 Ruru Guo1
Ruru Guo1 Wei Liu
Wei Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Med. , 13 December 2022
Sec. Ophthalmology
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.1042588
Purpose: To explore the clinical phenotype and genetic defects of families with congenital aniridia.
Methods: Four Chinese families with aniridia were enrolled in this study. The detailed ocular presentations of the patients were recorded. Whole exome sequencing (BGI MGIEasy V4 chip) was used to detect the gene mutation. Sanger sequencing was performed to validate the potential pathogenic variants, and segregation analysis was performed on all available family members.
Results: By whole exome sequencing and Sanger sequencing, three recurrent mutations (c.112del, p.Arg38Glyfs*16; c.299G > A, p.Trp100* and c.718C > T, p.Arg240*) and one novel mutation (c.278_281del, p.Glu93Alafs*30) of PAX6 were identified. All the mutations were co-segregated with the phenotype in the families. We also observed spontaneous anterior lens capsule rupture in aniridia for the first time.
Conclusion: We report spontaneous anterior lens capsule rupture as a novel phenotype of aniridia and three recurrent mutations and one novel mutation of PAX6 in families with aniridia. Our results expanded the phenotype and genotype spectra of aniridia and can help us better understand the disease.
Aniridia is a rare, bilateral, congenital panocular disorder that causes complete or partial absence of the iris and iris hypoplasia. The incidence of aniridia ranges from 1 in 64,000 to 1 in 96,000 population, with no known preference for race or sex (1–3). Aniridia can be accompanied by a range of other ocular anomalies, including corneal abnormalities, cataract, ectopia lentis, glaucoma, strabismus, refractive error, ptosis, microphthalmia, foveal hypoplasia, and optic nerve hypoplasia. Non-ocular sensory and neurological deficits can also be present, including hearing difficulties, reduced olfaction, and Wilms tumor, aniridia, genitourinary anomalies, and intellectual disability (WAGR) syndrome (3). About two-thirds of aniridia cases are familial and inherited as an autosomal dominant trait, with complete penetrance and variable expressivity (4, 5). Most aniridia cases are associated with mutations in the pair box 6 (PAX6) gene at chromosome 11p13.
PAX6 was firstly proposed as the causative gene of congenital aniridia in 1991 by positional cloning (6, 7). Human PAX6 encodes a 422-amino acid transcriptional regulatory protein, which consists of two DNA-binding domains (a paired domain at the NH2 terminal, including 128 amino acids, and a homeodomain, including 61 amino acids) separated by a 79-amino acid linker region, and a transcriptional transactivation domain at the COOH terminus rich in proline, serine, and threonine (6, 7). PAX6 is expressed in early eye structures, the forebrain, the neural tube, and the pancreas and plays a crucial role in the development of the eye and central nervous system (8, 9). The majority of PAX6 mutations result in null alleles and consequent PAX6 haploinsufficiency and lead to aniridia. Other ocular abnormalities have also been associated with PAX6 changes. Isolated foveal hypoplasia has been described in few families with PAX6 missense mutations [p.Pro76Arg, p.Arg128Cys, p.Gly72Ser (10–12)] or premature termination codon (PTC) mutations [p.Pro346Aspfs*20, p.Tyr354Cysfs*8 (11)]. Microphthalmia and Peters anomaly are associated with several PAX6 mutations, most of which are missense, while anophthalmia is associated with homozygous PAX6 variants (13). With the advances in neuroimaging technologies, both anatomical (changes in the anterior commissure, posterior commissure, pineal gland, corpus callosum, optic chiasm, and olfactory bulb) and functional neuro-abnormalities (deficits in olfactory function, cognitive ability, and auditory interhemispheric transfer) have been described in individuals with aniridia (14), highlighting the crucial role of PAX6 in development of central nervous system. However, the exact phenotype-genotype correlation is still unclear.
In this study, we enrolled four Chinese families with aniridia to explore the genetic defects of congenital aniridia.
The study was approved by the ethics committee of Tianjin Medical University Eye Hospital and was in compliance with the regulations of the Declaration of Helsinki. Informed consent was obtained from the enrolled family members. The method of whole exome sequencing and data analysis is described in detail previously (15–17). Briefly, genomic DNA was extracted from venous blood samples according to the manufacturer’s standard procedure (MagPure Buffy Coat DNA Midi KF Kit, Magen, China) and sequenced on MGISEQ-2000 (PE100) using the BGI MGIEasy V4 chip, which contains exons of all human genes and their adjacent ±20 bp introns. Sanger sequencing was then performed to validate the potential pathogenic variants, and segregation analysis was performed on all available family members. The primers for Sanger sequencing are summarized in Supplementary Table 1. Additionally, the structures of the mutant and homomeric wild-type PAX6 were modeled by the SWISS-MODEL server1 and shown using a PyMOL Molecular Graphic system. The PAX6 PDB file (AF-P26367-F1-model_v1.pdb) was downloaded from the AlphaFold Protein Structure Database2 and was used as a template.
We enrolled four Chinese families with aniridia in this study.
The proband of family 1 was a 14-year-old girl. The detailed ocular presentations of the affected members in family 1 are summarized in Table 1. Briefly, 11 individuals in the family were affected and presented with complete aniridia, while four members had nystagmus. All the affected individuals had poor vision (ranging from finger counting to 0.12), and the intraocular pressure (IOP) was high in two patients. Lens abnormalities were detected in eight patients, including cortical cataract, nuclear cataract, total cataract, and lens ectopia. We also observed spontaneous anterior lens capsule (ALC) rupture in two of the three patients with total cataracts, which was never reported before in patients with aniridia. Unfortunately, the ocular anterior segment photographs are not available in this family and a schematic picture was drawn to illustrate the changes (Figure 1).

Table 1. Ocular manifestations of affected individuals in aniridia family 1.
