 J. Samael Rodríguez-Sanabria1†
J. Samael Rodríguez-Sanabria1† Rebeca Escutia-Gutiérrez1†
Rebeca Escutia-Gutiérrez1† Rebeca Rosas-Campos1
Rebeca Rosas-Campos1 Juan S. Armendáriz-Borunda1,2*
Juan S. Armendáriz-Borunda1,2* Ana Sandoval-Rodríguez1*
Ana Sandoval-Rodríguez1*- 1Department of Molecular Biology and Genomics, Institute for Molecular Biology in Medicine and Gene Therapy, CUCS, University of Guadalajara, Guadalajara, Mexico
- 2School of Medicine and Health Sciences, Tecnologico de Monterrey, Campus Guadalajara, Zapopan, Mexico
Metabolic-associated fatty liver disease (MAFLD) is characterized by hepatic steatosis accompanied by one of three features: overweight or obesity, T2DM, or lean or normal weight with evidence of metabolic dysregulation. It is distinguished by excessive fat accumulation in hepatocytes, and a decrease in the liver's ability to oxidize fats, the accumulation of ectopic fat, and the activation of proinflammatory pathways. Chronic damage will keep this pathophysiologic cycle active causing progression from hepatic steatosis to cirrhosis and eventually, hepatocarcinoma. Epigenetics affecting gene expression without altering DNA sequence allows us to study MAFLD pathophysiology from a different perspective, in which DNA methylation processes, histone modifications, and miRNAs expression have been closely associated with MAFLD progression. However, these considerations also faced us with the circumstance that modifying those epigenetics patterns might lead to MAFLD regression. Currently, epigenetics is an area of great interest because it could provide new insights in therapeutic targets and non-invasive biomarkers. This review comprises an update on the role of epigenetic patterns, as well as innovative therapeutic targets and biomarkers in MAFLD.
Introduction
Metabolic-associated fatty liver disease (MAFLD) is characterized by hepatic steatosis accompanied by one of three features: overweight or obesity, T2DM, or lean or normal weight with evidence of metabolic dysregulation (1).
MAFLD, as with the previous term NAFLD, represents the hepatic manifestation of a multisystem disorder, whose incidence is 20–30% in the western countries (2). Currently, there is no FDA-approved therapeutic agent for MALFD, and changes in diet and increase in physical activity are the first-in-line treatment of hepatic steatosis (3).
Gene expression is ultimately influenced by diverse epigenetic processes, including DNA methylation, histone modification, and expression of non-coding RNA molecules, like miRNAS. Epigenetic changes are reversible, and lifestyle and environmental exposure can define epigenetic patterns throughout life (4).
Epigenetic variations differ in the same individual among cell types and are associated with disease susceptibility by producing long-term changes in gene transcription (5). Alterations in hepatic epigenetics significantly contribute to MAFLD development by altering transcriptional networks implicated in redox homeostasis, peroxisome and mitochondria function, inflammation, insulin sensibility, and fat homeostasis. Most important epigenetic mechanisms implicated in the development of metabolic associated fatty liver disease are described in the next sections.
DNA Methylation
DNA methylation is the covalent addition of a methyl group to the C5 position of cytosine generating a 5-methylcytosine (5mC), usually in cytosine–guanine dinucleotides-rich regions known as CpG islands. In general, hypermethylation of CpG islands is associated with gene repression, since the methyl group may physically block binding of transcription factors to the DNA, or it can act as a binding site for transcriptional repressors such as histone deacetylases; whereas hypomethylation is permissive to transcription (6). DNA methylation is catalyzed by a family of enzymes called DNA methyltransferases (DNMTs) that transfer the methyl group from an S-adenyl methionine (SAM) to DNA (7). DNMT1 accounts for the recognition of the hemimethylated strand after a cell cycle. DNMT3a and DNMT3b are responsible for de novo methylation (8). The ten–eleven translocation (Tet) enzymes remove the methyl group in DNA (9).
DNA methylation is the most studied epigenetic mechanism in MAFLD. Detection of aberrant DNA methylation patterns could provide therapeutic targets and molecular tools for diagnosis and prediction of MAFLD (10). Several studies have analyzed genome-wide methylation changes associated with MAFLD, showing alterations in the methylation signature of many genes including regulatory loci for key metabolic and inflammatory pathways. For example, a study in humans using liver biopsies from obese patients with MAFLD showed methylation and expression differences in nine key enzymes implicated in intermediate metabolism and insulin signaling: pyruvate carboxylase (PC), ATP citratelyase (ACLY), phospholipase C-gamma-1 (PLCG1), insulin-like growth factor 1 (IGF1), insulin-like growth factor binding protein 2 (IGFBP2), and protein kinase C epsilon (PRKCE), putative polypeptide N-acetylgalactosaminyl-transferase-like protein 4 (GALNTL4), glutamate receptor delta-1 (GRID1), and inositol hexaphosphate kinase 3 (IP6K3) (11). A similar study founded that 41 genes responsible for lipid homeostasis were significantly and differentially methylated, including members of the APO family (lipid transport), genes involved in cholesterol transport like intracellular cholesterol transporter 1 (NPC1L1), acyl-CoA, sterol regulatory element binding transcription factor 1 (SREBF1), StAR-related lipid transfer domain containing 5 (STARD5), and solute carrier family 2 member 4 (SLC2A4) (12). Insulin resistance (IR) is part of the pathophysiology of MAFLD and its progression to NASH (13). An increased hepatic methylation of peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PPARGC1A) has been correlated with high plasma fasting insulin levels (r = 0.51, p < 0.01) and HOMA-IR (r = 0.58, p < 0.003) in patients with MAFLD (14, 15).
Diet and DNA Methylation
“Western diet” is characterized by excessive fat and sugar consumption and seems to contribute to MAFLD pathogenesis (16). Preclinical studies demonstrated that the consumption of high-fat diet alters DNA methylation of gene clusters (17) and induces hypermethylation in promoter regions of peroxisome proliferator-activated receptor alpha (PPARA) (18), whereas, high fructose induces hypermethylation of carnitine palmitoyltransferase 1A (CTP1A) and PPARA genes (19) and global hypomethylation of mitochondrial DNA (20). PPARA is a transcriptional regulator of genes involved in mitochondrial beta-oxidation, fatty acid transport, and hepatic production of glucose, and PPARA hypermethylation decreased its gene expression and induced fatty accumulation in the liver. On the other hand, peroxisome proliferator-activated receptor gamma (PPARG) is upregulated in diabetes, obesity, and MAFLD. Mice fed a high-fat diet (HFD) showed a reduction of the level of cytosine methylation Pparg promoter, DNMT activity, and induction of hepatic Pparg expression (21).
Furthermore, Wang et al. proposed a regulatory pathway for sugar leading to induction of lipid accumulation; Huh-7 cells administered with high-glucose showed a close relationship between an increase in nuclear 25-hydroxycholesterol and activation of DNMT1, which methylates cytosine of CpG in promoter regions, suppressing expression of genes involving in MAFLD diseases (22).
It is challenging to confirm these studies in humans; however, a human study examined the effect of lifestyle interventions on DNA-methylation. The participants received a regimen of either low-fat or Mediterranean-low carbohydrates for 18 months. At baseline, intrahepatic fat was inversely correlated with DNA-methylation in calcium release activated channel regulator 2A (CRACR2A), alpha-2-macroglobulin pseudogene 1 (A2MP1), and ARH/RhoGEF and pleckstrin domain protein 1 (FARP1) genes. In conclusion, patterns in DNA-methylation changed in A2MP1 gene after lifestyle interventions (23).
DNA methylation patterns can be modified also by bioactive food components. For example, methyl-group donors (B9, B12, methionine, betaine, and choline) are required for SAM synthesis in one-carbon metabolism. One-carbon metabolism comprises a series of interlinking metabolic pathways that include the methionine and folate cycles that are central to cellular function, providing methyl groups for the synthesis of DNA, polyamines, amino acids, creatine, and phospholipids (24). Several studies have demonstrated that CH3 deficiency in one-carbon metabolism is strongly associated with MAFLD (25). In animal models, a deficient methyl-donor diet is associated with reduced hepatic global DNA methylation and altered DNA-methylation patterns of lipid genes associated with fatty-liver-like phenotype such as ATP binding cassette subfamily A member 1 (Abca1), acetyl-CoA acetyltransferase 1 (Acat1), 1-acylglycerol-3-phosphate O acyltransferase 3 (Agpat3), and angiotensin II receptor type 1 (AGTR1) (26, 27). In contrast, dietary methyl donor-supplementation prevents liver fat accumulation by modifying the methylation of specific gene promoters like Srebf2, Agpat3, and estrogen receptor 1 (Esr1) (28). Recently, these results were corroborated in humans; hepatic global DNA methylation levels were significantly lower in patients with MAFLD than in the control group, and also among participants who were overweight. These data correlate negatively with histological disease severity. In addition, MAFLD group had a significant higher serum homocysteine concentration (an indicator of methyl donor–deficient diet). This suggests that global DNA methylation and serum one-carbon metabolites may be markers of MAFLD status or severity (29). In patients with type 2 diabetes, a correlation between a high number of hypomethylated CpG sites and reduced levels of folate in the circulation was found (30). Another study was conducted in obese patients, associated low folate intakes with lower calcium/calmodulin-dependent protein kinase 2 (CAMKK2) gene methylation and IR (31).
DNA Methylation as Predictive Biomarkers of Disease
DNA methylation in peripheral cells or ccf-DNA is a potential biomarker to diagnose MAFLD. Hypomethylation in promoters of protein kinase C epsilon (PRKCE) and SEC14 like lipid binding 3 (SEC14L3) is associated with MAFLD by genome-wide DNA methylation profiling in peripheral blood leukocytes (32). Ma et al. reported differential methylation in 22 CpG in genes like SLC7A11, CPT1A, SREBF1, zinc finger RNA binding protein 2 (ZFR2), and SLC9A3R1 associated with increase hepatic fat in European Ancestry participants (33). Similarly, in patients with histologically confirmed MAFLD, six differentially methylated CpG sites were identified in the Acyl-CoA synthetase long-chain family member 4 (ACSL4), cardiolipin synthase 1 (CRLS1), carnitine palmitoyltransferase 1A (CTP1A), single Ig and TIR domain containing (SIGIRR), single-stranded DNA binding protein 1 (SSBP1), and zinc finger protein 622 (ZNF622) genes compared with healthy controls (34). Nano et al. reported an association between DNA methylation in SLC7A11, SLC1A5, SLC43A1, phosphoglycerate dehydrogenase (PHGDH), psoriasis susceptibility 1 candidate 1 (PSORS1C1), SREBF1, and ankyrin repeat and sterile alpha motif domain containing 3 (ANKS3) with gamma-glutamyl transferase (GGT) levels; while DNA methylation in SLC7A11 was associated with alanine aminotransferase (ALT) (35). MAFLD may progress to advanced liver disease with the presence of fibrosis, a key histological determinant of long-term prognosis. An observational study compared liver biopsies from patients with mild vs. advanced fibrosis, identifying significant more methylation in gene regulatory regions of transforming growth factor beta 1 (TGFB1) and platelet-derived growth factor subunit A (PDGFA) in patients with mild fibrosis, whereas PPARA and PPARD showed considerably less methylation (36).
