 Meera Gupta1,2*
Meera Gupta1,2* Gabriel Orozco1,2Madhumati Rao3Roberto Gedaly1,2Hartmut H. Malluche3Javier A. Neyra3,4*
Gabriel Orozco1,2Madhumati Rao3Roberto Gedaly1,2Hartmut H. Malluche3Javier A. Neyra3,4*- 1Department of Surgery - Transplant Division, University of Kentucky, College of Medicine, Lexington, KY, United States
- 2Department of Surgery, University of Kentucky, Lexington, KY, United States
- 3Department of Internal Medicine - Nephrology, Bone and Mineral Metabolism Division, University of Kentucky, College of Medicine, Lexington, KY, United States
- 4Division of Nephrology, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, United States
Cardiovascular disease and mineral bone disorders are major contributors to morbidity and mortality among patients with chronic kidney disease and often persist after renal transplantation. Ongoing hormonal imbalances after kidney transplant (KT) are associated with loss of graft function and poor outcomes. Fibroblast growth factor 23 (FGF-23) and its co-receptor, α-Klotho, are key factors in the underlying mechanisms that integrate accelerated atherosclerosis, vascular calcification, mineral disorders, and osteodystrophy. On the other hand, kidney donation is also associated with endocrine and metabolic adaptations that include transient increases in circulating FGF-23 and decreases in α-Klotho levels. However, the long-term impact of these alterations and their clinical relevance have not yet been determined. This manuscript aims to review and summarize current data on the role of FGF-23 and α-Klotho in the endocrine response to KT and living kidney donation, and importantly, underscore specific areas of research that may enhance diagnostics and therapeutics in the growing population of KT recipients and kidney donors.
Introduction
Cardiovascular disease (CVD) and bone mineral disorders are important causes of mortality and morbidity among end-stage kidney disease (ESKD) patients (1). The endocrine adaptation to a decreased glomerular filtration rate (GFR) plays a central role in the cardiovascular and skeletal alterations observed in patients with chronic kidney disease—mineral bone disorder (CKD-MBD). Fibroblast growth factor 23 (FGF-23) and its coreceptor, α-Klotho, are key factors in phosphate homeostasis, and their dysregulation is an essential link between osteodystrophy, left ventricular hypertrophy, atherosclerosis, systemic inflammation, and renal fibrosis observed in patients with ESKD (2).
While the treatment of choice for ESKD is kidney transplant (KT), CVD continues to be a significant cause of morbidity and mortality after transplantation and is a contributing factor to graft failure and loss (3). Post kidney transplant patients (PKTP) have persistent alterations in bone mineral metabolism exacerbated by immunosuppressive therapy, which can lead to progression of bone disease and CVD (4). While the bone-vascular axis has been extensively studied and is significantly affected in patients with ESKD, the endocrine response to kidney transplantation and kidney donation is not yet fully understood. Among kidney donors, significant derangements of markers of bone metabolism, including serum α-Klotho, have been reported after nephrectomy (5–9). However, the clinical impact, if any, of these alterations in otherwise healthy living donors has not been determined.
In this manuscript, we aim to review and summarize current data on the role of FGF-23 and α-Klotho in the endocrine response to KT and living kidney donation. Importantly, we intend to underpin specific areas of research that may enhance diagnostics and therapeutics in the growing population of KT recipients and kidney donors.
FGF-23 and α-Klotho in Mineral Metabolism
FGF-23 is one of the few fibroblast growth factors that enters the systemic circulation and acts as a hormone (10). FGF-23 is synthesized by osteocytes and secreted in response to rising serum phosphate levels. Its secretion is also stimulated by parathyroid hormone (PTH), vitamin D, soluble α-Klotho, and pro-inflammatory cytokines including IL-1, IL-6, and TNFα (2, 11–13). All fibroblast growth factor receptors (FGF-R) have a low affinity for FGF-23, and the high-affinity interaction depends on the expression of the co-receptor, α-Klotho (14, 15).
The organs most impacted by FGF-23 are the kidneys and parathyroid glands (Figure 1). In the absence of disease, FGF-23 induces the degradation and decline in the synthesis of sodium-dependent phosphate transport protein 2A (NaPi2A) in the kidney (16). In doing so, FGF-23 directly decreases proximal tubular reabsorption of phosphate (17). Highlighting its physiological relevance, animal models with a deletion on FGF-23 or α-Klotho genes develop phosphate retention (18). FGF-23 also modifies mineral metabolism by decreasing calcitriol levels through two different mechanisms: it decreases calcitriol synthesis by downregulating 25-hydroxyvitamin D3 1α hydroxylase in the proximal tubular cells of the kidney, and it increases vitamin D inactivation by up-regulating vitamin D 24-hydroxylase (CYP24A1) expression (19, 20). The decrease in serum calcitriol contributes to PTH secretion despite a direct inhibition of the parathyroid glands by FGF-23 (20, 21). FGF-23 also promotes calcium absorption in the distal convoluted tubules, counterbalancing the inhibition of calcitriol synthesis (22). Notably, FGF-23 auto-regulates its effects by inhibiting the expression of α-Klotho (23). Hence, FGF-23 has a significant direct and indirect effect on phosphate and calcium metabolism.
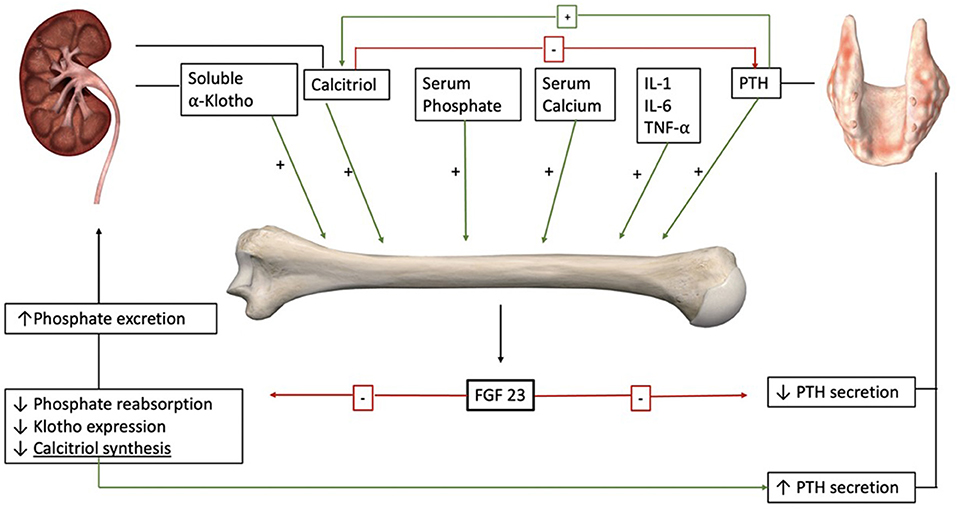
Figure 1. FGF-23 is secreted by osteocytes in response to different endocrine and mineral stimuli including PTH and calcitriol. The main targets of FGF-23 are the kidneys and the parathyroid glands. In the kidneys, it increases phosphate excretion and decreases calcitriol synthesis. Decreased levels of calcitriol increase PTH secretion. On the parathyroids glands, the direct effect of FGF-23 is inhibitory. Also, FGF-23 downregulates the expression of its co-receptor α-Klotho (anatomical images courtesy of Essential Anatomy @3D4Medical).
α-Klotho, discovered as a gene linked to aging, is predominantly expressed in the kidney, specifically in the distal tubules (24, 25). The α-Klotho gene encodes a 130 kDa single-pass transmembrane protein consisting of two extracellular domains (KL1 and KL2), a transmembrane domain, and a short cytoplasmic tail. Part of the extracellular domain of transmembrane α-Klotho is cleaved by proteases (26–28). A secreted form of α-Klotho has also been postulated from an alternatively spliced transcript (29, 30). Cleaved α-Klotho proteins are released into the circulation and are referred to as soluble α-Klotho. Soluble α-Klotho acts as an endocrine or paracrine factor affecting multiple distant organs such as the heart, lungs, bone, and brain (31–37). α-Klotho increases FGF-23 expression in the bone (36) and acts as a non-enzymatic scaffold protein that enhances FGF-23 signaling (38).
α-Klotho is an important regulator of mineral metabolism homeostasis: (1) it decreases renal phosphate reabsorption by acting as a co-receptor for FGF-23 binding to FGFR1 (15); (2) it directly promotes the internalization and degradation of the NaPi2a cotransporter in the renal proximal tubules (39); and (3) it suppresses vitamin D signaling (α-Klotho knockout mice can be rescued from a phenotype of soft tissue calcification by deletion of the CYP27B1 gene) (40–42). In addition, α-Klotho has a plethora of pleiotropic actions as an inhibitor of apoptosis, fibrosis, and cell senescence (43–46).
FGF-23 and α-Klotho in Chronic Kidney Disease and Its Complications
Patients with CKD have significantly reduced functional renal cell mass, leaving the residual renal parenchyma to maintain homeostasis by increasing phosphate excretion. FGF-23—through its phosphaturic effects— plays a crucial role in this response (2, 17) and is an early biomarker of CKD (47). As intact FGF-23 is cleared by the kidneys, a decreased glomerular filtration rate (GFR) also independently contributes to its rising levels (48–50). Among patients with CKD, sustained elevated levels of FGF-23 lead to constant inhibition in calcitriol synthesis, with a consequent increase in PTH secretion (51, 52). Because PTH stimulates the release of FGF-23 synthesis, a closed loop of stimulation is created. Supraphysiologic levels of FGF-23 are also perpetuated by downregulation of its co-receptor α-Klotho, resistance to its renal action, and further hyperphosphatemia (53–56). Remarkably, the compensation to impaired kidney function might promote further progression of CKD through different mechanisms: the phosphate hyperfiltration by functional nephrons leads to tubular damage (57), downregulation of α-Klotho promotes renal fibrosis through increasing TGFβ1 (44) and Wnt (58) signaling pathways, and lower α-Klotho levels might decrease its protective effect on the glomerular filtration barrier (59). The resulting endocrine profile that characterizes patients with CKD –high FGF-23 and PTH, with low calcitriol and α-Klotho—contributes to cardiovascular morbidity, systemic inflammation, dysregulation of bone metabolism, and anemia.
