 Mary Zhao
Mary Zhao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 03 January 2022
Sec. Ophthalmology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.802063
This article is part of the Research Topic Novel Diagnostic and Therapeutic Strategies for Retinal Diseases View all 42 articles
Pyroptosis is a gasdermin-mediated, pro-inflammatory form of cell death distinct from apoptosis. In recent years, increasing attention has shifted toward pyroptosis as more studies demonstrate its involvement in diverse inflammatory disease states, including retinal diseases. This review discusses how currently known pyroptotic cell death pathways have been implicated in models of age-related macular degeneration, diabetic retinopathy, and glaucoma. We also identify potential future therapeutic strategies for these retinopathies that target drivers of pyroptotic cell death. Presently, the drivers of pyroptosis that have been studied the most in retinal cells are the nucleotide-binding and oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome, caspase-1, and gasdermin D (GSDMD). Targeting these proteins may help us develop new drug therapies, or supplement existing therapies, in the treatment of retinal diseases. As novel mechanisms of pyroptosis come to light, including those involving other inflammatory caspases and members of the gasdermin protein family, more targets for pyroptosis-mediated therapies in retinal disease can be explored.
Cell death has long been a subject of interest in the study of retinal pathology. The role of programmed cell death (PCD) in retinal diseases is a particularly exciting avenue of research as the regulated nature of these cell death pathways implies that they can potentially be interrupted or manipulated by pharmacological interventions. Traditionally, apoptosis was equated to PCD because it was the most well-studied and well-characterized form of PCD. Research from the 1990's suggested that apoptosis was the main mechanism of regulated cell loss in retinal degeneration and this remained a popular view for most of the 21st century (1–3). The Nomenclature Committee on Cell Death now recognizes that there are other types of regulated cell death besides apoptosis–including necroptosis, ferroptosis, and pyroptosis (4). Necroptosis involves the activation of the pseudokinase mixed-lineage kinase domain-like protein (MLKL), receptor-interacting protein kinase 1 (RIPK1), and receptor-interacting protein kinase 3 (RIPK3) (5). Ferroptosis, as the name suggests, is iron-dependent and is driven by severe lipid peroxidation that results from loss of activity of the lipid repair enzyme glutathione peroxidase 4 (GPX4) (6). Finally, pyroptosis is characterized by membrane pore formation and rapid plasma membrane rupture caused by the binding of the N-terminal of gasdermin proteins to the inner leaflet of the plasma membrane (7–9). As the mechanisms of these cell death pathways have become clearer, more research has emerged supporting their involvement in retinal disease. A recent review summarized the role of necroptosis and ferroptosis in blinding eye disease (10); however, no such a review exists for pyroptosis. Here, we discuss the current understanding of pyroptosis, the research implicating pyroptotic cell death pathways in retinal diseases, and how this knowledge can be applied to identifying novel therapeutic approaches to retinopathies. Specifically, we will focus on the role of pyroptosis in the pathogenesis and potential treatment of three of the most prevalent retinal diseases–age-related macular degeneration, diabetic retinopathy, and glaucoma.
The term “pyroptosis” was first coined in 2001 by Cookson and Brennan from the Greek roots “pyro,” relating to fire or fever, and “ptosis” meaning falling, to describe pro-inflammatory PCD (11). This distinguished pyroptosis from apoptosis, which is non-inflammatory PCD. Initially, caspase-1 was believed to be the effector of pyroptosis after Salmonella-infected macrophages were found to undergo a caspase-1-dependent form of cell death that was associated with pore formation and was distinguishable from apoptosis (11–13). Later, this role shifted to gasdermin D (GSDMD) when it was discovered that the cleaved, N-terminal of GSDMD (GSDMD-N) could bind to and form pores in the cell membrane, leading to pyroptotic cell death (14). Interestingly, pore-forming activity is not exclusive to GSDMD-N; in fact, most gasdermins have an N-terminal pore-forming domain and have the ability to induce pyroptosis (7). This has led to our current understanding of pyroptosis as gasdermin-mediated cell death.
The gasdermin protein family includes gasdermin A/B/C/D/E (GSDMA/B/C/D/E) and DFNB59 (Pejvakin, PJVK) in humans. Gasdermin proteins share 45% sequence homology, and all members (except for Pejvakin) contain C-terminal and N-terminal domains (7, 15). The C-terminal domain is a repressor domain that, when linked to the N-terminal domain, auto-inhibits the N-terminal's cytotoxic activity (16, 17). Inflammatory caspases or granzymes can cleave inactive, full-length gasdermin and liberate its N-terminal domain (14, 18–20). When freed, the N-terminal can then bind to phosphoinositides or cardiolipin on the inner leaflet of the plasma membrane and form membrane pores characteristic of pyroptotic cell death. Pyroptosis is also associated with the release of pro-inflammatory cytokines IL-18 and IL-1β, through these approximately 18 nm-wide membrane pores (21). This adds an additional pathological stressor to cells that is not present with apoptotic cell death and is what earns pyroptosis its designation as a pro-inflammatory form of cell death (7, 8, 13, 15). It was previously thought that extracellular fluid also enters plasma membrane pores during pyroptosis, passively causing plasma membrane rupture (PMR) through oncotic cell swelling. However, PMR is actually an active event mediated by the cell-surface protein Ninjurin 1 (NINJ1) and has been proposed to occur after pyroptotic cell death and IL-18/IL-1β release (22).
Multiple different mechanisms can lead to gasdermin cleavage in pyroptosis. The two most well-studied mechanisms are the canonical and non-canonical inflammasome pathways. The canonical inflammasome pathway, leading to canonical pyroptosis, is mediated by caspase-1. Inflammasomes are multimeric protein complexes, composed of a central sensor protein, an adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase activation and recruitment domain), and pro-caspase-1. The most well-studied sensor proteins known to assemble canonical inflammasomes are NLRP1, NLRP3, NLRC4, AIM2, and pyrin (23, 24). Other proteins such as human NLRP2 and murine NLRP6 have also been implicated in inflammasome signaling (25, 26). These proteins respond to pathogen-associated and danger-associated molecular patterns (PAMPs and DAMPs), which causes “activation” (i.e., assembly) of the inflammasome. Pro-caspase-1 within the activated inflammasome undergoes autocatalytic cleavage into mature caspase-1, and mature caspase-1 can then cleave GSDMD to cause pyroptosis (14, 23, 27). Mature caspase-1 also has the ability to convert pro-IL-18 and pro-IL-1β into their mature forms that are released from membrane pores during pyroptosis (8, 12, 13). Intriguingly, inflammasomes can also be activated and release inflammatory cytokines without necessarily causing cell death through an unknown mechanism that may involve the Toll-IL-1R protein SARM (sterile alpha and HEAT armadillo motif-containing protein) (9, 28). The non-canonical pyroptosis pathway does not depend on caspase-1; rather, it is triggered by the direct binding of procaspase-4/5 in humans, or -11 in mice, to intracellular lipopolysaccharide (LPS). Like caspase-1, activated caspase-4/5/11 can then go on to cleave GSDMD to execute pyroptosis. However, these caspases cannot directly process pro-IL-18 and pro-IL-1β into their mature forms (15, 18, 29, 30).
Up until recently, the canonical and non-canonical inflammasome pathways leading to GSDMD activation were the only known pyroptotic pathways. However, in 2017, both in vitro and in vivo studies showed that pyroptosis could be induced by GSDME expression and cleavage of GSDME into GSDME-N by caspase-3 (31, 32). Furthermore, in 2018, it was found that GSDMD could also be cleaved by caspase-8 in mouse macrophages (33, 34). These findings were especially interesting because caspases 3 and 8 were previously associated with apoptosis and were not thought to be able to interact with gasdermins. Overall, these studies improve our understanding of pyroptosis as we now know that activation of other caspases besides caspases 1/4/5/11 and other gasdermins besides GSDMD can also cause pyroptotic cell death.
Major findings from studies investigating pyroptosis in age-related macular degeneration, diabetic retinopathy, and glaucoma are summarized in Tables 1–3, respectively.
