 Jiayi Wang
Jiayi Wang Lingyao Du
Lingyao Du Hong Tang1,2*
Hong Tang1,2*- 1Center of Infectious Diseases, West China Hospital of Sichuan University, Chengdu, China
- 2Division of Infectious Diseases, State Key Laboratory of Biotherapy and Center of Infectious Diseases, West China Hospital, Sichuan University, Chengdu, China
Chronic hepatitis B virus (CHB) infection remains a major global public health issue for which there is still lacking effective curative treatment. Interferon-α (IFN-α) and its pegylated form have been approved as an anti-HBV drug with the advantage of antiviral activity and host immunity against HBV infection enhancement, however, IFN-α treatment failure in CHB patients is a challenging obstacle with 70% of CHB patients respond poorly to exogenous IFN-α treatment. The IFN-α treatment response is negatively regulated by both viral and host factors, and the role of viral factors has been extensively illustrated, while much less attention has been paid to host negative factors. Here, we summarized evidence of host negative regulators and parameters involved in IFN-α therapy failure, review the mechanisms responsible for these effects, and discuss the possible improvement of IFN-based therapy and the rationale of combining the inhibitors of negative regulators in achieving an HBV cure.
Introduction
Chronic hepatitis B virus (CHB) infection affects more than 290 million people worldwide, with 1.5 million new infections each year, estimated by World Health Organization (1). The lifetime risk of developing hepatocellular carcinoma (HCC) and liver cirrhosis among HBV carriers ranges from 10 to 25% (2) and from 15 to 40% (3), respectively. HBV is a partially double-stranded circular DNA virus belonging to the Hepadnaviridae family (4), first discovered in an Australian aborigine (5). HBV has 10 genotypes (A-J) with nearly 40 sub-genotypes distributed in distinct areas, while the infectious in East Asia are most commonly HBV genotype B and C (3, 6). Current HBV treatment strategies can be categorized into 2 groups: interferons (IFN) and nucleos (t)ide analogs (NAs).
IFNs are a group of signaling proteins released by host cells in response to various pathogens, including viruses, bacteria, and parasites. IFN-α belongs to type I IFNs and has been approved as an anti-HBV therapy. Although with multiple adverse effects and inconvenient administration, IFN-α has the strength of a relatively short interval of treatment, without risk of drug resistance, higher rate of hepatitis B e antigen (HBeAg) seroconversion, and particularly hepatitis B surface antigen (HBsAg) seroclearance, which is unable to reach by current NAs administration (7, 8). Therefore, it is important to identify CHB patients who will benefit from treatment before the start of IFN-α-based therapy. Besides, IFN's efficacy is far less than satisfactory, with only one-third of HBeAg positive CHB patients achieved HBeAg seroconversion after the IFN-α therapy, and even less efficacy was observed in HBeAg negative patients (9). The molecular mechanisms responsible for the failure of IFN-α treatment are not well-understood, but evidence shows that both viral and host factors are involved. Viral factors include HBV genotype, mutations within the HBV genome, and baseline level of viral load. Individuals infected with HBV-C and D (comparing to HBV-A and B) (6, 10), HBV has mutations within the HBV pre-core and/or basal core promoter (PC and/or BCP) region (6, 11–13), and high baseline viral load are tend to be more resistance to IFN-α therapy. The molecular mechanisms underlying the virally mediated resistance to IFN-α have been summarized in another review (14). In addition to the viral factors, host factors play an equally important role in modulating the effectiveness of IFN-α therapy for CHB patients' treatment. Some host molecules function as negative regulators of IFN therapy by inhibiting IFN production or signaling pathways, while several host parameters provide essential information of liver function and are able to predict the efficacy of IFN-α therapy. Therefore, a thorough understanding of the mechanism responsible for host factor-mediated inhibition of IFN therapy is needed for providing therapeutic targets to improve the efficacy of IFN-α treatment in terms of HBV infection. In this review, we summarize host negative regulators that impair IFN-α therapy of HBV infection and host parameters that can predict the IFN-α therapy efficacy, review the underlying mechanisms, as well as discuss the potential therapeutic approaches for controlling HBV infection.
Classical IFN and ISGs Production Pathways in Response to HBV Infection and Antiviral Activity of IFN-α
Interferon (IFN) was originally discovered in 1957 by Isaacs and Lindenmann, and was named for their ability to interfere with viral replication (15, 16). Three interferon families are discovered—type I, II, and III. The type I IFN family encodes 13 partially homologous IFN-α subtypes in humans, IFN-β and several single gene products (IFN-ε, IFN-τ, IFN-κ, IFN-ω, IFN-δ, and IFN-ζ). The type IFN II family comprises a single gene product, IFN-γ, while the type III IFN family consists of IFN-λ1, IFN-λ2, IFN-λ3 (also known as IL-29, IL-28A and IL-28B, respectively), and IFN- λ4 (17, 18). Type I IFNs are secreted by almost all virus-infected cells including hepatocytes and by specialized blood lymphocytes, while the production of IFN-γ is restricted to immune cells, including natural killer (NK) cells, macrophages, and T cells. IFN-α and pegylated IFN-α have been approved for the treatment of chronic hepatitis B. IFN- α binds to its receptor (IFN-α/β-receptor, IFNAR) leading to the downstream signaling pathway and result in the expression of various IFN-stimulated genes (ISGs), which have multiple functions including anti-viral, anti-proliferation, anti-tumor, and immunomodulation. Some of these directly inhibit virus transcription and translation, others function as host immune modulators by NK cells activation, Dendritic cells (DCs) maturation, CD8+ T-cell augmentation, and B cell response (19). The antiviral effect against HBV infection of IFN was first known in 1976 by giving human fibroblast IFN to patients with HBsAg-positive chronic aggressive hepatitis (20).
The IFN response is initially induced by the recognition of HBV components. Multiple forms of nucleic acid are generated during the HBV life cycle, including double-strand relaxed circular DNA (rcDNA) and covalently closed circular DNA (cccDNA), single-strand RNAs, as well as double-strand RNAs, all of which could stimulate pattern recognition receptors (PRRs) on virus infection. Studies suggested that in the early phase of infection, HBV could activate some PRRs, which in turn stimulate the innate immune response to limit viral replication and clearance (21). For instance, Viral DNA is sensed by DEAD-box protein 41 (DDX41), cyclic GMP-AMP synthase (cGAS), and γ-IFN-inducible protein 16 (IFIT16), leading to the activation of stimulator of IFN genes (STING). RNA is recognized by either Toll-like receptor 3 (TLR3) or cytoplasmic sensors such as retinoic acid-inducible gene-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5), leading to its association with the mitochondrial antiviral signaling protein (MAVS). Both MAVS recruitment and STING activation lead to TANK-binding kinase 1 (TBK1) phosphorylation, which can activate IFN regulatory factor 3 (IRF3) and IRF7, two important transcriptional factors required for induction of type I IFN (22). In the HBV setting, Kupffer cells and liver parenchymal cells can recognize HBV components through intracellular PRRs such as TLRs and RIG-1-like receptors (RLRs), which induce the production of type I IFN (23). Several studies have also suggested TLR, RIG-I, STING, and myeloid differentiation primary response 88 (MyD88)-dependent pathway may participate in HBV-mediated IFN-induction in human cells (24, 25) and the pathways are meanwhile blocked by HBV polymerase in infected cells (26–28). In the MYD88-dependent pathway, MYD88 recruits a set of signal cascades such as MAPK and NF-κB through receptor-interacting serine/threonine protein kinase (RIPK/RIP) (29). After IFN-α was secreted by infected cells and blood cells, it will recruit in a pathway to induce ISGs expression.
The IFN-α signaling cascade is initiated through interactions with a multisubunit cell surface receptor consisting of two distinct receptor subunits, IFN-α receptor 1 (IFNAR1) and IFNAR2, leading to the activation of IFNAR-associated tyrosine kinases, Janus kinases 1 (JAK1) and tyrosine kinase 2 (Tyk2), which phosphorylate both IFNAR1 and IFNAR2 subunits, followed by the activation and phosphorylation of signal transducer and activator of transcription factors (STATs) in the canonical IFN signaling. STAT1 and STAT2 then form heterodimers and are joined by an Interferon regulatory factor 9 (IRF9) to form an active transcription factor complex known as IFN-stimulated gene factor 3 (ISGF3). ISGF3 translocates into the nucleus and binds to the IFN-stimulated response element (ISRE) to initiate transcription of ISGs to affect HBV replication or modulate host immune response (30). IFN-α exerts antiviral activity against HBV by both inducing antiviral gene products that inhibit viral replication in hepatocytes and by modulating the host immune system. Many ISGs are known to inhibit HBV replication at different steps, including inhibiting cccDNA transcription and HBV nucleocapsid formation, suppressing the activity of HBV enhancers, as well as control the HBV replication at the post-transcriptional level (31).
The other complex formed by STAT1 homodimers binds to the GAS motif and mainly active pro-inflammatory gene expression. After STAT1 homodimers bind to the GAS enhancer elements in the promoters of IFN-stimulate genes, genes encoding pro-inflammatory cytokines and apoptotic factors are induced (32). Type I IFN can also activate STAT3 homodimers and result in the production of both pro-inflammatory cytokines and anti-inflammatory cytokines (such as interleukin-10 (IL-10). In the non-canonical IFN signaling, IFN can induce a set of genes independent of STATs, including MAPKs and PI3K, to initiate ISGs transcription (33). In a STAT-independent manner, type I IFNs activate both p38, which is an upstream activator of several genes regulated by ISREs and GAS elements, and mammalian target of rapamycin (mTOR), which regulates mRNA translation (34).
Type I IFN can activate other important members of innate immunity, including natural killer (NK) cells and natural killer T (NKT) cells, recruiting them to the infected tissues and recognize infected hepatocytes. These immune cells secrete cytokines that promote intracellular HBV clearance and induce apoptosis. Pegylated IFN-α treatment enhanced recovery of memory T cells in CHB patients by down-regulating inhibitory receptors and up-regulating effector molecules (35). The antiviral effect against HBV of IFN and its immunomodulatory function have been finely summarized in several reviews (22, 31, 34).
Negative Regulators in IFN Induction and Signaling Pathway
Viruses could disrupt IFN responses by co-opting negative regulatory systems or use the antiviral system to their advantage (36), for example, HBV could hijack several ISGs either to facilitate its replication or impede innate immune response to HBV. Although viral component recognition by PRRs is essential for effective antiviral immune responses, the inflammatory immune response, including type I IFN production, has to be tightly regulated to prevent advert immune-related pathologies. Because activation of those inhibitory processes can cause IFN-α therapy suppression, understanding the molecular basis and identify related host negative regulators could provide targets for improving IFN-α treatment efficacy.
Studies showed that IFNAR1 and IFNAR2 in peripheral blood mononuclear cells and lymphocytes increased in CHB patients, but their level had a positive correlation with HBV-DNA in liver tissue (37). Several negative regulators mainly functioning to suppress IFNs expression or disturb the function or expression of IFNAR, thereby inhibit the JAK-STAT pathway and ISGs production (Figure 1). The related negative regulators are summarized in Table 1.
