 Jinzhi Wu
Jinzhi Wu Fan Yang
Fan Yang Xinlei Ma
Xinlei Ma Jin Lin
Jin Lin Weiqian Chen
Weiqian Chen- Division of Rheumatology, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, China
Rheumatoid arthritis is a chronic autoimmune disease that mainly affects the facet joints. Elderly-onset rheumatoid arthritis appears to exhibit symptoms similar to those of polymyalgia rheumatica, characterized by morning stiffness and pain in the shoulder and hip joints. Both diseases develop in the elderly, and it is sometimes challenging to distinguish them. Here, we identify the differences in pathogenesis between elderly-onset rheumatoid arthritis and polymyalgia rheumatica to assist with a clear differential diagnosis and effective early intervention.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease that affects synovial tissue and can lead to ongoing joint damage and irreversible disability (1). RA is most likely to affect the hands, feet, and knees. The most common clinical features of RA are pain, joint swelling, and stiffness. Rheumatoid factor (RF) and anti-citrullinated protein antibodies (ACPA) are two crucial antibodies in patients with RA and are used for diagnosis of the condition (2). Elderly-onset rheumatoid arthritis (EORA) is defined as RA with onset occurring when the patient is over 60 (3–5) or 65 (6) years of age, representing 10–33% (4) of cases of RA. Compared with early-onset rheumatoid arthritis, EORA is more evenly distributed across genders, and is associated with a more acute abrupt onset, larger proximal joint involvement (such as the knee and shoulder), higher erythrocyte sedimentation rate (ESR), higher likelihood of weight loss, lower RF positivity, and poorer disease outcomes (3–5, 7). Moderate to severe proliferative synovitis of the shoulder bursae is more often detected in EORA with PMR-like symptoms than in PMR (8). The pathogenesis of EORA may differ from that of early-onset rheumatoid arthritis. EORA also has a different proinflammatory cytokine profile, including higher IL-6 and lower TNF-α, and the susceptibility gene differs in the case of EORA (5). Finally, the geno–phenotype frequencies vary between EORA and early rheumatoid arthritis.
Since EORA exhibits polymyalgia rheumatica (PMR)-like symptoms, it is sometimes difficult to differentiate it from PMR, which is a chronic inflammatory rheumatic disorder commonly occurring in people over 50 years of age and usually with an acute onset. PMR may occur independently or with giant cell arteritis. The primary manifestations of PMR are musculoskeletal pain and morning stiffness in the shoulders and pelvic girdle. Due to a lack of specific antibodies such as RF and ACPA, diagnosis of PMR is generally based on clinical features and an increase in ESR and C-reactive protein (CRP). PMR responds dramatically to a low dose of glucocorticoids (9–12). Meanwhile, EORA responds well to a relatively high dose of glucocorticoids (GC) in the early disease phase.
As PMR and EORA have similar clinical manifestations and their response to glucocorticoid treatment is comparable, physicians should exercise care in distinguishing the two diseases. In this paper, we present a comprehensive review of the published literature and identify the differences in pathogenesis between EORA and PMR.
Genetic background
Multiple genes are involved in the pathogenic processes of RA and PMR. Thus far, approximately 100 loci related to the risk of RA have been identified, of which human leukocyte antigen (HLA)-DRB1 is the most important. HLA-DRB1 was believed several decades ago to be associated with the risk of RA, as proposed in the shared epitope (SE) hypothesis (13); this claim has been validated by several studies (14–16). HLA-DRB1 has been found to be linked to the T lymphocyte function of recognizing self-reactive peptides (17). HLA-DRB1 has also been linked to susceptibility to PMR; however, different phenotypic frequencies are involved. It has been reported that HLA-DRB1*01 occurs at a higher frequency in patients with EORA, while several studies have indicated that HLA-DRB1*04 and HLA-DRB1*13/14 are more closely related to PMR (18, 19).
Protein tyrosine phosphatase non-receptor (PTPN22) is another important gene that contributes to the development of RA by downregulating the activity of T cells, by preventing variant phosphatase from combining with recombinant C-Src tyrosine kinase (Csk). PTPN22 occurs at a higher frequency in RF-positive patients than in RF-negative patients (20). Although one study has found that the PTPN22 functional variant R620W may be a genetic susceptibility factor for GCA (21), the relationship between PTPN22 and PMR has not been investigated. Protein-arginine deiminase type-4 (PADI4) is a significant RA susceptibility gene in geriatric Asian individuals (22), but not in European individuals (23). CCR6 and TNFAIP3 have also been linked to RA, according to a genome-wide association study (GWAS) meta-analysis (15).
Immunosenescence
Immune senescence (also termed immunosenescence) is characterized by the immune dysfunction that occurs with aging. It has three main immunological manifestations: first, the immune response to endogenous and exogenous antigens is impaired; second, the immune protection effect is weakened, e.g., the antitumor effect declines or disappears; and third, the established immune memory response is affected, producing a decrease in or disappearance of the response to vaccination. In the process of immune aging, both the innate and the adaptive immune systems of the body will be affected. However, at present, research shows that aging has a greater impact on the adaptive immune system than on the innate immune system. Additionally, thymic involution is a crucial aspect of immunosenescence. Reduced thymus output and a decline in TCR diversity are the results of structural changes and functional thymus atrophy (24). An expansion of senescent CD4+CD28−T cells and CD8+CD28−T cells with shortened telomeres has been observed in RA patients (25–27). Moreover, T cell repertoire shrinks significantly as a result of the amplification of these clones (28). Levels of T cell receptor rearrangement excision circles (TRECs) in CD4+T cells, which can be used as a measure of the thymic output of T cells, have been found to be significantly lower in RA patients than in age-matched control donors (29). Immunosenescence may be a factor in the diminished thymus function observed in RA patients. Recently, a novel CD28−CD4+Foxp3+ subset of Treg cells (CD28−Treg) has been described in circulation in RA. These CD28−Treg cells exhibit a deficiency in control function compared to CD28+Treg cells and are associated with aging in both RA patients and healthy individuals (30). It has also been discovered that RA patients of all ages have an elevated rate of telomeric erosion. In addition, patients harboring the HLA-DRB1 gene tend to have shorter telomere lengths, which raises the possibility that HLA-DRB1 is related to accelerated telomere erosion in RA (31). Patients with PMR also exhibit higher levels of senescent T cells that express NKG2D, a hallmark of immunosenescence (32).
Innate immune response
The innate immune system, which includes neutrophils, dendritic cells, and NK cells as well as macrophages (monocytes), is crucial in the development of RA; in turn, it activates adaptive immunity to play a role in the disease. Although not as clear as in the case of RA, the role of the innate immune system in PMR has also been investigated.