Cardiovascular Diseases
Cardiovascular diseases cause more than half of all deaths among patients with ESKD, and they represent the leading cause of mortality in this vulnerable population (1). Among CKD patients older than 65 years, the prevalence of CVD is 65.8%, compared to 31.9% in patients of the same age group but with normal kidney function (60). Epidemiological studies have shown a direct association between FGF-23 levels, mortality, and risk of major cardiovascular events (61–63). Two major contributor mechanisms are left ventricular hypertrophy (LVH) and a pro-atherosclerotic milieu. LVH is associated with heart failure, changes in heart geometry, and cardiac arrhythmias, while atherosclerosis is associated with coronary, cerebrovascular, and peripheral artery diseases.
High circulating levels of FGF-23 directly induce LVH through the activation of FGFR4 in cardiac myocytes and recruitment of the PLCγ/Calcineurin/NFAT pathway independently of α- Klotho (64–66). FGFR4 is a potential therapeutic target for reducing cardiovascular risk in patients with CKD as pharmacological blockage of this receptor in animal models showed attenuated LVH, while loss of function (FGFR4−/−) protected mice from developing LVH (64).
The pro-atherosclerotic milieu characteristic of patients with CKD is related to alterations in the bone-kidney axis (60, 67). In the initial stages of CKD, increased secretion of FGF-23 helps maintain normal serum phosphate (47). As the GFR decreases, transient or continuous hyperphosphatemia leads to calcium phosphate precipitation and favors the formation of atherosclerotic plaques (68). There is a positive correlation between FGF-23 and vascular calcification in patients with CKD; however, it is still controversial if the association is independent of serum phosphate concentrations or a direct pro-atherosclerotic effect of FGF23 (69).
Aortic stiffness is another biomarker of cardiovascular risk that increases in patients with CKD. While atherosclerosis primarily affects the vascular intima, aortic stiffness represents degenerative changes in the extracellular matrix of the intima and media layers characterized by elastin fracture and collagen deposition (70). Smith et al. demonstrated that increased aortic stiffness among CKD patients is associated with elevated circulating calciprotein particles—which are formed by adsorption of calcium phosphate by Fetuin-A to prevent ectopic calcification (71, 72). Also, in-vitro studies have shown that hyperphosphatemia can trigger chronic inflammation, induce endothelial apoptosis and promote an osteoblastic phenotype in vascular smooth muscle cells (2, 73–75). These are other examples of how mineral metabolism affects the vascular system and increases cardiovascular risk in CKD.
Immune System and Inflammation
The systemic inflammation observed in patients with CKD contributes to their elevated morbidity and mortality (76–78). An association between renal function and inflammation is well-established, and eGFR is inversely correlated with circulating levels of pro-inflammatory cytokines (e.g., IL-1β, IL-6, TNF-α, and hs-CRP) (79). Factors that promote inflammation in CKD include increased production of cytokines, hyperphosphatemia, chronic and recurrent infections, acidosis and oxidative stress, altered metabolism of adipose tissue, and gut microbiota dysbiosis (80). FGF-23 directly induces liver secretion of inflammatory cytokines by targeting FGFR4 receptors in hepatocytes and activating PLCγ/Calcineurin/NFAT signaling pathway— independently of α-klotho. As an indicator of the therapeutic potential of this finding, blockage of FGFR4 prevented significant elevation in C-Reactive Protein (CRP) secretion by the liver in an animal model of CKD (76). Also, altered immune homeostasis increases the susceptibility to infections. FGF-23 impairs neutrophil recruitment by interfering with chemokine signaling through activation of FGFR2 (81). In an unfavorable feedback loop, FGF-23 increases hepatic secretion of cytokines, inflammatory cytokines stimulate the secretion of FGF-23 by osteocytes, and a predisposition to infections contributes to systemic inflammation.
Bone Metabolism
There is a wide range of bone disorders that affect patients with CKD. The spectrum includes disorders in bone turnover, impaired mineralization, and changes in bone volume (82, 83). These abnormalities cause decreased mechanical strength with increased risk of fractures (84, 85), and altered mineral metabolism with increased risk of ectopic calcification (86). Malluche et al. reported a series of 630 bone biopsies from patients with CKD on dialysis. With 418 cortical bone biopsies available, they found that 82% had altered bone turnover, 52% had high cortical porosity, cortical thickness was low in 44%, cancellous bone volume was altered in 69% of the cohort, and only 3% of patients had impaired mineralization. Notably, bone turnover was low among 58% of patients and high among 24% of patients (87). These data show the high prevalence and variety of bone alterations in patients with CKD. High bone turnover is related to elevated PTH, hyperphosphatemia, and increased cortical porosity. Conversely, low bone turnover is related to low calcium retention and ectopic calcification. Low calcium retention indicates impaired incorporation of calcium into the bone matrix with the consequent hypercalcemia after calcium loads (88).
Animal models have shown that FGF-23 also regulates bone mineralization through a phosphate-independent mechanism. Homozygous FGF-23 knock-out mice develop growth delay, low bone mineral density (BMD), skeletal malformations, increased bone fragility, and impaired osteoid mineralization with concomitant hyperphostemia (89). Furthermore, these skeletal abnormalities persisted in hypophosphatemic animal models (FGF-23 −/− / NaPi2a −/−) (90). On the other hand, in fetal rat calvaria cell cultures overexpressing FGF-23, there was also inhibition of bone mineralization (91). This apparent contradiction—where deficiency and exposure to FGF-23 lead to impaired bone mineralization—favors the possibility that FGF-23 acts as an autocrine/paracrine regulator of mineralization (92) and the inhibition of tissue-non-specific alkaline phosphatase activity (TNAP) by FGF-23 might play a role. TNAP hydrolyzes pyrophosphate and generates inorganic phosphate to enhance mineralization (93). FGF-23 can suppress TNAP activity, cause pyrophosphate accumulation, and inhibit mineralization. The deletion of FGF-23 in Hyp mice (a murine homolog for X-linked hypophosphatemia) rescued the suppressed TNAP activity in osteoblasts (93, 94). However, FGF-23 deficiency (Fgf−/− mice) increases TNAP activity, increases the release of osteopontin, and also leads to inhibition of mineralization. This inhibitory effect of osteopontin in bone mineralization can be attenuated with a low phosphate diet or by deletion of the NaPi2A transporter (92, 95).
Osteocytes and osteoblast synthesize α-Klotho (96). Mice with an osteocyte-specific deletion of the α-Klotho gene have increased osteoblast activity and increased bone volume with normal serum biochemistry. When the same deletion is introduced in mice models of CKD (5/6 nephrectomy and high-phosphate diet)—which present with elevated serum phosphate, PTH, and FGF-23—α-Klotho expression in the bone was significantly decreased and the pro-osteoblastic effect was no longer significant. This suggests that α-Klotho might act as a regulator of adequate bone mineralization rate when the systemic mineral homeostasis is preserved (92, 97). As described, the endocrine mechanisms affecting bone metabolism are complex and questions remain as to the role of FGF-23 and α-Klotho in the differential expression of bone disorders observed in patients with CKD.
Anemia
Epidemiological studies have found that FGF-23 is independently associated with anemia (98, 99), and animal studies have reported possible causal mechanisms. The administration of FGF-23 in wild-type mice rapidly decreases erythropoiesis while Knockout mice (FGF-23 −/−) have increased circulating erythropoietin and red blood cell number (100). Furthermore, Agoro et al. reported that inhibiting FGF-23 signaling rescues anemia and iron deficiency in a mice model of CKD by normalizing the hemoglobin levels, decreasing erythroid cell apoptosis, and increasing serum erythropoietin, iron, and ferritin. They also found an up-regulation on the liver synthesis of hepcidin that normalized after FGF-23 blocking and provided a possible link between FGF-23, inflammation, and anemia (101).
In summary, elevated serum FGF-23 is an early biomarker of impaired renal function that directly contributes to the morbidity observed in patients with CKD. Clinical and animal studies have shown that potential therapeutic benefits of targeting FGF-23 include attenuating left ventricular hypertrophy, decreasing the synthesis of pro-inflammatory cytokines by the liver, improving neutrophil recruitment, rescuing anemia by stimulating erythropoiesis, and improving iron deficiency by down-regulating the synthesis of hepcidin. Decreased circulating levels of α-klotho is also characteristic of patients with CKD and potential therapeutic benefits of targeting α-klotho-dependent actions include increasing circulating levels of calcitriol, decreasing renal fibrosis, and improving bone mineralization and osteoblast function.
Post Kidney Transplant Patients
Kidney transplantation is associated with a significant reduction in mortality compared to those with ESKD on dialysis. Wolfe et al. reported a 1.7-fold decrease in the annual death rate among patients with ESKD undergoing transplant compared to patients on waiting list (102). Notably, CVD persists as the leading cause of death after transplantation (60). According to the USRDS, the prevalence of CVD is 76.5% in patients on hemodialysis, 65% in patients on peritoneal dialysis, and 53.7% in patients with a functional kidney transplant (1). Life expectancy among kidney transplant recipients remains inferior compared to age-matched controls from the general population. For example, females who undergo kidney transplantation between 40 and 44 years old have a projected lifespan of more than 10 years shorter than similarly aged women from the general population (1). For this reason, it is paramount to identify and characterize the mechanisms underlying CVD in kidney transplant recipients.