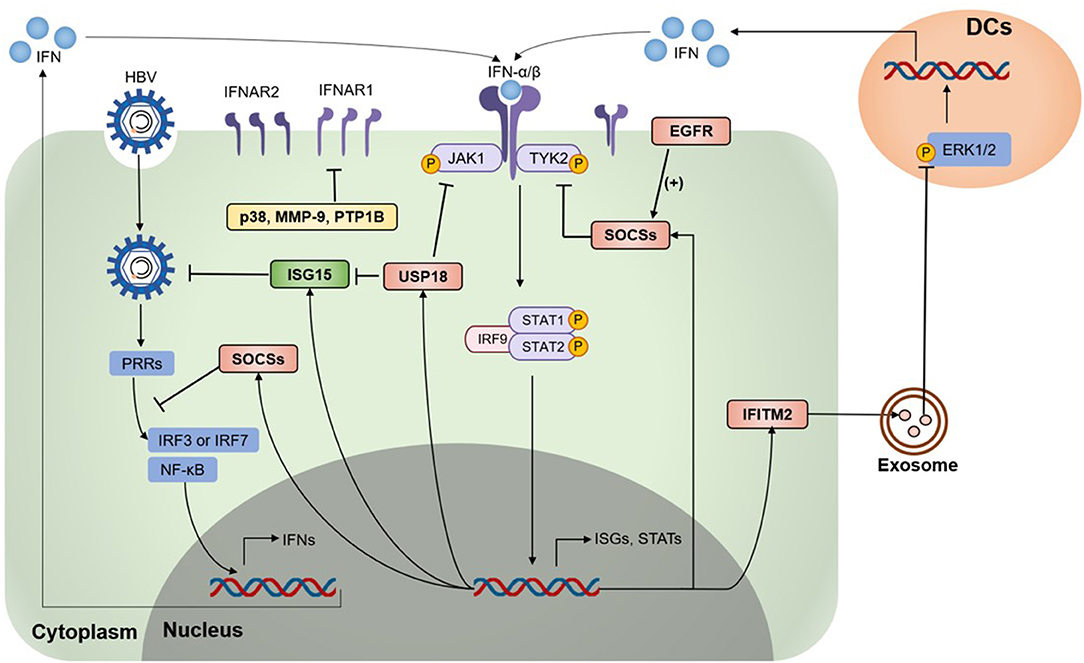
Figure 1. Type I IFN signaling is down-regulated by negative regulators. Various negative regulators cross-regulate the type I interferon (IFN) response, which modulates the expression levels and activation states of IFN signaling components. On HBV infection and invasion, pattern-recognition receptors (PRRs) recognize HBV and activate the downstream pathway to induce IFN expression. IFNAR1/2 recognize type I IFN and activate Janus kinases 1 (JAK1) and tyrosine kinase 2 (Tyk2), followed by the activation and phosphorylation of signal transducer and activator of transcription factors (STATs), leading to the expression of various IFN-stimulated genes (ISGs). Several ISGs were identified as negative regulators of the IFN signaling pathway, including the suppressor of cytokine signaling (SOCS) proteins, ubiquitin-specific protease 18 (USP18), Interferon-induced transmembrane protein 2 (IFITM2), p38 mitogen-activated kinases (p38 MAPKs), matrix metalloproteinase 9 (MMP-9), epidermal growth factor receptor (EGFR), protein tyrosine phosphatases 1 B (PTP1B), and some regulators awaiting exact mechanisms. HBV, hepatitis B virus; IRF3, IFN regulatory factor 3; IRF7, IFN regulatory factor 7; ERK1/2, extracellular signal-regulated kinase 1/2; DCs, dendritic cells.
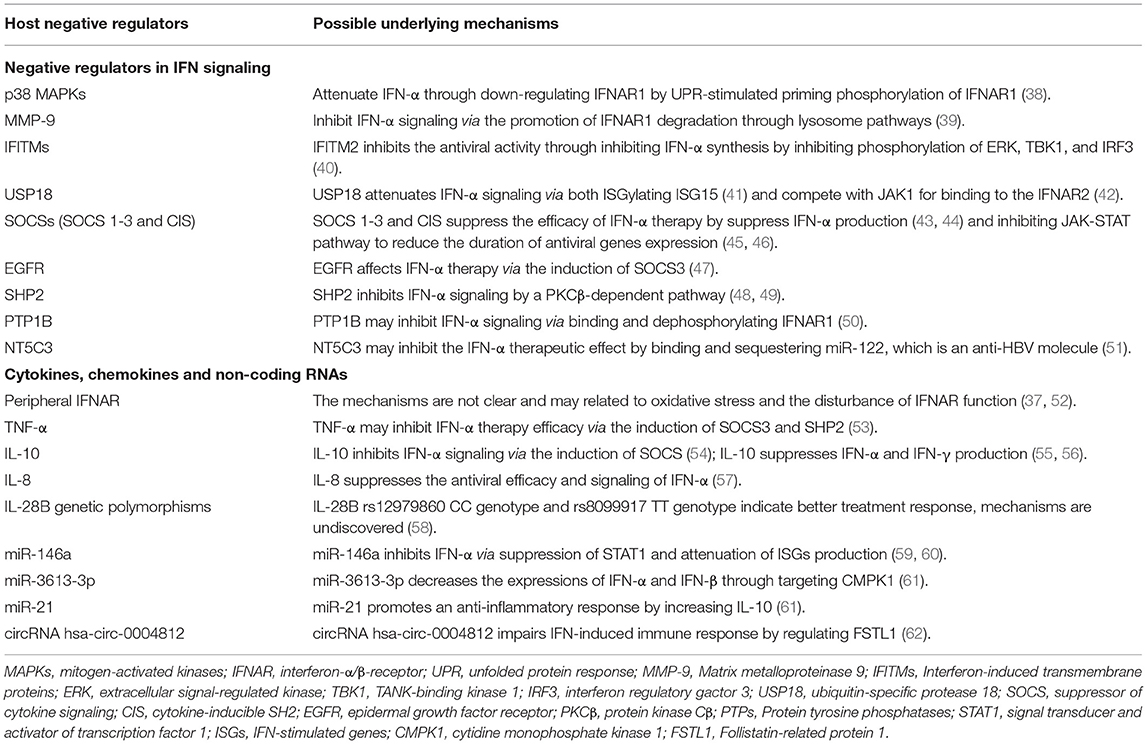
Table 1. Involvement and the possible underlying mechanisms of host negative regulators in the inhibition of IFN-α therapy.
p38 MAPKs
Mammalian p38 mitogen-activated kinases (MAPKs) are activated by cellular stresses and inflammatory cytokines and are critical for normal immune and inflammatory response (63). Several studies showed HBV infection could activate mitogen-activated protein kinases (MAPKs), including p38 MAPK kinases, c-Jun N-terminal kinase (JNK), and extracellular signal-regulated kinases (ERKs) (64–67). p38 MAPK, but not JNK or ERK1/2, was significantly phosphorylated in Huh7 cells after HBV infection and thereby promote intracellular HBV replication (68) with the involvement of STAT3 (67). p38 MAKP also acts as a negative regulator of type I IFN pathway through unfolded protein response (UPR)-stimulated priming phosphorylation of IFNAR1, ensuring IFNAR1 down-regulation and type I IFN signaling attenuation (38). Given this knowledge as well as the important role of HBV in p38 MAPKs production, we hypothesized that negative regulation of p38 MAPKs should stabilize IFNAR1 and improve IFN therapy efficacy against chronic HBV infection, which requires further investigation.
MMP-9
Matrix metalloproteinase 9 (MMP-9) is expressed in normal leukocyte and transformed cells, and is associated with IFNAR1 degradation (39). IFNAR1 degradation can be promoted by either ligand binding and cellular signaling induced ubiquitination-dependent pathway, or Ser532 phosphorylation and p38 kinase activity requiring ligand-independent way (69). MMP-9 can be activated by HBV in CHB patients (70, 71) and in turn facilitates HBV replication through attenuating IFN/JAK/STAT signaling, interacting with IFNAR1 to promoting its phosphorylation, ubiquitination, subcellular distribution, and degradation of IFNAR1 through lysosome pathways in Ser532 phosphorylation and p38 independent manner, as well as blocking its binding to IFN-α (39). However, a recent study found type I IFN strongly promotes the clearance of MMPs (72), thus, further study is needed to investigate the role of MMPs in the IFN-α therapy mediated antiviral efficacy against HBV infection.
IFITMs
Interferon-induced transmembrane proteins (IFITMs) are a family of small proteins that localize in the plasma and endolysosomal membranes (73) and were originally described based on their expression after IFN therapy (74). The human IFITM family comprises five members, including immune-related IFITM1, IFITM2, and IFITM3, as well as IFITM5 and IFITM10 with an unknown role in immunity (75). Brass et al. first described IFITM proteins as antiviral factors that target the early life cycle steps of several viruses (76). HCV infection could be restricted by IFITM proteins by targeting the endocytosed HCV virion for lysosomal degradation (77). However, our previous study indicated IFITM2 was shuttled by exosomes to DCs and inhibit IFN-α synthesis via inhibiting phosphorylation of extracellular signal-regulated kinase (ERK), TANK-binding kinase 1 (TBK1), and interferon regulatory factor 3 (IRF3), thereby blocking anti-HBV efficacy of exogenous IFN-α in chronic HBV infection (40). Thus, down-regulating IFITM2 may be a potential strategy to improve the therapeutic response to IFN-α treatment.
Most negative regulators function in the various biological processes in IFN production and signaling pathways, including inhibit IFN expression, disturb IFNAR function, and suppress the expression of ISGs which serve as anti-HBV molecules (Table 1).
USP18 and ISG15
Ubiquitin-specific protease 18 (USP18, also known as UBP43) and Interferon stimulated gene 15 (ISG15) have been studied extensively for their negative regulation of type I IFN signaling. Various studies showed that ISG15/USP18 signaling activation is involved in IFN therapy non-response in both HBV and HCV patients (78, 79). Responding to viral infection or IFN stimulation, both USP18 and ISG15 are strongly upregulated (80). USP18 belongs to the ubiquitin protease family, which is responsible for removing ubiquitin or ubiquitin-like proteins from their conjugated substrates (81). ISG15 is a ubiquitin-like modifier that binds other proteins in a process called ISGylation, and its antiviral activity was first observed using a recombinant chimeric Sindbis virus and IFNAR−/− mice model (82). ISG15 conjugated to various cellular substrates via an enzymatic cascade: E1 activating enzyme UbE1L (83), E2-conjugating enzyme UbCH8 (84, 85), and various E3 ligase (86, 87). ISG15 targets host and viral proteins and impacts diverse cellular pathways, including RNA splicing, chromatin remodeling/polymerase II transcription, cytoskeleton organization and regulation, stress responses, and translation mainly in a conjugation-dependent manner (88, 89). By deISGylating ISG15, USP18 was stabilized by unconjugated ISG15 via preventing its S-phase kinase-associated protein (SKP2)-induced ubiquitylation, therefore mediates downregulation of type I IFN signaling during HBV infection (41). Independent of its deconjugating activity, USP18 also competes with JAK1 for binding to the IFNAR2 and therefore impede IFN signaling (42). Because USP18 and ISG15 are inducible by viral factors, these inhibitory proteins may be important mediators in reducing IFN therapy efficacy by negatively regulating IFN-α signaling and host immune against chronic HBV infection.
