 Hye Eun Park
Hye Eun Park Hanna Oh
Hanna Oh Jea-Hyun Baek
Jea-Hyun Baek- School of Life Science, Handong Global University, Pohang, South Korea
Rheumatoid arthritis (RA) is a chronic autoimmune disease with a multifaceted etiology, which primarily affects and results in the deterioration of the synovium of patients. While the exact etiology of RA is still largely unknown, there is growing interest in the cytokine interleukin-34 (IL-34) as a driver or modulator of RA pathogenesis on the grounds that IL-34 is drastically increased in the serum and synovium of RA patients. Several studies have so far revealed the relationship between IL-34 levels and RA disease progression. Nevertheless, the significance and role of IL-34 in RA have remained ambiguous, as illustrated by two most recent studies, which reported contrasting effects of genetic IL-34 deletion in RA. Of note, IL-34 is a macrophage growth factor and is increasingly perceived as a master regulator of T-cell responses in RA via macrophage-dependent as well as T cell-intrinsic mechanisms. In this regard, several studies have demonstrated that IL-34 potentiates helper T-cell (Th) responses in RA, whereas studies also suggested that IL-34 alleviates synovial inflammation, potentially by inducing regulatory T-cells (Treg). Herein, we provide an overview of the current understanding of IL-34 involvement in RA and outline IL-34-mediated mechanisms in regulating T-cell responses in RA.
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease characterized by synovial inflammation attributed to an abundance of inflammatory cytokines and leukocytes, leading to the destruction of surrounding bone and cartilage (1–4). Symptoms of RA include joint pain, swelling and/or stiffness, and rheumatoid nodules under the skin (5). RA synovial inflammation involves self-reactive immune responses triggered by both innate and adaptive immune cells (e.g., neutrophils, macrophages, and T lymphocytes) (6–8). Notably, the inflamed synovium is continuously infiltrated by neutrophils, which are cleared by synovial macrophages (7–9). In general, activated macrophages may have versatile roles being ‘healers’ and ‘destroyers’ and are commonly divided into (1) classically (M1) and (2) alternatively activated macrophages (M2) being pro-inflammatory and anti-inflammatory cells, respectively (9). In the RA synovium, macrophages have been demonstrated to induce the proliferation of fibroblast-like synoviocytes (FLS), recruit monocytes, and activate T helper (Th) cells (e.g., Th17) (10, 11), collectively perpetuating the cycle of joint destruction in RA (12, 13). In contrast, other studies showed that macrophages counteract inflammation and clinical symptoms in RA (9). Overall, the pathological roles of macrophages in RA are enigmatic and need to be further clarified.
Interleukin-34 (IL-34) is a macrophage growth factor, which is detected at high levels in the synovial fluid (SF), synovial cells, and serum of RA patients (14, 15). Synovial IL-34 levels are implicated in leukocyte accumulation (16). IL-34 is also proposed as a biomarker of RA remission due to a strong correlation between IL-34 and the relapse of RA after the termination of the anti-rheumatic drug treatment (4, 17–23). While IL-34 presence in different states of RA pathology has been described, the immunological role of IL-34 has not been yet clearly delineated. Although systemic IL-34 was shown to exacerbate RA (11, 24), the deleterious role of IL-34 in RA has remained controversial through various studies including the latest finding suggesting that IL-34 is beneficial in the effector phase of RA (9). Whether IL-34 takes on either a pro- or anti-inflammatory role, is ambiguous and may depend on pathologic conditions as studies of certain diseases (e.g., acute kidney injury, inflammatory bowel disease, lupus nephritis, Sjögren's syndrome) characterized IL-34 as a pro-inflammatory factor (25–29), while other clinical settings (e.g., atopic dermatitis, autoimmune hepatitis, lung cancer, sepsis) identified IL-34 as a mediator of anti-inflammatory responses (30–33). Recently, accumulating evidence has suggested that IL-34-dependent pathologic mechanisms in RA involve T cells, and IL-34 substantially controls the nature of T-cell responses in RA. However, the current conceptualization of IL-34-mediated T-cell regulation in RA is as vague as the proposed immunological roles of IL-34 (9, 11, 34). Therefore, there is a strong need to amalgamate information surrounding IL-34 involvement in RA pathogenesis, especially regarding how IL-34 integrates into the mechanisms of T-cell activation noted in RA.
IL-34 in RA
IL-34 is a cytokine, which binds to multiple receptors. The colony-stimulating factor-1 receptor (CSF-1R) is the first identified and best-known receptor for IL-34. CSF-1R is expressed on the surfaces of mononuclear phagocytes (e.g., macrophages) and is activated by two known ligands: CSF-1 and IL-34 (Figure 1) (35). CSF-1R is crucial for the differentiation, polarization, activation, and maintenance of mononuclear phagocytes (2, 36–41). IL-34 and CSF-1 may have non-redundant roles in RA as studies have reported unique pathological properties of IL-34 that are independent of CSF-1 (25, 37). Interestingly, IL-34 is more drastically upregulated in human and murine RA as compared to CSF-1, underlining a predominant role of IL-34 in CSF-1R signaling during the pathogenesis of RA (9). Studies have investigated the effects of CSF-1R inhibition in RA. A study, using both human and murine RA models, reported suppression of RA inflammation when CSF-1 and IL-34 were simultaneously inhibited, while individual neutralization of either factor did not demonstrate beneficial effects in RA models (36). Nevertheless, numerous studies pointed to the direct association between IL-34 and RA pathology, although the effects of IL-34 on RA were not always consistent (2, 4, 9, 11, 19, 24, 41–44). For example, a very recent study demonstrated that the genetic deletion of IL-34 aggravates the effector phase of RA, implicating a protective role of IL-34 in RA (9), while another study showed that the genetic deletion of IL-34 mitigates RA pathology (11).