The innate immune system is activated when individuals carrying the RA risk gene are exposed to a risky environment (such as via smoking). By stimulating the production of cytokines, chemokines, and degradative enzymes, and causing joint destruction, macrophages play a crucial role in the progression of RA (33). There are two types of macrophages: inflammatory phenotype macrophages (M1) induced by lipopolysaccharide and interferon-γ, and anti-inflammatory macrophages (M2) induced by interleukin-4 (IL-4) or IL-13 (34). Increased levels of TNF-α and IL-1, both of which are typically released by M1 macrophages, have been observed in the synovium in RA (35). Recently, it has also been discovered that IL-1β can drive naïve CD4+ T cells to differentiate into osteoclastogenic regulatory T cells (O Tregs) in vivo. In contrast to the typical suppressive role played by Tregs, O Tregs can overexpress RANKL and cause bone erosion (36). The phagocytic function of macrophages diminishes in the aging population (37). Cytokine secretion is also significantly delayed, which results in defective responses to PAMPs and DAMPs by macrophages (37). However, no specific study has focused on macrophages or monocytes in EORA; thus, more data are urgently needed in this area. Anti-TNF-α therapy for RA has been found to reduce the formation of matrix metalloproteinase (MMP), activate leukocytes, reduce angiogenesis, and alleviate pain (1). TNF-α is higher in EORA patients than in PMR patients (although it is still higher in the latter group than in healthy controls) (38, 39); however, there is no difference in IL-1 levels between EORA and early-onset rheumatoid arthritis (39). Transforming growth factor-1 (macrophage-derived) and interleukin-1 (IL-1) have been found in the temporal artery in PMR (40). Meanwhile, the function of TNF-α in PMR is not fully understood (41). One study has reported observing no significant difference in TNF-α between PMR patients and HCs (42), while another has indicated that TNF-α is much higher in patients with PMR (38). Through measurement of methylome and transcriptome profiles, it has recently been demonstrated that a more proinflammatory phenotype of circulating CD14+ monocytes occurs in patients with GCA; the investigators also found that IL-11, a cytokine that can promote the development of Th17 cells, may activate GCA CD14+ monocytes (43). Monocyte levels are higher in patients with PMR (44). It can be assumed that CD14+ monocytes may also contribute to the inflammatory reaction in PMR.
Neutrophils are also crucial in the pathophysiology of RA. In the early stage, they migrate into the articular cavity, become activated, and exert their function in an inflammatory milieu. The ability of neutrophils to form neutrophil extracellular traps (NETs) actively triggers the immune response of RA via autoantigen production against citrullinated proteins (45). It is well known that the function of phagocyte and cytokine production in neutrophils is downregulated in older people (46). Neutrophils from patients with active EORA have been found to exhibit a higher capacity to migrate, but this is not the case in PMR. Neutrophils and monocytes with phagocytic activity have been found to be similar among PMR and EORA patients, and among age-matched HCs. In contrast, oxidative burst is significantly reduced in neutrophils and monocytes from patients with PMR or EORA; however, more data are essential to confirm this result (47). The number of neutrophils has been found to be higher in PMR than in controls, but this number falls following GC treatment, suggesting that the number of neutrophils may be related to the severity of the disease (44).
Toll-like receptors (TLRs) are involved in the recognition of pathogen-associated molecular patterns and the generation of antibodies. For instance, TLR-2 and TLR-4 play an essential role in the persistence of joint inflammation. In comparison to osteoarthritis, increased expression of TLR-2 and TLR-4 has been observed on the surface of monocyte cells from the synovium of RA patients. Both IL-12 and IL-18 can upregulate the production of TLR-2 and TLR-4 in the presence of IFN-γ. Furthermore, an increase in TLR-2/TLR-4 levels can influence downstream processes, such as by enhancing the production of TNF-α and IL-6, both of which promote bone erosion by inducing the secretion of RANKL (48, 49). However, blocking the TLR-4 pathway alone may not benefit patients with RA (50). TLR-7 has been found to be more abundant in circulating mononuclear cells from patients with PMR compared to healthy controls, but its ability to influence the production of proinflammatory cytokines from monocytes diminishes throughout the course of disease activity and reverses during the remission period (51).
IL-6 is another important proinflammatory cytokine in the pathogenesis of RA. It can cause bone destruction by stimulating osteoclast differentiation and suppressing osteoblast maturation, as well as by inducing Th17 differentiation and blocking Treg production (52). IL-6 has been found to be abundant in the synovial tissue and peripheral blood of RA patients, and also in the peripheral blood of EORA patients (53). Compared to methotrexate (MTX) treatment alone, a combination of MTX and tocilizumab effectively reduces the progression of joint deterioration (54). Patients with PMR have higher levels of circulating IL-6 when compared to patients with EORA (38). In the subacromial–subdeltoid bursa of patients with PMR, IL-6 has been found to be derived from various cells (endothelial cells, fibroblasts, and macrophages) (55). Additionally, serum IL-6 originating from CD14+ monocytes is significantly correlated with CRP, ESR levels, and disease activity (19, 56). A prospective longitudinal study has found that tocilizumab treatment significantly reduces polymyalgia rheumatica activity score (PMR-AS) (57), and another study has demonstrated that genetically proxied IL-6 receptor inhibition is associated with a lower risk of PMR (58).
Dendritic cells (DCs) exert their functions through antigen presentation and immune regulation. The nuclear factor kappa B (NF-B) pathway is activated after DCs recognize an inflammatory stimulus. As a result of this activation of the NF-B pathway, MHC class II expression has been found to increase, which primes T cells to be attracted by chemokines released by DCs in synovium or tissue in RA (59). Inflammatory DCs (infDCs), a newly discovered group of DCs, have been identified in RA and have been shown to induce the differentiation of Th17 cells from naïve CD4+T cells via secretion of IL-23 (60). However, there is evidence that the ability of DCs to engage in cross-presentation and uptake of microbes is reduced in the elderly population due to decreased migration and expression of costimulatory cytokines that are essential for T cell stimulation, which might constitute one of the primary defects in protection against infections in EORA (61, 62). In the temporal arteries of PMR patients, CD83+DCs may generate CCL19 and CCL21, and are essential for triggering and attracting T cells (63).