Transplant mineral and bone disorder (T-MBD) is a systemic condition characterized by endocrine alterations, vascular or other soft-tissue calcification, and abnormalities in bone metabolism (103, 104). Although there are significant differences with CKD-MBD, there is a continuum in some metabolic pathways conducive to atherosclerosis and bone disorders (104). There are several mechanisms involving the aforementioned biomarkers after kidney transplant with persistent interplay affecting mineral bone metabolism, cardiovascular disease, and load-bearing capacity.
After transplant, as renal function stabilizes, there is a tendency toward normalization of most biochemical indicators of ESKD. A recovered GFR allows excretion of excess serum phosphate and uremic toxins. High basal levels of PTH and FGF-23 make post-transplant hypophosphatemia and hypercalcemia a common finding (105–110). Transient hypophosphatemia is observed in more than 80% of transplant recipients early after surgery, and is related to appropriate allograft function and favorable outcomes (106). On the other hand, transient hypercalcemia is reported in up to 65% of transplants and is associated with poor outcomes (111–113). Hypercalcemia can compromise adequate graft function by acutely inducing vasoconstriction and, in the long-term, by favoring calcinosis and vascular calcification (114, 115).
In the first 3 months following surgery, there is a significant decrease in PTH, FGF-23, and calcium levels and an increase in serum phosphate and calcitriol toward normal levels (107). After this period, these changes normalize at a slower rate. Nonetheless, long-term endocrine alterations are common (106, 107, 109, 116). Among 50 patients undergoing kidney transplantation, Evenepoel et al. reported that 1 year after surgery, 57% had persistently high FGF-23, and 32% had high serum PTH levels. Mild to moderate hypophosphatemia was also present in 14% of patients (117). Elevated FGF-23 is associated with decreased post-transplant GFR (118, 119) and, and increased risk of mortality, graft loss (120, 121), and left ventricular wall strain (119, 121). The effects of FGF-23 in the cardiovascular system were also studied by Yilmaz et al. who reported that endothelium-dependent vasodilation improves after kidney transplantation, and it is independently associated with a decrease in FGF-23 and serum phosphorus (122). As mentioned above, high circulating levels of FGF-23 directly contribute to left ventricular hypertrophy (LVH) and anemia in patients with CKD. Decreases in FGF-23 might contribute to the reduction of LVH and improvements of ventricular contractility after kidney transplantation (123, 124). Baloglu et al. reported that hemoglobin levels are inversely associated with circulating FGF-23, however this association was no longer significant after kidney transplantation (125).
Serum α-Klotho levels increase after transplantation and might contribute to the transient hypophosphatemia by improving the sensitivity to FGF-23 (126, 127). Interestingly, Mizusaki et al. reported that serum α-Klotho was significantly higher in kidney transplant recipients receiving everolimus. They concluded that mTOR inhibition could be related to increased α-Klotho after kidney tranplantation (127), which underpins a new area of applicable α-Klotho therapeutics in kidney transplantation that needs further study. Higher serum α-Klotho has also been independently associated with improved GFR 1 year after transplant (128). Animal models have shown a reno-protective effect of α-Klotho that could play a role in protecting the renal allograft (129–131). Sugiura et al. reported that α-Klotho induction through gene transferring strategy mitigates apoptosis, histological damage and improves GFR in mice subject to renal ischemia-reperfusion injury. They concluded that the protective mechanism involved increases expression of heat shock protein 70 (129). The antifibrotic effect of α-Klotho through the inhibition of TGFβ1 and Wnt signaling pathways is also an important mechanism in kidney protection against injury (44, 58). Hu et al. studied mice after acute kidney injury and found that early treatment with α-Klotho prevents progression to CKD and significantly reduces uremic cardiomyopathy (37). As an indicator of renal function, α-Klotho activity varies depending on the quality of the transplanted kidney. Among kidneys from deceased donors, lower Kidney Donor Risk Index (KDRI) and age <50 years were associated with higher α-Klotho levels and better GFR after transplant (128, 132). In this context, α-Klotho represents a potentially useful biomarker and therapeutic agent in kidney transplantation.
Bone metabolism and load-bearing capacity are also greatly affected in kidney transplant recipients. The underlying mechanism is multifactorial and includes pre-existing renal osteodystrophy, reduced kidney function, and the effect of immunosuppressive therapy (133, 134). Among 68,814 kidney transplant recipients, Nikkel et al. reported a 22.5% incidence of fractures during the first 5 years after surgery (135). The first 6 months following transplant are associated with a rapid decrease in spine bone mineral density that continues to decline afterward but at a slower rate (136–138). It is well-established that immunosuppressive therapy with glucocorticoids induces osteopenia and is a contributing factor to osteodystrophy. Glucocorticoids are related to a decreased number and impaired function of osteoblasts. They also promote osteoclastic activity by up-regulating RANKL expression and down-regulating osteoprotegerin (139–141). Conversely, the effect of calcineurin inhibitors in the bone is more controversial and difficult to evaluate (141–143). While tacrolimus and cyclosporine have been linked to osteoporosis, population-based studies have failed to find an association with an increased risk of fractures (141, 143–146). Briner et al. conducted a clinical study that combined cyclosporine treatment with no or very low dose of glucocorticoids. They found that glucocorticoid-sparing immunosuppression after transplant prevented cancellous bone loss, and they did not observe any fractures during the 2 years follow-up (147). Also, Westeel et al. reported a series of 52 renal transplanted patients receiving low dose prednisone and cyclosporine. They concluded that cyclosporine, together with the decrease of prednisone dosage, contributes to a transient stimulation of bone remodeling at 6 months after transplant, which counterbalances the bone loss associated with prednisone therapy (142). In addition to immunosuppressive treatment, post-transplant hypophosphatemia is also independently associated with alterations in bone turnover, decreased osteoblast activity, defective mineralization, and osteoblast apoptosis (133, 148). In this regard, high PTH and FGF-23 levels are frequent after transplant and contribute to hypophosphatemia. Persistent tertiary hyperparathyroidism also leads to increased bone turnover, and high FGF-23 has been related to phosphate-independent skeletal abnormalities (89, 90, 149). Human studies of soluble α-Klotho and FGF-23 among kidney transplant recipients are summarized in Table 1.
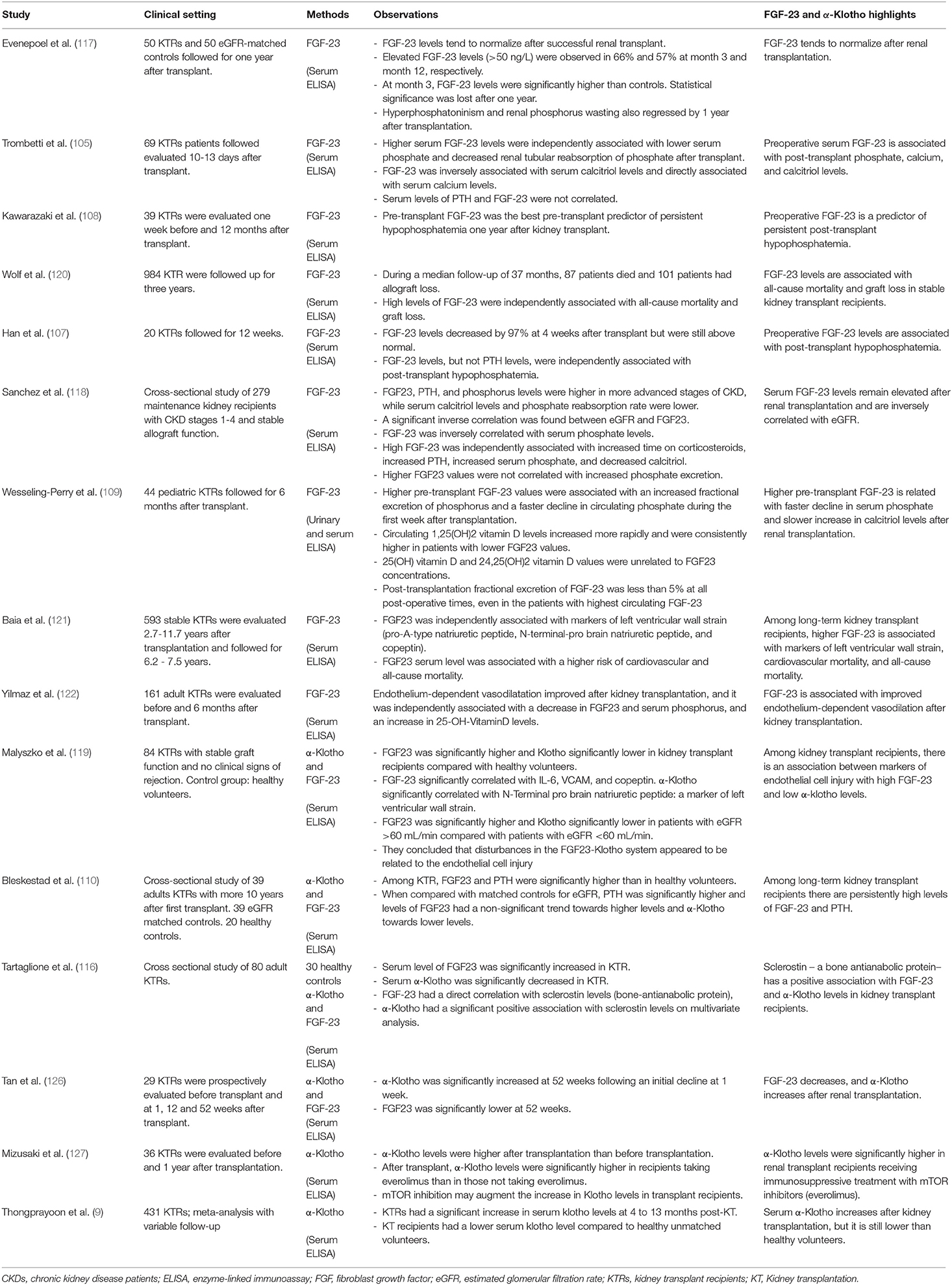
Table 1. Human studies of soluble α-Klotho and FGF-23 among kidney transplant recipients.
