 Paola Borgia1†
Paola Borgia1† Riccardo Papa1,2†
Riccardo Papa1,2† Matteo D'Alessandro1,3
Matteo D'Alessandro1,3 Roberta Caorsi2Giorgio Piaggio3
Roberta Caorsi2Giorgio Piaggio3 Andrea Angeletti3,4
Andrea Angeletti3,4 Isabella Ceccherini5
Isabella Ceccherini5 Gian Marco Ghiggeri3,4
Gian Marco Ghiggeri3,4 Marco Gattorno2*
Marco Gattorno2*- 1Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics and Maternal and Child Science (DINOGMI), Università degli Studi di Genova, Genoa, Italy
- 2Autoinflammatory Diseases and Immunodeficiencies Center, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 3Division of Nephrology, Dialysis and Transplantation, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 4Laboratory of Molecular Nephrology, IRCCS Istituto Giannina Gaslini, Genova, Italy
- 5Laboratorio di Genetica e Genomica delle Malattie Rare, IRCCS Istituto Giannina Gaslini, Genoa, Italy
Pyogenic arthritis, pyoderma gangrenosum and acne (PAPA) syndrome, and the proline-serine-threonine phosphatase-interacting protein 1 (PSTPIP1)-associated myeloid-related proteinemia inflammatory (PAMI) syndrome are two distinct clinical conditions caused by heterozygous mutations of the PSTPIP1 gene. While skin and joint involvements are shared by both conditions, PAMI is characterized by hepatosplenomegaly, pancytopenia, and growth failure. Kidney involvement is exceptional in PSTPIP1-mediated disorders. The two missense PSTPIP1 variants associated with PAMI syndrome are p.E250K and p.E257K. Long-term treatment with interleukin (IL)-1 inhibitors is effective to control inflammatory manifestations and is usually well-tolerated. We report a case of a patient carrying the PSTPIP1 p.E250K mutation who developed a late-onset kidney involvement despite a long treatment with canakinumab and anakinra. Kidney biopsy showed focal segmental glomerulosclerosis that was treated with tacrolimus (0.1 mg/kg/day in two doses). A literature revision with the aim to assess the proportion and type of kidney involvement in PAMI syndrome revealed that heterogeneous nephropathies may be part of the clinical spectrum. Our study supports the importance of a periodic diagnostic work-up, including kidney laboratory tests and kidney biopsy, in individuals affected with PAMI syndrome. Kidney and liver functions may be impaired regardless of anti-cytokines treatments and additional therapy approaches (i.e., multi-drugs, hematopoietic stem cell transplantation) should be carefully considered.
Introduction
Proline-serine-threonine phosphatase-interacting protein 1 (PSTPIP1) is a cytoskeleton adaptor protein mainly expressed in hematopoietic cells. Mutations of the PSTPIP1 gene were associated with a large group of inflammatory disorders collected under the term PSTPIP1-associated inflammatory diseases (PAID) (1, 2). The clinical spectrum of PAID ranges from a prevalent skin and joint involvement in case of pyogenic arthritis, pyoderma gangrenosum, and acne (pyoderma gangrenosum, acne, pyogenic arthritis, PAPA) syndrome, to more complex phenotypes involving several organs in case of the PSTPIP1-associated myeloid-related proteinemia inflammatory (PAMI) syndrome (3, 4).
The PAMI syndrome is due to the p.E250K and p.E257K missense mutations of the PSTPIP1 gene. Clinical manifestations may include cytopenia, recurrent infections, vasculitis-associated skin ulcers, hepatomegaly, splenomegaly, lymphadenopathy, and growth failure. High serum calprotectin and zinc concentration is the laboratory hallmark of the disease.
Steroid administration represents the cornerstone of treatment, resulting in effectiveness in about 55% of cases (5). In remaining cases, also the steroid-sparing drugs, such as the anti-cytokines biologics, may result not be completely effective in preventing the long-term complications. Hematopoietic stem cell transplantation has been recently proposed for the management of severe hematological manifestations (i.e., cytopenia) in complicated forms not responsive to conventional anti-cytokine treatments (6).
Here, we report a 22-year-old male previously included in a PAMI cohort (1), receiving chronic treatment with interleukin (IL)-1 inhibitor and presenting kidney involvement resulting in chronic kidney disease. We also present a literature review of this rare complication.
