 Salam Abbara1
Salam Abbara1 Jean-Benoit Monfort2
Jean-Benoit Monfort2 Léa Savey1Philippe Moguelet3
Léa Savey1Philippe Moguelet3 David Saadoun4Claude Bachmeyer1Olivier Fain5Benjamin Terrier6
David Saadoun4Claude Bachmeyer1Olivier Fain5Benjamin Terrier6 Zahir Amoura7Alexis Mathian7Laurent Gilardin8†David Buob3Chantal Job-Deslandre9Jean-François Dufour10Rebecca Sberro-Soussan11Gilles Grateau1
Zahir Amoura7Alexis Mathian7Laurent Gilardin8†David Buob3Chantal Job-Deslandre9Jean-François Dufour10Rebecca Sberro-Soussan11Gilles Grateau1 Sophie Georgin-Lavialle1,12*
Sophie Georgin-Lavialle1,12*- 1Département de Médecine Interne, Centre de Référence des Maladies Auto-Inflammatoires et des Amyloses d’Origine Inflammatoire (CEREMAIA), Hôpital Tenon, Sorbonne Université, AP-HP, Paris, France
- 2Département de Dermatologie, Hôpital Tenon, Sorbonne Université, AP-HP, Paris, France
- 3Département d’Anatomo-Pathologie, Hôpital Tenon, Sorbonne Université, AP-HP, Paris, France
- 4Département de Médecine Interne et Immunologie Clinique, Centre National de Référence Maladies Autoimmunes Systémiques Rares, Centre National de Référence Maladies Autoinflammatoires et Amylose Inflammatoire, INSERM UMR_S 959, Immunologie-Immunopathologie-Immunotherapie, i3 and Département Hospitalo-Universitaire Inflammation-Immunopathologie-Biothérapie i2B, Groupe Hospitalier Pitié-Salpêtrière, Sorbonne Université, AP-HP, Paris, France
- 5Service de Médecine Interne, Hôpital Saint-Antoine, Sorbonne Université, APHP, Paris, France
- 6Service de Médecine Interne, Centre de Référence Maladies Systémiques et Autoimmunes Rares d’Ile de France, Hôpital Cochin, Université Paris Cité, AP-HP, Paris, France
- 7Service de Médecine Interne 2, Institut E3M, Inserm UMRS, Centre d’Immunologie et des Maladies Infectieuses (CIMI-Paris), French National Referral Center for Systemic Lupus Erythematosus, Antiphospholipid Antibody Syndrome and Other Autoimmune Disorders, Groupement Hospitalier Pitié–Salpêtrière, Sorbonne Université, AP-HP, Paris, France
- 8Département de Médecine Interne et Immunologie Clinique, Groupe Hospitalier Pitié-Salpêtrière, Sorbonne Université, AP-HP, Paris, France
- 9Service de Pédiatrie, Immunologie, Hématologie et Rhumatologie, Centre de Référence pour les Rhumatismes Inflammatoires et les Maladies Auto-Immunes Systémique Rare de l’Enfant (RAISE), Hôpital Necker-Enfants Malades, AP-HP, Université Paris Cité, Paris, France
- 10Service Médecine Interne, Hôpital Nord-Ouest, Centre Hospitalier Villefranche sur Saône, Gleize, France
- 11Service de Transplantation Rénale Adulte, Hôpital Necker-Enfants Malades, AP-HP, Université Paris Cité, Paris, France
- 12INSERM U938, Centre de Recherche Saint-Antoine (CRSA), Paris, France
Objective: The frequency of vasculitis may be increased in patients with Familial Mediterranean Fever (FMF), according to several studies. Our aim was to assess the characteristics of French adult patients with both diseases.
Methods: Patients with vasculitis were selected from patients followed for FMF in the French JIR-cohort.
Results: Twenty-two patients were included [polyarteritis nodosa (PAN) n = 10, IgA vasculitis n = 8, unclassified vasculitis n = 2, granulomatosis with polyangiitis n = 1, and microscopic polyangiitis n = 1]. Pathogenic mutations in exon 10 were found in all 21 patients (96%) for which MEFV testing results were available, and 18 (82%) had two pathogenic mutations. Histology showed vasculitis in 59% of patients. Most patients with FMF-associated PAN were HBV-negative and had an inactive FMF before PAN onset, and 40% had a peri-renal or central nervous system bleeding. Most patients with FMF-associated IgA vasculitis had an active FMF before vasculitis onset, and 25% had digestive bleeding. Both patients with unclassified vasculitis had ischemic and/or hemorrhagic complications.
Conclusion: This study confirms the predominance of PAN and IgA vasculitis in patients with FMF and the high frequency of bleeding in FMF-associated PAN. FMF should be considered in case of persistent symptoms and/or inflammatory syndrome despite vasculitis treatment in Mediterranean patients.