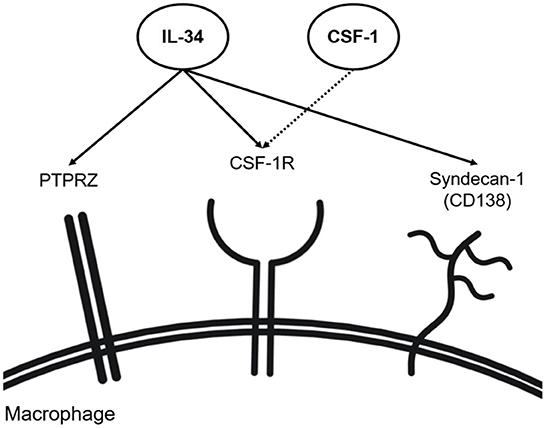
Figure 1. IL-34 receptors (including co-receptors). IL-34 has shown the capability of binding to CSF-1R alongside CSF-1 on macrophages. Independent of CSF-1, IL-34 has shown binding to syndecan-1 and PTPRZ found on the surface of macrophages.
IL-34 has been recently associated with metabolism in RA-associated macrophages. IL-34 may metabolically reprogram macrophages toward a hyper-glycolytic M1-like phenotype upregulating glucose transporter 1 (GLUT1), mammalian target of rapamycin (mTOR), and hypoxia-induced factor 1α (HIF-1α) in the RA synovium (11, 43); however, we need further clarification on whether this is a general mechanism of IL-34, applicable to any physiological or pathological condition.
CSF-1R is not only linked to macrophage activity in RA but is also expressed by RA FLS (4). Increased IL-6 production by the RA FLS was noted during stimulation with IL-34 in a CSF-1R-dependent manner (4). Also, synovial fibroblasts may conceivably function in the RA synovium through IL-34-mediated pathways as evidenced by a study showing that IL-34 supports synovial fibroblast survival by the activation of the STAT3-miR-21 axis (17).
Not only do FLS or synovial fibroblasts respond to exogenous IL-34 expressing relevant T-cell surface receptors, but it is also likely that they are the major source of IL-34 in RA SF. Indeed, in the presence of TNF-α, FLS upregulate the expression of IL-34 (20). Similarly, a different study showed that synovial fibroblasts increase the expression of IL-34 after exposure to TNF-α and IL-1β (45). Of note, synovial macrophages likely produce and provide TNF-α and IL-1β to synovial fibroblasts (46, 47). Synovial fibroblasts may, in turn, express IL-34 and ultimately stimulate Th17 cells to express IL-17 (48).
In addition to the activation of macrophages, T cells, and FLS, IL-34 may promote synovial inflammation by the initiation of osteoclastogenesis (43). Of note, osteoclastogenesis and osteoporosis are prominent in synovial inflammation (49–52). Indeed, the imbalance between M1 and M2 macrophages in RA patients has been linked to increased occurrence of osteoclastogenesis in RA patients (16). Importantly, IL-34 is known to directly promote osteoclast formation like CSF-1 (53) and may upregulate the expression of the receptor activator of nuclear factor (NF)-κB-ligand (RANKL) for the progression of osteoclastogenesis in RA (21, 53–55).
In addition to CSF-1R, syndecan-1 (CD138) and protein tyrosine phosphatase receptor type ζ1 (PTPRZ1) bind IL-34 (Figure 1). Syndecan-1 is a cell surface proteoglycan functioning as a co-receptor to various cell surface receptors (56) and, besides, a regulatory factor involved in cell adhesion and cell migration (49–52). Syndecan-1 is present in various human subsets of monocytes and macrophages (57). On macrophages, syndecan-1 enhances the activation of IL-34 receptors and increases their motility (58). In line with this, another study showed that the migration of syndecan-1-expressing monocytes and macrophages is induced with IL-34 and reversely inhibited by anti-syndecan-1 neutralizing antibodies (56). Syndecan-1 has been also associated with various diseases, such as aortic aneurysms (52), cancer (59), dermatitis (60), and multiple myeloma (61). Expression of both IL-34 and syndecan-1 was reported to be elevated in RA synovium (11, 43). As such, the syndecan-1 expression for the binding of IL-34 has recently emerged as a molecule of interest in RA. Although the exact mechanism of the interaction between IL-34 and syndecan-1 remains elusive, it was shown that the lack of syndecan-1 impairs macrophage differentiation, ultimately attenuating murine IL-34-induced arthritis (11, 43). Syndecan-1 was detected in the mononuclear infiltrates of the sub-lining layer in the synovium of RA patients, thus posing the possible role of syndecan-1 in RA pathophysiology in the migration and maintenance of mononuclear phagocytes in the synovium (62). A study detecting syndecan-1 mRNA expression within the joints in a murine RA model showed that syndecan-1 mRNA expression levels are highest in aging knee joints, implying its role in age-related RA (63). Besides impacting macrophage functions and synovial inflammation, syndecan-1 induces osteoclastogenesis, resulting in the degradation of joint and bone structures in RA patients. Syndecan-1 levels are positively correlated with levels of RANKL in murine serum while being negatively correlated with serum osteoprotegerin (OPG) (64). The study linked increased syndecan-1 levels with the progression of osteoclastogenesis, suggesting syndecan-1 as a novel modulator of the RANKL and OPG balance in RA.
Independently of CSF-1R, IL-34 signals via PTPRZ1 (65) (Figure 1). Human monocytes have been demonstrated to express PTPRZ1 (57). PTPRZ1 involvement has been linked to various inflammatory diseases such as inflammatory bowel disease (IBD) and lupus nephritis, where PTPRZ1 expression was found to correlate with IL-34, CSF1, and CSF-1R expression (9, 29, 66). The most recent study reported that IL-34-dependent PTPRZ1 activation ameliorates the effector phase of RA (9). In this study, PTPRZ signaling was shown to facilitate the removal of apoptotic neutrophils by macrophages preventing further escalation of synovial inflammation in RA conditions (9). However, the above-mentioned study (using the K/BxN-induced model) also reported a protective role of IL-34 in RA, contrasting with other studies (11, 43). The discrepancy may be due to the difference in how RA was induced (K/BxN-induced vs. collagen-induced murine RA model). Thus, further investigation on whether PTPRZ1 activation is protective throughout different models of RA is needed.
