 Yanchu Li
Yanchu Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Med. , 19 May 2022
Sec. Hematology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.840407
ALK-positive histiocytosis (APH) is a rare and recently described, solitary or generalized, histiocytic proliferative disorder with a characteristic gene translocation involving the fusion of the ALK gene at chromosome 2p23. To date, only 25 cases of APH have been reported. The patient presented with multiple nodules in the lung, liver, gallbladder, pancreas, kidney, and skin rashes, along with recurrent pancreatitis and cholecystitis. The histiocytes from the lesion were positive for CD68 and ALK and negative for S100 and CD1α. A reduced dose of the ALK inhibitor alectinib was administered rather than the standard dose of alectinib or chemotherapy because of recurrent pancreatitis, which has not been previously reported in APH cases. After 18 months of follow-up, the patient was maintained on alectinib, and a partial response (PR) was achieved.
ALK-positive histiocytosis (APH) is characterized by clonal proliferation of histiocytes and can present as either solitary or systemic. A series of ten cases with novel types of solitary (4/10) and systemic (6/10) histiocytosis was reported by Chan et al. (1, 2). A previous immunophenotypic study revealed that histiocytes were positive for ALK and histiocytic markers (CD68 and CD163), variably for S-100, but negative for CD1a, CD207, and BRAF-V600E (2, 3). Meanwhile, KIF5B-ALK and COL1A2-ALK gene fusions were identified via next-generation sequencing-based multiplex PCR (2), especially the primary KIF5B-ALK gene fusion, which was the most frequent (1, 4). However, no correlation has been observed between gene fusion type and disease localization or dissemination (2, 5).
Here, we report a case of aggressive systemic APH with severe recurrent pancreatitis. Furthermore, previously reported cases are reviewed.
The treatment timeline is shown in Figure 1. A 32-year-old Chinese man presented with obvious pain below the xiphoid and skin rashes. CT and MRI scan revealed multiple nodules in the pulmonary system, liver, pancreas, and kidneys, and polypoid lesions of the gallbladder (Figure 2A). The patient underwent laparoscopic cholecystectomy and liver biopsy. After cholecystectomy, recurrent pancreatitis occurred, and skin rashes in the body with multiple nodules developed (Figure 2). Therefore, a subsequent skin biopsy was performed. At the beginning of treatment, the patient also had hypoproteinemia (albumin 31.7 g/l), pancreatitis (lipase 151 IU/l, amylase 169 IU/l), and liver dysfunction. Liver enzymes, including glutamic-pyruvic transaminase, aspartate aminotransferase (AST), and glutamine transpeptidase (GGT), increased to 82 IU/L (normal range, <50 IU/L), 51 IU/L (normal range, <40 IU/L), and 146 IU/L (normal range, <60 IU/L), respectively. However, hemoglobin, white blood cells (WBCs), red blood cells (RBCs), thrombocytes (PLT), and conjugated/unconjugated bilirubin levels were normal (Figure 2).
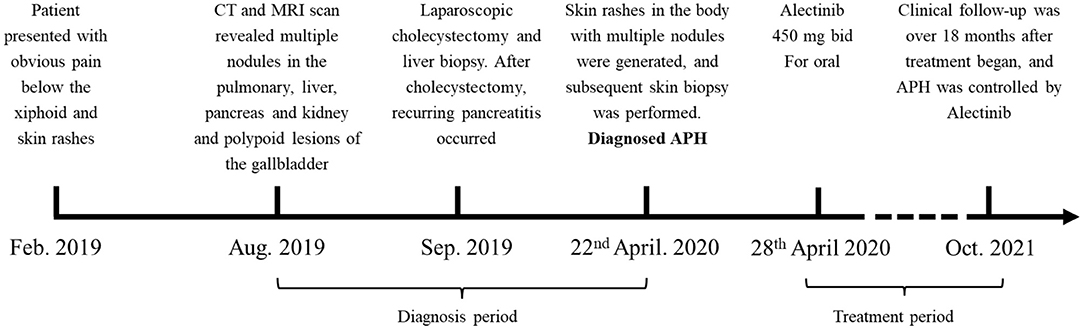
Figure 1. Treatment timeline. The patient suffered from APH since February 2019, and diagnosed in April 2020. After diagnosed, the patient was maintained on alectinib since the 28th April 2020. PR respond reached.
