
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med. , 25 November 2022
Sec. Gastroenterology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.1060244
This article is part of the Research Topic The Heart of NAFLD View all 5 articles
 Tomoki Yagai1*
Tomoki Yagai1* Takahisa Nakamura1,2,3,4*
Takahisa Nakamura1,2,3,4*Non-alcoholic fatty liver disease (NAFLD) is one of the most prevalent hepatic disorders that 20-30% of the world population suffers from. The feature of NAFLD is excess lipid accumulation in the liver, exacerbating multiple metabolic syndromes such as hyperlipidemia, hypercholesterolemia, hypertension, and type 2 diabetes. Approximately 20-30% of NAFLD cases progress to more severe chronic hepatitis, known as non-alcoholic steatohepatitis (NASH), showing deterioration of hepatic functions and liver fibrosis followed by cirrhosis and cancer. Previous studies uncovered that several metabolic regulators had roles in disease progression as key factors. Peroxisome proliferator-activated receptor alpha (PPARα) has been identified as one of the main players in hepatic lipid homeostasis. PPARα is abundantly expressed in hepatocytes, and is a ligand-dependent nuclear receptor belonging to the NR1C nuclear receptor subfamily, orchestrating lipid/glucose metabolism, inflammation, cell proliferation, and carcinogenesis. PPARα agonists are expected to be novel prescription drugs for NASH treatment, and some of them (e.g., Lanifibranor) are currently under clinical trials. These potential novel drugs are developed based on the knowledge of PPARα-activating target genes related to NAFLD and NASH. Intriguingly, PPARα is known to suppress the expression of subsets of target genes under agonist treatment; however, the mechanisms of PPARα-mediated gene suppression and functions of these genes are not well understood. In this review, we summarize and discuss the mechanisms of target gene repression by PPARα and the roles of repressed target genes on hepatic lipid metabolism, fibrosis and carcinogenesis related to NALFD and NASH, and provide future perspectives for PPARα pharmaceutical potentials.
Peroxisome proliferator-activated receptors (PPARs) are ligand-dependent nuclear receptors belonging to the NR1C nuclear receptor subfamily, involved in lipid/glucose metabolism, inflammation, cell proliferation, and carcinogenesis (1, 2). There are three PPAR isoforms, PPARα, PPARβ/δ, and PPARγ, each with different tissue distribution and expression patterns. PPARα is abundantly expressed in the energy-producing tissues such as the liver, heart, kidney, and brown adipose tissue, whereas PPARγ is mainly expressed in the adipose tissue and macrophages, and PPARβ/δ is more widely expressed compared with PPARα (3, 4). The intense interest in PPARα is driven in part to its activation by agonists that promote upregulation of target genes related to lipid catabolism, modulating microsomal, peroxisomal, and mitochondrial fatty acid oxidation, lipoprotein metabolism, triglyceride synthesis, and gluconeogenesis (5). In the liver, these target genes are significantly involved in the pathogenesis of liver steatosis, including non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) (6, 7). Specifically, PPARα activation may contribute to the prevention of NAFLD and NASH aggravation because the PPARα-activated target genes have roles in anti-inflammation and reduction of lipid accumulation in the liver. PPARα activation could, therefore, be a primary pharmaceutical target (8). PPARα agonists also repress target genes (9–11) but the contribution of these “repressed target genes” to NAFLD and NASH and the mechanisms involved are not well understood. In this review, we focus on the target genes repressed by PPARα and the repression mechanisms elucidated hitherto, and discuss the potential significance of PPARα as a transcriptional suppressor.
Peroxisome proliferator-activated receptor α was discovered in rodents in as primarily a carcinogen-responsible peroxisome proliferator (12). Peroxisome, a membrane-bound organelle in the cytoplasm of eukaryotic cells, performs key functions in multiple metabolic pathways such as purine catabolism, fatty acid β-oxidation, and phospholipid synthesis, in addition to the conversion of reactive oxygen species (13). PPARα is abundantly expressed in tissues metabolizing fatty acids such as liver, skeletal muscle, heart, and brown adipose tissue, in addition to inflammatory immune cells such as monocytes and macrophages (14–17). In hepatocytes, PPARα regulates peroxisomal and mitochondrial β-oxidation, lipid biogenesis and transport, cholesterol and glucose metabolism, and inflammation (18). Although PPARα protein is known to localize in the nucleus regardless of the activation state (19), in some cell types such as chondrocytes (20) and differentiated human macrophages (21), PPARα can also be found in the cytoplasm. The PPARα expression is regulated in transcriptional and post-transcriptional manners. HNF4α activates PPARα transcription by binding to the response element DR-1 in the promoter region (22), whereas COUP-TFII antagonizes the HNF4α transcriptional activity by competing with the binding to DR-1 in the promoter (23). A transcription factor KLF6 induces miR-10b that inhibits PPARα protein translation (24). A recent study showed that hepatic Argonaute 2 (Ago2) inhibits PPARα expression, suggesting that Ago2-mediated microRNA processing and RNA silencing have significant roles in PPARα repression (25).
PPARα has four structural domains, designed A/B, C, D, and E/F domains. The N-terminal A/B domain harbors a ligand-independent transcriptional activating function (AF-1). The C domain includes DNA binding domain (DBD) essential for binding to the PPAR response element (PPRE) in the target gene promoter/enhancer sites (26). The D domain is a hinge region that includes binding sites for co-repressors such as NCoR and SMRT. The E/F domains carry ligand binding domains (LBD) that harbor a relatively larger cavity of ligands compared with other nuclear receptors (17). The binding of agonists to LBD induces the conformation change, which results in the recruitment of transcriptional complexes with co-activators and subsequent transcriptional activation (27–29).
Free fatty acids (FFAs) have been identified as endogenous agonists for PPARα. The n-3 polyunsaturated fatty acids (PUFAs), such as Eicosapentaenoic acid (EPA) and Docosahexaenoic acid (DHA), in particular, have been shown to be potent agonism compared with other FFAs (30). Such endogenous agonists come from dietary nutrients when feeding or from adipose tissues during fasting. In addition to endogenous agonists, synthetic amphipathic carboxylic acids such as fibrates that are frequently used for the treatment of hyperlipidemia and hypercholesterolemia, are demonstrable PPARα selective agonists (5). The first fibrate drugs were developed during the 1960s-1980s, although PPARα was not identified as the direct molecular target at that time. Currently, several synthetic PPARα agonists developed have been used clinically and experimentally (e.g., Fenofibrate, Clofibrate, Bezafibrate, Gemfibrozil, Pemafibrate, Wy-14,643, GW9578, GW7647) (5).