Introduction
Familial Mediterranean fever (FMF) is the most common monogenic auto inflammatory disease, mainly affecting people from Mediterranean countries and associated with mutations in the MEFV gene (1). MEFV encodes pyrin, a protein expressed in neutrophils and monocytes and playing an important role in the innate immune response, resulting in the production of interleukin (IL)-1beta (2). Numerous reports suggest a higher frequency of vasculitis in patients with FMF compared to the general population (3–6). These frequencies could reach 2.7–7% for IgA vasculitis (3–5), 0.9–1.4% for polyarteritis nodosa (PAN) (4, 5), and 0.4% for Behçet disease (6). Moreover, the clinical characteristics of these vasculitis may differ between patients with FMF and the general population. Particularly, patients with FMF and PAN seem to have a higher incidence of peri-renal hematoma (7). Most studies arised from Turkey or the Middle East and data regarding European patients with vasculitis and FMF are lacking. In this nationwide French retrospective study, we report the main characteristics and outcomes of adult patients with FMF and vasculitis from the JIR-cohort.
Methods
Patients with vasculitis diagnosed according to international criteria (8–11) were identified among patients aged >18 years in the French JIR-cohort with FMF (12). The Juvenile Inflammatory Rheumatism (JIR) cohort is an international multicenter prospective data repository for patients with systemic inflammatory or rheumatological disease1 (13). The following data were collected: Socio-demographic (age, sex, and ethnicity), background (family history, and comorbidities), FMF characteristics (diagnostic criteria, age at symptoms onset and at diagnosis, clinical manifestations, age at the start of colchicine treatment, C Reactive Protein (CRP) levels during flares and follow-up, FMF control before the onset of vasculitis, treatments, and dose of colchicine at vasculitis diagnosis), MEFV gene testing results (14, 15), vasculitis characteristics (age at diagnosis, clinical manifestations, histological results, results of arteriography and/or CT or MRI angiography, treatment, and follow-up). FMF control was judged on the presence of flares, and monitoring of CRP ± SAA, when available. Data are described as median (first quartile—third quartile) for continuous variables and number (%) for categorical variables. This observational study was based on data extracted from the JIR-cohort, established by the National Commission on Informatics and Liberty (CNIL, authorization number N0: 914677). Patients consented to be included in the JIR-cohort and were informed that data collected in medical records might be used for research studies in accordance with privacy rules.
Results
