 Arthur J. L. Cooper
Arthur J. L. Cooper Thambi Dorai1,2
Thambi Dorai1,2 John T. Pinto
John T. Pinto Travis T. Denton
Travis T. Denton
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Med., 04 November 2022
Sec. Translational Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fmed.2022.1035335
This article is part of the Research TopicExperts' Opinion in Medicine 2022View all 5 articles
Many rapidly dividing cells, including various cancers, rely on L-glutamine (Gln) as a major energy/carbon/nitrogen source. For cancers this process is named “glutamine addiction” [e.g., (1–11)]. Almost all researchers studying glutamine addiction assume that only one major pathway contributes to this phenomenon. In this pathway, which is referred to here as the glutaminase I pathway (Figure 1), Gln is converted to L-glutamate (Glu) and ammonia by the action of a glutaminase, followed by conversion of Glu to α-ketoglutarate (KG) by a transaminase (aminotransferase) or by glutamate dehydrogenase (GDH). The KG thus formed can then enter the tricarboxylic acid (TCA) cycle. However, as outlined below, normal and cancerous tissues can utilize an alternative pathway (the glutaminase II pathway) for the formation of KG from Gln.
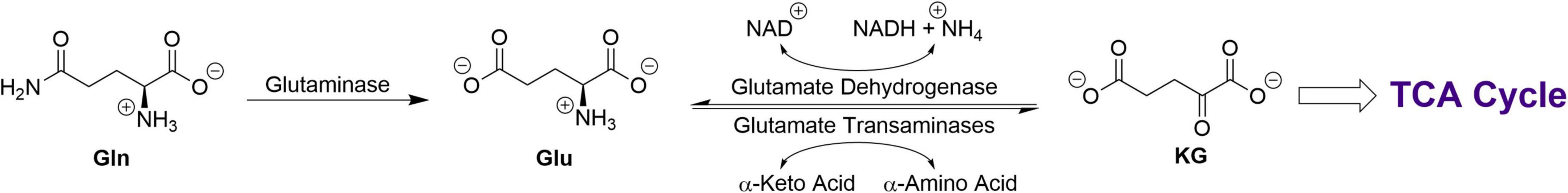
Figure 1. The glutaminase I pathway for the production of KG from Gln.
Rat liver glutaminase (Figure 1) was discovered in the 1940s and designated glutaminase I or (since it is activated by phosphate) phosphate-activated glutaminase (PAG) (12, 13). Subsequently, it was discovered that mammalian tissues contain two PAG isozymes, namely, a kidney type (KGA, GLS, GLS1) and its active splice variant (GAC) and a liver type (LGA, GLS2) and its active splice variant (GAB) (14–16).
Subsequently, Meister et al. showed that rat liver possesses a Gln transaminase (GT) that catalyzes the transamination of Gln to α–ketoglutaramate (KGM) in the presence of an α-keto acid acceptor, which is coupled to an enzyme (ω–amidase; ωA) that catalyzes the hydrolysis of KGM to KG (Figure 2) (17, 18). This pathway was named the glutaminase II pathway and we have retained this nomenclature in our previous publications [e.g., (19, 20)]. Unfortunately, researchers have overlooked this pathway, possibly because of confusion in nomenclature between the glutaminase II pathway and glutaminase 2 isozyme. To obviate this problem, we now refer to the glutaminase II pathway as the glutamine transaminase—ω-amidase (GTωA) pathway. We propose here that KGM—a key intermediate of this pathway—facilitates glutamine addiction in cancer cells.
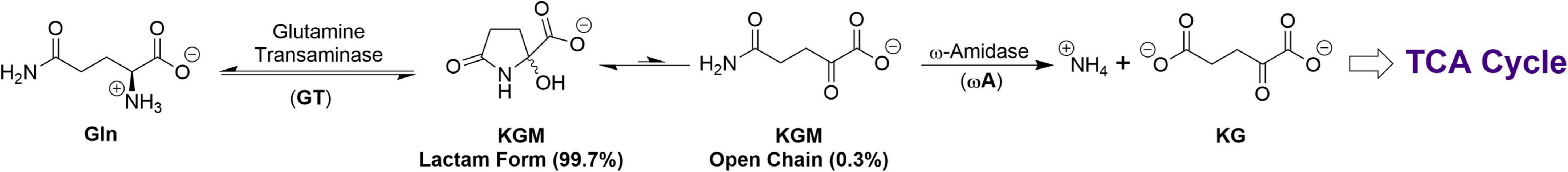
Figure 2. The GTωA pathway for production of KG from Gln. Note the relatively low concentration of the open-chain (substrate) form of KGM.
