 Anna Faivre
Anna Faivre Thomas Verissimo1,2
Thomas Verissimo1,2 David Legouis
David Legouis Sophie de Seigneux
Sophie de Seigneux
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 29 October 2021
Sec. Nephrology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.742072
This article is part of the Research Topic The Role of Mitochondria and Metabolic Rewiring in Kidney Diseases View all 7 articles
Acute and chronic kidney disease are responsible for large healthcare costs worldwide. During injury, kidney metabolism undergoes profound modifications in order to adapt to oxygen and nutrient shortage. Several studies highlighted recently the importance of these metabolic adaptations in acute as well as in chronic phases of renal disease, with a potential deleterious effect on fibrosis progression. Until recently, glucose metabolism in the kidney has been poorly studied, even though the kidney has the capacity to use and produce glucose, depending on the segment of the nephron. During physiology, renal proximal tubular cells use the beta-oxidation of fatty acid to generate large amounts of energy, and can also produce glucose through gluconeogenesis. In acute kidney injury, proximal tubular cells metabolism undergo a metabolic shift, shifting away from beta-oxidation of fatty acids and gluconeogenesis toward glycolysis. In chronic kidney disease, the loss of fatty acid oxidation is also well-described, and data about glucose metabolism are emerging. We here review the modifications of proximal tubular cells glucose metabolism during acute and chronic kidney disease and their potential consequences, as well as the potential therapeutic implications.
Renal disease encompasses acute and chronic lesions altering renal physiological function. Chronic kidney disease (CKD) is characterized by an alteration of kidney structure and/or function lasting for more than 3 months (1). Acute kidney injury (AKI) is defined by a sudden increase in serum creatinine or decrease in urine output (2, 3). AKI is recognized as one of the main factors contributing to CKD progression (4, 5) and is associated with significant mortality (6). Although they represent two different diseases by definition, AKI and CKD share many common traits and are very much interrelated (5). Understanding the pathophysiology of both acute and chronic renal injury is mandatory to address the unmet medical need. Tubular cells are important players in AKI and in the progression of CKD. Metabolic modifications and mitochondrial dysfunctions of these cells are key to fibrosis development (7).
The kidney displays the second highest metabolic rate (>400 kcal/kg tissue/day) (8) after the heart. It uses ~7% of the total body daily energy despite its relatively low weight (9). The high energetic needs of the kidney are due on one hand to the transport and active reabsorption of nutrients and electrolytes in the tubules and on the other hand to the active secretion of unneeded compounds (10). Depending on the segment of the nephron, tubular cells use different substrates such as glucose, amino acids, fatty acids, or ketone bodies to produce energy through fatty acid oxidation (FAO) and glycolysis (11, 12). Glycolysis is the process that converts glucose into pyruvate through 10 enzymatic reactions; pyruvate is subsequently converted into acetyl-CoA. FAO is the breakdown of fatty acids at the mitochondrial level that also generates acetyl-CoA. Acetyl-CoA is then used as substrate for the tricarboxylic acid cycle (TCA) and the electron transport chain to produce adenosine triphosphate (ATP). The kidney is also able to perform gluconeogenesis (similarly to the liver), a metabolic pathway that produces glucose from non-hexose precursors. The kidney is therefore both an important consumer of energy and a producer of glucose and participates in systemic glucose metabolism (13).
Proximal tubular cells (PTCs) are the cells that produce and consume most of the energy in the kidney (14). Under physiological condition, their ability to generate ATP relies mostly on the oxidative phosphorylation of acetyl-CoA from FAO (12). Oxidative phosphorylation allows the production of energy through the transfer of electrons from nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH) to oxygen, finally leading to ATP production. Besides, PTCs possess the ability to produce energy from a wide range of other substrates, including glutamine, lactate, pyruvate, acetate, citrate, or ketone bodies. PTCs are able to regulate glucose homeostasis by the de novo glucose production through gluconeogenesis but also by the reabsorption of large amounts of glucose, mostly via SGLT2 on the apical membrane of S1-2 segments and in a lesser extent via SGLT1 in the S3 segment. After reabsorption, glucose is released in the blood by glucose transporters (GLUT) (15). However, PTCs are almost unable to use glucose as an energetic substrate, except for the S3 segment (12). This specificity is due to the close localization of the S3 segments to the outer medulla and hence to its strong exposure to changes in O2 levels. The complete breakdown of fatty acids is depends on the presence of oxygen and S3 cells therefore have to rely on other energy sources than FAO in case of low oxygen supply (16). On the contrary, the cells in the more distal segments of the nephron display enhanced glycolytic capacity and progressively lose their capacity to perform gluconeogenesis and oxidative phosphorylation. Besides, they can use less varied substrates than the PTCs (10) (Figure 1). Finally, because of their passive blood filtration process glomerular, endothelial and mesangial cells display low rates of oxidative phosphorylation (17). They have a basic glycolytic metabolism, with the ability to perform either aerobic or anaerobic glycolysis, thus producing less energy but with greater efficiency when O2 supply is low (18, 19). As a result, they are less sensitive to changes in O2 levels than PTCs.
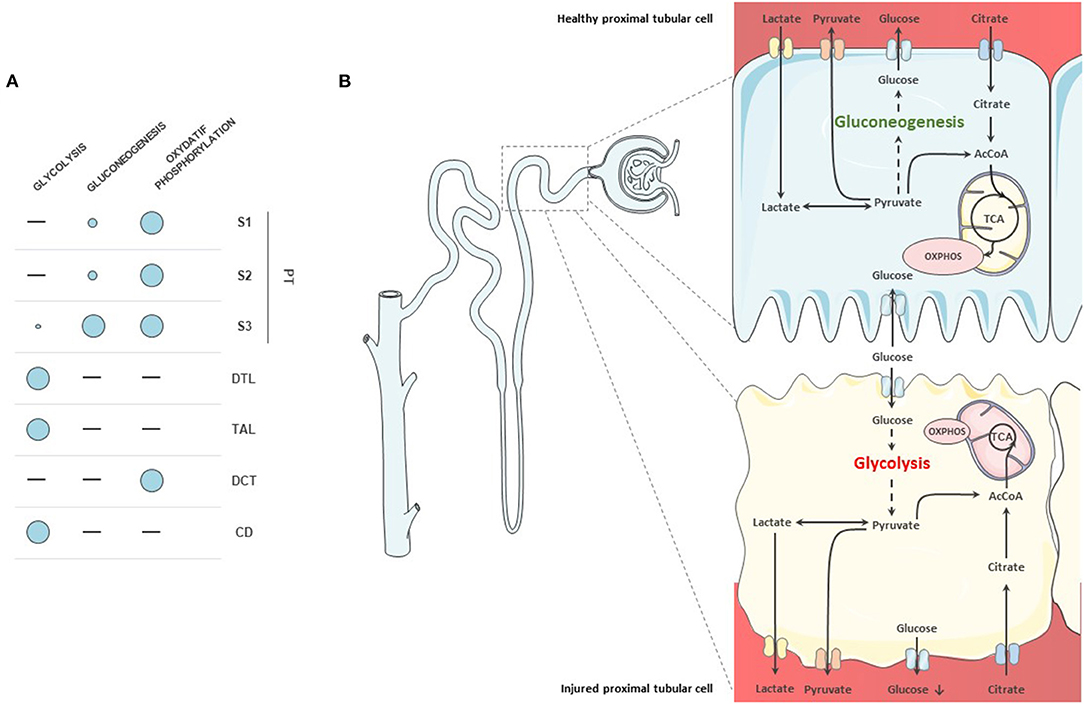
Figure 1. Metabolism of nephron segments. (A) Overview of metabolic capacities of tubular cells. Proximal tubular cells do not display glycolytic capacity, except for the S3 segment. They rely on oxidative phosphorylation for their metabolism. They are also able to produce glucose through gluconeogenesis. (B) Proximal tubule cells in health and disease. In healthy proximal tubular cells, glycolysis is almost absent although the cells reabsorb glucose. Glucose is produced through gluconeogenesis from pyruvate and released into the circulation. In diseased cells, fatty acid oxidation (FAO) decreases and cells start to use glucose as a substrate. Gluconeogenic abilities are also diminished in injured proximal tubular cells.