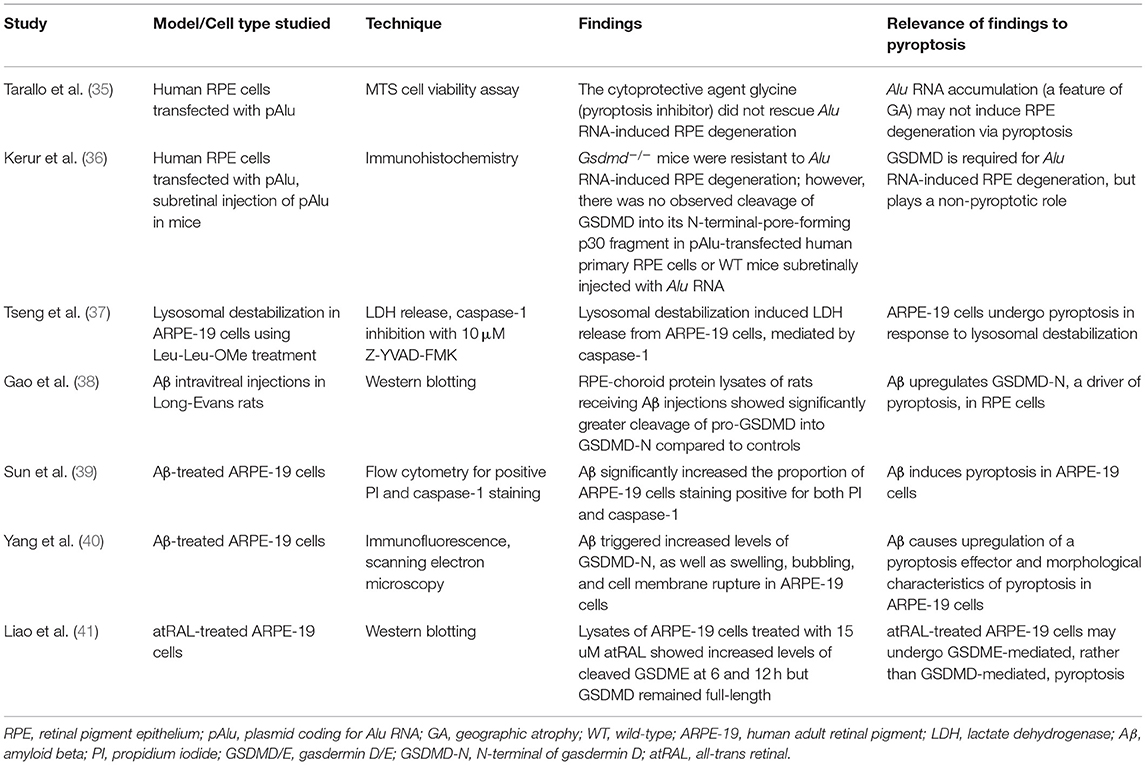
Table 1. Studies investigating pyroptosis in AMD.
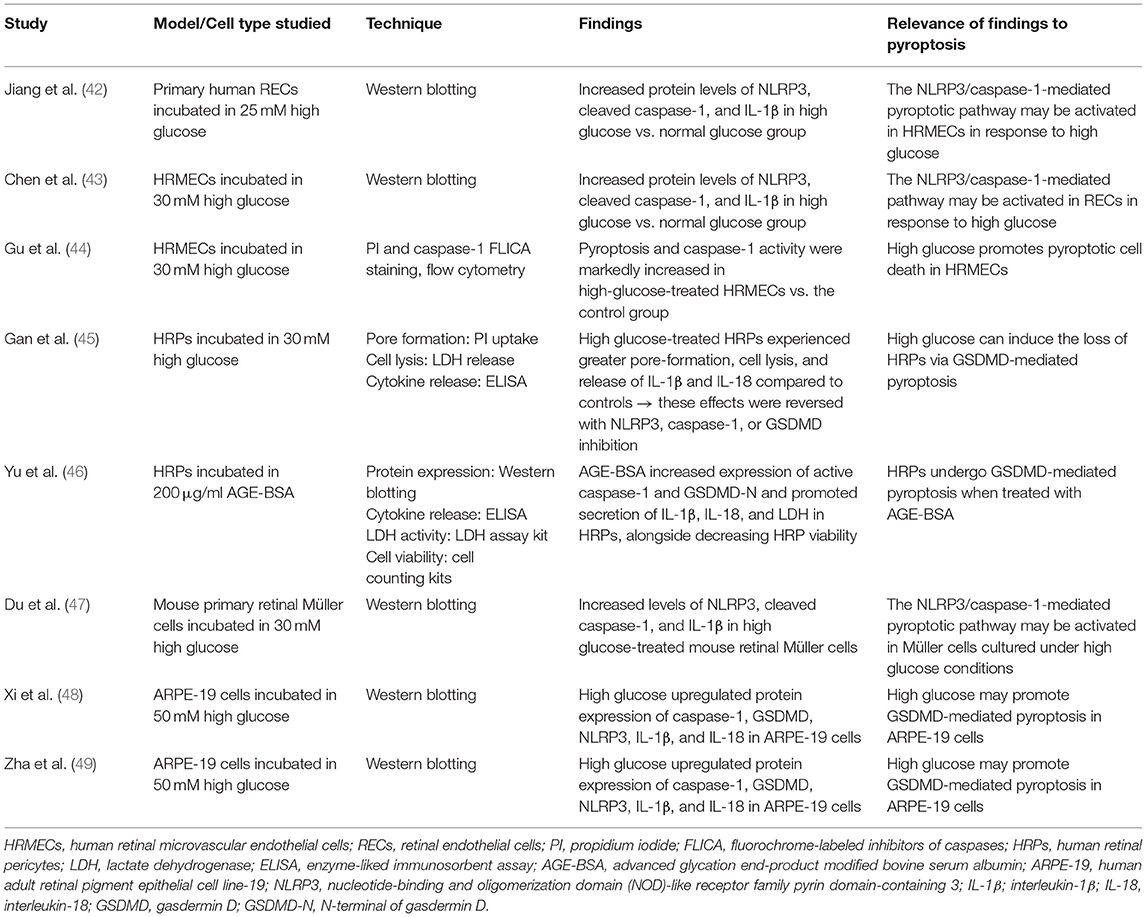
Table 2. Studies investigating pyroptosis in DR.
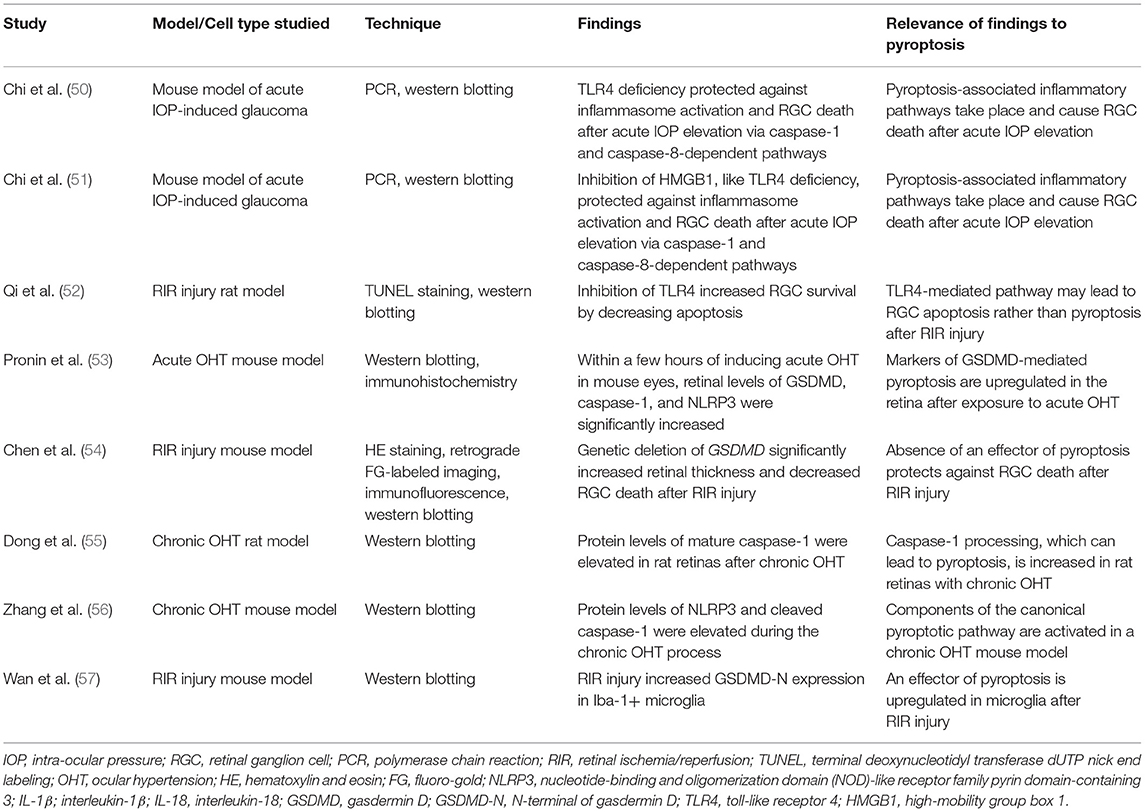
Table 3. Studies investigating pyroptosis in glaucoma.