Among 406 patients with FMF in the French JIR-cohort, 22 had vasculitis and were included (82% men). Most patients had a PAN (n = 10, 46%) or an IgA vasculitis (n = 8, 36%). The characteristics of FMF and of vasculitis in patients with PAN or IgA vasculitis are described in Table 1. Other patients had ANCA-associated vasculitis (n = 2 and 9%) or an unclassified vasculitis (n = 2 and 9%). The median ages at FMF and vasculitis diagnosis were 11.5 (7–22) and 22 (16.5–36.5) years, respectively. At least one episode of bleeding (renal, central nervous system, and pulmonary) or thrombosis complicated the vasculitis of 8 (36%) and 2 (9%) patients, respectively.
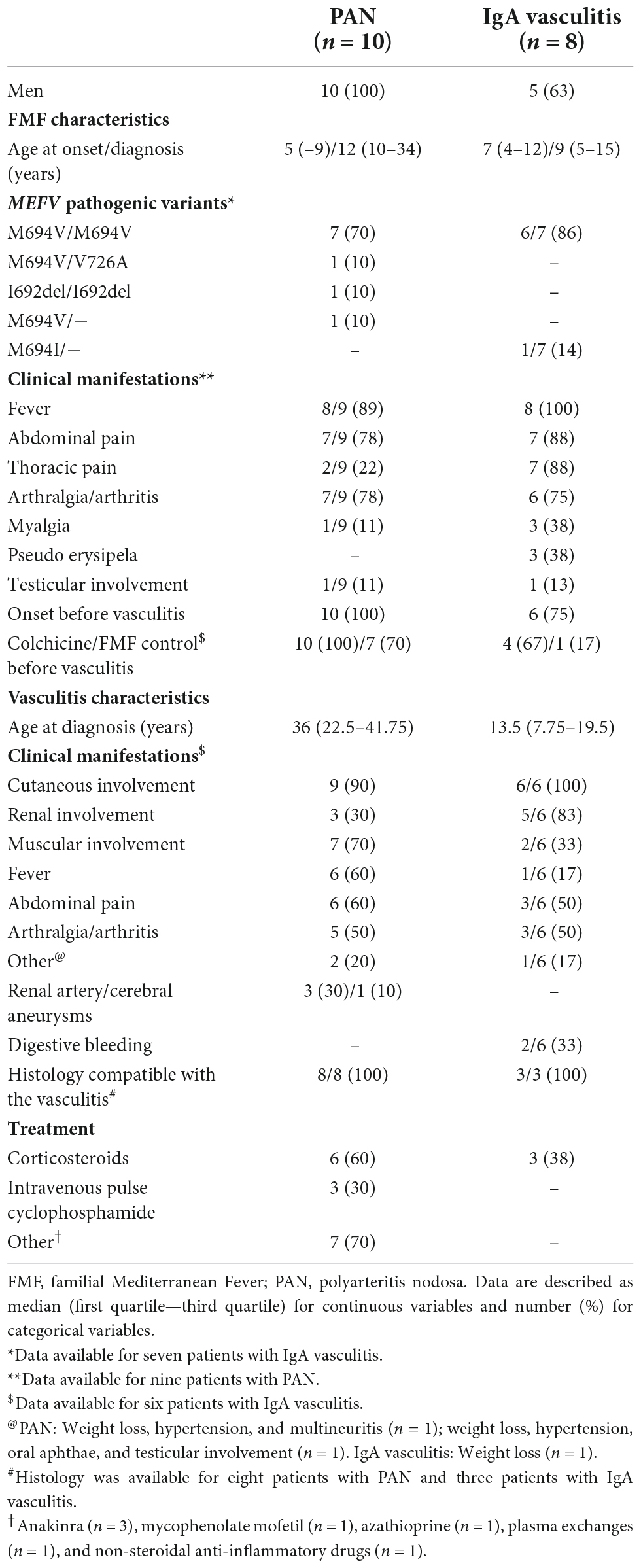
Table 1. Main characteristics of patients with FMF-associated PAN and IgA vasculitis.
Polyarteritis nodosa and familial Mediterranean fever (n = 10)
Symptoms of FMF appeared during youth in most cases, with a median age at diagnosis of 11.5 (10–34) years. Three patients had a family history of FMF. FMF and AA amyloidosis were diagnosed concomitantly in one patient. MEFV testing results were available for all patients; 9/10 had two pathogenic mutations. All patients had ethnicities at risk of FMF (Jewish, n = 6; Turkish, n = 2; Arab, n = 1; and Armenian, n = 1). In all patients, FMF preceded vasculitis and low-dose colchicine was prescribed [median dose 1 (1–1) mg/day]. FMF was controlled in 8/10 patients at vasculitis onset.
Most patients had an HBV-negative PAN (n = 9/10), with a median age at diagnosis of 36 (22.5–41.75) years. PAN was introduced by cutaneous signs (n = 6/10), peri-renal hematomas (n = 3/10), or multineuritis (n = 1/10). The main clinical manifestations of PAN were cutaneous (n = 9/10) or muscular involvement (n = 7/10), fever (n = 6/10), abdominal pain (n = 6/10), and arthralgia (n = 5/10). Cutaneous manifestations included subcutaneous nodules (n = 5/10, Figure 1), asymptomatic erythematous papules of the limbs (n = 3/10), purpura (n = 2/10), a pigmented livedo (n = 1/10), and an infiltrated, migrating, erythematous, pruritic annular rash (n = 1/10). No patient was tested for ADA2 deficiency. Histology was available for 8/10 patients and was compatible with PAN (Figure 1). Renal artery aneurysms were identified in 3/10 patients, and 4/10 patients had at least one episode of bleeding. Corticosteroids were administered to 6/10 patients. Notably, 3/10 patients with predominantly cutaneous involvement received anakinra, an IL-1-receptor antagonist, resulting in a rapid resolution of the clinical manifestations of PAN for all three patients, with resolution of the biological inflammatory syndrome for two patients. Treatment of PAN led to partial remission for all three patients with PAN introduced by a peri-renal hematoma. Despite treatment, these three patients had occasional flare-ups of febrile abdominal or joint pain, with an episode of purpura in one patient and an episode of erythema nodosum in another, but no patient had recurrent bleeding over a follow-up period of 3, 9, and 17 years. Follow-up was rarely longer than 1 year for the other patients. At last follow-up, 8/10 patients were taking colchicine at a median dose of 1 (1–2) mg/day, and 7/10 patients had a controlled FMF.