In this review, we will primarily focus on the altered metabolism of PTCs during renal disease. Although several metabolic pathways are interrelated, we here mainly discuss glucose metabolism modifications during kidney disease and its potential local and systemic consequences.
PTCs are the largest group of tubular cells and are at highest risk of being exposed to hypoxia and other types of injury (20). The importance of these cells in the pathophysiological process of AKI and CKD is now well-established and the specific influence of PTC metabolic alterations in the pathological processes of renal disease has received increasing attention. Among metabolic alterations, mitochondrial dysfunction (21–27) and FAO downregulation (21, 28–30) have been described and already been reviewed (31–36). We here describe in more details the modifications of glucose metabolism in PTCs that occurs during acute and chronic injuries.
Glycolysis is a 10 steps pathway that leads to the production of pyruvate (Figure 2). The first step is catalyzed by hexokinases, which phosphorylates glucose into glucose-6-phosphate (G6P). Glucose phosphate isomerase rearranges G6P into fructose 6-phosphate. Fructose can also be phosphorylated and enter the pathway at this stage (37). Fructose 6-phosphate is then transformed in fructose 1,6-bisphosphate by phosphofructokinase 1, an ATP-dependent reaction and a key regulatory point of the pathway (38, 39). After several steps leading to phosphoenolpyruvate, the pyruvate kinase catalyzes the last reaction, which is also a regulatory point (40). Depending on O2 availability, pyruvate is transported into the mitochondria to enter the TCA cycle or in the absence of oxygen, pyruvate is converted into lactate (41). The regulating enzymes of glycolysis are hexokinases, phosphofructokinases, and pyruvate kinases. These three enzymes are activated by the AMP/ADP ratio. Phosphofructokinase is additionally activated by fructose-2,6-bisphosphate and pyruvate kinase by fructose-1,6-bisphosphate. Hexokinase is inhibited by glucose-6-phosphate, phosphofructokinase, ATP, and citrate. Likewise, pyruvate kinase is inhibited by ATP, acetyl-CoA, and alanine (42). Insulin, stimulation of glucose uptake and epinephrine enhance glycolysis (43, 44). Among negative regulators of the pathway are glucagon, cortisol and growth hormone (42, 45, 46).
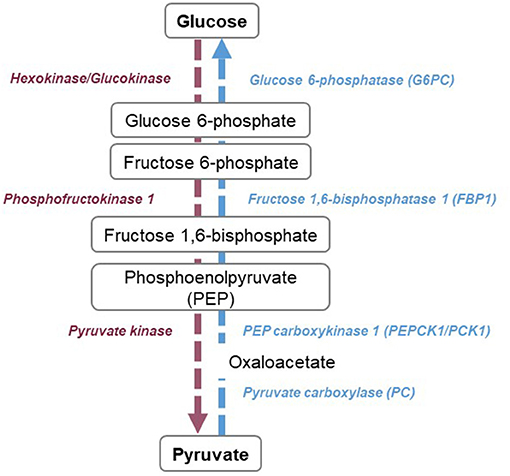
Figure 2. Overview of glycolysis and gluconeogenesis pathways. Glycolysis (purple) consists of 10 reactions, leading to the production of pyruvate. Three steps are considered as rate-limiting, catalyzed by hexokinases or glucokinases, phosphofructokinase 1, and pyruvate kinase. Gluconeogenesis (blue) is the reversed steps of glycolysis, with four specific reactions catalyzed by glucose-6-phosphatase, fructose 1,6-bisphosphatase, phosphoenolpyruvate carboxykinase 1, and pyruvate carboxylase.
Physiologically, in conditions of high oxygen supply, fatty acids are mainly taken up by PTCs and used for oxidative phosphorylation to generate ATP. However, AKI is usually associated with a decrease in oxygen supply or relative oxygen deficiency in PTCs. PTCs are hence forced to use glycolysis instead of FAO. This is well-described in different models of AKI, which have shown pyruvate depletion, glycolysis intermediates accumulation and glycolytic enzymes upregulation during this condition (27, 47–51) even at late stages post-reperfusion (27). In chronic disease, analysis of metabolomics and RNA sequencing data from human databases have demonstrated an upregulation of glycolysis, showing a specific metabolic pattern of CKD patients (52, 53). A similar shift toward glycolysis has been observed in obstructive mouse models, with an increased lactate to pyruvate ratio (54).
Gluconeogenesis is a major kidney metabolic pathway, specifically present in healthy PTCs that produces glucose from non-hexose substrates such as lactate, pyruvate, glycerol, or amino acids (55) (Figure 2). In the kidney, lactate is the main substrate of gluconeogenesis and accounts for around 50% of glucose production. It is followed by glutamine (20%) and glycerol (10%) (56, 57). The conversion of pyruvate into glucose is the central pathway of gluconeogenesis. For this, 10 enzymatic reactions are necessary, seven of which are reversible and three that are irreversible. Phosphoenolpyruvate carboxykinase 1 (PCK1) catalyzes the formation of phosphoenolpyruvate from oxaloacetate, fructose-1,6-bisphosphatase 1 (FBP1) catalyzes the hydrolysis of fructose 1,6-biphosphate to fructose 6-phosphate and Glucose 6-phosphatase catalyzes glucose 6-phosphate to D-glucose. Gluconeogenesis plays a crucial role in glycemic homeostasis (58). Indeed, during fasting, once glycogen stores becomes depleted, the body will increasingly rely on endogenous glucose production. Although the liver has for a long time been considered as the sole source of glucose production, the kidneys are responsible for 40% of de novo glucose production during the fasted state (13, 59, 60). Renal gluconeogenesis is positively regulated by stress hormones such as hydrocortisone, epinephrine, and norepinephrine (56, 57, 61, 62), which could be linked to a regulation of PCK/Pyruvate Carboxylase (PC) activity (63–65). Insulin on the other hand downregulates renal gluconeogenesis (66), which could be due to PCK1/G6Pc regulation through phosphorylation (66, 67) but also to the reduction of substrates availability (glycerol and glutamine) (68, 69) or their redirection to the oxidative pathways (13, 68). Gluconeogenesis is regulated by factors implicated in global metabolic regulation, such as peroxisome proliferator-activated receptor alpha (PPARα) (70), Forkhead Box O1(FOXO1) (71) and hepatocyte nuclear factor 4 alpha (HNF4α) but also by glucose levels, through the modification of the NAD+/NADH ratio which causes an indirect downregulation of PCK1 at mRNA level (66). Acidosis also seems to play a role, with the induction of PCK1 at the mRNA and enzymatic activity level (72, 73) and hence increased renal glucose production.
The regulation of gluconeogenesis during renal pathology was not well-studied until recently. In AKI, a downregulation of gluconeogenesis enzymes was shown in single-cell analysis of mouse AKI as well as in RNA sequencing data from post-transplant AKI patients (74). Alterations of lactate clearance and glucose production were observed using renal catheterism data and rodent experimental models. In a cohort of intensive care unit patients, alterations of metabolism were associated with mortality, underlying the importance of renal glucose metabolism at a systemic level. Overall, gluconeogenesis decreases during AKI and leads to systemic metabolic alterations. In CKD, our preliminary results show similar alterations of gluconeogenesis are observed in a stage specific manner.
Altogether, a metabolic switch from FAO and gluconeogenesis to glycolysis occurs in PTCs during AKI and CKD. To date, the exact cause of this metabolic switch remains unknown. Initially, the switch constitutes a protective mechanism allowing PTCs to maintain energy production in case of low oxygen supply: HIF activation indeed enhances glycolysis through the stimulation of several enzymes of the pathway (hexokinases, glyceraldehyde-3-phosphate dehydrogenase, enolases, phosphofructokinases) and through the activation of glucose transporters (GLUT1-3) (75–77). In later stages of renal disease, inflammation and TGFβ activation could also play an important role in the persistence of the metabolic switch (78, 79); the induction of glycolysis was reproduced by IL-β and c-myc signaling activation (32, 52). In addition, the expression of co-regulators of FAO and glycolysis such as HNF4α or estrogen-related receptor alpha (ESRRA) are modified during AKI (74). Although it is initially cytoprotective, the persistence of the switch may be maladaptive and is associated with a worse kidney prognosis (21, 80, 81). In the second part of this review, we will detail the potential consequences of such metabolic modifications.