A previous study has demonstrated that PPARG promoter hypermethylation correlated with severe fibrosis in liver biopsies (37), and more recently Hardy et al. found a similar degree of hypermethylation in the PPARG promoter in plasma ccf-DNA and hepatocyte-rich tissue captured by laser capture microdissection, suggesting that plasma DNA methylation of PPARG could potentially be used as a noninvasive method to determinate liver fibrosis severity in patients with MAFLD (38). Also, hypomethylation in a branched chain amino acid transaminase 1 (BCAT1) has been reported inversely associated with fibrosis degree (39). Hypomethylation of fibroblast growth factor receptor 2 (FGFR2), caspase 1 (CASP1), and hypermethylation of methionine adenosyltransferase 1A (MAT1A) were associated with advanced MAFLD in a study of Murphy et al. (40). Parvin beta variant 1 (PARVB) (hypomethylated in CpG26) and patatin like phospholipase domain containing (PNPLA3) (hypermethylated in CpG99) have also been associated with MAFLD (41). Figure 1 describes differential DNA methylation patterns associated with MAFLD, some of them proposed as biomarkers.
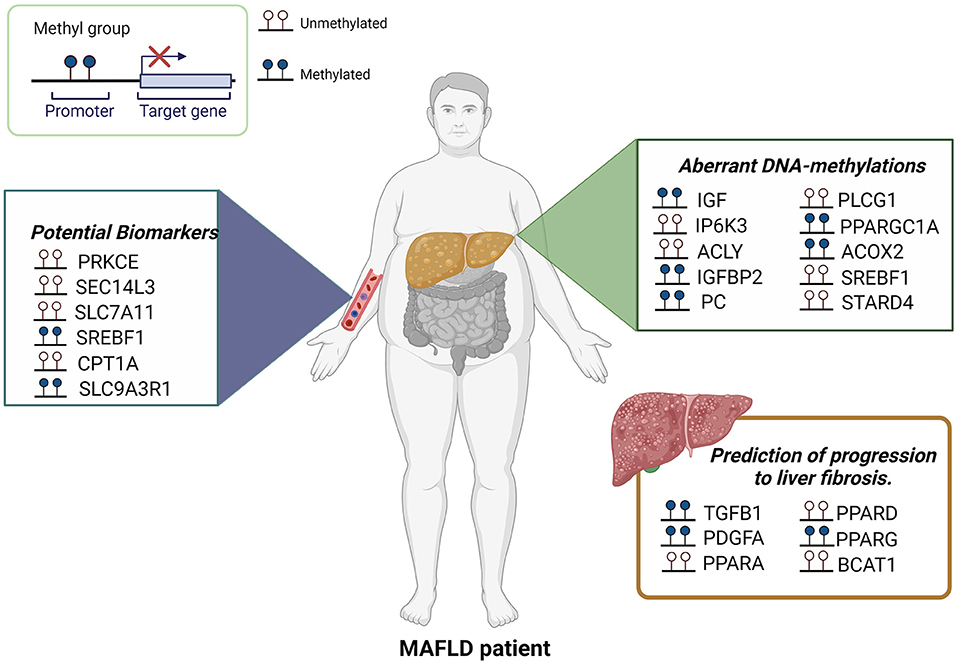
Figure 1. Aberrant DNA-methylations in MAFLD. Several studies using liver biopsies, ccf-DNA or peripheral blood leukocytes have been shown differential DNA methylation patterns associated with MAFLD. Certain CpGs also represent attractive biomarkers for MAFLD and the prediction of progression to fibrosis.
Histone Modifications in MAFLD
Histones are a family of basic proteins whose positive charges allow them to associate with DNA in the nucleus and help them condense it into a chromatin. The basic structural unit of chromatin, the nucleosome (42), is formed by a pair of each H2a, H2b, H3, and H4 histones, an octamer (43). These histones are small globular proteins containing an N-terminal tail that can undergo acetylation, methylation, phosphorylation, SUMOylation, ubiquitination, or ADP-ribosylation. Multiple histones modifying enzymes can carry out more than 60 chemical histone-modifications that affect specific DNA binding sites, causing transcription activation or silencing of specific genes (44).
Lysine acetylation or methylation in the N-terminal tail stands out as the histone modifications with greatest repercussion in gene expression (45). Acetylation is mediated by histone acetyltransferases (HAT) and is usually associated with active gene transcription due to its ability to decompress chromatin. For this reaction, acetyl CoA acts as a cofactor, and subsequently HAT catalyzes the transfer of an acetyl group to the epsilon-amino group of lysine (46), neutralizing the positive charge of lysine and weakening histone and DNA interactions (47). In the opposite way, histone deacetylases (HDAC) remove acetyl groups from lysine and thus restores the compacted form of chromatin (48).
On the other hand, histone methylation in residues in the N-terminal tail of histones causes silencing of chromatin and the inactivation of transcription. However, in particular cases, methylation of histone activates gene transcription and is associated with the initiation of chromatin remodeling (49). The precise effect of methylation is linked to the specific residue where the reaction takes place. The methylation process is carried out by histone methyltransferases (HTM), which have the ability to add one, two, or three methyl groups to lysine or arginine residues of histones. Histone demethylases (HDM) have the ability to remove methyl groups from histone, thus beginning the remodeling of chromatin toward a decompressed or active state. HDMs have been classified into two classes, the FAD-dependent amino oxidases (LSD) and the jumonji C demethylase (JMJD) (50). Imbalance in histone modifications causes a disproportion in transcriptional activity associated with the development of diseases such as type 2 diabetes mellitus, obesity, and consequently MAFLD (51). Main enzymes involved in histone modifications that are implicated in MAFLD development are enlisted in Figure 2.
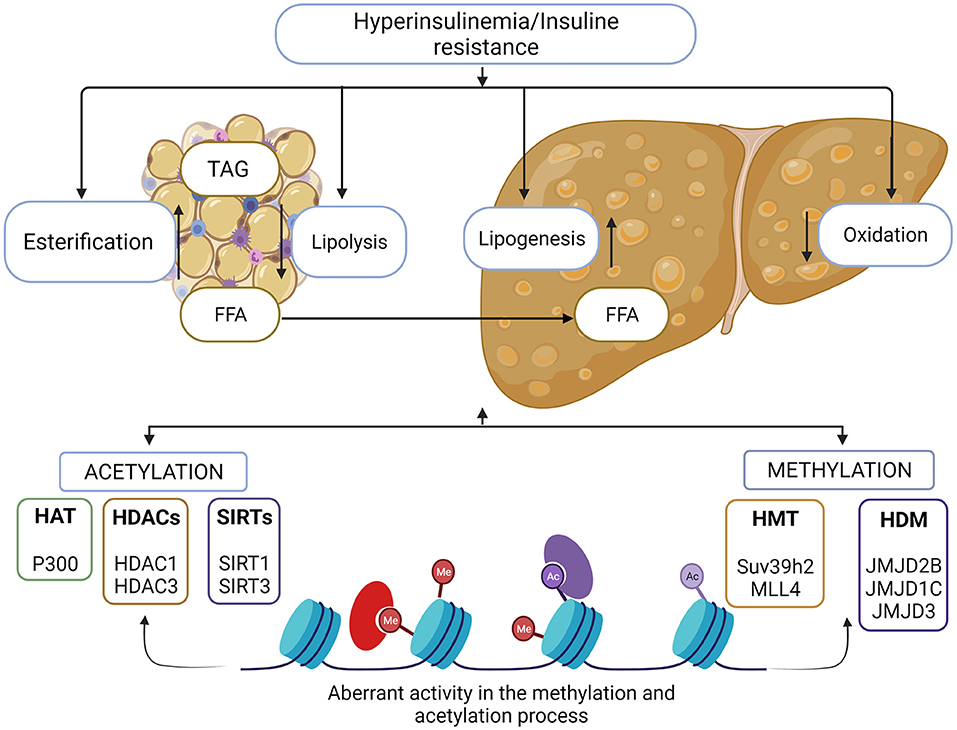
Figure 2. Histone modifications in MAFLD. Metabolic imbalance in lipids and carbohydrates are involved in the development of MAFLD. Adipose tissue storage and insulin resistance triggers the accumulation of free fatty acids in the liver and liver homeostasis is lost. There is a direct association between aberrant chromatin modifications and this metabolic imbalance. Histone methylation and acetylation process, allows the activation of genes associated with the lipogenic and inflammatory process, as well as the reduction in expression of the genes involved in the oxidation of fatty acids, enzymes responsible for these events are possible therapeutic target for MAFLD control.