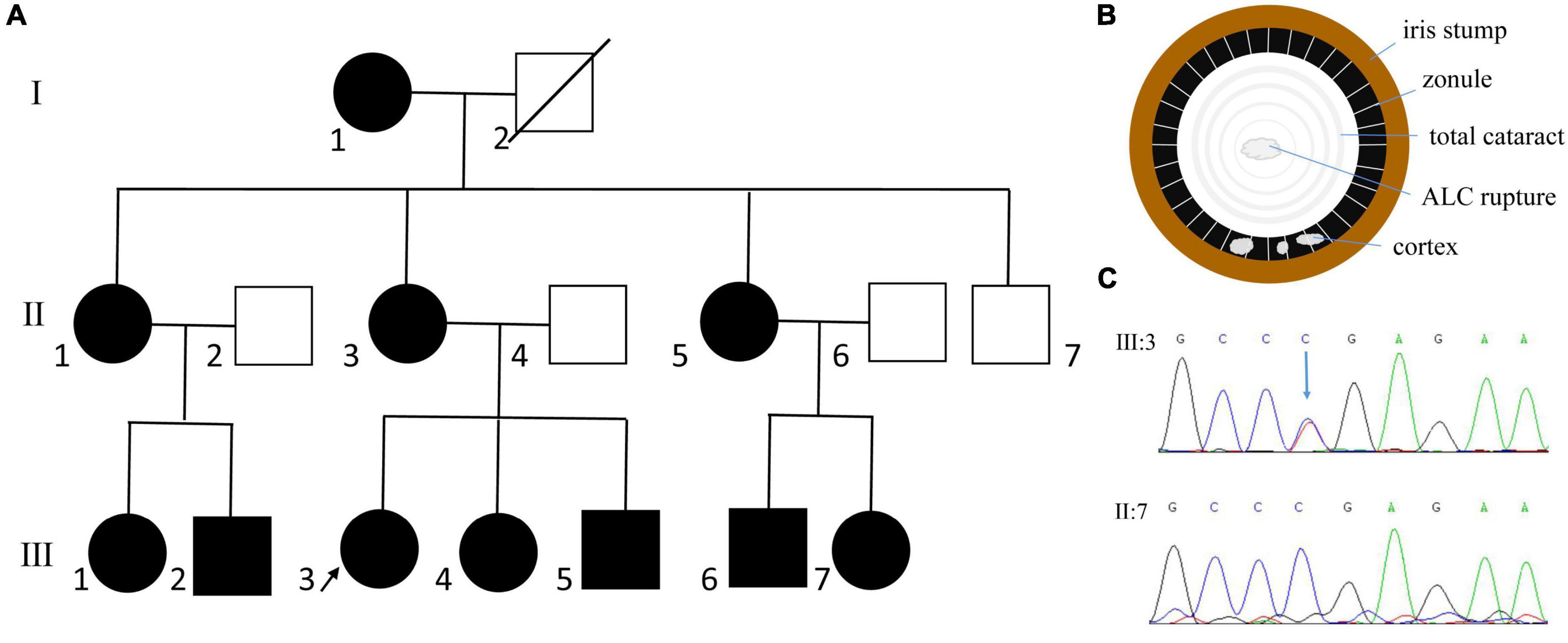
Figure 1. Clinical and genetic evaluation of family 1. (A) Pedigree map of family 1. The arrow indicates the proband. The circles and squares represent females and males, respectively. White and black denotes unaffected and affected individuals, respectively. (B) Schematic picture showing anterior lens capsule (ALC) rupture in patient with total cataract. (C) Sanger sequencing showing a previously reported nonsense mutation of PAX6 (c.718C > T, p.Arg240*).
By whole exome sequencing and Sanger sequencing, a previously reported nonsense mutation in PAX6 (c.718C > T, p.Arg240*) was detected (Figure 1). It was detected in all the affected individuals but not in the unaffected individuals in this family, indicating that the mutation was co-segregated with the phenotype.
The proband was a 30-year-old woman, and she had complained of blurred vision for more than 20 years. She was diagnosed with aniridia, nystagmus, congenital cataract (posterior polar cataract), and lens ectopia at the age of 15 years. At that time, her visual acuities were 0.12 and 0.1; IOP, 17.8 and 18.1 mmHg; axial lengths, 24.02 and 23.70 mm; central anterior chamber depths, 1.53 and 1.54 mm; flat K values, 39.5 and 39.0 D; steep K values, 40.5 and 40.0 D; endothelium cell densities, 3677.2/mm2 and 3780.2/mm2 in the right eye and left eyes, respectively. The patient accepted cataract extraction and intraocular lens implantation in 2007 for the right eye (Morcher GmbH Type 67G, + 22D) and in 2009 for the left eye (Morcher GmbH Type 67G, + 23D). After surgery, her vision improved to 0.2 in both eyes. In 2017, the patient complained of blurred vision again. On presentation, her vision was 0.1 and light perception, and the IOP was 29.1 mmHg in the right eye and 33.2 mmHg in the left eye, respectively. Slit-lamp microscopy (SLM) revealed aniridia and nystagmus, and both eyes were pseudophakic (Figure 2). A fundoscopy examination could not be performed because of severe nystagmus. The patient was given three anti-glaucoma medications to control the IOP. The patient’s mother had similar ocular presentations. The patient’s daughter was 3 months old and had nystagmus and aniridia in both eyes. The patient’s brother was found to have ectropion of the iris pigment epithelium in the right eye and iris coloboma in the left eye (Figure 2). He had normal vision and IOP in both eyes.