The transition from gluconeogenesis and FAO to glycolysis can have positive or detrimental consequences. Impaired glucose metabolism has been closely linked with mitochondrial dysfunction and a global decrease in energy production (27). The metabolic shift could also directly impact epithelial-mesenchymal transition (EMT) and fibrogenesis and therefore enhance CKD progression. Finally, the accumulation of metabolic precursors may influence renal disease (Figure 3).
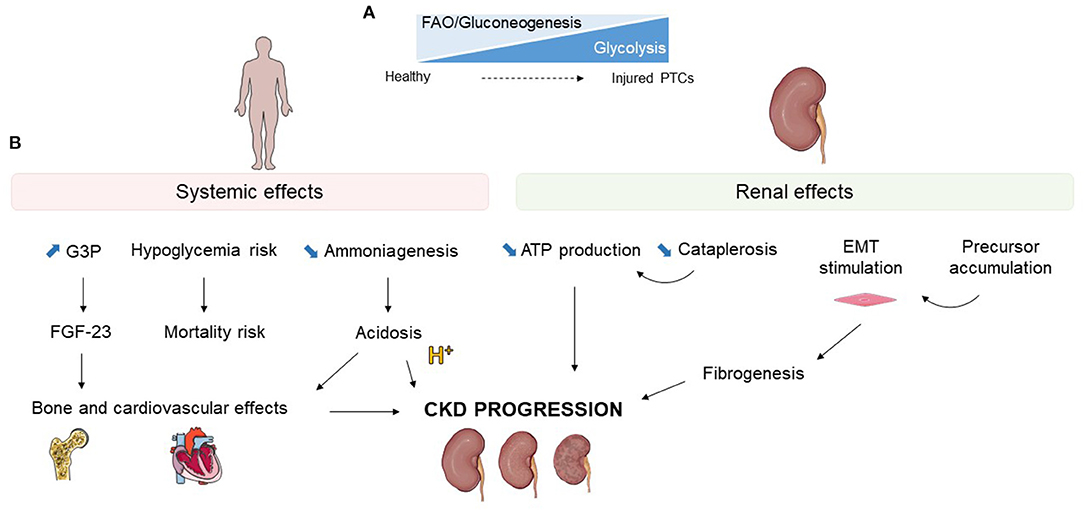
Figure 3. Effects of glucose metabolism alterations during kidney injury. (A) Schematic representation of the loss of fatty acid oxidation (FAO) and gluconeogenesis with upregulation of glycolysis occurring in injured proximal tubular cells (PTCs). (B) Systemic and renal potential consequences of glucose metabolism modifications during kidney injury. G3P, glycerol-3-phosphate; FGF-23, fibrobastic growth factor 23; EMT, epithelial mesenchymal transition; CKD, chronic kidney disease.
As described earlier, when oxygen supply decreases, PTCs start using mostly anaerobic glycolysis to meet their energy demand. This change results in a decrease in ATP production and thus a diminished energy availability. Indeed, compared with the complete metabolism of fatty acids, which generates 106 ATP, aerobic glycolysis produces 36 or 38ATP and anaerobic only only 2 ATP (14, 82). Enhanced glycolysis may in addition have a detrimental effect on mitochondrial function (83). Phosphokinase mutase 2 (PKM2), one of the glycolytic enzymes, may for example promote mitochondrial fusion (84). Inhibition of gluconeogenesis also influences the TCA cycle. Indeed, gluconeogenesis plays a role in cataplerosis, a crucial pathway for mitochondrial function. Cataplerosis consists of the removal of TCA cycle intermediates and is necessary to maintain the cycle's function (85). The gluconeogenic enzyme PCK1/2 catalyzes the reaction of oxaloacetate to phosphoenolpyruvate, a major reaction of the cataplerotic pathway (86). Downregulation of gluconeogenesis enzymes could for this reason lead to impaired cataplerosis and hence impact the TCA cycle (86), as has been shown in other organs (87).
Thus, although enhancing glycolysis and decreasing gluconeogenesis may initially spare oxygen and maintain ATP production, on the longer term these regulations may lead to further blockade of the TCA cycle and mitochondrial dysfunction, which will affect global energy production and therefore PTC function.
Modifications of tubular metabolism is associated with CKD progression and fibrosis. For instance, the decreased ability to perform FAO is associated with kidney fibrosis progression and reversing this loss by different approaches appears nephroprotective (21, 24, 29, 88). Regarding glucose metabolic enzymes, PKM, hexokinases, phosphofructokinase 1, and enolase are able to promote EMT via different mechanisms, as reviewed elsewhere (89). In cancer research, FBP1 expression, a key gluconeogenetic enzyme, was inversely linked with Snail activation, a major activator of EMT, whereas FBP1 overexpression protected from EMT (90).
Enhancing glycolytic capacity induces a reprogramming of the somatic cells that promotes tubular regeneration (91, 92) and spares more valuable substrates such as fatty acids or amino acids which could then be used for cell regeneration (93). Increased glycolysis also generates more NADPH and glutathione, thus decreasing oxidative stress (48, 94). Similarly, decreasing gluconeogenesis may spare kidney energy in conditions of stress, hence promoting cell survival. Altogether, glycolysis improves PTC survival initially. Nevertheless, the persistence of glycolysis after the acute phase could be detrimental for kidney function. Given this dual impact of glycolysis on PTCs, interventional studies with glycolysis modulation yielded mitigated results. In PTCs, an activation of glycolysis through the inhibition of TP53-inducible glycolysis and apoptosis regulator (TIGAR) is protective in ischemic AKI (93). On the contrary, decreasing glycolysis through the knockout of Nod-like receptor (NLR) family member X1 (NLRX1) was shown to be detrimental (95). Inhibition of PKM through the S-nitroso-CoA Reductase system or fructokinase blockade were also protective during AKI (96, 97). SGLT2 inhibitors have shown the ability to suppress aberrant glycolysis in proximal tubules (98) and stimulate gluconeogenesis (99). Even though some associations between SLGT2 inhibitors and AKI were reported in the US Food and Drug Administration Adverse Event Report System (100), subsequent meta-analysis of large clinical studies indicate a favorable safety profile (101) or even a reduction of the AKI risk (102–104). In preclinical models, some data indicate a protective role (105, 106). Finally, despite many confounding factors, stabilization of HIF, a key glycolysis promoter, is protective in AKI (91, 92, 107).
In diabetic kidney disease, the switch to anaerobic glycolysis is well-described (108) and associated with CKD progression (109). In other CKD models, a reduction of glycolysis appears in contrast to be rather protective overall. PKM activation was shown to be favorable in a diabetic kidney disease model (109) but induced interstitial fibrosis in other models (110). Fructokinase blockade was also protective in diabetic nephropathy (111, 112). Glycolysis inhibitors (shikonin and 2-deoxyglucose) demonstrated attenuated fibrosis in an obstructive CKD model (113). In the same model, deletion of Tuberous sclerosis complex 1, a key regulator of glycolysis, induced glycolysis and enhanced fibrosis (114). In diabetic mice models, the suppression of SIRT3, a major mitochondrial enzyme involved in central metabolism activating many oxidative pathway, was stimulated the fibrogenic kidney pathway (80). Glycolysis inhibition with 2-deoxyglucose alleviated the phenotype (80, 115). As described before, SGLT2 inhibitors seem to decrease glycolysis and enhance gluconeogenesis (98, 99); their major protective effect in CKD is well-established and could partially rely on the modulation of the metabolic switch. In the polycystic model of CKD, characterized by marked cellular proliferation, numerous studies observed that glycolysis blockade is nephroprotecive (115–117).
