 Teruhiko Yoshida
Teruhiko Yoshida Khun Zaw Latt
Khun Zaw Latt Jurgen Heymann
Jurgen Heymann Jeffrey B. Kopp
Jeffrey B. Kopp
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med. , 26 October 2021
Sec. Nephrology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.762901
This article is part of the Research Topic Molecular Mechanisms of Proteinuria, Volume II View all 16 articles
African-Americans have a three-fold higher rate of chronic kidney disease compared to European-Americans. Much of this excess risk is attributed to genetic variants in APOL1, encoding apolipoprotein L1, that are present only in individuals with sub-Saharan ancestry. Although 10 years have passed since the discovery of APOL1 renal risk variants, the mechanisms by which APOL1 risk allele gene products damage glomerular cells remain incompletely understood. Many mechanisms have been reported in cell culture models, but few have been demonstrated to be active in transgenic models. In this narrative review, we will review existing APOL1 transgenic models, from flies to fish to mice; discuss findings and limitations from studies; and consider future research directions.
APOL1 genetic variants are an important cause of kidney disease, affecting individuals who have sub-Saharan African ancestry (1). Apolipoprotein L1 is a component of the innate immune system. It is produced primarily by the liver but also by brain, kidney, and other tissues. APOL1 is a component of HDL particles, which serve as delivery platforms for multiple proteins related to defense against infectious disease. When African trypanosomes circulating in blood ingest HDL particles, APOL1 is released from the HDL particles and trafficked to the lysosomes where it forms ion channels, thereby killing the parasite and thus preventing African sleeping sickness.
Trypanosomes developed a defense protein, serum resistance antigen (SRA), that binds APOL1 and prevents trypanocide (2). In response to the evolutionary pressure, human APOL1 genetic variants, APOL1-G1 and APOL1-G2, arose that interfere with SRA binding. The APOL1 variants thereby restore trypanosomal killing but do so at the cost of increased risk for chronic kidney disease. This parallels the relationship between malaria and sickle cell hemoglobin, whereby the latter protects to some extent against malaria, particularly severe malaria, and consequently has been evolutionarily selected for in Mediterranean and sub-Saharan Africa regions (3). However, this protection comes at the cost of a disabling disorder, sickle cell anemia. While the molecular mechanism leading to sickle cell anemia is well understood, this has not been the case for the involvement of APOL1 in glomerular injury.
Histologic manifestations of APOL1 kidney disease including focal segmental glomerulosclerosis, collapsing glomerulopathy, arterionephrosclerosis (also termed hypertension-associated kidney disease), and advanced forms of lupus nephritis (4–6). Other manifestations of APOL1 kidney disease include shorter renal allograft survival among recipients with two APOL1 risk alleles and a rare but concerning gradual loss of kidney function among living kidney donors (7, 8). These risks are generally confined to those who carry two APOL1 renal risk alleles (high risk genotype). The one setting where there is an increased risk associated with carriage of one APOL1 risk allele is HIV-associated collapsing glomerulopathy in South Africa (9).
Current therapies for chronic kidney disease are less effective at preventing progressive loss of kidney function in individuals with APOL1 high risk genotype, compared to other forms of glomerular disease, even when there may be a reduction in proteinuria in a particular patient. APOL1 may not be an essential protein, as one individual who is genetically null for APOL1 appears phenotypically normal (10). However, it is only one case report and we obviously need further confirmation to understand function and necessity of APOL1. Further, APOL1 is unique to humans, gorillas, and baboons (11). Interestingly, the gene is absent from the genome of chimpanzees and bonobos, our nearest neighbors in evolutionary terms; this remains a puzzle. Taken together, these observations suggest that APOL1-targeted therapy for APOL1-associated kidney disease may be well-tolerated, except in regions with endemic African trypanosomiasis. One such approach is to suppress APOL1 protein levels; this is now investigated in clinical trials (12, 13). Other therapeutic approaches are also needed, and experimental animal models will be of great utility in testing the efficacy of such therapies, as well as improving understanding of APOL1 disease pathogenesis.
Criteria that are frequently applied when animal models are considered in the study of human disease include similarity of the model to humans with respect to physiology, anatomy and genetics. But others such as the level at which a model system can be manipulated, controlled, and examined and the scalability so to achieve statistical power are equally important.
Mouse models are the most frequently used animal models and have been widely explored to investigate APOL1 kidney disease, even though mice do not have an ortholog to the human APOL1 gene. The mouse apolipoprotein L family includes 12 genes and one pseudogene (Apol6, Apol7a, Apol7b, Apol7c, Apol7e, Apol8, Apol9a, Apol9b, Apol10a, Apol10b, Apol11a, Apol11b, and pseudogene Apol10c) clustered on chromosome 15 (14).