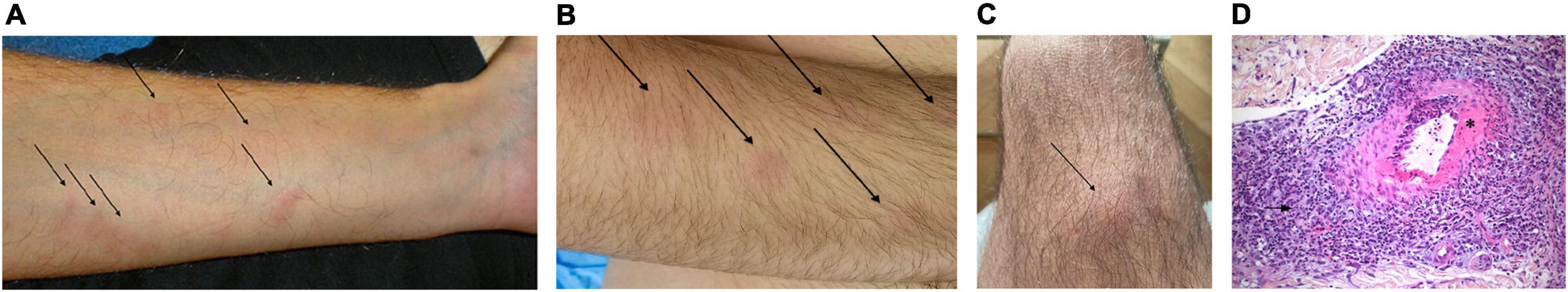
Figure 1. Picture of lesion and pathology slide in patients with PAN and FMF (A–C) subcutaneous nodules. (D) Skin biopsy (Hematoxylin, eosin, and saffron staining; magnification × 200): Deep dermal small artery vasculitis with intimal fibrinoid necrosis (*) and perivascular massive polymorphous inflammation of the adventice (arrow).
IgA vasculitis and familial Mediterranean fever (n = 8)
The median age at FMF diagnosis was 9 (4.5–15) years. MEFV testing results were available for 7/8 patients: Six had two pathogenic mutations, and One had a single pathogenic mutation. One patient’s ethnicity was unknown; the others belonged to an ethnic group at risk of FMF (Jewish, n = 6 and Arab, n = 1). Five patients had a family history of FMF, while two and one patients had a family history of AA amyloidosis and Behçet disease, respectively. In 6/8 patients, the onset of FMF preceded that of vasculitis by a median interval of 4 (3–7) years. Only 4/6 patients were prescribed colchicine before vasculitis onset [median dose 2 (1.75–2) mg/day], and FMF was controlled in only 1/6 patients.
The median age at IgA vasculitis diagnosis was 13.5 (7.75–19.5) years. Most patients had a single episode (n = 5/8, 63%). Three patients had two (n = 1/8, 13%) or three (n = 2/8, 25%) episodes of vasculitis. Clinical manifestations of IgA vasculitis were available for 6/8 patients. The main ones were purpura (n = 6/6), renal involvement (n = 5/6, 83%), abdominal pain (n = 3/6, 50%), arthritis (n = 2/6, 33%), and muscular involvement (n = 2/6, 33%; pain n = 1 and myositis n = 1). For two patients, the vasculitis was complicated by digestive bleeding. Histology was rarely available (n = 3/8) and showed vasculitis (n = 3/3) and IgA deposits (n = 2/3). Three patients were treated with corticosteroids.
The median follow-up time after diagnosis of IgA vasculitis was 12.5 (0–26.5) years. At last follow-up, all patients were taking colchicine, at a median dose of 1.5 (1–2) mg/day, and half of the patients had an active FMF.
Unclassified vasculitis and familial Mediterranean fever (n = 2)
One patient had a controlled FMF since his childhood, with two pathogenic mutations of MEFV (M694V/R761H). He developed a systemic vasculitis with a thrombosis of the superior mesenteric vein at age 36. Another patient presented with symptoms of FMF at age 13, with a homozygous M694V mutation. In the same year, he developed a vasculitis of vessels of all sizes. Despite treatments, he presented over the years several ischemic (stroke n = 1) or bleeding (bilateral peri-renal hematomas n = 1, testicular bleeding n = 1, and intra-alveolar hemorrhage n = 1) episodes, leading to his death at age 29. Both patients had two pathogenic mutations of MEFV.
ANCA-associated vasculitis and familial Mediterranean fever (n = 2)
One patient had a FMF since the age of seven, with a heterozygous M694V mutation. He presented at age 17 with a constrictive pericarditis, arthromyalgia, an axonal sensory neuropathy on electromyography, erythema nodosum, a skin biopsy showing arteritis with thrombosis, and positive ANCA without specificity; the patient did not present renal involvement. He received corticotherapy and azathioprine. Few years later, he developed two unexplained episodes of stroke. The late positivity of MPO-ANCA antibodies at age 27 led to the diagnosis of ANCA-associated vasculitis. Persistence of fluctuating arthromyalgia, infiltrated papules with thrombosing and inflammatory vasculitis on biopsy, positive MPO-ANCA antibodies (maximum 33 IU/mL) and elevated CRP levels up to 35 mg/L led to the introduction of rituximab. Given the persistence of cutaneous-articular signs, elevated CRP levels, treatment with anakinra was initiated and resulted in resolution of the clinical manifestations.