We have suggested that the GTωA pathway salvages α-keto acids arising through non-specific transamination of L-amino acids by converting them back to the original L-amino acid (19, 20). Indeed, recent work supports this idea (21). We have also suggested that the GTωA pathway may be involved in cellular/intracellular transport of α-keto/L-amino acids (19, 20). Finally, we have emphasized the importance of the GTωA pathway in closing the methionine salvage pathway (MSP) (19, 20). During polyamine synthesis, carbon 1 (C1) of methionine is converted to CO2, C2-C4 are incorporated into polyamines and the sulfur and methyl are incorporated into 5′-methylthioadenosine (MTA). In the MSP, this sulfur and methyl group are incorporated into α-keto-γ-methiolbutyrate (KMB). The last step of the MSP is transamination of KMB to L-methionine. An important amine donor in this process is Gln (19, 20 and references quoted therein). Indeed, a homolog of mammalian glutamine transaminase K (GTK) closes the MSP in bacteria and plants (22). Moreover, KMB is an excellent substrate of two mammalian Gln transaminases, namely GTK and L (GTL) (19, 20).
KG can be generated through the GTωA pathway under anoxic conditions, provided α-keto acids are available for transamination with Gln. This is a major advantage to dividing cancer cells, where the availability of oxygen may be limited. But, there is also another advantage. Polyamines are important for both normal and neoplastic cell function and replication (e.g., 23). Closure of the MSP is depicted in Figure 3. Thus, the GTωA pathway is important in cancer both for closing the MSP (Figure 3) and for providing anaplerotic KG, even under anoxic conditions (Figure 2).
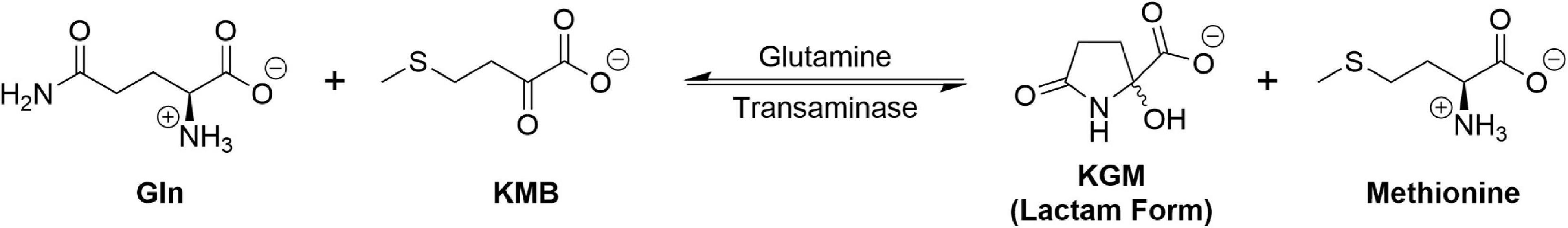
Figure 3. Closure of the MSP by transamination of KMB with Gln. For simplicity, the open-chain form of KGM is omitted since it represents only 0.3% of total KGM (see Figure 2).
Meister et al. partially purified a Gln transaminase from rat liver and showed that it possessed wide α-keto acid substrate specificity (17, 18). Subsequently, Cooper and Meister showed that rat tissues contain two Gln transaminases, namely a liver type (GTL) and a kidney type (GTK) (24, 25). Both isozymes were shown to exhibit broad α-keto/L-amino acid substrate specificity (24, 25). This has led to some redundant/confusing nomenclature in the scientific literature. For example, GTK and GTL are identical to enzymes other researchers have named kynurenine aminotransferase 1 (KAT1/KYAT1) and kynurenine aminotransferase 3 (KAT3/KYAT3), respectively [see the discussion in 19, 20]. However, Li et al. have shown that, of all the L-amino acids tested (including L-kynurenine), the highest catalytic efficiency for human GTK/KAT1 and mouse GTL/KAT3 is with Gln (26, 27). Moreover, the average concentration of Gln in human tissues is ∼9 mM [calculated from data in Cruzat et al. (28)], whereas that of L-kynurenine is ∼22 μM in rat liver, with lower concentrations in brain, lung and spleen (29). Human KAT2 (KYAT2, α-aminoadipate aminotransferase) exhibits some activity toward Gln (30). Moreover, Gln transaminase and ω-amidase activities are present in every rat tissue examined (31, 32). Taken together, the findings indicate that Gln transamination in mammalian tissues is extensive.