SOCSs
In addition to USP18 and ISG15, the suppressor of cytokine signaling (SOCS) proteins and cytokine-inducible SH2 (CIS) also play important roles in downregulating IFN-α signaling, particularly the JAK-STAT signaling pathway (90), and suppress type I IFN production. To date, SOCS/CIS family includes eight proteins (SOCS 1-7 and CIS) which sharing similar structures: a central Src-homology 2 (SH2) domain, a C-terminal SOCS box, and a highly variable N-terminal region (91). The SOCS box functions to recruit an E3 ubiquitin ligase complex, so the SOCS box-containing proteins can act as substrate-recognition modules to mediate the polyubiquitination and subsequent degradation of substrate proteins by the 26S proteasome (92). Responding to IFN signaling and cytokine stimulation, SOCS 1-3 and CIS proteins are induced and negatively regulate the JAK-STAT-mediated cytokine signal responsible for its production in a classic negative feedback loop (30, 45), while SOCS4-7 primarily focus on regulating the growth factor receptor signal (93, 94). SOCS1 and SOCS3, the most widely studied proteins in this family, possess a kinase inhibitory region (KIR) at the N-terminal region that inhibits JAK tyrosine kinase activity by acting as a pseudo-substrate (95). At the same time, SOCS1 is related to IFNAR1 specific signals, thereby abrogating tyrosine phosphorylation of STAT1 and reducing the duration of antiviral genes expression (46). Apart from regulating the IFN-α signaling pathway, SOCS proteins can suppress type I IFN production by binding and degrading key molecules in the TLR signaling pathway, including myeloid differentiation factor 88 (MYD88)-adaptor-like (Mal) protein (43) and IRF7 (44). HBV increased SOCS1 and SOCS3 expression and resulting in sustained STAT3 activation (96, 97). Thus, the SOCS family, especially SOCS1 and SOCS3, inhibit both type I IFN production and IFN-α signaling pathway in HBV infection and provides targets for improving IFN-α-mediated antiviral effect.
EGFR
Epidermal growth factor (EGF) receptor (EGFR) is a transmembrane protein that belongs to the ErbB family of receptors and is well-studied for its association with a number of cancers. EGFR has recently been shown to be a host entry cofactor triggering HBV internalization (98). Inhibition of EGFR activity enhances the antiviral efficacy of IFN-α by both inducing SOCS3 expression to reduce IFN-α-induced STAT3 activity and enhancing STAT1-mediated antiviral response. Moreover, EGFR inhibitors inhibited the replication, antigen syntheses, and cccDNA reservoir of HBV (47). Thus, EGFR inhibitors could be an important candidate that facilitates IFN-α therapy against HBV infection.
PTPs
Protein tyrosine phosphatases, known as PTPs, is a large family that encodes enzymes that are divided into the classical, phosphotyrosine (pTyr)-specific phosphatases and the dual specificity phosphatases (DSPs) (99), regulating the phosphorylation state of many important signaling molecules. The overexpression of HBx or NF-κB led to increased SHP2 expression, which is a ubiquitously expressed PTP, via NF-κB binding to the Shp2 promoter (100). SHP2, encoded by Ptpn11, containing two SH2 domains and acts as a negative regulator of IFN-induced STAT activation in a protein kinase Cβ (PKCβ)-dependent pathway (48, 49). PTP1B is another PTP protein and is capable of binding and dephosphorylating IFNAR1 and thereby suppress type I IFN signaling in human cells. PTP1B inhibitors robustly augmented the type I IFN-mediated antiviral effects against HCV (50). However, the knowledge on PTP1B expression and its function needs further study in chronic HBV infection. Currently, it is estimated that as many as 125 PTP genes are in the human genome, and most of them have not been identified (101). Thus, additional studies are required to identify PTP proteins that suppress the IFN-α signal pathway and therefore attenuate therapy in chronic HBV infection.
Other Molecules
Apart from the above-mentioned molecules, other molecules involved in the down-regulation of IFN therapy efficacy include NEIL3, TDG, and NT5C3, but the exact biological mechanisms underlying the influence of these molecules in IFN-α treatment response remain to be determined. It is reported that positive response to IFN-α treatment in CHB patients is associated with a lower level of NEIL3 and thymine DNA glycosylase (TDG), two intra-hepatic base excision repair (BER) genes (102), but the underlying mechanism is unknown. MiR-122, a miRNA that has been demonstrated to inhibit HBV replication by directly targeting the HBV pre-genomic RNA sequence or by indirectly modulating HO-1 and CCNG1/p53 pathways (103), could be sequestered by an ISG, NT5C3 (51). Therefore, NT5C3 could efficiently inhibit miR-122 and suppress IFN therapeutic efficacy. However, the function of NT5C3 and miR-122 need further confirmation in clinical research. In addition, numerous molecules have been shown to negatively regulate type I IFN signaling, including DCST1, PIAS1, and so on (32, 104), although further studies are needed to clarify their function in HBV infection.
Role of Cytokines, Chemokines, and miRNAs in Negative Regulation of IFN-α Therapy in CHB Infection
Peripheral IFNAR
In general, the anti-HBV activity of type I IFN is mediated through binding to their unique receptors, following by JAK-STAT pathway activation and ultimately ISGs production. Cell-surface IFNAR consists of 2 major subunits: IFNAR1 and IFNAR2. However, soluble IFNAR was reported as an inhibitory factor of IFN therapy in both HBV and HCV infection (52, 105). The expression of IFNAR1 and IFNAR2 in lymphocytes and monocytes was increased in CHB patients which was negatively correlated to plasma glutathione S-transferase (GST), a class of cytosolic enzyme participated in the protection of cell from reactive oxygen species (ROS), suggesting that oxidative stress play an important role in the upregulation of IFNAR in CHB patients (37, 52). Oxidative stress could modulate protein misfolding and then disrupt the biological protein conformation. Therefore, oxidative stress may influence IFNAR function, further studies are required to find out the mechanism.
TNF-α
TNF is a pleiotropic cytokine that exerts homeostatic and pathogenic bioactivities (106). Elevated levels of TNF-α are observed in serum and hepatocytes of patients with acute or chronic hepatitis B (107), possibly through ERKs and NF-κB pathway (108). TNF-α may play a role in downregulating HBV replication in hepatocytes by non-cytopathic mechanisms and synergizing with IFN in suppressing viral replication (109, 110). On the other hand, TNF-α was reported as a negative regulator during IFN-α production determined by HBsAg (55). It is also reported that the injections of synthetic TNF-α could inhibit IFN-α-induced signals in the liver and both SOCS3 and SHP2 contributes to the inhibitory effect (53). However, anti-TNF-α-agents may lead to enhanced HBV replication and cause reactivation of HBV infection in HBsAg carrier and Occult carrier (anti-HBc+) patients (111). Consequently, the ultimate impact of TNFα-mediated effects on IFN therapy against HBV is a question that remains to be resolved.
IL-10
Interleukin 10 (IL-10), also known as cytokine synthesis inhibitory factor (CSIF), is a pleiotropic cytokine with anti-inflammatory and immunosuppressive activities. HBcAg stimulated IL-10 production by CD4+ and CD8+ cells, as well as by monocytes from CHB patients, which serves as a viral strategy to downregulate the host immune response and allow viral persistence in the host (112). HBsAg inhibited the production of IFN-α by pDCs through the induction of monocytes that secreted IL-10 (55). IL-10 also suppresses NK cell IFN-γ production without altering cytotoxicity or death-ligand expression (56). In addition, SOCS1 and SOCS3 are both produced in response to IL-10, and both function as negative regulators during JAK-STAT signaling (54). On the other hand, evidence suggests that heterogeneity in the promoter region of the IL-10 gene has a role in determining the initial response of HBV infection to IFN-α therapy. IL-10 variants were more frequent among virologically sustained response (SR) compared with non-responders (NR) to IFN-α-2b. Carriage of the−592A allele,−592A/A genotype, and−1082/1819/-592 ATA haplotype was associated with SR (113).
IL-8
Interleukin 8 (IL-8), or chemokine (C-X-C motif) ligand 8 (CXCL8) is a pro-inflammatory chemokine produced by various cell types to recruit leukocytes to the sites of infection or tissue injury (114). It has been demonstrated that serum IL-8 amounts is elevated during HBV reactivation, and HBeAg-negative patients had significantly higher levels of IL-8 transcripts in the liver than HBeAg-positive patients (115). Accumulating evidence indicates that IL-8 may contribute to counteracting IFN-α antiviral action (116, 117). During the IFN challenge, IL-8 expression induced by HBV can impair the ability of endogenous IFN-α to inhibit early stages of viral replication, thus facilitate viral persistence, and can also contribute to the poor response to IFN-α treatment (57). IL-8 is induced by HBV through various mechanisms, but the mechanism underlying the IL-8 affecting the anti-HBV function of IFN-α therapy still needs further discussion.
IL-28B
Interleukin-28B (IL-28B), also known as IFN-λ3, has been reported by numerous studies that its genetic polymorphisms are related to the therapeutic efficacy of PEG-IFN-α. Sonneveld et al. reported the proportions of IL-28B genotypes were 77, 19, and 5% for AA/AG/GG at rs12980275 and also for CC/CT/TT at rs12979860, respectively (118). rs12980275 genotype AA and rs12979860 genotype CC were associated with a higher probability of HBeAg seroconversion (118). A meta-analysis summarized that IL-28B rs12979860 CC genotype and rs8099917 TT genotype indicated a better treatment response than non-CC and non-TT genotypes for PEG-IFN-α in patients with CHB (58).
miRNAs
MicroRNAs (miRNAs) are a class of non-coding small RNAs that play important roles in modulating gene expression mainly by base-pairing to the 3' translational region (3' UTR) of mRNAs. miRNAs have been reported to be widely involved in antiviral immunity and some studies show miRNA plays a role in HBV-host interaction. HBV could change the expression profiles of cellular miRNA and escape host immune clearance (119). It was shown that nearly 90% of the total miRNAs in the liver are comprised of ~10 miRNAs and miR-122 is the major contribution with 50–70% of the miRNA profile (120). Zhang et al. identified 11 response-related miRNAs, including let-7a, miR-30a, miR-1290, miR-106b, miR-1224-5p, miR-939, miR-1281, miR-198, let-7f, miR-22, and miR-638, by performing miRNA microarray analysis of plasma samples and liver biopsy samples from CHB patients who received IFN therapy and using the OneR feature ranking and incremental feather selection method to ultimately establish a prediction model for predicting the initial response of HBV patients receiving IFN injections (121). Not all of the 11 miRNAs in the predicted model is negatively associated with the IFN-α therapy efficacy. Results obtained by Tan et al. suggest that the aberrant expression of miRNAs was associated with different therapy outcomes, and monitoring the fluctuation patterns of miRNAs was important for predicting the IFN-α therapeutic effects. They also identified 11 miRNAs to be associated with the effect of HBV therapy, namely, let-7d-5p, let-7f-5p, let-7i-5p, miR-122-5p, miR-126-3p, miR-1307-3p, miR-181a-5p, miR-21-5p, miR-425-5p, miR-652-3p, and miR-320a-3p, with 9 miRNAs significantly declined after therapy in the responder group: let-7d-5p, let-7f-5p, let-7i-5p, miR-126-3p, miR-1307-3p, miR-181a-5p, miR-21-5p, miR-425-5p, and miR-652-3p (122).
miRNAs could modulate the IFN-α therapy effect in multiple processes, including type I IFN production, IFN signaling pathway, as well as ISGs transcription and translation. The effect of miRNA on IFN and its signaling pathway has been well-illustrated in many reviews (123, 124), indicating that miR-130a, let-7b, miR-26a, miR-24, miR-146a, and many other miRNAs could down-regulate the effect of IFN therapy. However, only a few miRNAs are studied extensively in CHB patients. HBV infection promotes miR-146a transcription, which suppressed STAT1 and ultimately attenuated the production of type I IFN-induced antiviral factors, as well as impairing T cell function, resulting in IFN resistance and immune defects during chronic HBV infection (59, 60). miR-3613-3p was upregulated in the CHB patients suppressed IFN-induced anti-HBV immune response by targeting cytidine monophosphate kinase 1 (CMPK1) (61), a kinase responsible for the metabolism of CMP, UMP, and deoxycytidine analogs (125), and ultimately decrease the relative expressions of IFN-α and IFN-β. miR-21 has been reported to be associated with the effect of HBV therapy (122) and promote an anti-inflammatory response by increasing IL-10 production through down-regulating programmed cell death 4 (PDCD4) (126). But the detailed mechanism of miR-21 affecting the anti-HBV effect of IFN-α therapy still needs further investigation.