A primary aim of the APOL1 transgenic mouse studies has been to characterize the molecular mechanism of kidney damage induced by APOL1 renal risk alleles (G1 and G2) and to examine effects of human APOL1 renal risk allele and non-risk allele (G0) in vivo. Several approaches have been taken to reproduce APOL1-associated human disease in mouse models, including various expression systems, gene promoters, gene variants, mouse background strain, and interventions that impose kidney stress or damage. Here we review published work in this area and consider what is missing or incomplete and what possible future approaches might be taken.
With regards to generating or selecting a transgenic mouse model, it is important to consider aspects of transgene expression such as tissue specificity of expression and regulation of expression. There are many available transgene promoters and enhancers. The transgene may be inserted randomly or may be inserted into a selected genetic locus. Random insertion is simpler, but tissue expression may be influenced by proximity to strong enhancer elements; this potential problem can be avoided by adding locus control regions to the transgene, but at the cost of a larger and more complex transgene (15). Further, the genetic background of the mouse strain must be considered, as outcomes may vary with genetic background.
In 2016, Bruggeman et al. reported the first transgenic APOL1 mouse model, using the mouse Nphs1 promotor (16). Both APOL1-G0 and -G2 mice showed decreased podocyte density and preeclampsia. Using the mouse Nphs1 promoter, which is highly specific within kidney for podocytes, allowed targeting of APOL1-transgene expression to the cell where APOL1 is expressed in human kidneys and from where it is thought to drive APOL1-mediated kidney disease. One limitation was that even though these investigators showed that APOL1 expression affected podocyte function, this effect was not specific to the renal risk alleles but also to common variant APOL1-G0.
Beckerman et al. took a different approach, using a Tet-on activatable overexpression system to generate APOL1 transgenic mice (17). They showed that mice with Nphs1 promotor-driven APOL1-G1 and -G2 overexpression developed global and segmental glomerulosclerosis, compared with -G0 mice, which had no kidney phenotype. In individual mice, albuminuria levels correlated with APOL1 expression levels. Mechanistically, reduced autophagy resulted in pyroptosis i.e., inflammatory cell death. Similarly, Kumar and others used the Tet-On3G inducible system to investigate APOL1 functions and showed that APOL1 risk alleles de-stabilized the junction complex, which contributes to cell-cell contact and cell-matrix adhesion (18). One limitation to all these transgenic mice is the difficulty in relating transgene expression levels to those seen in humans.
In another approach, Okamoto et al. studied human APOL1 gene locus transgenic mice, where the transgene is a 47 kb human cDNA that contains sequence from chromosome 22, included within a bacterial artificial chromosome (BAC). This BAC contains the exons and intron of APOL1, as well as portions of APOL2 and MYH9. This approach facilitates studies of gene regulation using physiologic stimuli, such as interferon or agents that promote interferon expression (19). The APOL1-G2 variant mouse showed significantly higher albuminuria compared to APOL1-G0 mice, following exposure to a combination of interferon-γ (to stimulate APOL1 gene transcription), basic fibroblast growth factor, and puromycin aminonucleoside (to induce podocyte injury). Importantly, the study showed that APOL1-variant mRNA activated protein kinase R and that this activation contributed to the observed podocyte injury.
To replicate physiological APOL1 gene regulation and expression levels as seen in humans, Aghajan et al. used 32 Mb fosmid DNA vectors in the generation of transgenic mice (20). The authors used interferon-γ injections to induce transient albuminuria, which was most prominent in APOL1-G1 mice. They demonstrated that pre-treatment with an anti-sense oligonucleotide targeting APOL1 could block the induction of albuminuria. This proof-of-concept study indicated that anti-sense oligonucleotides might have a therapeutic role in APOL1 nephropathy. However, many steps remain toward a clinical application of this technology.
McCarthy et al. recently reported the results of APOL1 BAC transgenic mice that received via hydrodynamic tail vein injection an IFN-γ-expressing pCpGfree plasmid that lacks CpG motifs to achieve sustained IFN-γ levels in the animals. They showed robust induction of proteinuria and glomerulosclerosis in G1/G1 and G2/G2 mice but not in G0/G0 mice. They also showed that the heterozygous mice (G0/G1 or G0/G2) had greater proteinuria response than hemizygous mice (G1/- or G2/-), suggesting that APOL1-G0 does not rescue -G1 or -G2 allele toxicity. Further, mice with a multicopy G2 transgene (G2multi/G2multi) showed the greatest proteinuria response with worst prognosis, supporting the recessive nature of APOL1-nephropathy and the notion that disease is a function of the expression level of APOL1 risk variant (21).
Ryu et al. used the BAC/APOL1 mouse model described above and showed that reduced cholesterol efflux and concomitant cholesterol accumulation may contribute to APOL1 nephropathy (22). In 2018, Kumar et al. generated a TetOn3G-APOL1 mouse model and identified a dual feedback loop in glomerular parietal epithelial cells, in which APOL1 suppresses miR-193a expression and miR-193a suppresses APOL1 expression (23).