Studies over the past decades elucidated gene transcription mechanisms of PPARα (2, 5, 31–33). Retinoid X receptor alpha (RXRα), a nuclear receptor belonging to the NR2B subfamily, is an obligate heterodimer partner for PPARα. RXR family consists of three distinct members, known as RXRα, RXRβ, and RXRγ (34, 35). RXRα (NR2B1) was the first identified RXR that is abundantly expressed in the liver. While RXRα is activated by the endogenous agonist 9-cis retinoid acid, ligand-independent RXRα forms a transcription complex with PPARα for the transcriptional activation. One of the significant physiological roles of PPARα is as a transcription factor activating target gene expressions (36–39). When a selective agonist binds to the LBD of PPARα, PPARα can bind to PPRE sites via heterodimerization with RXRα. PPRE sites consist of direct repeat type 1 (DR-1), which is a tandem repeat of recognition motif 5′-AGGTCA-3′ separated by a single nucleotide (40). PPARα binds to the 5′ extended half-site of the response element, whereas RXRα binds to the 3′ half-site (41, 42). Although PPARα can bind to PPRE without agonists, the interaction is not stable because of the chromatin condensed state (27–29). Furthermore, the lack of an agonist inhibits transcriptional activity of PPARα as co-repressors, such as NCOR1 and SMRT, are bound (43). Binding of the agonists PPARα releases bound co-repressors as PPARα conformation changes, and with co-activator recruitment of components, such as CBP1/P300, SRC-1, and PGC1, target gene transcription is activated (44). The binding of co-activator CBP1 is known to induce HAT activity, resulting in chromatin remodeling, which opens condensed genomic DNA to exposed PPRE sites and allows access of PPARα/RXRα heterodimer to the PPRE tightly (45). Taken together, a canonical function of PPARα is to induce target gene transcription by forming the transcription complex with RXRα and other transcriptional co-activators when the agonist binds and modifies the conformation of PPARα.
In addition to transcriptional activation, transcriptomic studies in the PPARα-activated cells and tissues indicate that there are numerous target genes whose expression is repressed by PPARα, and these repressed genes have significant roles in various homeostasis (10), although the molecular mechanisms of how PPARα suppresses target genes remain poorly studied. Gene repression mechanisms have been reported for other nuclear receptors, such as the Thyroid receptor and Glucocorticoid receptor (46), from which it is deduced that the repression mechanisms can be classified as Trans-acting, Cis-acting, or indirect manner.
Several studies reported that the activated PPARα directly binds to transcription factors and interferes the transcriptional activity. The protein-protein interaction-mediated transcription repression is known as trans-acting repression (Figure 1A). It has been elucidated that PPARα has roles in the repression of hepatic inflammation by inhibiting Activator protein 1 (AP-1) and Nuclear factor-κB (NF-κB) pathways through trans-repression. Ligand-activated PPARα directly binds to a NF-κB component p65 and the N-terminus JNK-responsive part of c-Jun, resulting in the prevention of the unique response element binding of NF-κB and AP-1 (47). Bougarne et al. uncovered that the PPARα interference of p65 is synergistically induced with trans-repression by GR binding to p50 (48). Another study involving mice carrying mutant PPARα lacking the DBD region showed significant suppression of chronic liver inflammation by NF-κB and AP-1 pathway, suggesting that NF-κB and AP-1 suppression by PPARα is independent of the DNA binding, and that PPARα directly interacts with NF-κB and AP-1 (49). Since NF-κB and AP-1 pathways regulate the expression of pro-inflammatory cytokines, these findings suggest that PPARα may exert its anti-inflammatory effects by suppressing these pathways in the liver. Previous studies reported that saturated fatty acids activate JNK in hepatocytes (50, 51), whereas the hepatic JNK is required for AP-1 activation and NCOR1 expression (52). As NCOR1 is a potent co-repressor for PPARα, these findings suggest that the JNK-NCOR1 axis reciprocally affects the PPARα anti-inflammatory effect. In addition to NF-κB and AP-1, it was revealed that activated PPARα also binds to GRIP1/TIF2, which is a co-activator of C/EBPβ. The PPARα’s interaction with GRIP1/TIF2 results in interference of C/EBPβ binding to the response element (53). Blanquart et al. reported that the protein kinase C pathway-mediated phosphorylation of C/EBPβ at Ser179 and Ser230 residues suppresses C/EBPβ activity in the fibrinogen-β promoter (54). Oka et al. reported that PPARα and SIRT1 form a heterodimer and bind to ERR-responsive elements, leading to competitive inhibition of ERR pathway related to mitochondria respiration (55, 56). Several studies reported HNF4α inhibition by PPARα. Shin et al showed that PPARα activation decreases HNF4α protein but not mRNA, resulting in transcriptional inhibition of the HNF4α target gene ACMSD (57). Another study showed that an HNF4α target gene, Gls2, is significantly repressed by PPARα with HNF4α protein degradation (58). Recently, it was reported that PPARα/RXRα heterodimer binds to HNF4α and promotes ubiquitination, resulting in the HNF4α protein degradation and repression of the HNF4α target gene Sds promoter activity (59). Of note, Leuenberger et al reported that SUMOylated but not naïve PPARα interacts with a transcription factor GABPα and represses the transcriptional activity in Cyp7b1 promoter, indicating that PPARα is subject to protein modifications for transcriptional activity (60). Altogether, these studies suggest that direct interactions between PPARα and transcription factors may affect numerous metabolic and inflammatory pathways through gene repression.