Some miRNAs exhibit an immune-modulating or IFN therapy enhancing effect in HBV infection. HBeAg could elevate miR-212-3p expression in macrophages, and thereby decrease the production of cytokines through targeting MAPK1 (127). Higher pretreatment plasma miR-301a-3p and miR-145-5p levels were observed in CHB patients who achieved combined response or HBsAg loss following peg-IFN/NA therapy compared to non-responders (120). miR-155 enhances innate antiviral immunity against HBV through promoting JAK/STAT signaling pathway by targeting SOCS1 and mildly inhibits HBV infection in human hepatoma cells (128). In conclusion, miRNAs play an important role in regulating the efficiency of IFN therapy in CHB patients, and could act as predictive factors of IFN therapy outcome, or be used for discovery for better treatment strategies. But the mechanism underlying these miRNAs calls for more attention.
LncRNAs and circRNAs
Long non-coding RNAs (lncRNAs) are a subset of non-coding RNAs >200 nucleotides in length and have recently been suggested to act as both positive and negative regulators in viral infections utilizing various mechanisms, mediated by their specific sequences or structural motifs that bind DNA, RNA, or protein (129). Recent study suggests that HBx-LINE1, a hybrid RNA transcript of the human LINE1 and the HBV-encoded X gene can serve as a sponge for sequestering miR-122 and may therefore promote HBV expression and replication, but whether HBx-LINE1 function during IFN-α treatment is still unknown (130). On the other hand, lncRNA#32 has been reported to be positively associated with type I IFN signaling in HBV infection by regulating APOBEC3A and APOBEC3G expression (131). Given the important and complicated roles of lncRNAs in IFN anti-HBV treatment, more extensive studies are required to develop a more comprehensive profile of lncRNAs in HBV infection and IFN therapy.
Circular RNAs (circRNAs) are closed long non-coding RNAs and can bind miRNA and act as a miRNA sponge to regulate gene expression, they may also play a role in negatively regulating IFN therapy outcomes. Among top ten circRNAs significantly up-regulated in CHB compared to normal controls, circRNA has-circ-0004812 impairs IFN-induced immune response by sponging miR-1287-5p to regulate Follistatin-related protein (FSTL) 1 in chronic hepatitis B (62, 132). Several circRNAs have been demonstrated to function in HBV infection, including hsa_circ_0005389, hsa_circ_0000038, hsa_circ_0000650, hsa_circ_4099, hsa_circ_0000976, hsa_circ_0007750, hsa_circ_0027089, and hsa_circ_100338, but it is still unknown whether they function in IFN-α therapy of HBV infection (133, 134). Therefore, due to presently poor understanding of circRNAs expression, function and regulation, further studies are desired to identify the mechanisms behind different circRNA regulation patterns associated with IFN-α treatment in HBV infection.
Host Clinical Parameters In Negatively Regulation of IFN-α Therapy
Patients' clinical characteristics are parameters that provide direct information of liver function and have been used for predicting the efficacy of IFN-α therapy. The appearance or upregulation of several parameters is associated with a lower response rate in IFN-α treatment (Table 2). Published data have also demonstrated that HBV serum markers, including HBsAg, HBeAg, and HBV DNA, are baseline predictors of IFN therapy response, which has been summarized in several reviews (153, 154).
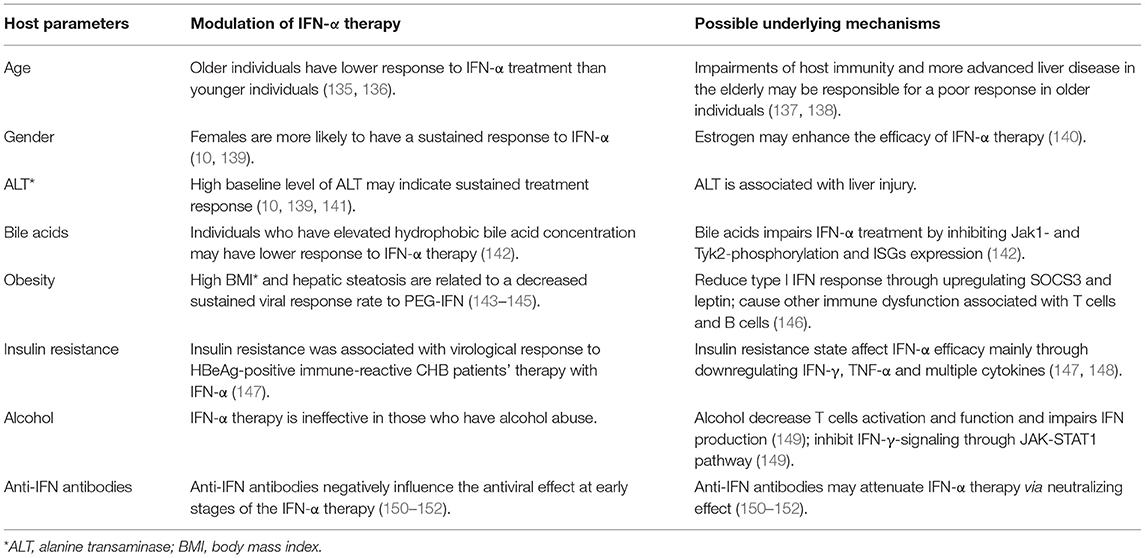
Table 2. Involvement of host parameters in reflecting the IFN-α therapy efficacy in CHB patients.
Age
Patient age is a host factor that is related to IFN-α therapy responsiveness in Chronic HBV infection. Generally, it is believed that younger individuals are more likely to develop a sustained virological response to IFN-α treatment than older individuals. Bonino et al. reported that patients under 40 years of age had higher combined response rates than patients over 40 years of age, regardless of which treatment they received (with or without lamivudine) (p < 0.024; OR: 1.3 per 10-year decrease; 95% CI: 1.0–1.7), indicating that aging imparts a negative influence in the IFN-α treatment efficacy (135). Similar results were showed in other clinical researches (136). Aging enhances the vulnerability to liver injury mainly by increasing oxidative stress and inflammatory response, accelerating cellular senescence, as well as suppressing mitochondrial function (137). A retrospective study showed that age parameters were also associated with significant histological abnormalities in patients with persistently normal ALT levels (PNALT) (155). Moreover, impairments of cellular, humoral, and innate immunity in the elderly may be another explanation responsible for decreased virological response to IFN-α therapy in older patients (138).
Gender and Hormone
Gender is another important host factor associated with IFN-mediated antiviral efficacy against HBV infection. It is summarized that females are more likely to achieve HBeAg seroconversion after IFN therapy comparing to males (10, 139), which may be an effect of sex hormones. Androgen-androgen receptor (AR) complex activates HBV RNA transcription through binding to the 2 response elements inside Enh I, while estrogen and/or its receptor suppressed HBV replication (156). The expression level of estrogen receptors (ESRs) in the cytosol of peripheral blood mononuclear cells (PBMCs) was significantly lower in patients with CHB than in healthy controls, and ESR1 mRNA expression at week 4 of PEG IFN-α treatment was a better indicator for treatment response although the underlying mechanism is still unknown (140). Further studies are required to clarify the effect of sex hormones in IFN-α therapy.
ALT Levels
Alanine transaminase (ALT) is a transaminase enzyme and is clinically measured as a biomarker of liver injury. ALT is associated with significant histological characteristics to some extent in HBeAg-positive and -negative patients and its normalization are related to favorable long-term CHB infection treatment outcomes. Several studies indicated that a high baseline ALT level can be a predictor of sustained treatment response (10, 139, 141).
Bile Acids
Bile acids are steroid acids and were found to be related to the IFN-α treatment. Elevated hydrophobic bile acid concentration impairs IFN-α treatment in patients with CHB by inhibiting Jak1- and Tyk2-phosphorylation, resulting in a decreased mRNA and protein expression of ISGs, including myxovirus resistance protein A (MxA) and dsRNA-activated protein kinase (PKR) (142). The uptake of bile acid into hepatocytes is induced by Na+-taurocholate cotransporting polypeptide (NTCP), which also plays an essential role in HBV entry into the target cells. HBV infection may promote the expression of genes involved in lipid and bile acid metabolism by binding to NTCP and limits its function, thus promoting compensatory BA synthesis (157).
Obesity and Hepatic Steatosis
High body mass index (BMI) or obese status is related to the presence of steatosis in CHB patients. Clinical researches showed that the occurrence of hepatic steatosis is significantly high in CHB patients, decreasing the sustained viral response (SVR) rates to PEG-IFN (143–145). Recent studies indicated that obesity attenuates and prolongs the type I IFN response that shows antiviral inefficacy. Obese individuals have a high level of blood leptin, resulting in the upregulation of the SOCS3. The elevated leptin and SOCS3 levels could reduce type I IFN response and cause other immune dysfunction associated with T cells and B cells in people with obesity (146).
Insulin Resistance
The relationship between HBV infection and insulin resistance (IR) has not been completely elucidated, but it is reported that HBV infection was associated with increased IR (158). Researches illustrated that IR was associated with virological response to HBeAg-positive immune-reactive CHB patients' therapy with IFN-α mainly through downregulation of IFN-γ, TNF-α and multiple cytokines (147, 148). On the other hand, IFN-α was reported to be a catastrophic feature of the diabetic islet microenvironment, although the conclusion is controversial (159, 160). Therefore, the relationship of diabetes and IFN-α treatment is complicated and need further study.
Alcohol
Alcohol abuse causes rapid progression of liver disease in HBV infected patients and allows HBV to persist chronically mainly by increasing HBV replication, inducing oxidative stress, and suppressing the immune response. As reported, alcohol reduces the number of T-cells and changes the ratio of T-cell types, resulting in decreased T-cell activation and function, thereby impair IFN production (149). On the other hand, the combination of HBV and ethanol metabolites impairs IFN-γ-signaling through the JAK-STAT1 pathway (149). Moreover, alcohol and HBV synergistically promote hepatic steatosis (161), which has been reported as another negative host factor of IFN therapy against HBV infection.