Histone Acetylation
During IR or DM2 the risk to develop non-alcoholic fatty liver increases due to inflammatory factors; where nuclear factor enhancing kappa of activated B cells (NFkB) or elements of carbohydrate metabolism that affect lipogenesis like element binding protein carbohydrate response (ChREBP) stand out. These factors are upregulated by some HAT (52). P300, a member of the HAT family, is a transcriptional regulator that plays a very important role modifying NFkB pathway. It has been shown that inhibition of p300 improves MAFLD in mice and restores biochemical parameters, decreases activity of genes involved in lipogenesis, and therefore, the aberrant activity of p300 favors MAFLD development (53). One of the main factors that is altered by p300 is ChREBP, a protein essential for the accumulation of fat in the liver. Bricambert et al. corroborated the interaction of these two molecules, activating or inhibiting kinase inducible by serine/threonine kinase 2 (SIK2), an element that regulates the activity of p300. In HepG2 cells and mice, SIK2 inhibited p300 activity by direct phosphorylation, and therefore also decreased the lipogenesis mediated by ChREBP. SIK2 depletion caused an overexpression of p300 increasing lipogenesis and causing insulin resistance, hepatic steatosis and inflammation (54). HDACs have 4 families (class I, IIa, IIb, and IV) that differ in structure, enzymatic function, and location. HDACs play an important role in the development of MAFLD, some with more evidence than others. For example, HDAC1, a member of the class I family-depleted HepG2 cells decreased sterol regulatory element binding protein (SREBP1c) as well as, liver tissue of P50 NFkB-subunit KO mice (55). In addition to HDAC1, the activity of HDAC3 has also been evaluated in MAFLD and in obesity and insulin resistance. HDAC3 regulates hepatic lipid metabolism in the opposite way to HDAC1. HDAC3 is an important lipid homeostatic regulator in the liver, and its loss leads to severe hepatic steatosis in mice (56). It is key to highlight that HDAC3 also has direct interaction with molecules that participate in the development of hepatic steatosis, such as SREBP1, key molecule in the lipogenic process (57). In addition, HDAC3 has a specific role in the circadian pattern of hepatic lipogenesis, a dysregulation in this cycle mediated by SREBP1 increases the lipogenic process (58, 59).
A well-known group of deacetylases are silent information regulatory proteins (SIRTs), also known as Sirtuins. SIRTs are members of the class III HDAC family and use NAD+ as a cofactor. They can interact with histones as well as non-histone proteins and have gained interest in metabolic diseases since they are involved in lipid homeostasis, oxidative stress, and insulin resistance, all events implicated in MAFLD development. Sirtuin family has 7 members characterized by their structure, enzymatic function, and localization. SIRT 1, 2, 3, 6, and 7 are mainly found in the nucleus, SIRT1 and SIRT2 are also in the cytoplasm, and SIRT4 and SIRT5 in mitochondria (60).
The sirtuins with greatest association to nonalcoholic fatty liver disease development are SIRT1 and SIRT3. Recent evidence showed that SIRT1 is an important piece in lipid homeostasis in the liver, and it is an agonist ligand of peroxisome proliferator-activated receptor alpha (PPAR), promoting oxidative activity in lipids. Sandoval-Rodriguez and Monroy-Ramirez et al. used synthetic inhibitors and activators of SIRT1 and PPARA in cultured HepG2 cells, demonstrating positive feedback between both proteins, which leads to the fact that a decrease in SIRT1 favors the development of MAFLD in part due to decrease in PPARA activity (61). The effect of SIRT1 on lipid metabolism has been an important part of the discussion of whether it could function as a therapeutic target for MAFLD. The activation of SIRT1 during MALFD decreases lipids and TGs accumulation in the liver, decreasing inflammation and lipogenic process (62). For its part, SIRT3 is also important in MAFLD. Mice deficient in SIRT3 and fed with HFD increased lipid levels in the liver, promoting development of MAFLD. SIRT3 deficiency leads to less DNA binding activity in PPARA, thereby decreasing the production of molecules activated by PPARA; promoting fatty acids oxidative status (63). In addition, regarding oxidative stress and mitochondrial damage, events involved in hepatic steatosis, SIRT3 deficiency increased oxidative stress and activation of caspase-9 pathway. However, overexpression of SIRT3 decreases reactive oxygen species and promotes the activation of the ERK-CREB-Bnip3 pathway improving mitophagy (63).
Histone Methylation
Transcription silencing is linked with a compacted state of chromatin, generally, associated with methylation of histone tail. Histone 3 lysine 9 (H3K9) has been associated with the development of MAFLD, and the aberrant activity of some methyltransferases have been associated with this process (51).
Histone–lysine N-methyltransferase SUV39H2, an enzyme capable of adding mono, di, and trimethylated labels to H3K9, has a fundamental role in the activation of inflammatory pathways. Also, it can reduce the activity of SIRT1 causing NASH progress. SUV39H2 activity was analyzed in KO mice fed a HFD, and they developed hepatic steatosis of less severity compared with the wild type for this enzyme (64).
In addition, there is also a relationship between the development of hepatic steatosis and methylation of histone 3 lysine 4 (H3K4) by myeloid/lymphoid or mixed-lineage leukemia 4 (MLL4) methyltransferase. It was shown that in overnutrition conditions, MLL4 provokes H3K4 methylation facilitating interaction with targets of peroxisome proliferator-activated receptor gamma 2 (PPARy2), which promotes lipogenesis (65).
On the other hand, the activity of methylases has also been studied in MAFLD. Clear evidence of the direct effect of JMJD2B on histone mark H3K9 has demonstrated the importance of this enzyme in the lipogenic process during MAFLD, with the interaction of PPARG2 and the ligand-activated liver X receptor alpha (LXRa). JMJD2B removes the trimethylated and dimethylated marks, leaving the monomethylated mark of H3K9, causing activation of PPARG2 and its target genes increasing the hepatic lipogenic process (66). The same situation occurs with Liver X receptor alpha (LXRA). It has been shown that the overexpression of JMJD2B increases the activity of this receptor, inducing intracellular accumulation of triglycerides and thus MAFLD development (67).
Another molecule that has a demethylase function and that has been associated with the progression of MAFLD is JMJD1C. In the same way, the interaction of this enzyme with the histone mark H3K9, removing repressive marks, promotes the transcription of genes, inducing lipogenesis and accumulation of hepatic fatty acids. It has been shown that the mammalian Target of Rapamycin (mTOR) complex phosphorylates JMJD1C, allowing interaction with upstream stimulatory factor 1 (USF1), a molecule that activates lipogenic genes and is associated with familial hyperlipidemia (68).
On the contrary, the activity of JMJD3 has been associated with the disease improvement; it removes the repressive mark of histone 3 lysine 27 (H3K27) leaving it in its dimethylated form (H3K27me2), promoting chromatin remodeling, and in turn, working together with SIRT1, to promote PPARA activation. This evidence was obtained in fasting conditions, and genes involved in gluconeogenesis pathway had no relevant activity, but these facts open up the possibility of a new therapeutic target (69).
microRNAs
microRNAs (miRNAs) are single-stranded non-coding RNAs of 18–25 nucleotides long that can regulate gene expression at posttranscriptional level by inhibiting translation or inducing degradation of target mRNAs through complementary base-pairing (70). miRNAs account for 1–5% of the human genome and regulate at least 50% of protein coding genes in mammals (71). To date, more than 2,800 human miRNAs have been registered in the miRBase 22.1, which are predicted to regulate up to 60% of the human genes. About 50% of miRNAs are transcribed from protein coding genes, mostly intragenic regions particularly introns and few exons. The other half are intergenic, transcribed independently, and regulated by their own promoters. Each miRNA can regulate several target genes, and vice versa, and each target gene can be regulated by various miRNAs, explaining why miRNAs can play crucial functions in essentially all biological processes and in all cell types (72). Evidence have demonstrated that miRNAs are implicated as important mediators in metabolic diseases including obesity, DM2, metabolic syndrome, and metabolic associated fatty liver disease (MAFLD) (73–75). Figure 3 summarizes upregulated miRNAs involved in pathogenesis and development of MAFLD.
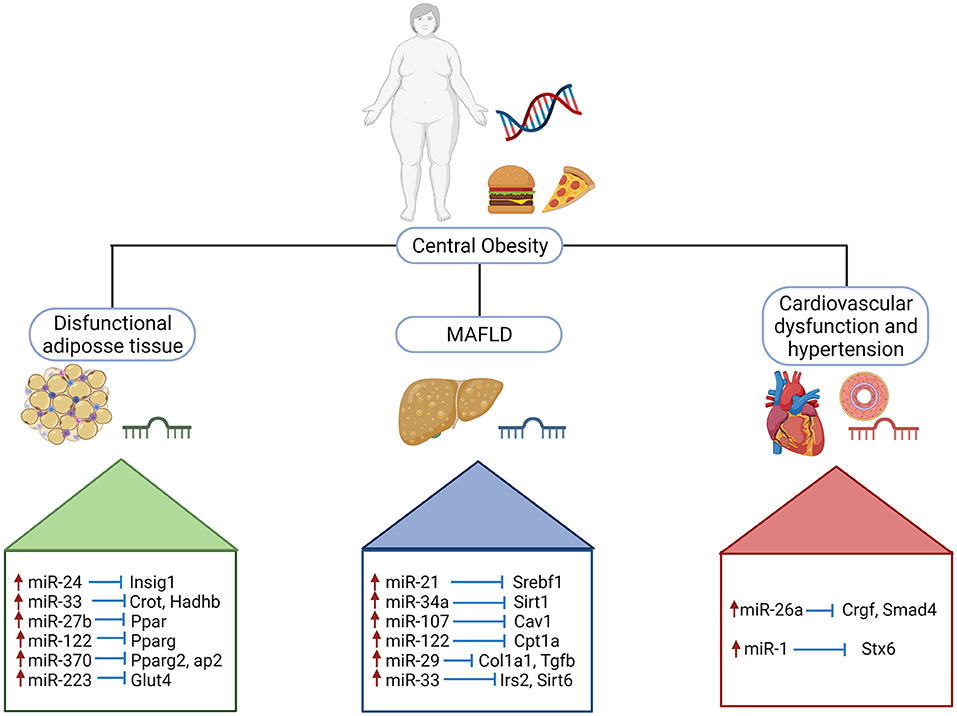
Figure 3. Upregulated miRNAs involved in pathogenesis and development of MAFLD. Schematic illustration of selected miRNAs shows the impact on the stimulatory effect on target genes implicated in obesity, metabolic dysregulation and cardiovascular dysfunction. Insig 1, insulin induced gene 1; Crot, carnitine O-octanoyltransferase; Hadhb, hydroxyacyl-coa dehydrogenase trifunctional multienzyme complex subunit beta; Ppar, peroxisome proliferator activated receptor; Pparg, peroxisome proliferator activated receptor gamma; Pparg2, peroxisome proliferator activated receptor gamma 2; aP2, activating protein 2; Glut4, glucose transporter type 4; Srebf1, sterol regulatory element binding transcription factor; Sirt1; sirtuin 1; Cav1, caveolin 1; Cpt1a, carnitine palmitoyltransferase 1A; Col1a1, collagen type I alpha 1 chain; Tgfb, transforming growth factor beta; Irs2, insulin receptor substrate 2; Sirt6, sirtuin 6; Crgf, teratocarcinoma-derived growth factor 1; Smad4, SMAD Family Member 4; Stx6, syntaxin 6.