Some models have involved dual-transgenic mice. Bruggeman et al. (24) studied the well-characterized Tg26 model of HIV-associated nephropathy, in which the presence of the six regulatory and accessory genes of HIV-1, under the control of viral long terminal repeats (LTR), develop FSGS and/or collapsing glomerulopathy (25). They reported that among Nphs1.APOL1 × Tg26 dual transgenic mice, APOL1-G0 × Tg26 mice showed less podocyte loss in compared with APOL1-G2 × Tg26 or Tg26 mice alone (24). These data suggest the intriguing idea that APOL-G0 protein might have some trophic or protective effect on podocytes, perhaps one that only manifests in certain injury settings.
Ge et al. studied a triple transgenic model, BAC/APOL1 × podocin-rtTA × TRE/NFATc1nuc mouse model (26). These mice manifest elevated levels of triglycerides and cholesterol in kidney, as well as glomerulosclerosis. The authors suggested that APOL1 risk variant expression increases the susceptibility to lipid-mediated podocyte injury, leading to mitochondrial dysfunction.
Another injury pathway was identified by Wakashin et al., who reported on a CAG-APOL1-B3 mouse model expressing the APOL1-B3 protein isoform under control of a CMV early enhancer/chicken β-actin promoter. The APOL1-B3 isoform, in contrast to the better characterized APOL1-A isoform, lacks a signal sequence for targeting to the secretory pathway and this results in cytosolic expression (27). These mice manifested podocyte injury and elevated IL-1β production for the G2 but not for the G0 variant. Further, APOL1-B3 interacted with NACHT, LRR, and PYD domains-containing protein 12 (NLRP12), a key regulator of toll-like receptor signaling. Through these pathways, APOL1-B3 and its risk variants seem to enhance inflammatory signaling in podocytes.
APOL1 is expressed most abundantly in liver, and this is the source of most plasma APOL1 (28). Targeted expression of APOL1 risk variants in murine livers by hydrodynamic gene delivery induced liver injury, demonstrating the cytotoxicity of the variants (11). Human data from kidney transplant studies suggest that only kidney-expressed, and not APOL1 expressed in other tissues has the potential to injure kidneys (29–31).
As shown above, mouse models have contributed in many ways to the understanding of molecular mechanisms of APOL1 kidney injury. Mouse kidney physiology shares many similarities to that of humans and diverse transgenic approaches are available, even though mice lack APOL1orthologs. Future research using existing and new mouse models will help to identify which mechanisms found in vitro are likely to be the relevant in vivo and relevant to human disease and may contribute to novel therapeutic approaches.
The zebrafish (Danio rerio) is the second most employed animal model in APOL1 research. Zebrafish express a protein with considerable homology to human APOL1 and this, together with its genetic tractability, has made it a useful model to study APOL1 function (32). Anderson et al. reported that translational suppression or CRISPR/Cas9 genome editing of apol1 in zebrafish embryos results in podocyte loss and glomerular filtration defects (32). They also showed that complementation with the APOL1-G0 allele rescued the phenotype, but complementation with APOL-G1 or -G2 did not. Thus, the gene encoding APOL1 appears essential to glomerular function in fish, whereas this may not be the case in humans. A human subject who developed an unusual trypanosomal infection was found to be lacking in the APOL1 gene but nevertheless had normal kidney function (10). However, the uniqueness of this intriguing finding warrants a cautious interpretation of the role of APOL1 or the lack of APOL1.
Kotb et al. confirmed that apol1 knockdown causes podocyte damage in zebrafish (33). Olabisi et al. investigated zebrafish with G0, G1, or G2 APOL1 variants, expressed using the Gal4-UAS system in podocytes or endothelial cells, using podocin and Flk (a receptor for vascular endothelial growth factor) promoters, respectively (34). Transgenic expression of APOL1 G1 and G2, compared to the G0 allele, was associated with histologic abnormalities in zebrafish glomeruli but renal function remained normal. Bundy et al. investigated molecular pathways in zebrafish podocytes, using pathway analysis of differentially expressed transcripts, and showed enrichment for transcripts characterized by autophagy associated-terms, implicating autophagy pathways in APOL1 G2-associated kidney dysfunction (35).
The zebrafish model recapitulates certain aspects of molecular mechanisms acting downstream of human APOL1 risk alleles, but the functional changes and the effects seen by knocking down APOL1 seem contradictory to observations in human subjects. Consequently, the specificities of zebrafish-APOL1 interactions have to be kept in mind and carefully further explored to advance our understanding of this animal model.