Anti-IFN Antibodies
Endogenous anti-IFN antibodies have been reported to be stimulated after the treatment with IFN. Multiple studies indicated that anti-IFN antibodies may negate the antiviral effects of IFN-α by neutralizing the effect (150–152). Anti-IFN was significantly more likely to develop in patients who received lower doses (2.5 or 5 MU/m2) of IFN than in those who received a higher dose (10 MU/m2) (151). Another study showed that the anti-IFN activity may negatively influence the effect of the IFN therapy on CHB patients at the early stages of the therapy, but the appearance of the anti-IFN activity at the end of a long-term IFN therapy has no significant influence on the treatment outcome (152).
Concluding Remarks
Improving the efficacy of IFN-α for chronic HBV infection as well as understanding the mechanisms underlying HBV- and host-mediated resistance to IFN-α therapy remains a challenge. Identifying and inhibiting the pathways or molecules that negatively regulate IFN-α signaling and therapy, as well as finding host parameters that predict the response of patients with CHB to antiviral therapy are obvious ways to improve the efficacy of IFN-α. Diversity negative factors function at many levels of IFN signaling: the recognition of HBV by the host immune system, the production of IFNs, the recognition of IFNs by their receptors, and the JAK-STAT signaling pathway.
IFN-α therapy prescript with inhibitors of negative regulators is a potential therapeutic strategy for achieving a clinical cure. However, the concentration, duration, and tissue specificity of the negative regulators are important parameters in their synergistic effect, and we need to know more. We may need further investigation to identify an appropriate level and ratio among the inhibitors of different regulators to suitably inhibit the negative regulators, limit the toxicity, and enable a return to homeostasis in a majority of CHB patients. For negative regulators with different duration of effects, for example, before, during, and after the IFN-α therapy, the inhibition strategy may be appropriate and desirable to be flexible. Inhibitors are better to function at the same biological procedure during the IFN-α therapy, so that enhancing its anti-HBV effect accurately. For negative regulators mainly expressed in the liver tissue, the effect of inhibition should be limited in the liver to minimum side effects.
Negative host factors also serve as predictive factors for predicting the IFN-α therapy treatment outcome of CHB patients before and during the therapy. Host parameters including age, gender, ALT level, and BMI have been recommended in guidelines and widely used in deciding whether to administer IFN-α therapy for CHB patients. miRNA and cytokines are fluctuating factors important for predicting the IFN-α therapeutic effects that worth and easy to be monitored. Some negative factors only saw upregulation in liver cells with the procedure of liver biopsy, thus, these molecules should be put last on the list of predictors.
In addition to the host negative factors mentioned above, host genetic factors can provide unexpected regulators of IFN-α therapy and influence the therapeutic effects in complicated manners, which has been well-summarized in other reviews (162, 163). Single nucleotide polymorphisms (SNPs) could also serve as potential markers that predict hepatitis B patient response (164). When combined with basic clinical parameters and other genetic and epigenetic markers, more reliable treatment indexes can be developed and ultimately applied to the clinic.
Thus, as discussed above, IFN therapy needs more novel and reliable biomarkers to improve management, and novel combination strategies to substantially increase the efficacy of treatment of CHB patients.
Author Contributions
JW, LD, and HT contributed to the design of the review. JW and LD searched the database and wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation (Grant No. 82172254) and 1.3.5 project for disciplines of excellence. West China Hospital, Sichuan University (Grant No. ZYGD20009).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. World Health Organization. Hepatitis B. (2021). Available online at: https://www.who.int/news-room/factsheets/detail/hepatitis-b (accessed September 25, 2021).
2. Sagnelli E, Macera M, Russo A, Coppola N, Sagnelli C. Epidemiological and etiological variations in hepatocellular carcinoma. Infection. (2020) 48:7–17. doi: 10.1007/s15010-019-01345-y
3. Tang LSY, Covert E, Wilson E, Kottilil S. Chronic Hepatitis B Infection: A Review. JAMA. (2018) 319:1802–13. doi: 10.1001/jama.2018.3795
4. Tsukuda S, Watashi K. Hepatitis B virus biology and life cycle. Antiviral Res. (2020) 182:104925. doi: 10.1016/j.antiviral.2020.104925
5. Blumberg BS, Alter HJ. A new antigen in leukemia sera. Jama. (1965) 191:541–6. doi: 10.1001/jama.1965.03080070025007
6. Rajoriya N, Combet C, Zoulim F, Janssen HLA. How viral genetic variants and genotypes influence disease and treatment outcome of chronic hepatitis B. Time for an individualised approach? J Hepatol. (2017) 67:1281–97. doi: 10.1016/j.jhep.2017.07.011
7. Yeh M-L, Huang J-F, Dai C-Y, Yu M-L, Chuang W-L. Pharmacokinetics and pharmacodynamics of pegylated interferon for the treatment of hepatitis B. Expert Opin Drug Metabo Toxicol. (2019) 15:779–85. doi: 10.1080/17425255.2019.1678584
8. Perrillo R. Benefits and risks of interferon therapy for hepatitis B. Hepatology. (2009) 49(5 Suppl):S103–11. doi: 10.1002/hep.22956
9. Lok AS, McMahon BJ, Brown RS Jr, Wong JB, Ahmed AT, et al. Antiviral therapy for chronic hepatitis B viral infection in adults: A systematic review and meta-analysis. Hepatology. (2016) 63:284–306. doi: 10.1002/hep.28280
10. Buster EH, Hansen BE, Lau GK, Piratvisuth T, Zeuzem S, Steyerberg EW, et al. Factors that predict response of patients with hepatitis B e antigen-positive chronic hepatitis B to peginterferon-alfa. Gastroenterology. (2009) 137:2002–9. doi: 10.1053/j.gastro.2009.08.061
11. Tseng TC, Yu ML, Liu CJ, Lin CL, Huang YW, Hsu CS, et al. Effect of host and viral factors on hepatitis B e antigen-positive chronic hepatitis B patients receiving pegylated interferon-α-2a therapy. Antiviral Ther. (2011) 16:629–37. doi: 10.3851/IMP1841
12. Jansen L, Welkers MRA, van Dort KA, Takkenberg RB, Lopatin U, Zaaijer HL, et al. Viral minority variants in the core promoter and precore region identified by deep sequencing are associated with response to peginterferon and adefovir in HBeAg negative chronic hepatitis B patients. Antiviral Res. (2017) 145:87–95. doi: 10.1016/j.antiviral.2017.07.013
13. Yang HC, Chen CL, Shen YC, Peng CY, Liu CJ, Tseng TC, et al. Distinct evolution and predictive value of hepatitis B virus precore and basal core promoter mutations in interferon-induced hepatitis B e antigen seroconversion. Hepatology. (2013) 57:934–43. doi: 10.1002/hep.26121
14. Guo P. Suppression of interferon-mediated antiviral immunity by hepatitis B virus: an overview of research progress. Scand J Immunol. (2013) 78:230–7. doi: 10.1111/sji.12086
15. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc London Series B Biol Sci. (1957) 147:258–67. doi: 10.1098/rspb.1957.0048
16. Isaacs A, Lindenmann J, Valentine RC. Virus interference. II. Some properties of interferon. Proc R Soc London Series B Biol Sci. (1957) 147:268–73. doi: 10.1098/rspb.1957.0049
17. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
18. Mesev EV, LeDesma RA, Ploss A. Decoding type I and III interferon signalling during viral infection. Nat Microbiol. (2019) 4:914–24. doi: 10.1038/s41564-019-0421-x
19. Gibbert K, Schlaak JF, Yang D, Dittmer U. IFN-alpha subtypes: distinct biological activities in anti-viral therapy. Br J Pharmacol. (2013) 168:1048–58. doi: 10.1111/bph.12010
20. Desmyter J, De Groote J, Desmet VJ, Billiau A, Ray MB, Bradburne AF, et al. Administration of human fibroblast interferon in chronic hepatitis-B infection. Lancet. (1976) 2:645–7. doi: 10.1016/S0140-6736(76)92460-0
21. Lucifora J, Durantel D, Testoni B, Hantz O, Levrero M, Zoulim F. Control of hepatitis B virus replication by innate response of HepaRG cells. Hepatology. (2010) 51:63–72. doi: 10.1002/hep.23230
22. Tan G, Song H, Xu F, Cheng G. When Hepatitis B virus meets interferons. Front Microbiol. (2018) 9:1611. doi: 10.3389/fmicb.2018.01611
23. Seki S, Habu Y, Kawamura T, Takeda K, Dobashi H, Ohkawa T, et al. The liver as a crucial organ in the first line of host defense: the roles of Kupffer cells, natural killer (NK) cells and NK1. 1 Ag+ T cells in T helper 1 immune responses. Immunol Rev. (2000) 174:35–46. doi: 10.1034/j.1600-0528.2002.017404.x
24. Suzuki T, Oshiumi H, Miyashita M, Aly HH, Matsumoto M, Seya T. Cell type-specific subcellular localization of phospho-TBK1 in response to cytoplasmic viral DNA. PLoS ONE. (2013) 8:e83639. doi: 10.1371/journal.pone.0083639
25. Yu S, Chen J, Wu M, Chen H, Kato N, Yuan Z. Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKϵ and DDX3. J Gene Virol. (2010) 91:2080–90. doi: 10.1099/vir.0.020552-0
26. Yang Y, Han Q, Hou Z, Zhang C, Tian Z, Zhang J. Exosomes mediate hepatitis B virus (HBV) transmission and NK-cell dysfunction. Cell Mol Immunol. (2017) 14:465–75. doi: 10.1038/cmi.2016.24
27. Leong CR, Oshiumi H, Suzuki T, Matsumoto M, Seya T. Nucleic acid sensors involved in the recognition of HBV in the liver-specific in vivo transfection mouse models-pattern recognition receptors and sensors for HBV. Med Sci (Basel). (2015) 3:16–24. doi: 10.3390/medsci3020016
28. Kumar M, Jung SY, Hodgson AJ, Madden CR, Qin J, Slagle BL. Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J Virol. (2011) 85:987–95. doi: 10.1128/JVI.01825-10