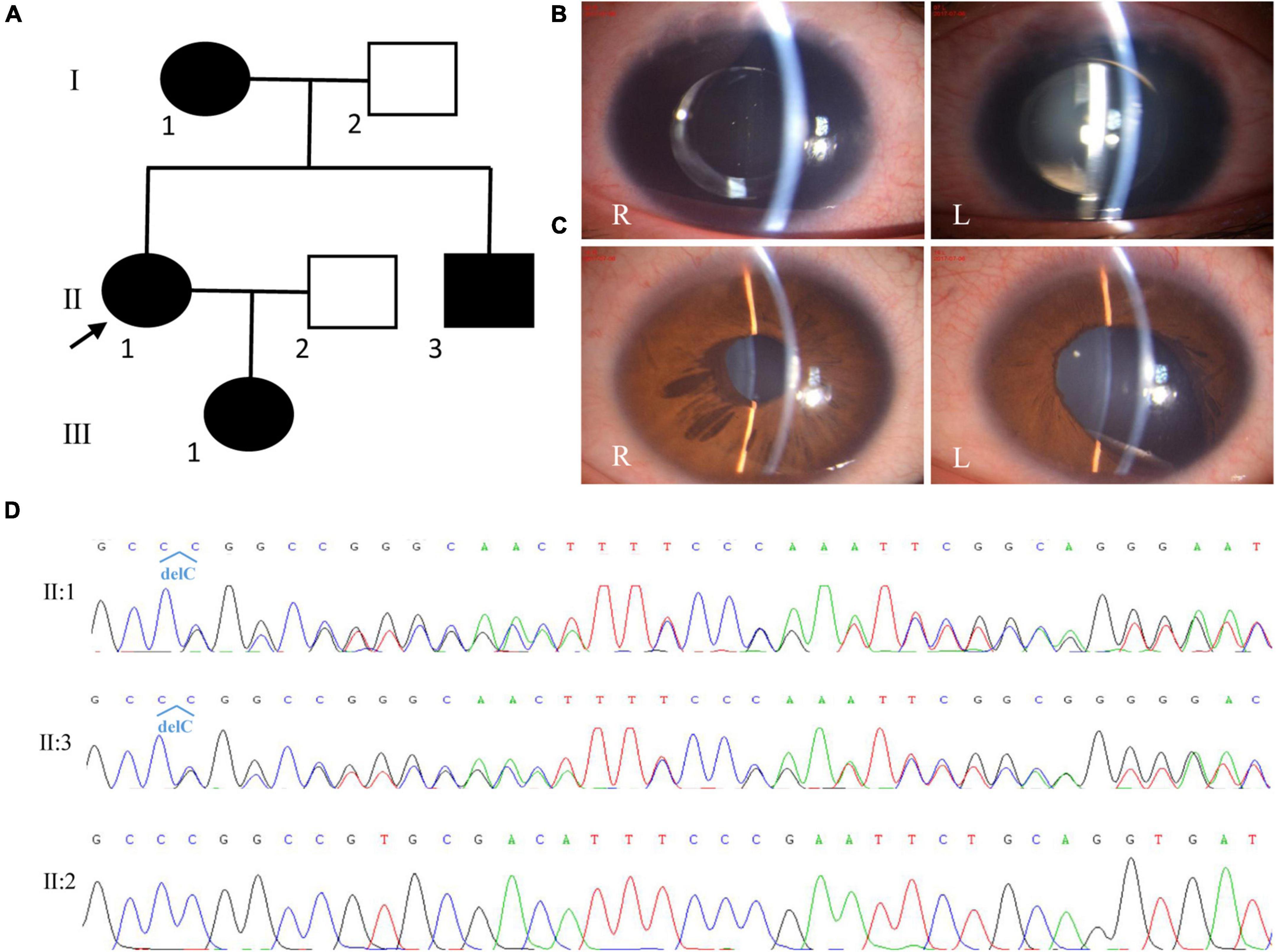
Figure 2. Clinical and genetic evaluations of family 2. (A) Pedigree map of family 2. (B) Anterior segment photograph showing complete aniridia and the intraocular lens in both eyes of the proband. (C) Anterior segment photograph showing ectropion of the iris pigment epithelium in the right eye (R) and iris coloboma in the left eye (L) of the proband’s brother. (D) Sanger sequencing showing a previously reported frame-shift mutation in PAX6 (c.112del, p.Arg38Glyfs*16).
By whole exome sequencing and Sanger sequencing, a previously reported frame-shift mutation in PAX6 (c.112del, p.Arg38Glyfs*16) was detected in the proband, her mother, and her brother (Figure 2). No blood sample was available from her daughter.
The proband of family 3 was a 34-year-old woman, and her medical records were reviewed. On presentation, her visual acuity was hand motion and 0.12, and her IOP was 12.7 mmHg in the right eye and 13.5 mmHg in the left eye. SLM revealed nystagmus, aniridia, cataracts (total cataract in the right eye and cortical cataract in the left eye), and lens ectopia in both eyes (Figure 3). In the right and left eyes, the axial lengths were 24.07 and 23.26 mm; central corneal thicknesses, 624 and 636 μm; endothelium cell densities, 3968.8/mm2 and 3275.1/mm2; flat K values, 40.45 and 40.21 D; steep K values, 43.12 and 42.15 D; corneal diameters, 11.45 and 11.24 mm, respectively. Ultrasonographic biomicroscopy revealed severe iris hypoplasia in both eyes (Figure 3). The patient accepted sequential cataract extraction of both eyes in 2009, without intraocular lens implantation. After that, the IOPs of both eyes increased (29.5 mmHg in the right eye and 26.6 mmHg in the left eye), and glaucomatous optic neuropathy occurred (cup-to-disc ratio: 0.8). In 2012, the patient accepted Ahmed glaucoma valve implantation for the right eye. One year later, the IOP in both eyes increased again, ranging from 20 to 30 mmHg, under three anti-glaucoma medications. The patient’s mother also had nystagmus, aniridia, cataracts, and glaucoma in both eyes.
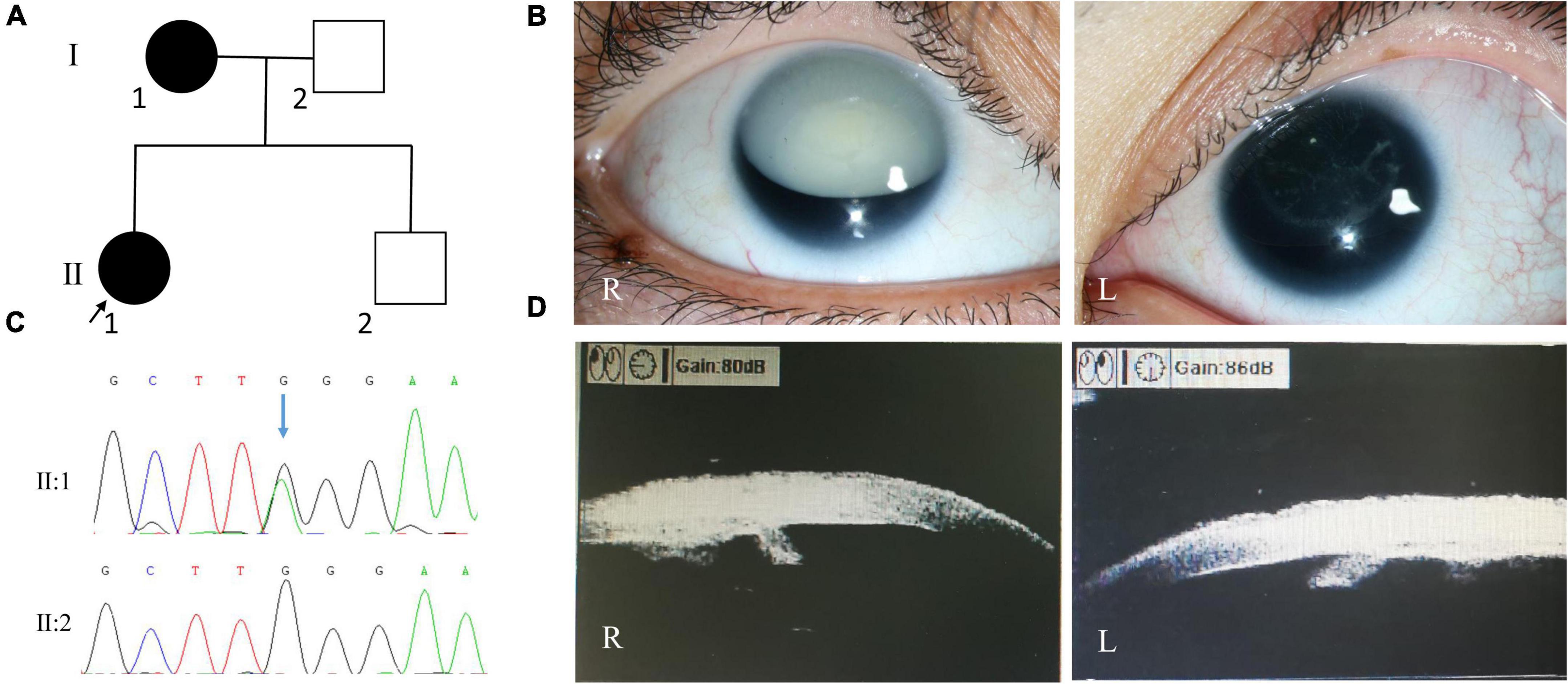
Figure 3. Clinical and genetic evaluations of family 3. (A) Pedigree map of family 3. (B) Anterior segment photograph showing white cataract of the right eye, cortical cataract of the left eye, and lens ectopia of both eyes in the proband. (C) Sanger sequencing showing a recurrent PAX6 nonsense mutation (c.299G > A, p.Trp100*). (D) Ultrasonographic biomicroscopy image showing severe iris hypoplasia in both eyes of the proband.
