 Michael J. Caplan
Michael J. Caplan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med., 31 January 2022
Sec. Nephrology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.753418
This article is part of the Research TopicThe Role of Mitochondria and Metabolic Rewiring in Kidney DiseasesView all 7 articles
Autosomal Dominant Polycystic Kidney Disease is a genetic disease that causes dramatic perturbations of both renal tissue architecture and of a multitude of cellular signaling pathways. The relationship between the products of the genes whose mutations cause polycystic kidney disease and these signaling pathways remains difficult to determine. It is clear, however, that cellular metabolism is dramatically altered in cells that are affected by polycystic kidney disease mutations. Adenosine monophosphate-stimulated protein kinase is a master regulator of cellular energy use and generation pathways whose activity appears to be perturbed in cells affected by polycystic kidney disease. Furthermore, modulation of this enzyme's activity may constitute a promising approach for the development of new therapeutics for polycystic kidney disease.
The clinical presentation of Autosomal Dominant Polycystic Kidney Disease (ADPKD) is most notable for the massive enlargement of the kidneys that affected individuals often experience over the space of decades (1). This dramatic transformation is produced through the development of numerous fluid-filled cysts that derive from nephron epithelial cells and whose expansion compresses and damages the surrounding normal parenchyma, resulting in end stage renal disease in ~50% of ADPKD patients. ADPKD is the most common life-threatening genetic disorder, affecting ~1:1,000 individuals independent of race, ethnicity, and gender. Mutations in either of two genes, PKD1 and PKD2, account for ~95% of cases of ADPKD (2). Consistent with the condition's autosomal dominant mode of inheritance, patients generally carry one wild type and one mutant allele of PKD1 or PKD2. While the mechanisms that initiate cyst formation remain the subject of investigation and debate, there is strong evidence that the stochastic acquisition of “second hit” mutations in the wild type PKD1 or PKD2 allele can play an important role in catalyzing the processes that lead to cyst development (3–5). According to this model, an individual epithelial cell in which a second hit mutation occurs becomes mitotically active and the proliferation that follows populates the wall of the resultant cyst. These cells also acquire a secretory phenotype, which contributes to the cyst's expanding fluid volume (6).
While the disruption of kidney architecture that ADPKD produces is anything but subtle, the mechanisms that connect mutation of the PKD1 and PKD2 genes to this profound structural disturbance remain obscure. The precise physiological functions of polycystin-1 (PC1) and polycystin-2 (PC2), the proteins encoded by PKD1 and PKD2 respectively, have yet to be elucidated, more than two decades after these genes were identified. Similarly, the processes through which the absence of functional PC1 or PC2 leads to the formation of cysts have not been completely unraveled. The mystery that shrouds the biological roles of PC1 and PC2 is not attributable to a lack of evidence for candidate signaling pathways in which they may participate. On the contrary, efforts to understand the cellular and molecular basis of ADPKD pathogenesis have been complicated in part by the overabundance of cellular processes that appear to be connected to, regulated by or responsible for governing some aspect of polycystin protein activity. It is clear that perturbing the expression of PC1 and PC2 extends ripples of downstream effects through a multiplicity of effectors (7, 8). It is not clear, however, which of these effectors are most proximate to the polycystin proteins themselves nor is it understood whether or how alterations in the messages communicated by one or more of these effectors can initiate cyst formation.
The absence of a causative link between the polycystin proteins and a single, direct and definitive explanation for cystic disease has been an impediment to the development of targeted therapeutics. Efforts to discover highly specific and efficacious drugs typically begin with assays that have evolved from a deep understanding of the relationship between a target and a disease. In the case of ADPKD such efforts have, of necessity, focused on intervening in processes that contribute to cyst formation, proliferation or expansion, even if the connections between those processes and the polycystin proteins are not clearly established. Thus, while the V2R vasopressin receptor is not thought to be an immediate upstream or downstream consort of the polycystin proteins, its influence on the elevated cAMP levels that clearly contribute to cystogenesis is exploited by Tolvaptan, the first drug that has been approved for the treatment of ADPKD (9, 10). Similarly, while the mechanisms that account for elevated mTOR activity in ADPKD are not fully understood, very convincing pre-clinical data from animal studies demonstrating that mTOR inhibition ameliorates cystic disease served as the basis for human trials of this approach (11–21). Until such time as a consensus understanding of the physiological roles of the polycystin proteins emerges, it is likely that progress in the development of new ADPKD therapeutics will resemble these efforts to target molecules and pathways that contribute to the cystic phenotype even if the biological basis of their connections to the functions of the polycystin proteins is obscure.