IL-34 and Th17 cells
The positive correlation between the severity of RA and the number of synovial macrophages has been well-documented (67, 68) (Table 1). Certain alleles of class II human leukocyte antigen (HLA) antigen (e.g., DRB1-β chain) are associated with RA via increased disease occurrence and inflammatory activity (72, 73). This is strong evidence for the involvement of T helper (Th) cells in the pathogenesis of RA. Although the mechanisms by which Th cell activation occurs in the RA microenvironment are yet to be fully understood, a positive correlation between Th population levels and RA disease severity has been noted (74). Of the Th subsets, a subset that is predominantly associated with RA pathogenesis is the T helper 17 (Th17) cell.

Table 1. Effects of IL-34 on T lymphocyte activation.
Previously, researchers have pinpointed Th17 cell differentiation, recruitment, and activation as a driver of the inflammatory response in RA pathogenesis (75). The impact of Th17 cells in RA pathogenesis is supported by the increased numbers of Th17 cells in RA patients as compared to healthy people (76). Earlier studies have shown a bias for Th17 cell polarization of naïve CD4+ T cells, which impose an inflammatory response in the surrounding environment (77). In addition, Th17 cells are being identified as osteoclastogenic subsets in autoimmune arthritis through cytokine secretion and RANKL expression (77).
Exploring the therapeutic potential of Th17 cell inhibition in RA has been studied previously, where IL-25 was found to suppress Th17 immune responses for attenuation of RA in a collagen-induced model (78). An in vitro study found that IL-34-induced macrophages possessed the capacity of converting certain memory CD4+ T cells into CCR4+CCR6+CD161+ Th17 cells, supporting the possibility that Th17 cells observed in RA disease conditions are activated through IL-34-induced macrophages (41). Congruently, a different study demonstrated that IL-34 participates in the differentiation of peripheral blood mononuclear cells (PBMC) to Th17 cells in RA patients (2). Stimulation of PBMC from RA patients with IL-34 showed an increased frequency of Th17 cells with upregulated expression of IL-17 (19). Moreover, such effects of IL-34 found in RA patients went unnoted from samples of healthy controls, supporting the notion that IL-34 effects on Th17 cell differentiation could be RA-specific (2). Human monocytes (THP-1) activated by IL-34 led to the secretion of IL-6 increasing Th17 cell numbers in RA (42). In line with this, IL-34 expression has shown involvement in Th1 and Th17 cell polarization through the expansion of certain macrophage populations in murine models (43). The increase of Th17 cell numbers has also been possible through IL-34-stimulated IL-6 secretion by RA FLS, such results are supported by the attenuation of Th17 cell production by IL-34-dependent RA FLS through the administration of IL-6 antagonists (4, 44). Similarly, myeloid-related protein, also highly abundant in RA SF, has been found to upregulate IL-6 production by FLS promoting Th17 differentiation (79). There is also evidence that IL-17 directly acts on FLS. Increased proliferation of RA FLS and inhibition of RA FLS apoptosis by IL-34 was neutralized when IL-17 inhibitor plumbagin was introduced (44). Th17 cells and IL-17 may participate in RA FLS survival by inhibiting the mitochondrial pathway of apoptosis (80). These collective results suggest a positive correlation between IL-34 and Th17 cell inflammatory response in RA.
A collagen-induced murine arthritis model study with additional IL-34 injection led to increased TNF-α and IL-17 mRNA expression in the synovial tissues, indicating a proinflammatory role for IL-34 via IL-17 production entailing aggravation of arthritis (24). This regulation of IL-17 production by IL-34 could also play a role in osteoclastogenesis (55). The same study reported that the enhanced expression of RANKL and OPG by FLS and PBMCs of RA patients by IL-34 was secondhandedly mediated by IL-17. This is in line with the known function of IL-17 as an osteoclastogenic factor for subsequent osteoporosis (81).
IL-34 and Tregs
Currently, researchers have also been investigating the potential of IL-34 in the regulation of balancing different T-cell populations, with Tregs being proposed as another T-cell subset mediated by IL-34 stimulation (Table 1). Tregs are a subpopulation of T cells that maintain homeostasis and self-tolerance through the suppression of different immune responses, such as those induced by Th17 cells (82). Tregs are known to be critical for the prevention of autoimmunity, as observed in RA, possibly by inhibiting the cytokine production by other pro-inflammatory T-cell subsets, as such the immunomodulatory role of Tregs has been studied in different clinical conditions (70, 82, 83). Concerning the activity of RA Th17 cells, Treg-mediated suppression of Th17 activity was studied in an in vitro would healing model and a murine autoimmune neuroinflammation model (84, 85). Naïve and memory Tregs were able to suppress Th17 cells ex vivo, and also in the wound-healing model, Tregs suppressed the Th17 cell antifibrotic effects (84). In an experimental multiple sclerosis model, Tregs limited the access of Th17 cells to APCs and suppressed Th17 cell calcium signaling (85). While Treg involvement in the anti-inflammatory activity and resolution of RA can be inferred as higher numbers of Tregs and their inhibitory activity have been detected in clinical remission states of RA (86), reports have observed the immunoregulatory role of Tregs to be compromised in RA patients (86).
Recently, the imbalance between Th17 and Treg populations, which would potentiate an inflammatory response in the surrounding microenvironment, is being studied as one determinant of disease progression. A study reported an increased frequency of peripheral Th17 cells and elevated Th17 cytokines, such as IL-17, IL-23, IL-6, and TNF-α in RA patients compared to healthy individuals (87). This contrasted with the significant decrease in both the frequency of Tregs and respective Treg-related cytokine [e.g., transforming growth factor-beta (TGF-β)] levels (11, 87). Meyer et al. (11) found that the TGF-β expression in the synovium is reduced in IL-34 and syndecan-1 KO mice, indicating that the synovial TGF-β expression is IL-34-dependent (11). Likewise, an investigation of Th1, Th17, and Treg populations in the bone marrow of RA patients reported a significant increase in Th1 and Th17 cell frequencies, and, in parallel, a significant decrease in Tregs (88). Reestablishing the balance between Treg and Th17 cell populations in RA by inhibiting NLRP3 inflammasome has exhibited promising results, which alludes to a potential therapeutic role of IL-34 for rebalancing Treg and Th17 cell populations (89). The amelioration of the proinflammatory response in a murine collagen-induced arthritis model was conducted through IL-12 suppression for suppression of Th17 cells and increase of the Treg population, which provides support for Treg population expansion in the treatment of RA (90).