miRNAs in Obesity
Several miRNAs including miR-27b, miR-33, miR-34a, miR-122, and miR-223 are important regulators in fatty acid metabolism and cholesterol biosynthesis in the liver (76). Specifically, miR-33 plays a key role in cholesterol homeostasis thought suppression of sterol regulatory element-binding protein 1 (SREBP1), high density lipoprotein formation, fatty acid oxidation, and insulin signaling (77).
miR-27b-3p exert regulatory effects in lipid metabolism and is altered in dyslipidemia (78). In high-fat diet model of obesity, miR-27b-3p suppress adipose tissue browning. Due to this key role in promoting body fat accumulation miR-27b-3p should be further explored as a potential target for the treatment of central obesity and linked diseases (79).
miR-122 is the most abundant miRNA in the liver, and has a key role in liver metabolism, cholesterol biosynthesis, fatty acid synthesis, and oxidation (80). It should be noted that miR-122 was the first miRNA to be associated with metabolic regulation (81). Long JK et al. found that miR-122 promoted hepatic lipogenesis inhibiting LKB1/AMPK pathway by targeting SIRT1 in HepG2 and Huh-7 cells cultured with free fatty acids (FFA) (82). miR-122 was downregulated in steatotic-FFA-induced hepatocytes, and nonalcoholic steatohepatitis mice model using streptozotocin and HFD (STZ- HFD). Besides, miR-122 showed an important role in hepatic triglyceride accumulation reducing YY1 mRNA stability causing upregulation in FXR-SHP signaling (83).
miR-34a has been reported as a probable tumor suppressor in numerous types of cancers (84). miR-34a is upregulated in MAFLD and is an essential regulator of lipid metabolism (85). In a work by Ding et al., miR-34a levels were increased in L02 cells transfected with miR-34a inhibitor and C57BL/6 mice injected with a miR-34a inhibitor. Ppara and Sirt1, which are target genes of miR-34a, were downregulated after miR-34a inhibitory treatment, provoking triacylglycerides, liver index, and activated-AMPK pathway decrease (86). In adipose tissue it has been reported that miR-34a expression gradually increases as dietary obesity develops. In miR-34a–KO mice glucose intolerance, insulin resistance, and systemic inflammation were present in epidydimal white adipose tissue (epiWAT).
Interestingly, increased miR-34a expression causes adipose inflammation principally by reduced expression of Klf4, resulting in suppressive effects on M2 macrophages polarization. Besides, it was found that high expression of miR-34a in visceral fat of overweight/obese patients correlated negatively with diminished Klf4 (87).
miR-33 is a key regulator of lipid metabolism by targeting genes involved in cholesterol uptake and efflux in the liver, fatty acid metabolism Cpt1, Crot, Hadhb, insulin signaling IRS2 and mitochondrial function Ampk, Pgc1a (88–90). miR-223 could inhibit cholesterol biosynthesis in mice through negative regulation of the 3-hydroxy-3-methylglutaryl-CoA synthase 1 (Hmgcs1) and the sterol-C4-methyloxidase-like protein (Sc4mol). Besides, miR-223 decreased high-density lipoprotein-cholesterol (HDL-C) uptake by targeting the scavenger receptor class B member 1 causing ABCA1 expression increase that rise cholesterol efflux (91). Otherwise, miR-223 targets include inflammatory and oncogenic genes like CXCL10 and TAZ, data obtained in hepatocytes of high fat diet fed mice and in NASH patient livers. Therefore, miR-223 could protect against NASH development 322 (92).
A recent study by Zhang et al. reported that overexpression of miR-802 downregulates insulin transcription and secretion, as well as impairs glucose tolerance, suggesting a role of miR-802 in the development of obesity-associated β cell dysfunction (93). Several studies have reported that miR-221 is upregulated in adipose tissue from obese patients (94, 95). Peng et al. suggested that miR-221 promotes white adipose tissue inflammation and reduces insulin sensitivity in obesity while suppressing SIRT1 (96).
miRNAs in Metabolic Alterations
Some miRNAs are crucial in MAFLD progression and metabolic alterations including waist circumference, blood pressure, serum triglycerides, and HOMA levels (97).
During adipogenesis, miR-425 expression is controlled by Pparg in adipocytes. miR-425 overexpression resulted in a proliferation reduction of 3T3-L1 preadipocytes, but accelerated cellular adipogenic differentiation. miR-425 also influences adipogenesis inhibiting its target gene Mapk14,a negative regulator of adipogenesis (98).
miR-107 is known to regulate insulin sensitivity in mouse models mainly by altering liver metabolism. miR-107 has a key role in lipid metabolism, inhibiting CDK6 expression and its downstream targets, reducing adipogenesis in preadipocytes. Besides, it has been proposed that miR-107 promotes ectopic fatty acid accumulation and reduced glucose tolerance since miR-107 decreased glucose uptake and triglycerides synthesis in mature adipocytes (99). In a work carried out by Okamoto et al., serum miR-379 expression was upregulated in patients with MAFLD compared with healthy individuals. Serum levels of miR-379 showed positive correlations with alkaline phosphatase, total cholesterol, low- density-lipoprotein cholesterol, and non-high-density lipoprotein cholesterol levels in patients with early stage MAFLD (100).
miR-126a has been validated as a biomarker in obesity and related metabolic disease in women by Vonhögen et al. Thus, circulating levels of miR-216a are predictive factors for obesity. Interestingly, they found the obesity predisposition locus, the miR-216a gene that includes CpG islands with differential DNA methylation levels among obese and non-obese children, and is related with differential circulating miR-216a plasma levels in obese and non-obese women (101).
Remarkably, Lin et al. demonstrated that miR-144 targets Foxo1, thus reducing its expression and inhibiting its promotional effect on adiponectin, thereby alleviating the inhibitory effect of adiponectin on adipogenesis in an experimental model (102). A study performed by Komaya et al. reported that miR-33b showed high expression in the liver, and its expression was increased in response to cholesterol overload, using genetically modified mice, miR-33 knockout mice, and miR-33b Knock in mice; as a result, miR-33b showed increased atherogenic potential (103).
Basic and clinical evidence has shed light on the association between MAFLD and cardiovascular diseases (CVD) (104); in this context, increased plasma miR-1 was found to be associated with myocardial steatosis and it has been suggested to be a biomarker for diabetic cardiomyopathy (105).
A recent work carried out by Jiang et al. reported that miR-1 expression was increased in liver tissues and primary hepatocytes derived from a diet-induced obese mice, as well as, selective increase of miR-1 expression in EVs derived from steatotic hepatocytes (106). Several studies have shown that miR-26a is highly associated to cardiovascular diseases. Zhang et al. reported that miR-26a prevented blood pressure elevation and inhibited myocardial fibrosis using hypertensive animal models (107). Figure 3 schematizes crucial miRNAs involved in pathogenesis and development of MAFLD, considering key parameters such as obesity, type 2 diabetes mellitus, and metabolic alterations (hypertension, high level of triglycerides and cholesterol, and HOMA). Table 1 lists the miRNAs implicated in crucial key process in development of MAFLD and their potential target genes.
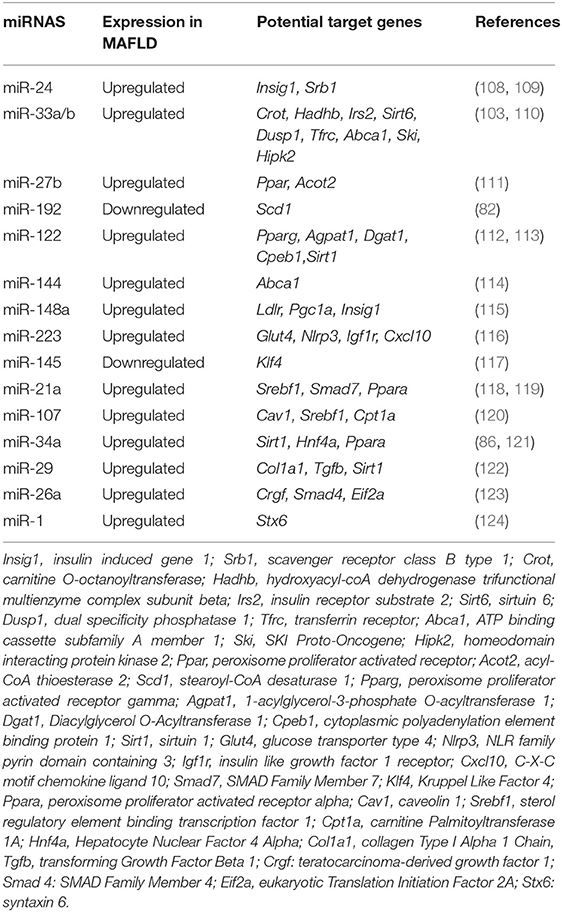
Table 1. miRNAs implicated in crucial key process in MAFLD and their potential target genes.
Clinical Trials Involving miRNAs for Hepatic Diseases
In the last decade, various miRNA-based therapeutics have been tested in different clinical trials. The first anti-miRNA drug for the treatment of hepatitis C is a locked nucleic acid (LNA) that inhibits miR-122, called Miravisen. Miravirsen inhibits miR-122 biogenesis and repressed HCV infection. miR-122 has a critical role in the life cycle of HCV due to the fact that miR-122 binds to two target sites (S1and S2) at the 5' end of the HCV genome, forming an oligomeric miR-122–HCV complex that protects the HCV genome from nucleolytic degradation or from host innate immune responses. Besides, at least three additional target sites in 3'-untranslated region of HCV genome have not been of functional importance (125). Currently, anti-miR-122 safety and effectiveness is being evaluated in a phase II clinical trial (126).
RG-101 is another novel anti-miR 122 for the treatment of hepatitis C virus. It is an N-acetylgalactosamine (GalNAc)-conjugated oligonucleotide. RG-101 repressed replication of HCV genotypes 1a and 1b in replicon systems. However, the precise mechanism of HCV suppression by RG-101 is not yet identified (127). Currently, RG-101 has reached the phase 1B clinical trial (128, 129).