When considered through the lens of this rather broadminded approach to target identification, the cellular energy sensor and regulator adenosine monophosphate-stimulated protein kinase (AMPK) comes into focus as an extremely interesting candidate for ADPKD drug development (8). As its name implies, the kinase activity of AMPK is stimulated when AMP levels rise (22). This occurs when ATP levels fall, thanks to the function of the ATP:AMP transphosphorylase, which converts two molecules of ADP into one ATP and one AMP. As befits a master regulator of cellular energy utilization and generation, activation of AMPK leads to inhibition of energy-intensive processes and upregulation of energy generating processes. Among the energy-intensive pursuits that are shut down by AMPK are several that are relevant to processes that drive cyst expansion in ADPKD. Fluid secretion into the ADPKD cyst lumen depends upon active electrolyte transport that requires, at least in part, the participation of the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel (23, 24). AMPK-mediated phosphorylation of CFTR inhibits its channel activity, thus reducing energy-consuming transepithelial ion fluxes (25, 26). By phosphorylating the tuberin protein (TSC2), AMPK stimulates the GTPase-activating protein (GAP) activity of the TSC1/2 tuberous sclerosis complex (27). This in turn leads to conversion of the small GTP binding protein Rheb from its GTP bound form to its GDP bound form, which results in the inhibition of the mTORC1 complex and its kinase activity. Inhibition of mTOR suppresses protein synthesis and cell proliferation (28), both of which are energy-demanding processes that contribute to ADPKD cyst formation. Interestingly the chromosomal location of the TSC2 gene is extremely close to the 3' end of the PKD1 gene. Mutations that compromise the expression of both the TSC2 and PC1 proteins produce ADPKD with a severe, early onset course, reinforcing the importance of TSC2 and the mTOR pathway that it regulates in the development of the cystic phenotype (29). Finally, in at least some cell types activation of AMPK may reduce cAMP levels, since AMPK phosphorylates and activates the cAMP degrading enzyme phosphodiesterase 4B (30) (Figure 1). It is also worth noting that cilia, which play an important role in pathways associated with polycystin-related signaling, also exert influence on the regulation of AMPK activity (31, 32). Because of its potential to decrease the contributions of three key pathways to cyst expansion, Takiar et al. proposed in 2011 that AMPK could constitute a novel and accessible target for ADPKD therapeutic development (33).
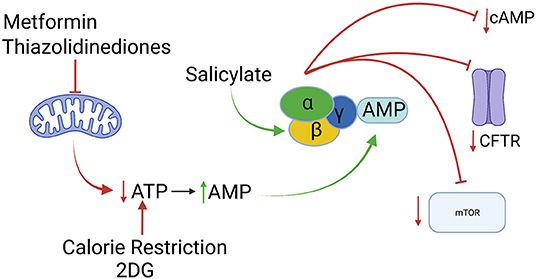
Figure 1. Activation of the AMPK enzyme, depicted as a heterotrimer of α, β and γ subunits, can result in inhibition of of the mTOR pathway, of CFTR-mediated fluid secretion and of cAMP accumulation. AMPK activity can be stimulated indirectly by interventions that reduce ATP production [metformin, thiazolidinediones, 2-deoxy-D-glucose (2DG), calorie restriction] or directly (salicylate). All of interventions slow disease progression in animal models of ADPKD. Created with Biorender.com.
AMPK is a heterotrimer, composed of an α-subunit that possesses the kinase activity, a γ-subunit that binds to AMP and a β-subunit that serves as the structural connection between α and γ. There are two isoforms each of the α and the β-subunits, and three isoforms of the γ-subunit (34). Renal epithelial cells predominantly express the α1 and β1-subunit isoform proteins, as well as both the γ1 and γ2 isoforms of the γ-subunit protein (35). Interaction with AMP induces a conformational change in the γ-subunit that is communicated to the α-subunit, as a result of which the α-subunit becomes a substrate for triggering phosphorylation by LKB1 or CAMKK, both of which can serve as upstream activators of the AMPK kinase (36). This phosphorylation, which occurs at Thr172, can be detected by western blotting using a phospho-specific antibody directed against phospho-AMPK (pAMPK) and the signal obtained in such assays is widely used as a surrogate for the level of AMPK kinase activity.