IL-34 has been proposed as a Treg-targeting cytokine in a study revolving around the transplant tolerance activity of IL-34-expanded Tregs (69). In line with this, a most recently published study reported that IL-34 deficiency impairs the suppressive function of CD4+ Tregs in rats and makes mice more prone to autoimmunity (71). Human macrophage exposure to IL-34 was verified to increase the numbers of CD8+ and CD4+ FOXP3+ Tregs. These specific Treg populations, as compared to Tregs not expanded via IL-34 addition, demonstrated improved suppressive action against anti-graft immune responses (69). Additionally, a study of graft-versus-host disease identified highly suppressive human Tregs producing IL-34 for their immunosuppressive activity (70). Notwithstanding, a study noted that IL-34 stimulation of PBMC isolated from RA patients did not affect differentiation or cytokine secretion of Tregs, despite IL-34 exposure of RA PBMCs increasing Th17 populations and IL-17 expression levels (2). Such results demonstrate the need for further investigation into the mechanisms by which IL-34 engages in the differentiation, proliferation, and activation of Tregs in RA.
Discussion
RA is an inflammatory autoimmune disease with a multifaceted etiology. While certain aspects of RA etiology and pathogenesis are becoming better understood, there is still much to learn for the development of improved therapeutic approaches. When observing the inflammatory response of the RA synovium, a positive correlation between macrophages and RA disease severity is well-observed (7, 8). Additionally, different subsets of T cells are being attributed as RA biomarkers, whether due to their up- or down-regulation (87). Furthermore, these immune cells are shown to participate in RA-associated osteoclastogenesis (16, 91).
Increased levels of IL-34 are being detected in the serum and SF of RA patients (4, 17–23). As reports of IL-34 involvement in macrophage and T-cell polarization are being released, IL-34 presents itself as a promising therapeutic target for immune response and osteoclastogenesis regulation in RA. However, the mechanisms, by which IL-34 either aggravates or mitigates RA, are not clearly understood. Notably, two recent independent studies using IL-34 knockout mice showed contrasting effects of IL-34 on RA (9, 11). Such conflicting observations may have resulted from different pathological mechanisms given that one study used K/BxN serum injection (autoantibody transfer) (9) and the other collagen immunization (11) to induce RA in mice, recapitulating hypersensitivity reactions type II/III (antibody-mediated) and type IV (cell-mediated), respectively. As IL-34 is highly prominent in RA patients, there are growing expectations for its potential as biomarker and therapeutic target in RA. IL-34 is a promising biomarker (92). However, as RA etiology and pathogenesis are not uniform in humans, continuous investigations into the precise roles of IL-34-induced synovial macrophages in T-cell responses and careful and thorough assessment of IL-34 as a target in RA subsets are needed for future therapeutic strategies.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study was funded by the 2022 University Innovation Support Project at Handong Global University led by the Ministry of Education of the South Korea and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education of the South Korea (2021R111A3059820).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
APC, antigen-presenting cell; CSF-1, colony-stimulating factor-1; FLS, fibroblast-like synoviocytes; IL-34, interleukin-34; Janus protein tyrosine kinase; OPG, osteoprotegerin; PBMC, peripheral blood mononuclear cells; PTPRZ1, protein tyrosine phosphatase receptor type ζ1; RA, rheumatoid arthritis; RANK, receptor activator of nuclear factor-κB; RANKL, RANK ligand; SF, synovial fluid; Th, T helper; Treg, regulatory T cell; TNF, tumor necrosis factor.
References
1. Baek JH. Interleukin-34: regulator of T lymphocytes in rheumatoid arthritis. Biomed J Sci Tech Res. (2019) 20:15062–6. doi: 10.26717/BJSTR.2019.20.003458
2. Li X, Lei Y, Gao Z, Zhang B, Xia L, Lu J, et al. Effect of IL-34 on T helper 17 cell proliferation and IL-17 secretion by peripheral blood mononuclear cells from rheumatoid arthritis patients. Sci Rep. (2020) 10:22239. doi: 10.1038/s41598-020-79312-z
3. McInnes IB, Buckley CD, Isaacs JD. Cytokines in rheumatoid arthritis – shaping the immunological landscape. Nat Rev Rheumatol. (2016) 12:63–8. doi: 10.1038/nrrheum.2015.171
4. Wang B, Ma Z, Wang M, Sun X, Tang Y, Li M, et al. IL-34 upregulated Th17 production through increased IL-6 expression by rheumatoid fibroblast-like synoviocytes. Mediators Inflamm. (2017) 2017:1567120. doi: 10.1155/2017/1567120
5. Bullock J, Rizvi SAA, Saleh AM, Ahmed SS, Do DP, Ansari RA, et al. Rheumatoid arthritis: a brief overview of the treatment. Med Princ Pract. (2018) 27:501–7. doi: 10.1159/000493390