Whole exome sequencing revealed a recurrent PAX6 nonsense mutation (c.299G > A, p.Trp100*; Figure 3). Sanger sequencing revealed that the patient’s mother also carried this mutation, while her father and brother did not.
The proband of family 4 was a 66-year-old man. He complained of bilateral ocular pain for 1 month. He was diagnosed with aniridia when he was 6 years old. On presentation, his visual acuity was hand motion and no light perception, and his IOP was 35.7 mmHg in the right eye and 55.2 mmHg in the left eye. SLM revealed corneal neovascularization, aniridia, and nystagmus in both eyes (Figure 4). A dense nuclear cataract was found in the right eye, and severe corneal opacity was found in the left eye, which made the fundus invisible. Keratoplasty and cataract surgery of the right eye was suggested, but was refused by the patient. The patient was administered anti-glaucoma medications to control the IOP in both eyes and was still under follow-up.
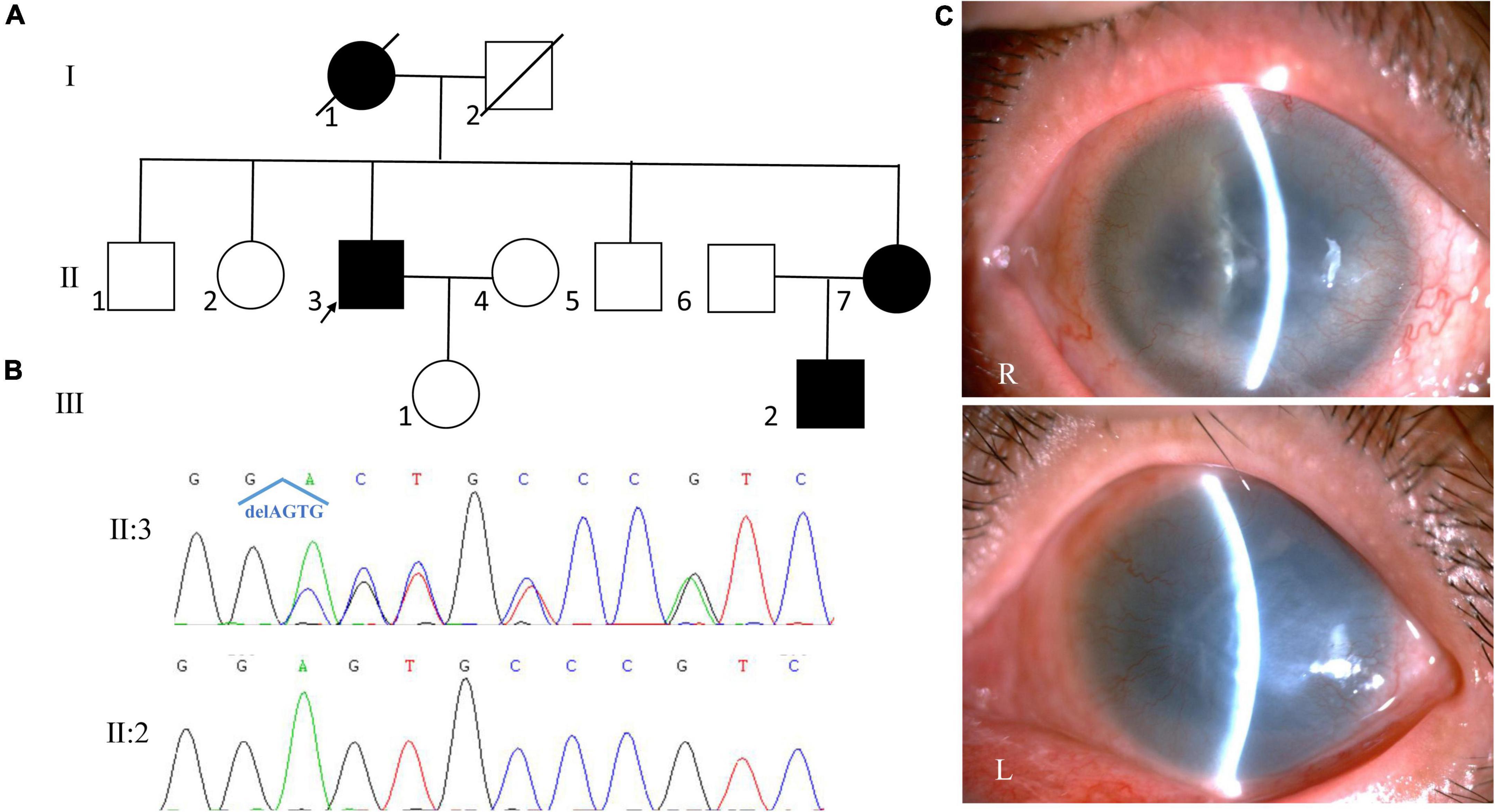
Figure 4. Clinical and genetic evaluations of family 4. (A) Pedigree map of family 4. (B) Sanger sequencing showing a novel frame-shift mutation of PAX6 (c.278_281del, p.Glu93Alafs*30). (C) Anterior segment photograph of the proband showing corneal neovascularization, aniridia in both eyes, a dense nuclear cataract in the right eye and severe corneal opacity in the left eye.