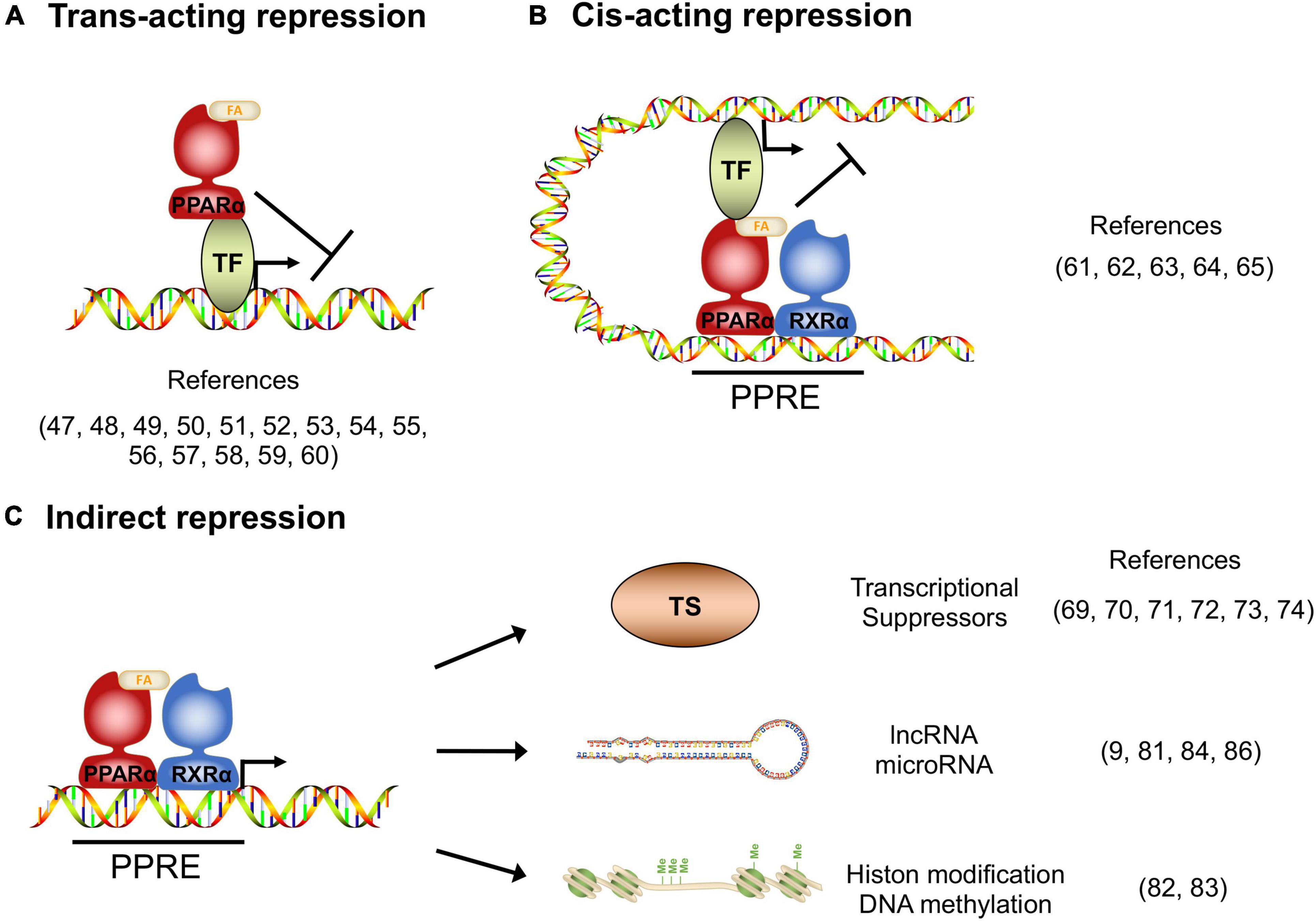
Figure 1. Transcriptional repression manners by PPARα. (A) Trans-acting repression manner by direct interaction between PPARα and the target transcription factors (TF). (B) Cis-acting repression manner depending on PPARα binding to the PPAR responsive element (PPRE). (C) Indirect repression manner, upregulating transcriptional suppressors, long non-coding RNAs, microRNAs and epigenetic modulators.
Previous studies revealed that DNA-bound PPARα could prevent the transcription of target genes in a number of different systems. The repression manner based on protein-DNA interaction is known as cis-acting repression (Figure 1B). Mogilenko et al. uncovered that transcriptional activity in complement C3 promoter is inhibited by physical interactions between PPRE-bound PPARα and p65. They showed that PPARα binding to PPRE is not limited only to transcriptional activation but repression (61). ChIP-chip analysis by van der Meer et al. proposed that PPARα binding to PPRE near STAT response elements interferes with the STAT1 and STAT3 transcriptional activation in the target gene promoters such as STARD13 and TOX3 in HepG2 cells (62). You et al reported that activated PPARα binds to a PPRE located on the Glut-1 promoter, resulting in the inhibition of transcriptional activity and cancer cell proliferation (63). A recent study supported that the Glut-1 inhibition by PPARα contributes to tumor growth and chemo-resistance (64). Zhang et al showed that PPARα activation by fenofibrate recruits NCOR and associated HDAC to the INFγ gene locus, resulting in the repression of IFNγ expression in mouse T cells (65). Although the functional detail of “repressive PPRE” as a transcriptional silencer is still controversial, these studies clearly suggest that PPARα has cis-element-dependent gene repression mechanisms.
Several studies have reported that PPARα had mechanisms of transcriptional inhibition not only by direct interaction but also indirectly through involvement/regulation of other transcriptional regulators, long non-coding RNA (lncRNA), microRNA and epigenetic modulators (Figure 1C). Previous studies uncovered that Cyp7a1 and Cyp27a1 expression are repressed by PPARα agonism in human and rodent cells (66–68). These cytochrome P450 proteins have significant roles in bile acid synthesis, resulting in a decline in the output of bile acids and an increase in cholesterol secretion. It was reported that PPARα agonism increases a nuclear receptor Rev-erbα expression, and Rev-erbα inhibits Apoa1 gene transcription in rodents (69–71). The PPARα-activated Rev-erbα also represses Apoc3 gene expression by binding to the enhancer/promoter region (72–74). As genetic deletion of Rev-erbα leads to hepatic steatosis in mice, PPARα-mediated Rev-erbα induction appears to exert a protective role in the development of NAFLD, at least in part, by suppressing the expression of Apoa1 and Apoc3 that control lipid transport in hepatocytes (75). Makled et al. reported that a pan-agonist for PPARα/γ (a.k.a. Saroglitazar) downregulates pro-fibrotic gene expressions such as TGF-β1 and PDGF-BB, followed by the downstream target gene repressions such as PAI-1 and Smad-3 in rat liver fibrosis model (76, 77). The results suggest that PPARα (and also PPARγ) can transcriptionally repress the TGF-β signaling pathway. Bansal et al identified that the AF-1 domain of activated PPARα directly binds to the kinase domain of TAK-1 protein and prevents phosphorylation. Phosphorylation of TAK-1 is a molecule switch of the TGF-β signal cascade, indicating that the inhibition of phosphorylation results in the prevention of the TGF-β pathway (78). Brocker et al unveiled that hepatic PPARα directly upregulates a lncRNA Gm15441. Gm15441 expression suppresses its antisense transcript encoding TXNIP, resulting in inflammasome activation, CASP1 cleavage and proinflammatory IL-1β maturation (9). These findings suggest that PPARα regulates the expression of lincRNAs relevant to the development of steatohepatitis. Although precise mechanisms are still unclear, it was reported that PPARα activation represses Oleate-inducible Fatp1 expression, attenuating total free fatty acid and triglyceride accumulation in macrophages (79). Triglyceride accumulation is related to macrophage activation (80), suggesting that the Fatp1 repression by PPARα is related to inflammation. Furthermore, a recent study unveiled that the let-7 microRNA family is significantly repressed by PPARα agonism and the let-7 microRNA prevented RXRα ubiquitination through RNF8 mRNA decay. RXRα degradation results in the inhibition of the transcriptional activity of the PPARα/RXRα complex, indicating that PPARα - let-7 microRNA - RNF8 - RXRα axis is a negative feedback loop in the hepatic lipid metabolism (81).
