 Yuanji Dong
Yuanji Dong Jixin Zhong
Jixin Zhong Lingli Dong
Lingli Dong- Department of Rheumatology and Immunology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Interleukin-33 (IL-33) is a nuclear factor mainly expressed in barrier epithelium, endothelial cells, and fibroblast reticular cells. Some inflammatory cells also express IL-33 under certain conditions. The important role of IL-33 in allergic reactions, helminth infection, cancer, tissue fibrosis, chronic inflammation, organ transplantation, and rheumatic immune diseases has been extensively studied in recent years. IL-33 primarily activates various circulating and tissue-resident immune cells, including mast cell, group 2 innate lymphoid cell (ILC2), regulatory T cell (Treg), T helper 2 cell (Th2), natural killer cell (NK cell), and macrophage. Therefore, IL-33 plays an immunomodulatory role and shows pleiotropic activity in different immune microenvironments. The IL-33/serum stimulation-2 (ST2) axis has been shown to have a detrimental effect on rheumatoid arthritis, systemic lupus erythematosus, and other rheumatic diseases. Interestingly, IL-33 also plays a protective role in the repair of barrier epithelium and the activation of Tregs. Therefore, the role of IL-33/ST2 depends on the underlying pathological conditions in rheumatic diseases. This review focuses on the dual role of the IL-33/ST2 axis in rheumatic diseases.
Introduction
Interleukin-33 (IL-33), a member of the IL-1 family, was first discovered in human tissues in 2003 and was originally defined as a nuclear factor of high endothelial venules (NF-HEV) (1). In 2005, Schmitz et al. reported that the C-terminal (amino acids from 112 to 270) of NF-HEV exhibited an IL-1-like three-dimensional folding and induced a type 2 immune response through binding to its receptor serum stimulation-2 (ST2) (2). In 2006, the identity between IL-33 and NF-HEV and its role as a chromatin-related nuclear factor was further confirmed (3). IL-33 is produced by various cell types such as endothelial cells, epithelial cells, macrophages, fibroblasts, adipose progenitor cells, and dendritic cells. Under conditions of cell damage, necrosis, necroptosis, stress, and virus infection, it is released as a pro-inflammatory factor and activates different types of immune cells (4–7). The role of IL-33 in type 2 immune diseases has been extensively studied in allergic reactions, asthma, and parasitic infections (6). However, it is well-known that HEV is involved in the activation and mobilization of lymphocytes, indicating that IL-33 may also be involved in chronic inflammation (8, 9). Rheumatic diseases are chronic inflammatory disorders in which the immune system attacks itself and organs of the body. As an incurable condition so far, it brings a heavy burden to individuals and society (10). A growing number of studies have demonstrated a critical role of the IL-33/ST2 axis in rheumatic diseases, including systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), primary Sjögren's syndrome (pSS), systemic sclerosis (SSc), psoriatic arthritis (PsA), gout, IgG4-related diseases, and ankylosing spondylitis (AS), indicating a promising potential for IL-33/ST2-targeting therapy in rheumatic disease (10).
Biology of IL-33
The Distribution and Function of IL-33
Unlike some cytokines, which have classical secretion patterns, IL-33 is normally localized in the nucleus (11, 12). Although the localization of IL-33 in the cytoplasm has been reported in the literature, the results were not obtained under normal conditions. This ectopic expression was observed in murine cell line NIH3T3 that expressed tetracysteine-labeled human IL-33 by genetic engineering. Because the cysteine residues can change the folding of IL-33 and the fluorescence staining has not been tested by knockout, the results of cytoplasmic localization need to be treated with caution (13). The N-terminal domain of IL-33 shows evolutionary conservation and is closely related to the nuclear location of IL-33. The N-terminal domain of IL-33 was initially thought to contain homologous domain-like structures bound to deoxyribonucleic acid (DNA), but this has not been confirmed. In fact, IL-33 binds to DNA via protein–protein interactions. Through the tight hairpin structure of the chromatin-binding protein, it is combined with the acid pocket formed by histone 2A (H2A) and histone 2B (H2B) (1, 14). Although IL-33 is located in the nucleus, it does not appear to regulate the expression of genes. The nuclear localization of IL-33 seems to regulate the activity of IL-33 as a cytokine (15, 16). IL-33 is released outside of the cell and has a variety of immunological effects. Initially, researchers believed that the full-length IL-33 should be processed to be biologically active, and in the next few years, it was considered to be activated by caspase-1 and inflammasome, similar to IL-1β and IL-18. However, in 2009, Girard et al. reported that full-length IL-33 could interact with ST2 and activate nuclear factor-kappa B (NF-κB) activity to induce cytokine production (17, 18). Meanwhile, further studies also found the inflammatory protease hydrolysis site of IL-33. An 18- to 21-kilodalton (kDa) mature form can be produced when IL-33 is cleaved, with its biological activity level increased by 10–30 times (17, 19, 20). Although inflammatory proteases are able to convert IL-33 into a more active mature form, they may also result in the inactivation of IL-33 through protein degradation. This degradation has been observed in chymotrypsin. In addition, the endogenous caspase-3 can cleave at the DGVDG site in the C-terminal IL-1-like domain of IL-33 to inactivate IL-33. This structure is specific to IL-33, indicating that IL-33 is strictly regulated in the process of apoptosis (21). There was evidence showing that recombinant caspase-3 and caspase-7 could cleave IL-33 in vitro. Caspase-1 had no direct effect on IL-33 but could inactivate IL-33 by activating caspase-7 (22). In addition, when IL-33 was released into the extracellular microenvironment, it was quickly inactivated by the formation of two disulfide bonds. The oxidation of cysteine residues resulted in conformational changes and subsequent reduction in binding affinity to ST2. This regulation mechanism occurred much faster than protein degradation (23). Therefore, after a 2-h exposure to allergens, no biologically active IL-33 could be detected in the alveolar lavage fluid (23). Furthermore, IL-33 was found to accumulate in several models within a few hours after release and was not detectable after 6 h (24–26). These also reflect that IL-33 is a short-acting protein, and its biological role in vivo is precisely regulated.