Whole exome sequencing and Sanger sequencing revealed a novel frame-shift mutation in PAX6 (c.278_281del, p.Glu93Alafs*30; Figure 4). The variant was detected by further Sanger sequencing in all affected patients enrolled in this study (III:2, III:3) but not in the unaffected family members (III:4, III:5, IV:2). The variant was co-segregated with the disease in family members and was not found in NCBI dbSNP, HapMap, 1000 human genome dataset, the database of 100 healthy Chinese adults, and ClinVar, suggesting that the variant may be the pathogenic mutation in this family.
The location of the four mutations detected in this study was highlighted in Figure 5.
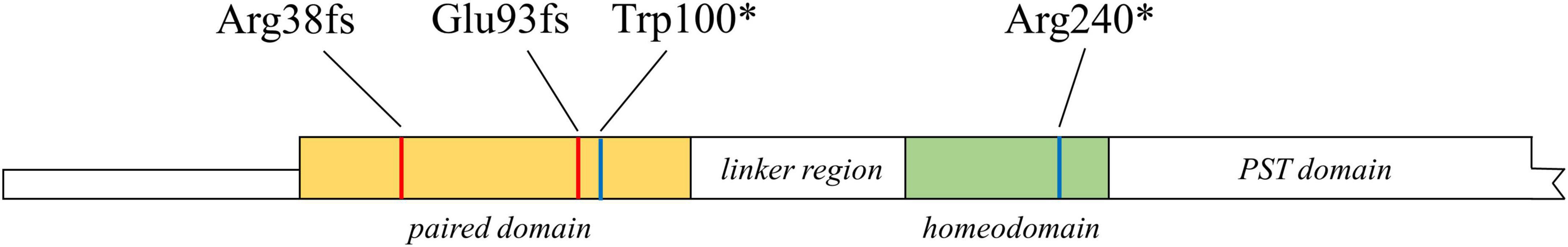
Figure 5. Schematic diagram of the location of the four mutations detected in this study in PAX6 protein. PST, proline, serine, and threonine. *Means a stop codon.
Compared with the wild-type PAX6 protein, all four mutations detected in this study were predicted to produce a truncated protein and interfere with DNA binding (Figure 6).
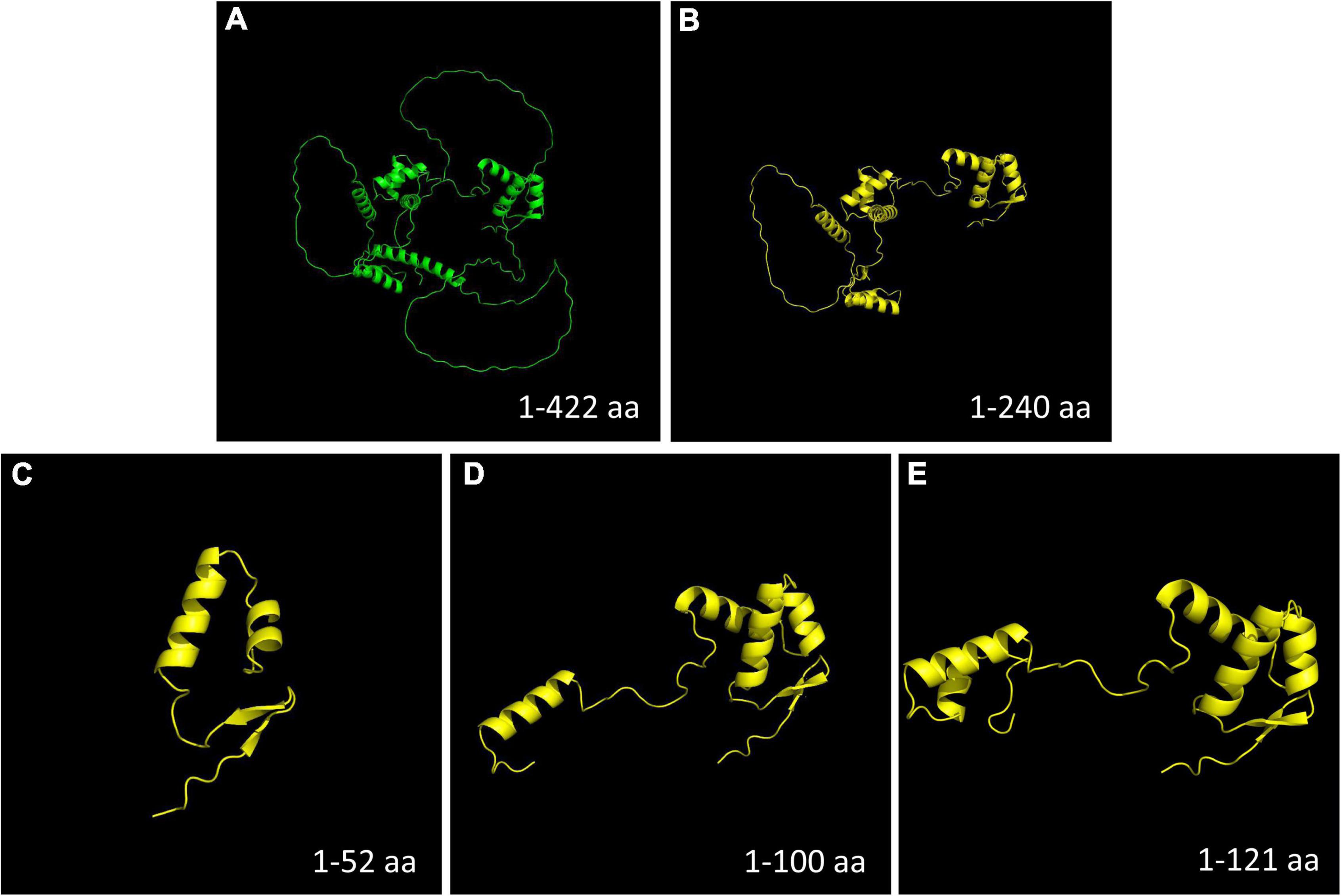
Figure 6. Protein model construction. Compared with the wild-type PAX6 protein, all the four mutations detected in this study were predicted to produce a truncated protein. (A) Wild-type PAX6 protein. (B) Mutant PAX6 protein of family 1 (c.718C > T, p.Arg240*). (C) Mutant PAX6 protein of family 2 (c.112del, p.Arg38Glyfs*16). (D) Mutant PAX6 protein of family 3 (c.299G > A, p.Trp100*). (E) Mutant PAX6 protein of family 4 (c.278_281del, p.Glu93Alafs*30). Aa, amino acids.