At an early stage of RA, natural killer (NK) cells can be found in synovial tissue; these have been found to trigger the differentiation of osteoclasts. Specifically, NK cells promote the formation of osteoclasts by expressing cell-surface receptor activator of NF-κB ligand (RANKL) and a low level of macrophage colony-stimulating factor (M-CSF) (64). However, it has also been noted that NK cells stimulated by IL-15 might play a protective role by killing osteoclasts through contact-dependent pathways (65). CD56+NK cells have been observed in RA and may enhance the ability of CD14+ monocytes to secrete TNF (33). Reduced numbers of circulating CD56dim cells have been found in patients with recently diagnosed RA, but this is not the case for the CD56bright subset. This decrease has been found to result from cell apoptosis induced by CD16 signaling following interaction with IgG complexes (66). On the other hand, the enrichment of CD56bright cells in synovial fluid and tissue may be the primary reason for the longstanding inflammatory process in late-stage RA. Moreover, this subset of NK cells in the ordinary age-related population has been found to be shifted from CD56dim to CD56bright. Reduced activity is observed in the functions of cytotoxicity and cytokine secretion, as well as migration (67). In the peripheral blood of PMR patients (36), NK cell numbers have been shown to be reduced (44), although the role of these cells in the disease remains unclear.
Adaptive immune response
T and B cells move into the synovial membrane after being activated by the innate system, leading to persistent inflammation and causing further joint destruction. Multiple subsets of T cells are involved in PMR; however, the role of B cells remains unclear.
T cells
Various effector subsets divided out from activated CD4+T cells play a pivotal role in autoimmune diseases. Th1 cells have been found to be marked by the release of IFN-γ and Th2 cells by IL-4, an anti-inflammation cytokine. Patients with RA have high levels of IFN-γ and low levels of IL-4 in their synovium, indicating that Th1 cells predominate in the disease (68). In contrast, patients with PMR have been shown to have lower levels of Th1 in their peripheral blood (69). However, in synovial fluid and bursal tissue, an increase in Th1 cells rather than Th17 cells and an increase in IFN-γ rather than IL-17 has been observed in PMR (70).
Tregs are a subset of T cells that can modulate the immune response and maintain self-tolerance; an imbalance between Treg and Th17 may result in autoimmune diseases (71). In comparison to healthy controls, patients with RA have a lower incidence of Tregs and higher incidence of Th17 cells in their peripheral blood, which suggests the involvement of an imbalance between Tregs and Th17 at the onset of the disease. In addition, when cultured in vivo, effector memory T cells (EMTs) have been found to induce more Th17 cells compared with central T cells and naïve T cells in RA patients, indicating that the amplification of Th17 cells in RA patients may arise through differentiation from EMTs (72). According to a recent study, MaR1 can effectively ameliorate the progression of RA by upregulating mRi21 to correct the Treg/Th17 imbalance (73). The frequency of Treg cells is increased by MTX and biological DMARDs; this alters the Treg/Th17 balance and improves the prognosis of RA patients. Patients with RA can also benefit from JAK inhibitor therapy, although this does not alter the Treg/Th17 balance (74). In patients with PMR, Treg cells are only mildly altered, while levels of Th17 cells are significantly elevated in the peripheral blood and have been found to decrease following glucocorticoid treatment. CD161+CD4+T lymphocytes, which are thought to be precursors of Th17 cells, are similar in PMR patients and healthy controls, but they may produce more IL-17 in vitro. These strands of evidence underline the claim that the pathophysiology of PMR may be influenced by Treg/Th17 imbalance (69).
In patients with RA, natural killer group 2 member D (NKG2D) has been found to be expressed on CD4+CD28−T cells in peripheral blood and synovial tissue (75). One study has demonstrated a dose-dependent reduction of TNF-α after 24 h when RA synovial membrane cells are cultured with anti-NKG2D monoclonal antibody. Furthermore, clinical scores (with disease severity being assessed by paw thickness) have been found to improve significantly when mice with collagen-induced arthritis receive an intraperitoneal injection of CX5, a monoclonal anti-NKG2D antibody, indicating that anti-NKG2D therapy has a protective effect against joint damage. The authors of this study have also investigated the effects of this type of therapy on NK cells' killing capacity, discovering that anti-NKG2D therapy reduces the amount of IL-17 in CD4+ T cells (76, 77). PMR patients exhibit higher expression of NKG2D (which is upregulated by antigenic stimulation and pro-inflammatory cytokines) in circulating CD4+CD28−T cells and CD8+T cells compared with HCs. NKG2D-expressing T cells may also contribute to chronic inflammation in PMR by enhancing the secretion of IFN-γ (32).
Follicular helper T (Tfh) cells, which are effector T cells that help to activate B cells, can be detected in the circulating blood in patients with RA. CD4+CXCR5+PD-1hiT cells have been found to be involved in RA via inducement of B cells to produce more antibodies and an increase in levels of cytokine IL-21. Tph cells have also been found to have the ability to convert B cells into plasma cells. The presence of Tph cells in the synovium in RA suggests that T–B interaction can occur locally in RA. During disease remission, Tfh rather than Tph levels have been found to decrease, indicating the role of the former as a possible marker for RA activity (78, 79). No significant difference has been observed in the alternation of circulating Tfh cells among patients with PMR (80).
Th22 cells, differentiated from CD4+T cells induced by IL-6 and TNF-α, are characterized by high levels of secretion of IL-22 (81); they are found in abundance in peripheral blood in RA, and have been found to migrate to synovia via CCL28. When cultured with monocytes, Th17, Th1, and Th2 cells do not appear to produce as many osteoclasts as Th22 cells do. Th22 is involved in the bone destruction that occurs in RA via enhancement of the formation of osteoclasts through IL-22 (82). IL-22 has been observed in the inflamed temporal arteries occurring in GCA (83), and may also be involved in the pathogenesis of PMR.
B cells
In addition to producing antibodies and participating in humoral immune responses, B cells can be transformed into plasma cells, which can act as antigen-presenting cells by processing and presenting antigens. The generation of the autoantibodies RF and ACPA are currently the best-known functions of B cells in RA. Although RA can be diagnosed with greater specificity via ACPA, it is still unclear whether APCA contributes to RA by stimulating osteoclastogenesis. ACPA enhances the interaction of CD147 and ITGB1, which in turn activates the PI3K/Akt/NFB pathway, resulting in the synthesis of NOD-like NLRP3 inflammasome and IL-1 (84). Anti-cyclic citrullinated peptide (CCP) titers have been found to be 65% positive in patients with EORA, while they are negative in PMR. The anti-CCP antibody can therefore function as an important marker for the differential diagnosis from EORA to PMR with good sensitivity and specificity, especially for EORA patients with PMR-like symptoms. Similarly, RF has been found to be positive at 66.7% in patients with EORA and 7% in PMR, and RF titers are much higher in EORA than in PMR (423 ± 112 vs. 35.3 ± 2.4, normal range 0–22IU/L). High levels of RF may thus be helpful in distinguishing EORA from PMR (85). Although a substantial correlation has been observed between RF levels and disease activity, and RF positivity is associated with poorer prognosis, the role of RF in the etiology of RA is as yet unknown (86). On the other hand, lower levels of RF in EORA are associated with poorer outcomes, whereas no specific antibodies marking PMR disease activity or diagnosis have yet been discovered.