Another patient had recurrent sinusitis with an alveolar hemorrhage and positive ANCA (type not specified), leading to the diagnosis of granulomatosis with polyangiitis. She was treated with oral corticosteroids and intravenous pulse cyclophosphamide. The persistence of a biological inflammatory syndrome despite treatment with intravenous pulse cyclophosphamide, then methotrexate, then mycophenolate mofetil, led to the diagnosis of FMF with a homozygous M694V mutation, which symptoms had occurred during childhood.
Discussion
We describe the main clinical and genetical characteristics of 22 French adult patients with both FMF and vasculitis. The most frequent vasculitis were PAN (46%) and IgA vasculitis (36%), similar to previous reports from Mediterranean countries (4, 5, 7, 16). Despite possible overlaps between vasculitis and FMF clinical manifestations, all patients fulfilled the Tel Hashomer FMF criteria (12). A recent review pointed the paucity of genetic data for described patients with FMF and vasculitis (7). In this study, pathogenic mutations in exon 10 were found in all 21 patients for which MEFV testing results were available, and 18 had two pathogenic mutations (15).
Polyarteritis nodosa was the most frequent vasculitis. FMF preceded PAN in all cases and was mostly controlled with low-dose colchicine treatment. Age at diagnosis of PAN (median, 36 years) was midway between patients with PAN and FMF described in a recent review (7), and those with idiopathic PAN (17). The prevalence of peri-renal hematoma in FMF-associated PAN could reach 50% (7, 18, 19) in the literature. Along this line, they affected 30% of our patients, and introduced the disease in all cases. Moreover, cutaneous manifestations were particularly frequent and heralded the disease in 60% of cases. Note, only one patient had livedo, unlike the classic cutaneous manifestations of PAN. Other findings were consistent with a literature review describing less weight loss, peripheral neuropathy, cardiac involvement, and more abdominal pain in patients with FMF-associated PAN (7). However, the proportions of joint and CNS signs were closer to that in idiopathic PAN (17). Overall, all these signs should raise the suspicion of PAN in a patient with FMF, and clinicians should be vigilant for the high risk of peri-renal bleeding. In this study, treatment of PAN was standard, except for three patients who received anakinra, a recombinant IL-1-receptor antagonist, resulting in resolution of clinical signs. Its efficacy suggests that IL1 may play a role in the pathophysiological mechanisms associated with vasculitis in these patients.
IgA vasculitis was the second most identified vasculitis in our cohort. FMF symptoms preceded vasculitis in 75% of patients; their FMF was mostly uncontrolled despite high colchicine levels, indicating active disease. These results may suggest a link between active FMF, an ongoing inflammatory state, and the triggering of IgA vasculitis (7). As such, clinicians should consider IgA vasculitis when they see purpura suggestive of small- or medium-vessel vasculitis in a patient with FMF. Our patients were older at the time of diagnosis of their vasculitis, had more renal and muscular involvement, and less fever, than has been described in IgA vasculitis associated or not with FMF (7). The prevalence of renal involvement in our series (83%) was close to that described by Audemard Verger et al. (70%) in their review of adult patients with IgA vasculitis (20). Abbara et al. reported an increased rate of intussusception (9%) in FMF-associated IgA vasculitis, which we did not observe in our cohort (7). However, 33% of patients presented digestive bleeding, which may be related to undiagnosed intussusception. Moreover, a low rate of IgA deposits was reported in FMF-associated IgA vasculitis (23%) (7). In this study, when histology was available, IgA deposits were present in 67% of cases, a rate like that described in patients with IgA vasculitis in the general population.
We described two patients with ANCA-associated vasculitis. In both patients, the diagnosis and management of FMF and vasculitis were challenging, given the overlap of FMF and vasculitis signs. Thus, although the association of FMF and ANCA-associated vasculitis could be fortuitous, we invite clinicians to evoke FMF in case of persistent compatible symptoms and/or unexplained chronic biological inflammatory syndrome, especially if the patient is of Mediterranean origin.
Few patients with FMF and unclassified vasculitis have been described so far (7). Almost half of them developed ischemic or bleeding complications. In this study, we described two patients with unclassified vasculitis; both developed such complications.
Behçet disease (BD) was proposed to be more prevalent in patients with FMF in a study by Schwartz et al. (6). BD shares clinical characteristics with FMF (21, 22). MEFV has been evaluated in several studies as a potential pathogenic gene of BD. There are contradictory results as some studies have shown an association of BD and FMF genes, whereas others did not (23–26). We did not identify any patient with both diseases, which could be due to the retrospective nature of the study, the absence of association between both diseases, or the low incidence of Behçet disease in France. In their literature review, Abbara et al. did not find a higher prevalence of BD in patients with FMF (7).