Another miRNA-based therapeutic, a GalNAc conjugated anti-miR 103/107, called RG-125 (AZD4076) is an insulin sensitizer to treat patients with metabolic diseases such as type 2 diabetes and NASH. It has been reported that RG-125 normalized glucose tolerance and improved HOMA-IR in obese-diet induced mice compared with the control group. RG-125 treatment also reversed the extreme hyperglycemia that develops with age in db/db mice (130). Table 2 lists the clinical trials using miRNAs-based drugs registered in the ClinicalTrials.gov web site (August 2021) for liver diseases.
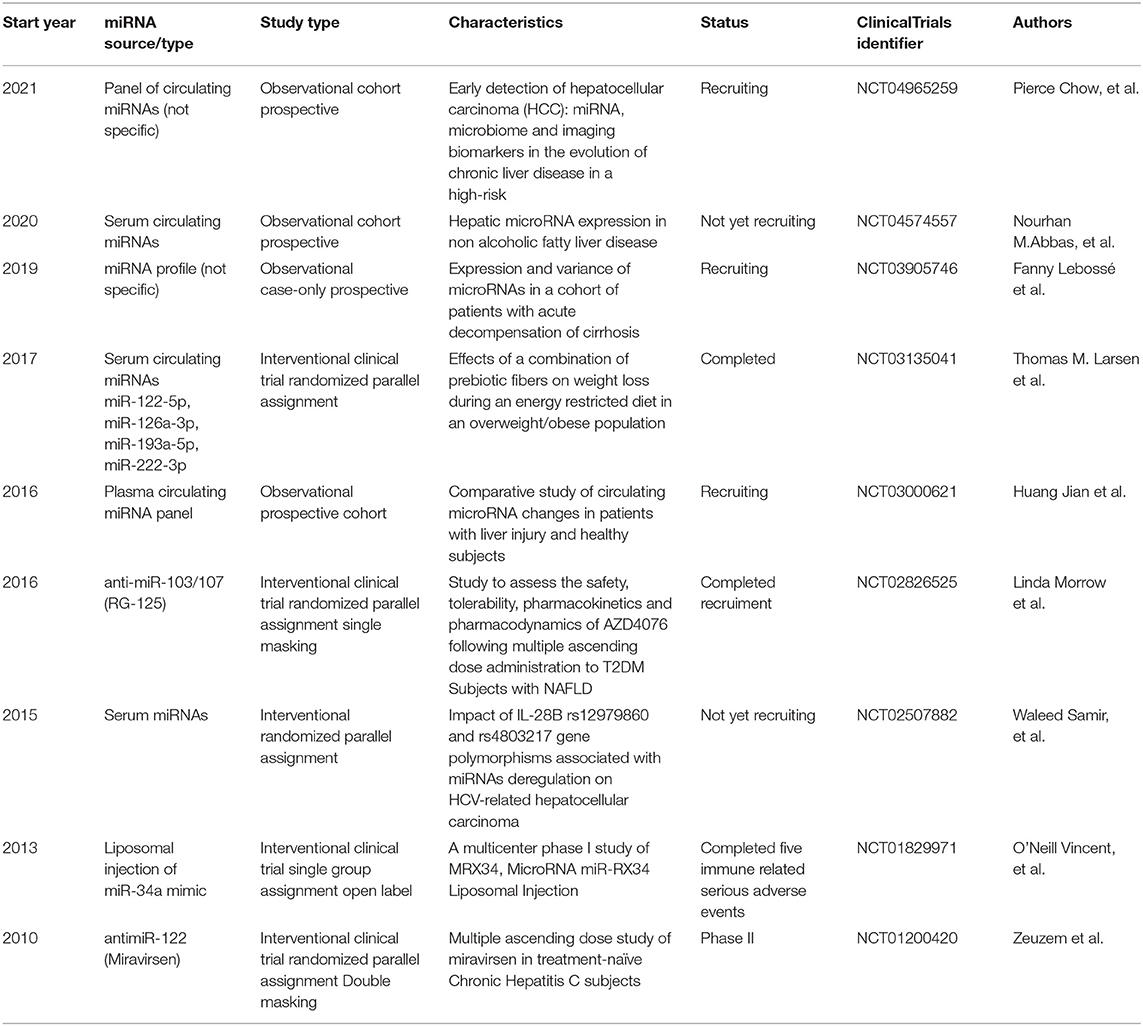
Table 2. Clinical trials using miRNAs for hepatic diseases.
Conclusion and Perspectives
Metabolic dysfunction associated fatty liver disease is currently a global health problem, epidemically associated to obesity, metabolic syndrome, and type II diabetes mellitus. MAFLD development and progression involves several genetic and environmental factors including epigenetics. Epigenetics includes an extensive amount of events such as methylation in CpGs, chemical modification of histones, and posttranscriptional gene regulation by the modification of mRNA stability through short noncoding RNAs such as miRNAs. In latest years, epigenetic modifications in DNA and histone have been studied as essential mechanisms that modify the development of liver diseases including MAFLD. Hence the dysregulation of epigenetic modifications has a critical role in MAFLD progression since it regulates the expression and activity of various genes implicated in lipid metabolism, insulin resistance, DNA repair, and inflammatory process that enhance the pathogenesis of MAFLD (10, 131). Currently, it has been demonstrated that miRNAs involved in lipid synthesis, fatty acid, and glucose catabolism and inflammation are dysregulated in MAFLD being useful as biomarkers (132). Moreover, it has been suggested that precise methylation patterns in DNA may be used as a predictor or diagnostic for MAFLD progression (133). Besides, the crucial paper of numerous micronutrients seems necessary to maintain DNA methylation homeostasis, as they act as cofactors of a variety of enzymes involved in DNA methylation, synthesis, and repair (134). To date, no therapeutic strategy is approved for the treatment of MAFLD, and lifestyle modifications, physical exercise, and weight loss account as the keystone therapeutics for patients with MAFLD. Certainly, a profound understanding of the molecular mechanisms related to gene expression, epigenetic modifications, and environment interactions ought to be a main concern for future studies. Overall, further basic research is necessary to improve mechanistic knowledge of the epigenetic processes and their interactions, their dysregulation in MAFLD, and the molecular and cellular response to epigenetic-based therapies. These studies together with clinical trials will enhance epigenetic-based personalized medicine. In conclusion, research in this area is in constant advance; however, there is still more to study to increase our understanding in MAFLD.
Author Contributions
JR-S and RE-G contributed to planning, bibliographic revision, writing of the manuscript, and figures design. RR-C contributed to the writing of the manuscript and literature review. JA-B contributed to figures design and writing and revising of the manuscript. AS-R was responsible for the manuscript planning and revising. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Fondo de Desarrollo Científico de Jalisco (FODECIJAL) Grant 7941-2019 awarded to JA-B.
Conflict of Interest
JA-B is a consultant for CellPharma Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. (2020) 73:202–9. doi: 10.1016/j.jhep.2020.03.039
2. Sayiner M, Koenig A, Henry L, Younossi ZM. Epidemiology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in the United States and the rest of the World. Clin Liver Dis. (2016) 20:205–14. doi: 10.1016/j.cld.2015.10.001
3. Conlon BA, Beasley JM, Aebersold K, Jhangiani SS, Wylie-Rosett J. Nutritional management of insulin resistance in nonalcoholic fatty liver disease (NAFLD). Nutrients. (2013) 5:4093–114. doi: 10.3390/nu5104093
4. Nilsson EE, Sadler-Riggleman I, Skinner MK. Environmentally induced epigenetic transgenerational inheritance of disease. Environ Epigenet. (2018) 4:dvy016. doi: 10.1093/eep/dvy016
5. Beekman M, Nederstigt C, Suchiman HE, Kremer D, van der Breggen R, Lakenberg N, et al. Genome-wide association study (GWAS)–identified disease risk alleles do not compromise human longevity. Proc Natl Acad Sci USA. (2010) 107:18046–9. doi: 10.1073/pnas.1003540107
6. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. (2003) 33:245–54. doi: 10.1038/ng1089
7. Angeloni A, Bogdanovic O. Enhancer DNA methylation: implications for gene regulation. Essays Biochem. (2019) 63:707–15. doi: 10.1042/EBC20190030
8. Gujar H, Weisenberger DJ, Liang G. The roles of human DNA methyltransferases and their isoforms in shaping the epigenome. Genes. (2019) 10:172. doi: 10.3390/genes10020172
9. Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. (2016) 30:733–50. doi: 10.1101/gad.276568.115
10. Hyun J, Jung Y, DNA. Methylation in nonalcoholic fatty liver disease. Int J Mol Sci. (2020) 21:8138. doi: 10.3390/ijms21218138
11. Ahrens M, Ammerpohl O, von Schönfels W, Kolarova J, Bens S, Itzel T, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. (2013) 18:296–302. doi: 10.1016/j.cmet.2013.07.004
12. Mwinyi J, Boström AE, Pisanu C, Murphy SK, Erhart W, Schafmayer C, et al. NAFLD is associated with methylation shifts with relevance for the expression of genes involved in lipoprotein particle composition. Biochim Biophys Acta Mol Cell Biol Lipids. (2017) 1862:314–23. doi: 10.1016/j.bbalip.2016.12.005
13. Khan RS, Bril F, Cusi K, Newsome PN. Modulation of insulin resistance in nonalcoholic fatty liver disease. Hepatology. (2019) 70:711–24. doi: 10.1002/hep.30429
14. Sookoian S, Rosselli MS, Gemma C, Burgueño AL, Fernández Gianotti T, Castaño GO, et al. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: impact of liver methylation of the peroxisome proliferator-activated receptor γ coactivator 1α promoter. Hepatology. (2010) 52:1992–2000. doi: 10.1002/hep.23927
15. Santos JL, Krause BJ, Cataldo LR, Vega J, Salas-Pérez F, Mennickent P, et al. PPARGC1A gene promoter methylation as a biomarker of insulin secretion and sensitivity in response to glucose challenges. Nutrients. (2020) 12:2790. doi: 10.3390/nu12092790
16. Oddy WH, Herbison CE, Jacoby P, Ambrosini GL, O'Sullivan TA, Ayonrinde OT, et al. The Western dietary pattern is prospectively associated with nonalcoholic fatty liver disease in adolescence. Am J Gastroenterol. (2013) 108:778–85. doi: 10.1038/ajg.2013.95
17. Yoon A, Tammen SA, Park S, Han SN, Choi SW. Genome-wide hepatic DNA methylation changes in high-fat diet-induced obese mice. Nutr Res Pract. (2017) 11:105–13. doi: 10.4162/nrp.2017.11.2.105
18. Li YY, Tang D, Du YL, Cao CY, Nie YQ, Cao J, et al. Fatty liver mediated by peroxisome proliferator-activated receptor-α DNA methylation can be reversed by a methylation inhibitor and curcumin. J Dig Dis. (2018) 19:421–30. doi: 10.1111/1751-2980.12610
19. Ohashi K, Munetsuna E, Yamada H, Ando Y, Yamazaki M, Taromaru N, et al. High fructose consumption induces DNA methylation at PPARα and CPT1A promoter regions in the rat liver. Biochem Biophys Res Commun. (2015) 468:185–9. doi: 10.1016/j.bbrc.2015.10.134
20. Yamazaki M, Munetsuna E, Yamada H, Ando Y, Mizuno G, Murase Y, et al. Fructose consumption induces hypomethylation of hepatic mitochondrial DNA in rats. Life Sci. (2016) 49:146–52. doi: 10.1016/j.lfs.2016.02.020
21. Hajri T, Zaiou M, Fungwe TV, Ouguerram K, Besong S. Epigenetic regulation of peroxisome proliferator-activated receptor gamma mediates high-fat diet-induced non-alcoholic fatty liver disease. Cells. (2021) 10:1355. doi: 10.3390/cells10061355
22. Wang Y, Chen L, Pandak WM, Heuman D, Hylemon PB, Ren S. High glucose induces lipid accumulation via 25-hydroxycholesterol DNA-CpG methylation. iScience. (2020) 23:101102. doi: 10.1016/j.isci.2020.101102