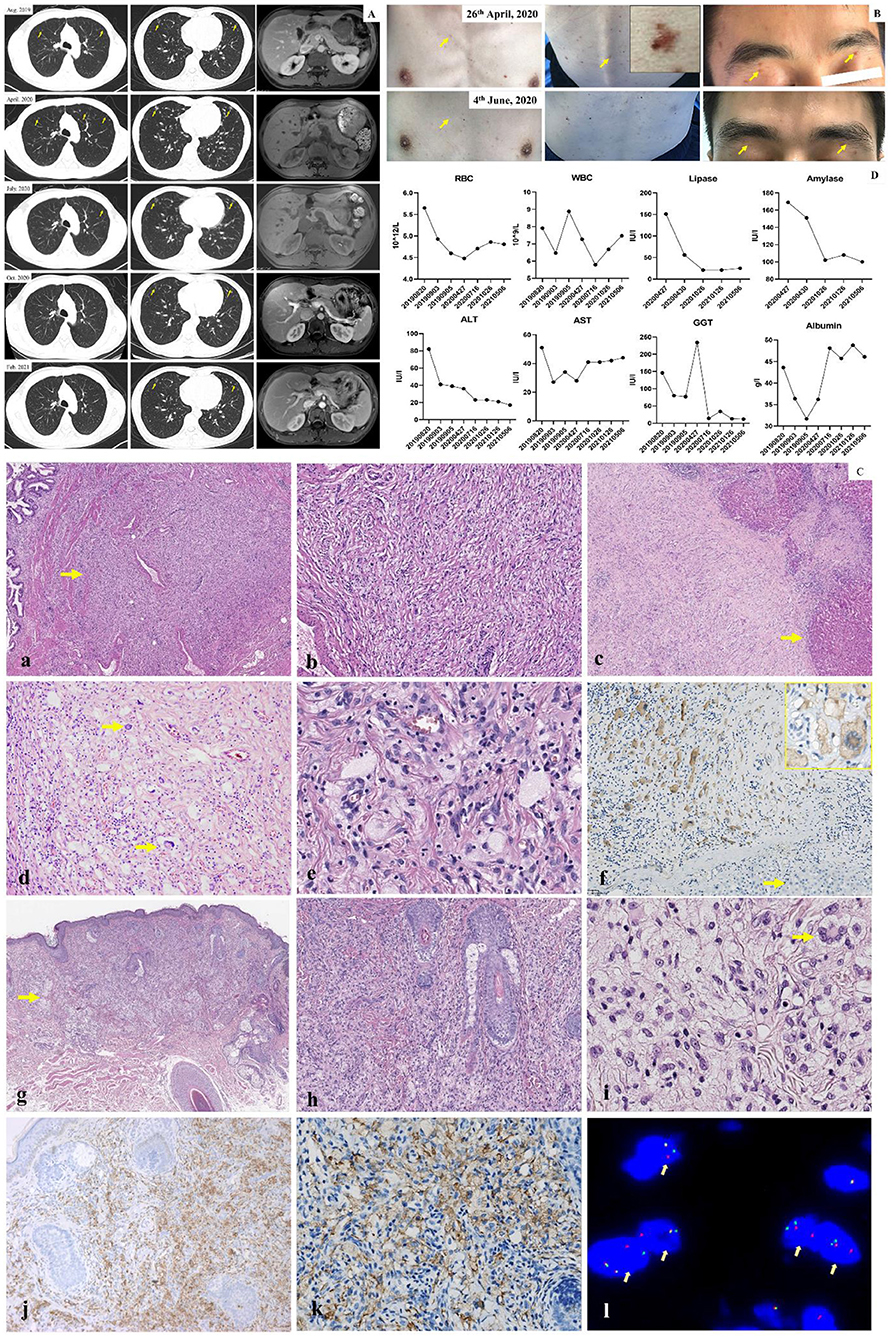
Figure 2. (A) CT and MRI scan of lung and abdomen. CT and MR images showed pulmonary nodule (yellow arrow), liver, kidney and polypoid lesion of gallbladder from August 2019 to February 2021. The multinodules resolved significantly following alectinib treatment. (B) Observation of skin lesion. Before treatment, the multiskin rash and nodule were observed (yellow arrow) on chest wall, back and face; After 1 month treatment, the multiskin rash and nodule were dramatically improved. (C) Pathology. (a) Nonencapsulated gallbladder lesion (arrow) with ill defined margin (H, E, 4x). (b) Gallbladder lesion of fascicular spindle cells, foamy histiocytes and inflammatory cells (H&E, 20x). (c) adjacent to liver capsule with infiltration of the liver parenchyma (arrow) (H&E, original magnification 4x). (d) Liver lesion of numerous foamy histiocytes admixed with Touton-type giant cells (arrow), spindle cells and inflammatory cells (H&E, 20x). (e) The histiocytes had small dark nuclei to medium-sized folded vesicular nuclei with fine chromatin and small to prominent nucleoli (H&E, 40x). (f) ALK expression with a cytoplasmic staining pattern in histiocyes and Touton-type giant cells (inset). Hepatocytes (arrow) were negative for ALK (20x, inset 40x). (g) Nonencapsulated skin lesion (arrow) with ill defined margin (H&E, 4x). (h) Xanthogranuloma-like appearance of the skin lesion (H&E, 10x). (i) Classic and characteristic moderate- to large-sized foamy histocytes with folded nuclei and small to prominent nucleoli of the liver lesion. Note also a Touton-type giant cells (arrow) (H&E, 40x). (j) CD68 expression in histiocytes of the skin lesion (20x). (k) ALK expression in histocytes of the skin lesion (40x). (l) Break-apart fluorescence in situ hybridization (FISH) assay for ALK gene rearrangements showed positive result with separations of the green and red signals (arrow, 100x). (D) Routine blood test, liver function and lipase/amylase test. No severe liver function, gastrointestinal and hematological toxicity over Grade II (CTCAE 3.0) were observed. For liver function, the levels of AST, ALT, GGT, and albumin were 28 U/l (0–40 U/l), 36 U/l (0–50 U/l), 234 IU/l (<60 IU/l), and 36.2 g/l (40–55 μmol/l), respectively. For routine blood test, WBC and RBC were 7.27 × 109/l (3.6–9.5 × 109/l) and 4.48 × 1012/l (4.3–5.8 × 1012/l), respectively. For lipase/amylase test, the lipase and amylase were 151 IU/l (13–60 IU/l) and 169 IU/l (344–135 IU/l), respectively. The recurrent pancreatitis was simultaneously cured and was accompanied by lipase/amylase normalization.