The GTωA pathway was shown to be present in freshly isolated human kidney tissue (33–35). Moreover, Darmaun et al. investigated the turnover kinetics of [15N]Glu, [2-15N]Gln and [5-15N]Gln administered to adult human male volunteers and concluded that Gln transamination in humans may be extensive (36).
Both GTK/KAT1 and GTL/KAT3 possess mitochondrial leader sequences (37, 38)—alternative splicing results in activity in both cytosolic and mitochondrial compartments. ω-Amidase (annotated as Nit2 in the human genome) RNA has been detected in all sixteen human tissues investigated, with highest levels in liver and kidney (39). ω-Amidase activity has also been detected in cytosolic and mitochondrial fractions of rat tissues (19, 20 and references cited therein). The presence of a mitochondrial GTωA pathway is predicted to be advantageous to human cells, especially to rapidly dividing cancer cells. Thus, as noted above, anaplerotic KG can be generated from Gln under anaerobic conditions as long as the tumor is supplied with α-keto acid substrates of the Gln transaminases.
Meister presented evidence that open-chain KGM, in solution, is in equilibrium with a cyclic (lactam) form (2-hydroxy-5-oxoproline, Figure 2) (40). This observation was verified by Hersh who showed that, at neutral pH, the equilibrium favors the lactam (99.7%) over the open-chain form (0.3%) (41) (Figure 2). In fact, in the NMR spectrum of pure KGM, the open chain form (the actual substrate of ω-amidase) is undetectable (42). (Nevertheless, unless otherwise stated, the designation KGM herein is the sum total of open-chain and lactam forms).
A major reason why the GTωA pathway has been little studied is possibly because KGM is not available commercially and, until recently, could only be synthesized enzymatically. This procedure involves oxidation of Gln (solution neutralized with NaOH) by snake venom L-amino acid oxidase in the presence of oxygen and catalase (40). The generated KGM is separated from NH4+, Na+ and unreacted Gln on a Dowex 50[H+] column. The KGM eluted with water can be lyophilized to the free acid or converted to sodium or barium salts (40). This procedure has been repeated in our laboratory (43) and by others (44). However, Gln, at neutral pH, slowly cyclizes to 5-oxoproline (5-OP), with the elimination of ammonia (45), and this occurs during the enzymatic reaction to contaminate the final KGM product. Similarly, hydrolysis of KGM to KG occurs upon prolonged incubation. Thus, the enzymatic reaction must be carried out as quickly as possible. Nevertheless, KGM, prepared by the enzymatic procedure, invariably contains ∼1 to a few percent 5-OP (43, 44) and traces of KG.
In order to overcome problems associated with enzymatic synthesis and to scale-up production, a group led by one of us (TTD) recently published a procedure for the organic synthesis of KGM that is not contaminated with either KG or 5-OP (42). After this article was published, an article came to our attention in which the authors oxidized Gln at neutral pH with ammonium vanadate (NH4VO3) to a complex isolated in 65% yield as [V2O3 (2-hydroxy-5-oxoproline)2(2,2′-bipyridine)2]-7H2O (46). The possibility that cleanup of this complex will provide an alternative, convenient synthetic route to KGM lactam is currently being considered. We anticipate that a convenient source of KGM will stimulate interest in the GTωA pathway and especially the role of ω-amidase and its substrate KGM in cancer biology.
Rat liver ω-amidase has a broad pH optimum (∼5.0–9.5), with α-ketosuccinamate (KSM, the α-keto acid analog of asparagine) as the substrate, yet the pH optimum with the next higher homolog of KSM (i.e., KGM) is much narrower, with an apparent pH optimum of ∼9.0 and very little activity at pH values below 7.5 (40). Hersh showed that the rate of interconversion between lactam and open-chain forms of KGM is base (OH–)-catalyzed and, thus, favors faster rate of open-chain formation of KGM at high pH values (41). At pH values ≥ 8.5, the rate of interconversion is so rapid that it is unlikely to be rate-limiting for enzyme activity. Thus, ω-amidase activity measurements with KGM as substrate are usually conducted at pH values of 8.5–9.0. Since KSM does not cyclize, there is no pH limitation to enzyme activity over the pH range of 5.0–9.5.
Due to the very low relative concentration of open-chain KGM compared to its lactam, at pH 7.0–7.2, this compound is expected to accumulate to measurable steady state concentrations in mammalian tissues in this pH range, even though ω-amidase activity is inherently high. Indeed, normal concentrations of KGM in several rat tissues are 6 μM to 216 μM (47, 48).