Case Presentation
A 4-year-old Caucasian boy was referred to our unit for recurrent episodes of asymmetrical polyarthritis at the large joints of the lower limbs (Table 1). Family history was negative. Since he was 6 months, physical examination revealed mild hepatomegaly and splenomegaly. Laboratory tests showed microcytic anemia (hemoglobin 11 g/dL, mean corpuscular volume 70 fL), neutropenia (780 neutrophils/mm3), and the bone marrow analysis revealed dyserythropoiesis. Joint aspirations showed sterile but purulent fluid. Partial control of the symptoms was achieved with multiple intra-articular steroid injections.

Table 1. Characteristics of the patient.
Since 8 years of age, severe pyoderma gangrenosum started at the periorbital and periungual area [refer to Figure 1B of reference (1)]. Local treatments with antibiotics and steroids were ineffective, and surgical exportation was needed. Due to the very early onset of atypical inflammatory skin and joint involvement and the evidence of high serum levels of zinc and calprotectin (113 μmol/L, normal <50, and 14 μg/mL, normal <0.5, respectively), the Sanger sequencing analysis of the PSTPIP1 gene was performed, revealing the heterozygous c.748G>A, p.E250K mutation. Therefore, the diagnosis of PAMI syndrome was confirmed.
Since 10 years of age, only the inflammatory skin manifestations persisted: biweekly infusion of human monoclonal anti-tumor necrosis factor (TNF)α antibody adalimumab resulted ineffectively. Since 13 years of age, severe nodulocystic acne has also developed in the face. Oral colchicine was administered but early stopped after 2 weeks due to abdominal pain. IL-1 receptor 1 (IL1R1) antagonist (anakinra) was then attempted, resulting in a prompt clinical improvement of skin lesions. Due to poor patient's compliance, after 2 years, anakinra was replaced with a monthly infusion of the fully human monoclonal anti-IL-1beta antibody canakinumab (7).
At 15 years of age, the weight and height were 48 kg and 147 cm, respectively, revealing a growth failure (10th and <3rd percentile based on Tanner's growth charts). The low serum IGF-1 levels and the abnormal provocative test with arginine confirmed a growth hormone deficiency: the recombinant growth hormone therapy was proposed but refused by the family. After that, the patient performed a blood and urine test every 6 months. At the age of 18, the patient was transferred to a local adult rheumatology unit. Due to administrative issues, the supply of IL-1 monoclonal antibody by the local health system was not regular, leading to transient flares of the skin manifestations, treated with an oral steroid.
At the age of 19 years, the patient was seen for a long-term follow-up visit in our center. Laboratory tests showed new onset of proteinuria in the nephrotic range (4.5 g/daily), microhematuria, and hyperuricemia (9 mg/dL), with normal kidney function (creatininemia 0.8 mg/dl, estimated glomerular filtration rate 129 ml/min/1.73 m2). Mild hypoalbuminemia (3,340 mg/dl) and slightly elevated acute phase reactants (C-reactive protein 1.30 mg/dl, serum amyloid A 42.6 mg/L) were also reported. The patient was referred to a local adult nephrologist for competence. Needle kidney biopsy revealed focal segmental glomerulosclerosis (FSGS) without evidence of amyloid deposition. Canakinumab was discontinued, and prednisone (30 mg daily) was started. After 5 months, proteinuria was still in the nephrotic range (4.3 g/daily) with worsening of renal function (creatininemia 1.5 mg/dL, eGFR 63 ml/min/1.73 m2). Tacrolimus (maintaining serum range of 4–6 ng/dl) was started, and canakinumab restarted due to worsening of the skin inflammatory manifestations (Figure 1A), resulting in a prompt clinical improvement of skin lesions (Figure 1B). One year later, due to the persistence of proteinuria in the nephrotic range (3.2 g/daily) with stable renal function (creatininemia 1.7 mg/dL, eGFR 56 ml/min/1.73 m2), a second kidney biopsy was performed in our center, showing interstitial fibrosis (Figures 1C–F). Thus, tacrolimus was discontinued, prednisone gradually tapered, and he is still receiving canakinumab at the dose of 150 mg monthly.