Perturbations in cellular energetics are features of a wide variety of diseases, including diabetes and cancer. Because of its central role as a regulator of the cell's energy economy, AMPK has been viewed as a potentially useful tool that could possibly be brought to bear to correct these perturbations and thus to treat the diseases that cause them (37). In order for the useful properties of AMPK to be exploited for therapeutic purposes, it is necessary to identify safe and well-tolerated treatments that effectively induce AMPK kinase activation. Fortunately, this can be achieved through several different direct and indirect approaches.
Although AMPK was first proposed and explored as a target for ADPKD therapeutic development in 2011 (33), the first demonstration that AMPK activity might be inappropriately suppressed in the context of ADPKD was provided in a landmark paper by Rowe et al. (38). These investigators discovered that cells that lack expression of PC1 exhibit a profound alteration in their metabolic processes. Even in the presence of oxygen, the highly proliferative cells that lack PC1 prefer to generate ATP via glycolysis. This behavior resembles the Warburg effect, which is characteristic of many types of highly proliferative tumor cells (39). In the case of the cells that lack PC1 expression, this metabolic dysregulation leads not only to a suppression of oxidative phosphorylation in favor of glycolysis, but also to a surprisingly large increase in the rate of glycolysis and ATP production. The levels of ATP in cells that lack PC1 are sufficiently elevated to result in the suppression of the activity of AMPK, which is normally triggered when ATP levels fall. This behavior was observed both in cultured cells and in renal tissue for an ADPKD mouse model (38). In an effort to exploit the metabolic vulnerability created by this switch to glycolysis, these investigators treated mice with 2-deoxy-D-glucose (2DG), which is a competitive inhibitor of an early step in glycolysis. Treatment with 2DG slowed the progression of cystic disease and, consistent with its capacity to interrupt ATP production in the glycolysis-dependent ADPKD cells, it also led to the activation of AMPK (38, 40). Thus, it is tempting to hypothesize that the beneficial effects produced by 2DG in the context of cystic disease are referenceable at least in part to its stimulatory effect on AMPK.
Calorie restriction is another indirect but simple and very well-tolerated intervention that leads to AMPK activation. In the context of ADPKD, Warner et al. showed that food restriction dramatically slowed cyst development in a mouse model of ADPKD (41). This effect was “dose dependent,” in that the degree of cystic disease suppression was well-correlated with the extent of the food restriction, which ranged from 10 to 40% in this study. Perhaps most striking was the observation that the imposition of calorie restriction was able to at least partially reverse established cystic disease. While many mechanisms could contribute to the suppression of cystic disease by food restriction, it is interesting to note that food restriction might be expected to produce AMPK activation since it should reduce the availability of the substrate molecules that are required to fuel cellular energy production. In fact, in renal tissue from food restricted animals AMPK activity is substantially increased, as evidenced by increased levels of pAMPK. In addition, the activity of the mTOR pathway is reduced in these animals, consistent with increased AMPK activation. Thus, it is possible that some or all of the influence of food restriction on the course of cystic disease progression is attributable to its capacity to activate AMPK.