Age-related macular degeneration (AMD) is the most common cause of irreversible vision loss among the elderly in the developed world, and is projected to affect 288 million people globally by 2040 (58). AMD is a neurodegenerative disease; accumulation of drusen deposits results in progressive degeneration of photoreceptors and retinal pigment epithelium (RPE), primarily in the macula. Clinically, AMD can present as a spectrum of disease phenotypes, with the severity of the disease depending on drusen size. Earlier stages of AMD are defined by the presence of medium-sized drusen deposits and do not present with vision loss. As drusen grow in size and number, atrophy of photoreceptors, RPE, and choriocapillaris and scotoma development can occur. These features are characteristic of a late stage of AMD called geographic atrophy (GA or “dry AMD”). Large drusen also increase the risk of developing neovascular AMD, in which new, abnormal vessels form and invade the outer retina, subretinal space, or subRPE space. Exudative or “wet AMD” occurs when these new vessels rupture and leak exudates, causing fluid accumulation/hemorrhage and severe central vision loss if left untreated (59, 60).
In 2011, Kaneko and colleagues discovered that reduction of the RNase DICER1 led to accumulation of Alu RNA, non-coding RNA transcripts expressed by the Alu retrotransposon, in RPE from human donor eyes with GA. Alu RNA accumulation, in turn, resulted in RPE degeneration in both humans and mice (61). A year later, the same group found that Alu RNA did not induce RPE degeneration in Nlrp3−/− or Casp1−/− mice. This suggested that the canonical NLRP3/caspase-1-dependent pyroptotic pathway may be involved in RPE degeneration in AMD. However, glycine, a cytoprotective agent that attenuates pyroptosis, did not rescue Alu RNA-induced RPE degeneration in the same study. The authors concluded that while NLRP3 and caspase-1 are critical for Alu RNA-induced RPE degeneration, Alu RNA does not induce RPE degeneration via pyroptosis (35). Gsdmd−/− mice, like Nlrp3−/− and Casp1−/− mice, were shown to be resistant to Alu RNA-induced RPE degeneration in a study by Kerur et al. (36), but there was no observed cleavage of GSDMD into its N-terminal pore-forming domain. Furthermore, reconstituting Gsdmd−/− mice with a GSDMD mutant unable to undergo cleavage (pGSDMD-D276A) restored susceptibility to Alu RNA-induced RPE degeneration. Full-length GSDMD cannot induce pyroptosis; thus, it must exert its effects on Alu RNA-induced RPE toxicity through another mechanism. Administration of mature IL-18 to Gsdmd−/− mice restored Alu RNA-induced RPE degeneration and led to the appearance of annexin V positive, propidium iodide (PI) negative staining RPE cells. This suggested that GSDMD plays a role in Alu RNA-induced cytotoxicity via IL-18-dependent apoptosis, rather than pyroptosis, in RPE (36). While the above studies do not support that RPE cells undergo pyroptotic cell death in response to Alu RNA, they do identify NLRP3, caspase-1, and full-length GSDMD as potential therapeutic targets for AMD, particularly for GA.
Lysosomal destabilization, which can result from drusen accumulation and trigger inflammasome activation, has also been studied for its potential to cause pyroptosis in AMD. Lysosomal destabilization with Leu-Leu-OMe treatment induced IL-1β and LDH release from ARPE-19 cells, mediated by caspase-1. These findings indicate that lysosomal destabilization leads to a caspase-1-dependent, pro-inflammatory, and lytic form of cell death in RPE, characteristic of pyroptosis (37). Caspase-1 inhibition may also therefore be a worthwhile therapeutic strategy for AMD treatment.
Additional support for RPE pyroptosis in AMD comes from research on amyloid beta (Aβ), a component of drusen. After the NLRP3 inflammasome was activated by repeated intravitreal injections of Aβ into the eyes of Long-Evans rats, RPE-choroid protein lysates from Aβ-injected animals showed significantly increased levels of GSDMD-N and decreased levels of full-length GSDMD (38). This supported that GSDMD-mediated pyroptosis can be activated in RPE cells and that NLRP3 and GDSMD-N are possible targets for AMD therapy. In another study using Aβ-induced ARPE-19 cells as a model for AMD, Baicalin was found to alleviate Aβ-induced pyroptosis detected by flow cytometry for positive PI and caspase-1 labeling (39). The protective action of Baicalin was mediated by upregulating miRNA-223, which had been previously found to reduce the expression of NLRP3 (62). Baicalin's anti-pyroptotic effects were reversed by miRNA-223 knockdown, whereas adding MCC950 (an NLRP3 inhibitor) once again reduced pyroptosis (39). Lycium Barbarum Polysaccharides (LBP), present in Goji berries, also rescued Aβ-induced reduction of RPE cell viability at low (3.5 mg/L) and high (14 mg/L) doses via attenuation of pyroptosis. Aβ triggered increased levels of GSDMD-N and caused morphological changes in RPE characteristic of pyroptosis, both of which were reversed by LBP treatment (40). As such, inhibiting pyroptosis using Baicalin or LBP may potentially be therapeutic for AMD.
There is also evidence that GSDME-mediated, rather than GSDMD-mediated, pyroptosis occurs in RPE in the all-trans retinal (atRAL) model of AMD. atRAL is generated during the visual (retinoid) cycle and can accumulate in visual cycle anomalies, causing RPE atrophy in AMD. Cleavage of GSDME was detected at 6 and 12 h in lysates of atRAL-treated ARPE-19 cells but GSDMD remained intact, suggesting that atRAL triggers pyroptosis in ARPE-19 cells by activating the caspase-3/GSDME pathway of pyroptosis (41). Research on GSDME-mediated pyroptosis in retinal cells is sparse and further study is required to see if this pathway can be targeted for the treatment of AMD.
Diabetic retinopathy (DR) is a leading cause of preventable vision loss in working-age adults and can be broadly classified into two clinical stages: non-proliferative diabetic retinopathy (NPDR) and proliferative diabetic retinopathy (PDR) (63). Early in NPDR, retinal pericytes that support retinal capillaries are lost, causing capillary occlusion and increased vascular permeability. On fundoscopy, intra-retinal hemorrhages, microaneurysms, and exudates called “cotton wool spots” may be observed in NPDR. NPDR can eventually lead to PDR, in which vascular endothelial growth factor (VEGF) promotes neovascularization in the retina. These newly formed vessels have leaky tight junctions that can result in vitreous hemorrhage or tractional retinal detachment (TRD), and cause vision loss. Another cause of vision loss in DR is diabetic macular edema (DME), where the macula becomes thickened due to breakdown of the blood-retina barrier (BRB) (64). DR is primarily considered a microvascular disease and breakdown of the BRB is key to this disease state. Maintenance of the BRB depends on the functioning of an interdependent network of cells–including endothelial cells that make up the inner BRB, supportive Müller cells and pericytes, and RPE cells which form the outer BRB (65).