Microscopically, as shown in Figure 2C, lesions in both the gallbladder and liver were not encapsulated and had ill-defined margins. The lesion of the gallbladder and liver were shown in Figure 2Ca–c. Both lesions were composed of slender bland spindle cells with a fascicular growth pattern and were mixed with numerous moderate- to large-sized foamy histiocytes, a few Touton-type giant cells, lymphocytic infiltrates, eosinophils, and occasional plasma cells (Figure 2Cd). The histiocytes had small dark nuclei to medium-sized folded vesicular nuclei with fine chromatin and small to prominent nucleoli (Figure 2Ce). No mitosis or necrosis was observed in either of the lesions. Immunohistochemically, the lesion cells showed cytoplasmic staining for ALK (Anti ALK clone OTI1H, 1:100, Zhongshan Golden Bridge Biotechnology, Beijing, China) (Figure 2Cf), and the histiocytes were positive for CD68 (CD68-PGM-1, Prediluted, Zhongshan Golden Bridge Biotechnology, Beijing, China) but negative for CD1α (CD1α-EP80, 1:200, Zhongshan Golden Bridge Biotechnology, Beijing, China), S-100 protein (S100-4C4.9, 1:300, MXB Biotechnology, Fuzhou, China), and Langerin (Langerin-12D6, Prediluted, MXB Biotechnology, Fuzhou, China). Spindle cells were positive for SMA (SMA-UMAB237, 1:200; Zhongshan Golden Bridge Biotechnology, Beijing, China). BRAF V600E and V600K mutations, tested via PCR and Sanger sequencing, were negative. Break-apart fluorescence in situ hybridization (FISH) assay for ALK gene rearrangements revealed split signals in 30% of the analyzed nuclei. Thus, the initial diagnosis of an inflammatory myofibroblastic tumor was made, which seemed to be inconsistent with the involvement of multiple organs in the patient. The skin biopsy taken subsequently revealed a xanthogranuloma-like lesion (Figure 2Cg,h), composed of large foamy histiocytes and Touton-type giant cells (Figure 2Ci), similar to those seen in the liver and gallbladder lesions. The histiocytes were positive for CD68 (Figure 2Cj), CD163, and ALK (Figure 2Ck) but negative for the S-100 protein. FISH assays were positive for ALK rearrangements (Figure 2Cl). This, a final diagnosis of APH was made.