Calorie restriction and 2DG deny cells the high energy substrates that they require to produce ATP, thus lowering ATP levels, raising AMP levels and producing AMPK activation. A number of insulin-sensitizing drugs that are widely used to treat Type 2 diabetes, the metabolic syndrome and polycystic ovary syndrome, also reduce ATP levels, elevate AMP levels and produce consequent AMPK activation. These drugs do not act by reducing the availability of high energy substrates but instead by interfering with the capacity of mitochondria to carry out the oxidative phosphorylation that liberates the energy embedded in those substrates. Metformin has been used for this purpose for more than 60 years. The precise mechanism through which metformin produces reduced ATP levels and increased AMP levels is not entirely clear, but it is generally thought that metformin inhibits the mitochondrial electron transport chain by blocking the function of Complex I (42). It is also important to note that metformin appears to affect numerous cellular processes and that therapeutic doses of metformin administered to treat Type 2 diabetes may not be sufficient to incite AMPK activation in vivo (30, 37, 43–46). It is likely, therefore, that its insulin-sensitizing and hypoglycemic effects are not directly related to its capacity to activate AMPK. Thiazolidinediones are another class of insulin-sensitizing drugs that produce multiple effects, among which are both inhibition of mitochondrial Complex I and AMPK activation (47, 48). It has recently been shown that, at least in the liver, the elevated AMP produced by metformin treatment can act to inhibit adenylate cyclase and thus prevent hormone-stimulated increases in cAMP (44). It is possible, therefore, that in addition to possessing the potential to activate AMPK, metformin may also have the ability to suppress cAMP levels. Thus, at least in theory, metformin may be able to influence three pathways that are thought to be critical to cyst progression. Two of these pathways (mTOR and CFTR) could be suppressed by indirect metformin-induced activation of AMPK, while the third (cAMP) could be the product of indirect metformin-induced elevation of AMP and consequent inhibition of adenylate cyclase or of direct stimulatory effects of AMPK on the activity of phosphodiesterase 4B.
Because of its well-established safety profile and its possible capacity to inhibit cystogenesis through AMPK activation, Takiar et al. tested the effects of metformin treatment in cell culture systems that recapitulate aspects of ADPKD-related processes as well as in two rapidly progressing orthologous ADPKD mouse models (33). In all of these experimental settings, metformin treatment inhibited pathways associated with cyst development and slowed the progression of cystic disease. These results have since been recapitulated in other animal models of renal cystic disease, including zebrafish (49), mice (50) and pigs (51). Metformin also slowed hepatic cyst development in a rodent model of polycystic liver disease (52). It is important to note that one study employing an orthologous mouse model of ADPKD did not detect a suppressive effect of metformin treatment on cyst development (53). It will be important to understand the basis for these discrepant results, since illuminating differences in mouse strain susceptibilities, dose administration, pharmacokinetics, etc. might reveal important factors that should be considered if metformin were to be developed as a therapeutic for patients with ADPKD. At the other extreme, the data obtained with metformin and 2DG, alone or in combination, in the pig system was especially striking, since each of these treatments impressively suppressed the dramatic cystic pathology that is characteristic of this highly orthologous model of ADPKD (51). Preclinical studies have tested two thiazolidinediones, pioglitazone and rosiglitazone, in non-orthologous rodent models of PKD and found that both compounds reduced the progression of cystic disease (54–56).
The well-understood safety profiles of metformin, pioglitazone and rosiglitazone, coupled with the accumulated promising pre-clinical data, has inspired the initiation of several small clinical trials designed to assess the safety of these compounds in ADPKD patients (57–59). Two of these have recently concluded and demonstrated that both metformin and pioglitazone are safe and well-tolerated in ADPKD patients (57, 60, 61). The TAME metformin trial employed the relatively high doses of the drug that are administered to treat polycystic ovary syndrome (60). Neither study was adequately powered to provide an assessment of efficacy. Thus, a determination as to whether metformin or thiazolidinediones can produce clinical benefit for ADPKD patients will await the results of future larger trials. One such large trial, administered through the University of Queensland in Australia, is currently enrolling 1,164 patients with rapidly progressing ADPKD. This Phase 3 study, “Implementation of Metformin theraPy to Ease Decline of Kidney Function in Polycystic Kidney Disease” (IMPEDE-PKD), will test the efficacy of 2 years of treatment with a slow-release form of metformin (61). The results of this study, which is expected to conclude in 2026, should provide a valuable assessment of metformin's promise as an ADPKD therapeutic.
The interventions that have been discussed up to this point achieve AMPK activation indirectly by interfering with cellular energy production. There are also a number of AMPK-interacting compounds whose administration produces direct effects on this enzyme's kinase activity. Salicylate, the active ingredient in aspirin, physically binds to the AMPK β-subunit and induces phosphorylation-independent AMPK activation (37, 62, 63). A recent very promising study showed that administration of salsalate, which is metabolized into salicylate, produced impressive suppression of cystic disease in an orthologous mouse model of ADPKD (53). In light of its very long history as a component of the pharmacopeia and its excellent safety profile, these observations should hopefully be translatable into a human clinical trial in the near future.