Drosophila melanogaster, the common fruit fly, is a model system that offers ready access to genetic manipulation, enabling studies of protein function and protein-protein interaction. In 2017, two reports presented findings on effects of APOL1 expression on nephrocytes in Drosophila (36, 37). These models exploit structural and functional similarities of Drosophila pericardial nephrocytes to those of human podocytes and proximal tubular cells. The accessibility of Drosophila nephrocytes facilitates high resolution in vivo analysis of renal cells (36).
Fu et al. reported that ubiquitous expression of human APOL1-G0 or -G1 variants in Drosophila induced lethal phenotypes, with -G1 being more toxic than -G0. Expressing the APOL1 transgene in nephrocytes impaired the acidification of organelles. Kruzel-Davila et al. reported that ubiquitous expression of the human APOL1-G1 and -G2 variants caused near-complete lethality in the flies, with no effect observed for -G0 (37). These effects are more severe than those seen in humans, perhaps due to higher expression levels in the flies or the particular pattern of tissue expression, but this study does lend support to a gain-of-dysfunction model for APOL1 variant cell toxicity. These authors also observed differential toxicity of the APOL1 risk alleles compared with non-risk alleles including disruption of endolysosomal processes.
The fly models have proven useful in showing a lethal phenotype and endolysosomal disruption by APOL1 risk alleles, and to identify interacting partners of APOL1. These models to address specific mechanisms that may be shared by nephrocytes and podocytes, but these two cell types have important differences that limit definitive conclusions.
Several factors should be considered when designing or selecting an animal model system that can most effectively and efficiently address the selected research questions.
Promoter selection is of great importance, as it determines the specificity, kinetics, and dose of transgene expression. These parameters determine the types of questions that can be addressed. In the kidney, APOL1-mediated cytotoxicity is particularly prominent in podocytes. Consequently, many transgenic mouse studies have selected podocyte-specific promoters to interrogate podocyte-specific function of APOL1. However, APOL1 is not only expressed by podocytes but also expressed by renal endothelial cells (38). This finding warrants the application of endothelial cell-specific animal models to specifically investigate the effects of APOL1 on endothelial cells. It is true that APOL1 expression is still a controversial topic as Ma et al. reported the presence of APOL1 protein in kidney tubular cells suggesting a function of APOL1 there (39). Further, several studies have explored systemic expression of APOL1 to interrogate unbiased, systemic function of APOL1. Considering APOL1's ubiquitous expression in many organs plus its presence on HDL particles circulating in the bloodstream, systemic models offer avenues to uncover the remaining unknowns in APOL1 functions.
Establishing a robust transgene phenotype is critical in generating a persuasive animal model. In human APOL1 nephropathy, APOL1 variant risk status appears to be insufficient for disease onset and a second factor, generally increased APOL1 expression, appears to be required. Interventions to increase transgene expression are also required in several APOL1 transgenic animal models to increase gene expression. IFN-γ activates the APOL1 promoter (19, 20) and doxycycline activates the Tet-On promoter (17, 18). Puromycin aminonucleoside and/or basic fibroblast growth factor have been used to induce podocyte injury (19). Identifying effective interventions that result in overt phenotypes remain challenging.
Mouse background strain can have a substantial effect on susceptibility to disease or response to a disease-promoting intervention. A common background strains for transgenic mice is FVB/N, due to the relative ease of injecting the large pronuclei in the zygote. This strain has also proven very useful in kidney studies, as other strains including strains 129S1 or B6 turned out to be more resistant to kidney injury.
In Table 1, we summarize the models that have been reported. Although a range of mechanisms have been documented for in vitro models that investigate APOL1 function, only limited number of mechanisms have been identified and confirmed in animal models. We also summarized major animal models in Figure 1. Animal models come with a variety of limitations. Designing and generating an animal model for complex human genetic diseases is challenging especially if specific physiology or environmental factors are contributing elements. The APOL gene family arose in primates and diversified rapidly, by gene duplication, on the one hand, and by gene loss and pseudogenization on the other hand. This gene family is absent from all the commonly used research animal species, including mice. This raised the potential concern that other molecules that co-evolved in primates to interact with APOL1 and perhaps regulate or alter its activity would be missing in mice and other laboratory animals. Although modern molecular biology methods allow a gene or genomic fragments containing gene and extended flanking regions to be introduced in these model systems, it remains a challenge to decipher how the transgene expression affects the transgenic animal and how these effects relate to pathological processes and especially their kinetics observed in human subjects. The duty remains to carefully explore how far a model can inform on the human setting.