KGM was shown to accumulate in the cerebrospinal fluid (CSF) of hyperammonemic patients with hepatic encephalopathy (HE) (49, 50). KGM also accumulates in the urine of hyperammonemic patients with (1) disorders of the urea cycle (51, 52) and (2) citrin deficiency (53). Evidently, KGM is a biomarker for hyperammonemia associated with several disorders. The increase in KGM may be related to the hyperammonemia-induced increase in Gln concentrations, leading to increased Gln transamination (49, 52).
Many cancer cells convert glucose to lactate even in the presence of adequate oxygen (Warburg effect). This phenomenon is associated with the acidification of the microenvironment (54). However, recent evidence suggests a reverse pH gradient in tumors (55). The pH optimum of GTK and GTL is ∼8.0 to 9.0 (26, 27). Thus, an alkaline environment will favor Gln transamination, whereas an acidic environment will have the reverse effect. Additionally, it is predicted that lower pH values will favor higher steady state concentrations of KGM. Thus, concentrations of KGM throughout the tumor will be affected, in part, by Gln transaminase activity, ω-amidase activity and pH gradients. Moreover, ammonia generated from Gln is a tumor promoter (56). However, the relationship between ammonia and KGM concentrations is uncertain. Whether KGM is a cancer biomarker remains to be established.
Interestingly, in a study from Johns Hopkins University (JHU) it was reported that KGM could be detected by 1H NMR in patient-derived pancreatic cancer (JHU094) orthotopic tumors in nude mice (57). Moreover, the KGM tumor concentration increased in mice in which GLS1 was inhibited, most likely as a result of increased Gln transamination in the face of inhibited GLS1 activity (57). This suggests a metabolic shift in cancer cells from reliance on the glutaminase I pathway to the GTωA pathway. The JHU group also showed that the median survival time was significantly increased in nude mice carrying a human pancreatic cancer xenograft in which GTK was knocked down, compared to controls carrying xenografts expressing normal GTK activity (57).
Thul et al. demonstrated that GTK (annotated as KYAT1/CCBL1), GTL (annotated as KYAT3/CCBL2) and ω-amidase (annotated as Nit2) occur in several cancer cell lines, including A-431, U-251MG and U2OS (58). The human protein atlas1 also lists the presence of the protein and mRNA for GTK, GTL and ω-amidase in many human cancers. Below, we provide additional information on the GTωA pathway in cancers.
Medulloblastoma is the most common malignant cancer in children and is associated with a poor prognosis [(59) and references cited therein]. Pham et al. investigated the isotopomer patterns in L-glutamate generated from L-[U-13C]Gln and deduced that the D425MED orthotopic human Myc-amplified medulloblastoma tumors in nude mice preferentially utilize the GTωA pathway to maintain glutamine addiction (59). Moreover, KYAT (i.e., the gene for GTK/KAT1), and its mRNA, are upregulated in medulloblastoma, compared to other pediatric brain tumors, in the Children’s Brain Tumor Network/KidsFirst Pediatric Brain Tumor Atlas RNAseq dataset (59).
Pan et al. (60) and Zhang et al. (61) reported an increase in GLS1 expression with increasing aggressiveness of human prostate cancer cells. We also found this to be the case (62). This is consistent with GLS1 playing a prominent role in the anaplerotic supply of KG in human prostate cells. We also showed the importance of the GTωA pathway in this process in several human cancers. Thus, immunohistochemistry studies from our laboratory showed that GTK/ω-amidase are well represented in human bladder, pancreatic and prostate cancers (19, 20, 62, 63). An especially intriguing finding is the discovery that specific activities of glutamine transaminase(s) and ω-amidase are relatively high in rat prostate (62). Moreover, as was found to be the case with GLS1, we showed that the amount of GTK and ω-amidase protein in prostate cancer cells, in culture, increases with increasing aggressiveness of the cells (62). These findings are consistent with the ability of the prostate to produce polyamines and large amounts of citrate that are excreted into the seminal fluid (references cited in 62). The coupling of the MSP to the GTωA pathway (Figure 3) not only allows methionine to be regenerated following polyamine biosynthesis, but also allows for the anaplerotic replenishment of citrate carbon lost in the semen. Thus, both GLS1 and enzymes of the GTωA pathway are upregulated and adaptive to tumor cell environment in human prostate cancer and can contribute to KG anaplerosis.