29. Jiang X, Kanda T, Wu S, Nakamura M, Miyamura T, Nakamoto S, et al. Regulation of microRNA by hepatitis B virus infection and their possible association with control of innate immunity. World J Gastroenterol. (2014) 20:7197–206. doi: 10.3748/wjg.v20.i23.7197
30. Gao B, Hong F, Radaeva S. Host factors and failure of interferon-alpha treatment in hepatitis C virus. Hepatology. (2004) 39:880–90. doi: 10.1002/hep.20139
31. Pei RJ, Chen XW, Lu MJ. Control of hepatitis B virus replication by interferons and Toll-like receptor signaling pathways. World J Gastroenterol. (2014) 20:11618–29. doi: 10.3748/wjg.v20.i33.11618
32. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. (2014) 14:36–49. doi: 10.1038/nri3581
33. Chen K, Liu J, Cao X. Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J Autoimmun. (2017) 83:1–11. doi: 10.1016/j.jaut.2017.03.008
34. Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. (2012) 12:125–35. doi: 10.1038/nri3133
35. Liu YZ, Hou FQ, Ding P, Ren YY, Li SH, Wang GQ. Pegylated interferon α enhances recovery of memory T cells in e antigen positive chronic hepatitis B patients. Virol J. (2012) 9:274. doi: 10.1186/1743-422X-9-274
36. Taylor KE, Mossman KL. Recent advances in understanding viral evasion of type I interferon. Immunology. (2013) 138:190–7. doi: 10.1111/imm.12038
37. Meng F, Wang J, Ge J, Fan X, Wang B, Han L, et al. Alteration of interferon-α/β receptors in chronic hepatitis B patients. J Clin Immunol. (2011) 31:521–32. doi: 10.1007/s10875-011-9518-6
38. Bhattacharya S, Qian J, Tzimas C, Baker DP, Koumenis C, Diehl JA, et al. Role of p38 protein kinase in the ligand-independent ubiquitination and down-regulation of the IFNAR1 chain of type I interferon receptor. J Biol Chem. (2011) 286:22069–76. doi: 10.1074/jbc.M111.238766
39. Chen J, Xu W, Chen Y, Xie X, Zhang Y, Ma C, et al. Matrix metalloproteinase 9 facilitates hepatitis B virus replication through binding with type I interferon (IFN) receptor 1 to repress IFN/JAK/STAT signaling. J Virol. (2017) 91:e01824–16. doi: 10.1128/JVI.01824-16
40. Shi Y, Du L, Lv D, Li H, Shang J, Lu J, et al. Exosomal interferon-induced transmembrane protein 2 transmitted to dendritic cells inhibits interferon alpha pathway activation and blocks anti-hepatitis B virus efficacy of exogenous interferon alpha. Hepatology. (2019) 69:2396–413. doi: 10.1002/hep.30548
41. Vuillier F, Li Z, Commere P-H, Dynesen LT, Pellegrini S. USP18 and ISG15 coordinately impact on SKP2 and cell cycle progression. Sci Rep. (2019) 9:4066. doi: 10.1038/s41598-019-39343-7
42. Malakhova OA, Kim K, Luo JK, Zou W, Kumar KS, Fuchs SY, et al. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. (2006) 25:2358–67. doi: 10.1038/sj.emboj.7601149
43. Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, et al. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. (2006) 7:148–55. doi: 10.1038/ni1299
44. Yu CF, Peng WM, Schlee M, Barchet W, Eis-Hübinger AM, Kolanus W, et al. SOCS1 and SOCS3 target IRF7 degradation to suppress TLR7-mediated type I IFN production of human plasmacytoid dendritic cells. J Immunol. (2018) 200:4024–35. doi: 10.4049/jimmunol.1700510
45. Yoshimura A, Suzuki M, Sakaguchi R, Hanada T, Yasukawa H. SOCS, inflammation, and autoimmunity. Front Immunol. (2012) 3:20. doi: 10.3389/fimmu.2012.00020
46. Fenner JE, Starr R, Cornish AL, Zhang J-G, Metcalf D, Schreiber RD, et al. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat Immunol. (2006) 7:33–9. doi: 10.1038/ni1287
47. Gan CJ, Li WF, Li CN, Li LL, Zhou WY, Peng XM. EGF receptor inhibitors comprehensively suppress hepatitis B virus by downregulation of STAT3 phosphorylation. Biochem Biophys Rep. (2020) 22:100763. doi: 10.1016/j.bbrep.2020.100763
48. Du Z, Shen Y, Yang W, Mecklenbrauker I, Neel BG, Ivashkiv LB. Inhibition of IFN-α signaling by a PKC- and protein tyrosine phosphatase SHP-2-dependent pathway. Proc Natl Acad Sci USA. (2005) 102:10267. doi: 10.1073/pnas.0408854102
49. You M, Yu DH, Feng GS. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol Cell Biol. (1999) 19:2416–24. doi: 10.1128/MCB.19.3.2416
50. Carbone CJ, Zheng H, Bhattacharya S, Lewis JR, Reiter AM, Henthorn P, et al. Protein tyrosine phosphatase 1B is a key regulator of IFNAR1 endocytosis and a target for antiviral therapies. Proc Natl Acad Sci USA. (2012) 109:19226–31. doi: 10.1073/pnas.1211491109
51. Hao J, Jin W, Li X, Wang S, Zhang X, Fan H, et al. Inhibition of alpha interferon (IFN-α)-induced microRNA-122 negatively affects the anti-hepatitis B virus efficiency of IFN-α. J Virol. (2013) 87:137–47. doi: 10.1128/JVI.01710-12
52. Zhao J, Fan YC, Sun FK, Zhao ZH, Wang LY, Hu LH, et al. Peripheral type I interferon receptor correlated with oxidative stress in chronic hepatitis B virus infection. J Interferon Cytokine Res. (2013) 33:405–14. doi: 10.1089/jir.2012.0153
53. Hong F, Nguyen V-A, Gao B. Tumor necrosis factor α attenuates interferon-α signaling in the liver: involvement of SOCS3 and SHP2 and implication in resistance to interferon therapy. FASEB J. (2001) 15:1595–7. doi: 10.1096/fj.00-0908fje
54. Carey AJ, Tan CK, Ulett GC. Infection-induced IL-10 and JAK-STAT: A review of the molecular circuitry controlling immune hyperactivity in response to pathogenic microbes. Jak-stat. (2012) 1:159–67. doi: 10.4161/jkst.19918
55. Shi B, Ren G, Hu Y, Wang S, Zhang Z, Yuan Z. HBsAg inhibits IFN-alpha production in plasmacytoid dendritic cells through TNF-alpha and IL-10 induction in monocytes. PLoS ONE. (2012) 7:e44900. doi: 10.1371/journal.pone.0044900
56. Peppa D, Micco L, Javaid A, Kennedy PTF, Schurich A, Dunn C, et al. Blockade of immunosuppressive cytokines restores NK cell antiviral function in chronic Hepatitis B Virus Infection. PLoS Pathog. (2010) 6:e1001227. doi: 10.1371/journal.ppat.1001227
57. Pollicino T, Bellinghieri L, Restuccia A, Raffa G, Musolino C, Alibrandi A, et al. Hepatitis B virus (HBV) induces the expression of interleukin-8 that in turn reduces HBV sensitivity to interferon-alpha. Virology. (2013) 444:317–28. doi: 10.1016/j.virol.2013.06.028
58. Ying SY, Hu YR, Gao GS, Lou KH, Huang Z. Interleukin-28B polymorphisms predict the efficacy of peginterferon alpha in patients with chronic hepatitis B: A meta-analysis. Front Med. (2021) 8:691365. doi: 10.3389/fmed.2021.691365
59. Hou ZH, Han QJ, Zhang C, Tian ZG, Zhang J. miR146a impairs the IFN-induced anti-HBV immune response by downregulating STAT1 in hepatocytes. Liver Int. (2014) 34:58–68. doi: 10.1111/liv.12244
60. Wang S, Zhang X, Ju Y, Zhao B, Yan X, Hu J, et al. MicroRNA-146a feedback suppresses T cell immune function by targeting Stat1 in patients with chronic hepatitis B. J Immunol. (2013) 191:293–301. doi: 10.4049/jimmunol.1202100
61. Zhao Y, Yu Y, Ye L. MiR-3613-3p impairs IFN-induced immune response by targeting CMPK1 in chronic hepatitis B. Infect Genet Evolut. (2019) 74:103919. doi: 10.1016/j.meegid.2019.103919
62. Zhang L, Wang Z. Circular RNA hsa_circ_0004812 impairs IFN-induced immune response by sponging miR-1287-5p to regulate FSTL1 in chronic hepatitis B. Virol J. (2020) 17:40. doi: 10.1186/s12985-020-01314-0
63. Cuenda A, Rousseau S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta. (2007) 1773:1358–75. doi: 10.1016/j.bbamcr.2007.03.010
64. Wang WH, Gregori G, Hullinger RL, Andrisani OM. Sustained activation of p38 mitogen-activated protein kinase and c-Jun N-terminal kinase pathways by hepatitis B virus X protein mediates apoptosis via induction of Fas/FasL and tumor necrosis factor (TNF) receptor 1/TNF-alpha expression. Mol Cell Biol. (2004) 24:10352–65. doi: 10.1128/MCB.24.23.10352-10365.2004
65. Dai S, Chen X, Yu Y, Zang G, Tang Z. Immunization with lentiviral vector-modified dendritic cells encoding ubiquitinated hepatitis B core antigen promotes Th1 differentiation and antiviral immunity by enhancing p38 MAPK and JNK activation in HBV transgenic mice. Mol Med Rep. (2018) 18:4691–9. doi: 10.3892/mmr.2018.9487
66. Chen Z, Li YX, Fu HJ, Ren YL, Zou L, Shen SZ, et al. Hepatitis B virus core antigen stimulates IL-6 expression via p38, ERK and NF-kappaB pathways in hepatocytes. Cell Physiol Biochem. (2017) 41:91–100. doi: 10.1159/000455954
67. Yang Q, Zhang Q, Zhang X, You L, Wang W, Liu W, et al. HoxA10 facilitates SHP-1-catalyzed dephosphorylation of p38 MAPK/STAT3 to repress hepatitis B virus replication by a feedback regulatory mechanism. J Virol. (2019) 93:e01607–18. doi: 10.1128/JVI.01607-18
68. Chang WW, Su IJ, Lai MD, Chang WT, Huang W, Lei HY. Suppression of p38 mitogen-activated protein kinase inhibits hepatitis B virus replication in human hepatoma cell: the antiviral role of nitric oxide. J Viral Hepatitis. (2008) 15:490–7. doi: 10.1111/j.1365-2893.2008.00968.x
69. Qian J, Zheng H, Huangfu WC, Liu J, Carbone CJ, Leu NA, et al. Pathogen recognition receptor signaling accelerates phosphorylation-dependent degradation of IFNAR1. PLoS Pathog. (2011) 7:e1002065. doi: 10.1371/journal.ppat.1002065
70. Chung TW, Lee YC, Kim CH. Hepatitis B viral HBx induces matrix metalloproteinase-9 gene expression through activation of ERK and PI-3K/AKT pathways: involvement of invasive potential. FASEB J. (2004) 18:1123–5. doi: 10.1096/fj.03-1429fje
71. Li Y, Liu H, Xu L. Expression of MMP-9 in different degrees of chronic hepatitis B and its correlation with inflammation. Exp Ther Med. (2018) 16:4136–40. doi: 10.3892/etm.2018.6673
72. Rahman A-NMI, Yamashita S, Islam MR, Fujihara T, Yamaguchi H, Kawahara M, et al. Type-I interferon regulates matrix metalloproteinases clearance of the bovine endometrial spheroid. Animal Sci J. (2020) 91:e13350. doi: 10.1111/asj.13350