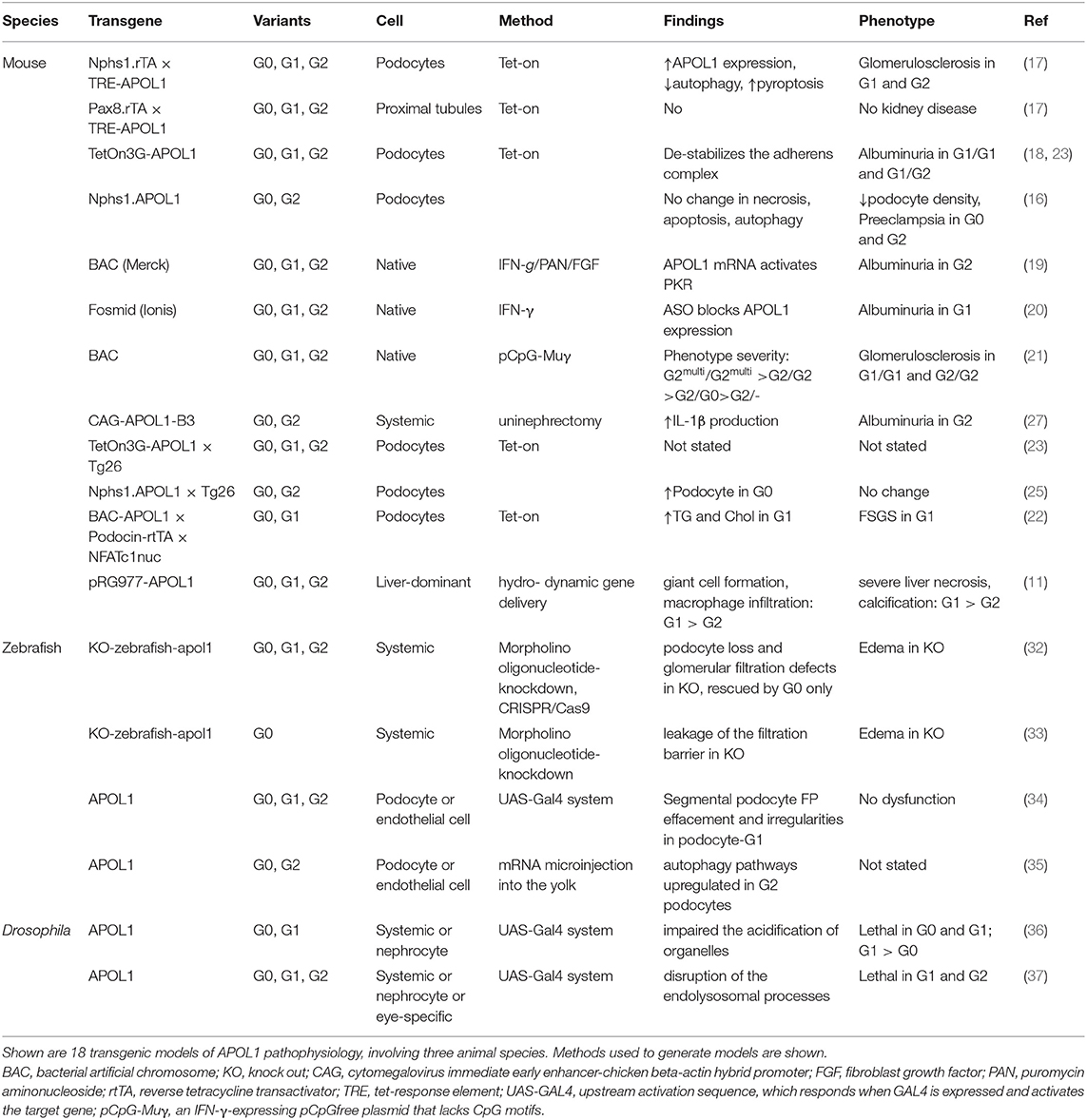
Table 1. Summary of APOL1 animal models.