6. Weyand CM. New insights into the pathogenesis of rheumatoid arthritis. Rheumatology. (2000) 39:3–8. doi: 10.1093/oxfordjournals.rheumatology.a031491
7. Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR, et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat Med. (2020) 26:1295–306. doi: 10.1038/s41591-020-0939-8
8. Udalova IA, Mantovani A, Feldmann M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat Rev Rheumatol. (2016) 12:472–85. doi: 10.1038/nrrheum.2016.91
9. González-Sánchez HM, Baek JH, Weinmann-Menke J, Ajay AK, Charles JF, Noda M, et al. IL-34 and protein-tyrosine phosphatase receptor type-zeta-dependent mechanisms limit arthritis in mice. Lab Invest. (2022) 14:1–3. doi: 10.1038/s41374-022-00772-0
10. Paoletti A, Rohmer J, Ly B, Pascaud J, Rivière E, Seror R, et al. Monocyte/macrophage abnormalities specific to rheumatoid arthritis are linked to miR-155 and are differentially modulated by different TNF inhibitors. J Immunol. (2019) 203:1766–75. doi: 10.4049/jimmunol.1900386
11. Meyer A, Sienes R, Zanotti B, van Raemdonck K, Palasiewicz K, Mass DP, et al. Dysregulation of IL-34 ligation to SDC-1 mitigates collagen-induced arthritis. Cell Mol Immunol. (2022) 8:1–3. doi: 10.1038/s41423-022-00886-x
12. Yang X, Chang Y, Wei W. Emerging role of targeting macrophages in rheumatoid arthritis: focus on polarization, metabolism and apoptosis. Cell Prolif. (2020) 53:e12854. doi: 10.1111/cpr.12854
13. Kinne RW, Bräuer R, Stuhlmüller B, Palombo-Kinne E, Burmester GR. Macrophages in rheumatoid arthritis. Arthritis Res. (2000) 2:189–202. doi: 10.1186/ar86
14. Llorente I, García-Castañeda N, Valero C, González-Álvaro I, Castañeda S. Osteoporosis in rheumatoid arthritis: dangerous liaisons. Front Med. (2020) 7:601618. doi: 10.3389/fmed.2020.601618
15. Tomizawa T, Ito H, Murata K, Hashimoto M, Tanaka M, Murakami K, et al. Distinct biomarkers for different bones in osteoporosis with rheumatoid arthritis. Arthritis Res Ther. (2019) 21:174. doi: 10.1186/s13075-019-1956-1
16. Fukui S, Iwamoto N, Takatani A, Igawa T, Shimizu T, Umeda M, et al. M1 and M2 monocytes in rheumatoid arthritis: a contribution of imbalance of M1/M2 monocytes to osteoclastogenesis. Front Immunol. (2017) 8:1958. doi: 10.3389/fimmu.2017.01958
17. Yang S, Jiang S, Wang Y, Tu S, Wang Z, Chen Z. Interleukin 34 upregulation contributes to the increment of MicroRNA 21 expression through STAT3 activation associated with disease activity in rheumatoid arthritis. J Rheumatol. (2016) 43:1312–9. doi: 10.3899/jrheum.151253
18. Zhang F, Ding R, Li P, Ma C, Song D, Wang X, et al. Interleukin-34 in rheumatoid arthritis: potential role in clinical therapy. Int J Clin Exp Med. (2015) 8:7809–15.
19. Tian Y, Shen H, Xia L, Lu J. Elevated serum and synovial fluid levels of interleukin-34 in rheumatoid arthritis: possible association with disease progression via interleukin-17 production. J Interferon Cytokine Res. (2013) 33:398–401. doi: 10.1089/jir.2012.0122
20. Hwang SJ, Choi B, Kang SS, Chang JH, Kim YG, Chung YH, et al. Interleukin-34 produced by human fibroblast-like synovial cells in rheumatoid arthritis supports osteoclastogenesis. Arthritis Res Ther. (2012) 14:R14. doi: 10.1186/ar3693
21. Moon SJ, Hong YS, Ju JH, Kwok SK, Park SH, Min JK. Increased levels of interleukin 34 in serum and synovial fluid are associated with rheumatoid factor and anticyclic citrullinated peptide antibody titers in patients with rheumatoid arthritis. J Rheumatol. (2013) 40:1842–9. doi: 10.3899/jrheum.130356
22. Chang SH, Choi BY, Choi J, Yoo JJ, Ha YJ, Cho HJ, et al. Baseline serum interleukin-34 levels independently predict radiographic progression in patients with rheumatoid arthritis. Rheumatol Int. (2015) 35:71–9. doi: 10.1007/s00296-014-3056-5
23. Selim ZI, Rashad SM, Abdelaziz MM, Salah S, Fouad DM, Hetta HF. Interleukin-34 in the serum and synovial fluid of rheumatoid arthritis patients: relation to disease activity and radiographic damage. Aktuelle Rheumatologie. (2019) 44:65–70. doi: 10.1055/a-0606-4982
24. Zhang L, Cui M, Ding L, Xia L, Lu J, Shen H. Interleukin-34 aggravates the severity of arthritis in collagen-induced arthritis mice by inducing interleukin-17 production. J Interferon Cytokine Res. (2018) 38:221–5. doi: 10.1089/jir.2017.0095
25. Baek JH, Zeng R, Weinmann-Menke J, Valerius MT, Wada Y, Ajay AK, et al. IL-34 mediates acute kidney injury and worsens subsequent chronic kidney disease. J Clin Invest. (2015) 125:3198–214. doi: 10.1172/JCI81166
26. Ciccia F, Alessandro R, Rodolico V, Guggino G, Raimondo S, Guarnotta C, et al. IL-34 is overexpressed in the inflamed salivary glands of patients with Sjogren's syndrome and is associated with the local expansion of pro-inflammatory CD14(bright)CD16+ monocytes. Rheumatology. (2013) 52:1009–17. doi: 10.1093/rheumatology/kes435
27. Piao C, Wang X, Peng S, Guo X, Zhao H, He L, et al. IL-34 causes inflammation and beta cell apoptosis and dysfunction in gestational diabetes mellitus. Endocr Connect. (2019) 8:1503–12. doi: 10.1530/EC-19-0436