A previous review discussed modes of retinal cell death in DR (66). Only Müller cell loss in diabetes was outlined to show characteristics of pyroptosis while other retinal cells including endothelial cells and pericytes were thought to primarily undergo apoptosis or necrosis. More recent studies have found signs of possible pyroptotic cell death in many types of retinal cells in DR models including endothelial cells, pericytes, Müller cells, and RPE. Endothelial cells line the retinal microvasculature and comprise the highly selective inner BRB (65). NLRP3/caspase-1 activation and IL-1β release have been recorded in retinal endothelial cells (RECs) in various in vivo and in vitro models of DR (42, 43, 67). Pyroptotic cell death and caspase-1 activity were markedly increased in human retinal microvascular endothelial cells (HRMECs) incubated in 30 mM high glucose compared to controls (44). Pyroptosis was identified in this study using PI/caspase-1 fluorochrome inhibitor (FLICA) staining and flow cytometry. This suggests that canonical pyroptosis may take place in RECs and targeting NLRP3 and caspase-1 may be a treatment strategy to prevent their loss in DR. Retinal pericytes provide structural support to retinal vessel walls and regulate the expression of tight junctions in adjacent endothelial cells (65). A study published in 2020 showed that silencing GSDMD inhibits IL-1β and IL-18 release, decreases pore formation, and decreases lysis of human retinal pericytes exposed to 30 mM high glucose (45). Another study using advanced glycation endproducts modified bovine serum albumin (AGE-BSA) to simulate the DR environment found increased expression of active caspase-1 and GSDMD-N as well as increased secretion of IL-1β, IL-18, and lactate dehydrogenase (LDH) in human retinal pericytes (HRPCs) alongside decreased HRPC viability (46). Thus, pyroptotic pericyte loss may occur in DR and blocking caspase-1 and GSDMD can potentially preserve pericyte viability. Müller cells are the principal glial cells of the retina and, because of their innate role in mediating neuroinflammation, have long been speculated to participate in pyroptosis (66). Protein levels of NLRP3, ASC, cleaved caspase-1 and cleaved IL-1β were increased by 30 mM high glucose in mouse primary retinal Müller cells. Furthermore, NLRP3 antagonism with the inhibitor drug MCC950 downregulated high glucose-induced upregulation of pro-angiogenic factors including VEGF (47). This implicated activation of the NLRP3 inflammasome pathway in Müller cells in DR and provided support that NLRP3 specifically plays a role in late-stage neovascularization. Finally, while the RPE (part of the outer BRB) is not traditionally viewed to play a central role in the pathophysiology of DR, ARPE-19 cells have recently been found to undergo pyroptotic cell death under stimulation with 50mM glucose, which increased expression of pyroptosis-associated proteins NLRP3, caspase-1, and GSDMD (48, 49). Overall, the NLRP3/caspase-1/GSDMD canonical pyroptotic pathway appears to play a role in the loss of endothelial cells, pericytes, Müller cells, and RPE in cell culture, and in animal and human models for DR. However, few studies have directly demonstrated that the effector of pyroptosis, GSDMD-N, is activated in DR. Future studies aimed at GSDMD-N are needed to evaluate its potential to be a target for DR therapy.
Glaucoma is a group of ocular diseases characterized by the progressive loss of retinal ganglion cells (RGCs), the neurons that communicate visual information from the retina to the brain (68). It is another leading cause of irreversible blindness worldwide and is projected to affect 112 million individuals aged 40–80 by 2040 (69). Various risk factors for glaucoma have been identified–the most notable being elevated intraocular pressure (IOP) and age–but the exact molecular mechanisms that link these risk factors to RGC loss are still under investigation. Past research has demonstrated that RGCs die by apoptosis (70). However, recent studies have implicated inflammasomes, caspase-1, and GSDMD in acute and chronic models of glaucoma, suggesting that apoptosis is not the only form of cell death involved in glaucomatous RGC loss.
In a mouse model for acute elevated IOP-induced glaucoma, NLRP1, NLRP3, ASC, and caspase-1 levels were rapidly upregulated in the retina after ischemic injury. Knockdown of the gene encoding toll-like receptor 4 (TLR4) using TLR4−/− mice reduced inflammasome production and RGC death after ischemic injury (50). TLR4 deficiency therefore seems to protect against RGC death through the inactivation of canonical inflammasomes and may be a potential treatment strategy for acute glaucoma. Chi et al. (50) also showed that caspase-8 is the link between TLR4 and NLRP1/NLRP3 activation. As discussed previously, caspase-8 is traditionally thought of as an initiator of apoptosis but has also been found to play non-apoptotic roles (71). In support of this idea, this study demonstrated that inhibition of caspase-8 significantly reduced levels of NLRP1, NLRP3, ASC, caspase-1, and IL-1β, and also attenuated IOP-induced RGC death. Interestingly, inhibition of caspase-8 completely suppressed production of IL-1β while inhibition of caspase-1 only partially suppressed production of IL-1β. Therefore, therapeutic strategies targeting caspase-8 may be more effective at preventing inflammation in acute glaucoma than those targeting caspase-1. A year later, the same authors found that high-mobility group box 1 (HMGB1), an endogenous ligand of TLR4, is also involved in the above pathway. Inhibition of HMGB1 suppressed the production of NLRP3, ASC, activated caspase-1, activated caspase-8, and IL-1β, and also decreased RGC death after acute IOP elevation similarly to TLR4 (51). Thus, blocking HMGB1 is another way to target TLR4-induced inflammasome pathways in the treatment of acute glaucoma. A study using a retinal ischemia/reperfusion (RIR) injury rat model provided further support for TLR4-induced activation of NLRP3 and also found that inhibition of TLR4 decreased loss of RGCs. However, the type of cell death studied and detected to occur in these RGCs was apoptosis rather than pyroptosis (52). Aside from acute ischemic injury, inflammasomes are also involved in RGC loss from optic nerve crush injury. Following partial optic nerve crush (pONC) in mice, NLRP3 was upregulated at the site of injury and then propagated to the optic nerve head (ONH) and the entire retina within 1 day. Furthermore, NLRP3−/− mice experienced delayed RGC somal loss for 1 week and similarly delayed/decreased axon loss (72). These findings are in congruence with those from previous studies and support that NLRP3 is important for inflammation and RGC death in models of acute glaucoma, making it a worthwhile target for therapy.
The above studies implicated drivers of canonical pyroptosis in glaucomatous RGC death, but they did not study the effector of canonical pyroptosis, GSDMD. Pronin et al. (53) found that within a few hours of inducing acute ocular hypertension (OHT) in mouse eyes, retinal levels of GSDMD, in addition to activated caspase-1 and NLRP3, were significantly increased. RGCs were also shown to be the first cell type in the ganglion cell layer (GCL) to significantly express cleaved GSDMD after acute OHT injury (53). Taken together, these findings suggest that after acute elevation of IOP, inflammasomes are activated in the retina and caspase-1 cleaves GSDMD to potentially trigger pyroptosis in RGCs. In a mouse RIR injury model, intravitreal injection of a Casp1 inhibitor (Z-YVAD-fmk) markedly reduced cleavage of IL-1β and GSDMD, and restored RGC numbers during RIR injury. Furthermore, genetic deletion of GSDMD significantly increased retinal thickness and decreased RGC death after RIR injury (54). Therefore, both caspase-1 inhibition and knockdown of GSDMD expression are possible strategies to therapeutically attenuate RGC death in acute glaucoma. This study also reconciled the previously discovered role of caspase-8 in elevated IOP-induced RGC death with findings from other disease states that caspase-8 can cleave GSDMD, by showing that Casp8 silencing in mice significantly lowered levels of cleaved GSDMD protein after RIR injury (refer to Supplementary Figure 1 for a summary of proposed caspase-8-mediated apoptotic and pyroptotic pathways in acute glaucoma) (33, 34, 54).