The function of B lymphocytes has been further investigated since the discovery that anti-CD20 treatment has a positive impact in terms of preventing the progression of RA. Fc-receptor-like-4F (FcRL4) B cells, a subpopulation of memory B cells that express RANKL, have been discovered in synovial tissue in RA. It has also been shown that TNF-α, which can cause bone deterioration, is produced at a significantly higher rate when FcRL4 B cells are present (49). Another subset of B cells, namely CD21−/lowB cells, may also contribute to joint damage by expressing RANKL and proinflammatory cytokines (87). One study has found that dominant BCR clones may be found in the peripheral blood of those at risk of developing RA. In particular, at-risk patients with more than five dominant BCR clones found in their peripheral blood are more likely to develop RA. Once RA has manifested, these BCR clones move to synovial tissue. However, because the at-risk individuals who participated in this study were under 60, it is unclear whether BCR contributes to the development of EORA (88).
Recently, increased expression of CXCL9 and CXCL13 has been observed in the blood of drug-naïve patients with PMR. Baseline CXCL13 expression is correlated with the disease activity index of PMR. Conversely, circulating CXCR3+ and CXCR5+ switched memory B cells have been found to be decreased, and these are inversely associated with CXCL9 and CXCL13. This study has demonstrated that the CXCL9–CXCR3 and CXCL13–CXCR5 axes play an important role in patients with PMR. Changes in the chemokine and chemokine receptor pathways contribute to the homing and organization of B-cells in the vessel wall (89). B cells are reduced in circulating blood (44) and are rarely found in the synovial fluid in PMR (70). In comparison to controls, patients with PMR have been found to have considerably fewer CD19+ B cells and CD24highCD38high transitional B cells; the levels of these increase in response to tocilizumab therapy (90).
Conclusions
There are few published papers involving a head-to-head comparison between EORA and PMR; hence, we have been unable to draw a clear conclusion on the difference in pathogenesis between these two similar diseases (Figure 1). However, EORA and PMR have different genetic backgrounds, in that HLA-DRB1*01 is associated with EORA, whereas HLA-DRB1*04 and HLA-DRB1*13/14 are more closely related to PMR. Given that these two diseases both affect elderly patients, the potential involvement of immunosenescence in their pathophysiology requires further investigation. Predominant macrophages and T cells have been found to be present in the synovial tissue of patients with PMR, while few neutrophils and no B cells, natural killer cells, or gamma/delta T cells are observed in PMR (91). In contrast, NK cells are present in the synovial fluids of RA patients, and a strong correlation with disease severity has been observed (92). Innate immune cells, such as M1 macrophages and neutrophils, are crucial in the development of EORA, and monocytes and neutrophils may function as proinflammatory cells that can trigger the “fire” of PMR. Following innate immune activation, T cells and B cells move to the synovial membrane via chemokines, generating a chronic inflammatory response that further damages the joint. Levels of both Th1 and Th17 cells are increased in both of the two diseases. However, Tfh cells are increased in EORA, but not altered in PMR. B cells are highly activated in EORA, with the ability to produce APCA or RF, but B cells are decreased in PMR; the function of B cells is still unknown. TNF-α, IL-6, and IL-1β are all increased in EORA, although IL-6 and IL-1β are also elevated in PMR.
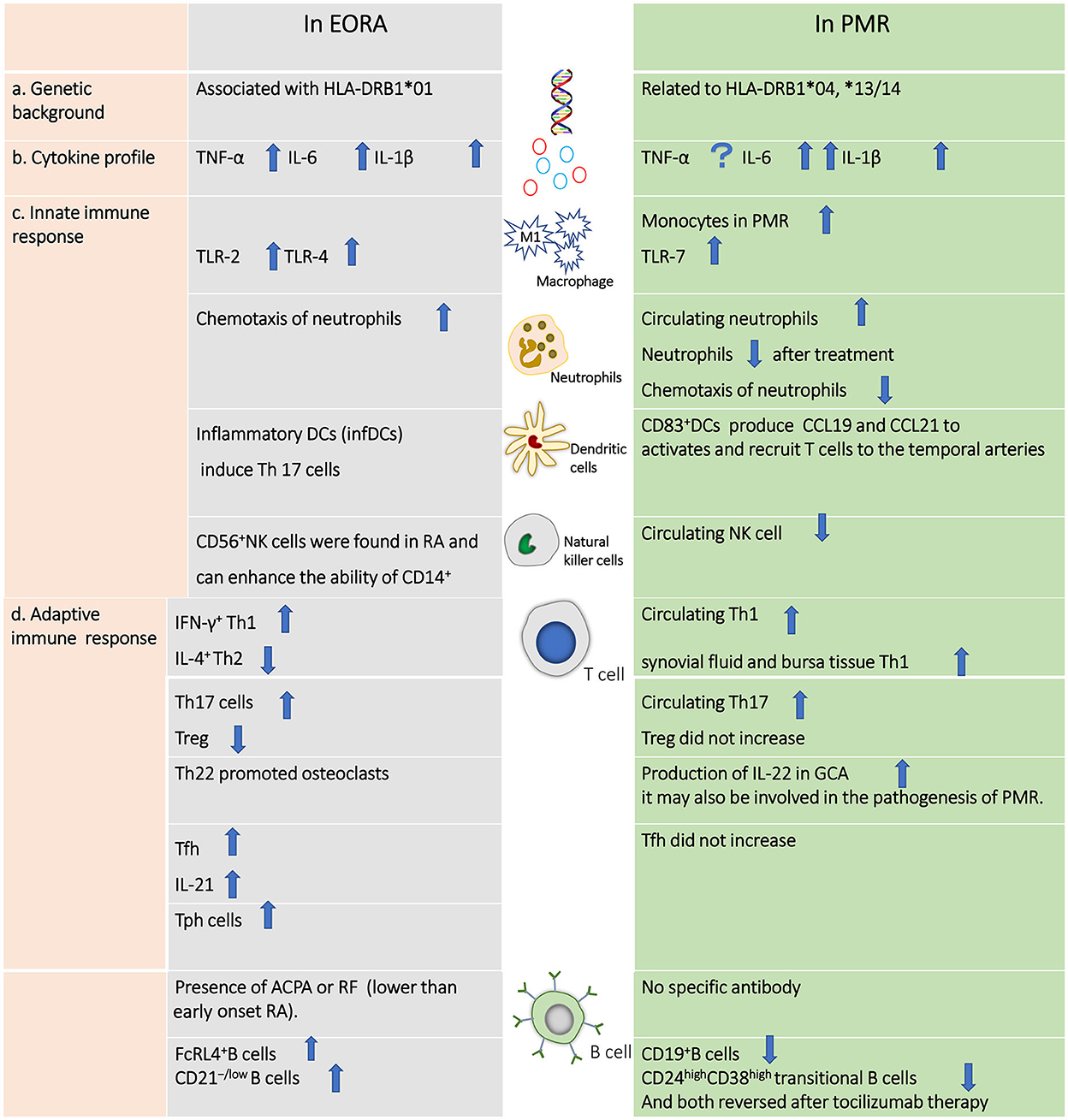
Figure 1. Differences in the pathogenesis of elderly-onset rheumatoid arthritis vs. polymyalgia rheumatic.