Besides vasculitis, patients with FMF could have an increased prevalence of various immune-mediated conditions, including spondyloarthritis (27, 28), psoriasis (29), hidradenitis suppurativa (30, 31), juvenile idiopathic arthritis (32, 33), multiple sclerosis (34), and inflammatory bowel disease (35, 36). The pathogenic mechanism of these associations remains unknown, particularly the role of MEFV mutations (37–42).
Conclusion
In conclusion, this multicenter retrospective study confirms the predominant coexistence of IgA vasculitis and PAN with FMF in French multi-ethnic patients. The presence of a high frequency of bleeding in patients with FMF and PAN, IgA vasculitis, and unclassified vasculitis is intriguing. Whether FMF increases the frequency of bleeding, or whether those vasculitis are FMF-related and represent a severe form of FMF, especially in patients with two pathogenic mutations, is unknown.
Data availability statement
The original contributions presented in this study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
This observational study was based on data extracted from the JIR-cohort, an international multicenter data repository established by the National Commission on Informatics and Liberty (CNIL, authorization number N°: 914677). Patients consented to be included in the JIR-cohort and were informed that data collected in medical records might be used for research study in accordance with privacy rules.
Author contributions
SA wrote the first draft of the manuscript, collected the data, and performed the statistical analysis. SA, SG-L, GG, CB, OF, JB-M, and DS contributed to the conception and design of the study. SG-L and GG supervised the study. All authors contributed to the manuscript revision, read, and approved the submitted version.
Acknowledgments
We thank Pr Olivier Benveniste, Pr Claire Lejeunne, and Pr Patrice Cacoub for helping us search and collect data from patients followed in their departments. We thank Pr Luc Mouthon for helping us search for patients through the interrogation of the register of the French vasculitis study group. We also thank the following geneticists who performed MEFV sequencing: Camille Louvrier, Serge Amselem, Irina Giurgea, Laurence Cuisset, Isabelle Jeru, and Isabelle Touitou.
Conflict of interest
SG-L and GG received honoraria as speakers or occasional consultants for the SOBI and Novartis laboratories (5000 euros).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
1. Ben-Chetrit E, Levy M. Familial Mediterranean fever. Lancet. (1998) 351:659–64. doi: 10.1016/S0140-6736(97)09408-7
2. Werts C, Girardin SE, Philpott DJ. TIR, CARD and PYRIN: three domains for an antimicrobial triad. Cell Death Differ. (2006) 13:798–815. doi: 10.1038/sj.cdd.4401890
3. Flatau E, Kohn D, Schiller D, Lurie M, Levy E. Schönlein-henoch syndrome in patients with familial Mediterranean fever. Arthritis Rheum. (1982) 25:42–7. doi: 10.1002/art.1780250107
4. Ozdogan H, Arisoy N, Kasapçapur O, Sever L, Calişkan S, Tuzuner N, et al. Vasculitis in familial Mediterranean fever. J Rheumatol. (1997) 24:323–7.
5. Tekin M, Yalçinkaya F, Tümer N, Cakar N, Koçak H, Ozkaya N, et al. Familial Mediterranean fever–renal involvement by diseases other than amyloid. Nephrol Dial Transplant. (1999) 14:475–9. doi: 10.1093/ndt/14.2.475
6. Schwartz T, Langevitz P, Zemer D, Gazit E, Pras M, Livneh A. Behçet’s disease in familial Mediterranean fever: characterization of the association between the two diseases. Semin Arthritis Rheum. (2000) 29:286–95.
7. Abbara S, Grateau G, Ducharme-Bénard S, Saadoun D, Georgin-Lavialle S. Association of vasculitis and familial Mediterranean fever. Front Immunol. (2019) 10:763. doi: 10.3389/fimmu.2019.00763
8. Mills JA, Michel BA, Bloch DA, Calabrese LH, Hunder GG, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Henoch-Schönlein purpura. Arthritis Rheum. (1990) 33:1114–21. doi: 10.1002/art.1780330809
9. Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, Brik R, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: final classification criteria. Ann Rheum Dis. (2010) 69:798–806. doi: 10.1136/ard.2009.116657
10. Lightfoot RW, Michel BA, Bloch DA, Hunder GG, Zvaifler NJ, McShane DJ, et al. The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa. Arthritis Rheum. (1990) 33:1088–93.
11. Leavitt RY, Fauci AS, Bloch DA, Michel BA, Hunder GG, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum. (1990) 33:1101–7.