The bulk of the research on pyroptosis in glaucoma has been done on acute models, as outlined above. Few studies have looked at the role of pyroptosis in chronic glaucoma. In human donor eyes of chronic glaucoma patients, various inflammasome components, including NLRP3 and caspase-1, were found to be upregulated along with significant cleaved caspase-1 expression in the retina. These early findings suggested that caspase-1 is activated by inflammasome assembly in chronic glaucomatous human retinas (73). NLRP3 and cleaved caspase-1 protein levels were also elevated in the retina of rodent models of chronic OHT (55, 56). These studies implicated P2X7 as the upstream activator of NLRP3. P2X7 receptors are nonselective cation channel receptors that contribute to inflammation in the central nervous system and are activated by ATP (74). Activation of the P2X7 receptor with an agonist (BzATP) increased expression of NLRP3, Casp-1, and ASC in rat retinal microglia. Inhibition of the above pathway using the P2X7 inhibitor A438079 or the NLRP3 inhibitor MCC950 decreased microglial activation and protected against RGC death (56). Thus, inhibiting the P2X7-NLRP3 pathway may be a therapeutic strategy for reducing microglial activation and subsequent RGC death in chronic glaucoma. Research on inflammatory signaling in glaucoma pathogenesis has also identified other ion channels located at the surface of RGCs, such as Transient Receptor Potential Vanilloid isoform 4 (TRPV4) and Pannexin-1 (Panx1), that act as potential sensors and effectors of mechanical strain, ischemia, and inflammatory responses. These signaling pathways are also associated with RGC axonal injury and cell death and can be further explored for potential interactions with inflammasome pathways in chronic glaucoma (75).
NLRP3 and caspase-1 were shown to be increased in the retina in the chronic glaucoma models above. However, whether these pyroptosis inducers and the pyroptosis effector GSDMD are expressed in neurons and RGCs specifically is still debated. There is evidence from other disease states that neurons express NLRP3. Functional inflammasomes and caspase-1 activity were present in cultured human CNS neurons and NLRP3 expression was detected in mesencephalic neurons in a Parkinson's disease model (76–78). However, in glaucomatous human donor eyes, cleaved caspase-1 was more prominent in non-ganglion cells (Brn-3-negative glial cells) (73). This favored that RGCs may undergo cell death through a glial-cell mediated inflammatory pathway. Pronin et al. (53), discussed above, demonstrated the upregulation of NLRP3 inflammasome in RGCs and astrocytes in acute OHT. Zhang et al. (56) from above also supported a glial cell-mediated inflammatory pathway by showing increased expression of inflammasome components in rat retinal microglia rather than RGCs. Our recent research using chronic glaucoma mouse model DBA/2J demonstrated an age-dependent upregulation of NLRP3 in RGCs and a concomitant increase in intraocular pressure (79). RIR injury in mice increased GSDMD-N expression in Iba-1+ microglia, suggesting that microglia undergo pyroptosis in response to RIR injury. On the other hand, RGCs in this study were found to undergo apoptosis (57). All in all, these controversies suggest that inflammation in the glaucomatous eye consists of multiple levels of responses that, at present, we do not fully understand. Neuronal cells including RGCs, possibly perturbed by age-related and/or IOP-induced inflammatory stress, activate glial cells by releasing DAMPs and PAMPs, which could further result in the release of pro-inflammatory cytokines and contribute to neurotoxicity and loss of RGCs. Alternatively, sensors on RGCs may respond to ischemia and inflammatory stress and lead to the remodeling of axons and cell death (50, 80). More research is needed, particularly in chronic models of glaucoma, to determine how pyroptosis fits into this inflammatory picture and whether pyroptotic drivers are appropriate therapeutic targets for glaucoma.
Potential novel therapeutic targets for AMD, DR, and glaucoma have been highlighted throughout this review and are summarized in Table 4. In brief, majority of the suggested strategies target the canonical, NLRP3/caspase-1/GSDMD-mediated pyroptotic cell death pathway. A few studies also supported targeting caspase-3/GSDME and caspase-8/GSDMD pathways in AMD and glaucoma. Aside from the possibility of using these targets to develop new drugs for retinal diseases, targeting pyroptotic cell death pathways can also have an impact on existing therapies for retinal diseases, namely anti-VEGF. Anti-VEGF therapy is the mainstay of treatment for ocular angiogenic disease processes including AMD and DR (81). Studies have shown that NLRP3 inflammasome-mediated pathways can also affect angiogenesis in AMD and DR, and this evidence will be reviewed below. Targeting these pathways may be an alternative strategy to anti-VEGF treatment or enhance the therapeutic effect of existing anti-VEGF treatments.

Table 4. Potential pyroptotic targets for AMD, DR, and glaucoma therapy.
We know that inflammasome activation and release of inflammatory cytokines are associated with pyroptotic cell death; thus, we may expect that inhibiting these factors would have a protective effect in retinal disease. On the contrary, Nlrp3−/− and IL-18−/− mice showed significantly more choroidal neovascularization (CNV) development and subretinal hemorrhage compared to wild-type (WT) mice in a laser-induced model of wet AMD. Furthermore, intravitreal injections of IL-18-neutralizing antibodies after laser-induced CNV resulted in significantly increased CNV development in WT mice, suggesting that IL-18 may protect against CNV through the downregulation of VEGF. Indeed, treatment with IL-18 significantly decreased the amount of VEGF secreted by human ARPE-19 cells as well as mouse brain microvascular endothelial cells (82). Therefore, NLRP3 could be used as a protective agent against AMD and delivering IL-18 to the eye may have a therapeutic effect on CNV progression by decreasing VEGF. The latter was also supported by another study that found that deficiency of IL-18 significantly increased the number of CNV lesions in VEGF-Ahyper mice (83). In 2014, Doyle et al. (84) further demonstrated that IL-18 injection would be safe to use in human eyes. They did not find any measurable cell death, changes in cell morphology, or compromise of plasma membrane integrity even when hyper-physiological doses of recombinant human IL-18 were applied to human ARPE-19 cells and native human RPE cells from three donors. Interestingly, Doyle et al. (84) also showed that IL-18 could enhance the CNV-attenuating effects of anti-VEGF therapy when applied in tandem as an intravitreal injection or systemically via a single subcutaneous dose. CNV volume was most significantly reduced when intravitreal injection of DMS1529 (mouse anti-VEGF) was combined with either intravitreal or subcutaneous administration of GSK (mouse IL-18) in C57BL/6J mice after laser-induced CNV. Systemic IL-18 therapy was also effective at reducing CNV volume alone–subcutaneous administration of GSK at a dose of 0.1 or 1.0 mg/kg 1 day before, and on each day after, laser-induced CNV both significantly attenuated CNV and CNV-induced permeability with no observable adverse effects (84). This shows the potential of using intravitreal or subcutaneous IL-18 separately or as an adjunct to existing anti-VEGF therapies to treat wet AMD pathology.
While the above research is promising, it has been met with some controversy. Tarallo et al. (35) found the opposite effect of IL-18 where IL-18 neutralization protected against RPE death in a mouse model for GA and IL-18 levels were significantly greater in human eyes with GA than in healthy controls. This implies that IL-18 is cytotoxic and may signify that IL-18 plays different roles in wet vs. dry AMD. IL-18 levels were also found to be significantly elevated in the serum of AMD patients compared to healthy controls, suggesting that higher systemic levels of IL-18 are associated with AMD diagnosis (85). This opposes the above suggestion that systemic injection of IL-18 can be therapeutic for AMD. Furthermore, while the studies by Doyle and colleagues suggested that NLRP3 could be used as a protective agent in AMD, other studies have found that NLRP3 activation/consequent increase in active IL-1β is pro-angiogenic and promotes VEGF-induced AMD pathologies (83). Nucleoside reverse transcriptase inhibitors (NRTIs) such as stavudine (d4T) also reduced CNV volume in a laser-induced mouse model of CNV via blockade of a P2X7-induced pathway of inflammasome activation (86). In DR, studies have proposed that the pro-inflammatory events associated with NLRP3 activity cause breakdown of the BRB and subsequent neovascular response leading to PDR (87). Inhibition of caspase-1 with minocycline prevented acellular capillary development in STZ-induced diabetic and galactosemia mouse models (88). Elevated protein expression of NLRP3, caspase-1, and inflammatory cytokines was found in the proliferative membranes of human donor eyes with PDR compared to healthy controls (89). Similar results were seen in vitreous fluid samples of DR patients, especially in PDR eyes with TRD and active neovessel formation (90, 91). Finally, NLRP3 inhibition with MCC950 downregulated high glucose-induced upregulation of pro-angiogenic factors including VEGF (47). In sum, NLRP3-mediated inflammatory pathways are involved in angiogenic disease processes in AMD and DR, but further research is required to resolve the debate over whether its role is deleterious or beneficial.