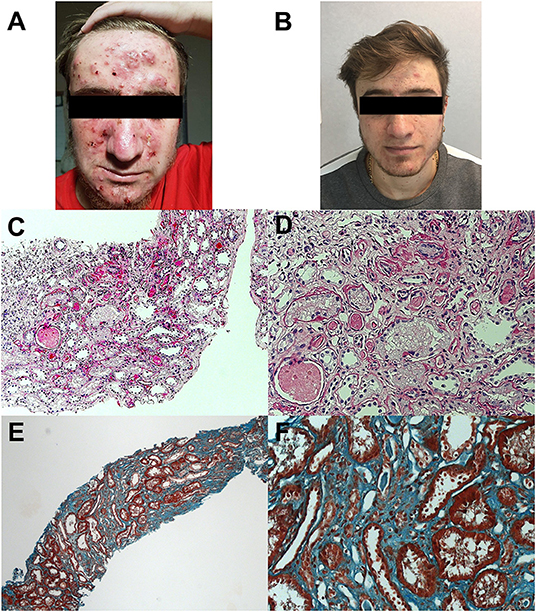
Figure 1. Skin manifestations and kidney histology of the patient. Photos show cystic acne before (A) and after (B) rechallenge of canakinumab. Histology analysis shows interstitial fibrosis with interstitial infiltration by mononuclear cells and tubular atrophy, classical and with aspects of thyroidization; tubular lumens are dilated and with flaking material; epithelium of some tubules appears vacuolated; segmental hyalinosis of arteriole is also present (C,D; periodic acid–Schiff staining; original magnification × 50 and × 200, respectively). Diffuse interstitial fibrosis (E,F; trichrome staining; original magnification at × 50 and × 200, respectively).
The acute phase reactants were almost completely normalized (Table 1). Of note, mild anemia and neutropenia persist together with hepatosplenomegaly.
Discussion
In the context of the clinical spectrum of PSTPIP1-related disorders, renal involvement is reported only in PAMI syndrome. All the English articles found in the PubMed database by querying MEDLINE with the keywords “PSTPIP1” or “PAPA” or “PAMI” were revised. We found 179 articles, among them, only seven articles reported patients with p.E250K or p.E257K mutation, of which four did not report a kidney involvement. In total, only five subjects with the PAMI-associated PSTPIP1 mutations displayed a renal involvement with at least three different phenotypes: glomerular vasculitis in three subjects, tubule-interstitial infiltration, probably related to a systemic inflammatory state, in one subject, and glomerular calprotectin deposition in the remaining one. No evidence of amyloid or immune complex deposition has been reported. Thus, kidney involvement in PAMI seems to present with heterogeneous manifestations and is linked with specific variants of the PSTPIP1 gene. Of note, 2/6 (33%) patients in Table 2 developed end-stage liver cirrhosis, leading them to liver transplant.

Table 2. Reported cases of kidney involvement in PSTPIP1-associated inflammatory diseases.
The triad of vasculitis, cytopenia, and lymphoproliferation described in PAMI can be related to specific ligands of the PSTPIP1 protein. In fact, PSTPIP1 is not expressed primitively in the kidney, whereas it is highly expressed in hematopoietic cells. The protein is able to bind with immune-related proteins, like the cytosolic protein tyrosine phosphatase (PTP-PEST), the Wiskott–Aldrich syndrome protein (WASP), the c-Abl kinase (ABL), the CD2, and the Fas ligand, probably with the aim to counteract the cytotoxic cell functioning (10) (Figure 2).
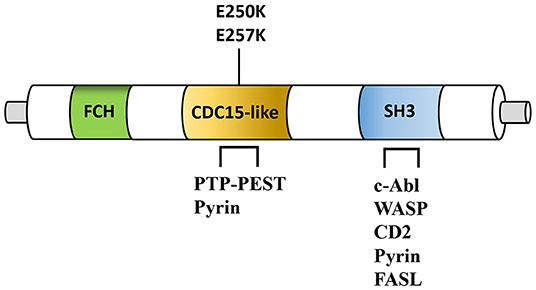
Figure 2. Simplified structure of PSTPIP1 protein. Fes/CIP4 homology domain (FCH), CDC15-like adaptor protein (CDC15-like), SRC Homology 3 Domain (SH3) are shown. Note the position of the PAMI-associated mutations (E250K and E257K). PSTPIP1 interacting proteins include the protein-tyrosine phosphatase (PTP-PEST), pyrin, the Wiskott–Aldrich syndrome protein (WASP), the c-Abl kinase, CD2, and the Fas ligand (FASL). PSTPIP1, proline-serine-threonine phosphatase-interacting protein 1.