Several target genes repressed by PPARα are related to cancer progression and tumor growth. A study unveiled that intestinal PPARα upregulated RB1 expression in mouse colon, resulting in the repression of DNMT1 and PRMT6. DNMT1 and PRMT6 contribute to the inhibition of tumor suppressor genes such as Cdkn1a and Cdkn1b via DNA methylation and histone H3R2 dimethylation (82). Another study reported that hepatic PPARα upregulates a transcription factor E2F8 in mice. The E2F8 increases Uhrf1 expression, contributing to DNA methylation in the Cdh1 promoter and the inhibition of expression (83). CDH1 represses proto-oncogene Myc expression through the Wnt pathway, suggesting that the PPARα-CDH1 pathway may enhance tumor growth. Shah et al. uncovered that PPARα agonism repressed at least twelve microRNA expressions in mouse liver. Especially the repressed target gene let-7C microRNA targets Myc mRNA and decays the stability (84). These studies indicate that rodent PPARα has multiple roles in the promotion of carcinogenesis. It is consistent with previous publications showing that long-term activation of rodent PPARα induces carcinogenesis (5). In contrast, Shi et al suggested that PPARα activation represses E2F1 transcriptional activity and the target gene expressions via the p21 pathway, modulating transcriptional complexes of E2F1 and pRB in human glioma cells (85). In human glioma cells, another study showed that PPARα upregulated miR-214 expression, resulting in E2F2 mRNA decay and inhibition of cell proliferation (86). These studies suggest that PPARα-repressed target genes contribute to the inhibition of cancer progression and tumor growth in humans.
Studies about repressed target genes of PPARα are not sufficiently understood compared with those of the activated target genes. However, it has become obvious that one of the critical functions of PPARα is to exert transcriptional suppression of its target genes through multiple mechanisms (Table 1). The repressed target genes include various master regulators related to inflammation, fibrosis, and carcinogenesis, which contribute to, at least in part, the physiological roles of PPARα and the beneficial effects of PPARα agonist treatment. Given that PPARα represses the major pro-inflammatory transcriptional regulators, NF-κB and AP-1 pathways, the mechanisms of PPARα-mediated gene suppression may play a significant role in exacerbating hepatic inflammation (87). In addition, PPARα represses other transcriptional regulators/pathways, such as GRIP1/TIF2, HNF4α, IFNγ and TGF-β, that are related to lipid metabolism and inflammation, which appear to contribute to the beneficial effects of PPARα activation in hepatocytes. When PPARα is active as a transcription activator, the activated PPARα generally forms a transcriptional activation complex with co-activators, and the physical contact with the transcription factors accelerates their transcriptional activities. Conversely, the molecular mechanisms of how PPARα suppresses the target gene transcription and whether PPARα requires to form a specific transcriptional suppression complex to be a transcriptional suppressor remain to be elucidated. Although several previous studies identified PPREs located near repressed target gene promoter/enhancer, the sequential and positional differences between activating and repressing PPREs are still unclear. In addition, PPARα-mediated gene repression and activation occur at approximately the same time upon agonist treatment. As several epigenetic repression mechanisms have been shown in rodent cancer models and inflammation, the differences in epigenetic modifications in PPREs may be involved in the regulation of gene repression or activation of PPARα’s target genes. Identifications of the elements that distinguish PPRE enhancers from silencers would drastically advance our knowledge of PPARα biology. In addition, PPARα protein modifications, including SUMOylation and phosphorylation, may also impact PPARα-mediated gene repression and activation. One study demonstrated that SUMOylation of PPARα accelerates trans-acting repression (60), whereas another showed that post-translational phosphorylation of PPARα has a significant role in the trans-repression (54). Although the detail of mechanisms needs further analyses, such protein modifications could be related to the binding affinity of PPARα to the PPRE enhancer or silencer, or the other repressed target genes. At present, one PPARα agonist (a.k.a. Pemafibrate) and three pan-PPAR agonists (a.k.a. Lanifibranor, Pioglitazone, and Saroglitazar) are under clinical trials as drugs for NASH treatment respectively (88–91). Novel insights into the mechanisms will help the process of current clinical trials.
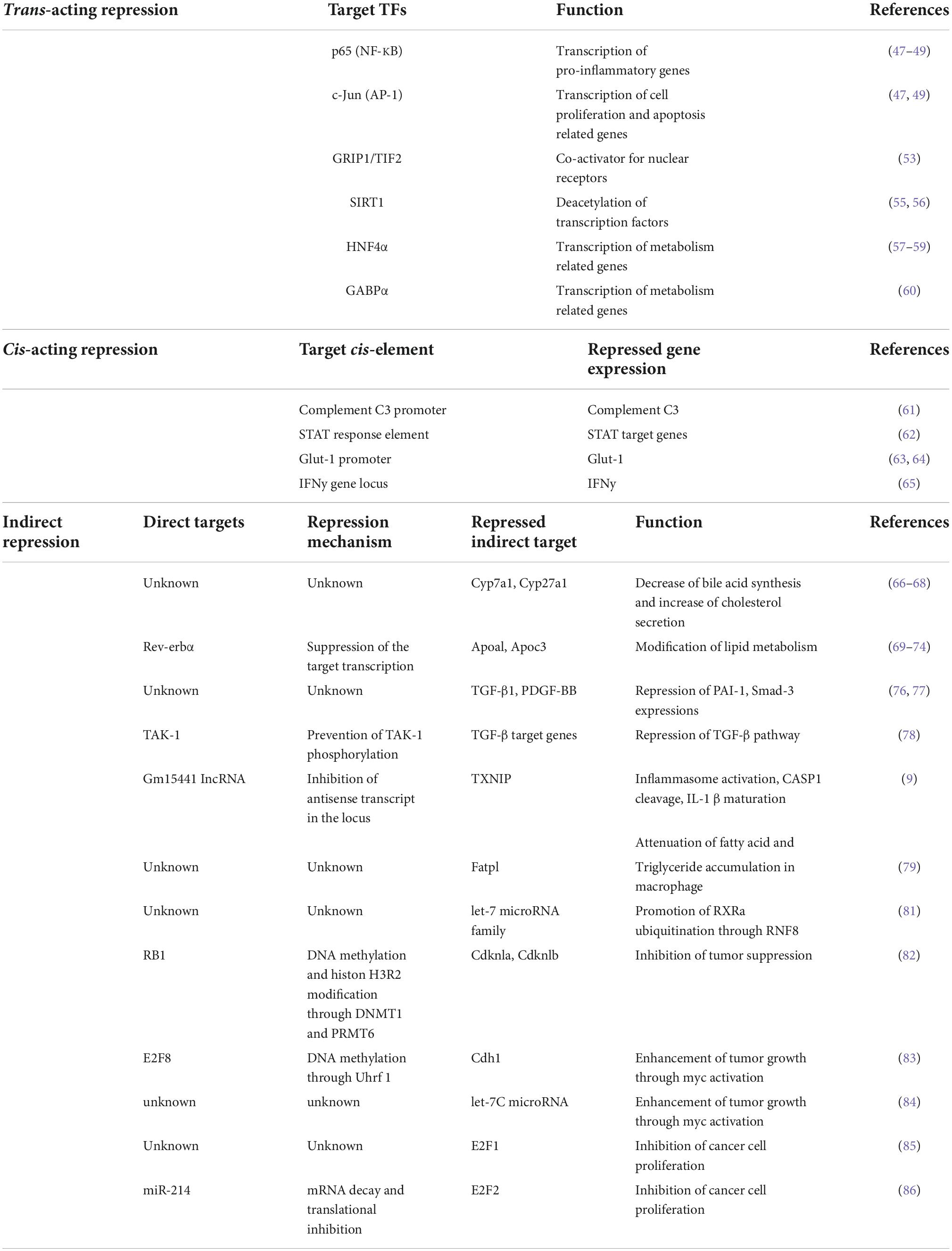
Table 1. Summary of repressed target genes of PPARα.