23. Yaskolka Meir A, Keller M, Müller L, Bernhart SH, Tsaban G, Zelicha H, et al. Effects of lifestyle interventions on epigenetic signatures of liver fat: central randomized controlled trial. Liver Int. (2021) 41:2101–11. doi: 10.1111/liv.14916
24. Anderson OS, Sant KE, Dolinoy DC. Nutrition and epigenetics: an interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J Nutr Biochem. (2012) 23:853–9. doi: 10.1016/j.jnutbio.2012.03.003
25. Da Silva RP, Eudy BJ, Deminice R. One-carbon metabolism in fatty liver disease and fibrosis: one-carbon to rule them all. J Nutr. (2020) 150:994–1003. doi: 10.1093/jn/nxaa032
26. Bakir MB, Salama MA, Refaat R, Ali MA, Khalifa EA, Kamel MA. Evaluating the therapeutic potential of one-carbon donors in nonalcoholic fatty liver disease. Eur J Pharmacol. (2019) 847:72–82. doi: 10.1016/j.ejphar.2019.01.039
27. Chen G, Broséus J, Hergalant S, Donnart A, Chevalier C, Bolaños-Jiménez F, et al. Identification of master genes involved in liver key functions through transcriptomics and epigenomics of methyl donor deficiency in rat: relevance to nonalcoholic liver disease. Mol Nutr Food Res. (2015) 59:293–302. doi: 10.1002/mnfr.201400483
28. Cordero P, Campion J, Milagro FI, Martinez JA. Transcriptomic and epigenetic changes in early liver steatosis associated to obesity: effect of dietary methyl donor supplementation. Mol Genet Metab. (2013) 110:388–95. doi: 10.1016/j.ymgme.2013.08.022
29. Lai Z, Chen J, Ding C, Wong K, Chen X, Pu L, et al. Association of hepatic global DNA methylation and serum one-carbon metabolites with histological severity in patients with NAFLD. Obesity. (2020) 28:197–205. doi: 10.1002/oby.22667
30. Nilsson E, Matte A, Perfilyev A, de Mello VD, Käkelä P, Pihlajamäki J, et al. Epigenetic alterations in human liver from subjects with type 2 diabetes in parallel with reduced folate levels. J Clin Endocrinol Metab. (2015) 100:E1491–1501. doi: 10.1210/jc.2015-3204
31. Ramos-Lopez O, Samblas M, Milagro FI, Zulet MA, Mansego ML, Riezu-Boj JI, et al. Association of low dietary folate intake with lower CAMKK2 gene methylation, adiposity, and insulin resistance in obese subjects. Nutr Res. (2018) 50:53–62. doi: 10.1016/j.nutres.2017.11.007
32. Pan X, Wu Y, Peng H, Cai X, Hu Z, Lin X, et al. Genome-wide DNA methylation profiling in nonalcoholic fatty liver reveals predictive aberrant methylation in PRKCE and SEC14L3 promoters. Dig Liver Dis. (2021) 7:S1590–8658. doi: 10.1016/j.dld.2021.05.013
33. Ma J, Nano J, Ding J, Zheng Y, Hennein R, Liu C, et al. A peripheral blood DNA methylation signature of hepatic fat reveals a potential causal pathway for nonalcoholic fatty liver disease. Diabetes. (2019) 68:1073–83. doi: 10.2337/DB18-1193
34. Wu J, Zhang R, Shen F, Yang R, Zhou D, Cao H, et al. Altered DNA methylation sites in peripheral blood leukocytes from patients with simple steatosis and nonalcoholic steatohepatitis (NASH). Med Sci Monit. (2018) 24:6946–67. doi: 10.12659/MSM.909747
35. Nano J, Ghanbari M, Wang W, de Vries PS, Dhana K, Muka T, et al. Epigenome-wide association study identifies methylation sites associated with liver enzymes and hepatic steatosis. Gastroenterology. (2017) 153:1096–106. doi: 10.1053/j.gastro.2017.06.003
36. Zeybel M, Hardy T, Robinson SM, Fox C, Anstee QM, Ness T, et al. Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin Epigenetics. (2015) 7:25. doi: 10.1186/s13148-015-0056-6
37. Hardy T, Zeybel M, Day CP, Dipper C, Masson S, McPherson S, et al. Plasma DNA methylation: a potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut. (2017) 1321:1328. doi: 10.1136/gutjnl-2016-311526
38. Wegermann K, Henao R, Diehl AM, Murphy SK, Abdelmalek MF, Moylan CA. Branched chain amino acid transaminase 1 (BCAT1) is overexpressed and hypomethylated in patients with non-alcoholic fatty liver disease who experience adverse clinical events: A pilot study. PLoS ONE. (2018) 13:e0204308. doi: 10.1371/journal.pone.0204308
39. Murphy SK, Yang H, Moylan CA, Pang H, Dellinger A, Abdelmalek MF, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology. (2013) 1076:87. doi: 10.1053/j.gastro.2013.07.047
40. Kitamoto T, Kitamoto A, Ogawa Y, Honda Y, Imajo K, Saito S, et al. Targeted-bisulfite sequence analysis of the methylation of CpG islands in genes encoding PNPLA3, SAMM50, and PARVB of patients with non-alcoholic fatty liver disease. J Hepatol. (2015) 494:502. doi: 10.1016/j.jhep.2015.02.049
41. Kouzarides T. Chromatin modifications and their function. Cell. (2007) 693:705. doi: 10.1016/j.cell.2007.02.005
42. Liu L, Jin G, Zhou X. Modeling the relationship of epigenetic modifications to transcription factor binding. Nucleic Acids Res. (2015) 3873:85. doi: 10.1093/nar/gkv255
43. Berger SL. The complex language of chromatin regulation during transcription. Nature. (2007) 407:12. doi: 10.1038/nature05915
44. Jenuwein T, Allis CD. Translating the histone code. Science. (2001) 1074:80. doi: 10.1126/science.1063127
45. Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. (2007) 981:9. doi: 10.1158/1541-7786.MCR-07-0324
46. Tammen SA, Friso S, Choi SW. Epigenetics: the link between nature and nurture. Mol Aspects Med. (2013) 753:64. doi: 10.1016/j.mam.2012.07.018
47. Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Annu Rev Biochem. (2011) 473:99. doi: 10.1146/annurev-biochem-061809-175347
48. Fazary AE, Ju YH, Abd-Rabboh HSM. How does chromatin package DNA within nucleus and regulate gene expression? Int J Biol Macromol. (2017) 862:881. doi: 10.1016/j.ijbiomac.2017.03.165
49. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. (2011) 381:95. doi: 10.1038/cr.2011.22
50. Yuan GC. Linking genome to epigenome. Wiley Interdiscip Rev Syst Biol Med. (2012) 297:309. doi: 10.1002/wsbm.1165
51. Lee J, Kim Y, Friso S, Choi SW. Epigenetics in non-alcoholic fatty liver disease. Mol Aspects Med. (2017) 78:88. doi: 10.1016/j.mam.2016.11.008
52. Lee J, Song JH, Chung MY, Lee JH, Nam TG, Park JH, et al. 3,4-dihydroxytoluene, a metabolite of rutin, suppresses the progression of nonalcoholic fatty liver disease in mice by inhibiting p300 histone acetyltransferase activity. Acta Pharmacol Sin. (2021) 1449:1460. doi: 10.1038/s41401-020-00571-7
53. Bricambert J, Miranda J, Benhamed F, Girard J, Postic C, Dentin R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J Clin Invest. (2010) 4316:31. doi: 10.1172/JCI41624
54. Guo Y, Zhang X, Zhao Z, Lu H, Ke B, Ye X, et al. NF- κ B/HDAC1/SREBP1c pathway mediates the inflammation signal in progression of hepatic steatosis. Acta Pharm Sin B. (2020) 825:836. doi: 10.1016/j.apsb.2020.02.005
55. Sun Z, Miller RA, Patel RT, Chen J, Dhir R, Wang H, et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med. (2012) 934:42. doi: 10.1038/nm.2744
56. Papazyan R, Sun Z, Kim YH, Titchenell PM, Hill DA, Lu W, et al. Physiological suppression of lipotoxic liver damage by complementary actions of HDAC3 and SCAP/SREBP. Cell Metab. (2016) 863:874. doi: 10.1016/j.cmet.2016.10.012
57. Gallego-Durán R, Romero-Gómez M. Epigenetic mechanisms in non-alcoholic fatty liver disease: An emerging field. World J Hepatol. (2015) 2497:502. doi: 10.4254/wjh.v7.i24.2497
58. Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, et al. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science. (2011) 1315:9. doi: 10.1126/science.1198125
59. Nassir F, Ibdah JA. Sirtuins and nonalcoholic fatty liver disease. World J Gastroenterol. (2016) 10084:10092. doi: 10.3748/wjg.v22.i46.10084
60. Sandoval-Rodriguez A, Monroy-Ramirez HC, Meza-Rios A, Garcia-Bañuelos J, Vera-Cruz J, Gutiérrez-Cuevas J, et al. Pirfenidone is an agonistic ligand for PPARα and improves NASH by activation of SIRT1/LKB1/pAMPK. Hepatol Commun. (2020) 434:449. doi: 10.1002/hep4.1474
61. Wang LF, Wang XN, Huang CC, Hu L, Xiao YF, Guan XH, et al. Inhibition of NAMPT aggravates high fat diet-induced hepatic steatosis in mice through regulating Sirt1/AMPKα/SREBP1 signaling pathway. Lipids Health Dis. (2017) 16:82. doi: 10.1186/s12944-017-0464-z