73. Smith SE, Weston S, Kellam P, Marsh M. IFITM proteins-cellular inhibitors of viral entry. Curr Opin Virol. (2014) 4:71–7. doi: 10.1016/j.coviro.2013.11.004
74. Friedman RL, Manly SP, McMahon M, Kerr IM, Stark GR. Transcriptional and posttranscriptional regulation of interferon-induced gene expression in human cells. Cell. (1984) 38:745–55. doi: 10.1016/0092-8674(84)90270-8
75. Zhao X, Li J, Winkler CA, An P, Guo JT. IFITM genes, variants, and their roles in the control and pathogenesis of viral infections. Front Microbiol. (2018) 9:3228. doi: 10.3389/fmicb.2018.03228
76. Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. (2009) 139:1243–54. doi: 10.1016/j.cell.2009.12.017
77. Narayana SK, Helbig KJ, McCartney EM, Eyre NS, Bull RA, Eltahla A, et al. The Interferon-induced transmembrane proteins, IFITM1, IFITM2, and IFITM3 inhibit hepatitis C virus entry. J Biol Chem. (2015) 290:25946–59. doi: 10.1074/jbc.M115.657346
78. Li Y, Li S, Duan X, Liu B, Yang C, Zeng P, et al. Activation of endogenous type I IFN signaling contributes to persistent HCV infection. Rev Med Virol. (2014) 24:332–42. doi: 10.1002/rmv.1795
79. Xiao C, Qin B, Chen L, Liu H, Zhu Y, Lu X. Preactivation of the interferon signalling in liver is correlated with nonresponse to interferon alpha therapy in patients chronically infected with hepatitis B virus. J Viral Hepatitis. (2012) 19:e1–10. doi: 10.1111/j.1365-2893.2011.01471.x
80. François-Newton V, Magno de Freitas Almeida G, Payelle-Brogard B, Monneron D, Pichard-Garcia L, Piehler J, et al. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon α response. PLoS ONE. (2011) 6:e22200. doi: 10.1371/journal.pone.0022200
81. Honke N, Shaabani N, Zhang DE, Hardt C, Lang KS. Multiple functions of USP18. Cell Death Dis. (2016) 7:e2444. doi: 10.1038/cddis.2016.326
82. Lenschow DJ, Giannakopoulos NV, Gunn LJ, Johnston C, O'Guin AK, Schmidt RE, et al. Identification of interferon-stimulated gene 15 as an antiviral molecule during Sindbis virus infection in vivo. J Virol. (2005) 79:13974–83. doi: 10.1128/JVI.79.22.13974-13983.2005
83. Yuan W, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. (2001) 20:362–71. doi: 10.1093/emboj/20.3.362
84. Kim KI, Giannakopoulos NV, Virgin HW, Zhang DE. Interferon-inducible ubiquitin E2, Ubc8, is a conjugating enzyme for protein ISGylation. Mol Cell Biol. (2004) 24:9592–600. doi: 10.1128/MCB.24.21.9592-9600.2004
85. Zhao C, Beaudenon SL, Kelley ML, Waddell MB, Yuan W, Schulman BA, et al. The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN-alpha/beta-induced ubiquitin-like protein. Proc Natl Acad Sci USA. (2004) 101:7578–82. doi: 10.1073/pnas.0402528101
86. Dastur A, Beaudenon S, Kelley M, Krug RM, Huibregtse JM. Herc5, an interferon-induced HECT E3 enzyme, is required for conjugation of ISG15 in human cells. J Biol Chem. (2006) 281:4334–8. doi: 10.1074/jbc.M512830200
87. Zou W, Zhang DE. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J Biol Chem. (2006) 281:3989–94. doi: 10.1074/jbc.M510787200
88. Zhao C, Denison C, Huibregtse JM, Gygi S, Krug RM. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc Natl Acad Sci USA. (2005) 102:10200–5. doi: 10.1073/pnas.0504754102
89. Morales DJ, Lenschow DJ. The antiviral activities of ISG15. J Mol Biol. (2013) 425:4995–5008. doi: 10.1016/j.jmb.2013.09.041
90. Xie J, Wang M, Cheng A, Jia R, Zhu D, Liu M, et al. The role of SOCS proteins in the development of virus- induced hepatocellular carcinoma. Virol J. (2021) 18:74. doi: 10.1186/s12985-021-01544-w
91. Chen XP, Losman JA, Rothman P. SOCS proteins, regulators of intracellular signaling. Immunity. (2000) 13:287–90. doi: 10.1016/S1074-7613(00)00028-5
92. Linossi EM, Nicholson SE. The SOCS box-adapting proteins for ubiquitination and proteasomal degradation. IUBMB life. (2012) 64:316–23. doi: 10.1002/iub.1011
93. Kario E, Marmor MD, Adamsky K, Citri A, Amit I, Amariglio N, et al. Suppressors of cytokine signaling 4 and 5 regulate epidermal growth factor receptor signaling. J Biol Chem. (2005) 280:7038–48. doi: 10.1074/jbc.M408575200
94. Nicholson SE, Metcalf D, Sprigg NS, Columbus R, Walker F, Silva A, et al. Suppressor of cytokine signaling (SOCS)-5 is a potential negative regulator of epidermal growth factor signaling. Proc Natl Acad Sci USA. (2005) 102:2328–33. doi: 10.1073/pnas.0409675102
95. Kershaw NJ, Murphy JM, Lucet IS, Nicola NA, Babon JJ. Regulation of Janus kinases by SOCS proteins. Biochem Soc Transact. (2013) 41:1042–7. doi: 10.1042/BST20130077
96. Du L-y, Cui Y-l, Chen E-q, Cheng X, Liu L, Tang H. Correlation between the suppressor of cytokine signaling-1 and 3 and hepatitis B virus: possible roles in the resistance to interferon treatment. Virol J. (2014) 11:51. doi: 10.1186/1743-422X-11-51
97. Koeberlein B, Hausen Az, Bektas N, Zentgraf H, Chin R, Toan NL, et al. Hepatitis B virus overexpresses suppressor of cytokine signaling-3 (SOCS3) thereby contributing to severity of inflammation in the liver. Virus Res. (2010) 148:51–9. doi: 10.1016/j.virusres.2009.12.003
98. Iwamoto M, Saso W, Sugiyama R, Ishii K, Ohki M, Nagamori S, et al. Epidermal growth factor receptor is a host-entry cofactor triggering hepatitis B virus internalization. Proc Natl Acad Sci USA. (2019) 116:8487. doi: 10.1073/pnas.1811064116
99. Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. (2006) 7:833–46. doi: 10.1038/nrm2039
100. Kang HJ, Chung D-H, Sung CO, Yoo SH, Yu E, Kim N, et al. SHP2 is induced by the HBx-NF-κB pathway and contributes to fibrosis during human early hepatocellular carcinoma development. Oncotarget. (2017) 8:27263–76. doi: 10.18632/oncotarget.15930
101. Alonso A, Pulido R. The extended human PTPome: a growing tyrosine phosphatase family. FEBS J. (2016) 283:1404–29. doi: 10.1111/febs.13600
102. Li Y, Xia Y, Han M, Chen G, Zhang D, Thasler WE, et al. IFN-α-mediated base excision repair pathway correlates with antiviral response against hepatitis B virus infection. Sci Rep. (2017) 7:12715. doi: 10.1038/s41598-017-13082-z
103. Qiu L, Fan H, Jin W, Zhao B, Wang Y, Ju Y, et al. miR-122-induced down-regulation of HO-1 negatively affects miR-122-mediated suppression of HBV. Biochem Biophys Res Commun. (2010) 398:771–7. doi: 10.1016/j.bbrc.2010.07.021
104. Arimoto K-I, Miyauchi S, Stoner SA, Fan J-B, Zhang D-E. Negative regulation of type I IFN signaling. J Leukocyte Biol. (2018) 103:1099–116. doi: 10.1002/JLB.2MIR0817-342R
105. Mizukoshi E, Kaneko S, Kaji K, Terasaki S, Matsushita E, Muraguchi M, et al. Serum levels of soluble interferon Alfa/Beta receptor as an inhibitory factor of interferon in the patients with chronic hepatitis C. Hepatology. (1999) 30:1325–31. doi: 10.1002/hep.510300516
106. Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. (2016) 12:49–62. doi: 10.1038/nrrheum.2015.169
107. Fuchs I, Abu-Shakra M, Sikuler E. Hepatitis B and C reactivation with tumor necrosis factor inhibitors: synopsis and interpretation of screening and prophylaxis recommendations. Israel Med Assoc J: IMAJ. (2013) 15:303–7.
108. Lu HZ, Zhou JH. Hepatitis B virus X protein up-regulates tumor necrosis factor-α expression in cultured mesangial cells via ERKs and NF-κB pathways. Asian Pacific J Trop Biomed. (2013) 3:217–22. doi: 10.1016/S2221-1691(13)60053-2
109. Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. (1996) 4:25–36. doi: 10.1016/S1074-7613(00)80295-2
110. Schlaak JF, Tully G, Löhr HF, Gerken G, Meyer zum Büschenfelde KH. HBV-specific immune defect in chronic hepatitis B (CHB) is correlated with a dysregulation of pro- and anti-inflammatory cytokines. Clin Experi Immunol. (1999) 115:508–14. doi: 10.1046/j.1365-2249.1999.00812.x
111. Lopetuso LR, Mocci G, Marzo M, D'Aversa F, Rapaccini GL, Guidi L, et al. Harmful effects and potential benefits of anti-tumor necrosis factor (TNF)-α on the liver. Int J Mol Sci. (2018) 192199. doi: 10.3390/ijms19082199
112. Hyodo N, Nakamura I, Imawari M. Hepatitis B core antigen stimulates interleukin-10 secretion by both T cells and monocytes from peripheral blood of patients with chronic hepatitis B virus infection. Clin Experi Immunol. (2004) 135:462–6. doi: 10.1111/j.1365-2249.2003.02376.x
113. Wang S, Huang D, Sun S, Ma W, Zhen Q. Interleukin-10 promoter polymorphism predicts initial response of chronic hepatitis B to interferon alfa. Virol J. (2011) 8:28. doi: 10.1186/1743-422X-8-28
114. David JM, Dominguez C, Hamilton DH, Palena C. The IL-8/IL-8R axis: a double agent in tumor immune resistance. Vaccines. (2016) 4:22. doi: 10.3390/vaccines4030022
115. Dunn C, Brunetto M, Reynolds G, Christophides T, Kennedy PT, Lampertico P, et al. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J Experi Med. (2007) 204:667–80. doi: 10.1084/jem.20061287
116. Lee C-M, Yen Y-H, Hung C-H, Lu S-N, Wang J-H, Wang J-C, et al. Liver interleukin-8 messenger RNA expression and interferon sensitivity-determining region mutations relate to treatment response in hepatitis C 1b. Antiviral Ther. (2011) 16:825. doi: 10.3851/IMP1868
117. Girard S, Shalhoub P, Lescure P, Sabile A, Misek DE, Hanash S, et al. An altered cellular response to interferon and up-regulation of interleukin-8 induced by the hepatitis C viral protein NS5A uncovered by microarray analysis. Virology. (2002) 295:272–83. doi: 10.1006/viro.2002.1373
118. Sonneveld MJ, Wong VW, Woltman AM, Wong GL, Cakaloglu Y, Zeuzem S, et al. Polymorphisms near IL28B and serologic response to peginterferon in HBeAg-positive patients with chronic hepatitis B. Gastroenterology. (2012) 142:513–20.e1. doi: 10.1053/j.gastro.2011.11.025
119. Wei YF, Cui GY, Ye P, Chen JN, Diao HY. MicroRNAs may solve the mystery of chronic hepatitis B virus infection. World J Gastroenterol. (2013) 19:4867–76. doi: 10.3748/wjg.v19.i30.4867