In sum, the timing of glycolysis induction may be the reason of these contradictory results (27). Glycolysis enhancement and gluconeogenesis loss in PTCs during AKI may promote cell survival and proliferation in an initial phase but their persistence could favor fibrosis progression in the kidney in a more chronic phase. The type of kidney injury could also explain the difference in experimental results.
Metabolic pathway blockade leads to the accumulation of precursors, which may overload the cells and contribute to inflammation and fibrosis. Among these precursors, some also have direct toxic effects; blocking FAO will for example increase intracellular concentrations of fatty acids. This results in a toxic environment, impacting ATP production and inducing apoptosis (14). Flux analysis of metabolites indicate that the kidney also plays a major role in clearing circulating citrate and lactate (118, 119); in the case of gluconeogenesis blockade, as observed in AKI, a decrease in lactate clearance occurs (74). Although some biases exist, lactate clearance has been associated with mortality in AKI (120), as well as metabolic alterations (74). Recent papers indicate a potential toxic role of tubule-derived lactate, with increased fibrogenesis (121). Indeed, the inhibition of lactate production via glycolysis inhibition was able to reduce the activation of fibroblasts in a model of folic acid-induced AKI. In CKD, the effects of gluconeogenesis decrease are less described but can be inferred from enzymatic deficits. Deficiency in glucose-6-phosphatase (G6PC), a major gluconeogenesis enzyme, has been extensively studied in humans with glycogen storage disease type I. These patients are especially prone to CKD development, due to lipid and glycogen accumulation in the kidneys and the activation of the renin-angiotensin system. G6PC knock-out mice also develop renal cysts. G6PC deficiency seems to be linked with the loss of polycystic kidney genes and HNF1B (122).
Besides its potential effects on the outcome of kidney disease, modifying renal glucose metabolism has a systemic impact. Alteration of tubular cell metabolism may also imply release of new metabolites that can affect unexpected systemic functions (Figure 3).
In a fasting state, once glycogen stores are depleted, gluconeogenesis becomes increasingly important and represents up to 90% of total glucose production after 40 h of fasting in order to maintain normoglycemia (123). In fasting conditions, the kidney is able to provide up to 40% of systemic glucose (13, 59, 60).
As demonstrated recently, the enhancement in glycolysis and decrease in gluconeogenesis in AKI leads to a negative renal glucose balance and increases hypoglycemia risk (74). The role of glucose production of the kidney has probably as much importance in CKD as in AKI. In the ACCORD study, diabetic patients with low renal function displayed a higher risk of hypoglycemia and mortality (124). Similarly in another study, both diabetic and non-diabetic patients with CKD had an increased risk of in-hospital hypoglycemia (125). Although not clearly described or studied in CKD, the enhanced risk could be partly related to loss of gluconeogenesis.
In the kidney, gluconeogenesis uses lactate as its main substrate. In renal arteriovenous catheterization experiments, impaired renal gluconeogenesis due to AKI induced a decrease in glucose production and lactate clearance. The increased lactate and lower glucose levels were associated with higher mortality in a retrospective cohort of critically ill patients (74).
Changes in glucose metabolism impacts acid-base regulation as illustrated by the close correlation between gluconeogenesis and ammoniagenesis. To counterbalance acidosis, the kidney generates ammonia, mainly from glutamine deamination, which forms α-ketoglutarate (α-KG) and NH4+ via the ammoniagenesis pathway (126, 127). After its uptake by PTCs, glutamine is catalyzed into glutamate. Glutamate is then converted to α-ketoglutarate, the carbon skeleton of glutamine. α-KG is further metabolized and is ultimately transformed into glucose, through the gluconeogenic pathway (128). Renal ammoniagenesis and gluconeogenesis are hence two closely interdependent pathways. Despite the ability of all segments to produce ammonia, metabolic acidosis will only enhance ammoniagenesis in the proximal segments S1 and S2 (127), a process which is altered in AKI and CKD. As the complete metabolism of glutamine requires PCK1 activity (129), gluconeogenesis alterations may participate in the loss of ammoniagenesis abilities of PTCs and hence decrease their defense against metabolic acidosis.
Local changes of metabolism lead to modification of arterial and venous kidney metabolite profile. Fibroblast growth factor 23 (FGF23) is a protein implicated in vitamin D metabolism and phosphatemia regulation. FGF23 levels increase with any type of kidney injury; in AKI, in early and late CKD, and could be linked with cardiovascular mortality (130–132). Despite that, the regulatory factors of FGF23 during AKI and CKD are poorly described. A recent study identified glycerol-3-phosphate (G3P) renal venous production as the most predictive factor for FGF23 elevation in AKI, which was confirmed experimentally (133). In human and rodent with AKI, G3P levels increased rapidly, a process mediated by glycerol-3-phosphate acyltransferase and lysophosphatidic acid (LPA) (133). G3P can arise from three mechanisms, one being the glycolytic pathway via the conversion of dihydroxyacetone phosphate in G3P glucose (134). As glycolysis increases during AKI, it may explain these results and outlines the importance of renal glucose metabolism during AKI and its potential systemic effects. A second pathway for G3P production is from glycerol, which is indirectly linked to gluconeogenesis as PCK1 is involved in glycerol production. As glycolysis and gluconeogenesis are also dysregulated during CKD, similar effects could be expected in this condition. Therefore, kidney metabolic switches may impact pathways as diverse as mineral metabolism and heart hypertrophy, both processes being regulated by FGF23 (135). This example shows how local metabolic change in PTCs can influence very diverse systemic pathways, many of which are probably still unknown to this date.
Metabolic adaptation of renal cells during acute or sustained injury is a complex mechanism. The metabolic switch from fatty acid oxidation and gluconeogenesis toward glycolysis is part of a cytoprotective stress mechanism. However, persistence of this metabolic profile is associated with fibrogenesis. Glucose metabolism impairment could have a broad impact, ranging from local to systemic consequences. Interestingly, renal glucose metabolism is amenable to therapeutic interventions.
AF reviewed the literature, drafted the manuscript, and drew the figures. TV reviewed the literature, edited the manuscript, and drew the figures. HA reviewed the literature and edited the manuscript. DL edited the manuscript and the figures. SS designed and supervised the project, edited the manuscript and the figures. All authors contributed to the article and approved the submitted version.