Several researchers have suggested that glutaminase inhibitors [e.g., BPTES (bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide] or analogs] alone, or in combination with other drugs, may be useful anti-cancer agents [e.g., (64–66)]. Unfortunately, blocking Gln anaplerosis by utilizing the highly potent allosteric GLS inhibitor CB-839 as a monotherapeutic intervention, has not been clinically successful. Nevertheless, there are numerous, active ongoing clinical trials utilizing a combination of CB-839 with FDA approved chemotherapies (e.g., www.clinicaltrials.gov identifiers NCT02861300, NCT03798678). We suggest that, once GLS1 is inhibited, the cancer cells will begin to rely more heavily on the GTωA pathway for the generation of KG, making ω-amidase a valid therapeutic target. This emphasizes the urgency for the development of ω-amidase inhibitors and their utilization either alone, or in combination with GLS inhibitors. It has been suggested that GTK inhibitors, either alone or in combination with a GLS1 inhibitor, may be alternative and effective anti-cancer agents (57, 59). While we agree with this premise, we propose that a potent, selective inhibitor of ω-amidase may be even more useful as an anti-cancer agent. Unfortunately, no such inhibitor currently exists. When Duffy et al. infused KGM into the CSF of rats, only minor neurological symptoms were noted, and only at relatively enormous concentrations (49). Thus, it is possible that inhibition of ω-amidase and accumulation of KGM may have only minimal (patho) physiological effects on normal tissues, whereas removal of a source of anaplerotic KG may be deleterious to tumors. We strongly urge cancer researchers to consider the importance of the GTωA pathway in cancer cell survival and prioritize the development of an ω-amidase inhibitor as a new weapon in the fight against cancer.
AJLC wrote the first draft. All authors contributed to the work quoted and approved the final draft.
TTD would like to acknowledge the WSU CPPS for startup funding that has led to the study of ω-amidase.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Li T, Le A. Glutamine metabolism in cancer. Adv Exp Med Biol. (2018) 1063:13–32. doi: 10.1007/978-3-319-77736-8_2
2. Delgir S, Bastami M, Ilkhani K, Safi A, Seif F, Alivand MR. The pathways related to glutamine metabolism, glutamine inhibitors and their implication for improving the efficiency of chemotherapy in triple-negative breast cancer. Mutat Res Rev Mutat Res. (2021) 787:108366. doi: 10.1016/j.mrrev.2021.108366
3. Milano SK, Huang Q, Nguyen TT, Ramachandran S, Finke A, Kriksunov I, et al. New insights into the molecular mechanisms of glutaminase C inhibitors in cancer cells using serial room temperature crystallography. J Biol Chem. (2022) 298:101535. doi: 10.1016/j.jbc.2021.101535
4. Pallett LJ, Dimeloe S, Sinclair LV, Byrne AJ, Schurich A. A glutamine ‘tug-of-war’: targets to manipulate glutamine metabolism for cancer immunotherapy. Immunother Adv. (2021) 1:ltab010. doi: 10.1093/immadv/ltab010
5. Yu W, Yang X, Zhang Q, Sun L, Yuan S, Xin Y. Targeting GLS1 to cancer therapy through glutamine metabolism. Clin Transl Oncol. (2021) 23:2253–68. doi: 10.1007/s12094-021-02645-2
6. Yang R, Ying G, Li B. Potential of electron transfer and its application in dictating routes of biochemical processes associated with metabolic reprogramming. Front Med. (2021) 15:679–92. doi: 10.1007/s11684-021-0866-1
7. Martins F, Gonçalves LG, Pojo M, Serpa J. Take advantage of glutamine anaplerosis, the kernel of the metabolic rewiring in malignant gliomas. Biomolecules. (2020) 10:1370. doi: 10.3390/biom10101370
8. Obara-Michlewska M, Szeliga M. Targeting glutamine addiction in gliomas. Cancers (Basel). (2020) 12:310. doi: 10.3390/cancers12020310
9. Bernfeld E, Foster DA. Glutamine as an essential amino acid for KRas-driven cancer cells. Trends Endocrinol Metab. (2019) 30:357–68. doi: 10.1016/j.tem.2019.03.003
10. Hoerner CR, Chen VJ, Fan AC. The ‘Achilles Heel’ of metabolism in renal cell carcinoma: glutaminase inhibition as a rational treatment strategy. Kidney Cancer. (2019) 3:15–29. doi: 10.3233/KCA-180043
11. Vanhove K, Derveaux E, Graulus GJ, Mesotten L, Thomeer M, Noben JP, et al. Glutamine addiction and therapeutic strategies in lung cancer. Int J Mol Sci. (2019) 20:252. doi: 10.3390/ijms20020252
12. Errera M, Greenstein JP. Phosphate-activated glutaminase in kidney and other tissues. J Biol Chem. (1949) 178:495–502.