Another direct, but potentially more problematic, intervention involves the administration of chemical compounds that can mimic the capacity of AMP to bind to the AMPK γ-subunit and initiate the consequent conformational changes that are prerequisites for activating phosphorylation of the α-subunit by LKB1 or CAMKK. One such compound, AICAR, has been used extensively in research studies that have helped to elucidate the physiological consequences of AMPK activation in vitro and in vivo (64). MK-8722, a compound that employs this same mechanism to achieve AMPK activation, was tested as a potential treatment for diabetes and diabetic sequelae (65, 66). Administration of this compound to rodent and monkey models produced impressive improvements in glucose tolerance and also ameliorated aspects of diabetic nephropathy. Importantly, however, exposure to this compound produced cardiac hypertrophy. As a result, concerns have been raised as to the safety of chronic administration of direct AMPK activators that share this mechanism of action.
It is not clear whether the activity of AMPK is directly regulated by some aspect of polycystin protein function. It is clear, however, that AMPK modulates the activity of several pathways that participate in ADPKD pathogenesis. Furthermore, a number of metabolism-related therapeutic approaches that have shown promise in preclinical models of ADPKD induce AMPK activation and may achieve their beneficial effects in slowing cyst development through their influence on AMPK (8). A great deal of work remains to be done to determine whether and how AMPK responds to polycystin-related signals. It is clear, however, that AMPK is an interesting and attractive potential target for the development of safe and effective ADPKD therapeutics.
MC wrote the manuscript and is entirely responsible for its content.
Our research work on metabolism in ADPKD was supported by NIH RC2 grant DK120534.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The author wishes to thank members of his laboratory, past and present, for sharing their valuable insights and creativity. The author is especially grateful to Drs. Vinita Takiar and Patty Seo-Mayer who were the key drivers of our group's contributions to the topics addressed in this article.
1. Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. (2009) 60:321–37. doi: 10.1146/annurev.med.60.101707.125712
2. Takiar V, Caplan MJ. Polycystic kidney disease: pathogenesis and potential therapies. Biochim Biophys Acta. (2010) 1812:1337–43. doi: 10.1016/j.bbadis.2010.11.014
3. Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. (1996) 87:979–87. doi: 10.1016/S0092-8674(00)81793-6
4. Tan AY, Zhang T, Michaeel A, Blumenfeld J, Liu G, Zhang W, et al. Somatic mutations in renal cyst epithelium in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. (2018) 29:2139–56. doi: 10.1681/ASN.2017080878
5. Zhang Z, Bai H, Blumenfeld J, Ramnauth AB, Barash I, Prince M, et al. Detection of PKD1 and PKD2 somatic variants in autosomal dominant polycystic kidney cyst epithelial cells by whole-genome sequencing. J Am Soc Nephrol. (2021) 32:3114-29. doi: 10.1681/ASN.2021050690
6. Chapin HC, Caplan MJ. The cell biology of polycystic kidney disease. J Cell Biol. (2010) 191:701–10. doi: 10.1083/jcb.201006173
7. Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest. (2014) 124:2315–24. doi: 10.1172/JCI72272
8. Padovano V, Podrini C, Boletta A, Caplan MJ. Metabolism and mitochondria in polycystic kidney disease research and therapy. Nat Rev Nephrol. (2018) 14:678–87. doi: 10.1038/s41581-018-0051-1
9. Gattone VH 2nd, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med. (2003) 9:1323–6. doi: 10.1038/nm935
10. Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. (2012) 367:2407–18. doi: 10.1056/NEJMoa1205511
11. Becker JU, Opazo Saez A, Zerres K, Witzke O, Hoyer PF, Schmid KW, et al. The mTOR pathway is activated in human autosomal-recessive polycystic kidney disease. Kidney Blood Press Res. (2010) 33:129–38. doi: 10.1159/000314380
12. Belibi F, Ravichandran K, Zafar I, He Z, Edelstein CL. mTORC1/2 and rapamycin in female Han:SPRD rats with polycystic kidney disease. Am J Physiol Renal Physiol. (2011) 300:F236–44. doi: 10.1152/ajprenal.00129.2010
13. Novalic Z AM. van der Wal AM, Leonhard WN, Koehl G, Breuning MH, Geissler EK, de Heer E, Peters DJ. Dose-dependent effects of sirolimus on mTOR signaling and polycystic kidney disease. J Am Soc Nephrol. (2012) 23:842–53. doi: 10.1681/ASN.2011040340
14. Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. (2010) 363:820–9. doi: 10.1056/NEJMoa0907419
15. Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A. (2006) 103:5466–71. doi: 10.1073/pnas.0509694103
16. Shillingford JM, Piontek KB, Germino GG, Weimbs T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol. (2010) 21:489–97. doi: 10.1681/ASN.2009040421
17. Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant. (2006) 21:598–604. doi: 10.1093/ndt/gfi181
18. Walz G, Budde K, Mannaa M, Nurnberger J, Wanner C, Sommerer C, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. (2010) 363:830–40. doi: 10.1056/NEJMoa1003491
19. Wu M, Wahl PR, Le Hir M, Wackerle-Men Y, Wuthrich RP, Serra AL. Everolimus retards cyst growth and preserves kidney function in a rodent model for polycystic kidney disease. Kidney Blood Press Res. (2007) 30:253–9. doi: 10.1159/000104818
20. Zafar I, Belibi FA, He Z, Edelstein CL. Long-term rapamycin therapy in the Han:SPRD rat model of polycystic kidney disease (PKD). Nephrol Dial Transplant. (2009) 24:2349–53. doi: 10.1093/ndt/gfp129
21. Zafar I, Ravichandran K, Belibi FA, Doctor RB, Edelstein CL. Sirolimus attenuates disease progression in an orthologous mouse model of human autosomal dominant polycystic kidney disease. Kidney Int. (2010) 78:754–61. doi: 10.1038/ki.2010.250
22. Hardie DG AMP-activated/SNF1 AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. (2007) 8:774–85. doi: 10.1038/nrm2249
23. Brill SR, Ross KE, Davidow CJ, Ye M, Grantham JJ, Caplan MJ. Immunolocalization of ion transport proteins in human autosomal dominant polycystic kidney epithelial cells. Proc Natl Acad Sci U S A. (1996) 93:10206–11. doi: 10.1073/pnas.93.19.10206
24. Davidow CJ, Maser RL, Rome LA, Calvet JP, Grantham JJ. The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int. (1996) 50:208–18. doi: 10.1038/ki.1996.304
25. Hallows KR, McCane JE, Kemp BE, Witters LA, Foskett JK. Regulation of channel gating by AMP-activated protein kinase modulates cystic fibrosis transmembrane conductance regulator activity in lung submucosal cells. J Biol Chem. (2003) 278:998–1004. doi: 10.1074/jbc.M210621200
26. King JD Jr, Fitch AC, Lee JK, McCane JE, Mak DO, Foskett JK, et al. AMP-activated protein kinase phosphorylation of the R domain inhibits PKA stimulation of CFTR. Am J Physiol Cell Physiol. (2009) 297:C94–101. doi: 10.1152/ajpcell.00677.2008
27. Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. (2004) 6:91–9. doi: 10.1016/j.ccr.2004.06.007
28. Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. (2005) 17:596–603. doi: 10.1016/j.ceb.2005.09.009
29. Brook-Carter PT, Peral B, Ward CJ, Thompson P, Hughes J, Maheshwar MM, et al. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease–a contiguous gene syndrome. Nat Genet. (1994) 8:328–32. doi: 10.1038/ng1294-328