At the beginning, the patient was not provided specific treatment, which not only resulted in the rapid and continuous increase of both lesion size and the number of nodules in the involved organs (Figure 2) but also caused recurrent pancreatitis and body weight loss of approximately 15 kg. However, because of recurrent pancreatitis, chemotherapeutic agents and ALK inhibitors that increase the risk of pancreatitis, such as ceritinib and crizotinib, could not be used for treatment. Thus, the ALK inhibitor alectinib (450 mg bid) was used, then this patient was maintained on alectinib. After treatment, the multiple nodules in the pulmonary system, liver, pancreas, kidneys, as well as skin rashes were dramatically improved (Figure 2), and partial response was achieved. Clinical follow-up was conducted after treatment initiation, and revealed that APH progression was inhibited by alectinib. The recurrent pancreatitis was simultaneously cured and was accompanied by lipase/amylase normalization (Figure 2D). Meanwhile, according to the Common Terminology Criteria for Adverse Events (CTCAE) criteria, no severe liver function, gastrointestinal. or hematological side effects were observed beyond grade II.
The patient was still followed-up and remained on alectinib treatment. There was no evidence of APH recurrence, and the pancreatitis was cured and had no recurrence.
APH is a rare disease; the first publication on APH was in 2008 (1). Currently, classification systems of histiocytosis have evolved into five groups, which are defined based on the cell of origin, molecular mutations, and clinical behavior (5). However, APH has not yet been included in the WHO classification of tumors (6). Recently, only 26 cases, including this study, have been reported under the term “APH” (1–4, 7–17).
The pathogenesis of this condition is yet to be fully elucidated. The general information, histological and clinical characters of the 26 patients is shown in Tables 1, 2. The male-to-female ratio was 9:17, and systemic and solitary cases were equal. The median age of the patients was 15 years. Regarding race, 6 patients were Caucasian, 8 were Asian, including 4 Chinese patients (1, 2, 8), and the other 12 were not stated. Histologically, the tumor histiocytes were large, with irregularly folded, lobulated, or clefted nuclei, fine chromatin, and abundant eosinophilic or foamy cytoplasm, sometimes with emperipolesis (3, 10). In some cases, the tumor cells displayed a spindle appearance with a fascicular to storiform growth pattern (3). Touton-type giant cells and lymphocytic infiltrates were observed. Immunohistochemically, the histiocytes were positive for ALK and histiocytic markers (CD68 and CD163), variable for S-100 and negative for CD1a, CD207, and BRAF-V600E. The staining pattern for ALK can be cytoplasmic, membranous, and perinuclear or cytoplasmic dot-like (14). ALK rearrangement was identified in all APH cases, and KIF5B-ALK was the most frequent gene translocation, and the detailed information on the gene types of the 26 patients is shown in Table 2.