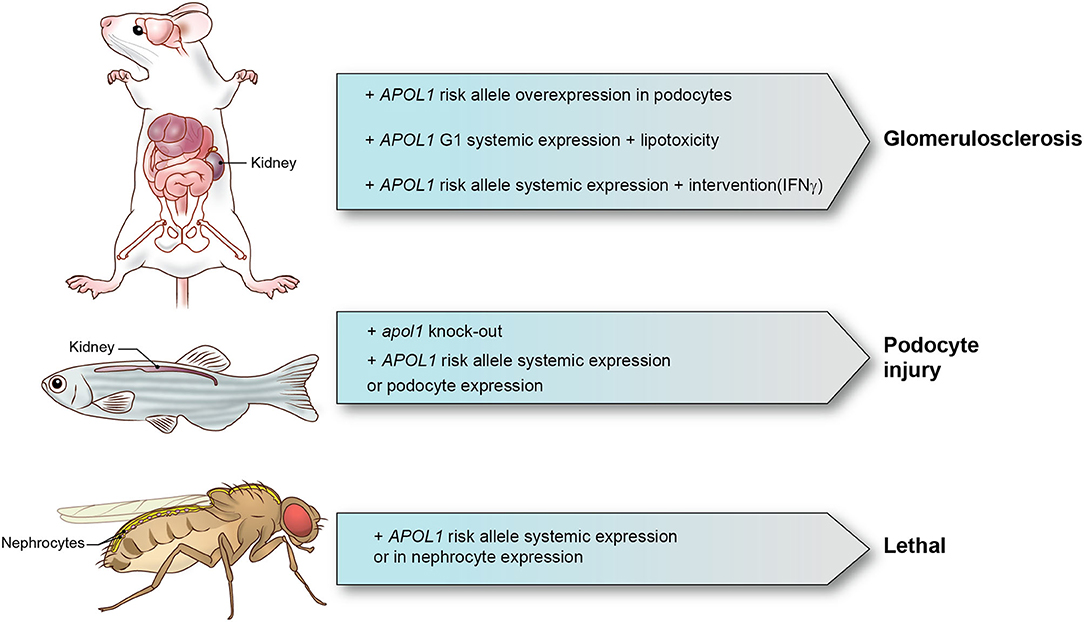
Figure 1. APOL1 animal models and phenotypes. Representative APOL1 animal models with interventions and phenotypes are shown.
In patients, APOL1 kidney disease appears to follow a two-hit model, whereby a genetic predisposition (presence of two APOL1 kidney risk alleles) is coupled with factors that increase APOL1 gene expression (interferon being the most well-documented). Given an increase in APOL1 gene expression, it remains unclear to what extent peak magnitude of expression vs. area under the curve of expression is more important in inducing glomerular injury. Also, there may be factors other than interferon that increase gene expression or alternatively synergize with what might otherwise be sub-pathogenic APOL1 expression levels to induce kidney injury. Future animal studies will be required to understand the relationship between the kinetics of gene expression and glomerular injury.
Animal studies offer the opportunity to examine what other factors, beyond APOL1 variant expression level, might alter the timing and degree of glomerular injury. These might include age, sex, body weight and various co-morbidities. Single cell methods that provide unprecedented insights into otherwise not recognizable changes in per-cell based transcriptomes may also shed light into molecular mechanisms by APOL1, together with novel animal models. Further APOL1 animal studies will be warranted to better understand to which factors have to be paid more attention in the population carrying APOL1 risk alleles.
TY reviewed the literature and drafted the manuscript. KL and JK provided comments. TY, JH, and JK edited the manuscript. All authors read and approved the final draft.
This work was supported by the NDDK Intramural Research Program, NIH, Bethesda, MD.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Genovese G, Tonna SJ, Knob AU, Appel GB, Katz A, Bernhardy AJ, et al. A risk allele for focal segmental glomerulosclerosis in African Americans is located within a region containing APOL1 and MYH9. Kidney Int. (2010) 78:698–704. doi: 10.1038/ki.2010.251
2. Zoll S, Lane-Serff H, Mehmood S, Schneider J, Robinson CV, Carrington M, et al. The structure of serum resistance-associated protein and its implications for human African trypanosomiasis. Nat Microbiol. (2018) 3:295–301. doi: 10.1038/s41564-017-0085-3
3. Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, et al. Sickle cell disease. Nat Rev Dis Primers. (2018) 4:18010. doi: 10.1038/nrdp.2018.10
4. Larsen CP, Beggs ML, Saeed M, Ambruzs JM, Cossey LN, Messias NC, et al. Histopathologic findings associated with APOL1 risk variants in chronic kidney disease. Mod Pathol. (2015) 28:95–102. doi: 10.1038/modpathol.2014.92
5. Kopp JB, Winkler CA, Zhao X, Radeva MK, Gassman JJ, D'Agati VD, et al. Clinical features and histology of apolipoprotein L1-Associated nephropathy in the FSGS clinical trial. J Am Soc Nephrol. (2015) 26:1443–8. doi: 10.1681/ASN.2013111242
6. Blazer AD, Clancy RM. ApoL1 and the immune response of patients with systemic lupus erythematosus. Curr Rheumatol Rep. (2017) 19:13. doi: 10.1007/s11926-017-0637-9
7. Doshi MD, Ortigosa-Goggins M, Garg AX, Li L, Poggio ED, Winkler CA, et al. APOL1 genotype and renal function of black living donors. J Am Soc Nephrol. (2018) 29:1309–16. doi: 10.1681/ASN.2017060658
8. Freedman BI, Julian BA, Pastan SO, Israni AK, Schladt D, Gautreaux MD, et al. Apolipoprotein L1 gene variants in deceased organ donors are associated with renal allograft failure. Am J Transplant. (2015) 15:1615–22. doi: 10.1111/ajt.13223
9. Schwimmer JA, Markowitz GS, Valeri A, Appel GB. Collapsing glomerulopathy. Semin Nephrol. (2003) 23:209–18. doi: 10.1053/snep.2003.50019
10. Johnstone DB, Shegokar V, Nihalani D, Rathore YS, Mallik L, Ashish, et al. APOL1 null alleles from a rural village in India do not correlate with glomerulosclerosis. PLoS ONE. (2012) 7:e51546. doi: 10.1371/journal.pone.0051546
11. Thomson R, Genovese G, Canon C, Kovacsics D, Higgins MK, Carrington M, et al. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci U S A. (2014) 111:E2130–9. doi: 10.1073/pnas.1400699111
12. A First-in-human Study to Assess the Safety Tolerability Pharmacokinetics and Pharmacodynamics of AZD2373 After Single Dose Administration in Healthy Male Subjects of African Ancestry. Available onlline at: https://ClinicalTrials.gov/show/NCT04269031. (acessed August 1, 2021).