AF was the recipient of a grant from the Swiss National Science Foundation (SNSF 323530_191224) and from the Carlos et Elsie de Reuter Founding. SS was supported by grants from the Swiss National Science Foundation (SNSF PP00P3-187186/1), Jules Thorn Foundation and NCCR Kidney.ch.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Levey AS, Coresh J, Greene T, Stevens LA, Zhang Y, Hendriksen S, et al. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann Intern Med. (2006) 145:247. doi: 10.7326/0003-4819-145-4-200608150-00004
2. Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P, Acute Dialysis Quality Initiative Workgroup. Acute renal failure - definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care Lond Engl. (2004) 8:R204–212. doi: 10.1186/cc2872
3. Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet Lond Engl. (2012) 380:756–66. doi: 10.1016/S0140-6736(11)61454-2
4. Legouis D, Galichon P, Bataille A, Chevret S, Provenchère S, Boutten A, et al. Rapid occurrence of chronic kidney disease in patients experiencing reversible acute kidney injury after cardiac surgery. Anesthesiology. (2017) 126:39–46. doi: 10.1097/ALN.0000000000001400
5. Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. (2014) 371:58–66. doi: 10.1056/NEJMra1214243
6. Hoste EAJ, Bagshaw SM, Bellomo R, Cely CM, Colman R, Cruz DN, et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med. (2015) 41:1411–23. doi: 10.1007/s00134-015-3934-7
7. Qi R, Yang C. Renal tubular epithelial cells: the neglected mediator of tubulointerstitial fibrosis after injury. Cell Death Dis. (2018) 9:1126. doi: 10.1038/s41419-018-1157-x
8. Tian Z, Liang M. Renal metabolism and hypertension. Nat Commun. (2021) 12:963. doi: 10.1038/s41467-021-21301-5
9. Wang Z, Ying Z, Bosy-Westphal A, Zhang J, Schautz B, Later W, et al. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure1234. Am J Clin Nutr. (2010) 92:1369–77. doi: 10.3945/ajcn.2010.29885
10. Scholz H, Boivin FJ, Schmidt-Ott KM, Bachmann S, Eckardt K-U, Scholl UI, et al. Kidney physiology and susceptibility to acute kidney injury: implications for renoprotection. Nat Rev Nephrol. (2021) 17:335–49. doi: 10.1038/s41581-021-00394-7
11. Rabelink TJ, Giera M. New insights into energy and protein homeostasis by the kidney. Nat Rev Nephrol. (2019) 15:596–8. doi: 10.1038/s41581-019-0192-x
12. Uchida S, Endou H. Substrate specificity to maintain cellular ATP along the mouse nephron. Am J Physiol-Ren Physiol. (1988) 255:F977–83. doi: 10.1152/ajprenal.1988.255.5.F977
13. Gerich JE, Meyer C, Woerle HJ, Stumvoll M. Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes Care. (2001) 24:382–91. doi: 10.2337/diacare.24.2.382
14. Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol. (2017) 13:629–46. doi: 10.1038/nrneph.2017.107
15. Alsahli M, Gerich JE. Renal glucose metabolism in normal physiological conditions and in diabetes. Diabetes Res Clin Pract. (2017) 133:1–9. doi: 10.1016/j.diabres.2017.07.033
16. Kessler G, Friedman J. Metabolism of fatty acids and glucose. Circulation. (1998) 98:1350a–3. doi: 10.1161/circ.98.13.1350/a
17. Pollak MR, Quaggin SE, Hoenig MP, Dworkin LD. The glomerulus: the sphere of influence. Clin J Am Soc Nephrol CJASN. (2014) 9:1461–9. doi: 10.2215/CJN.09400913
18. Guder WG, Ross BD. Enzyme distribution along the nephron. Kidney Int. (1984) 26:101–11. doi: 10.1038/ki.1984.143
19. Wirthensohn G, Guder WG. Renal substrate metabolism. Physiol Rev. (1986) 66:469–97. doi: 10.1152/physrev.1986.66.2.469
20. Chevalier RL. The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am J Physiol Ren Physiol. (2016) 311:F145–61. doi: 10.1152/ajprenal.00164.2016
21. Kang HM, Ahn SH, Choi P, Ko Y-A, Han SH, Chinga F, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. (2015) 21:37–46. doi: 10.1038/nm.3762
22. Frazier R, Mehta R, Cai X, Lee J, Napoli S, Craven T, et al. Associations of fenofibrate therapy with incidence and progression of CKD in patients with type 2 diabetes. Kidney Int Rep. (2018) 4:94–102. doi: 10.1016/j.ekir.2018.09.006
23. Rutkowski JM, Wang ZV, Park ASD, Zhang J, Zhang D, Hu MC, et al. Adiponectin promotes functional recovery after podocyte ablation. J Am Soc Nephrol. (2013) 24:268–82. doi: 10.1681/ASN.2012040414
24. Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. (2016) 531:528–32. doi: 10.1038/nature17184
25. Poyan Mehr A, Tran MT, Ralto KM, Leaf DE, Washco V, Messmer J, et al. De novo NAD+ biosynthetic impairment in acute kidney injury in humans. Nat Med. (2018) 24:1351–9. doi: 10.1038/s41591-018-0138-z
26. Katsyuba E, Mottis A, Zietak M, De Franco F, van der Velpen V, Gariani K, et al. De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature. (2018) 563:354–9. doi: 10.1038/s41586-018-0645-6
27. Lan R, Geng H, Singha PK, Saikumar P, Bottinger EP, Weinberg JM, et al. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol. (2016) 27:3356–67. doi: 10.1681/ASN.2015020177
28. Bataille A, Galichon P, Chelghoum N, Oumoussa BM, Ziliotis M-J, Sadia I, et al. Increased fatty acid oxidation in differentiated proximal tubular cells surviving a reversible episode of acute kidney injury. Cell Physiol Biochem. (2018) 47:1338–51. doi: 10.1159/000490819
29. Lakhia R, Yheskel M, Flaten A, Quittner-Strom EB, Holland WL, Patel V. PPARα agonist fenofibrate enhances fatty acid β-oxidation and attenuates polycystic kidney and liver disease in mice. Am J Physiol Renal Physiol. (2018) 314:F122–31. doi: 10.1152/ajprenal.00352.2017
30. Chen L, Vasoya RP, Toke NH, Parthasarathy A, Luo S, Chiles E, et al. HNF4 regulates fatty acid oxidation and is required for renewal of intestinal stem cells in mice. Gastroenterology. (2020) 158:985–99.e9. doi: 10.1053/j.gastro.2019.11.031
31. Simon N, Hertig A. Alteration of fatty acid oxidation in tubular epithelial cells: from acute kidney injury to renal fibrogenesis. Front Med. (2015) 2:52. doi: 10.3389/fmed.2015.00052
32. Quinn GZ, Dhillon P, Susztak K. It takes two to tango: the role of dysregulated metabolism and inflammation in kidney disease development. Semin Nephrol. (2020) 40:199–205. doi: 10.1016/j.semnephrol.2020.01.010
33. Jang H-S, Noh MR, Kim J, Padanilam BJ. Defective mitochondrial fatty acid oxidation and lipotoxicity in kidney diseases. Front Med. (2020) 7:65. doi: 10.3389/fmed.2020.00065
34. Reidy KJ, Ross MJ. Re-energizing the kidney: targeting fatty acid metabolism protects against kidney fibrosis. Kidney Int. (2021) 100:742–4. doi: 10.1016/j.kint.2021.06.010
35. Galvan DL, Green NH, Danesh FR. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. (2017) 92:1051–7. doi: 10.1016/j.kint.2017.05.034
36. Che R, Yuan Y, Huang S, Zhang A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am J Physiol-Ren Physiol. (2014) 306:F367–78. doi: 10.1152/ajprenal.00571.2013
37. Berg JM, Tymoczko JL, Stryer L. The glycolytic pathway is tightly controlled. In: Biochemistry, 5th Ed. New York, NY: W H Freeman (2002).