Kidney Donors
In 2020, living kidney donors represented 23% of the total transplanted kidney allografts in the US. The long-term mortality after kidney donation has proven to be similar to the general population or matched controls in several studies (150–152). However, the innocuity of kidney donation is the subject of greater debate. O'Keeffe et al. conducted a meta-analysis that included 118,426 living kidney donors with an average follow-up of one to 24 years. They did not find evidence of increased mortality, cardiovascular diseases, or diabetes among donors compared to controls (153). Nonetheless, they found that donors had an increased risk of ESKD and pre-eclampsia. Muzaale et al. conducted a cohort study involving 96,217 kidney donors and found that 15 years after donation, the risk of ESKD increased from 0.04% in matched non-donors to 0.31% in kidney donors (154). There are scarce data available evaluating the outcomes after donation beyond a follow-up of 20 years. Based on OPTN data as of 2020, 26.5% of kidney donors were under 35 years old. In this regard, an increasing population is expected to live longer than 40 years after donation (155), and the long-term metabolic consequences of unilateral nephrectomy in otherwise healthy individuals are not clearly understood.
As discussed above, α-Klotho is primarily synthesized in the kidneys, and it has a reno-protective effect through different mechanisms. Shortly after kidney donation, α-Klotho levels decrease and remain lower than baseline 1 year after surgery (5–8, 156). However, one study that evaluated serum α-Klotho 5 years after donation did not find a significant difference compared to healthy controls (157). The long-term consequences of changes in α-Klotho levels in response to nephrectomy remain unknown. Further, the scope of the possible therapeutic benefit of increasing α-Klotho levels to prevent the development of ESKD after nephrectomy has not been explored in humans.
Other parameters of the bone-kidney axis are also altered after donation. There is a decrease in GFR, phosphate, and calcitriol synthesis, and an increase in PTH that persists 1 year after surgery (5). However, there is conflicting information about the changes in FGF-23 after donation. Some authors have reported that FGF-23 remains elevated 1 year after donation (6, 158), while others did not find significant long-term changes (5, 157, 159). A meta-analysis conducted by Thongprayoon et al. reported no significant change in FGF-23 1 year after kidney donation. Notably, they also found that serum α-Klotho remained lower than baseline 1 year after nephrectomy among 56 donors (9). As such, there is limited available data considering the clinical relevance of alterations in FGF-23 and α-Klotho after kidney donation (159). Human studies of soluble α-Klotho and FGF-23 among kidney donors are summarized in Table 2.
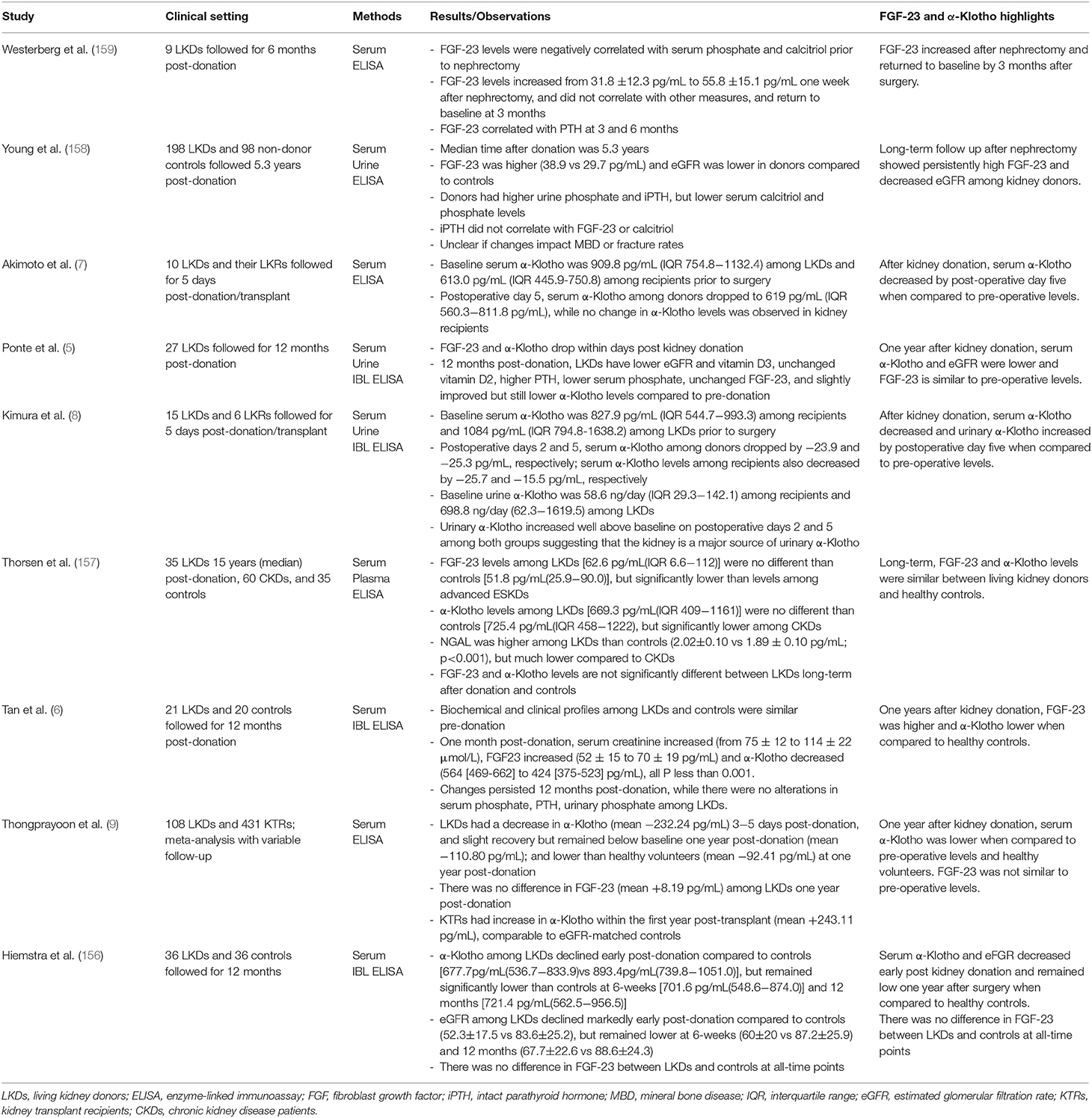
Table 2. Human studies of soluble α-Klotho and FGF-23 among living donors.
The understanding of long-term adaptations to kidney donation is still the subject of intense investigation. Living kidney donors are essential to close the gap between supply and kidney allograft demand, and sustaining their long-term health is important. Further research and education are necessary to support the kidney health of kidney donors and ensure the maintenance of their endocrine, cardiovascular, and bone mineral health.
Conclusion
FGF-23 and α-Klotho are integral endocrine factors that link bone-mineral metabolism, kidney function, and cardiovascular health. Among patients with CKD, this multi-looped axis is significantly impaired and translates into greater cardiovascular morbidity, mortality, and bone disorders. Kidney transplantation partially restores α-Klotho activity and FGF-23 levels. However, persistently high FGF-23 is associated with increased mortality and graft loss, and low circulating α-Klotho is associated with decreased kidney function after transplant. Notably, the metabolic bone and vascular alterations observed post kidney transplant are not fixed and instead are dynamic, influenced by organ quality, kidney recovery, immunosuppression, recipient comorbidities, and associated treatments. This highlights the concept that patients continue to need personalized medical support after transplant to ensure normalization of endocrine and bone-vascular axes indirectly affected by CKD. On the other hand, while some authors have reported that kidney donors have lower α-Klotho levels and elevated FGF-23 that persist 1 year after surgery, the scarce available data are inconclusive regarding the clinical relevance of these findings.
Author Contributions
MG, GO, and JN contributed to conception and design of the study. MG and GO wrote the first draft of the manuscript. JN wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Figure adapted from anatomical images provided by Essential Anatomy. Many thanks to 3D4Medical.
References
1. System USRD. 2020 Usrds Annual Data Report: Epidemiology of Kidney Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (2020).