28. Franzè E, Monteleone I, Cupi ML, Mancia P, Caprioli F, Marafini I, et al. Interleukin-34 sustains inflammatory pathways in the gut. Clin Sci. (2015) 129:271–80. doi: 10.1042/CS20150132
29. Wada Y, Gonzalez-Sanchez HM, Weinmann-Menke J, Iwata Y, Ajay AK, Meineck M, et al. IL-34-Dependent intrarenal and systemic mechanisms promote lupus nephritis in MRL-Fas lpr mice. J Am Soc Nephrol. (2019) 30:244–59. doi: 10.1681/ASN.2018090901
30. Esaki H, Ewald DA, Ungar B, Rozenblit M, Zheng X, Xu H, et al. Identification of novel immune and barrier genes in atopic dermatitis by means of laser capture microdissection. J Allergy Clin Immunol. (2015) 135:153–63. doi: 10.1016/j.jaci.2014.10.037
31. Lin X, Luo H, Yan X, Song Z, Gao X, Xia Y, et al. Interleukin-34 ameliorates survival and bacterial clearance in polymicrobial sepsis. Crit Care Med. (2018) 46:e584–90. doi: 10.1097/CCM.0000000000003017
32. Liu Y, Liu H, Zhu J, Bian Z. Interleukin-34 drives macrophage polarization to the M2 phenotype in autoimmune hepatitis. Pathol Res Pract. (2019) 215:152493. doi: 10.1016/j.prp.2019.152493
33. Baghdadi M, Wada H, Nakanishi S, Abe H, Han N, Putra WE, et al. Chemotherapy-induced IL34 enhances immunosuppression by tumor-associated macrophages and mediates survival of chemoresistant lung cancer cells. Cancer Res. (2016) 76:6030–42. doi: 10.1158/0008-5472.CAN-16-1170
34. Yang H, Luo Y, Lai X. Il-34 regulates MAPKs, PI3K/Akt, JAK and NF-κ B pathways and induces the expression of inflammatory factors in RA-FLS. Clin Exp Rheumatol. (2022) 40:1779–88. doi: 10.55563/clinexprheumatol/6t1d4i
35. Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol. (2014) 6:a021857. doi: 10.1101/cshperspect.a021857
36. Garcia S, Hartkamp LM, Malvar-Fernandez B, van Es IE, Lin H, Wong J, et al. Colony-stimulating factor (CSF) 1 receptor blockade reduces inflammation in human and murine models of rheumatoid arthritis. Arthritis Res Ther. (2016) 18:75. doi: 10.1186/s13075-016-0973-6
37. Greter M, Lelios I, Pelczar P, Hoeffel G, Price J, Leboeuf M, et al. Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity. (2012) 37:1050–60. doi: 10.1016/j.immuni.2012.11.001
38. Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. (2012) 13:753–60. doi: 10.1038/ni.2360
39. Wei S, Nandi S, Chitu V, Yeung YG Yu W, Huang M, et al. Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. J Leukoc Biol. (2010) 88:495–505. doi: 10.1189/jlb.1209822
40. Boulakirba S, Pfeifer A, Mhaidly R, Obba S, Goulard M, Schmitt T, et al. IL-34 and CSF-1 display an equivalent macrophage differentiation ability but a different polarization potential. Sci Rep. (2018) 8:256. doi: 10.1038/s41598-017-18433-4
41. Foucher ED, Blanchard S, Preisser L, Garo E, Ifrah N, Guardiola P, et al. IL-34 induces the differentiation of human monocytes into immunosuppressive macrophages antagonistic effects of GM-CSF and IFNγ. PLoS ONE. (2013) 8:e56045. doi: 10.1371/journal.pone.0056045
42. Wang B, Tang Y, Sun X, Ouyang X, Li H, Wei J, et al. Increased IL-6 expression on THP-1 by IL-34 stimulation up-regulated rheumatoid arthritis Th17 cells. Clin Rheumatol. (2018) 37:127–37. doi: 10.1007/s10067-017-3746-y
43. Van Raemdonck K, Umar S, Palasiewicz K, Volin MV, Elshabrawy HA, Romay B, et al. IL-34 reprograms glycolytic and osteoclastic RA macrophages via Syndecan-1 and M-CSFR. Arthritis Rheumatol. (2021) 73:2003–14. doi: 10.1002/art.41792
44. Li X, Lei Y, Gao Z, Wu G, Gao W, Xia L, et al. IL-34 affects fibroblast-like synoviocyte proliferation, apoptosis and function by regulating IL-17. Sci Rep. (2021) 11:16378. doi: 10.1038/s41598-021-95839-1
45. Chemel M, Le Goff B, Brion R, Cozic C, Berreur M, Amiaud J, et al. Interleukin 34 expression is associated with synovitis severity in rheumatoid arthritis patients. Ann Rheum Dis. (2012) 71:150–4. doi: 10.1136/annrheumdis-2011-200096
46. Yoshitomi H. Regulation of immune responses and chronic inflammation by fibroblast-like synoviocytes. Front Immunol. (2019) 10:1395. doi: 10.3389/fimmu.2019.01395
47. Tu J, Hong W, Zhang P, Wang X, Körner H, Wei W. Ontology and function of fibroblast-like and macrophage-like synoviocytes: how do they talk to each other and can they be targeted for rheumatoid arthritis therapy? Front Immunol. (2018) 9:1467. doi: 10.3389/fimmu.2018.01467
48. Clavel G, Thiolat A, Boissier MC. Interleukin newcomers creating new numbers in rheumatology: IL-34 to IL-38. Joint Bone Spine. (2013) 80:449–53. doi: 10.1016/j.jbspin.2013.04.014
49. Pasqualon T, Lue H, Groening S, Pruessmeyer J, Jahr H, Denecke B, et al. Cell surface syndecan-1 contributes to binding and function of macrophage migration inhibitory factor (MIF) on epithelial tumor cells. Biochim Biophys Acta. (2016) 1863:717–26. doi: 10.1016/j.bbamcr.2016.02.003
50. Kouwenberg M, Rops A., Bakker-van Bebber M, Diepeveen L, Götte M, Hilbrands L, et al. Role of syndecan-1 in the interaction between dendritic cells and T cells. PLoS ONE. (2020) 15:e0230835. doi: 10.1371/journal.pone.0230835