38. Li X, Gu J, Zhou Q. Review of aerobic glycolysis and its key enzymes – new targets for lung cancer therapy. Thorac Cancer. (2015) 6:17–24. doi: 10.1111/1759-7714.12148
39. Wu R. Rate-limiting factors in glycolysis and inorganic orthophosphate transport in rat liver and kidney slices. J Biol Chem. (1965) 240:2373–81. doi: 10.1016/S0021-9258(18)97333-3
40. Hance AJ, Robin ED, Simon LM, Alexander S, Herzenberg LA, Theodore J. Regulation of glycolytic enzyme activity during chronic hypoxia by changes in rate-limiting enzyme content. Use of monoclonal antibodies to quantitate changes in pyruvate kinase content. J Clin Invest. (1980) 66:1258–64. doi: 10.1172/JCI109977
41. Herzig S, Raemy E, Montessuit S, Veuthey J-L, Zamboni N, Westermann B, et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science. (2012) 337:93–6. doi: 10.1126/science.1218530
42. Locasale JW. New concepts in feedback regulation of glucose metabolism. Curr Opin Syst Biol. (2018) 8:32–8. doi: 10.1016/j.coisb.2017.11.005
43. James JH, Wagner KR, King JK, Leffler RE, Upputuri RK, Balasubramaniam A, et al. Stimulation of both aerobic glycolysis and Na(+)-K(+)-ATPase activity in skeletal muscle by epinephrine or amylin. Am J Physiol. (1999) 277:E176–86. doi: 10.1152/ajpendo.1999.277.1.E176
44. Raz I, Katz A, Spencer MK. Epinephrine inhibits insulin-mediated glycogenesis but enhances glycolysis in human skeletal muscle. Am J Physiol-Endocrinol Metab. (1991) 260:E430–5. doi: 10.1152/ajpendo.1991.260.3.E430
45. Qaid MM, Abdelrahman MM. Role of insulin and other related hormones in energy metabolism—a review. Cogent Food Agric. (2016) 2:1267691. doi: 10.1080/23311932.2016.1267691
46. Wang Z, Iwasaki Y, Zhao LF, Nishiyama M, Taguchi T, Tsugita M, et al. Hormonal regulation of glycolytic enzyme gene and pyruvate dehydrogenase kinase/phosphatase gene transcription. Endocr J. (2009) 56:1019–30. doi: 10.1507/endocrj.K09E-178
47. Waltz P, Carchman E, Gomez H, Zuckerbraun B. Sepsis results in an altered renal metabolic and osmolyte profile. J Surg Res. (2016) 202:8–12. doi: 10.1016/j.jss.2015.12.011
48. Smith JA, Stallons LJ, Schnellmann RG. Renal cortical hexokinase and pentose phosphate pathway activation through the EGFR/Akt signaling pathway in endotoxin-induced acute kidney injury. Am J Physiol Renal Physiol. (2014) 307:F435–444. doi: 10.1152/ajprenal.00271.2014
49. Zager RA, Johnson ACM, Becker K. Renal cortical pyruvate depletion during AKI. J Am Soc Nephrol. (2014) 25:998–1012. doi: 10.1681/ASN.2013070791
50. Dickman KG, Mandel LJ. Differential effects of respiratory inhibitors on glycolysis in proximal tubules. Am J Physiol-Ren Physiol. (1990) 258:F1608–15. doi: 10.1152/ajprenal.1990.258.6.F1608
51. Hato T, Friedman AN, Mang H, Plotkin Z, Dube S, Hutchins GD, et al. Novel application of complementary imaging techniques to examine in vivo glucose metabolism in the kidney. Am J Physiol Renal Physiol. (2016) 310:F717–25. doi: 10.1152/ajprenal.00535.2015
52. Lemos DR, McMurdo M, Karaca G, Wilflingseder J, Leaf IA, Gupta N, et al. Interleukin-1β activates a MYC-dependent metabolic switch in kidney stromal cells necessary for progressive tubulointerstitial fibrosis. J Am Soc Nephrol. (2018) 29:1690–705. doi: 10.1681/ASN.2017121283
53. Kwan B, Fuhrer T, Zhang J, Darshi M, Van Espen B, Montemayor D, et al. Metabolomic markers of kidney function decline in patients with diabetes: evidence from the Chronic Renal Insufficiency Cohort (CRIC) study. Am J Kidney Dis. (2020) 76:511–20. doi: 10.1053/j.ajkd.2020.01.019
54. Niles DJ, Gordon JW, Huang G, Reese S, Adamson EB, Djamali A, et al. Evaluation of renal metabolic response to partial ureteral obstruction with hyperpolarized 13 C MRI. NMR Biomed. (2018) 31:3846. doi: 10.1002/nbm.3846
55. Legouis D, Faivre A, Cippà PE, de Seigneux S. Renal gluconeogenesis: an underestimated role of the kidney in systemic glucose metabolism. Nephrol Dial Transplant. (2020) gfaa302. doi: 10.1093/ndt/gfaa302
56. Meyer C, Stumvoll M, Dostou J, Welle S, Haymond M, Gerich J. Renal substrate exchange and gluconeogenesis in normal postabsorptive humans. Am J Physiol-Endocrinol Metab. (2002) 282:E428–34. doi: 10.1152/ajpendo.00116.2001
57. Stumvoll M, Chintalapudi U, Perriello G, Welle S, Gutierrez O, Gerich J. Uptake and release of glucose by the human kidney. Postabsorptive rates and responses to epinephrine. J Clin Invest. (1995) 96:2528–33. doi: 10.1172/JCI118314
58. Schoolwerth AC, Smith BC, Culpepper RM. Renal gluconeogenesis. Miner Electrolyte Metab. (1988) 14:347–61.
59. Petersen KF, Price T, Cline GW, Rothman DL, Shulman GI. Contribution of net hepatic glycogenolysis to glucose production during the early postprandial period. Am J Physiol-Endocrinol Metab. (1996) 270:E186–91. doi: 10.1152/ajpendo.1996.270.1.E186
60. Chandramouli V, Ekberg K, Schumann WC, Kalhan SC, Wahren J, Landau BR. Quantifying gluconeogenesis during fasting. Am J Physiol-Endocrinol Metab. (1997) 273:E1209–15. doi: 10.1152/ajpendo.1997.273.6.E1209
61. McGuinness OP, Fugiwara T, Murrell S, Bracy D, Neal D, O'Connor D, et al. Impact of chronic stress hormone infusion on hepatic carbohydrate metabolism in the conscious dog. Am J Physiol. (1993) 265:E314–322. doi: 10.1152/ajpendo.1993.265.2.E314
62. Silva P, Ross B, Spokes K. Competition between sodium reabsorption and gluconeogenesis in kidneys of steroid-treated rats. Am J Physiol. (1980) 238:F290–295. doi: 10.1152/ajprenal.1980.238.4.F290
63. Filsell OH, Jarrett IG, Taylor PH, Keech DB. Effects of fasting, diabetes and glucocorticoids on gluconeogenic enzymes in the sheep. Biochim Biophys Acta. (1969) 184:54–63. doi: 10.1016/0304-4165(69)90098-1
64. Flores H, Alleyne GA. Phosphoenolpyruvate carboxykinase of kidney. Subcellular distribution and response to acid-base changes. Biochem J. (1971) 123:35–9. doi: 10.1042/bj1230035
65. Joseph PK, Subrahmanyam K. Evaluation of the rate-limiting steps in the pathway of glucose metabolism in kidney cortex of normal, diabetic, cortisone-treated and growth hormone-treated rats. Biochem J. (1972) 128:1293–301. doi: 10.1042/bj1281293
66. Sasaki M, Sasako T, Kubota N, Sakurai Y, Takamoto I, Kubota T, et al. Dual regulation of gluconeogenesis by insulin and glucose in the proximal tubules of the kidney. Diabetes. (2017) 66:2339–50. doi: 10.2337/db16-1602
67. Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. (2004) 117:421–6. doi: 10.1016/S0092-8674(04)00452-0
68. Meyer C, Dostou J, Nadkarni V, Gerich J. Effects of physiological hyperinsulinemia on systemic, renal, and hepatic substrate metabolism. Am J Physiol. (1998) 275:F915–921. doi: 10.1152/ajprenal.1998.275.6.F915
69. Cersosimo E, Garlick P, Ferretti J. Insulin regulation of renal glucose metabolism in humans. Am J Physiol. (1999) 276:E78–84. doi: 10.1152/ajpendo.1999.276.1.E78
70. Kersten S, Stienstra R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie. (2017) 136:75–84. doi: 10.1016/j.biochi.2016.12.019
71. Fatima K, Mathew S, Faheem M, Mehmood T, Yassine HM, Al Thani AA, et al. The dual specificity role of transcription factor FOXO in type 2-diabetes and cancer. Curr Pharm Des. (2018) 24:2839–48. doi: 10.2174/1381612824666180911114210