In this review, we have outlined the role of pyroptosis as a gasdermin-mediated inflammatory form of PCD in three common retinal diseases–age-related macular degeneration, diabetic retinopathy, and glaucoma. In AMD, GSDMD-mediated pyroptosis appears to occur in RPE when triggered by lysosomal destabilization or Aβ while GSDME-mediated pyroptosis occurs in the atRAL model of AMD. The research on pyroptosis in DR is in more preliminary stages, with most of its evidence for pyroptosis being limited to inflammasome activation rather than gasdermin activation in endothelial cells, pericytes, Müller cells, and RPE. Finally, there is support for gasdermin involvement in RGC loss in acute glaucoma, but evidence in chronic glaucoma models remains in its infancy. All in all, as our understanding of pyroptosis has grown and evolved, there is more support for its involvement in retinal disease. However, there are still many limitations in our understanding of pyroptosis in retinal disease that must be addressed.
Firstly, the involvement of pyroptosis in retinal disease does not exclude the occurrence of other forms of PCD such as apoptosis, ferroptosis, and necroptosis. There is substantial evidence for the involvement of these other PCD pathways in retinal disease as well (10, 66, 92). Further research is needed to uncover how different forms of PCD interact with each other in the retina and what factors ultimately determine the type of PCD an individual retinal cell will succumb to in pathological states. This information is vital in the development of therapies targeting PCD. If we target a form of PCD that is not the primary mode of cell death naturally occurring in AMD, DR, or glaucoma, then such treatments for these diseases may be ineffective. Or, if blocking one form of PCD such as apoptosis causes another, more inflammatory cell death mechanism to occur, we could potentially do more harm. A limited number of studies have provided some insight into how different types of PCD may be linked. Jiang et al. (93) showed that the caspase-3/GSDME pathway can result in either apoptosis or pyroptosis, depending on the expression level of GSDME. GSDME may therefore be the link between PCD pathways we have been looking for and provide an explanation for why we have been able to identify both apoptotic and pyroptotic mechanisms in retinal disease. On the other hand, Kayagaki et al. (22) identified that NINJ1 plays a potent role in causing plasma membrane rupture and DAMP-release not only following pyroptosis, but also during apoptosis and necrosis. Therefore, targeting NINJ1 could be a downstream therapeutic strategy that suppresses propagation of the cell death-associated inflammatory response regardless of its upstream mechanism (pyroptotic or otherwise).
We also need to be wary of the limitations in how we interpret the existing literature on pyroptosis in retinal disease. Because the essential role of gasdermin in pyroptosis was only recently established in 2015, earlier research on pyroptosis in retinal disease could only aim to identify inflammasome and caspase-1 activation in these diseases. We now know that activated inflammasomes can cause caspase-1 to cleave and release inflammatory cytokines without resulting in cell death (9, 28). Thus, we cannot assume that inflammasome activity, presence of mature caspase-1, and release of inflammatory cytokines in retinal cells necessarily means that pyroptosis is occurring in those cells. In addition, with the discovery that caspases 3 and 8 can activate GSDME and GSDMD (respectively) to mediate pyroptosis, we must also re-evaluate previous results suggesting that activation of these caspases in retinal cells represented apoptotic cell death. This also supports that there is significant overlap and a complex interplay between pyroptotic and apoptotic cell death that we do not currently understand. Many questions related to this require further study. For one, under what conditions do caspases 3 and 8 favor cleaving gasdermin over their usual apoptotic substrates? Furthermore, what other cell types and pathologies besides those already identified demonstrate caspase-3/8-mediated pyroptosis as opposed to the more well-known mechanisms of pyroptosis? GSDME-mediated pyroptosis is increasingly being demonstrated to play a role in cancer (94), but its involvement in neurodegenerative diseases including retinal diseases is still largely unexplored. There is also the question of if GSDMA/B/C-mediated pyroptosis has a role to play in retinal diseases. The gold standard for demonstrating the occurrence of pyroptosis should be the identification of cleaved N-terminal of gasdermin proteins in well-established models of retinal disease. More studies like this would provide a better foundation for us to determine if gasdermin-mediated therapy is a viable strategy for the treatment of retinal pathologies. Gasdermin-mediated therapies are currently being studied in tumor treatment (9), and potential translation of these therapies to retinal diseases is another area for future research.
MZ sourced and analyzed the referenced literature, conceived the structure of the manuscript, and wrote the manuscript. SL was a significant contributor in reviewing the drafts of the manuscript, adding to section “Pyroptosis and Glaucoma” of the manuscript, and creating supplementary figures. JM was a significant contributor in refining the topic of the review, conceiving the structure of the manuscript, reviewing the drafts of the manuscript, creating supplementary figures, and providing funding for the study. All authors have read and approved the final manuscript.
Authors acknowledge funding from Canadian Institutes of Health Research, Natural Science and Engineering Research Council, and National Institutes of Health Research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Authors wish to acknowledge Faculty of Medicine (UBC), Jing Cui, Eleanor To, Jeanne Xi, Gideon Obasanmi and Manjosh Uppal.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2021.802063/full#supplementary-material
1. Remé CE, Grimm C, Hafezi F, Wenzel A, Williams TP. Apoptosis in the retina: the silent death of vision. News Physiol Sci. (2000) 15:120–5. doi: 10.1152/physiologyonline.2000.15.3.120
2. Remé CE, Grimm C, Hafezi F, Marti A, Wenzel A. Apoptotic cell death in retinal degenerations. Prog Retin Eye Res. (1998) 17:443–64. doi: 10.1016/S1350-9462(98)00009-3
3. Portera-Cailliau C, Sung CH, Nathans J, Adler R. Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc Natl Acad Sci U S A. (1994) 91:974–8. doi: 10.1073/pnas.91.3.974
4. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-018-0102-y
5. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. (2015) 517:311–20. doi: 10.1038/nature14191
6. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. (2016) 26:165–76. doi: 10.1016/j.tcb.2015.10.014
7. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. (2016) 535:111–6. doi: 10.1038/nature18590
8. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
9. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. (2021) 6:1. doi: 10.1038/s41392-021-00507-5
10. Peng JJ, Song WT, Yao F, Zhang X, Peng J, Luo XJ, et al. Involvement of regulated necrosis in blinding diseases: focus on necroptosis and ferroptosis. Exp Eye Res. (2020) 191:107922. doi: 10.1016/j.exer.2020.107922
11. Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol. (2000) 38:31–40. doi: 10.1046/j.1365-2958.2000.02103.x
12. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. (2009) 7:99–109. doi: 10.1038/nrmicro2070
13. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. J Immunol. (2006) 202:1913–26. doi: 10.1111/j.1462-5822.2006.00751.x
14. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
15. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. (2017) 42:245–54. doi: 10.1016/j.tibs.2016.10.004
16. Liu Z, Wang C, Yang J, Zhou B, Yang R, Ramachandran R, et al. Crystal structures of the full-length murine and human gasdermin d reveal mechanisms of autoinhibition, lipid binding, and oligomerization. Immunity. (2019) 51:43–9. doi: 10.1016/j.immuni.2019.04.017
17. Kuang S, Zheng J, Yang H, Li S, Duan S, Shen Y, et al. Structure insight of GSDMD reveals the basis of GSDMD autoinhibition in cell pyroptosis. Proc Natl Acad Sci U S A. (2017) 114:10642–7. doi: 10.1073/pnas.1708194114
18. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. (2015) 526:666–71. doi: 10.1038/nature15541
19. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. (2020) 368:eaaz7548. doi: 10.1126/science.aaz7548
20. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. (2020) 579:415–20. doi: 10.1038/s41586-020-2071-9
21. Ruan J, Xia S, Liu X, Lieberman J, Wu H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature. (2018) 557:62–7. doi: 10.1038/s41586-018-0058-6
22. Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O'Rourke K, Li Q, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. (2021) 591:131–6. doi: 10.1038/s41586-021-03218-7
23. Place DE, Kanneganti TD. Recent advances in inflammasome biology. Curr Opin Immunol. (2018) 50:32–8. doi: 10.1016/j.coi.2017.10.011
24. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
25. Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia. (2013) 61:1113–21. doi: 10.1002/glia.22499
26. Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell. (2015) 163:1428–43. doi: 10.1016/j.cell.2015.10.048
27. Yerramothu P, Vijay AK, Willcox MDP. Inflammasomes, the eye and anti-inflammasome therapy. Eye. (2018) 32:491–505. doi: 10.1038/eye.2017.241
28. Carty M, Kearney J, Shanahan KA, Hams E, Sugisawa R, Connolly D, et al. Cell survival and cytokine release after inflammasome activation is regulated by the Toll-IL-1R protein SARM. Immunity. (2019) 50:1412–24. doi: 10.1016/j.immuni.2019.04.005
29. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A. (2016) 113:7858–63. doi: 10.1073/pnas.1607769113
30. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. (2014) 514:187–92. doi: 10.1038/nature13683
31. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. (2017) 547:99–103. doi: 10.1038/nature22393
32. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. (2017) 8:1–14. doi: 10.1038/ncomms14128
33. Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A. (2018) 115:E10888–97. doi: 10.1073/pnas.1809548115
34. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science. (2018) 362:1064–9. doi: 10.1126/science.aau2818
35. Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. (2012) 149:847–59. doi: 10.1016/j.cell.2012.03.036
36. Kerur N, Fukuda S, Banerjee D, Kim Y, Fu D, Apicella I, et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat Med. (2018) 24:50–61. doi: 10.1038/nm.4450
37. Tseng WA, Thein T, Kinnunen K, Lashkari K, Gregory MS, D'Amore PA, et al. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. (2013) 54:110–20. doi: 10.1167/iovs.12-10655
38. Gao J, Cui JZ, To E, Cao S, Matsubara JA. Evidence for the activation of pyroptotic and apoptotic pathways in RPE cells associated with NLRP3 inflammasome in the rodent eye. J Neuroinflammation. (2018) 15:13–5. doi: 10.1186/s12974-018-1062-3
39. Sun HJ, Jin XM, Xu J, Xiao Q. Baicalin alleviates age-related macular degeneration via miR-223/NLRP3-regulated pyroptosis. Pharmacology. (2020) 105:28–38. doi: 10.1159/000502614
40. Yang M, So KF, Lo ACY, Lam WC. The effect of lycium barbarum polysaccharides on pyroptosis-associated amyloid β(1-40) oligomers-induced adult retinal pigment epithelium 19 cell damage. Int J Mol Sci. (2020) 21:4658. doi: 10.3390/ijms21134658
41. Liao Y, Zhang H, He D, Wang Y, Cai B, Chen J, et al. Retinal pigment epithelium cell death is associated with NLRP3 inflammasome activation by all-trans retinal. Invest Ophthalmol Vis Sci. (2019) 60:3034–45. doi: 10.1167/iovs.18-26360
42. Jiang Y, Liu L, Curtiss E, Steinle JJ. Epac1 blocks NLRP3 inflammasome to reduce IL-1β in retinal endothelial cells and mouse retinal vasculature. Mediators Inflamm. (2017) 2017:2860956. doi: 10.1155/2017/2860956
43. Chen W, Zhao M, Zhao S, Lu Q, Ni L, Zou C, et al. Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: a novel inhibitory effect of minocycline. Inflamm Res Off J Eur Histamine Res Soc. (2017) 66:157–66. doi: 10.1007/s00011-016-1002-6
44. Gu C, Draga D, Zhou C, Su T, Zou C, Gu Q, et al. miR-590-3p Inhibits pyroptosis in diabetic retinopathy by targeting NLRP1 and inactivating the NOX4 signaling pathway. Invest Ophthalmol Vis Sci. (2019) 60:4215–23. doi: 10.1167/iovs.19-27825
45. Gan J, Huang M, Lan G, Liu L, Xu F. High glucose induces the loss of retinal pericytes partly via NLRP3-caspase-1-GSDMD-mediated pyroptosis. Biomed Res Int. (2020) 2020:1–12. doi: 10.1155/2020/4510628
46. Yu X, Ma X, Lin W, Xu Q, Zhou H, Kuang HY. Long noncoding RNA MIAT regulates primary human retinal pericyte pyroptosis by modulating miR-342–3p targeting of CASP1 in diabetic retinopathy. Exp Eye Res. (2020). doi: 10.1016/j.exer.2020.108300
47. Du J, Wang Y, Tu Y, Guo Y, Sun X, Xu X, et al. A prodrug of epigallocatechin-3-gallate alleviates high glucose-induced pro-angiogenic factor production by inhibiting the ROS/TXNIP/NLRP3 inflammasome axis in retinal Müller cells. Exp Eye Res. (2020) 196:108065. doi: 10.1016/j.exer.2020.108065
48. Xi X, Yang Y, Ma J, Chen Q, Zeng Y, Li J, et al. MiR-130a alleviated high-glucose induced retinal pigment epithelium (RPE) death by modulating TNF-α/SOD1/ROS cascade mediated pyroptosis. Biomed Pharmacother. (2020) 125:109924. doi: 10.1016/j.biopha.2020.109924
49. Zha X, Xi X, Fan X, Ma M, Zhang Y, Yang Y. Overexpression of METTL3 attenuates high-glucose induced RPE cell pyroptosis by regulating miR-25-3p/PTEN/Akt signaling cascade through DGCR8. Aging. (2020) 12:8137–50. doi: 10.18632/aging.103130
50. Chi W, Li F, Chen H, Wang Y, Zhu Y, Yang X, et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1β production in acute glaucoma. Proc Natl Acad Sci U S A. (2014) 111:11181–6. doi: 10.1073/pnas.1402819111
51. Chi W, Chen H, Li F, Zhu Y, Yin W, Zhuo Y. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-κB pathway in acute glaucoma. J Neuroinflammation. (2015) 12:1–12. doi: 10.1186/s12974-015-0360-2
52. Qi Y, Zhao M, Bai Y, Huang L, Yu W, Bian Z, et al. Retinal ischemia/reperfusion injury is mediated by tolllike receptor 4 activation of NLRP3 inflammasomes. Investig Ophthalmol Vis Sci. (2014) 55:5466–75. doi: 10.1167/iovs.14-14380
53. Pronin A, Pham D, An W, Dvoriantchikova G, Reshetnikova G, Qiao J, et al. Inflammasome activation induces pyroptosis in the retina exposed to ocular hypertension injury. Front Mol Neurosci. (2019) 12:36. doi: 10.3389/fnmol.2019.00036
54. Chen H, Deng Y, Gan X, Li Y, Huang W, Lu L, et al. NLRP12 collaborates with NLRP3 and NLRC4 to promote pyroptosis inducing ganglion cell death of acute glaucoma. Mol Neurodegener. (2020) 15:1–16. doi: 10.1186/s13024-020-00372-w
55. Dong L, Hu Y, Zhou L, Cheng X. P2X7 receptor antagonist protects retinal ganglion cells by inhibiting microglial activation in a rat chronic ocular hypertension model. Mol Med Rep. (2018) 17:2289–96. doi: 10.3892/mmr.2017.8137
