017) 28:1169–79. doi: 10.1089/hum.2017.025
142. Zhang W, Solanki M, Müther N, Ebel M, Wang J, Sun C, et al. Hybrid adeno-associated viral vectors utilizing transposase-mediated somatic integration for stable transgene expression in human cells. PLoS One. (2020) 15:e0228707. doi: 10.1371/journal.pone.0228707
143. Howard ZM, Dorn LE, Lowe J, Gertzen MD, Ciccone P, Rastogi N, et al. Micro-dystrophin gene therapy prevents heart failure in an improved Duchenne muscular dystrophy cardiomyopathy mouse model. JCI Insight. (2021) 6:e146511. doi: 10.1172/jci.insight.146511
144. Walton RT, Christie KA, Whittaker MN, Kleinstiver BP. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. (2020) 368:290–6. doi: 10.1126/science.aba8853
145. Nelson JW, Randolph PB, Shen SP, Everette KA, Chen PJ, Anzalone AV, et al. Engineered pegRNAs improve prime editing efficiency. Nat Biotechnol. (2021) 4:1–9.
146. Li C, Samulski RJ. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet. (2020) 21:255–72.
147. Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A. Duchenne muscular dystrophy. Nat Rev Dis Primers. (2021) 7:1–19. doi: 10.1093/med/9780199681488.003.0001
148. Olson EN. Toward the correction of muscular dystrophy by gene editing. Proc Natl Acad Sci USA. (2021) 118:e2004840117. doi: 10.1073/pnas.2004840117
149. Yuasa K, Ishii A, Miyagoe Y, Takeda S. [Introduction of rod-deleted dystrophin cDNA, delta DysM3, into mdx skeletal muscle using adenovirus vector]. Nihon Rinsho. (1997) 55: 3148–53.
150. Saccone V, Consalvi S, Giordani L, Mozzetta C, Barozzi I, Sandoná M, et al. HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev. (2014) 28:841–57. doi: 10.1101/gad.234468.113
Keywords: artificial chromosome, progenitor cell, CRISPR/Cas 9, Duchenne muscular dystrophy, dystrophin, exon skipping, gene replacement, read through treatment
Citation: Happi Mbakam C, Lamothe G and Tremblay JP (2022) Therapeutic Strategies for Dystrophin Replacement in Duchenne Muscular Dystrophy. Front. Med. 9:859930. doi: 10.3389/fmed.2022.859930
Received: 24 January 2022; Accepted: 01 March 2022;
Published: 28 March 2022.
Edited by:
Annarita Miccio, INSERM U1163 Institut Imagine, FranceReviewed by:
Fulvio Mavilio, University of Modena and Reggio Emilia, ItalyMario Amendola, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2022 Happi Mbakam, Lamothe and Tremblay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacques P. Tremblay, SmFjcXVlcy1QLlRyZW1ibGF5QGNyY2h1bC51bGF2YWwuY2E=