72. Burch HB, Narins RG, Chu C, Fagioli S, Choi S, McCarthy W, et al. Distribution along the rat nephron of three enzymes of gluconeogenesis in acidosis and starvation. Am J Physiol. (1978) 235:F246–253. doi: 10.1152/ajprenal.1978.235.3.F246
73. Tayek JA, Katz J. Glucose production, recycling, and gluconeogenesis in normals and diabetics: a mass isotopomer [U-13C]glucose study. Am J Physiol. (1996) 270:E709–717. doi: 10.1152/ajpendo.1996.270.4.E709
74. Legouis D, Ricksten S-E, Faivre A, Verissimo T, Gariani K, Verney C, et al. Altered proximal tubular cell glucose metabolism during acute kidney injury is associated with mortality. Nat Metab. (2020) 2:732–43. doi: 10.1038/s42255-020-0238-1
75. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. (2006) 3:187–97. doi: 10.1016/j.cmet.2006.01.012
76. Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. (1996) 271:32529–37. doi: 10.1074/jbc.271.51.32529
77. Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol. (2021) 599:23–37. doi: 10.1113/JP280572
78. Hua W, ten Dijke P, Kostidis S, Giera M, Hornsveld M. TGFβ-induced metabolic reprogramming during epithelial-to-mesenchymal transition in cancer. Cell Mol Life Sci. (2020) 77:2103–23. doi: 10.1007/s00018-019-03398-6
79. Soukupova J, Malfettone A, Hyroššová P, Hernández-Alvarez M-I, Peñuelas-Haro I, Bertran E, et al. Role of the transforming growth factor-β in regulating hepatocellular carcinoma oxidative metabolism. Sci Rep. (2017) 7:12486. doi: 10.1038/s41598-017-12837-y
80. Srivastava SP, Li J, Kitada M, Fujita H, Yamada Y, Goodwin JE, et al. SIRT3 deficiency leads to induction of abnormal glycolysis in diabetic kidney with fibrosis. Cell Death Dis. (2018) 9:1–14. doi: 10.1038/s41419-018-1057-0
81. Miguel V, Tituaña J, Herrero JI, Herrero L, Serra D, Cuevas P, et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J Clin Invest. (2021) 131:140695. doi: 10.1172/JCI140695
82. Zhang G, Darshi M, Sharma K. The warburg effect in diabetic kidney disease. Semin Nephrol. (2018) 38:111–20. doi: 10.1016/j.semnephrol.2018.01.002
83. Liu Y, Zhang Z, Wang J, Chen C, Tang X, Zhu J, et al. Metabolic reprogramming results in abnormal glycolysis in gastric cancer: a review. OncoTargets Ther. (2019) 12:1195–204. doi: 10.2147/OTT.S189687
84. Wu H, Yang P, Hu W, Wang Y, Lu Y, Zhang L, et al. Overexpression of PKM2 promotes mitochondrial fusion through attenuated p53 stability. Oncotarget. (2016) 7:78069–82. doi: 10.18632/oncotarget.12942
85. Yang J, Kalhan SC, Hanson RW. What is the metabolic role of phosphoenolpyruvate carboxykinase? J Biol Chem. (2009) 284:27025–9. doi: 10.1074/jbc.R109.040543
86. Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. (2002) 277:30409–12. doi: 10.1074/jbc.R200006200
87. Hakimi P, Johnson MT, Yang J, Lepage DF, Conlon RA, Kalhan SC, et al. Phosphoenolpyruvate carboxykinase and the critical role of cataplerosis in the control of hepatic metabolism. Nutr Metab. (2005) 2:33. doi: 10.1186/1743-7075-2-33
88. Hou X, Shen YH, Li C, Wang F, Zhang C, Bu P, et al. PPARalpha agonist fenofibrate protects the kidney from hypertensive injury in spontaneously hypertensive rats via inhibition of oxidative stress and MAPK activity. Biochem Biophys Res Commun. (2010) 394:653–9. doi: 10.1016/j.bbrc.2010.03.043
89. Georgakopoulos-Soares I, Chartoumpekis DV, Kyriazopoulou V, Zaravinos A. EMT factors and metabolic pathways in cancer. Front Oncol. (2020) 10:499. doi: 10.3389/fonc.2020.00499
90. Dong C, Yuan T, Wu Y, Wang Y, Fan TWM, Miriyala S, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. (2013) 23:316–31. doi: 10.1016/j.ccr.2013.01.022
91. Schley G, Klanke B, Schödel J, Forstreuter F, Shukla D, Kurtz A, et al. Hypoxia-inducible transcription factors stabilization in the thick ascending limb protects against ischemic acute kidney injury. J Am Soc Nephrol. (2011) 22:2004–15. doi: 10.1681/ASN.2010121249
92. Wang J, Biju MP, Wang M-H, Haase VH, Dong Z. Cytoprotective effects of hypoxia against cisplatin-induced tubular cell apoptosis: involvement of mitochondrial inhibition and p53 suppression. J Am Soc Nephrol. (2006) 17:1875–85. doi: 10.1681/ASN.2005121371
93. Kim J, Devalaraja-Narashimha K, Padanilam BJ TIGAR. regulates glycolysis in ischemic kidney proximal tubules. Am J Physiol Renal Physiol. (2015) 308:F298–308. doi: 10.1152/ajprenal.00459.2014
94. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. (2014) 39:347–54. doi: 10.1016/j.tibs.2014.06.005
95. Stokman G, Kors L, Bakker PJ, Rampanelli E, Claessen N, Teske GJD, et al. NLRX1 dampens oxidative stress and apoptosis in tissue injury via control of mitochondrial activity. J Exp Med. (2017) 214:2405–20. doi: 10.1084/jem.20161031
96. Zhou H-L, Zhang R, Anand P, Stomberski CT, Qian Z, Hausladen A, et al. Metabolic reprogramming by the S -nitroso-CoA reductase system protects against kidney injury. Nature. (2019) 565:96–100. doi: 10.1038/s41586-018-0749-z
97. Andres-Hernando A, Li N, Cicerchi C, Inaba S, Chen W, Roncal-Jimenez C, et al. Protective role of fructokinase blockade in the pathogenesis of acute kidney injury in mice. Nat Commun. (2017) 8:14181. doi: 10.1038/ncomms14181
98. Li J, Liu H, Takagi S, Nitta K, Kitada M, Srivastava SP, et al. Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubules. JCI Insight. (2020) 5:129034. doi: 10.1172/jci.insight.129034
99. Kim JH, Ko HY, Wang HJ, Lee H, Yun M, Kang ES. Effect of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on gluconeogenesis in proximal renal tubules. Diabetes Obes Metab. (2020) 22:373–82. doi: 10.1111/dom.13905
100. Perlman A, Heyman SN, Matok I, Stokar J, Muszkat M, Szalat A. Acute renal failure with sodium-glucose-cotransporter-2 inhibitors: analysis of the FDA adverse event report system database. Nutr Metab Cardiovasc Dis. (2017) 27:1108–13. doi: 10.1016/j.numecd.2017.10.011
101. Nadkarni GN, Ferrandino R, Chang A, Surapaneni A, Chauhan K, Poojary P, et al. Acute kidney injury in patients on SGLT2 inhibitors: a propensity-matched analysis. Diabetes Care. (2017) 40:1479–85. doi: 10.2337/dc17-1011
102. Neuen BL, Young T, Heerspink HJL, Neal B, Perkovic V, Billot L, et al. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: a systematic review and meta-analysis. Lancet Diabetes Endocrinol. (2019) 7:845–54. doi: 10.1016/S2213-8587(19)30256-6
103. Iskander C, Cherney DZ, Clemens KK, Dixon SN, Harel Z, Jeyakumar N, et al. Use of sodium–glucose cotransporter-2 inhibitors and risk of acute kidney injury in older adults with diabetes: a population-based cohort study. Can Med Assoc J. (2020) 192:E351–60. doi: 10.1503/cmaj.191283
104. Pasternak B, Wintzell V, Melbye M, Eliasson B, Svensson A-M, Franzén S, et al. Use of sodium-glucose co-transporter 2 inhibitors and risk of serious renal events: Scandinavian cohort study. BMJ. (2020) 29:m1186. doi: 10.1136/bmj.m1186