120. van der Ree MH, Jansen L, Kruize Z, van Nuenen AC, van Dort KA, Takkenberg RB, et al. Plasma MicroRNA levels are associated with hepatitis B e antigen status and treatment response in chronic hepatitis B patients. J Infect Dis. (2017) 215:1421–9. doi: 10.1093/infdis/jix140
121. Zhang X, Chen C, Wu M, Chen L, Zhang J, Zhang X, et al. Plasma microRNA profile as a predictor of early virological response to interferon treatment in chronic hepatitis B patients. Antiviral Ther. (2012) 17:1243. doi: 10.3851/IMP2401
122. Tan B, Liu M, Wang L, Wang J, Xiong F, Bao X, et al. Serum microRNAs predict response of patients with chronic hepatitis B to antiviral therapy. Int J Infect Dis. (2021) 108:37–44. doi: 10.1016/j.ijid.2021.05.015
123. Savan R. Post-transcriptional regulation of interferons and their signaling pathways. J Interferon Cytokine Res. (2014) 34:318–29. doi: 10.1089/jir.2013.0117
124. Sedger LM. microRNA control of interferons and interferon induced anti-viral activity. Mol Immunol. (2013) 56:781–93. doi: 10.1016/j.molimm.2013.07.009
125. Chu H, Han N, Xu J. CMPK1 regulated by miR-130b attenuates response to 5-FU treatment in gastric cancer. Front Oncol. (2021) 11:e637470. doi: 10.3389/fonc.2021.637470
126. Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O'leary JJ, Ruan Q, et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol. (2010) 11:141–7. doi: 10.1038/ni.1828
127. Chen W, Bian H, Xie X, Yang X, Bi B, Li C, et al. Negative feedback loop of ERK/CREB/miR-212-3p inhibits HBeAg-induced macrophage activation. J Cell Mol Med. (2020) 24:10935–45. doi: 10.1111/jcmm.15723
128. Su C, Hou Z, Zhang C, Tian Z, Zhang J. Ectopic expression of microRNA-155 enhances innate antiviral immunity against HBV infection in human hepatoma cells. Virol J. (2011) 8:354. doi: 10.1186/1743-422X-8-354
129. Liu W, Ding C. Roles of LncRNAs in viral infections. Front Cell Infect Microbiol. (2017) 7:205. doi: 10.3389/fcimb.2017.00205
130. Liang HW, Wang N, Wang Y, Wang F, Fu Z, Yan X, et al. Hepatitis B virus-human chimeric transcript HBx-LINE1 promotes hepatic injury via sequestering cellular microRNA-122. J Hepatol. (2016) 64:278–91. doi: 10.1016/j.jhep.2015.09.013
131. Nishitsuji H, Ujino S, Yoshio S, Sugiyama M, Mizokami M, Kanto T, et al. Long noncoding RNA #32 contributes to antiviral responses by controlling interferon-stimulated gene expression. Proc Natl Acad Sci USA. (2016) 113:10388. doi: 10.1073/pnas.1525022113
132. Zhou TC, Li X, Chen LJ, Fan JH, Lai X, Tang Y, et al. Differential expression profile of hepatic circular RNAs in chronic hepatitis B. J Viral Hepatitis. (2018) 25:1341–51. doi: 10.1111/jvh.12944
133. Mo Y, Liu Y, Lu A, Zhang H, Tang L. Role of circRNAs in viral infection and their significance for diagnosis and treatment (Review). Int J Mol Med. (2021) 47:4921. doi: 10.3892/ijmm.2021.4921
134. Xie H, Sun H, Mu R, Li S, Li Y, Yang C, et al. The role of circular RNAs in viral infection and related diseases. Virus Res. (2021) 291:198205. doi: 10.1016/j.virusres.2020.198205
135. Bonino F, Marcellin P, Lau GK, Hadziyannis S, Jin R, Piratvisuth T, et al. Predicting response to peginterferon alpha-2a, lamivudine and the two combined for HBeAg-negative chronic hepatitis B. Gut. (2007) 56:699–705. doi: 10.1136/gut.2005.089722
136. Kau A, Vermehren J, Sarrazin C. Treatment predictors of a sustained virologic response in hepatitis B and C. J Hepatol. (2008) 49:634–51. doi: 10.1016/j.jhep.2008.07.013
137. Kim IH, Kisseleva T, Brenner DA. Aging and liver disease. Curr Opin Gastroenterol. (2015) 31:184–91. doi: 10.1097/MOG.0000000000000176
138. Ginaldi L, Loreto MF, Corsi MP, Modesti M, De Martinis M. Immunosenescence and infectious diseases. Microbes Infect. (2001) 3:851–7. doi: 10.1016/S1286-4579(01)01443-5
139. Yeh ML, Peng CY, Dai CY, Lai HC, Huang CF, Hsieh MY, et al. Pegylated-interferon alpha therapy for treatment-experienced chronic hepatitis B patients. PLoS ONE. (2015) 10:e0122259. doi: 10.1371/journal.pone.0122259
140. Zhang T, Zhang Z, Zhang Y, Ye J, Li X. A lower PBMC estrogen receptor α gene expression in chronic hepatitis B Is Associated with a sustained virological response to pegylated interferon. J Interferon Cytokine Res. (2016) 36:120–8. doi: 10.1089/jir.2014.0223
141. Liver EAftSot. EASL 2017 clinical practice guidelines on the management of hepatitis B virus infection. J Hepatol. (2017) 67:370–98. doi: 10.1016/j.jhep.2017.03.021
142. Graf D, Haselow K, Munks I, Bode JG, Haussinger D. Inhibition of interferon-alpha-induced signaling by hyperosmolarity and hydrophobic bile acids. Biol Chem. (2010) 391:1175–87. doi: 10.1515/bc.2010.108
143. Ateş F, Yalniz M, Alan S. Impact of liver steatosis on response to pegylated interferon therapy in patients with chronic hepatitis B. World J Gastroenterol. (2011) 17:4517–22. doi: 10.3748/wjg.v17.i40.4517
144. Gong L, Liu J, Wang J, Lou GQ, Shi JP. Hepatic steatosis as a predictive factor of antiviral effect of pegylated interferon therapy in patients with hepatitis B. Transplant Proc. (2015) 47:2886–91. doi: 10.1016/j.transproceed.2015.10.023
145. Zhu Y, Yang Q, Lv F, Yu Y. The effect of hepatosteatosis on response to antiviral treatment in patients with chronic hepatitis B: a meta-analysis. Gastroenterol Res Practice. (2017) 2017:1096406. doi: 10.1155/2017/1096406
146. Tian Y, Jennings J, Gong Y, Sang Y. Viral infections and interferons in the development of obesity. Biomolecules. (2019) 9:726. doi: 10.3390/biom9110726
147. Wang X, Zhao Y-R, Liu H-L, Ma X-H, Zhang Y, Yi R-T. Insulin resistance predicts virological response to interferon-α in chronic hepatitis B patients. J Clin Gastroenterol. (2016) 50:452. doi: 10.1097/MCG.0000000000000452
148. Mahmoud MA, Ghareeb DA, Sahyoun HA, Elshehawy AA, Elsayed MM. In vivo interrelationship between insulin resistance and interferon gamma production: protective and therapeutic effect of berberine. Evid Based Complement Alternat Med. (2016) 2016:2039897. doi: 10.1155/2016/2039897
149. Ganesan M, Eikenberry A, Poluektova LY, Kharbanda KK, Osna NA. Role of alcohol in pathogenesis of hepatitis B virus infection. World J Gastroenterol. (2020) 26:883–903. doi: 10.3748/wjg.v26.i9.883
150. Mao Q, Luo K, Liu D, Zhang M, Hou J. Clinical significance of neutralizing anti-interferon antibodies in chronic hepatitis B patients treated with recombinant interferon-alpha. Chinese J Hepatol. (2004) 12:205–7.
151. Lok ASF, Lai CL, Leung EKY. Interferon antibodies may negate the antiviral effects of recombinant α-interferon treatment in patients with chronic hepatitis B virus infection. Hepatology. (1990) 12:1266–70. doi: 10.1002/hep.1840120603
152. Stancek D, Fuchsberger N, Oltman M, Schmeisser H, Kontsek P, Jahnová E, et al. Significance of anti-interferon-alpha2 and sICAM-1 activities in the sera of viral hepatitis B and C patients treated with human recombinant interferon-alpha2. Acta Virol. (2001) 45:287–92.
153. Woo ASJ, Kwok R, Ahmed T. Alpha-interferon treatment in hepatitis B. Ann Transl Med. (2017) 5:159. doi: 10.21037/atm.2017.03.69
154. Rijckborst V, Janssen HLA. The role of interferon in Hepatitis B therapy. Curr Hepat Rep. (2010) 9:231–8. doi: 10.1007/s11901-010-0055-1
155. Tan Y, Ye Y, Zhou X, Chen L, Wen D. Age as a predictor of significant fibrosis features in HBeAg-negative chronic hepatitis B virus infection with persistently normal alanine aminotransferase. PLoS ONE. (2015) 10:e0123452. doi: 10.1371/journal.pone.0123452
156. Tong S. Hepatitis B virus, a sex hormone-responsive virus. Gastroenterology. (2012) 142:696–9. doi: 10.1053/j.gastro.2012.02.036
157. Oehler N, Volz T, Bhadra OD, Kah J, Allweiss L, Giersch K, et al. Binding of hepatitis B virus to its cellular receptor alters the expression profile of genes of bile acid metabolism. Hepatology. (2014) 60:1483–93. doi: 10.1002/hep.27159
158. Ishizaka N, Ishizaka Y, Seki G, Nagai R, Yamakado M, Koike K. Association between hepatitis B/C viral infection, chronic kidney disease and insulin resistance in individuals undergoing general health screening. Hepatol Res. (2008) 38:775–83. doi: 10.1111/j.1872-034X.2008.00334.x
159. Newby BN, Mathews CE. Type I interferon is a catastrophic feature of the diabetic islet microenvironment. Front Endocrinol. (2017) 8:232. doi: 10.3389/fendo.2017.00232
160. Tai TY, Lu JY, Chen CL, Lai MY, Chen PJ, Kao JH, et al. Interferon-alpha reduces insulin resistance and beta-cell secretion in responders among patients with chronic hepatitis B and C. J Endocrinol. (2003) 178:457–65. doi: 10.1677/joe.0.1780457
161. Li ZM, Kong CY, Zhang SL, Han B, Zhang ZY, Wang LS. Alcohol and HBV synergistically promote hepatic steatosis. Ann Hepatol. (2019) 18:913–7. doi: 10.1016/j.aohep.2019.04.013
162. Zhang Z, Wang C, Liu Z, Zou G, Li J, Lu M. Host genetic determinants of hepatitis B virus infection. Front Genet. (2019) 10:696. doi: 10.3389/fgene.2019.00696
163. Wang L, Zou ZQ, Wang K. Clinical relevance of HLA gene variants in HBV infection. J Immunol Res. (2016) 2016:9069375. doi: 10.1155/2016/9069375
Keywords: chronic hepatitis B, interferon-α, negative regulators, host factors, non-response
Citation: Wang J, Du L and Tang H (2021) Suppression of Interferon-α Treatment Response by Host Negative Factors in Hepatitis B Virus Infection. Front. Med. 8:784172. doi: 10.3389/fmed.2021.784172
Received: 27 September 2021; Accepted: 03 November 2021;
Published: 24 November 2021.
Edited by:
Jian Wu, Zhejiang University, ChinaCopyright © 2021 Wang, Du and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Tang, aHRhbmc2MTk4QGhvdG1haWwuY29t
†These authors have contributed equally to this work