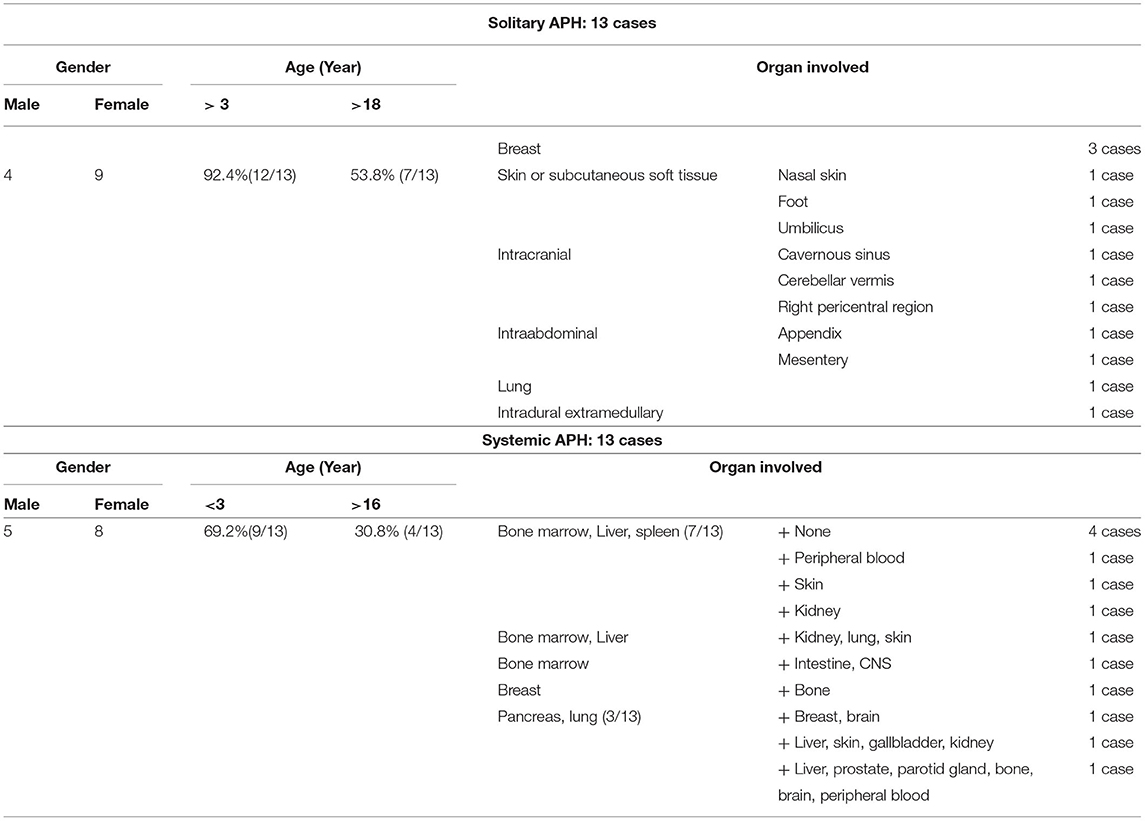
Table 1. Summary of clinicopathologic findings for patients with solitary and systemic APH.

Table 2. Summary of organ involvement, gene type, treatment and response for patients with APH.
Meanwhile, APH should be differentiated from other histiocytosis types, including juvenile xanthogranuloma (JXG), Erdheim-Chester disease (ECD), Langerhans cell histiocytosis, and Rosai-Dorfman disease (RDD) (18–20). The tumor histiocytes with marked nuclear foldings, which are not found in other histiocytosis types, could be a clue for APH (10), and an immunohistochemical profile featuring positivity to both CD68 (CD163) and ALK virtually excludes all the histiocytic neoplasms as well as all the other ALK-positive neoplasms, such as ALK-positive peripheral T cell lymphoma, ALK-positive B cell lymphoma, lung adenocarcinoma, inflammatory myofibroblastic tumor, and some melanocytic tumors of the Spitz lineage (1). Notably, some cases of histiocytosis with ALK gene rearrangement are reported under the diagnosis of ECD, JXG, atypical juvenile histiocytosis, or histiocytosis not otherwise specified (11, 12, 21), some of which have detailed clinicopathologic data displaying the lack of characteristic long bone or skin involvement for ECD or JXG, respectively (12). In addition, Huang et al. reports two cases of congenital/ early-onset RDD without any molecular detection for ALK, yet displaying an identical clinical presentation and proliferation in young children (10). These reported cases reveal that whether APH is a distinct entity is unclear and controversial. However, as illustrated by Chen (1, 2) and other researchers (3, 4, 7–16), APH differs significantly from ECD, JXG, and RDD in terms of clinical, morphological, and genetic aspects.