105. Kale A, Sankrityayan H, Anders H-J, Bhanudas Gaikwad A. Klotho: A possible mechanism of action of SGLT2 inhibitors preventing episodes of acute kidney injury and cardiorenal complications of diabetes. Drug Discov Today. (2021) 26:1963–71. doi: 10.1016/j.drudis.2021.04.007
106. Maayah ZH, Ferdaoussi M, Takahara S, Soni S, Dyck JRB. Empagliflozin suppresses inflammation and protects against acute septic renal injury. Inflammopharmacology. (2021) 29:269–79. doi: 10.1007/s10787-020-00732-4
107. Shu S, Wang Y, Zheng M, Liu Z, Cai J, Tang C, et al. Hypoxia and hypoxia-inducible factors in kidney injury and repair. Cells. (2019) 8. doi: 10.3390/cells8030207
108. Blantz RC. Phenotypic characteristics of diabetic kidney involvement. Kidney Int. (2014) 86:7–9. doi: 10.1038/ki.2013.552
109. Qi W, Keenan HA, Li Q, Ishikado A, Kannt A, Sadowski T, et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med. (2017) 23:753–62. doi: 10.1038/nm.4328
110. Yin X-N, Wang J, Cui L-F, Fan W-X. Enhanced glycolysis in the process of renal fibrosis aggravated the development of chronic kidney disease. Eur Rev Med Pharmacol Sci. (2018) 22:4243–51. doi: 10.26355/eurrev_201807_15419
111. Lanaspa MA, Ishimoto T, Cicerchi C, Tamura Y, Roncal-Jimenez CA, Chen W, et al. Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. J Am Soc Nephrol. (2014) 25:2526–38. doi: 10.1681/ASN.2013080901
112. Nakagawa T, Sanchez-Lozada LG, Andres-Hernando A, Kojima H, Kasahara M, Rodriguez-Iturbe B, et al. Endogenous fructose metabolism could explain the warburg effect and the protection of SGLT2 inhibitors in chronic kidney disease. Front Immunol. (2021) 12:694457. doi: 10.3389/fimmu.2021.694457
113. Ding H, Jiang L, Xu J, Bai F, Zhou Y, Yuan Q, et al. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am J Physiol Renal Physiol. (2017) 313:F561–75. doi: 10.1152/ajprenal.00036.2017
114. Cao H, Luo J, Zhang Y, Mao X, Wen P, Ding H, et al. Tuberous sclerosis 1 (Tsc1) mediated mTORC1 activation promotes glycolysis in tubular epithelial cells in kidney fibrosis. Kidney Int. (2020) 98:686–98. doi: 10.1016/j.kint.2020.03.035
115. Rowe I, Chiaravalli M, Mannella V, Ulisse V, Quilici G, Pema M, et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med. (2013) 19:488–93. doi: 10.1038/nm.3092
116. Chiaravalli M, Rowe I, Mannella V, Quilici G, Canu T, Bianchi V, et al. 2-Deoxy-d-glucose ameliorates PKD progression. J Am Soc Nephrol. (2016) 27:1958–69. doi: 10.1681/ASN.2015030231
117. Riwanto M, Kapoor S, Rodriguez D, Edenhofer I, Segerer S, Wüthrich RP. Inhibition of aerobic glycolysis attenuates disease progression in polycystic kidney disease. PLoS ONE. (2016) 11:e0146654. doi: 10.1371/journal.pone.0146654
118. Bartman CR, TeSlaa T, Rabinowitz JD. Quantitative flux analysis in mammals. Nat Metab. (2021) 3:896–908. doi: 10.1038/s42255-021-00419-2
119. Jang C, Hui S, Zeng X, Cowan AJ, Wang L, Chen L, et al. Metabolite exchange between mammalian organs quantified in pigs. Cell Metab. (2019) 30:594–606.e3. doi: 10.1016/j.cmet.2019.06.002
120. Passos R da H, Ramos JGR, Gobatto A, Mendonça EJB, Miranda EA, Dutra FRD, et al. Lactate clearance is associated with mortality in septic patients with acute kidney injury requiring continuous renal replacement therapy: a cohort study. Medicine. (2016) 95:e5112. doi: 10.1097/MD.0000000000005112
121. Shen Y, Jiang L, Wen P, Ye Y, Zhang Y, Ding H, et al. Tubule-derived lactate is required for fibroblast activation in acute kidney injury. Am J Physiol Renal Physiol. (2020) 318:F689–701. doi: 10.1152/ajprenal.00229.2019
122. Gjorgjieva M, Monteillet L, Calderaro J, Mithieux G, Rajas F. Polycystic kidney features of the renal pathology in glycogen storage disease type I: possible evolution to renal neoplasia. J Inherit Metab Dis. (2018) 41:955–63. doi: 10.1007/s10545-018-0207-y
123. Landau BR, Wahren J, Chandramouli V, Schumann WC, Ekberg K, Kalhan SC. Contributions of gluconeogenesis to glucose production in the fasted state. J Clin Invest. (1996) 98:378–85. doi: 10.1172/JCI118803
124. Action to Control Cardiovascular Risk in Diabetes Study Group, Gerstein HC, Miller ME, Byington RP, Goff DC Jr, Bigger JT, et al. Effects of intensive glucose lowering in Type 2 diabetes. N Engl J Med. (2008) 358:2545–59. doi: 10.1056/NEJMoa0802743
125. Papademetriou V, Lovato L, Doumas M, Nylen E, Mottl A, Cohen RM, et al. Chronic kidney disease and intensive glycemic control increase cardiovascular risk in patients with type 2 diabetes. Kidney Int. (2015) 87:649–59. doi: 10.1038/ki.2014.296
126. Chakravarty K, Cassuto H, Reshef L, Hanson RW. Factors that control the tissue-specific transcription of the gene for phosphoenolpyruvate carboxykinase-C. Crit Rev Biochem Mol Biol. (2005) 40:129–54. doi: 10.1080/10409230590935479
127. Weiner ID, Verlander JW. Renal ammonia metabolism and transport. Compr Physiol. (2013) 3:201–20. doi: 10.1002/cphy.c120010
128. Bürki R, Mohebbi N, Bettoni C, Wang X, Serra AL, Wagner CA. Impaired expression of key molecules of ammoniagenesis underlies renal acidosis in a rat model of chronic kidney disease. Nephrol Dial Transplant. (2015) 30:770–81. doi: 10.1093/ndt/gfu384
129. Moret C, Dave MH, Schulz N, Jiang JX, Verrey F, Wagner CA. Regulation of renal amino acid transporters during metabolic acidosis. Am J Physiol Renal Physiol. (2007) 292:F555–566. doi: 10.1152/ajprenal.00113.2006
130. Leaf DE, Wolf M, Waikar SS, Chase H, Christov M, Cremers S, et al. FGF-23 levels in patients with AKI and risk of adverse outcomes. Clin J Am Soc Nephrol. (2012) 7:1217–23. doi: 10.2215/CJN.00550112
131. Heine GH, Seiler S, Fliser D. FGF-23: the rise of a novel cardiovascular risk marker in CKD. Nephrol Dial Transplant. (2012) 27:3072–81. doi: 10.1093/ndt/gfs259
132. Marthi A, Donovan K, Haynes R, Wheeler DC, Baigent C, Rooney CM, et al. Fibroblast growth factor-23 and risks of cardiovascular and noncardiovascular diseases: a meta-analysis. J Am Soc Nephrol. (2018) 29:2015–27. doi: 10.1681/ASN.2017121334
133. Simic P, Kim W, Zhou W, Pierce KA, Chang W, Sykes DB, et al. Glycerol-3-phosphate is an FGF23 regulator derived from the injured kidney. J Clin Invest. (2020) 130:1513–26. doi: 10.1172/JCI131190
134. Festuccia WTL, Kawashita NH, Garofalo MR, Moura MF, Brito SRC, Kettelhut IC, et al. Control of glyceroneogenic activity in rat brown adipose tissue. Am J Physiol Regul Integr Comp Physiol. (2003) 285:R177–82. doi: 10.1152/ajpregu.00713.2002
Keywords: glycolysis, gluconeogenesis, metabolic shift, acute kidney injury, chronic kidney disease
Citation: Faivre A, Verissimo T, Auwerx H, Legouis D and de Seigneux S (2021) Tubular Cell Glucose Metabolism Shift During Acute and Chronic Injuries. Front. Med. 8:742072. doi: 10.3389/fmed.2021.742072
Received: 15 July 2021; Accepted: 11 October 2021;
Published: 29 October 2021.
Edited by:
Katalin Susztak, University of Pennsylvania, United StatesReviewed by:
Hee-Seong Jang, Icahn School of Medicine at Mount Sinai, United StatesCopyright © 2021 Faivre, Verissimo, Auwerx, Legouis and de Seigneux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie de Seigneux, U29waGllLmRlc2VpZ25ldXhAaGN1Z2UuY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.