Although the two diseases have similar symptoms, fine distinctions do exist. In PMR, there is a tendency for pain and stiffness to occur in the shoulder and pelvic girdle, while EORA primarily affects the large joints such as the knee, ankle, and sometimes the shoulder and hip joints. ACPA is increased in most patients with EORA, but not in patients with PMR. Therefore, the serum marker ACPA appears to be helpful in making a differential diagnosis of the two diseases. If ACPA is also negative, the presence of synovitis confirmed by ultrasound or MRI scan can confirm a diagnosis of EORA. Extracapsular changes such as bursitis, peritendinitis, capsulitis, or myofascial lesions, confirmed by ultrasound, MRI scan, or PET scan, suggest a diagnosis of PMR.
Author contributions
WC: conception and design of the study and final approval of the manuscript. JW, FY, XM, and JL: literature review and writing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by the National Natural Science Foundation of China (82171768) and by the National Key Research and Development Program of China (2022YFC3602000).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. van Delft MAM, Huizinga TWJ. An overview of autoantibodies in rheumatoid arthritis. J Autoimmun. (2020) 110:102392. doi: 10.1016/j.jaut.2019.102392
3. van Schaardenburg D, Breedveld FC. Elderly-onset rheumatoid arthritis. Semin Arthritis Rheum. (1994) 23:367–78. doi: 10.1016/0049-0172(94)90087-6
4. Soubrier M, Mathieu S, Payet S, Dubost J, Ristori JM. Elderly-onset rheumatoid arthritis. Joint Bone Spine. (2010) 77:290–6. doi: 10.1016/j.jbspin.2010.04.004
5. Serhal L, Lwin MN, Holroyd C, Edwards CJ. Rheumatoid arthritis in the elderly: characteristics and treatment considerations. Autoimmun Rev. (2020) 19:102528. doi: 10.1016/j.autrev.2020.102528
6. Kobak S, Bes C. An autumn tale: geriatric rheumatoid arthritis. Ther Adv Musculoskelet Dis. (2018) 10:3-11. doi: 10.1177/1759720X17740075
7. Yazici Y, Paget SA. Elderly-onset rheumatoid arthritis. Rheum Dis Clin North Am. (2000) 26:517–26. doi: 10.1016/S0889-857X(05)70154-X
8. Suzuki T, Yoshida R, Hidaka Y, Seri Y. Proliferative synovitis of the shoulder bursae is a key feature for discriminating elderly onset rheumatoid arthritis mimicking polymyalgia rheumatica from polymyalgia rheumatica. Clin Med Insights Arthritis Musculoskelet Disord. (2017) 10:1179544117745851. doi: 10.1177/1179544117745851
9. Nesher G. Polymyalgia rheumatic—diagnosis and classification. J Autoimmunity. (2014) 48–49:76–78. doi: 10.1016/j.jaut.2014.01.016
10. Kermani TA, Warrington KJ. Polymyalgia rheumatica. Lancet (London, England). (2013) 381:63–72. doi: 10.1016/S0140-6736(12)60680-1
11. Lundberg IE, Sharma A, Turesson C, Mohammad AJ. An update on polymyalgia rheumatica. J Int Med. (2022) 292:717–32. doi: 10.1111/joim.13525
12. Camellino D, Giusti A, Girasole G, Bianchi G, Dejaco C. Pathogenesis, diagnosis and management of polymyalgia. Rheumatica Drugs Aging. (2019) 36:1015–26. doi: 10.1007/s40266-019-00705-5
13. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. (1987) 30:1205–13. doi: 10.1002/art.1780301102
14. Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. (2010) 42:508−14. doi: 10.1038/ng.582
15. Okada Y, Terao C, Ikari K, Kochi Y, Ohmura K, et al. Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat Genet. (2012) 44:511–6. doi: 10.1038/ng.2231
16. Kang CP, Lee H, Ju H, Cho H, Kang C, Bae SC. A functional haplotype of the PADI4 gene associated with increased rheumatoid arthritis susceptibility in Koreans. Arthritis Rheumatol. (2006) 54:90–6. doi: 10.1002/art.21536
17. McInnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet (London, England). (2017) 389:2328–37. doi: 10.1016/S0140-6736(17)31472-1
18. Gonzalez-Gay MA, Hajeer AH, Dababneh A, Makki R, Garcia-Porrua C, Thomson W, et al. Seronegative rheumatoid arthritis in elderly and polymyalgia rheumatica have similar patterns of HLA association. J Rheumatol. (2001) 28:122–5.