12. Livneh A, Langevitz P, Zemer D, Zaks N, Kees S, Lidar T, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. (1997) 40:1879–85. doi: 10.1002/art.1780401023
13. Cabrera N, Lega J-C, Kassai B, Wouters C, Kondi A, Cannizzaro E, et al. Safety of biological agents in paediatric rheumatic diseases: a real-life multicenter retrospective study using the JIRcohorte database. Joint Bone Spine. (2019) 86:343–50. doi: 10.1016/j.jbspin.2018.08.003
14. Jéru I, Hentgen V, Cochet E, Duquesnoy P, Le Borgne G, Grimprel E, et al. The risk of familial Mediterranean fever in MEFV heterozygotes: a statistical approach. PLoS One. (2013) 8:e68431. doi: 10.1371/journal.pone.0068431
15. Shinar Y, Obici L, Aksentijevich I, Bennetts B, Austrup F, Ceccherini I, et al. Guidelines for the genetic diagnosis of hereditary recurrent fevers. Ann Rheum Dis. (2012) 71:1599–605. doi: 10.1136/annrheumdis-2011-201271
16. Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med. (1967) 43:227–53. doi: 10.1016/0002-9343(67)90167-2
17. Pagnoux C, Seror R, Henegar C, Mahr A, Cohen P, Le Guern V, et al. Clinical features and outcomes in 348 patients with polyarteritis nodosa: a systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French vasculitis study group database. Arthritis Rheum. (2010) 62:616–26. doi: 10.1002/art.27240
18. Glikson M, Galun E, Schlesinger M, Cohen D, Haskell L, Rubinow A, et al. Polyarteritis nodosa and familial Mediterranean fever: a report of 2 cases and review of the literature. J Rheumatol. (1989) 16:536–9.
19. Ozen S, Ben-Chetrit E, Bakkaloglu A, Gur H, Tinaztepe K, Calguneri M, et al. Polyarteritis nodosa in patients with familial Mediterranean Fever (FMF): a concomitant disease or a feature of FMF? Semin Arthritis Rheum. (2001) 30:281–7. doi: 10.1053/sarh.2001.19958
20. Audemard-Verger A, Terrier B, Dechartres A, Chanal J, Amoura Z, Le Gouellec N, et al. Characteristics and management of IgA vasculitis (Henoch-Schönlein) in adults: data from 260 patients included in a French multicenter retrospective survey. Arthritis Rheumatol. (2017) 69:1862–70. doi: 10.1002/art.40178
21. Direskeneli H. Autoimmunity vs autoinflammation in Behcet’s disease: do we oversimplify a complex disorder? Rheumatology. (2006) 45:1461–5. doi: 10.1093/rheumatology/kel329
22. Gul A. Behçets disease as an autoinflammatory disorder. Curr Drug Targets Inflamm Allergy. (2005) 4:81–3. doi: 10.2174/1568010053622894
23. Atagunduz P, Ergun T, Direskeneli H. MEFV mutations are increased in Behçet’s disease (BD) and are associated with vascular involvement. Clin Exp Rheumatol. (2003) 21:S35–7.
24. Ayesh S, Abu-Rmaileh H, Nassar S, Al-Shareef W, Abu-Libdeh B, Muhanna A, et al. Molecular analysis of MEFV gene mutations among Palestinian patients with Behçet’s disease. Scand J Rheumatol. (2008) 37:370–4. doi: 10.1080/03009740801998788
25. Dursun A, Durakbasi-Dursun HG, Zamani AG, Gulbahar ZG, Dursun R, Yakicier C. Genetic analysis of MEFV Gene pyrin domain in patients with Behçet’s disease. Mediators Inflamm. (2006) 2006:1–4. doi: 10.1155/MI/2006/41783
26. Imırzalıoglu N, Dursun A, Tastan B, Soysal Y, Yakıcıer MC. MEFV gene is a probable susceptibility gene for Behçet’s disease. Scand J Rheumatol. (2005) 34:56–8. doi: 10.1080/03009740510017931
27. Acer Kasman S, Duruöz MT. Spondyloarthritis in familial Mediterranean fever: a cohort study. Rheumatol Int. (2022) 42:1729–39. doi: 10.1007/s00296-022-05158-5
28. Georgin-Lavialle S, Stankovic Stojanovic K, Bachmeyer C, Sellam J, Abbara S, Awad F, et al. Spondyloarthritis associated with familial Mediterranean fever: successful treatment with anakinra. Rheumatology. (2017) 56:167–9. doi: 10.1093/rheumatology/kew290
29. Erden A, Batu ED, Seyhoğlu E, Sari A, Sönmez HE, Armagan B, et al. Increased psoriasis frequency in patients with familial Mediterranean fever. Ups J Med Sci. (2018) 123:57–61. doi: 10.1080/03009734.2017.1423425
30. Hodak E, Atzmony L, Pavlovsky L, Comaneshter D, Cohen AD. Hidradenitis suppurativa is associated with familial Mediterranean fever—A population-based study. J Investig Dermatol. (2017) 137:2019–21. doi: 10.1016/j.jid.2017.04.024
31. Abbara S, Georgin-Lavialle S, Stankovic Stojanovic K, Bachmeyer C, Senet P, Buob D, et al. Association of hidradenitis suppurativa and familial Mediterranean fever: a case series of 6 patients. Joint Bone Spine. (2017) 84:159–62. doi: 10.1016/j.jbspin.2016.02.021
32. Keleş I, Aydın G, Tosun A, İnal E, Keleş H, Orkun S. Familial Mediterranean fever and ankylosing spondylitis in a patient with juvenile idiopathic arthritis: a case report and review of the literature. Rheumatol Int. (2006) 26:846–51. doi: 10.1007/s00296-005-0080-5
33. Rozenbaum M, Rosner I. Severe outcome of juvenile idiopathic arthritis (JIA) associated with familial Mediterranean fever (FMF). Clin Exp Rheumatol. (2004) 22:S75–8.