2. Kuro OM. The klotho proteins in health and disease. Nat Rev Nephrol. (2019) 15:27–44. doi: 10.1038/s41581-018-0078-3
3. D'Marco L, Bellasi A, Mazzaferro S, Raggi P. Vascular calcification, bone and mineral metabolism after kidney transplantation. World J Transplant. (2015) 5:222–30. doi: 10.5500/wjt.v5.i4.222
4. Westenfeld R, Schlieper G, Woltje M, Gawlik A, Brandenburg V, Rutkowski P, et al. Impact of sirolimus, tacrolimus and mycophenolate mofetil on osteoclastogenesis–implications for post-transplantation bone disease. Nephrol Dial Transplant. (2011) 26:4115–23. doi: 10.1093/ndt/gfr214
5. Ponte B, Trombetti A, Hadaya K, Ernandez T, Fumeaux D, Iselin C, et al. Acute and long term mineral metabolism adaptation in living kidney donors: a prospective study. Bone. (2014) 62:36–42. doi: 10.1016/j.bone.2014.01.020
6. Tan SJ, Hewitson TD, Hughes PD, Holt SG, Toussaint ND. Changes in markers of mineral metabolism after living kidney donation. Transplant Direct. (2017) 3:e150. doi: 10.1097/TXD.0000000000000660
7. Akimoto T, Kimura T, Watanabe Y, Ishikawa N, Iwazu Y, Saito O, et al. The impact of nephrectomy and renal transplantation on serum levels of soluble klotho protein. Transplant Proc. (2013) 45:134–6. doi: 10.1016/j.transproceed.2012.07.150
8. Kimura T, Akimoto T, Watanabe Y, Kurosawa A, Nanmoku K, Muto S, et al. Impact of renal transplantation and nephrectomy on urinary soluble klotho protein. Transplant Proc. (2015) 47:1697–9. doi: 10.1016/j.transproceed.2015.06.025
9. Thongprayoon C, Neyra JA, Hansrivijit P, Medaura J, Leeaphorn N, Davis PW, et al. Serum klotho in living kidney donors and kidney transplant recipients: a meta-analysis. J Clin Med. (2020) 9:61834. doi: 10.3390/jcm9061834
10. Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. (2007) 27:3417–28. doi: 10.1128/MCB.02249-06
11. McKnight Q, Jenkins S, Li X, Nelson T, Marlier A, Cantley LG, et al. Il-1beta drives production of fgf-23 at the onset of chronic kidney disease in mice. J Bone Miner Res. (2020) 35:1352–62. doi: 10.1002/jbmr.4003
12. Han X, Li L, Yang J, King G, Xiao Z, Quarles LD. Counter-regulatory paracrine actions of Fgf-23 and 1,25(Oh)2 D in macrophages. FEBS Lett. (2016) 590:53–67. doi: 10.1002/1873-3468.12040
13. Lawrence T. The nuclear factor Nf-kappab pathway in inflammation. Cold Spring Harb Perspect Biol. (2009) 1:a001651. doi: 10.1101/cshperspect.a001651
14. Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. (2006) 281:6120–3. doi: 10.1074/jbc.C500457200
15. Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical Fgf receptor into a specific receptor for Fgf23. Nature. (2006) 444:770–4. doi: 10.1038/nature05315
16. Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, et al. Fgf23 decreases renal Napi-2a and Napi-2c expression and induces hypophosphatemia in vivo predominantly via Fgf receptor 1. Am J Physiol Renal Physiol. (2009) 297:F282–91. doi: 10.1152/ajprenal.90742.2008
19. Bai X, Miao D, Xiao S, Qiu D, St-Arnaud R, Petkovich M, et al. Cyp24 inhibition as a therapeutic target in Fgf23-mediated renal phosphate wasting disorders. J Clin Invest. (2016) 126:667–80. doi: 10.1172/JCI81928
20. Quarles LD. Skeletal secretion of Fgf-23 regulates phosphate and vitamin D metabolism. Nat Rev Endocrinol. (2012) 8:276–86. doi: 10.1038/nrendo.2011.218
21. Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R., Kuro-o M, Mohammadi M, Sirkis R, et al. The parathyroid is a target organ for Fgf23 in rats. J Clin Invest. (2007) 117:4003–8. doi: 10.1172/JCI32409
22. Quinn SJ, Thomsen AR, Pang JL, Kantham L, Brauner-Osborne H, Pollak M, et al. Interactions between calcium and phosphorus in the regulation of the production of fibroblast growth factor 23 in vivo. Am J Physiol Endocrinol Metab. (2013) 304:E310–20. doi: 10.1152/ajpendo.00460.2012
23. Marsell R, Krajisnik T, Goransson H, Ohlsson C, Ljunggren O, Larsson TE, et al. Gene expression analysis of kidneys from transgenic mice expressing fibroblast growth factor-23. Nephrol Dial Transplant. (2008) 23:827–33. doi: 10.1093/ndt/gfm672
24. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse Klotho gene leads to a syndrome resembling ageing. Nature. (1997) 390:45–51. doi: 10.1038/36285
25. Hu MC, Shi M, Zhang J, Addo T, Cho HJ, Barker SL, et al. Renal production, uptake, and handling of circulating alphaklotho. J Am Soc Nephrol. (2016) 27:79–90. doi: 10.1681/ASN.2014101030
26. Bloch L, Sineshchekova O, Reichenbach D, Reiss K, Saftig P, Kuro-o M, et al. Klotho is a substrate for alpha-, beta- and gamma-secretase. FEBS Lett. (2009) 583:3221–4. doi: 10.1016/j.febslet.2009.09.009
27. Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by Adam10 and Adam17. Proc Natl Acad Sci USA. (2007) 104:19796–801. doi: 10.1073/pnas.0709805104
28. Chen CD, Tung TY, Liang J, Zeldich E, Tucker Zhou TB, Turk BE, et al. Identification of cleavage sites leading to the shed form of the anti-aging protein Klotho. Biochemistry. (2014) 53:5579–87. doi: 10.1021/bi500409n
29. Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y. Identification of the human Klotho gene and its two transcripts encoding membrane and secreted Klotho protein. Biochem Biophys Res Commun. (1998) 242:626–30. doi: 10.1006/bbrc.1997.8019
30. Shiraki-Iida T, Aizawa H, Matsumura Y, Sekine S, Iida A, Anazawa H, et al. Structure of the mouse Klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Lett. (1998) 424:6–10. doi: 10.1016/S0014-5793(98)00127-6
31. Panesso MC, Shi M, Cho HJ, Paek J, Ye J, Moe OW, et al. Klotho has dual protective effects on cisplatin-induced acute kidney injury. Kidney Int. (2014) 85:855–70. doi: 10.1038/ki.2013.489
32. Cheng MF, Chen LJ, Niu HS, Yang TT, Lin KC, Cheng JT. Signals mediating Klotho-induced neuroprotection in hippocampal neuronal cells. Acta Neurobiol Exp. (2015) 75:60–71.
33. Sun S, Cheng B, Sun PG, Wu XH, Wu QQ, He P. Rtef-1 protects against oxidative damage induced by H2o2 in human umbilical vein endothelial cells through Klotho activation. Exp Biol Med. (2015) 240:1606–13. doi: 10.1177/1535370215587914
34. Ravikumar P, Ye J, Zhang J, Pinch SN, Hu MC, Kuro-o M, et al. Alpha-Klotho protects against oxidative damage in pulmonary epithelia. Am J Physiol Lung Cell Mol Physiol. (2014) 307:L566–75. doi: 10.1152/ajplung.00306.2013
35. Wang Y, Chen L, Huang G, He D, He J, Xu W, et al. Klotho sensitizes human lung cancer cell line to cisplatin via Pi3k/Akt pathway. PLoS ONE. (2013) 8:e57391. doi: 10.1371/journal.pone.0057391
36. Smith RC, O'Bryan LM, Farrow EG, Summers LJ, Clinkenbeard EL, Roberts JL, et al. Circulating alphaklotho influences phosphate handling by controlling Fgf23 production. J Clin Invest. (2012) 122:4710–5. doi: 10.1172/JCI64986
37. Hu MC, Shi M, Gillings N, Flores B, Takahashi M, Kuro OM, et al. Recombinant alpha-Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney Int. (2017) 91:1104–14. doi: 10.1016/j.kint.2016.10.034
38. Chen G, Liu Y, Goetz R, Fu L, Jayaraman S, Hu MC, et al. Alpha-Klotho is a non-enzymatic molecular scaffold for Fgf23 hormone signalling. Nature. (2018) 553:461–6. doi: 10.1038/nature25451
39. Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. (2010) 24:3438–50. doi: 10.1096/fj.10-154765
40. Ohnishi M, Nakatani T, Lanske B, Razzaque MS. Reversal of mineral ion homeostasis and soft-tissue calcification of Klotho knockout mice by deletion of vitamin D 1alpha-hydroxylase. Kidney Int. (2009) 75:1166–72. doi: 10.1038/ki.2009.24
41. Razzaque MS. Fgf23, Klotho and Vitamin D Interactions: what have we learned from in vivo mouse genetics studies? Adv Exp Med Biol. (2012) 728:84–91. doi: 10.1007/978-1-4614-0887-1_5
42. Neyra JA, Hu MC. Alphaklotho and chronic kidney disease. Vitam Horm. (2016) 101:257–310. doi: 10.1016/bs.vh.2016.02.007
43. Sugiura H, Yoshida T, Shiohira S, Kohei J, Mitobe M, Kurosu H, et al. Reduced Klotho expression level in kidney aggravates renal interstitial fibrosis. Am J Physiol Renal Physiol. (2012) 302:F1252–64. doi: 10.1152/ajprenal.00294.2011
44. Doi S, Zou Y, Togao O, Pastor JV, John GB, Wang L, et al. Klotho inhibits transforming growth factor-beta1 (Tgf-Beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem. (2011) 286:8655–65. doi: 10.1074/jbc.M110.174037