In this study, we enrolled four families with aniridia and identified three recurrent mutations (c.112del, p.Arg38Glyfs*16; c.299G > A, p.Trp100* and c.718C > T, p.Arg240*) and one novel mutation (c.278_281del, p.Glu93Alafs*30) of PAX6 by whole exome sequencing. We also found spontaneous ALC rupture in aniridia, which expanded the disease phenotype.
Although aniridia has a spectrum of ocular findings, cataract, glaucoma and keratopathy are main reasons for visual compromise in such patients. Cataracts may develop at any age and are usually progressive. The common morphological types of cataract include posterior polar, posterior subcapsular, and total cataract. Cataract surgery in aniridia is challenging, because of the poor ocular surface, corneal opacification, friable capsule and lens dislocation (18). Iris prosthesis implantation can also be considered in cataract surgery, although several complications may occur during or after the surgery. Glaucoma occurs in 46–70% of cases (19). Several possible mechanisms, including maldevelopment of the anterior chamber angle, absence of Schlemm’s canal, and obstruction of the angle with a shapeless, homogenous avascular tissue, are implicated in the development of glaucoma (18). Although medical therapy remains the first line of treatment, most patients with glaucoma eventually require surgical therapy, such as goniotomy, trabeculotomy, trabeculectomy, glaucoma drainage devices implantation and cyclodestructive procedures (19). However, the management of aniridic glaucoma remains a challenge. At present, there is a lack of prospective and randomized controlled studies on glaucoma treatment in aniridia, and the results from the existing studies are inconsistent. Aniridia-associated keratopathy (AAK), characterized by gradual corneal pannus and opacification, is another common cause of progressive vision loss. AAK is resulted from the breakdown of the limbal stem cell niche, impaired wound healing, and neural deterioration and can occur as early as 2 years of age (18). The management of AAK depends on its severity. In the early stages, supportive treatment includes artificial tear fluid without preservatives and autologous serum are generally recommended. When the keratopathy affects the visual axis, surgery intervention is often needed. Penetrating keratoplasty tends to fail eventually, owing to the underlying limbal stem cell deficiency. Even procedures attempting to restore or improve the limbal stem cell niche, such as amniotic membrane transplantation, limbal or keratolimbal allograft transplantation, cultivated limbal epithelial transplantation, cultivated oral mucosa epithelial transplantation, have a risk of surgery failure (18, 19). Keratoprosthesis is an option in treatment of end-stage AAK. Boston type 1 keratoprosthesis is reported to have good anatomical and functional long-term results in patients with AAK (20). Obviously, an appropriate individualized treatment regimen should be determined for patients with aniridia, in order to achieve the best chance of postoperative success with minimized complications.
According to the Human Gene Mutation Database (HGMD),3 over 750 PAX6 mutations were reported, with predicted premature truncations being the most common PAX6 mutations. Although so many mutations have been reported, the exact phenotype-genotype correlation of aniridia is not yet clear. However, patients with nonsense or frame-shift mutations that leading to the introduction of a PTC tend to present classical aniridia phenotype (21–23), which is in accordance with our findings. All four mutations detected in this study were truncating mutations, and all four families presented with typical aniridia. It has also been reported that missense mutations are usually associated with milder atypical aniridia phenotypes (21, 22), probably resulting from the prediction that the PAX6 proteins of missense mutations retain some of their functions (24). Nevertheless, a recent statistical analysis of the genotype-phenotype correlations in PAX6-associated aniridia revealed that missense mutations were associated with a severe aniridia phenotype similar to the truncating mutations, because some formally missense mutations could disrupt splicing and lead to nonsense-mediated decay (NMD) (25). Obviously, the exact genotype-phenotype correlations remain to be further elucidated.
Phenotypic variability is frequently reported in aniridia, even among members of the same family (13, 22, 26–30). In an analysis of 155 patients with aniridia, complete aniridia was observed in 78% patients, and foveal hypoplasia, cataract, nystagmus, keratopathy, and glaucoma was observed in 85, 80, 78, 58, and 26% patients, respectively (25), which is in accordance with other reports (23, 29). In our study, there were 18 patients with aniridia whose ocular presentations were available. Complete aniridia was the predominant ocular manifestation, which occurred in 17 of the 18 patients (94.4%). Cataract, nystagmus, glaucoma, lens ectopia, strabismus, ALC rupture, keratopathy and iris coloboma occurred in 66.7% (12/18), 55.6% (10/18), 33.3% (6/18), 22.2% (4/18), 11.1% (2/18), 11.1% (2/18), 5.6% (1/18) and 5.6% (1/18) of the patients, respectively. Generally, the incidences of each ocular presentation was comparable to that of other reports (23, 25, 29), except for keratopathy. The percentage of keratopathy was much smaller in our study, this may be resulted from that the enrolled patients were generally younger and aniridia-associated keratopathy is age-dependent (18). Our results also highlight the high phenotypic heterogeneity of aniridia (Table 2). In family 2, the proband presented with complete aniridia, while her brother, who carried the same mutation as the proband, was only found to have ectropion of the iris pigment epithelium in the right eye and iris coloboma in the left eye. In family 1, although all the patients presented with complete aniridia, nystagmus was present in only four members, and the lens abnormalities were variable among the affected individuals. We also observed spontaneous ALC rupture in the patients with aniridia who had total cataracts in family 1, which has not previously been reported. We think that the occurrence of ALC rupture in aniridia is reasonable because it has been discovered that the ALC of patients with aniridia is thinner and more fragile (31–33). Moreover, the intumescence of total cataracts can also facilitate the rupture of the thinner and fragile ALC. This phenotypic variability is not correlated with the location or the nature of the mutation (13). It is still far from clear how PAX6 mutations translate into variable expressivity among individuals with aniridia from the same or different families (13, 22).