13. Phase 2a Study of VX-147 in Adults With APOL1-mediated Focal Segmental Glomerulosclerosis. Available onlline at: https://ClinicalTrials.gov/show/NCT04340362. (acessed August 1, 2021).
14. Kreit M, Vertommen D, Gillet L, Michiels T. The interferon-inducible mouse apolipoprotein L9 and prohibitins cooperate to restrict theiler's virus replication. PLoS ONE. (2015) 10:e0133190. doi: 10.1371/journal.pone.0133190
15. Cattini PA, Jin Y, Jarmasz JS, Noorjahan N, Bock ME. Obesity and regulation of human placental lactogen production in pregnancy. J Neuroendocrinol. (2020) 32:e12859. doi: 10.1111/jne.12859
16. Bruggeman LA, Wu Z, Luo L, Madhavan SM, Konieczkowski M, Drawz PE, et al. APOL1-G0 or APOL1-G2 transgenic models develop preeclampsia but not kidney disease. J Am Soc Nephrol. (2016) 27:3600–10. doi: 10.1681/ASN.2015111220
17. Beckerman P, Bi-Karchin J, Park AS, Qiu C, Dummer PD, Soomro I, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med. (2017) 23:429–38. doi: 10.1038/nm.4287
18. Kumar V, Paliwal N, Ayasolla K, Vashistha H, Jha A, Chandel N, et al. Disruption of APOL1-miR193a axis induces disorganization of podocyte actin cytoskeleton. Sci Rep. (2019) 9:3582. doi: 10.1038/s41598-019-39376-y
19. Okamoto K, Rausch JW, Wakashin H, Fu Y, Chung JY, Dummer PD, et al. APOL1 risk allele RNA contributes to renal toxicity by activating protein kinase R. Commun Biol. (2018) 1:188. doi: 10.1038/s42003-018-0188-2
20. Aghajan M, Booten SL, Althage M, Hart CE, Ericsson A, Maxvall I, et al. Antisense oligonucleotide treatment ameliorates IFN-gamma-induced proteinuria in APOL1-transgenic mice. JCI Insight. (2019) 4:e126124. doi: 10.1172/jci.insight.126124
21. McCarthy GM, Blasio A, Donovan OG, Schaller LB, Bock-Hughes A, Magraner JM, et al. Recessive, gain-of-function toxicity in an APOL1 BAC transgenic mouse model mirrors human APOL1 kidney disease. Dis Model Mech. (2021) 14:dmm048952. doi: 10.1242/dmm.048952
22. Ryu JH, Ge M, Merscher S, Rosenberg AZ, Desante M, Roshanravan H, et al. APOL1 renal risk variants promote cholesterol accumulation in tissues and cultured macrophages from APOL1 transgenic mice. PLoS ONE. (2019) 14:e0211559. doi: 10.1371/journal.pone.0211559
23. Kumar V, Vashistha H, Lan X, Chandel N, Ayasolla K, Shoshtari SSM, et al. Role of apolipoprotein L1 in human parietal epithelial cell transition. Am J Pathol. (2018) 188:2508–28. doi: 10.1016/j.ajpath.2018.07.025
24. Bruggeman LA, Wu Z, Luo L, Madhavan S, Drawz PE, Thomas DB, et al. APOL1-G0 protects podocytes in a mouse model of HIV-associated nephropathy. PLoS ONE. (2019) 14:e0224408. doi: 10.1371/journal.pone.0224408
25. Kopp JB, Klotman ME, Adler SH, Bruggeman LA, Dickie P, Marinos NJ, et al. Progressive glomerulosclerosis and enhanced renal accumulation of basement membrane components in mice transgenic for human immunodeficiency virus type 1 genes. Proc Natl Acad Sci U S A. (1992) 89:1577–81. doi: 10.1073/pnas.89.5.1577
26. Ge M, Molina J, Ducasa GM, Mallela SK, Santos JV, Mitrofanova A, et al. APOL1 risk variants affect podocyte lipid homeostasis and energy production in focal segmental glomerulosclerosis. Hum Mol Genet. (2021) 30:182–97. doi: 10.1093/hmg/ddab022
27. Wakashin H, Heymann J, Roshanravan H, Daneshpajouhnejad P, Rosenberg A, Shin MK, et al. APOL1 renal risk variants exacerbate podocyte injury by increasing inflammatory stress. BMC Nephrol. (2020) 21:371. doi: 10.1186/s12882-020-01995-3