45. Bian A, Neyra JA, Zhan M, Hu MC. Klotho, stem cells, and aging. Clin Interv Aging. (2015) 10:1233–43. doi: 10.2147/CIA.S84978
46. Shi M, Flores B, Gillings N, Bian A, Cho HJ, Yan S, et al. Alphaklotho mitigates progression of Aki to Ckd through activation of autophagy. J Am Soc Nephrol. (2016) 27:2331–45. doi: 10.1681/ASN.2015060613
47. Isakova T, Wahl P, Vargas GS, Gutierrez OM, Scialla J, Xie H, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. (2011) 79:1370–8. doi: 10.1038/ki.2011.47
48. Zeng S, Querfeld U, Feger M, Haffner D, Hasan AA, Chu C, et al. Relationship between Gfr, Intact Pth, Oxidized Pth, non-oxidized Pth as well as Fgf23 in patients with Ckd. FASEB J. (2020) 34:15269–81. doi: 10.1096/fj.202000596R
49. Mace ML, Gravesen E, Hofman-Bang J, Olgaard K, Lewin E. Key role of the kidney in the regulation of fibroblast growth factor 23. Kidney Int. (2015) 88:1304–13. doi: 10.1038/ki.2015.231
50. Mace ML, Gravesen E, Nordholm A, Hofman-Bang J, Secher T, Olgaard K, et al. Kidney fibroblast growth factor 23 does not contribute to elevation of its circulating levels in uremia. Kidney Int. (2017) 92:165–78. doi: 10.1016/j.kint.2017.01.015
51. Imanishi Y, Inaba M, Nakatsuka K, Nagasue K, Okuno S, Yoshihara A, et al. Fgf-23 in patients with end-stage renal disease on hemodialysis. Kidney Int. (2004) 65:1943–6. doi: 10.1111/j.1523-1755.2004.00604.x
52. Gutierrez O, Isakova T, Rhee E, Shah A, Holmes J, Collerone G, et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol. (2005) 16:2205–15. doi: 10.1681/ASN.2005010052
53. Olauson H, Vervloet MG, Cozzolino M, Massy ZA, Urena Torres P, Larsson TE. New insights into the Fgf23-Klotho axis. Semin Nephrol. (2014) 34:586–97. doi: 10.1016/j.semnephrol.2014.09.005
54. Lim K, Lu TS, Molostvov G, Lee C, Lam FT, Zehnder D, et al. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation. (2012) 125:2243–55. doi: 10.1161/CIRCULATIONAHA.111.053405
55. Christov M, Neyra JA, Gupta S, Leaf DE. Fibroblast growth factor 23 and Klotho in Aki. Semin Nephrol. (2019) 39:57–75. doi: 10.1016/j.semnephrol.2018.10.005
56. Neyra JA, Hu MC, Moe OW. Klotho in clinical nephrology: diagnostic and therapeutic implications. Clin J Am Soc Nephrol. (2020) 16:162–76. doi: 10.2215/CJN.02840320
57. Haut LL, Alfrey AC, Guggenheim S, Buddington B, Schrier N. Renal toxicity of phosphate in rats. Kidney Int. (1980) 17:722–31. doi: 10.1038/ki.1980.85
58. Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. (2007) 317:803–6. doi: 10.1126/science.1143578
59. Kim JH, Xie J, Hwang KH, Wu YL, Oliver N, Eom M, et al. Klotho may ameliorate proteinuria by targeting Trpc6 channels in podocytes. J Am Soc Nephrol. (2017) 28:140–51. doi: 10.1681/ASN.2015080888
60. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. (2019) 139:e56–e528. doi: 10.1161/CIR.0000000000000659
61. Gutierrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. (2008) 359:584–92. doi: 10.1056/NEJMoa0706130
62. Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. J Am Med Assoc. (2011) 305:2432–9. doi: 10.1001/jama.2011.826
63. Parker BD, Schurgers LJ, Brandenburg VM, Christenson RH, Vermeer C, Ketteler M, et al. The associations of fibroblast growth factor 23 and uncarboxylated matrix gla protein with mortality in coronary artery disease: the heart and soul study. Ann Intern Med. (2010) 152:640–8. doi: 10.7326/0003-4819-152-10-201005180-00004
64. Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, Yanucil C, et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. (2015) 22:1020–32. doi: 10.1016/j.cmet.2015.09.002
65. Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, et al. Fgf23 induces left ventricular hypertrophy. J Clin Invest. (2011) 121:4393–408. doi: 10.1172/JCI46122
66. Gutierrez OM, Januzzi JL, Isakova T, Laliberte K, Smith K, Collerone G, et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation. (2009) 119:2545–52. doi: 10.1161/CIRCULATIONAHA.108.844506
67. Seifert ME, Hruska KA. The kidney-vascular-bone axis in the chronic kidney disease-mineral bone disorder. Transplantation. (2016) 100:497–505. doi: 10.1097/TP.0000000000000903
68. Zhou C, Shi Z, Ouyang N, Ruan X. Hyperphosphatemia and cardiovascular disease. Front Cell Dev Biol. (2021) 9:644363. doi: 10.3389/fcell.2021.644363
69. Turan MN, Kircelli F, Yaprak M, Sisman AR, Gungor O, Bayraktaroglu S, et al. Fgf-23 levels are associated with vascular calcification, but not with atherosclerosis, in hemodialysis patients. Int Urol Nephrol. (2016) 48:609–17. doi: 10.1007/s11255-016-1231-1
70. Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. (2005) 25:932–43. doi: 10.1161/01.ATV.0000160548.78317.29
71. Jirak P, Stechemesser L, More E, Franzen M, Topf A, Mirna M, et al. Clinical implications of fetuin-A. Adv Clin Chem. (2019) 89:79–130. doi: 10.1016/bs.acc.2018.12.003
72. Smith ER, Ford ML, Tomlinson LA, Rajkumar C, McMahon LP, Holt SG. Phosphorylated fetuin-a-containing calciprotein particles are associated with aortic stiffness and a procalcific milieu in patients with pre-dialysis Ckd. Nephrol Dial Transplant. (2012) 27:1957–66. doi: 10.1093/ndt/gfr609
73. Valdivielso JM, Rodriguez-Puyol D, Pascual J, Barrios C, Bermudez-Lopez M, Sanchez-Nino MD, et al. Atherosclerosis in chronic kidney disease: more, less, or just different? Arterioscler Thromb Vasc Biol. (2019) 39:1938–66. doi: 10.1161/ATVBAHA.119.312705
74. Di Marco GS, Hausberg M, Hillebrand U, Rustemeyer P, Wittkowski W, Lang D, et al. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am J Physiol Renal Physiol. (2008) 294:F1381–7. doi: 10.1152/ajprenal.00003.2008
75. Guerin AP, London GM, Marchais SJ, Metivier F. Arterial stiffening and vascular calcifications in end-stage renal disease. Nephrol Dial Transplant. (2000) 15:1014–21. doi: 10.1093/ndt/15.7.1014
76. Singh S, Grabner A, Yanucil C, Schramm K, Czaya B, Krick S, et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. (2016) 90:985–96. doi: 10.1016/j.kint.2016.05.019
77. Kimmel PL, Phillips TM, Simmens SJ, Peterson RA, Weihs KL, Alleyne S, et al. Immunologic function and survival in hemodialysis patients. Kidney Int. (1998) 54:236–44. doi: 10.1046/j.1523-1755.1998.00981.x
78. Pereira BJ, Shapiro L, King AJ, Falagas ME, Strom JA, Dinarello CA. Plasma levels of Il-1 beta, Tnf alpha and their specific inhibitors in undialyzed chronic renal failure, capd and hemodialysis patients. Kidney Int. (1994) 45:890–6. doi: 10.1038/ki.1994.117
79. Gupta J, Mitra N, Kanetsky PA, Devaney J, Wing MR, Reilly M, et al. Association between albuminuria, kidney function, and inflammatory biomarker profile in Ckd in Cric. Clin J Am Soc Nephrol. (2012) 7:1938–46. doi: 10.2215/CJN.03500412
80. Mihai S, Codrici E, Popescu ID, Enciu AM, Albulescu L, Necula LG, et al. Inflammation-related mechanisms in chronic kidney disease prediction, progression, and outcome. J Immunol Res. (2018) 2018:2180373. doi: 10.1155/2018/2180373
81. Rossaint J, Oehmichen J, Van Aken H, Reuter S, Pavenstadt HJ, Meersch M, et al. Fgf23 signaling impairs neutrophil recruitment and host defense during Ckd. J Clin Invest. (2016) 126:962–74. doi: 10.1172/JCI83470
82. Malluche HH, Porter DS, Pienkowski D. Evaluating bone quality in patients with chronic kidney disease. Nat Rev Nephrol. (2013) 9:671–80. doi: 10.1038/nrneph.2013.198
83. Moe S, Drueke T, Cunningham J, Goodman W, Martin K, Olgaard K, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from kidney disease: improving global outcomes (Kdigo). Kidney Int. (2006) 69:1945–53. doi: 10.1038/sj.ki.5000414
84. Iseri K, Carrero JJ, Evans M, Fellander-Tsai L, Berg HE, Runesson B, et al. Incidence of fractures before and after dialysis initiation. J Bone Miner Res. (2020) 35:2372–80. doi: 10.1002/jbmr.4141
85. Ensrud KE, Lui LY, Taylor BC, Ishani A, Shlipak MG, Stone KL, et al. Renal function and risk of hip and vertebral fractures in older women. Arch Intern Med. (2007) 167:133–9. doi: 10.1001/archinte.167.2.133
86. Reiss AB, Miyawaki N, Moon J, Kasselman LJ, Voloshyna I, D'Avino R Jr, et al. Ckd, Arterial calcification, atherosclerosis and bone health: inter-relationships and controversies. Atherosclerosis. (2018) 278:49–59. doi: 10.1016/j.atherosclerosis.2018.08.046