19. Alegria GC, Boukhlal S, Cornec D, Devauchelle-Pensec V. The pathophysiology of polymyalgia rheumatica, small pieces of a big puzzle. Autoimmun Rev. (2020) 19:102670. doi: 10.1016/j.autrev.2020.102670
20. Begovich AB, Carlton V, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Human Genet. (2004) 75:330−7. doi: 10.1086/422827
21. Serrano A, Márquez A, Mackie SL, Carmona FD, Solans R, Miranda-Filloy JA, et al. Identification of the PTPN22 functional variant R620W as susceptibility genetic factor for giant cell arteritis. Ann Rheum Dis. (2013) 72:1882–6. doi: 10.1136/annrheumdis-2013-203641
22. Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. (2003) 34:395–402. doi: 10.1038/ng1206
23. Cantaert T, Coucke P, De Rycke L, Veys EM, De Keyser F, Baeten D. Functional haplotypes of PADI4: relevance for rheumatoid arthritis specific synovial intracellular citrullinated proteins and anticitrullinated protein antibodies. Ann Rheum Dis. (2005) 64:1316–20. doi: 10.1136/ard.2004.033548
24. Thomas R, Wang W, Su D-M. Contributions of age-related thymic involution to immunosenescence and inflammaging. Immunity Ageing. (2020) 17:2. doi: 10.1186/s12979-020-0173-8
25. Pawlik A, Ostanek L, Brzosko I, Brzosko M, Masiuk M, Machalinski B, et al. The expansion of CD4+CD28- T cells in patients with rheumatoid arthritis. Arthritis Res Therapy. (2003) 5:R210–3. doi: 10.1186/ar766
26. Warrington KJ, et al. CD4+,CD28- T cells in rheumatoid arthritis patients combine features of the innate and adaptive immune systems. Arthritis Rheum. (2001) 44:13–20. doi: 10.1002/1529-0131(200101)44:1<13::AID-ANR3>3.0.CO;2-6
27. Fessler J, Husic R, Schwetz R, Lerchbaum E, Aberer F, Fasching P, et al. Senescent T-cells promote bone loss in rheumatoid arthritis. Front Immunol. (2018) 9:95. doi: 10.3389/fimmu.2018.00095
28. Bauer ME. Accelerated immunosenescence in rheumatoid arthritis: impact on clinical progression. Immunity Ageing. (2020) 17:6. doi: 10.1186/s12979-020-00178-w
29. Koetz K, Bryl E, Spickschen K, O'Fallon WM, Goronzy JJ, Weyand CC. T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci USA. (2000) 97:9203–8. doi: 10.1073/pnas.97.16.9203
30. Fessler J, Raicht A, Husic R, Ficjan A, Schwarz C, Duftner C, et al. Novel senescent regulatory T-cell subset with impaired suppressive function in rheumatoid arthritis. Front Immunol. (2017) 8:300. doi: 10.3389/fimmu.2017.00300
31. Steer SE, Williams FMK, Kato B, Gardner JP, Norman PJ, Hall MA, et al. Reduced telomere length in rheumatoid arthritis is independent of disease activity and duration. Ann Rheum Dis. (2007) 66:476–80. doi: 10.1136/ard.2006.059188
32. Dejaco C, Duftner C, Al-Massad J, Wagner AD, Park J-K, Fessler J, et al. NKG2D stimulated T-cell autoreactivity in giant cell arteritis and polymyalgia rheumatica Ann Rheum Dis. (2013) 72:1852–9. doi: 10.1136/annrheumdis-2012-201660
33. Edilova MI, Akram A, Abdul-Sater AA. Innate immunity drives pathogenesis of rheumatoid arthritis. Biomed J. (2021) 44:172–82. doi: 10.1016/j.bj.2020.06.010
34. Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. (2016) 17:34–40. doi: 10.1038/ni.3324
35. Udalova IA, Mantovani A, Feldmann M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat Rev Rheumatol. (2016) 12:472–85. doi: 10.1038/nrrheum.2016.91
36. Levescot A, Chang M, Schnell J, Maney N, Yan J, Bonet M, et al. IL-1β-driven osteoclastogenic Tregs accelerate bone erosion in arthritis. J Clini Investig. (2021) 131:18. doi: 10.1172/JCI141008
37. Aprahamian T, Takemura Y, Goukassian D, Walsh K. Ageing is associated with diminished apoptotic cell clearance in vivo. Clin Exp Immunol. (2008) 152:448–55. doi: 10.1111/j.1365-2249.2008.03658.x
38. Cutolo M, Montecucco CM, Cavagna L, Caporali R, Capellino S, Montagna P, et al. Serum cytokines and steroidal hormones in polymyalgia rheumatica and elderly-onset rheumatoid arthritis. Ann Rheum Dis. (2006) 65:1438–43. doi: 10.1136/ard.2006.051979
39. Chen D-Y, Hsieh T-Y, Chen Y-M, Hsieh C-W, Lan J-L, Lin F-J. Proinflammatory cytokine profiles of patients with elderly-onset rheumatoid arthritis: a comparison with younger-onset disease. Gerontology. (2009) 55:250–8. doi: 10.1159/000164393
40. Weyand CM, Hicok KC, Hunder GG, Goronzy JJ. Tissue cytokine patterns in patients with polymyalgia rheumatica and giant cell arteritis. Annals Int Med. (1994) 121:484–91. doi: 10.7326/0003-4819-121-7-199410010-00003
41. Hysa E, Gotelli E, Sammorì S, Cimmino MA, Paolino S, Pizzorni C, et al. Immune system activation in polymyalgia rheumatica: which balance between autoinflammation and autoimmunity? A systematic review. Autoimmun Rev. (2022) 21:102995. doi: 10.1016/j.autrev.2021.102995
42. Alvarez-Rodríguez L, Lopez-Hoyos M, Mata C, Marin MJ, Calvo-Alen J, Blanco R, Aurrecoechea E, Ruiz-Soto M, Martínez-Taboada VM. Circulating cytokines in active polymyalgia rheumatic. Ann Rheum Dis. (2010) 69:263–9. doi: 10.1136/ard.2008.103663
43. Estupiñán-Moreno E, Ortiz-Fernández L, Li T, Hernández-Rodríguez J, Ciudad L, Andrés-León E, et al. Methylome and transcriptome profiling of giant cell arteritis monocytes reveals novel pathways involved in disease pathogenesis and molecular response to glucocorticoids. Ann Rheum Dis. (2022) 81:9. doi: 10.1136/annrheumdis-2022-222156
44. van Sleen Y, Graver JC, Abdulahad WH, van der Geest KSM, Boots AMH, Sandovici M, et al. Leukocyte dynamics reveal a persistent myeloid dominance in giant cell arteritis and polymyalgia rheumatica. Front Immunol, (2019) 10:1981. doi: 10.3389/fimmu.2019.01981
45. Croia C, Bursi R, Sutera D, Petrelli F, Alunno A, Puxeddu I. One year in review 2019: pathogenesis of rheumatoid arthritis. Clin Exp Rheumatol. (2019) 37:347–57.