13. Greenstein JP, Carter CE. Influence of α-keto acids on the desamidation of amino acid amides. J Natl Cancer Inst. (1946) 7:57–60.
14. Nguyen TT, Ramachandran S, Hill MJ, Cerione RA. High-resolution structures of mitochondrial glutaminase C tetramers indicate conformational changes upon phosphate binding. J Biol Chem. (2022) 298:101564. doi: 10.1016/j.jbc.2022.101564
15. Masisi BK, El Ansari R, Alfarsi L, Rakha EA, Green AR, Craze ML. The role of glutaminase in cancer. Histopathology. (2020) 76:498–508. doi: 10.1111/his.14014
16. Matés JM, Campos-Sandoval JA, Márquez J. Glutaminase isoenzymes in the metabolic therapy of cancer. Biochim Biophys Acta Rev Cancer. (2018) 1870:158–64. doi: 10.1016/j.bbcan.2018.07.007
17. Meister A, Tice SV. Transamination from glutamine to α-keto acids. J Biol Chem. (1950) 187:173–87.
18. Meister A, Sober HA, Tice SV, Fraser PE. Transamination and associated deamidation of asparagine and glutamine. J Biol Chem. (1952) 197:319–30.
19. Dorai T, Pinto JT, Denton TT, Krasnikov BF, Cooper AJL. The metabolic importance of the glutaminase II pathway in normal and cancerous cells. Anal Biochem. (2022) 644:114083. doi: 10.1016/j.ab.2020.114083
20. Cooper AJL, Shurubor YI, Dorai T, Pinto JT, Isakova EP, Deryabina YI, et al. ω-Amidase: an underappreciated, but important enzyme in L-glutamine and L-asparagine metabolism; relevance to sulfur and nitrogen metabolism, tumor biology and hyperammonemic diseases. Amino Acids. (2015) 48:1–20. doi: 10.1007/s00726-015-2061-7
21. Caligiore F, Zangelmi E, Vetro C, Kentache T, Dewulf JP, Veiga-da-Cunha M, et al. Human cytosolic transaminases: side activities and patterns of discrimination towards physiologically available alternative substrates. Cell Mol Life Sci. (2022) 79:421. doi: 10.1007/s00018-022-04439-3
22. Ellens KW, Richardson LG, Frelin O, Collins J, Ribeiro CL, Hsieh YF, et al. Evidence that glutamine transaminase and ω-amidase potentially act in tandem to close the methionine salvage cycle in bacteria and plants. Phytochemistry. (2015) 113:160–9. doi: 10.1016/j.phytochem.2014.04.012
23. Holbert CE, Cullen MT, Casero RA Jr., Stewart TM. Polyamines in cancer: integrating organismal metabolism and antitumour immunity. Nat Rev Cancer. (2022) Epub ahead of print. doi: 10.1038/s41568-022-00473-2
24. Cooper AJL, Meister A. Isolation and properties of highly purified glutamine transaminase. Biochemistry. (1972) 11:661–71.
25. Cooper AJL, Meister A. Isolation and properties of a new glutamine transaminase from rat kidney. J Biol Chem. (1974) 249:2554–61.
26. Han Q, Li J, Li J. pH dependence, substrate specificity and inhibition of human kynurenine aminotransferase I. Eur J Biochem. (2004) 271:4804–14. doi: 10.1111/j.1432-1033.2004.04446.x
27. Han Q, Robinson H, Cai T, Tagle DA, Li J. Biochemical and structural properties of mouse KAT III. Mol Cell Biol. (2009) 29:784–93. doi: 10.1128/MCB.01272-08
28. Cruzat V, Macedo Rogero M, Noel Keane K, Curi P, Newsholme R. Glutamine: metabolism and immune function, supplementation and clinical translation. Nutrients. (2018) 10:1564. doi: 10.3390/nu10111564
29. Ohta Y, Kubo H, Yashiro K, Ohashi K, Tsuzuki Y, Wada N, et al. Effect of water-immersion restraint stress on tryptophan catabolism through the kynurenine pathway in rat tissues. J Physiol Sci. (2017) 67:361–72. doi: 10.1007/s12576-016-0467-y
30. Han Q, Cai T, Tagle DA, Robinson H, Li J. Substrate specificity and structure of human aminoadipate aminotransferase/kynurenine aminotransferase II. Biosci Rep. (2008) 28:205–15. doi: 10.1042/BSR20080085
31. Cooper AJL, Meister A. Comparative studies of glutamine transaminases from rat tissues. Comp Biochem Physiol. (1981) 69B:137–45.