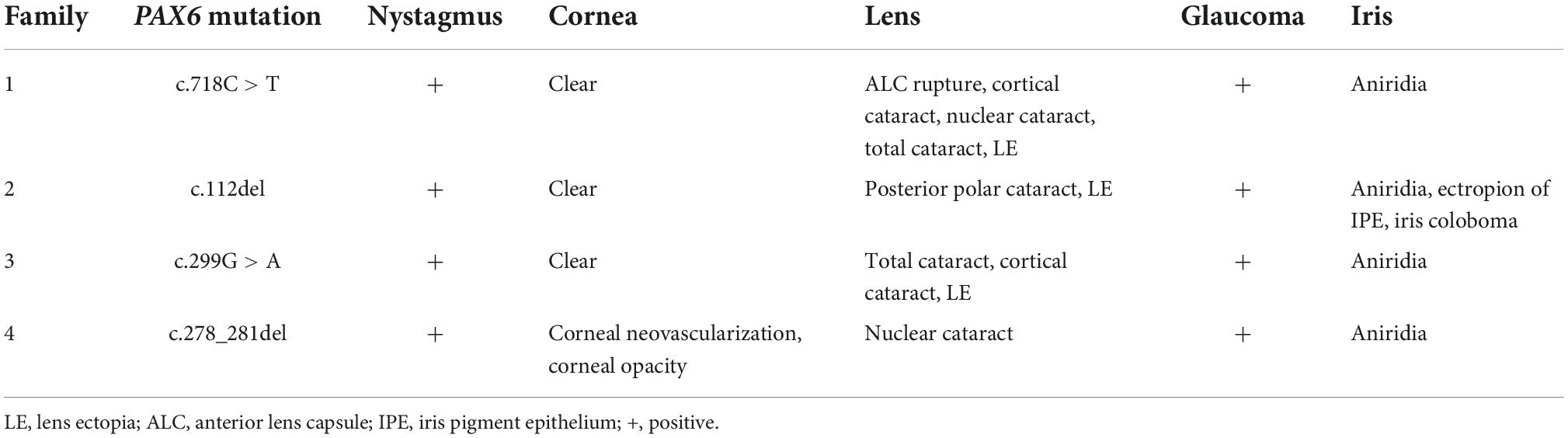
Table 2. Phenotypes and genotypes of the families with aniridia.
Thus far, most PAX6 pathogenic mutations are PTC mutations, including nonsense, frame-shift, and splice-site mutations, which are predicted to produce truncated proteins (Figure 6). It is now accepted that mutations leading to PTC located up to 50 bp upstream of the last exon of PAX6 will give rise to NMD (34). NMD is a process by which abnormal mRNAs resulting from a PTC are degraded before large quantities of truncated proteins are produced (35–37). This is considered a protective mechanism by which abnormal aggregation of truncated protein products in the cell can be prevented. This mechanism illustrates the etiology of aniridia in the families enrolled in this study. All four mutations detected in our study were PTC mutations located before the last 50 bp upstream of the last exon junction and would theoretically activate NMD, which will produce a mutant null allele, resulting in haploinsufficiency (loss of function of one copy). The mRNA transcribed from a single functional allele will lead to a 50% reduction in the PAX6 protein level, which is insufficient to trigger the transcription of its downstream target genes, and consequently, hinder normal eye development and lead to aniridia (38–41).
For the first time, we report spontaneous ALC rupture in aniridia and detected three recurrent mutations (c.112del, p.Arg38Glyfs*16; c.299G > A, p.Trp100*; and c.718C > T, p.Arg240*) and one novel mutation (c.278_281del, p.Glu93Alafs*30) of PAX6 in families with aniridia. Our results expanded the phenotype and genotype spectra of aniridia and can help us better understand the disease.
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Ethics Committee of Tianjin Medical University Eye Hospital. Written informed consent to participate in this study was provided by the participants or their legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
WL and JJ designed and supervised the study. RG and XZ collected the data. RG drafted the manuscript. WL, RG, AL, and JJ analyzed the data. All authors read and approved the final manuscript.
This study was supported by General Project of Natural Science Foundation of Xinjiang Uygur Autonomous Region (2020D01A06); Open Project of Tianjin Key Laboratory of Retinal Functions and Diseases (2020tjswmq003); Youth Special Fund of Clinical Research of Tianjin Medical University Eye Hospital (2020QN02); and Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-037A).
We would like to thank the patients and their family members for their participation in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2022.1042588/full#supplementary-material
IOP, intraocular pressure; SLM, slit-lamp microscopy; ALC, anterior lens capsule; NMD, nonsense-mediated decay; PTC, premature termination codon.
2. Edén U, Iggman D, Riise R, Tornqvist K. Epidemiology of aniridia in Sweden and Norway. Acta Ophthalmol. (2008) 86:727–9. doi: 10.1111/j.1755-3768.2008.01309.x
3. Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. (2012) 20:1011–7. doi: 10.1038/ejhg.2012.100
4. Valenzuela A, Cline R. Ocular and nonocular findings in patients with aniridia. Can J Ophthalmol. (2004) 39:632–8. doi: 10.1016/S0008-4182(04)80028-0
6. Ton C, Hirvonen H, Miwa H, Weil M, Monaghan P, Jordan T, et al. Positional cloning and characterization of a paired box- and homeobox-containing gene from the aniridia region. Cell. (1991) 67:1059–74. doi: 10.1016/0092-8674(91)90284-6
7. Glaser T, Walton D, Maas R. Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat Genet. (1992) 2:232–9. doi: 10.1038/ng1192-232
8. van Heyningen V, Williamson K. PAX6 in sensory development. Hum Mol Genet. (2002) 11:1161–7. doi: 10.1093/hmg/11.10.1161
9. Simpson T, Price D. Pax6: a pleiotropic player in development. Bioessays. (2002) 24:1041–51. doi: 10.1002/bies.10174
10. Azuma N, Nishina S, Yanagisawa H, Okuyama T, Yamada M. PAX6 missense mutation in isolated foveal hypoplasia. Nat Genet. (1996) 13:141–2. doi: 10.1038/ng0696-141
11. Hingorani M, Williamson K, Moore A, van Heyningen V. Detailed ophthalmologic evaluation of 43 individuals with PAX6 mutations. Invest Ophthalmol Vis Sci. (2009) 50:2581–90. doi: 10.1167/iovs.08-2827
12. Thomas S, Thomas M, Andrews C, Chan W, Proudlock F, McLean R, et al. Autosomal-dominant nystagmus, foveal hypoplasia and presenile cataract associated with a novel PAX6 mutation. Eur J Hum Genet. (2014) 22:344–9. doi: 10.1038/ejhg.2013.162
13. Dubey S, Mahalaxmi N, Vijayalakshmi P, Sundaresan P. Mutational analysis and genotype-phenotype correlations in southern Indian patients with sporadic and familial aniridia. Mol Vis. (2015) 21:88–97.
14. Grant M, Bobilev A, Branch A, Lauderdale J. Structural and functional consequences of PAX6 mutations in the brain: implications for aniridia. Brain Res. (2021) 1756:147283.