51. Altemeier WA, Schlesinger SY, Buell CA, Brauer R, Rapraeger AC, Parks WC, et al. Transmembrane and extracellular domains of syndecan-1 have distinct functions in regulating lung epithelial migration and adhesion. J Biol Chem. (2012) 287:34927–35. doi: 10.1074/jbc.M112.376814
52. Xiao J, Angsana J, Wen J, Smith SV, Park PW, Ford ML, et al. Syndecan-1 displays a protective role in aortic aneurysm formation by modulating T cell-mediated responses. Arterioscler Thromb Vasc Biol. (2012) 32:386–96. doi: 10.1161/ATVBAHA.111.242198
53. Chen Z, Buki K, Vääräniemi J, Gu G, Väänänen HK. The critical role of IL-34 in osteoclastogenesis. PLoS ONE. (2011) 6:e18689. doi: 10.1371/journal.pone.0018689
54. Baud'Huin M, Renault R, Charrier C, Riet A, Moreau A, Brion R, et al. Interleukin-34 is expressed by giant cell tumours of bone and plays a key role in RANKL-induced osteoclastogenesis. J Pathol. (2010) 221:77–86. doi: 10.1002/path.2684
55. Cui MY Li X, Lei YM, Xia LP, Lu J, Shen H. Effects of IL-34 on the secretion of RANKL/OPG by fibroblast-like synoviocytes and peripheral blood mononuclear cells in rheumatoid arthritis. Eur Cytokine Netw. (2019) 30:67–73. doi: 10.1684/ecn.2019.0428
56. Segaliny AI, Brion R, Mortier E, Maillasson M, Cherel M, Jacques Y, et al. Syndecan-1 regulates the biological activities of interleukin-34. Biochim Biophys Acta. (2015) 1853:1010–21. doi: 10.1016/j.bbamcr.2015.01.023
57. Bézie S, Freuchet A, Sérazin C, Salama A, Vimond N, Anegon I, et al. IL-34 Actions on FOXP3. Front Immunol. (2020) 11:1496. doi: 10.3389/fimmu.2020.01496
58. Angsana J, Chen J, Smith S, Xiao J, Wen J, Liu L, et al. Syndecan-1 modulates the motility and resolution responses of macrophages. Arterioscler Thromb Vasc Biol. (2015) 35:332–40. doi: 10.1161/ATVBAHA.114.304720
59. Saleh ME, Gadalla R, Hassan H, Afifi A, Götte M, El-Shinawi M, et al. The immunomodulatory role of tumor Syndecan-1 (CD138) on ex vivo tumor microenvironmental CD4+ T cell polarization in inflammatory and non-inflammatory breast cancer patients. PLoS ONE. (2019) 14:e0217550. doi: 10.1371/journal.pone.0217550
60. Jaiswal AK, Sadasivam M, Archer NK, Miller RJ, Dillen CA, Ravipati A, et al. Syndecan-1 regulates psoriasiform dermatitis by controlling homeostasis of IL-17-producing γδ T cells. J Immunol. (2018) 201:1651–61. doi: 10.4049/jimmunol.1800104
61. Yu T, Chaganty B, Lin L, Xing L, Ramakrishnan B, Wen K, et al. VIS832, a novel CD138-targeting monoclonal antibody, potently induces killing of human multiple myeloma and further synergizes with IMiDs or bortezomib in vitro and in vivo. Blood Cancer J. (2020) 10:110. doi: 10.1038/s41408-020-00378-z
62. Patterson AM, Cartwright A, David G, Fitzgerald O, Bresnihan B, Ashton BA, et al. Differential expression of syndecans and glypicans in chronically inflamed synovium. Ann Rheum Dis. (2008) 67:592–601. doi: 10.1136/ard.2006.063875
63. Salminen-Mankonen H, Säämänen AM, Jalkanen M, Vuorio E, Pirilä L. Syndecan-1 expression is upregulated in degenerating articular cartilage in a transgenic mouse model for osteoarthritis. Scand J Rheumatol. (2005) 34:469–74. doi: 10.1080/03009740500304338
64. Timmen M, Hidding H, Götte M, Khassawna TE, Kronenberg D, Stange R. The heparan sulfate proteoglycan Syndecan-1 influences local bone cell communication via the RANKL/OPG axis. Sci Rep. (2020) 10:20510. doi: 10.1038/s41598-020-77510-3
65. Nandi S, Cioce M, Yeung YG, Nieves E, Tesfa L, Lin H, et al. Receptor-type protein-tyrosine phosphatase ζ is a functional receptor for interleukin-34. J Biol Chem. (2013) 288:21972–86. doi: 10.1074/jbc.M112.442731
66. Zwicker S, Bureik D, Bosma M, Martinez GL, Almer S, Boström EA. Receptor-type protein-tyrosine phosphatase ζ and colony stimulating factor-1 receptor in the intestine: cellular expression and cytokine- and chemokine responses by interleukin-34 and colony stimulating factor-1. PLoS ONE. (2016) 11:e0167324. doi: 10.1371/journal.pone.0167324
67. Ohno H, Uemura Y, Murooka H, Takanashi H, Tokieda T, Ohzeki Y, et al. The orally-active and selective c-Fms tyrosine kinase inhibitor Ki20227 inhibits disease progression in a collagen-induced arthritis mouse model. Eur J Immunol. (2008) 38:283–91. doi: 10.1002/eji.200737199
68. Paniagua RT, Chang A, Mariano MM, Stein EA, Wang Q, Lindstrom TM, et al. c-Fms-mediated differentiation and priming of monocyte lineage cells play a central role in autoimmune arthritis. Arthritis Res Ther. (2010) 12:R32. doi: 10.1186/ar2940
69. Bézie S, Picarda E, Ossart J, Tesson L, Usal C, Renaudin K, et al. IL-34 is a Treg-specific cytokine and mediates transplant tolerance. J Clin Invest. (2015) 125:3952–64. doi: 10.1172/JCI81227
70. Bézie S, Meistermann D, Boucault L, Kilens S, Zoppi J, Autrusseau E, et al. Expanded human non-cytotoxic CD8 + CD45RC low/− tregs efficiently delay skin graft rejection and GVHD in humanized mice. Front Immunol. (2017) 8:2014. doi: 10.3389/fimmu.2017.02014
71. Freuchet A, Salama A, Bézie S, Tesson L, Rémy S, Humeau R, et al. IL-34 deficiency impairs FOXP3+ Treg function in a model of autoimmune colitis and decreases immune tolerance homeostasis. Clin Transl Med. (2022) 12:e988. doi: 10.1002/ctm2.988