32. Cooper AJL. Glutamine aminotransferases and ω-amidases. In: E Kvamme editor. Glutamine and Glutamate in Mammals. (Vol. 1), (Boca Raton, FL: CRC Press, Inc) (1988). p. 33–52.
33. Bourke E, Fine A, Scott JM. Glutaminase II pathway in human kidney. Nat New Biol. (1971) 233:249–50. doi: 10.1038/newbio233249a0
34. Bourke E, Fine A, Scott J. Mechanism of ammoniagenesis in human kidney. Biochem J. (1971) 125:94. doi: 10.1042/bj1250094pa
35. Fine A, Scott J, Bourke E. Studies on the glutamine aminotransferase-ω-amidase pathway in human kidney in vitro. J Lab Clin Med. (1972) 80:591–7.
36. Darmaun D, Matthews DE, Bier DM. Glutamine and glutamate kinetics in humans. Am J Physiol. (1986) 251:E117–26. doi: 10.1152/ajpendo.1986.251.1.E117
37. Malherbe P, Alberati-Giani D, Köhler C, Cesura AM. Identification of a mitochondrial form of kynurenine aminotransferase/glutamine transaminase K from rat brain. FEBS Lett. (1995) 367:141–4. doi: 10.1016/0014-5793(95)00546-l
39. Lin CH, Chung MY, Chen WB, Chien CH. Growth inhibitory effect of the human NIT2 gene and its allelic imbalance in cancers. FEBS J. (2007) 274:2946–56. doi: 10.1111/j.1742-4658.2007.05828.x
40. Meister A. Preparation of enzymatic reactions of the keto analogues of asparagine and glutamine. J Biol Chem. (1953) 200:571–89.
41. Hersh LB. Rat liver ω-amidase. Purification and properties. Biochemistry. (1971) 10:2884–91. doi: 10.1021/bi00791a014
42. Shen D, Kruger L, Deatherage T, Denton TT. Synthesis of α-ketoglutaramic acid. Anal Biochem. (2020) 607:113862. doi: 10.1016/j.ab.2020.113862
43. Krasnikov BF, Nostramo R, Pinto JT, Cooper AJL. Assay and purification of ω-amidase/Nit2, a ubiquitously expressed putative tumor suppressor, that catalyzes the deamidation of the α-keto acid analogues of glutamine and asparagine. Anal Biochem. (2009) 391:144–50. doi: 10.1016/j.ab.2009.05.025
44. Nikulin M, Drobot V, Švedas V, Krasnikov BF. Preparative biocatalytic synthesis of α-ketoglutaramate. Int J Mol Sci. (2021) 22:12748. doi: 10.3390/ijms222312748
45. Bray HG, James SP, Raffan IM, Thorpe WV. The enzymic hydrolysis of glutamine and its spontaneous decomposition in buffer solutions. Biochem J. (1949) 44:625–7.
46. Deng L, Zhou ZH. Spontaneous conversions of glutamine, histidine and arginine into α-hydroxycarboxylates with NH4VO3 or V2O5. Dalton Trans. (2020) 49:11921–30. doi: 10.1039/d0dt02150d
47. Duffy TE, Cooper AJL, Meister A. Identification of α-ketoglutaramate in rat liver, kidney, and brain. Relationship to glutamine transaminase and ω-amidase activities. J Biol Chem. (1974) 249:7603–6.
48. Shurubor YI, Cooper AJL, Isakova EP, Deryabina YI, Beal MF, Krasnikov BF. HPLC determination of α-ketoglutaramate [5-amino-2,5-dioxopentanoate] in biological samples. Anal Biochem. (2016) 494:52–4. doi: 10.1016/j.ab.2015.11.003
49. Duffy TE, Vergara F, Plum F. α-Ketoglutaramate in hepatic encephalopathy. Res Publ Assoc Res Nerv Ment Dis. (1974) 53:39–52.