46. Montgomery RR, Shaw AC. Paradoxical changes in innate immunity in aging: recent progress and new directions. J Leukoc Biol. (2015) 98:937–43. doi: 10.1189/jlb.5MR0315-104R
47. Álvarez-Rodríguez L, López-Hoyos M, Calvo-Alén J, Aurrecoechea E, Villa I, Martínez-Taboada VM. Phagocyte dysfunction in polymyalgia rheumatica and other age-related, chronic, inflammatory conditions. J Leukocyte Biol. (2013) 94:1071–8. doi: 10.1189/jlb.0113034
48. Radstake TRDJ, Roelofs MF, Jenniskens YM, Oppers-Walgreen B, van Riel PLCM, Barrera P, et al. Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-gamma. Arthritis Rheumat. (2004) 50:3856–65. doi: 10.1002/art.20678
49. Yeo L, Lom H, Juarez M, Snow M, Buckley CD, Filer A, et al. Expression of FcRL4 defines a pro-inflammatory, RANKL-producing B cell subset in rheumatoid arthritis. Ann Rheum Dis. (2015) 74:928–35. doi: 10.1136/annrheumdis-2013-204116
50. Monnet E, Choy EH, McInnes I, Kobakhidze T, de Graaf K, Jacqmin P, et al. Efficacy and safety of NI-0101, an anti-toll-like receptor 4 monoclonal antibody, in patients with rheumatoid arthritis after inadequate response to methotrexate: a phase II study. Ann Rheum Dis, (2020) 79:316–23. doi: 10.1136/annrheumdis-2019-216487
51. Rodríguez LÁ, López-Hoyos M, Mata C, Fontalba A, Alen JC, Marín MJ, et al. Expression and function of toll-like receptors in peripheral blood mononuclear cells of patients with polymyalgia rheumatica and giant cell arteritis. Ann Rheum Dis. (2011) 70:1677–83. doi: 10.1136/ard.2010.140194
52. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
53. Takeuchi T, Yoshida H, Tanaka S. Role of interleukin-6 in bone destruction and bone repair in rheumatoid arthritis. Autoimmun Rev. (2021) 20:102884. doi: 10.1016/j.autrev.2021.102884
54. Kremer JM, Blanco R, Brzosko M, Burgos-Vargas R, Halland A-M, Vernon E, et al. Tocilizumab inhibits structural joint damage in rheumatoid arthritis patients with inadequate responses to methotrexate: results from the double-blind treatment phase of a randomized placebo-controlled trial of tocilizumab safety and prevention of structural joint damage at one year. Arthritis Rheumat. (2011) 63:609–21. doi: 10.1002/art.30158
55. Jiemy WF, Zhang A, Boots AMH, Heeringa P, Sandovici M, Diepstra A, et al. Expression of interleukin-6 in synovial tissue of patients with polymyalgia rheumatica. Ann Rheum Dis. (2022). doi: 10.1136/ard-2022-222873 [Epub ahead of print].
56. van der Geest KS, Abdulahad WH, Rutgers A, Horst G, Bijzet J, Arends S, Roffel MP, Boots AM, Brouwer E. Serum markers associated with disease activity in giant cell arteritis and polymyalgia rheumatic. Rheumatology (Oxford, England). (2015) 54:1397–402. doi: 10.1093/rheumatology/keu526
57. Devauchelle-Pensec V, Berthelot JM, Cornec D, Renaudineau Y, Marhadour T, Jousse-Joulin S, et al. Efficacy of first-line tocilizumab therapy in early polymyalgia rheumatica: a prospective longitudinal study. Ann Rheum Dis. (2016) 75:1506–10. doi: 10.1136/annrheumdis-2015-208742
58. Zhao SS, Gill D. Genetically proxied IL-6 receptor inhibition and risk of polymyalgia rheumatica. Ann Rheum Dis. (2022). doi: 10.1136/annrheumdis-2022-222578
59. Wehr P, Purvis H, Law S-C, Thomas R. Dendritic cells, T cells and their interaction in rheumatoid arthritis. Clin Exp Immunol. (2019) 196:12–27. doi: 10.1111/cei.13256
60. Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity. (2013) 38:336–48. doi: 10.1016/j.immuni.2012.10.018
61. Cumberbatch M, Dearman RJ, Kimber I. Influence of ageing on Langerhans cell migration in mice: identification of a putative deficiency of epidermal interleukin-1beta. Immunology. (2002) 105:466–77. doi: 10.1046/j.1365-2567.2002.01381.x
62. Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, et al. Loss of phagocytic and antigen cross-presenting capacity in aging dendritic cells is associated with mitochondrial dysfunction. J Immunol (Baltimore, Md.: 1950). (2015) 195:2624–32. doi: 10.4049/jimmunol.1501006
63. Ma-Krupa W, Jeon M-S, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J Exp Med. (2004) 199:173–83. doi: 10.1084/jem.20030850
64. Söderström K, Stein E, Colmenero P, Purath U, Müller-Ladner U, de Matos CT, et al. Natural killer cells trigger osteoclastogenesis and bone destruction in arthritis. Proc Natl Acad Sci USA. (2010) 107:13028–33. doi: 10.1073/pnas.1000546107
65. Fathollahi A, Samimi LN, Akhlaghi M, Jamshidi A, Mahmoudi M, Farhadi E. The role of NK cells in rheumatoid arthritis Inflammation Research: Official. Inflamm Res. (2021) 70:1063–73. doi: 10.1007/s00011-021-01504-8
66. Chalan P, Bijzet J, Kroesen B-J, Boots AMH, Brouwer E. Altered natural killer cell subsets in seropositive arthralgia and early rheumatoid arthritis are associated with autoantibody status. J Rheumatol. (2016) 43:1008–16. doi: 10.3899/jrheum.150644
67. Manser AR, Uhrberg M. Age-related changes in natural killer cell repertoires: impact on NK cell function and immune surveillance. Cancer Immunol Immunother. (2016) 65:417–26. doi: 10.1007/s00262-015-1750-0