Mutations associated with PAID decrease the binding activity of the protein to PTP-PEST (11), suggesting that gain and loss of function should be considered. Furthermore, despite T cells do not show a prevalent involvement in PAID lesions, the mild lymphoproliferation described in some patients with PAMI may be related to a T-cell activation defect caused by abnormal CD2 binding activity or absence of the Fas ligand, may be due to its sequestration into lysosomes. Moreover, WASP and ABL are actin-related proteins and the cytopenia and vasculitis of PAMI patients may be secondary to their dysfunction, as reported in patients with actin remodeling defects (12, 13). Thus, PAMI syndrome represents the most severe form of the PAID spectrum, and its clinical manifestations may be secondary also to actin remodeling defect and lymphocytes hyperactivation, other than the high IL-1 production as for others PAID.
As reported, kidney biopsy was negative for amyloid deposition (14), in line with previous reports (Table 2), and the histological findings do not support calprotectin deposition as recently described (9). Therefore, other pathogenic mechanisms causing kidney lesions should be investigated. In the first kidney biopsy, a diagnosis of FSGS was reported. The pathogenic mechanism of FSGS remains still poorly understood (15). Recent findings demonstrated the role of the IL-1 pathway as a possible pathogenic mechanism in proteinuric disease, supporting therefore the administration of treatments blocking the IL-1β/IL-1R1 signaling to delay the development of sclerotic lesions (16). In the second kidney biopsy, performed despite stable renal function and proteinuria, a diffused tubule-interstitial fibrosis was revealed, probably due to tacrolimus administration, thus discontinued. Notably, our patient developed proteinuria after irregular administration of IL-1 inhibitors: scarce adherence to IL-1 receptor antagonist (anakinra) was admitted, as above reported and, due to local administrative issues, the supplying of canakinumab was rather inconstant. At this stage, it is difficult to determine the real IL-1 dependence of the renal manifestation.
As reported in Table 2, in patient 4, colchicine was effective in reversing proteinuria. A multi-drug approach may control various manifestations of PAMI by silencing the concomitant defects of different components of the immune system. Furthermore, it remains a matter of speculation that the kidney involvement of other pyrin-related autoinflammatory disorders may not be due only to amyloid deposition. More studies are required to investigate the mechanisms of renal involvement and the possible role of anti-IL drugs against these manifestations.
In conclusion, PAMI syndrome is a rare inflammatory disorder and the most severe phenotype among PAID, characterized by alterations of various immune system agents. The severe kidney involvement of our patient is, according to our best knowledge, the first subjected to a documented histological modification after anti-IL-1 treatment.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee Genoa, Italy. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
PB, RP, AA, GG, and MG conceptualized the manuscript. PB and RP conducted the literature review and drafted the manuscript. MD'A, RC, and GP were involved in the clinical care of the patient. IC performed the genetic analysis. All authors read and approved the final version of the submitted manuscript.
Funding
The study was supported with public funds granted by the Italian Ministry of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank the patient and his family. Several authors of the present paper are members of the European Reference Network for Rare Immunodeficiency, Autoinflammatory and Autoimmune Diseases—Project ID No 739543.