Both authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
This work was supported by the National Institute of Health (NIH) R01 DK123181 (TN), R21 OD031906 (TN), and P30DK078392 for the Digestive Disease Research Core Center in Cincinnati, and JSPS KAKENHI 20H03445 (TN), 20K16152 (TY), a grant-in-aid of ONO Medical Research Foundation (TY) and Takeda Science Foundation (TY).
We thank Saki Okamoto for assistance with the studies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat Rev Cancer. (2012) 12:181–95. doi: 10.1038/nrc3214
2. Wang Y, Nakajima T, Gonzalez FJ, Tanaka N. Ppars as metabolic regulators in the liver: lessons from liver-specific Ppar-null mice. Int J Mol Sci. (2020) 21:2061. doi: 10.3390/ijms21062061
3. Cabrero A, Laguna JC, Vázquez M. Peroxisome proliferator-activated receptors and the control of inflammation. Curr Drug Targets Inflamm Allergy. (2002) 1:243–8. doi: 10.2174/1568010023344616
4. Grygiel-Górniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications–a review. Nutr J. (2014) 13:17. doi: 10.1186/1475-2891-13-17
5. Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B, et al. Molecular actions of Pparα in lipid metabolism and inflammation. Endocr Rev. (2018) 39:760–802. doi: 10.1210/er.2018-00064
6. Greco D, Kotronen A, Westerbacka J, Puig O, Arkkila P, Kiviluoto T, et al. Gene expression in human NAFLD. Am J Physiol Gastrointest Liver Physiol. (2008) 294:G1281–7. doi: 10.1152/ajpgi.00074.2008
7. Wang R, Wang X, Zhuang L. Gene expression profiling reveals key genes and pathways related to the development of non-alcoholic fatty liver disease. Ann Hepatol. (2016) 15:190–9. doi: 10.5604/16652681.1193709
8. Kersten S, Stienstra R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie. (2017) 136:75–84. doi: 10.1016/j.biochi.2016.12.019
9. Brocker CN, Kim D, Melia T, Karri K, Velenosi TJ, Takahashi S, et al. Long non-coding Rna Gm15441 attenuates hepatic inflammasome activation in response to Ppara agonism and fasting. Nat Commun. (2020) 11:5847. doi: 10.1038/s41467-020-19554-7
10. Janssen AW, Betzel B, Stoopen G, Berends FJ, Janssen IM, Peijnenburg AA, et al. The impact of Pparα activation on whole genome gene expression in human precision cut liver slices. BMC Genomics. (2015) 16:760. doi: 10.1186/s12864-015-1969-3
11. Yan T, Luo Y, Yan N, Hamada K, Zhao N, Xia Y, et al. Intestinal peroxisome proliferator-activated receptor A -fatty acid-binding protein 1 axis modulates nonalcoholic steatohepatitis. Hepatology. (2022) Epub ahead of print. doi: 10.1002/hep.32538
12. Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. (1990) 347:645–50. doi: 10.1038/347645a0
13. Islinger M, Cardoso MJ, Schrader M. Be different–the diversity of peroxisomes in the animal kingdom. Biochim Biophys Acta. (2010) 1803:881–97. doi: 10.1016/j.bbamcr.2010.03.013
14. Feige JN, Gelman L, Michalik L, Desvergne B, Wahli W. From molecular action to physiological outputs: peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog Lipid Res. (2006) 45:120–59. doi: 10.1016/j.plipres.2005.12.002
15. Hamblin M, Chang L, Fan Y, Zhang J, Chen YE. Ppars and the cardiovascular system. Antioxid Redox Signal. (2009) 11:1415–52. doi: 10.1089/ars.2008.2280
16. Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of Ppar alpha in energy metabolism and vascular homeostasis. J Clin Invest. (2006) 116:571–80. doi: 10.1172/jci27989
17. Pyper SR, Viswakarma N, Yu S, Reddy JK. Pparalpha: energy combustion, hypolipidemia, inflammation and cancer. Nucl Recept Signal. (2010) 8:e002. doi: 10.1621/nrs.08002
18. Rakhshandehroo M, Knoch B, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. (2010) 2010:612089. doi: 10.1155/2010/612089
19. Feige JN, Gelman L, Tudor C, Engelborghs Y, Wahli W, Desvergne B. Fluorescence Imaging reveals the nuclear behavior of peroxisome proliferator-activated receptor/retinoid X receptor heterodimers in the absence and presence of ligand. J Biol Chem. (2005) 280:17880–90. doi: 10.1074/jbc.M500786200
20. Bordji K, Grillasca JP, Gouze JN, Magdalou J, Schohn H, Keller JM, et al. Evidence for the presence of peroxisome proliferator-activated receptor (Ppar) alpha and gamma and retinoid Z receptor in cartilage. Ppar gamma activation modulates the effects of interleukin-1beta on rat chondrocytes. J Biol Chem. (2000) 275:12243–50. doi: 10.1074/jbc.275.16.12243
21. Chinetti G, Griglio S, Antonucci M, Torra IP, Delerive P, Majd Z, et al. Activation of proliferator-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J Biol Chem. (1998) 273:25573–80. doi: 10.1074/jbc.273.40.25573
22. Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2a1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol. (2001) 21:1393–403. doi: 10.1128/mcb.21.4.1393-1403.2001
23. Pineda Torra I, Jamshidi Y, Flavell DM, Fruchart JC, Staels B. Characterization of the human pparalpha promoter: identification of a functional nuclear receptor response element. Mol Endocrinol. (2002) 16:1013–28. doi: 10.1210/mend.16.5.0833
24. Bechmann LP, Vetter D, Ishida J, Hannivoort RA, Lang UE, Kocabayoglu P, et al. Post-transcriptional activation of Ppar alpha by Klf6 in hepatic steatosis. J Hepatol. (2013) 58:1000–6. doi: 10.1016/j.jhep.2013.01.020
25. Bhattacharjee J, Borra VJ, Salem ESB, Zhang C, Murakami K, Gill RK, et al. Hepatic ago2 regulates Pparα for oxidative metabolism linked to glycemic control in obesity and post bariatric surgery. Endocrinology. (2021) 162:bqab007. doi: 10.1210/endocr/bqab007