68. Kidd P. Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev. (2003) 8:223–46.
69. Samson M, Audia S, Fraszczak J, Trad M, Ornetti P, Lakomy D, et al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum. (2012) 64:3788–98. doi: 10.1002/art.34647
70. Reitsema RD, Jiemy WF, Wekema L, Boots AMH, Heeringa P, Huitema MG, et al. Contribution of pathogenic T helper 1 and 17 cells to bursitis and tenosynovitis in polymyalgia rheumatica. Front Immunol. (2022) 13:943574. doi: 10.3389/fimmu.2022.943574
71. Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmunity Rev. (2014) 13:668–77. doi: 10.1016/j.autrev.2013.12.004
72. Jiang X, Wang S, Zhou C, Wu J, Jiao Y, Lin L, et al. Comprehensive TCR repertoire analysis of CD4(+) T-cell subsets in rheumatoid arthritis. J Autoimmun. (2020) 109:102432. doi: 10.1016/j.jaut.2020.102432
73. Jin S, Chen H, Li Y, Zhong H, Sun W, Wang J, et al. Maresin 1 improves the Treg/Th17 imbalance in rheumatoid arthritis through miR-21. Ann Rheum Dis. (2018) 77:1644–52. doi: 10.1136/annrheumdis-2018-213511
74. Meyer A, Wettikind P, Kotschenreuther K, Schiller J, TresckowJ, Haak T, et al. Regulatory T cell frequencies in patients with rheumatoid arthritis are increased by conventional and biological DMARDs but not by JAK inhibitors. Ann Rheum Dis. (2021) 80:e196. doi: 10.1136/annrheumdis-2019-216576
75. Veronika Groh AB, El-Gabalawy H, Lee Nelson J, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. PANS. (2003) 100:9452–7. doi: 10.1073/pnas
76. Andersson AK, Sumariwalla PF, McCann FE, Amjadi P, Chang C, McNamee K, et al. Blockade of NKG2D ameliorates disease in mice with collagen-induced arthritis: a potential pathogenic role in chronic inflammatory arthritis. Arthritis Rheum. (2011) 63:2617–29. doi: 10.1002/art.30460
77. Killock D. Experimental arthritis: NKG2D: a potential therapeutic target in RA? Nat Rev Rheumatol. (2011) 7:438. doi: 10.1038/nrrheum.2011.91
78. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. (2017) 542:110–4. doi: 10.1038/nature20810
79. Lu J, Wu J, Xia X, Peng H, Wang S. Follicular helper T cells: potential therapeutic targets in rheumatoid arthritis. CMLS. (2021) 78:5095–106. doi: 10.1007/s00018-021-03839-1
80. Matsumoto K, Suzuki K, Yoshimoto K, Seki N, Tsujimoto H, Chiba K, et al. Significant association between clinical characteristics and changes in peripheral immuno-phenotype in large vessel vasculitis. Arthritis Res Therapy. (2019) 21:304. doi: 10.1186/s13075-019-2068-7
81. Jiang Q, Yang G, Xiao F, Xie J, Wang S, Lu L, et al. Role of Th22 cells in the pathogenesis of autoimmune diseases. Front Immunol. (2021) 12:688066. doi: 10.3389/fimmu.2021.688066
82. Zhang L, Li J-M, Liu X-G, Ma D-X, Hu N-W, Li Y-G, et al. Elevated Th22 cells correlated with Th17 cells in patients with rheumatoid arthritis. J Clini Immunol. (2011) 31:606–14. doi: 10.1007/s10875-011-9540-8
83. Zerbini A, Muratore F, Boiardi L, Ciccia F, Bonacini M, Belloni L, et al. Increased expression of interleukin-22 in patients with giant cell arteritis. Rheumatology (Oxford, England). (2018) 57:64–72. doi: 10.1093/rheumatology/kex334
84. Dong X, Zheng Z, Lin P, Fu X, Li F, Jiang J, et al. ACPAs promote IL-1β production in rheumatoid arthritis by activating the NLRP3 inflammasome. Cellular Mol Immunol. (2020) 17:261–71. doi: 10.1038/s41423-019-0201-9
85. Lopez-Hoyos M, de Alegria CR, Blanco R, Crespo J, Peña M, Rodriguez-Valverde V, et al. Clinical utility of anti-CCP antibodies in the differential diagnosis of elderly-onset rheumatoid arthritis and polymyalgia rheumatica. Rheumatology (Oxford, England). (2004) 43:655–7. doi: 10.1093/rheumatology/keh143
86. Volkov M, van Schie KA, van der Woude D. Autoantibodies and B Cells: The ABC of rheumatoid arthritis pathophysiology. Immunol Rev. (2020) 294:148–63. doi: 10.1111/imr.12829
87. Thorarinsdottir K, Camponeschi A, Johnsson C, Onnhelm K, Nilsoon J, Forslind J, et al. CD21 B cells associate with joint damage in rheumatoid arthritis patients. Scandinavian J Immunol. (2019) 90:e12792. doi: 10.1111/sji.12792
88. Tak PP, Doorenspleed M, Hair M, Klarenbeek PL, Beers-Tas M, Kampen A, et al. Dominant B cell receptor clones in peripheral blood predict onset of arthritis in individuals at risk for rheumatoid arthritis. Ann Rheum Dis. (2017) 76:1924–30. doi: 10.1136/annrheumdis-2017-211351
89. Graver JC, Abdulahad W, van der Geest KSM, Heeringa P, Boots AMH, Brouwer E, et al. Association of the CXCL9-CXCR3 and CXCL13-CXCR5 axes with B-cell trafficking in giant cell arteritis and polymyalgia rheumatica. J Autoimmun. (2021) 123:102684. doi: 10.1016/j.jaut.2021.102684
90. Alegria GC, Devauchelle-Pensec V, Renaudineau Y, Saraux A, Pers J-O, Cornec D. Correction of abnormal B-cell subset distribution by interleukin-6 receptor blockade in polymyalgia rheumatica. Rheumatology (Oxford). (2017) 56:1401–6. doi: 10.1093/rheumatology/kex169
91. Meliconi R, Pulsatelli L, Uguccioni M, Salvarani C, Macchioni P, Melchiorri C, et al. Leukocyte infiltration in synovial tissue from the shoulder of patients with polymyalgia rheumatica Quantitative analysis and influence of corticosteroid treatment. Arthritis Rheum. (1996) 39:1199–207. doi: 10.1002/art.1780390719
Keywords: elderly-onset rheumatoid arthritis, polymyalgia rheumatica, rheumatoid arthritis, pathogenesis, inflammation
Citation: Wu J, Yang F, Ma X, Lin J and Chen W (2023) Elderly-onset rheumatoid arthritis vs. polymyalgia rheumatica: Differences in pathogenesis. Front. Med. 9:1083879. doi: 10.3389/fmed.2022.1083879
Received: 29 October 2022; Accepted: 28 December 2022;
Published: 12 January 2023.
Edited by:
Cong-Qiu Chu, Oregon Health and Science University, United StatesReviewed by:
Dai Lie, Sun Yat-sen Memorial Hospital, ChinaWenfeng Tan, Nanjing Medical University, China
Copyright © 2023 Wu, Yang, Ma, Lin and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin Lin,  bGluamluemp1QHpqdS5lZHUuY24=; Weiqian Chen, Y3dxNjc4QHpqdS5lZHUuY24=
bGluamluemp1QHpqdS5lZHUuY24=; Weiqian Chen, Y3dxNjc4QHpqdS5lZHUuY24=
†These authors have contributed equally to this work