References
1. Holzinger D, Fassl SK, de Jager W, Lohse P, Röhrig UF, Gattorno M, et al. Single amino acid charge switch defines clinically distinct proline-serine-threonine phosphatase-interacting protein 1 (PSTPIP1)–associated inflammatory diseases. J Allergy Clin Immunol. (2015) 136:1337–45. doi: 10.1016/j.jaci.2015.04.016
2. Huang X, Xu M, Dai S, Wang M, Zheng H, Zeng K, et al. Rare cases of PAMI syndrome in both father and son with the same missense mutation in PSTPIP1 gene and literature review. J Dermatol. (2021) 48:519–28. doi: 10.1111/1346-8138.15706
3. Boursier G, Piram M, Rittore C, Sarrabay G, Touitou I. Phenotypic associations of PSTPIP1 sequence variants in PSTPIP1-associated autoinflammatory diseases. J Invest Dermatol. (2021) 141:1141–7. doi: 10.1016/j.jid.2020.08.028
4. Genovese G, Moltrasio C, Garcovich S, Marzano AV. PAPA spectrum disorders. G Ital Dermatol Venereol. (2020) 155:542–50. doi: 10.23736/S0392-0488.20.06629-8
5. Holzinger D, Roth J. Alarming consequences-autoinflammatory disease spectrum due to mutations in proline-serine-threonine phosphatase-interacting protein 1. Curr Opin Rheumatol. (2016) 28:550–9. doi: 10.1097/BOR.0000000000000314
6. Laberko A, Burlakov V, Maier S, Abinun M, Skinner R, Kozlova A, et al. HSCT is effective in patients with PSTPIP1-associated myeloid-related proteinemia inflammatory (PAMI) syndrome. J Allergy Clin Immunol. (2021) 148:250–5.e1. doi: 10.1016/j.jaci.2020.11.043
7. Omenetti A, Carta S, Caorsi R, Finetti M, Marotto D, Lattanzi B, et al. Disease activity accounts for long-term efficacy of IL-1 blockers in pyogenic sterile arthritis pyoderma gangrenosum and severe acne syndrome. Rheumatology. (2016) 55:1325–35. doi: 10.1093/rheumatology/kew031
8. Lindwall E, Singla S, Davis WE, Quinet RJ. Novel PSTPIP1 gene mutation in a patient with pyogenic arthritis, pyoderma gangrenosum and acne (PAPA) syndrome. Semin Arthritis Rheum. (2015) 45:91–3. doi: 10.1016/j.semarthrit.2015.02.012
9. Dai P, Furlong T, Gracie G, Huang ML, Yang T, Wu KHC, et al. Autoinflammation masquerading as autoimmunity in an adult with heterozygous p.E250K PSTPIP1 mutation. J Clin Immunol. (2019) 39:519–22. doi: 10.1007/s10875-019-00646-z
10. Smith JE, Allantaz F, Bennett L, Zhang D, Gao X, Wood G, et al. Clinical, molecular, and genetic characteristics of PAPA syndrome: a review. Curr Genomics. (2010) 11:519–27. doi: 10.2174/138920210793175921
11. Wise CA, Gillum JD, Seidman CE, Lindor NM, Veile R, Bashiardes S, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet. (2002) 11:961–9. doi: 10.1093/hmg/11.8.961
12. Papa R, Penco F, Volpi S, Gattorno M. Actin remodeling defects leading to autoinflammation and immune dysregulation. Front Immunol. (2021) 11:604206. doi: 10.3389/fimmu.2020.604206
13. Starnes TW, Bennin DA, Bing X, Eickhoff JC, Grahf DC, Bellak JM, et al. The F-BAR protein PSTPIP1 controls extracellular matrix degradation and filopodia formation in macrophages. Blood. (2014) 123:2703–14. doi: 10.1182/blood-2013-07-516948
14. Papa R, Lachmann HJ. Secondary, AA, amyloidosis. Rheum Dis Clin North Am. (2018) 44:585–603. doi: 10.1016/j.rdc.2018.06.004
15. Fogo AB. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol. (2015) 11:76–87. doi: 10.1038/nrneph.2014.216
Keywords: PSTPIP1, PAPA syndrome, PAMI syndrome, kidney, IL-1 inhibitors
Citation: Borgia P, Papa R, D'Alessandro M, Caorsi R, Piaggio G, Angeletti A, Ceccherini I, Ghiggeri GM and Gattorno M (2021) Kidney Involvement in PSTPIP1 Associated Inflammatory Diseases (PAID): A Case Report and Review of the Literature. Front. Med. 8:759092. doi: 10.3389/fmed.2021.759092
Received: 15 August 2021; Accepted: 27 September 2021;
Published: 29 October 2021.
Edited by:
Xu-jie Zhou, Peking University First Hospital, ChinaReviewed by:
Angelo Valerio Marzano, University of Milan, ItalyDavid Andrew Fulcher, Australian National University, Australia
Copyright © 2021 Borgia, Papa, D'Alessandro, Caorsi, Piaggio, Angeletti, Ceccherini, Ghiggeri and Gattorno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marco Gattorno, bWFyY29nYXR0b3Jub0BnYXNsaW5pLm9yZw==
†These authors have contributed equally to this work and share first authorship