The clinical features of the previously reported cases of APH are summarized in Table 2. Surgery is the typical treatment for localized APH. A total of 92.3% (12/13) of the localized cases underwent surgical resection and were followed up from 1 month to 3.5 years after surgery, and no recurrence was found. One patient showing involvement of the cavernous sinus was treated with the ALK inhibitor crizotinib, and a complete response (CR) was reached 3 months later. No recurrence was observed for 6 months after follow-up. On the other hand, treatment for systemic APH mainly includes chemotherapy and/or ALK inhibitors (8, 16). Thus, 8 patients received chemotherapy, of which 37.5% (3/8) received chemotherapy alone, 50.0% (4/8) received steroid therapy, and 12.5% (1/8) was treated with the PD-1 inhibitor pembrolizumab. Moreover, previous clinical studies have shown an encouraging response of the cavernous sinus, pancreas, and lung lesions to ALK inhibitor treatment (2, 8); thus, even if chemotherapy is not effective, PR and CR might be achieved by receiving anti-ALK therapy as second-line therapy. More recently, ibrutinib, a Bruton tyrosine kinase (BTK) inhibitor, was shown to be effective in and was able to achieve CR from rare APH cases with concurrent CLL/SLL.
For this systemic APH patient with severe recurrent pancreatitis, not only was the patient unable to tolerate the standard treatment; the patient's disease also had an aggressive biological behavior and progressed rapidly. The ALK inhibitor alectinib, which did not induce pancreatitis, was selected, and a 25% reduced dose was used (12, 14).
To our knowledge, this is the first report of a patient with systemic and aggressive APH with severe recurrent pancreatitis. Because of recurrent pancreatitis, the treatment strategy is different from common APH cases since the disease is more aggressive. Thus, in summary, we indicated that APH is not just an indolent disease and that those complex complications represent APH as an aggressive disease. Moreover, the original report was published 13 years ago, but APH entities were likely under-reported.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
YJ and YL contributed to the study conception and design. YL, CS, YW, and MH performed material preparation, data collection, and analysis. YL, XX, and JL wrote the first draft of the manuscript. All authors commented on previous versions of the manuscript and read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chan JK, Lamant L, Algar E, Delsol G, Tsang WY, Lee KC, et al. Alk+ Histiocytosis: a novel type of systemic histiocytic proliferative disorder of early infancy. Blood. (2008) 112:2965–8. doi: 10.1182/blood-2008-03-147017
2. Chang KTE, Tay AZE, Kuick CH, Chen H, Algar E, Taubenheim N, et al. Alk-Positive histiocytosis: an expanded clinicopathologic spectrum and frequent presence of Kif5b-Alk fusion. Mod Pathol. (2019) 32:598–608. doi: 10.1038/s41379-018-0168-6
3. Zhu Y, Fan J, Pan H, Huang B, Wu Y, Shi H, et al. Alk-Positive histiocytosis of the umbilicus with Kif5b-Alk fusion: a case report and review of the literature. Hum Pathol: Case Rep. (2021) 24:1–5. doi: 10.1016/j.ehpc.2021.200504
4. Tran TAN CK, Kuick CH, Goh JY, Chang CC. Local Alk-positive histiocytosis with unusual morphology and novel Trim33-Alk gene fusion. Int J Surg Pathol. (2021) 29:543–9. doi: 10.1177/1066896920976862
5. Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. (2016) 127:2672–81. doi: 10.1182/blood-2016-01-690636
6. Bruneau J, Molina TJ. WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. Springer International Publishing (2019). doi: 10.1007/978-3-319-95309-0_3817