56. Zhang Y, Xu Y, Sun Q, Xue S, Guan H, Ji M. Activation of P2X7R- NLRP3 pathway in retinal microglia contribute to retinal ganglion cells death in chronic ocular hypertension (COH). Exp Eye Res. (2019) 188:107771. doi: 10.1016/j.exer.2019.107771
57. Wan P, Su W, Zhang Y, Li Z, Deng C, Li J, et al. LncRNA H19 initiates microglial pyroptosis and neuronal death in retinal ischemia/reperfusion injury. Cell Death Differ. (2020) 27:176–91. doi: 10.1038/s41418-019-0351-4
58. Wong WL, Su X, Li X, Cheung CMG, Klein R, Cheng CY, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Heal. (2014) 2:e106–16. doi: 10.1016/S2214-109X(13)70145-1
59. Mitchell P, Liew G, Gopinath B, Wong TY. Age-related macular degeneration. Lancet. (2018) 392:1147–59. doi: 10.1016/S0140-6736(18)31550-2
60. Fleckenstein M, Keenan TDL, Guymer RH, Chakravarthy U, Schmitz-Valckenberg S, Klaver CC, et al. Age-related macular degeneration. Nat Rev Dis Prim. (2021) 7:1. doi: 10.1038/s41572-021-00265-2
61. Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, et al. DICER1 deficit induces alu RNA toxicity in age-related macular degeneration. Nature. (2011) 471:325–32. doi: 10.1038/nature09830
62. Hu Z, Lv X, Chen L, Gu X, Qian H, Fransisca S, et al. Protective effects of microRNA-22-3p against retinal pigment epithelial inflammatory damage by targeting NLRP3 inflammasome. J Cell Physiol. (2019) 234:18849–57. doi: 10.1002/jcp.28523
63. Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet. (2010) 376:124–36. doi: 10.1016/S0140-6736(09)62124-3
64. Wang W, Lo ACY. Diabetic retinopathy: pathophysiology and treatments. Int J Mol Sci. (2018) 19:6. doi: 10.3390/ijms19061816
65. Lechner J, O'Leary OE, Stitt AW. The pathology associated with diabetic retinopathy. Vision Res. (2017) 139:7–14. doi: 10.1016/j.visres.2017.04.003
66. Feenstra DJ, Yego EC, Mohr S. Modes of retinal cell death in diabetic retinopathy. J Clin Exp Ophthalmol. (2013) 04:1–16. doi: 10.4172/2155-9570.1000298
67. Jiang Y, Steinle JJ. Epac1 inhibits PKR to reduce NLRP3 inflammasome proteins in retinal endothelial cells. J Inflamm Res. (2019) 12:153–9. doi: 10.2147/JIR.S210441
68. Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA. (2014) 311:1901–11. doi: 10.1001/jama.2014.3192
69. Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng C-Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology. (2014) 121:2081–90. doi: 10.1016/j.ophtha.2014.05.013
70. Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res. (2012) 31:152–81. doi: 10.1016/j.preteyeres.2011.11.002
71. Nagata S, Tanaka M. Programmed cell death and the immune system. Nat Rev Immunol. (2017) 17:333–40. doi: 10.1038/nri.2016.153
72. Puyang Z, Feng L, Chen H, Liang P, Troy JB, Liu X. Retinal ganglion cell loss is delayed following optic nerve crush in NLRP3 knockout mice. Sci Rep. (2016) 6:6–13. doi: 10.1038/srep20998
73. Yang X, Luo C, Cai J, Powell DW, Yu D, Kuehn MH, et al. Neurodegenerative and inflammatory pathway components linked to TNF-α/TNFR1 signaling in the glaucomatous human retina. Invest Ophthalmol Vis Sci. (2011) 52:8442–54. doi: 10.1167/iovs.11-8152
74. Stokes L, Spencer SJ, Jenkins TA. Understanding the role of P2X7 in affective disorders—are glial cells the major players? Front Cell Neurosci. (2015) 9:1–6. doi: 10.3389/fncel.2015.00258
75. KriŽaj D, Ryskamp DA, Tian N, Tezel G, Mitchell CH, Slepak VZ, et al. From Mechanosensitivity to inflammatory responses: new players in the pathology of glaucoma. Curr Eye Res. (2014) 39:105–19. doi: 10.3109/02713683.2013.836541
76. Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, et al. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. (2015) 22:1676–86. doi: 10.1038/cdd.2015.16
77. von Herrmann KM, Salas LA, Martinez EM, Young AL, Howard JM, Feldman MS, et al. NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson's disease. npj Park Dis. (2018) 4:1. doi: 10.1038/s41531-018-0061-5
78. Cheng X, Xu S, Zhang C, Qin K, Yan J, Shao X. The BRCC3 regulated by Cdk5 promotes the activation of neuronal NLRP3 inflammasome in Parkinson's disease models. Biochem Biophys Res Commun. (2020) 522:647–54. doi: 10.1016/j.bbrc.2019.11.141
79. Li S, Gao J, Cui JZ, Matsubara JA. NLRP3 inflammasome contributes to retinal ganglion cell (RGC) death during glaucoma pathogenesis in DBA/2J mouse model. Invest Ophthalmol Vis Sci. (2019) 60:3790–3790. doi: 10.14288/1.0394326
80. Lu W, Albalawi F, Beckel JM, Lim JC, Laties AM, Mitchell CH. The P2X7 receptor links mechanical strain to cytokine IL-6 up-regulation and release in neurons and astrocytes. J Neurochem. (2017) 141:436–48. doi: 10.1111/jnc.13998
81. Tah V, Orlans HO, Hyer J, Casswell E, Din N, Sri Shanmuganathan V, et al. Anti-VEGF therapy and the retina: an update. J Ophthalmol. (2015) 2015:627674. doi: 10.1155/2015/627674
82. Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med. (2012) 18:791–8. doi: 10.1038/nm.2717
83. Marneros AG. NLRP3 inflammasome blockade inhibits VEGF-A-induced age-related macular degeneration. Cell Rep. (2013) 4:945–58. doi: 10.1016/j.celrep.2013.08.002
84. Doyle SL, Ozaki E, Brennan K, Humphries MM, Mulfaul K, Keaney J, et al. IL-18 attenuates experimental choroidal neovascularization as a potential therapy for wet age-related macular degeneration. Sci Transl Med. (2014) 6:230ra44. doi: 10.1126/scitranslmed.3007616
85. Weaver C, Cyr B, de Rivero Vaccari JC, de Rivero Vaccari JP. Inflammasome proteins as inflammatory biomarkers of age-related macular degeneration. Transl Vis Sci Technol. (2020) 9:1–9. doi: 10.1167/tvst.9.13.27
86. Fowler BJ, Gelfand BD, Kim Y, Kerur N, Tarallo V, Hirano Y, et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science. (2014) 346:1000–3. doi: 10.1126/science.1261754
87. Chaurasia SS, Lim RR, Parikh BH, Wey YS, Tun BB, Wong TY, et al. The NLRP3 inflammasome may contribute to pathologic neovascularization in the advanced stages of diabetic retinopathy. Sci Rep. (2018) 8:2847-z. doi: 10.1038/s41598-018-21198-z
88. Vincent JA, Mohr S. Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes. (2007) 56:224–30. doi: 10.2337/db06-0427
89. Zhang Y, Lv X, Hu Z, Ye X, Zheng X, Ding Y, et al. Protection of Mcc950 against high-glucose-induced human retinal endothelial cell dysfunction. Cell Death Dis. (2017) 8:e2941. doi: 10.1038/cddis.2017.308
90. Loukovaara S, Piippo N, Kinnunen K, Hytti M, Kaarniranta K, Kauppinen A. NLRP3 inflammasome activation is associated with proliferative diabetic retinopathy. Acta Ophthalmol. (2017) 95:803–8. doi: 10.1111/aos.13427
91. Chen H, Zhang X, Liao N, Mi L, Peng Y, Liu B, et al. Enhanced expression of NLRP3 inflammasome-related inflammation in diabetic retinopathy. Invest Ophthalmol Vis Sci. (2018) 59:978–85. doi: 10.1167/iovs.17-22816
92. Telegina DV, Kozhevnikova OS, Kolosova NG. Molecular mechanisms of cell death in retina during development of age-related macular degeneration. Adv Gerontol. (2017) 7:17–24. doi: 10.1134/S2079057017010155
93. Jiang M, Qi L, Li L, Li Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. (2020) 6:1. doi: 10.1038/s41420-020-00349-0
Keywords: pyroptosis, cell death, NLRP3, caspase-1, GSDMD, age-related macular degeneration, diabetic retinopathy, glaucoma
Citation: Zhao M, Li S and Matsubara JA (2022) Targeting Pyroptotic Cell Death Pathways in Retinal Disease. Front. Med. 8:802063. doi: 10.3389/fmed.2021.802063
Received: 26 October 2021; Accepted: 29 November 2021;
Published: 03 January 2022.
Edited by:
Hetian Lei, Shenzhen Eye Hospital, ChinaReviewed by:
Youssef Aachoui, University of Arkansas for Medical Sciences, United StatesCopyright © 2022 Zhao, Li and Matsubara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mary Zhao, zhaomary@student.ubc.ca
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.