72. Seidl C, Koch U, Buhleier T, Frank R, Möller B, Markert E, et al. HLA-DRB1*04 subtypes are associated with increased inflammatory activity in early rheumatoid arthritis. Br J Rheumatol. (1997) 36:941–4. doi: 10.1093/rheumatology/36.9.941
73. Weyand CM, Hicok KC, Conn DL, Goronzy JJ. The influence of HLA-DRB1 genes on disease severity in rheumatoid arthritis. Ann Intern Med. (1992) 117:801–6. doi: 10.7326/0003-4819-117-10-801
74. Li S, Yin H, Zhang K, Wang T, Yang Y, Liu X, et al. Effector T helper cell populations are elevated in the bone marrow of rheumatoid arthritis patients and correlate with disease severity. Sci Rep. (2017) 7:4776. doi: 10.1038/s41598-017-05014-8
75. Kang KY, Kim YK Yi H, Kim J, Jung HR, Kim IJ, et al. Metformin downregulates Th17 cells differentiation and attenuates murine autoimmune arthritis. Int Immunopharmacol. (2013) 16:85–92. doi: 10.1016/j.intimp.2013.03.020
76. Shen H, Goodall JC, Hill Gaston JS. Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis Rheum. (2009) 60:1647–56. doi: 10.1002/art.24568
77. Yang Z, Shen Y, Oishi H, Matteson EL, Tian L, Goronzy JJ, et al. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med. (2016) 8:331ra38. doi: 10.1126/scitranslmed.aad7151
78. Liu D, Cao T, Wang N, Liu C, Ma N, Tu R, et al. IL-25 attenuates rheumatoid arthritis through suppression of Th17 immune responses in an IL-13-dependent manner. Sci Rep. (2016) 6:36002. doi: 10.1038/srep36002
79. Lee DG, Woo JW, Kwok SK, Cho ML, Park SH. MRP8 promotes Th17 differentiation via upregulation of IL-6 production by fibroblast-like synoviocytes in rheumatoid arthritis. Exp Mol Med. (2013) 45:e20. doi: 10.1038/emm.2013.39
80. Kim EK, Kwon JE, Lee SY, Lee EJ, Kim DS, Moon SJ, et al. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis. (2017) 8:e2565. doi: 10.1038/cddis.2016.490
81. Song L, Tan J, Wang Z, Ding P, Tang Q, Xia M, et al. Interleukin-17A facilitates osteoclast differentiation and bone resorption via activation of autophagy in mouse bone marrow macrophages. Mol Med Rep. (2019) 19:4743–52. doi: 10.3892/mmr.2019.10155
82. Kondelková K, Vokurková D, Krejsek J, Borská L, Fiala Z, Ctirad A. regulatory T cells (TREG) and their roles in immune system with respect to immunopathological disorders. Acta Medica. (2010) 53:73–7. doi: 10.14712/18059694.2016.63
83. Khan MA. Regulatory T cells mediated immunomodulation during asthma: a therapeutic standpoint. J Transl Med. (2020) 18:456. doi: 10.1186/s12967-020-02632-1
84. Crome SQ, Clive B, Wang AY, Kang CY, Chow V, Yu J, et al. Inflammatory effects of ex vivo human Th17 cells are suppressed by regulatory T cells. J Immunol. (2010) 185:3199–208. doi: 10.4049/jimmunol.1000557
85. Othy S, Jairaman A, Dynes JL, Dong TX, Tune C, Yeromin AV, et al. Regulatory T cells suppress Th17 cell Ca2+ signaling in the spinal cord during murine autoimmune neuroinflammation. Proc Natl Acad Sci U S A. (2020) 117:20088–99. doi: 10.1073/pnas.2006895117
86. Kanjana K, Chevaisrakul P, Matangkasombut P, Paisooksantivatana K, Lumjiaktase P. Inhibitory activity of FOXP3+ regulatory T cells reveals high specificity for displaying immune tolerance in remission state rheumatoid arthritis. Sci Rep. (2020) 10:19789. doi: 10.1038/s41598-020-76168-1
87. Niu Q, Cai B, Huang ZC, Shi YY, Wang LL. Disturbed Th17/Treg balance in patients with rheumatoid arthritis. Rheumatol Int. (2012) 32:2731–6. doi: 10.1007/s00296-011-1984-x
88. Wang T, Li S, Yang Y, Zhang K, Dong S, Wang X, et al. T helper 17 and T helper 1 cells are increased but regulatory T cells are decreased in subchondral bone marrow microenvironment of patients with rheumatoid arthritis. Am J Transl Res. (2016) 8:2956–68.
89. Jin S, Sun S, Ling H, Ma J, Zhang X, Xie Z, et al. Protectin DX restores Treg/Th17 cell balance in rheumatoid arthritis by inhibiting NLRP3 inflammasome via miR-20a. Cell Death Dis. (2021) 12:280. doi: 10.1038/s41419-021-03562-6
90. Lee SY, Moon SJ, Moon YM, Seo HB Ryu JG, Lee AR, et al. A novel cytokine consisting of the p40 and EBI3 subunits suppresses experimental autoimmune arthritis via reciprocal regulation of Th17 and Treg cells. Cell Mol Immunol. (2022) 19:79–91. doi: 10.1038/s41423-021-00798-2
91. Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. (2006) 203:2673–82. doi: 10.1084/jem.20061775
Keywords: rheumatoid arthritis, macrophage, T-cell, IL-34, inflammation
Citation: Park HE, Oh H and Baek J-H (2022) Interleukin-34-regulated T-cell responses in rheumatoid arthritis. Front. Med. 9:1078350. doi: 10.3389/fmed.2022.1078350
Received: 24 October 2022; Accepted: 14 November 2022;
Published: 30 November 2022.
Edited by:
Zoltan Szekanecz, University of Debrecen, HungaryReviewed by:
Gyorgy Nagy, Semmelweis University, HungaryFerenc Boldizsar, University of Pécs, Hungary
Copyright © 2022 Park, Oh and Baek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jea-Hyun Baek, amJhZWtAaGFuZG9uZy5lZHU=