7. Andolina JR. Stalking Histiocytosis: Alk-Positive histiocytosis identified through peripheral blood smear. Acta Haematol. (2021) 144:241–3. doi: 10.1159/000509924
8. Bai Y, Sun W, Niu D, Yang X, Diao X, Yu Y, et al. Localized Alk-positive histiocytosis in a Chinese woman: report of a case in the lung with a novel Eml4-Alk rearrangement. Virchows Arch. (2021) 479:1079–83. doi: 10.1007/s00428-021-03092-8
9. Gupta GKXL, Pack SD, Jones JB, Pittaluga S, Raffeld M, Jaffe ES. Alk-Positive histiocytosis with Kif5b-Alk fusion in an adult female. Haematologica. (2019) 104:e534–e6. doi: 10.3324/haematol.2019.230094
10. Huang H, Gheorghe G, North PE, Suchi M. Expanding the phenotype of Alk-positive histiocytosis: a report of 2 cases. Pediatr Dev Pathol. (2018) 21:449–55. doi: 10.1177/1093526617740784
11. Jaber OI, Jarrah DA, Hiasat M, Hussaini MA. Alk-Positive histiocytosis: A case report and literature review. Turkish J Pathol. (2021) 37:172–7. doi: 10.5146/tjpath.2020.01507
12. Kashima J YM, Jimbo K, Izutsu K, Ushiku T, Yonemori K, Yoshida A. Alk-Positive histiocytosis of the breast: a clinicopathologic study highlighting spindle cell histology. Am J Surg Pathol. (2021) 45:347–55. doi: 10.1097/PAS.0000000000001567
13. Lucas C-HG, Gilani A, Solomon DA, Liang X, Maher OM, Chamyan G, et al. Alk-Positive histiocytosis with Kif5b-Alk fusion in the central nervous system. Acta Neuropathol. (2019) 138:335–7. doi: 10.1007/s00401-019-02027-7
14. Qiu L, Weitzman SP, Nastoupil LJ, Williams MD, Medeiros LJ, Vega F. Disseminated Alk-positive histiocytosis with Kif5b-Alk fusion in an adult. Leuk Lymphoma. (2021) 62:1234–8. doi: 10.1080/10428194.2020.1861273
15. Swain F, Williams B, Barbaro P. Alk-Positive histiocytosis with peripheral blood histiocytes: a case report. Acta Haematol. (2021) 144:218–21. doi: 10.1159/000508524
16. Syrykh C, Ysebaert L, Pericart S, Evrard SM, Meggetto F, Kanoun S, et al. Alk-Positive histiocytosis associated with chronic lymphocytic leukaemia/small lymphocytic lymphoma: a multitarget response under ibrutinib. Virchows Arch. (2021) 478:779–83. doi: 10.1007/s00428-020-02937-y
17. Ross JS, Ali SM, Fasan O, Block J, Pal S, Elvin JA, et al. Alk fusions in a wide variety of tumor types respond to Anti-Alk targeted therapy. Oncologist. (2017) 22:1444–50. doi: 10.1634/theoncologist.2016-0488
18. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 Revision of the world health organization classification of lymphoid neoplasms. Blood. (2016) 127:2375–90. doi: 10.1182/blood-2016-01-643569
19. Diamond EL, Durham BH, Haroche J, Yao Z, Ma J, Parikh SA, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov. (2016) 6:154–65. doi: 10.1158/2159-8290.CD-15-0913
20. Estrada-Veras JI, O'Brien KJ, Boyd LC, Dave RH, Durham B, Xi L, et al. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv. (2017) 1:357–66. doi: 10.1182/bloodadvances.2016001784
Keywords: case report, ALK-positive histiocytosis, pancreatitis, ALK inhibitor, distinct entity
Citation: Li Y, Shi C, Wu Y, He M, Xia X, Liu J and Jiang Y (2022) Case Report: Rare Systemic and Aggressive ALK-Positive Histiocytosis With Recurrent Pancreatitis Treating by Alectinib. Front. Med. 9:840407. doi: 10.3389/fmed.2022.840407
Received: 13 January 2022; Accepted: 25 April 2022;
Published: 19 May 2022.
Edited by:
Gerardo Ferrara, Ospedale Generale Provinciale Macerata, ItalyCopyright © 2022 Li, Shi, Wu, He, Xia, Liu and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Jiang, jiang_yu@scu.edu.cn
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.