50. Vergara F, Plum F, Duffy TE. α-Ketoglutaramate: increased concentrations in the cerebrospinal fluid of patients in hepatic coma. Science. (1974) 183:81–3. doi: 10.1126/science.183.4120.81
51. Kuhara T, Inoue Y, Ohse M, Krasnikov BF, Cooper AJL. Urinary 2-hydroxy-5-oxoproline, the lactam form of α-ketoglutaramate, is markedly increased in urea cycle disorders. Anal Bioanal Chem. (2011) 400:1843–51. doi: 10.1007/s00216-011-4688-x
52. Cooper AJL, Kuhara T. α-Ketoglutaramate: an overlooked metabolite of glutamine and a biomarker for hepatic encephalopathy and inborn errors of the urea cycle. Metab Brain Dis. (2014) 29:991–1006. doi: 10.1007/s11011-013-9444-9
53. Kuhara T, Ohse M, Inoue Y, Cooper AJL. A GC/MS-based metabolomic approach for diagnosing citrin deficiency. Anal Bioanal Chem. (2011) 400:1881–94. doi: 10.1007/s00216-011-4766-0
54. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. (2004) 4:891–9. doi: 10.1038/nrc1478
55. Koltai T. Targeting the pH paradigm at the bedside: a practical approach. Int J Mol Sci. (2020) 21:9221. doi: 10.3390/ijms21239221
56. Cheng C, Geng F, Li Z, Zhong Y, Wang H, Cheng X, et al. Ammonia stimulates SCAP/Insig dissociation and SREBP-1 activation to promote lipogenesis and tumour growth. Nat Metab. (2022) 4:575–588. doi: 10.1038/s42255-022-00568-y
57. Udupa S, Nguyen S, Hoang G, Nguyen T, Quinones A, Pham K, et al. Upregulation of the glutaminase ii pathway contributes to glutamate production upon glutaminase II inhibition in pancreatic cancer. Proteomics. (2019) 19:e1800451. doi: 10.1002/pmic.201800451
58. Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, et al. A subcellular map of the human proteome. Science. (2017) 356:6340.
59. Pham K, Hanaford AR, Poore BA, Maxwell MJ, Sweeney H, Parthasarathy A, et al. Comprehensive metabolic profiling of myc-amplified medulloblastoma tumors reveals key dependencies on amino acid, tricarboxylic acid and hexosamine pathways. Cancers. (2022) 14:1311. doi: 10.3390/cancers14051311
60. Pan T, Gao L, Wu G, Shen G, Xie S, Wen H, et al. Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem Biophys Res Commun. (2015) 456:452–8. doi: 10.1016/j.bbrc.2014.11.105
61. Zhang J, Mao SY, Guo YD, Wu Y, Yao XD, Huang Y. Inhibition of GLS suppresses proliferation and promotes apoptosis in prostate cancer. Biosci Rep. (2019) 39:BSR20181826. doi: 10.1042/BSR20181826
62. Dorai T, Dorai B, Pinto JT, Grasso M, Cooper AJL. High levels of glutaminase II pathway enzymes in normal and cancerous prostate suggest a role in ‘glutamine addiction’. Biomolecules. (2019) 10:2. doi: 10.3390/biom10010002
63. Cooper AJL, Dorai T, Dorai B, Krasnikov BF, Li J, Hallen A, et al. Role of glutamine transaminases in nitrogen, sulfur, selenium and 1-carbon metabolism: glutamine transaminases in normal and cancer cells. In: R Rajendram, VR Preedy, VB Patel editors. Glutamine in Clinical Nutrition (Nutrition and Health Series). (New York, NY: Humana Press) (2015). p. 37–54.
64. Zhao Y, Feng X, Chen Y, Selfridge JE, Gorityala S, Du Z, et al. 5-Fluorouracil enhances the antitumor activity of the glutaminase inhibitor CB-839 against PIK3CA-mutant colorectal cancers. Cancer Res. (2020) 80:4815–27. doi: 10.1158/0008-5472.CAN-20-0600
65. Harding JJ, Telli ML, Munster PN, Le MH, Molineaux C, Bennett MK, et al. Safety and tolerability of increasing doses of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase, in solid tumors. J Clin Oncol. (2015) 33:2512.
Keywords: ω-amidase, glutaminase II pathway, glutamine transaminase K, glutamine transaminases, α-ketoglutaramate, α-ketoglutarate, methionine salvage pathway, nitrogen homeostasis
Citation: Cooper AJL, Dorai T, Pinto JT and Denton TT (2022) α-Ketoglutaramate—A key metabolite contributing to glutamine addiction in cancer cells. Front. Med. 13:1035335. doi: 10.3389/fmed.2022.1035335
Received: 02 September 2022; Accepted: 10 October 2022;
Published: 04 November 2022.
Edited by:
Victoria Bunik, Lomonosov Moscow State University, RussiaReviewed by:
Frank Gaunitz, University Hospital Leipzig, GermanyCopyright © 2022 Cooper, Dorai, Pinto and Denton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Travis T. Denton, dHJhdmlzLmRlbnRvbkB3c3UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.