62. Barroso E, Rodríguez-Rodríguez R, Zarei M, Pizarro-Degado J, Planavila A, Palomer X, et al. SIRT3 deficiency exacerbates fatty liver by attenuating the HIF1α-LIPIN 1 pathway and increasing CD36 through Nrf2. Cell Commun Signal. (2020) 18:147. doi: 10.1186/s12964-020-00640-8
63. Li R, Xin T, Li D, Wang C, Zhu H, Zhou H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: the role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. (2018) 229:243. doi: 10.1016/j.redox.2018.07.011
64. Fan Z, Li L, Li M, Zhang X, Hao C, Yu L, et al. The histone methyltransferase Suv39h2 contributes to nonalcoholic steatohepatitis in mice. Hepatology. (2017) 1904:1919. doi: 10.1002/hep.29127
65. Kim DH, Kim J, Kwon JS, Sandhu J, Tontonoz P, Lee SK, et al. Critical roles of the histone methyltransferase MLL4/KMT2D in murine hepatic steatosis directed by ABL1 and PPARγ2. Cell Rep. (2016) 1671:1682. doi: 10.1016/j.celrep.2016.10.023
66. Kim JH, Jung DY, Nagappan A, Jung MH. Histone H3K9 demethylase JMJD2B induces hepatic steatosis through upregulation of PPARγ2. Sci Rep. (2018) 8:13734. doi: 10.1038/s41598-018-31953-x
67. Kim JH, Jung DY, Kim HR, Jung MH. Histone H3K9 demethylase JMJD2B plays a role in LXRα-dependent lipogenesis. Int J Mol Sci. (2020) 21:8313. doi: 10.3390/ijms21218313
68. Viscarra JA, Wang Y, Nguyen HP, Choi YG, Sul HS. Histone demethylase JMJD1C is phosphorylated by mTOR to activate de novo lipogenesis. Nat Commun. (2020) 11:796. doi: 10.1038/s41467-020-14617-1
69. Seok S, Kim YC, Byun S, Choi S, Xiao Z, Iwamori N, et al. Fasting-induced JMJD3 histone demethylase epigenetically activates mitochondrial fatty acid β-oxidation. J Clin Invest. (2018) 3144:3159. doi: 10.1172/JCI97736
70. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. (2004) 116:281–97. doi: 10.1016/s0092-8674(04)00045-5
71. Rajewsky N. microRNA target predictions in animals. Nat Genet. (2006) 38:S8–13. doi: 10.1038/ng1798
72. O'Brien J, Hayder H, Zayed Y, Peng C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol. (2018) 9:402. doi: 10.3389/fendo.2018.00402
73. Szabo G, Csak T. Role of microRNAs in NAFLD/NASH. Dig Dis Sci. (2016) 61:1314–24. doi: 10.1007/s10620-015-4002-4
74. Baffy G. MicroRNAs in nonalcoholic fatty liver disease. J Clin Med. (2015) 4:1977–88. doi: 10.3390/jcm4121953
75. Escutia-Gutiérrez R, Rodríguez-Sanabria JS, Monraz-Méndez CA, García-Bañuelos J, Santos-García A, Sandoval-Rodríguez A, et al. Pirfenidone modifies hepatic miRNAs expression in a model of MAFLD/NASH. Sci Rep. (2021) 11:11709. doi: 10.1038/s41598-021-91187-2
76. Landrier JF, Derghal A, Mounien L. MicroRNAs in obesity and related metabolic disorders. Cells. (2019) 8:859. doi: 10.3390/cells8080859
77. Horie T, Nishino T, Baba O, Kuwabara Y, Nakao T, Nishiga M, et al. MicroRNA-33 regulates sterol regulatory element-binding protein 1 expression in mice. Nat Commun. (2013) 4:2883. doi: 10.1038/ncomms3883
78. Vickers KC, Shoucri BM, Levin MG, Wu H, Pearson DS, Osei-Hwedieh D, et al. MicroRNA-27b is a regulatory hub in lipid metabolism and is altered in dyslipidemia. Hepatology. (2013) 57:533–42. doi: 10.1002/hep.25846
79. Yu J, Lv Y, Wang F, Kong X, Di W, Liu J, et al. MiR-27b-3p inhibition enhances browning of epididymal fat in high-fat diet induced obese mice. Front Endocrinol. (2019) 10:38. doi: 10.3389/fendo.2019.00038
80. Bandiera S, Pfeffer S, Baumert TF, Zeisel MB. miR-122 a key factor and therapeutic target in liver disease. J Hepatol. (2015) 62:448–57. doi: 10.1016/j.jhep.2014.10.004
81. Lewis AP, Jopling CL. Regulation and biological function of the liver-specific miR-122. Biochem Soc Trans. (2010) 38:1553–7. doi: 10.1042/BST0381553
82. Long JK Dai W, Zheng YW, Zhao SP. miR-122 promotes hepatic lipogenesis via nhibiting the LKB1/AMPK pathway by targeting Sirt1 in non-alcoholic fatty liver disease. Mol Med. (2019) 25:26. doi: 10.1186/s10020-019-0085-2
83. Wu GY, Rui C, Chen JQ, Sho E, Zhan SS, Yuan XW, et al. MicroRNA-122 inhibits lipid droplet formation and hepatic triglyceride accumulation via Yin Yang 1. Cell Physiol Biochem. (2017) 44:1651–64. doi: 10.1159/000485765
84. Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. (2007) 26:731–43. doi: 10.1016/j.molcel.2007.05.017
85. Yamada H, Suzuki K, Ichino N, Ando Y, Sawada A, Osakabe K, et al. Associations between circulating microRNAs (miR-21, miR-34a, miR-122 and miR-451) and non-alcoholic fatty liver. Clin Chim Acta. (2013) 424:99–103. doi: 10.1016/j.cca.2013.05.021
86. Ding J, Li M, Wan X, Jin X, Chen S, Yu C, et al. Effect of miR-34a in regulating steatosis by targeting PPARα expression in nonalcoholic fatty liver disease. Sci Rep. (2015) 5:13729. doi: 10.1038/srep13729
87. Pan Y, Hui X, Hoo RLC, Ye D, Chan CYC, Feng T, et al. Adipocyte-secreted exosomal microRNA-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. J Clin Invest. (2019) 129:834–49. doi: 10.1172/JCI123069
88. Marquart TJ, Allen RM, Ory DS, Baldán A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci USA. (2010) 107:12228–32. doi: 10.1073/pnas.1005191107
89. Dávalos A, Goedeke L, Smibert P, Ramírez CM, Warrier NP, Andreo U, et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci USA. (2011) 108:9232–7. doi: 10.1073/pnas.1102281108
90. Price NL, Zhang X, Fernández-Tussy P, Singh AK, Burnap SA, Rotllan N, et al. Loss of hepatic miR-33 improves metabolic homeostasis and liver function without altering body weight or atherosclerosis. Proc Natl Acad Sci USA. (2021) 118:e2006478118. doi: 10.1073/pnas.2006478118
91. Vickers KC, Landstreet SR, Levin MG, Shoucri BM, Toth CL, Taylor RC, et al. MicroRNA-223 coordinates cholesterol homeostasis. Proc Natl Acad Sci USA. (2014) 111:14518–23. doi: 10.1073/pnas.1215767111
92. He Y, Hwang S, Cai Y, Kim SJ, Xu M, Yang D, et al. MicroRNA-223 ameliorates nonalcoholic steatohepatitis and cancer by targeting multiple inflammatory and oncogenic genes in hepatocytes. Hepatology. (2019) 70:1150–67. doi: 10.1002/hep.30645
93. Zhang F, Ma D, Zhao W, Wang D, Liu T, Liu Y, et al. Obesity-induced overexpression of miR-802 impairs insulin transcription and secretion. Nat Commun. (2020) 11:1822. doi: 10.1038/s41467-020-15529-w
94. Ortega FJ, Moreno M, Mercader JM, Moreno-Navarrete JM, Fuentes-Batllevell N, Sabater M, et al. Inflammation triggers specific microRNA profiles in human adipocytes and macrophages and in their supernatants. Clin Epigenetics. (2015) 7:49. doi: 10.1186/s13148-015-0083-3
95. Meerson A, Traurig M, Ossowski V, Fleming JM, Mullins M, Baier LJ. Human adipose microRNA-221 is upregulated in obesity and affects fat metabolism downstream of leptin and TNF-α. Diabetologia. (2013) 56:1971–9. doi: 10.1007/s00125-013-2950-9
96. Peng J, Zhou Y, Deng Z, Zhang H, Wu Y, Song T, et al. miR-221 negatively regulates inflammation and insulin sensitivity in white adipose tissue by repression of sirtuin-1 (SIRT1). J Cell Biochem. (2018) 119:6418–28. doi: 10.1002/jcb.26589
97. Rovira-Llopis S, Díaz-Rúa R, Grau-Del Valle C, Iannantuoni F, Abad-Jimenez Z, Bosch-Sierra N, et al. Characterization of Differentially Expressed Circulating miRNAs in Metabolically Healthy versus Unhealthy Obesity. Biomedicines. (2021) 9:321. doi: 10.3390/biomedicines9030321
98. Qi R, Wang J, Wang Q, Qiu X, Yang F, Liu Z, et al. MicroRNA-425 controls lipogenesis and lipolysis in adipocytes. Biochim Biophys Acta Mol Cell Biol Lipids. (2019) 1864:744–55. doi: 10.1016/j.bbalip.2019.02.007
99. Ahonen MA, Haridas PAN, Mysore R, Wabitsch M, Fischer-Posovszky P, Olkkonen VM. miR-107 inhibits CDK6 expression, differentiation, and lipid storage in human adipocytes. Mol Cell Endocrinol. (2019) 479:110–6. doi: 10.1016/j.mce.2018.09.007
100. Okamoto K, Koda M, Okamoto T, Onoyama T, Miyoshi K, Kishina M, et al. Serum miR-379 expression is related to the development and progression of hypercholesterolemia in non-alcoholic fatty liver disease. PLoS ONE. (2020) 15:e0219412. doi: 10.1371/journal.pone.0219412