30. Johanns M, Lai YC, Hsu MF, Jacobs R, Vertommen D, Van Sande J, et al. AMPK antagonizes hepatic glucagon-stimulated cyclic AMP signalling via phosphorylation-induced activation of cyclic nucleotide phosphodiesterase 4B. Nat Commun. (2016) 7:10856. doi: 10.1038/ncomms10856
31. Boehlke C, Kotsis F, Patel V, Braeg S, Voelker H, Bredt S, et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol. (2010) 12:1115–22. doi: 10.1038/ncb2117
32. Orhon I, Dupont N, Zaidan M, Boitez V, Burtin M, Schmitt A, et al. Primary-cilium-dependent autophagy controls epithelial cell volume in response to fluid flow. Nat Cell Biol. (2016) 18:657–67. doi: 10.1038/ncb3360
33. Takiar V, Nishio S, Seo-Mayer P, King JD Jr, Li H, Zhang L, et al. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc Natl Acad Sci U S A. (2011) 108:2462–7. doi: 10.1073/pnas.1011498108
34. Davies SP, Hawley SA, Woods A, Carling D, Haystead TA, Hardie DG. Purification of the AMP-activated protein kinase on ATP-gamma-sepharose and analysis of its subunit structure. Eur J Biochem. (1994) 223:351–7. doi: 10.1111/j.1432-1033.1994.tb19001.x
35. Tain YL, Hsu CN. AMP-activated protein kinase as a reprogramming strategy for hypertension and kidney disease of developmental origin. Int J Mol Sci. (2018) 19:1744. doi: 10.3390/ijms19061744
36. Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. (2007) 100:328–41. doi: 10.1161/01.RES.0000256090.42690.05
37. Hardie DG. AMPK: a target for drugs and natural products with effects on both diabetes and cancer. Diabetes. (2013) 62:2164–72. doi: 10.2337/db13-0368
38. Rowe I, Chiaravalli M, Mannella V, Ulisse V, Quilici G, Pema M, et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med. (2013) 19:488–93. doi: 10.1038/nm.3092
39. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
40. Chiaravalli M, Rowe I, Mannella V, Quilici G, Canu T, Bianchi V, et al. 2-Deoxy-d-Glucose Ameliorates PKD Progression. J Am Soc Nephrol. (2016) 27:1958–69. doi: 10.1681/ASN.2015030231
41. Warner G, Hein KZ, Nin V, Edwards M, Chini CC, Hopp K, et al. Food restriction ameliorates the development of polycystic kidney disease. J Am Soc Nephrol. (2016) 27:1437–47. doi: 10.1681/ASN.2015020132
42. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. (2001) 108:1167–74. doi: 10.1172/JCI13505
43. Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. (2002) 51:2420–5. doi: 10.2337/diabetes.51.8.2420
44. Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. (2013) 494:256–60. doi: 10.1038/nature11808
45. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. (2017) 60:1577–1585. doi: 10.1007/s00125-017-4342-z
46. Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. (2010) 120:2355–69. doi: 10.1172/JCI40671
47. Brunmair B, Staniek K, Gras F, Scharf N, Althaym A, Clara R, et al. Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes. (2004) 53:1052–9. doi: 10.2337/diabetes.53.4.1052
48. LeBrasseur NK, Kelly M, Tsao TS, Farmer SR, Saha AK, Ruderman NB, et al. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am J Physiol Endocrinol Metab. (2006) 291:E175–81. doi: 10.1152/ajpendo.00453.2005
49. Chang MY, Ma TL, Hung CC, Tian YC, Chen YC, Yang CW, et al. Metformin Inhibits Cyst Formation in a Zebrafish Model of Polycystin-2 Deficiency. Sci Rep. (2017) 7:7161. doi: 10.1038/s41598-017-07300-x
50. Wang J, Chin D, Poon C, Mancino V, Pham J, Li H, et al. Oral delivery of metformin by chitosan nanoparticles for polycystic kidney disease. J Control Release. (2021) 329:1198–209. doi: 10.1016/j.jconrel.2020.10.047
51. Lian X, Wu X, Li Z, Zhang Y, Song K, Cai G, et al. The combination of metformin and 2-deoxyglucose significantly inhibits cyst formation in miniature pigs with polycystic kidney disease. Br J Pharmacol. (2019) 176:711–24. doi: 10.1111/bph.14558