26. Lee WS, Kim J. Peroxisome proliferator-activated receptors and the heart: lessons from the past and future directions. PPAR Res. (2015) 2015:271983. doi: 10.1155/2015/271983
27. Dowell P, Peterson VJ, Zabriskie TM, Leid M. Ligand-induced peroxisome proliferator-activated receptor alpha conformational change. J Biol Chem. (1997) 272:2013–20. doi: 10.1074/jbc.272.3.2013
28. Juge-Aubry CE, Gorla-Bajszczak A, Pernin A, Lemberger T, Wahli W, Burger AG, et al. Peroxisome proliferator-activated receptor mediates cross-talk with thyroid hormone receptor by competition for retinoid x receptor. Possible role of a leucine zipper-like heptad repeat. J Biol Chem. (1995) 270:18117–22. doi: 10.1074/jbc.270.30.18117
29. Xu HE, Lambert MH, Montana VG, Plunket KD, Moore LB, Collins JL, et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA. (2001) 98:13919–24. doi: 10.1073/pnas.241410198
30. Clarke SD. Nonalcoholic steatosis and steatohepatitis. I. Molecular mechanism for polyunsaturated fatty acid regulation of gene transcription. Am J Physiol Gastrointest Liver Physiol. (2001) 281:G865–9. doi: 10.1152/ajpgi.2001.281.4.G865
31. Fruchart JC, Hermans MP, Fruchart-Najib J. Selective peroxisome proliferator-activated receptor alpha modulators (Spparmα): new opportunities to reduce residual cardiovascular risk in chronic kidney disease? Curr Atheroscler Rep. (2020) 22:43. doi: 10.1007/s11883-020-00860-w
32. Sinha RA, Rajak S, Singh BK, Yen PM. Hepatic lipid catabolism via Pparα-lysosomal crosstalk. Int J Mol Sci. (2020) 21:2391. doi: 10.3390/ijms21072391
33. Yang Z, Roth K, Agarwal M, Liu W, Petriello MC. The transcription factors Crebh, Ppara, and foxo1 as critical hepatic mediators of diet-induced metabolic dysregulation. J Nutr Biochem. (2021) 95:108633. doi: 10.1016/j.jnutbio.2021.108633
34. Allenby G, Bocquel MT, Saunders M, Kazmer S, Speck J, Rosenberger M, et al. Retinoic acid receptors and retinoid X receptors: interactions with endogenous retinoic acids. Proc Natl Acad Sci USA. (1993) 90:30–4. doi: 10.1073/pnas.90.1.30
35. Mangelsdorf DJ, Borgmeyer U, Heyman RA, Zhou JY, Ong ES, Oro AE, et al. Characterization of three Rxr genes that mediate the action of 9-Cis retinoic acid. Genes Dev. (1992) 6:329–44. doi: 10.1101/gad.6.3.329
36. Barbier O, Fontaine C, Fruchart JC, Staels B. Genomic and non-genomic interactions of Ppar alpha with xenobiotic-metabolizing enzymes. Trends Endocrinol Metab. (2004) 15:324–30. doi: 10.1016/j.tem.2004.07.007
37. Barbier O, Torra IP, Duguay Y, Blanquart C, Fruchart JC, Glineur C, et al. Pleiotropic actions of peroxisome proliferator-activated receptors in lipid metabolism and atherosclerosis. Arterioscler Thromb Vasc Biol. (2002) 22:717–26. doi: 10.1161/01.atv.0000015598.86369.04
38. Bocher V, Pineda-Torra I, Fruchart JC, Staels B. Ppars: transcription factors controlling lipid and lipoprotein metabolism. Ann N Y Acad Sci. (2002) 967:7–18. doi: 10.1111/j.1749-6632.2002.tb04258.x
39. Etgen GJ, Mantlo N. Ppar ligands for metabolic disorders. Curr Top Med Chem. (2003) 3:1649–61. doi: 10.2174/1568026033451673
40. Gervois P, Chopin-Delannoy S, Fadel A, Dubois G, Kosykh V, Fruchart JC, et al. Fibrates increase human rev-erb alpha expression in liver via a novel peroxisome proliferator-activated receptor response element. Mol Endocrinol. (1999) 13:400–9. doi: 10.1210/mend.13.3.0248
41. IJpenberg A, Jeannin E, Wahli W, Desvergne B. Polarity and specific sequence requirements of peroxisome proliferator-activated receptor (Ppar)/retinoid X receptor heterodimer binding to DNA. A functional analysis of the malic enzyme gene Ppar response element. J Biol Chem. (1997) 272:20108–17. doi: 10.1074/jbc.272.32.20108
42. Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. (2008) 454:470–7. doi: 10.1038/nature07202
43. Kang Z, Fan R. Pparα and Ncor/Smrt corepressor network in liver metabolic regulation. Faseb J. (2020) 34:8796–809. doi: 10.1096/fj.202000055RR
44. Scarpulla RC. Metabolic control of mitochondrial biogenesis through the Pgc-1 family regulatory network. Biochim Biophys Acta. (2011) 1813:1269–78. doi: 10.1016/j.bbamcr.2010.09.019
45. Tahri-Joutey M, Andreoletti P, Surapureddi S, Nasser B, Cherkaoui-Malki M, Latruffe N. Mechanisms mediating the regulation of peroxisomal fatty acid beta-oxidation by Pparα. Int J Mol Sci. (2021) 22:8969. doi: 10.3390/ijms22168969
46. Santos GM, Fairall L, Schwabe JW. Negative regulation by nuclear receptors: a plethora of mechanisms. Trends Endocrinol Metab. (2011) 22:87–93. doi: 10.1016/j.tem.2010.11.004
47. Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, et al. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors Nf-Kappab and Ap-1. J Biol Chem. (1999) 274:32048–54. doi: 10.1074/jbc.274.45.32048
48. Bougarne N, Paumelle R, Caron S, Hennuyer N, Mansouri R, Gervois P, et al. Pparalpha blocks glucocorticoid receptor alpha-mediated transactivation but cooperates with the activated glucocorticoid receptor alpha for transrepression on Nf-Kappab. Proc Natl Acad Sci USA. (2009) 106:7397–402. doi: 10.1073/pnas.0806742106
49. Pawlak M, Baugé E, Bourguet W, De Bosscher K, Lalloyer F, Tailleux A, et al. The transrepressive activity of peroxisome proliferator-activated receptor alpha is necessary and sufficient to prevent liver fibrosis in mice. Hepatology. (2014) 60:1593–606. doi: 10.1002/hep.27297
50. Holzer RG, Park EJ, Li N, Tran H, Chen M, Choi C, et al. Saturated fatty acids induce C-Src clustering within membrane subdomains, leading to Jnk activation. Cell. (2011) 147:173–84. doi: 10.1016/j.cell.2011.08.034