101. Vonhögen IGC, Mohseni Z, Winkens B, Xiao K, Thum T, Calore M, et al. Circulating miR-216a as a biomarker of metabolic alterations and obesity in women. Noncoding RNA Res. (2020) 5:144–52. doi: 10.1016/j.ncrna.2020.08.001
102. Lin W, Tang Y, Zhao Y, Zhao J, Zhang L, Wei W, et al. MiR-144-3p targets FoxO1 to reduce its regulation of adiponectin and promote adipogenesis. Front Genet. (2020) 11:603144. doi: 10.3389/fgene.2020.603144
103. Koyama S, Horie T, Nishino T, Baba O, Sowa N, Miyasaka Y, et al. Identification of differential roles of microRNA-33a and−33b during atherosclerosis progression with genetically modified mice. J Am Heart Assoc. (2019) 8:e012609. doi: 10.1161/JAHA.119.012609
104. Ismaiel A, Dumitrascu DL. Genetic predisposition in metabolic-dysfunction-associated fatty liver disease and cardiovascular outcomes—systematic review. Eur J Clin Invest. (2020) 50:e13331. doi: 10.1111/eci.13331
105. De Gonzalo-Calvo D, van der Meer RW, Rijzewijk LJ, Smit JW, Revuelta-Lopez E, Nasarre L, et al. Serum microRNA-1 and microRNA-133a levels reflect myocardial steatosis in uncomplicated type 2 diabetes. Sci Rep. (2017) 7:47. doi: 10.1038/s41598-017-00070-6
106. Jiang F, Chen Q, Wang W, Ling Y, Yan Y, Xia P. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J Hepatol. (2020) 72:156–66. doi: 10.1016/j.jhep.2019.09.014
107. Zhang W, Wang Q, Feng Y, Chen X, Yang L, Xu M, et al. MicroRNA-26a protects the heart against hypertension-induced myocardial fibrosis. J Am Heart Assoc. (2020) 9:e017970. doi: 10.1161/JAHA.120.017970
108. Wang M, Li L, Liu R, Song Y, Zhang X, Niu W, et al. Obesity-induced overexpression of miRNA-24 regulates cholesterol uptake and lipid metabolism by targeting SR-B1. Gene. (2018) 668:196–203. doi: 10.1016/j.gene.2018.05.072
109. Xu D, Wang Z, Xia Y, Shao F, Xia W, Wei Y. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature. (2020) 580:530–35. doi: 10.1038/s41586-020-2183-2
110. Murata Y, Yamashiro T, Kessoku T, Jahan I, Usuda H, Tanaka T, et al. Up-regulated microRNA-27b promotes adipocyte differentiation via induction of acyl-CoA thioesterase 2 expression. BioMed Res Int. (2019). 2019:2916243. doi: 10.1155/2019/2916243
111. Liu XL, Cao HX, Wang BC, Xin FZ, Zhang RN, Zhou D, et al. miR-192-5p regulates lipid synthesis in non-alcoholic fatty liver disease through SCD-1. World J Gastroenterol. (2017) 23:8140–51. doi: 10.3748/wjg.v23.i46.8140
112. Baranova A, Maltseva D, Tonevitsky A. Adipose may actively delay progression of NAFLD by releasing tumor-suppressing, anti-fibrotic miR-122 into circulation. Obes Rev. (2019) 20:108–18. doi: 10.1111/obr.12765
113. Jin X, Gao J, Zheng R, Yu M, Ren Y, Yan T, et al. Antagonizing circRNA_002581-miR-122-CPEB1 axis alleviates NASH through restoring PTEN-AMPK-mTOR pathway regulated autophagy. Cell Death Dis. (2020) 11:123. doi: 10.1038/s41419-020-2293-7
114. de Aguiar Vallim TQ, Tarling EJ, Kim T, Civelek M, Baldán Á, Esau C, et al. MicroRNA-144 regulates hepatic ATP binding cassette transporter A1 and plasma high-density lipoprotein after activation of the nuclear receptor farnesoid X receptor. Circ Res. (2013) 112:1602–12. doi: 10.1161/CIRCRESAHA.112.300648
115. Goedeke L, Rotllan N, Canfrán-Duque A, Aranda JF, Ramírez CM, Araldi E, et al. MicroRNA-148a regulates LDL receptor and ABCA1 expression to control circulating lipoprotein levels. Nat Med. (2015) 21:1280–9. doi: 10.1038/nm.3949
116. Hou X, Yin S, Ren R, Liu S, Yong L, Liu Y, et al. Myeloid cell-specific IL-6 signaling promotes miR-223-enriched exosome production to attenuate NAFLD-associated fibrosis. Hepatology. (2020) 74:116–132. doi: 10.1002/hep.31658
117. Men R, Wen M, Zhao M, Dan X, Yang Z, Wu W, et al. MircoRNA-145 promotes activation of hepatic stellate cells via targeting krüppel-like factor 4. Sci Rep. (2017) 7:40468. doi: 10.1038/srep40468
118. Dattaroy D, Pourhoseini S, Das S, Alhasson F, Seth RK, Nagarkatti M, et al. Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. (2015) 308:G298–312. doi: 10.1152/ajpgi.00346.2014
119. Loyer X, Paradis V, Hénique C, Vion A-C, Colnot N, Guerin CL, et al. Liver microRNA-21 is overexpressed in non-alcoholic steatohepatitis and contributes to the disease in experimental models by inhibiting PPARα expression. Gut. (2016) 65:1882–94. doi: 10.1136/gutnl-2014-308883
120. Gracia A, Fernández-Quintela A, Miranda J, Eseberri I, González M, Portillo MP. Are miRNA-103, miRNA-107 and miRNA-122 involved in the prevention of liver steatosis induced by resveratrol? Nutrients. (2017) 9:360. doi: 10.3390/nu9040360
121. Xu Y, Zalzala M, Xu J, Li Y, Yin L, Zhang Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat Commun. (2015) 6:7466. doi: 10.1038/ncomms8466
122. Hung YH, Kanke M, Kurtz CL, Cubitt RL, Bunaciu RP, Zhou L, et al. MiR-29 regulates de novo lipogenesis in the liver and circulating triglyceride levels in a Sirt1-dependent manner. Front Physiol. (2019) 10:1367. doi: 10.3389/fphys.2019.01367/full
123. Xu H, Tian Y, Tang D, Zou S, Liu G, Song J, et al. An endoplasmic reticulum stress-microrna-26a feedback circuit in NAFLD. Hepatology. (2021) 73:1327–45. doi: 10.1002/hep.31428
124. Jiang F, Chen Q, Wang W, Ling Y, Yan Y, Xia P. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J Hepatol. (2020) 72:156–66. doi: 10.1016/j.jhep.2019.09.014.
125. Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. (2013) 368:1685–94. doi: 10.1056/NEJMoa1209026
126. Gebert LF, Rebhan MA, Crivelli SE, Denzler R, Stoffel M, Hall J. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. (2014) 42:609–21. doi: 10.1093/nar/gkt852
127. Lima JF, Cerqueira L, Figueiredo C, Oliveira C, Azevedo NF. Anti-miRNA oligonucleotides: a comprehensive guide for design. RNA Biol. RNA Biol. (2018) 15:338–937352. doi: 10.1080/15476286.2018.1445959
128. Schult P, Roth H, Adams RL, Mas C, Imbert L, Orlik C, et al. microRNA-122 amplifies hepatitis C virus translation by shaping the structure of the internal ribosomal entry site. Nat Commun. (2018) 9:2613. doi: 10.1038/s41467-018-05053-3
129. Van der Ree MH, de Vree JM, Stelma F, Willemse S, van der Valk M, Rietdijk S, et al. Safety, tolerability, and antiviral effect of RG-101 in patients with chronic hepatitis C: a phase 1B, double-blind, randomised controlled trial. Lancet. (2017) 389:709–17. doi: 10.1016/S0140-6736(16)31715-9
130. Monga I, Kumar M. Computational resources for prediction and analysis of functional miRNA and their targetome. Methods Mol Biol. (2019) 1912:215–50. doi: 10.1007/978-1-4939-8982-9_9
131. Fu S, Yu M, Tan Y, Liu D. Role of histone deacetylase on nonalcoholic fatty liver disease. Expert Rev Gastroenterol Hepatol. (2021) 15:353–61. doi: 10.1080/17474124.2021.1854089
132. López-Pastor AR, Infante-Menéndez J, Escribano Ó, Gómez-Hernández A. miRNA dysregulation in the development of non-alcoholic fatty liver disease and the related disorders type 2 diabetes mellitus and cardiovascular disease. Front Med (Lausanne). (2020) 7:527059. doi: 10.3389/fmed.2020.527059
133. Buzova D, Maugeri A, Liguori A, Napodano C, Lo Re O, Oben J, et al. Circulating histone signature of human lean metabolic-associated fatty liver disease (MAFLD). Clin Epigenetics. (2020) 12:126. doi: 10.1186/s13148-020-00917-2
Keywords: MAFLD, NASH, DNA methylation, histone modification, miRNAs
Citation: Rodríguez-Sanabria JS, Escutia-Gutiérrez R, Rosas-Campos R, Armendáriz-Borunda JS and Sandoval-Rodríguez A (2022) An Update in Epigenetics in Metabolic-Associated Fatty Liver Disease. Front. Med. 8:770504. doi: 10.3389/fmed.2021.770504
Received: 03 September 2021; Accepted: 02 December 2021;
Published: 11 January 2022.
Edited by:
Alessandro Granito, University of Bologna, ItalyReviewed by:
Rohitash Jamwal, University of Rhode Island, United StatesJosep Jimenez-Chillaron, Sant Joan de Déu Research Institute (IRSJD), Spain
Copyright © 2022 Rodríguez-Sanabria, Escutia-Gutiérrez, Rosas-Campos, Armendáriz-Borunda and Sandoval-Rodríguez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan S. Armendáriz-Borunda, YXJtZGJvQGdtYWlsLmNvbQ==; Ana Sandoval-Rodríguez, c29sZWRhZC5zYW5kb3ZhbEBhY2FkZW1pY29zLnVkZy5teA==
†These authors share first authorship