52. Sato Y, Qiu J, Hirose T, Miura T, Sato Y, Kohzuki M, et al. Metformin slows liver cyst formation and fibrosis in experimental model of polycystic liver disease. Am J Physiol Gastrointest Liver Physiol. (2021) 320:G464–73. doi: 10.1152/ajpgi.00120.2020
53. Leonhard WN, Song X, Kanhai AA, Iliuta IA, Bozovic A, Steinberg GR, et al. Salsalate, but not metformin or canagliflozin, slows kidney cyst growth in an adult-onset mouse model of polycystic kidney disease. EBioMedicine. (2019) 47:436–445. doi: 10.1016/j.ebiom.2019.08.041
54. Muto S, Aiba A, Saito Y, Nakao K, Nakamura K, Tomita K, et al. Pioglitazone improves the phenotype and molecular defects of a targeted Pkd1 mutant. Hum Mol Genet. (2002) 11:1731–42. doi: 10.1093/hmg/11.15.1731
55. Raphael KL, Strait KA, Stricklett PK, Baird BC, Piontek K, Germino GG, et al. Effect of pioglitazone on survival and renal function in a mouse model of polycystic kidney disease. Am J Nephrol. (2009) 30:468–73. doi: 10.1159/000242432
56. Yoshihara D, Kurahashi H, Morita M, Kugita M, Hiki Y, Aukema HM, et al. PPAR-gamma agonist ameliorates kidney and liver disease in an orthologous rat model of human autosomal recessive polycystic kidney disease. Am J Physiol Renal Physiol. (2011) 300:F465–74. doi: 10.1152/ajprenal.00460.2010
57. Blazer-Yost BL, Bacallao RL, Erickson BJ, LaPradd ML, Edwards ME, Sheth N, et al. A randomized phase 1b cross-over study of the safety of low-dose pioglitazone for treatment of autosomal dominant polycystic kidney disease. Clin Kidney J. (2021) 14:1738–46. doi: 10.1093/ckj/sfaa232
58. Seliger SL, Abebe KZ, Hallows KR, Miskulin DC, Perrone RD, Watnick T, et al. A Randomized clinical trial of metformin to treat autosomal dominant polycystic kidney disease. Am J Nephrol. (2018) 47:352–360. doi: 10.1159/000488807
59. Seliger SL, Watnick T, Althouse AD, Perrone RD, Abebe KZ, Hallows KR, et al. Baseline characteristics and patient-reported outcomes of ADPKD patients in the multicenter TAME-PKD clinical trial. Kidney. (2020) 1:1363–72. doi: 10.34067/KID.0004002020
60. Perrone RD, Abebe KZ, Watnick T, Althouse A, Hallows KR, Lalama CM, et al. Primary results of the randomized trial of metformin administration in polycystic kidney disease (TAME PKD). Kidney Int. (2021) 100:684–96. doi: 10.1016/j.kint.2021.06.013
61. Ong ACM, Gansevoort RT. TAMEing ADPKD with metformin: safe and effective? Kidney Int. (2021) 100:513–5. doi: 10.1016/j.kint.2021.07.021
62. Hardie DG, Ross FA, Hawley SA. AMP-activated protein kinase: a target for drugs both ancient and modern. Chem Biol. (2012) 19:1222–36. doi: 10.1016/j.chembiol.2012.08.019
63. Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. (2012) 336:918–22. doi: 10.1126/science.1215327
64. Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. (1995) 229:558–65. doi: 10.1111/j.1432-1033.1995.0558k.x
65. Zhou X, Muise ES, Haimbach R, Sebhat IK, Zhu Y, Liu F, et al. PAN-AMPK Activation improves renal function in a rat model of progressive diabetic nephropathy. J Pharmacol Exp Ther. (2019) 371:45–55. doi: 10.1124/jpet.119.258244
Keywords: Autosomal Dominant Polycystic Kidney Disease, adenosine monophosphate-stimulated protein kinase, metabolism, mTOR, CFTR, metformin
Citation: Caplan MJ (2022) AMPK and Polycystic Kidney Disease Drug Development: An Interesting Off-Target Target. Front. Med. 9:753418. doi: 10.3389/fmed.2022.753418
Received: 04 August 2021; Accepted: 10 January 2022;
Published: 31 January 2022.
Edited by:
Alessandra Boletta, San Raffaele Hospital (IRCCS), ItalyReviewed by:
Albert Ong, The University of Sheffield, United KingdomCopyright © 2022 Caplan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael J. Caplan, bWljaGFlbC5jYXBsYW5AeWFsZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.