51. Min RWM, Aung FWM, Liu B, Arya A, Win S. Mechanism and therapeutic targets of C-Jun-N-terminal kinases activation in nonalcoholic fatty liver disease. Biomedicines. (2022) 10:2035. doi: 10.3390/biomedicines10082035
52. Vernia S, Cavanagh-Kyros J, Garcia-Haro L, Sabio G, Barrett T, Jung DY, et al. The Pparα-Fgf21 hormone axis contributes to metabolic regulation by the hepatic Jnk signaling pathway. Cell Metab. (2014) 20:512–25. doi: 10.1016/j.cmet.2014.06.010
53. Gervois P, Vu-Dac N, Kleemann R, Kockx M, Dubois G, Laine B, et al. Negative regulation of human fibrinogen gene expression by peroxisome proliferator-activated receptor alpha agonists via inhibition of Ccaat Box/enhancer-binding protein beta. J Biol Chem. (2001) 276:33471–7. doi: 10.1074/jbc.M102839200
54. Blanquart C, Mansouri R, Paumelle R, Fruchart JC, Staels B, Glineur C. The protein kinase c signaling pathway regulates a molecular switch between transactivation and transrepression activity of the peroxisome proliferator-activated receptor alpha. Mol Endocrinol. (2004) 18:1906–18. doi: 10.1210/me.2003-0327
55. Oka S, Alcendor R, Zhai P, Park JY, Shao D, Cho J, et al. Pparα-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the Err transcriptional pathway. Cell Metab. (2011) 14:598–611. doi: 10.1016/j.cmet.2011.10.001
56. Oka S, Zhai P, Alcendor R, Park JY, Tian B, Sadoshima J. Suppression of Err targets by a Pparα/Sirt1 complex in the failing heart. Cell Cycle. (2012) 11:856–64. doi: 10.4161/cc.11.5.19210
57. Shin M, Kim I, Inoue Y, Kimura S, Gonzalez FJ. Regulation of mouse hepatic alpha-amino-beta-carboxymuconate-epsilon-semialdehyde decarboxylase, a key enzyme in the tryptophan-nicotinamide adenine dinucleotide pathway, by hepatocyte nuclear factor 4alpha and peroxisome proliferator-activated receptor alpha. Mol Pharmacol. (2006) 70:1281–90. doi: 10.1124/mol.106.026294
58. Velázquez-Villegas LA, Charabati T, Contreras AV, Alemán G, Torres N, Tovar AR. Pparα downregulates hepatic glutaminase expression in mice fed diets with different protein: carbohydrate ratios. J Nutr. (2016) 146:1634–40. doi: 10.3945/jn.116.232868
59. Tobón-Cornejo S, Vargas-Castillo A, Leyva-Martínez A, Ortíz V, Noriega LG, Velázquez-Villegas LA, et al. Pparα/Rxrα downregulates amino acid catabolism in the liver via interaction with hnf4α promoting its proteasomal degradation. Metabolism. (2021) 116:154705. doi: 10.1016/j.metabol.2021.154705
60. Leuenberger N, Pradervand S, Wahli W. Sumoylated Pparalpha mediates sex-specific gene repression and protects the liver from estrogen-induced toxicity in mice. J Clin Invest. (2009) 119:3138–48. doi: 10.1172/jci39019
61. Mogilenko DA, Kudriavtsev IV, Shavva VS, Dizhe EB, Vilenskaya EG, Efremov AM, et al. Peroxisome proliferator-activated receptor α positively regulates complement C3 expression but inhibits tumor necrosis factor α-mediated activation of C3 gene in mammalian hepatic-derived cells. J Biol Chem. (2013) 288:1726–38. doi: 10.1074/jbc.M112.437525
62. van der Meer DL, Degenhardt T, Väisänen S, de Groot PJ, Heinäniemi M, de Vries SC, et al. Profiling of promoter occupancy by pparalpha in human hepatoma cells via chip-chip analysis. Nucleic Acids Res. (2010) 38:2839–50. doi: 10.1093/nar/gkq012
63. You M, Jin J, Liu Q, Xu Q, Shi J, Hou Y. Pparα promotes cancer cell glut1 transcription repression. J Cell Biochem. (2017) 118:1556–62. doi: 10.1002/jcb.25817
64. Gou Q, Dong C, Jin J, Liu Q, Lu W, Shi J, et al. Pparα agonist alleviates tumor growth and chemo-resistance associated with the inhibition of glucose metabolic pathway. Eur J Pharmacol. (2019) 863:172664. doi: 10.1016/j.ejphar.2019.172664
65. Zhang MA, Ahn JJ, Zhao FL, Selvanantham T, Mallevaey T, Stock N, et al. Antagonizing peroxisome proliferator-activated receptor α activity selectively enhances Th1 immunity in male mice. J Immunol. (2015) 195:5189–202. doi: 10.4049/jimmunol.1500449
66. Marrapodi M, Chiang JY. Peroxisome proliferator-activated receptor alpha (Pparalpha) and agonist inhibit cholesterol 7alpha-hydroxylase gene (Cyp7a1) transcription. J Lipid Res. (2000) 41:514–20.
67. Patel DD, Knight BL, Soutar AK, Gibbons GF, Wade DP. The effect of peroxisome-proliferator-activated receptor-alpha on the activity of the cholesterol 7 alpha-hydroxylase gene. Biochem J. (2000) 351:747–53.
68. Post SM, Duez H, Gervois PP, Staels B, Kuipers F, Princen HM. Fibrates suppress bile acid synthesis via peroxisome proliferator-activated receptor-alpha-mediated downregulation of cholesterol 7alpha-hydroxylase and sterol 27-hydroxylase expression. Arterioscler Thromb Vasc Biol. (2001) 21:1840–5. doi: 10.1161/hq1101.098228
69. Berthou L, Duverger N, Emmanuel F, Langouët S, Auwerx J, Guillouzo A, et al. Opposite regulation of human versus mouse apolipoprotein a-i by fibrates in human apolipoprotein a-i transgenic mice. J Clin Invest. (1996) 97:2408–16. doi: 10.1172/jci118687
70. Vu-Dac N, Chopin-Delannoy S, Gervois P, Bonnelye E, Martin G, Fruchart JC, et al. The nuclear receptors peroxisome proliferator-activated receptor alpha and rev-erb alpha mediate the species-specific regulation of apolipoprotein a-I expression by fibrates. J Biol Chem. (1998) 273:25713–20. doi: 10.1074/jbc.273.40.25713
71. Vu-Dac N, Schoonjans K, Laine B, Fruchart JC, Auwerx J, Staels B. Negative regulation of the human apolipoprotein a-I promoter by fibrates can be attenuated by the interaction of the peroxisome proliferator-activated receptor with its response element. J Biol Chem. (1994) 269:31012–8.