87. Malluche HH, Mawad HW, Monier-Faugere MC. Renal osteodystrophy in the first decade of the new millennium: analysis of 630 bone biopsies in black and white patients. J Bone Miner Res. (2011) 26:1368–76. doi: 10.1002/jbmr.309
88. Kurz P, Monier-Faugere MC, Bognar B, Werner E, Roth P, Vlachojannis J, et al. Evidence for abnormal calcium homeostasis in patients with adynamic bone disease. Kidney Int. (1994) 46:855–61. doi: 10.1038/ki.1994.342
89. Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, et al. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. (2004) 23:421–32. doi: 10.1016/j.matbio.2004.09.007
90. Sitara D, Kim S, Razzaque MS, Bergwitz C, Taguchi T, Schuler C, et al. Genetic evidence of serum phosphate-independent functions of Fgf-23 on bone. PLoS Genet. (2008) 4:e1000154. doi: 10.1371/journal.pgen.1000154
91. Wang H, Yoshiko Y, Yamamoto R, Minamizaki T, Kozai K, Tanne K, et al. Overexpression of fibroblast growth factor 23 suppresses osteoblast differentiation and matrix mineralization in vitro. J Bone Miner Res. (2008) 23:939–48. doi: 10.1359/jbmr.080220
92. Moyses RMA, Dusso A. Is osteocyte Klotho bad for bone health? Kidney Int. (2017) 92:540–3. doi: 10.1016/j.kint.2017.04.012
93. Vimalraj S. Alkaline phosphatase: structure, expression and its function in bone mineralization. Gene. (2020) 754:144855. doi: 10.1016/j.gene.2020.144855
94. Murali SK, Andrukhova O, Clinkenbeard EL, White KE, Erben RG. Excessive osteocytic Fgf23 secretion contributes to pyrophosphate accumulation and mineralization defect in Hyp mice. PLoS Biol. (2016) 14:e1002427. doi: 10.1371/journal.pbio.1002427
95. Yuan Q, Jiang Y, Zhao X, Sato T, Densmore M, Schuler C, et al. Increased osteopontin contributes to inhibition of bone mineralization in Fgf23-deficient mice. J Bone Miner Res. (2014) 29:693–704. doi: 10.1002/jbmr.2079
96. Rhee Y, Bivi N, Farrow E, Lezcano V, Plotkin LI, White KE, et al. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone. (2011) 49:636–43. doi: 10.1016/j.bone.2011.06.025
97. Komaba H, Kaludjerovic J, Hu DZ, Nagano K, Amano K, Ide N, et al. Klotho expression in osteocytes regulates bone metabolism and controls bone formation. Kidney Int. (2017) 92:599–611. doi: 10.1016/j.kint.2017.02.014
98. Mehta R, Cai X, Hodakowski A, Lee J, Leonard M, Ricardo A, et al. Fibroblast growth factor 23 and anemia in the chronic renal insufficiency cohort study. Clin J Am Soc Nephrol. (2017) 12:1795–803. doi: 10.2215/CJN.03950417
99. Nam KH, Kim H, An SY, Lee M, Cha MU, Park JT, et al. Circulating fibroblast growth factor-23 levels are associated with an increased risk of anemia development in patients with nondialysis chronic kidney disease. Sci Rep. (2018) 8:7294. doi: 10.1038/s41598-018-25439-z
100. Coe LM, Madathil SV, Casu C, Lanske B, Rivella S, Sitara D. Fgf-23 is a negative regulator of prenatal and postnatal erythropoiesis. J Biol Chem. (2014) 289:9795–810. doi: 10.1074/jbc.M113.527150
101. Agoro R, Montagna A, Goetz R, Aligbe O, Singh G, Coe LM, et al. Inhibition of fibroblast growth factor 23 (Fgf23) signaling rescues renal anemia. FASEB J. (2018) 32:3752–64. doi: 10.1096/fj.201700667R
102. Wolfe RA, Ashby VB, Milford EL, Ojo AO, Ettenger RE, Agodoa LY, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med. (1999) 341:1725–30. doi: 10.1056/NEJM199912023412303
103. KDIGO Guidelines. Chapter 1: introduction and definition of Ckd–Mbd and the development of the guideline statements. Kidney Int. (2009) 76:S3–8. doi: 10.1038/ki.2009.189
104. Chapter 5: evaluation and treatment of kidney transplant bone disease. Kidney Int. (2009) 76113:S100–10.
105. Trombetti A, Richert L, Hadaya K, Graf JD, Herrmann FR, Ferrari SL, et al. Early post-transplantation hypophosphatemia is associated with elevated Fgf-23 levels. Eur J Endocrinol. (2011) 164:839–47. doi: 10.1530/EJE-10-1150
106. van Londen M, Aarts BM, Deetman PE, van der Weijden J, Eisenga MF, Navis G, et al. Post-transplant hypophosphatemia and the risk of death-censored graft failure and mortality after kidney transplantation. Clin J Am Soc Nephrol. (2017) 12:1301–10. doi: 10.2215/CJN.10270916
107. Han SY, Hwang EA, Park SB, Kim HC, Kim HT. Elevated fibroblast growth factor 23 levels as a cause of early post-renal transplantation hypophosphatemia. Transplant Proc. (2012) 44:657–60. doi: 10.1016/j.transproceed.2011.11.046
108. Kawarazaki H, Shibagaki Y, Fukumoto S, Kido R, Nakajima I, Fuchinoue S, et al. The relative role of fibroblast growth factor 23 and parathyroid hormone in predicting future hypophosphatemia and hypercalcemia after living donor kidney transplantation: a 1-year prospective observational study. Nephrol Dial Transplant. (2011) 26:2691–5. doi: 10.1093/ndt/gfq777
109. Wesseling-Perry K, Pereira RC, Tsai E, Ettenger R, Juppner H, Salusky IB. Fgf23 and mineral metabolism in the early post-renal transplantation period. Pediatr Nephrol. (2013) 28:2207–15. doi: 10.1007/s00467-013-2547-z
110. Bleskestad IH, Thorsen IS, Jonsson G, Skadberg O, Bergrem H, Goransson LG. Soluble Klotho and intact fibroblast growth factor 23 in long-term kidney transplant patients. Eur J Endocrinol. (2015) 172:343–50. doi: 10.1530/EJE-14-0457
111. Vezzoli G, Elli A, Palazzi P, Bertoni T, Scabini M, Quarto di Palo F, et al. High plasma ionized calcium with normal Pth and total calcium levels in normal-function kidney transplant recipients. Nephron. (1986) 42:290–4. doi: 10.1159/000183690
112. Stavroulopoulos A, Cassidy MJ, Porter CJ, Hosking DJ, Roe SD. Vitamin D status in renal transplant recipients. Am J Transplant. (2007) 7:2546–52. doi: 10.1111/j.1600-6143.2007.01978.x
113. Boom H, Mallat MJ, de Fijter JW, Paul LC, Bruijn JA, van Es LA. Calcium levels as a risk factor for delayed graft function. Transplantation. (2004) 77:868–73. doi: 10.1097/01.TP.0000116417.03114.87
114. Messa P, Cafforio C, Alfieri C. Calcium and phosphate changes after renal transplantation. J Nephrol. (2010) 23(Suppl.16):S175–81.
115. Torregrosa JV, Barros X. Management of hypercalcemia after renal transplantation. Nefrologia. (2013) 33:751–7. doi: 10.3265/Nefrologia.pre2013.Aug.11888
116. Tartaglione L, Pasquali M, Rotondi S, Muci ML, Leonangeli C, Farcomeni A, et al. Interactions of sclerostin with Fgf23, soluble Klotho and vitamin D in renal transplantation. PLoS ONE. (2017) 12:e0178637. doi: 10.1371/journal.pone.0178637
117. Evenepoel P, Meijers BK, de Jonge H, Naesens M, Bammens B, Claes K, et al. Recovery of hyperphosphatoninism and renal phosphorus wasting one year after successful renal transplantation. Clin J Am Soc Nephrol. (2008) 3:1829–36. doi: 10.2215/CJN.01310308
118. Sanchez Fructuoso AI, Maestro ML, Perez-Flores I, Valero R, Rafael S, Veganzones S, et al. Serum level of fibroblast growth factor 23 in maintenance renal transplant patients. Nephrol Dial Transplant. (2012) 27:4227–35. doi: 10.1093/ndt/gfs409
119. Malyszko J, Koc-Zorawska E, Matuszkiewicz-Rowinska J, Malyszko J. Fgf23 and Klotho in relation to markers of endothelial dysfunction in kidney transplant recipients. Transplant Proc. (2014) 46:2647–50. doi: 10.1016/j.transproceed.2014.09.015
120. Wolf M, Molnar MZ, Amaral AP, Czira ME, Rudas A, Ujszaszi A, et al. Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J Am Soc Nephrol. (2011) 22:956–66. doi: 10.1681/ASN.2010080894
121. Baia LC, Humalda JK, Vervloet MG, Navis G, Bakker SJ, de Borst MH, et al. Fibroblast growth factor 23 and cardiovascular mortality after kidney transplantation. Clin J Am Soc Nephrol. (2013) 8:1968–78. doi: 10.2215/CJN.01880213
122. Yilmaz MI, Sonmez A, Saglam M, Yaman H, Kilic S, Turker T, et al. Longitudinal analysis of vascular function and biomarkers of metabolic bone disorders before and after renal transplantation. Am J Nephrol. (2013) 37:126–34. doi: 10.1159/000346711
123. Parfrey PS, Harnett JD, Foley RN, Kent GM, Murray DC, Barre PE, et al. Impact of renal transplantation on uremic cardiomyopathy. Transplantation. (1995) 60:908–14. doi: 10.1097/00007890-199511000-00005
124. Zolty R, Hynes PJ, Vittorio TJ. Severe left ventricular systolic dysfunction may reverse with renal transplantation: uremic cardiomyopathy and cardiorenal syndrome. Am J Transplant. (2008) 8:2219–24. doi: 10.1111/j.1600-6143.2008.02407.x