15. Liu W, Guo R, Hao H, Ji J. Identification of a novel RHO heterozygous nonsense mutation in a Chinese family with autosomal dominant retinitis pigmentosa. BMC Ophthalmol. (2021) 21:360. doi: 10.1186/s12886-021-02125-9
16. Li D, Xu C, Huang D, Guo R, Ji J, Liu W. Identification and functional analysis of a novel missense mutation in GJA8, p.Ala69Thr. BMC Ophthalmol. (2020) 20:461. doi: 10.1186/s12886-020-01725-1
17. Guo R, Huang D, Ji J, Liu W. A novel mutation GJA8 NM_005267.5: c.124G>A, p.(E42K) causing congenital nuclear cataract. BMC Ophthalmol. (2022) 22:172. doi: 10.1186/s12886-022-02386-y
18. Tibrewal S, Ratna R, Gour A, Agarkar S, Dubey S, Ganesh S, et al. Clinical and molecular aspects of congenital aniridia – A review of current concepts. Indian J Ophthalmol. (2022) 70:2280–92. doi: 10.4103/ijo.IJO_2255_21
19. Landsend E, Lagali N, Utheim T. Congenital aniridia – A comprehensive review of clinical features and therapeutic approaches. Surv Ophthalmol. (2021) 66:1031–50. doi: 10.1016/j.survophthal.2021.02.011
20. Dyer A, De Faria A, Julio G, Álvarez de Toledo J, Barraquer R, de la Paz M. Long-term anatomical and functional survival of boston type 1 keratoprosthesis in congenital aniridia. Front Med. (2021) 8:749063. doi: 10.3389/fmed.2021.749063
21. Tzoulaki I, White I, Hanson I. PAX6 mutations: genotype-phenotype correlations. BMC Genet. (2005) 6:27. doi: 10.1186/1471-2156-6-27
22. Lima Cunha D, Arno G, Corton M, Moosajee M. The spectrum of PAX6 mutations and genotype-phenotype correlations in the eye. Genes. (2019) 10:1050. doi: 10.3390/genes10121050
23. Kit V, Cunha D, Hagag A, Moosajee M. Longitudinal genotype-phenotype analysis in 86 patients with PAX6-related aniridia. JCI Insight. (2021) 6:e148406. doi: 10.1172/jci.insight.148406
24. Lee H, Colby K. A review of the clinical and genetic aspects of aniridia. Semin Ophthalmol. (2013) 28:306–12.
25. Vasilyeva T, Marakhonov A, Voskresenskaya A, Kadyshev V, Käsmann-Kellner B, Sukhanova N, et al. Analysis of genotype-phenotype correlations in PAX6-associated aniridia. J Med Genet. (2021) 58:270–4. doi: 10.1136/jmedgenet-2019-106172
26. Miao Q, Ping X, Tang X, Zhang L, Zhang X, Cheng Y, et al. Experimental assessment of novel PAX6 splicing mutations in two Chinese families with aniridia. Gene. (2017) 630:44–8. doi: 10.1016/j.gene.2017.07.073
27. Vasilyeva T, Voskresenskaya A, Käsmann-Kellner B, Khlebnikova O, Pozdeyeva N, Bayazutdinova G, et al. Molecular analysis of patients with aniridia in Russian federation broadens the spectrum of PAX6 mutations. Clin Genet. (2017) 92:639–44. doi: 10.1111/cge.13019
28. Yahalom C, Blumenfeld A, Hendler K, Wussuki-Lior O, Macarov M, Shohat M, et al. Mild aniridia phenotype: an under-recognized diagnosis of a severe inherited ocular disease. Graefes Arch Clin Exp Ophthalmol. (2018) 256:2157–64. doi: 10.1007/s00417-018-4119-1
29. You B, Zhang X, Xu K, Xie Y, Ye H, Li Y. Mutation spectrum of PAX6 and clinical findings in 95 Chinese patients with aniridia. Mol Vis. (2020) 26:226–34.
30. Lim H, Kim D, Kim H. PAX6 aniridia syndrome: clinics, genetics, and therapeutics. Curr Opin Ophthalmol. (2017) 28:436–47. doi: 10.1097/ICU.0000000000000405
31. Liu W, Huang D, Guo R, Ji J. Pathological changes of the anterior lens capsule. J Ophthalmol. (2021) 2021:9951032. doi: 10.1155/2021/9951032
32. Lee H, Khan R, O’Keefe M. Aniridia: current pathology and management. Acta Ophthalmol. (2008) 86:708–15. doi: 10.1111/j.1755-3768.2008.01427.x
33. Schneider S, Osher R, Burk S, Lutz T, Montione R. Thinning of the anterior capsule associated with congenital aniridia. J Cataract Refract Surg. (2003) 29:523–5. doi: 10.1016/s0886-3350(02)01602-4
34. Celik A, Kervestin S, Jacobson A. NMD: at the crossroads between translation termination and ribosome recycling. Biochimie. (2015) 114:2–9. doi: 10.1016/j.biochi.2014.10.027
35. Baker K, Parker R. Nonsense-mediated mRNA decay: terminating erroneous gene expression. Curr Opin Cell Biol. (2004) 16:293–9.
36. Frischmeyer P, Dietz H. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. (1999) 8:1893–900. doi: 10.1093/hmg/8.10.1893
37. Khajavi M, Inoue K, Lupski J. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet. (2006) 14:1074–81. doi: 10.1038/sj.ejhg.5201649
38. Kokotas H, Petersen M. Clinical and molecular aspects of aniridia. Clin Genet. (2010) 77:409–20.
39. Singh S, Tang H, Lee J, Saunders G. Truncation mutations in the transactivation region of PAX6 result in dominant-negative mutants. J Biol Chem. (1998) 273:21531–41. doi: 10.1074/jbc.273.34.21531
40. Cvekl A, Sax C, Bresnick E, Piatigorsky J. A complex array of positive and negative elements regulates the chicken alpha A-crystallin gene: involvement of Pax-6, USF, CREB and/or CREM, and AP-1 proteins. Mol Cell Biol. (1994) 14:7363–76. doi: 10.1128/mcb.14.11.7363-7376.1994
Keywords: aniridia, PAX6, gene mutation, phenotype, genotype
Citation: Guo R, Zhang X, Liu A, Ji J and Liu W (2022) Novel clinical presentation and PAX6 mutation in families with congenital aniridia. Front. Med. 9:1042588. doi: 10.3389/fmed.2022.1042588
Received: 12 September 2022; Accepted: 29 November 2022;
Published: 13 December 2022.
Edited by:
Shida Chen, Zhongshan Ophthalmic Center, Sun Yat-sen University, ChinaReviewed by:
Li Huang, Zhongshan Ophthalmic Center, Sun Yat-sen University, ChinaCopyright © 2022 Guo, Zhang, Liu, Ji and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Liu, d2VpbGl1MDVAdG11LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.