72. Haubenwallner S, Essenburg AD, Barnett BC, Pape ME, DeMattos RB, Krause BR, et al. Hypolipidemic activity of select fibrates correlates to changes in hepatic apolipoprotein C-Iii expression: a potential physiologic basis for their mode of action. J Lipid Res. (1995) 36:2541–51.
73. Raspé E, Duez H, Mansén A, Fontaine C, Fiévet C, Fruchart JC, et al. Identification of rev-erb alpha as a physiological repressor of apoc-Iii gene transcription. J Lipid Res. (2002) 43:2172–9. doi: 10.1194/jlr.m200386-jlr200
74. Staels B, Vu-Dac N, Kosykh VA, Saladin R, Fruchart JC, Dallongeville J, et al. Fibrates downregulate apolipoprotein C-Iii expression independent of induction of peroxisomal acyl coenzyme a oxidase. A potential mechanism for the hypolipidemic action of fibrates. J Clin Invest. (1995) 95:705–12. doi: 10.1172/jci117717
75. Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, et al. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science. (2011) 331:1315–9. doi: 10.1126/science.1198125
76. Makled MN, El-Kashef DH. Saroglitazar attenuates renal fibrosis induced by unilateral ureteral obstruction via inhibiting Tgf-B/Smad signaling pathway. Life Sci. (2020) 253:117729. doi: 10.1016/j.lfs.2020.117729
77. Makled MN, Sharawy MH, El-Awady MS. The dual Ppar-A/Γ agonist saroglitazar ameliorates thioacetamide-induced liver fibrosis in rats through regulating leptin. Naunyn Schmiedebergs Arch Pharmacol. (2019) 392:1569–76. doi: 10.1007/s00210-019-01703-5
78. Bansal T, Chatterjee E, Singh J, Ray A, Kundu B, Thankamani V, et al. Arjunolic acid, a peroxisome proliferator-activated receptor α agonist, regresses cardiac fibrosis by inhibiting non-canonical Tgf-B signaling. J Biol Chem. (2017) 292:16440–62. doi: 10.1074/jbc.M117.788299
79. Ye G, Gao H, Wang Z, Lin Y, Liao X, Zhang H, et al. Pparα and Pparγ activation attenuates total free fatty acid and triglyceride accumulation in macrophages via the inhibition of fatp1 expression. Cell Death Dis. (2019) 10:39. doi: 10.1038/s41419-018-1135-3
80. Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS, et al. Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol. (2012) 92:829–39. doi: 10.1189/jlb.1111537
81. Yagai T, Yan T, Luo Y, Takahashi S, Aibara D, Kim D, et al. Feedback repression of Pparα signaling by Let-7 microrna. Cell Rep. (2021) 36:109506. doi: 10.1016/j.celrep.2021.109506
82. Luo Y, Xie C, Brocker CN, Fan J, Wu X, Feng L, et al. Intestinal Pparα protects against colon carcinogenesis via regulation of methyltransferases Dnmt1 and Prmt6. Gastroenterology. (2019) 157:744–59.e4. doi: 10.1053/j.gastro.2019.05.057
83. Aibara D, Takahashi S, Yagai T, Kim D, Brocker CN, Levi M, et al. Gene repression through epigenetic modulation by Ppara enhances hepatocellular proliferation. iScience. (2022) 25:104196. doi: 10.1016/j.isci.2022.104196
84. Shah YM, Morimura K, Yang Q, Tanabe T, Takagi M, Gonzalez FJ. Peroxisome proliferator-activated receptor alpha regulates a microrna-mediated signaling cascade responsible for hepatocellular proliferation. Mol Cell Biol. (2007) 27:4238–47. doi: 10.1128/mcb.00317-07
85. Shi Y, Tao T, Liu N, Luan W, Qian J, Li R, et al. Pparα, a predictor of patient survival in glioma, inhibits cell growth through the E2f1/Mir-19a feedback loop. Oncotarget. (2016) 7:84623–33. doi: 10.18632/oncotarget.13170
86. Gao Y, Han D, Sun L, Huang Q, Gai G, Wu Z, et al. Pparα regulates the proliferation of human glioma cells through mir-214 and E2f2. Biomed Res Int. (2018) 2018:3842753. doi: 10.1155/2018/3842753
87. Gehrke N, Hövelmeyer N, Waisman A, Straub BK, Weinmann-Menke J, Wörns MA, et al. Hepatocyte-specific deletion of Il1-Ri attenuates liver injury by blocking Il-1 driven autoinflammation. J Hepatol. (2018) 68:986–95. doi: 10.1016/j.jhep.2018.01.008
88. Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A randomized, controlled trial of the pan-Ppar agonist lanifibranor in NASH. N Engl J Med. (2021) 385:1547–58. doi: 10.1056/NEJMoa2036205
89. Gawrieh S, Noureddin M, Loo N, Mohseni R, Awasty V, Cusi K, et al. Saroglitazar, a Ppar-A/Γ agonist, for treatment of Nafld: a randomized controlled double-blind phase 2 trial. Hepatology. (2021) 74:1809–24. doi: 10.1002/hep.31843
90. Nakajima A, Eguchi Y, Yoneda M, Imajo K, Tamaki N, Suganami H, et al. Randomised clinical trial: pemafibrate, a novel selective peroxisome proliferator-activated receptor A modulator (Spparmα), versus placebo in patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther. (2021) 54:1263–77. doi: 10.1111/apt.16596
Keywords: NAFLD, NASH, PPARα, transcriptional suppressor, transcriptional suppression
Citation: Yagai T and Nakamura T (2022) Mechanistic insights into the peroxisome proliferator-activated receptor alpha as a transcriptional suppressor. Front. Med. 9:1060244. doi: 10.3389/fmed.2022.1060244
Received: 03 October 2022; Accepted: 08 November 2022;
Published: 25 November 2022.
Edited by:
Nicholas Chew, National University Heart Centre, Singapore (NUHCS), SingaporeReviewed by:
Shuibang Wang, National Institutes of Health (NIH), United StatesCopyright © 2022 Yagai and Nakamura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomoki Yagai, dG9tb2tpLnlhZ2FpLmEyQHRvaG9rdS5hYy5qcA==; Takahisa Nakamura, dGFrYWhpc2EubmFrYW11cmFAY2NobWMub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.