125. Baloglu I, Ozer H, Ozturk Y, Erdur MF, Tonbul HZ, Turkmen K. The relationship between Fgf23 and anemia in Hd and renal transplant patients. Int Urol Nephrol. (2021) 21:9. doi: 10.1007/s11255-021-02982-9
126. Tan SJ, Crosthwaite A, Langsford D, Obeysekere V, Ierino FL, Roberts MA, et al. Mineral adaptations following kidney transplantation. Transpl Int. (2017) 30:463–73. doi: 10.1111/tri.12925
127. Mizusaki K, Hasuike Y, Kimura T, Nagasawa Y, Kuragano T, Yamada Y, et al. Inhibition of the mammalian target of rapamycin may augment the increase in soluble Klotho levels in renal transplantation recipients. Blood Purif. (2019) 47(Suppl.2):12–8. doi: 10.1159/000496630
128. Kim SM, Kim SJ, Ahn S, Min S-I, Min S-K, Ha J. The potential role of klotho as a prognostic biomarker in deceased donor kidney transplantation. Transplantation. (2018) 102(Suppl.7S-1):S539–S. doi: 10.1097/01.tp.0000543389.60945.78
129. Sugiura H, Yoshida T, Mitobe M, Yoshida S, Shiohira S, Nitta K, et al. Klotho reduces apoptosis in experimental ischaemic acute kidney injury via Hsp-70. Nephrol Dial Transplant. (2010) 25:60–8. doi: 10.1093/ndt/gfp451
130. Sugiura H, Yoshida T, Tsuchiya K, Mitobe M, Nishimura S, Shirota S, et al. Klotho reduces apoptosis in experimental ischaemic acute renal failure. Nephrol Dial Transplant. (2005) 20:2636–45. doi: 10.1093/ndt/gfi165
131. Hu MC, Shi M, Zhang J, Quinones H, Kuro-o M, Moe OW. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int. (2010) 78:1240–51. doi: 10.1038/ki.2010.328
132. Deng G, Yang A, Wu J, Zhou J, Meng S, Zhu C, et al. The value of older donors' Klotho level in predicting recipients' short-term renal function. Med Sci Monit. (2018) 24:7936–43. doi: 10.12659/MSM.913274
133. Rojas E, Carlini RG, Clesca P, Arminio A, Suniaga O, De Elguezabal K, et al. The pathogenesis of osteodystrophy after renal transplantation as detected by early alterations in bone remodeling. Kidney Int. (2003) 63:1915–23. doi: 10.1046/j.1523-1755.2003.00938.x
134. Bellorin-Font E, Rojas E, Carlini RG, Suniaga O, Weisinger JR. Bone remodeling after renal transplantation. Kidney Int Suppl. (2003) 85:S125–8. doi: 10.1046/j.1523-1755.63.s85.30.x
135. Nikkel LE, Hollenbeak CS, Fox EJ, Uemura T, Ghahramani N. Risk of fractures after renal transplantation in the United States. Transplantation. (2009) 87:1846–51. doi: 10.1097/TP.0b013e3181a6bbda
136. Mikuls TR, Julian BA, Bartolucci A, Saag KG. Bone mineral density changes within six months of renal transplantation. Transplantation. (2003) 75:49–54. doi: 10.1097/00007890-200301150-00009
137. Julian BA, Laskow DA, Dubovsky J, Dubovsky EV, Curtis JJ, Quarles LD. Rapid loss of vertebral mineral density after renal transplantation. N Engl J Med. (1991) 325:544–50. doi: 10.1056/NEJM199108223250804
138. Brandenburg VM, Politt D, Ketteler M, Fassbender WJ, Heussen N, Westenfeld R, et al. Early rapid loss followed by long-term consolidation characterizes the development of lumbar bone mineral density after kidney transplantation. Transplantation. (2004) 77:1566–71. doi: 10.1097/01.TP.0000131990.13277.28
139. Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. (1998) 102:274–82. doi: 10.1172/JCI2799
140. Canalis E, Delany AM. Mechanisms of glucocorticoid action in bone. Ann N Y Acad Sci. (2002) 966:73–81. doi: 10.1111/j.1749-6632.2002.tb04204.x
141. Malluche HH, Monier-Faugere MC, Herberth J. Bone disease after renal transplantation. Nat Rev Nephrol. (2010) 6:32–40. doi: 10.1038/nrneph.2009.192
142. Westeel FP, Mazouz H, Ezaitouni F, Hottelart C, Ivan C, Fardellone P, et al. Cyclosporine bone remodeling effect prevents steroid osteopenia after kidney transplantation. Kidney Int. (2000) 58:1788–96. doi: 10.1046/j.1523-1755.2000.00341.x
143. Marcen R, Caballero C, Pascual J, Teruel JL, Tenorio M, Ocana J, et al. Lumbar bone mineral density in renal transplant patients on neoral and tacrolimus: a four-year prospective study. Transplantation. (2006) 81:826–31. doi: 10.1097/01.tp.0000203557.36884.e3
144. Ugur A, Guvener N, Isiklar I, Turan M, Erdal R, Haberal M. Osteoporosis after renal transplantation: single center experience. Transplantation. (2001) 71:645–9. doi: 10.1097/00007890-200103150-00011
145. Vautour LM, Melton LJ 3rd, Clarke BL, Achenbach SJ, Oberg AL, McCarthy JT. Long-term fracture risk following renal transplantation: a population-based study. Osteoporos Int. (2004) 15:160–7. doi: 10.1007/s00198-003-1532-y
146. Patel S, Kwan JT, McCloskey E, McGee G, Thomas G, Johnson D, et al. Prevalence and causes of low bone density and fractures in kidney transplant patients. J Bone Miner Res. (2001) 16:1863–70. doi: 10.1359/jbmr.2001.16.10.1863
147. Briner VA, Thiel G, Monier-Faugere MC, Bognar B, Landmann J, Kamber V, et al. Prevention of cancellous bone loss but persistence of renal bone disease despite normal 1,25 vitamin D levels two years after kidney transplantation. Transplantation. (1995) 59:1393–400. doi: 10.1097/00007890-199505270-00006
148. Bouquegneau A, Salam S, Delanaye P, Eastell R, Khwaja A. Bone disease after kidney transplantation. Clin J Am Soc Nephrol. (2016) 11:1282–96. doi: 10.2215/CJN.11371015
149. Kanaan N, Claes K, Devogelaer JP, Vanderschueren D, Depresseux G, Goffin E, et al. Fibroblast growth factor-23 and parathyroid hormone are associated with post-transplant bone mineral density loss. Clin J Am Soc Nephrol. (2010) 5:1887–92. doi: 10.2215/CJN.00950110
150. Lentine KL, Patel A. Risks and outcomes of living donation. Adv Chronic Kidney Dis. (2012) 19:220–8. doi: 10.1053/j.ackd.2011.09.005
151. Segev DL, Muzaale AD, Caffo BS, Mehta SH, Singer AL, Taranto SE, et al. Perioperative mortality and long-term survival following live kidney donation. J Am Med Assoc. (2010) 303:959–66. doi: 10.1001/jama.2010.237
152. Ibrahim HN, Foley R, Tan L, Rogers T, Bailey RF, Guo H, et al. Long-term consequences of kidney donation. N Engl J Med. (2009) 360:459–69. doi: 10.1056/NEJMoa0804883
153. O'Keeffe LM, Ramond A, Oliver-Williams C, Willeit P, Paige E, Trotter P, et al. Mid- and long-term health risks in living kidney donors: a systematic review and meta-analysis. Ann Intern Med. (2018) 168:276–84. doi: 10.7326/M17-1235
154. Muzaale AD, Massie AB, Wang MC, Montgomery RA, McBride MA, Wainright JL, et al. Risk of end-stage renal disease following live kidney donation. J Am Med Assoc. (2014) 311:579–86. doi: 10.1001/jama.2013.285141
155. Arias E, Tejada-Vera B, Ahmad F. Provisional Life Expectancy Estimates for January through June, 2020. Vital Statistics Rapid Release no 10. Hyattsville, MD: National Center for Health Statistics. (2020). doi: 10.15620/cdc:100392
156. Hiemstra TF, Smith JC, Lim K, Xu D, Kulkarni S, Bradley JA, et al. Effect of kidney donation on bone mineral metabolism. PLoS ONE. (2020) 15:e0235082. doi: 10.1371/journal.pone.0235082
157. Thorsen IS, Bleskestad IH, Jonsson G, Skadberg O, Goransson LG. Neutrophil gelatinase-associated lipocalin, fibroblast growth factor 23, and soluble klotho in long-term kidney donors. Nephron Extra. (2016) 6:31–9. doi: 10.1159/000450621
158. Young A, Hodsman AB, Boudville N, Geddes C, Gill J, Goltzman D, et al. Bone and mineral metabolism and fibroblast growth factor 23 levels after kidney donation. Am J Kidney Dis. (2012) 59:761–9. doi: 10.1053/j.ajkd.2011.09.019
Keywords: alpha-Klotho, FGF-23, kidney donation, kidney transplant, ESKD (end stage kidney disease), bone mineral density (BMD), cardiovascular disease, mineral bone disease
Citation: Gupta M, Orozco G, Rao M, Gedaly R, Malluche HH and Neyra JA (2022) The Role of Alterations in Alpha-Klotho and FGF-23 in Kidney Transplantation and Kidney Donation. Front. Med. 9:803016. doi: 10.3389/fmed.2022.803016
Received: 27 October 2021; Accepted: 03 February 2022;
Published: 06 May 2022.
Edited by:
Mostafa Abdelkhalek, Mansoura University, EgyptReviewed by:
Alexander Grabner, Duke University, United StatesDaniela Picciotto, San Martino Hospital (IRCCS), Italy
Copyright © 2022 Gupta, Orozco, Rao, Gedaly, Malluche and Neyra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meera Gupta, bWVlcmEuZ3VwdGFAdWt5LmVkdQ==; Javier A. Neyra, amF2aWVyLm5leXJhQHVreS5lZHU=