34. Elhani I, Dumont A, Vergneault H, Ardois S, Le Besnerais M, Levesque H, et al. Association between familial Mediterranean fever and multiple sclerosis: a case series from the JIR cohort and systematic literature review. Mult Scler Relat Disord. (2021) 50:102834. doi: 10.1016/j.msard.2021.102834
35. Beser OF, Cullu Cokugras F, Kutlu T, Erginoz E, Gulcu D, Kasapcopur O, et al. Association of familial Mediterranean fever in Turkish children with inflammatory bowel disease. Turk Arch Ped. (2014) 49:198–202. doi: 10.5152/tpa.2014.1998
36. Fidder HH, Chowers Y, Lidar M, Sternberg M, Langevitz P, Livneh A. Crohn disease in patients with familial Mediterranean fever. Medicine. (2002) 81:411–6. doi: 10.1097/00005792-200211000-00001
37. Villani A-C, Lemire M, Louis E, Silverberg MS, Collette C, Fortin G, et al. Genetic variation in the familial Mediterranean fever gene (MEFV) and risk for Crohn’s disease and ulcerative colitis. PLoS One. (2009) 4:e7154. doi: 10.1371/journal.pone.0007154
38. Akyuz F, Besisik F, Ustek D, Ekmekçi C, Uyar A, Pinarbasi B, et al. Association of the MEFV gene variations with inflammatory bowel disease in Turkey. J Clin Gastroenterol. (2013) 47:e23–7. doi: 10.1097/MCG.0b013e3182597992
39. Kelly G, Sweeney CM, Tobin A-M, Kirby B. Hidradenitis suppurativa: the role of immune dysregulation. Int J Dermatol. (2014) 53:1186–96. doi: 10.1111/ijd.12550
40. Masters SL, Lagou V, Jéru I, Baker PJ, Van Eyck L, Parry DA, et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci Transl Med. (2016) 8:332ra45. doi: 10.1126/scitranslmed.aaf1471
41. Touitou I, Magne X, Molinari N, Navarro A, Quellec AL, Picco P, et al. MEFV mutations in Behçet’s disease. Hum Mutat. (2000) 16:271–2. doi: 10.1002/1098-1004(200009)16:33.0.CO;2-A
Keywords: vasculitis, familial Mediterranean fever, polyarteritis nodosa, IgA vasculitis, pyrin, anti-neutrophil cytoplasmic antibody-associated vasculitis, Behçet syndrome
Citation: Abbara S, Monfort J-B, Savey L, Moguelet P, Saadoun D, Bachmeyer C, Fain O, Terrier B, Amoura Z, Mathian A, Gilardin L, Buob D, Job-Deslandre C, Dufour J-F, Sberro-Soussan R, Grateau G and Georgin-Lavialle S (2022) Vasculitis and familial Mediterranean fever: Description of 22 French adults from the juvenile inflammatory rheumatism cohort. Front. Med. 9:1000167. doi: 10.3389/fmed.2022.1000167
Received: 21 July 2022; Accepted: 11 October 2022;
Published: 28 October 2022.
Edited by:
Riccardo Papa, Giannina Gaslini Institute (IRCCS), ItalyReviewed by:
Giovanni Conti, Azienda Ospedaliera Universitaria Policlinico “G. Martino”, ItalyGiorgio Trivioli, University of Florence, Italy
Copyright © 2022 Abbara, Monfort, Savey, Moguelet, Saadoun, Bachmeyer, Fain, Terrier, Amoura, Mathian, Gilardin, Buob, Job-Deslandre, Dufour, Sberro-Soussan, Grateau and Georgin-Lavialle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie Georgin-Lavialle, c29waGllLmdlb3JnaW4tbGF2aWFsbGVAYXBocC5mcg==
†Present Address: Laurent Gilardin, Service de Médecine Interne, Hôpital Jean-Verdier, Bondy, France