28. Shukha K, Mueller JL, Chung RT, Curry MP, Friedman DJ, Pollak MR, et al. Most ApoL1 is secreted by the liver. J Am Soc Nephrol. (2017) 28:1079–83. doi: 10.1681/ASN.2016040441
29. Bruggeman LA, O'Toole JF, Ross MD, Madhavan SM, Smurzynski M, Wu K, et al. Plasma apolipoprotein L1 levels do not correlate with CKD. J Am Soc Nephrol. (2014) 25:634–44. doi: 10.1681/ASN.2013070700
30. Chang JH, Husain SA, Santoriello D, Stokes MB, Miles CD, Foster KW, et al. Donor's APOL1 risk genotype and “second hits” associated with de novo collapsing glomerulopathy in deceased donor kidney transplant recipients: a report of 5 cases. Am J Kidney Dis. (2019) 73:134–9. doi: 10.1053/j.ajkd.2018.05.008
31. Santoriello D, Husain SA, De Serres SA, Bomback AS, Crew RJ, Vasilescu ER, et al. Donor APOL1 high-risk genotypes are associated with increased risk and inferior prognosis of de novo collapsing glomerulopathy in renal allografts. Kidney Int. (2018) 94:1189–98. doi: 10.1016/j.kint.2018.06.024
32. Anderson BR, Howell DN, Soldano K, Garrett ME, Katsanis N, Telen MJ, et al. In vivo modeling implicates APOL1 in nephropathy: evidence for dominant negative effects and epistasis under anemic stress. PLoS Genet. (2015) 11:e1005349. doi: 10.1371/journal.pgen.1005349
33. Kotb AM, Simon O, Blumenthal A, Vogelgesang S, Dombrowski F, Amann K, et al. Knockdown of ApoL1 in zebrafish larvae affects the glomerular filtration barrier and the expression of nephrin. PLoS ONE. (2016) 11:e0153768. doi: 10.1371/journal.pone.0153768
34. Olabisi O, Al-Romaih K, Henderson J, Tomar R, Drummond I, MacRae C, et al. From man to fish: what can ebrafish tell us about ApoL1 nephropathy? Clin Nephrol. (2016) 86:114–8. doi: 10.5414/CNP86S116
35. Bundy JL, Anderson BR, Francescatto L, Garrett ME, Soldano KL, Telen MJ, et al. RNA sequencing of isolated cell populations expressing human APOL1 G2 risk variant reveals molecular correlates of sickle cell nephropathy in zebrafish podocytes. PLoS ONE. (2019) 14:e0217042. doi: 10.1371/journal.pone.0217042
36. Fu Y, Zhu JY, Richman A, Zhang Y, Xie X, Das JR, et al. APOL1-G1 in nephrocytes induces hypertrophy and accelerates cell death. J Am Soc Nephrol. (2017) 28:1106–16. doi: 10.1681/ASN.2016050550
37. Kruzel-Davila E, Shemer R, Ofir A, Bavli-Kertselli I, Darlyuk-Saadon I, Oren-Giladi P, et al. APOL1-Mediated cell injury involves disruption of conserved trafficking processes. J Am Soc Nephrol. (2017) 28:1117–30. doi: 10.1681/ASN.2016050546
38. Scales SJ, Gupta N, De Maziere AM, Posthuma G, Chiu CP, Pierce AA, et al. Apolipoprotein L1-Specific antibodies detect endogenous APOL1 inside the endoplasmic reticulum and on the plasma membrane of podocytes. J Am Soc Nephrol. (2020) 31:2044–4. doi: 10.1681/ASN.2019080829
Keywords: APOL1, animal model, CKD—chronic kidney disease, glomerular diseases, podocyte
Citation: Yoshida T, Latt KZ, Heymann J and Kopp JB (2021) Lessons From APOL1 Animal Models. Front. Med. 8:762901. doi: 10.3389/fmed.2021.762901
Received: 23 August 2021; Accepted: 30 September 2021;
Published: 26 October 2021.
Edited by:
Ilse Sofia Daehn, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Chris Larsen, Arkana Laboratories, United StatesCopyright © 2021 Yoshida, Latt, Heymann and Kopp. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teruhiko Yoshida, dGVydWhpa28ueW9zaGlkYUBuaWguZ292
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.