In both physical and pathological inflammatory conditions, the main cellular sources of IL-33 are not CD45+ hematopoietic cells. Endothelial cells, epithelial cells, fibroblasts, and myofibroblasts in humans and mice were demonstrated to be the main cells expressing IL-33 (27). In addition to the epithelial barrier tissue and lymphatic organs, IL-33 was abundantly expressed in the brain and eyes of mice and weakly expressed in visceral smooth muscle cells of the human gastrointestinal tract and urogenital tract. Although several studies suggested that CD45+ hematopoietic cells may be a source of IL-33 even a major source, stronger evidence is needed (28–34). In an IL-33 luciferin reporter mouse model, no IL-33 was detected in CD45+ hematopoietic cells (including macrophages, dendritic cells, T cells, B cells, eosinophil, and neutrophil) in the lung of the mice with allergic pneumonia (35). It cannot be ruled out that certain leukocyte subsets may produce low levels of functional IL-33, but more experiments are still needed to verify it.
IL-33 Signaling Pathway
The receptor ST2, also known as DER4, Fit-1, or T1, is one of the co-receptors of IL-33 and is mainly encoded by the IL-1RL1 gene. Before the discovery of IL-33, ST2 was considered as an orphan receptor, and now IL-33 is still the only ligand of ST2 (36, 37). Three isoforms of ST2 have been identified in humans, all of which are produced by alternative splicing: transmembrane receptor type (ST2L), soluble form (sST2), and variant ST2 (ST2V) (38–40). The soluble form acts as a decoy receptor to antagonize IL-33. When IL-33 binds to the transmembrane ST2 receptor, the membrane-anchored ST2 will recruit IL-1 receptor accessory protein (IL-1RAcP) to form a dimer, resulting in the dimerization of its intracellular domain. The adaptor protein myeloid differentiation protein 88 (MyD88) is recruited through the dimerization of Toll/IL-1 receptor (TIR) to activate downstream kinases. IL-1R-associated kinase 1 (IRAK1) and IL-1R-associated kinase 1 (IRAK4) and TNF receptor-associated factor 6 (TRAF6) are then activated, which ultimately leads to the activation of mitogen-activated protein kinases (MAPKs) and NF-κB transcription factor (41–44). This signal pathway is very similar to that of IL-1β and IL-18. Unlike IL-1RAcP, IL-33 stimulation induces a binding of ST2 with single immunoglobulin domain IL-1R-related molecule (SIGIRR) to form a complex, which can inhibit the formation of intracellular dimers and activate the ubiquitin–proteasome system (45) (Figure 1).
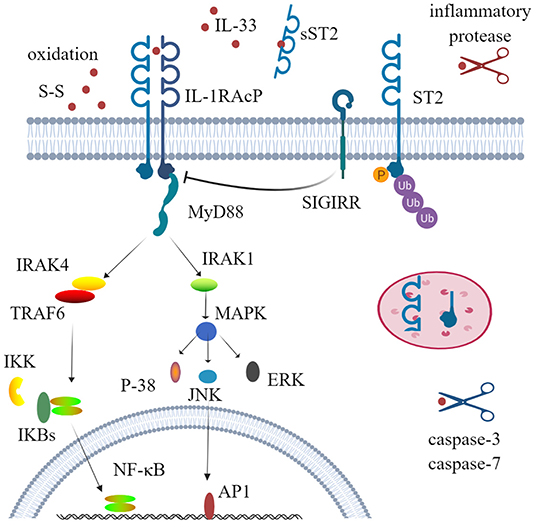
Figure 1. Physiological function and activity regulation of interleukin-33 (IL-33). IL-33 firstly binds to its transmembrane receptor serum stimulation-2 (ST2) to induce conformational changes; then ST2 interacts with IL-1RAcP and recruits the downstream adaptor MyD88, IRAK1, IRAK4, and TRAF6 via Toll/IL-1 receptor domains; and ultimately activates nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinases (ERK, p38, and JNK). This signal pathway can also be inhibited by phosphorylation and ubiquitylation of ST2 and SIGIRR, which disrupt ST2 and IL-1RAcP dimerization. Several mechanisms are involved to regulate the activity of IL-33. Processing by inflammatory proteases can greatly increase (up to 30-fold) cytokine activity, while caspase-3 and caspase-7 lead to cytokine inactivity. Decoy receptor soluble ST2 (sST2) and rapid oxidation of IL-33 are also crucial mechanisms to limit the activity of IL-33. SIGIRR, single immunoglobulin domain IL-1 receptor-related molecule; IRAK, IL-1R-associated kinase; TRAF6, TNF receptor-associated factor 6; ERK, extracellular signal-regulated kinase.
Effects of IL-33 on Tissue Cells and Innate Immune Cells
Many types of cells express IL-33 receptors, including epithelial cells, endothelial cells, fibroblasts, and osteoblasts. IL-33 activates extracellular signal-regulated kinase (ERK) and p38 MAPK signaling pathways in primary human lung epithelial and endothelial cells to produce IL-8, which are associated with chronic airway inflammation (46). In addition, the IL-33 receptor ST2 is expressed on a variety of innate immune cells. IL-33/ST2 activation in mast cells not only promotes the activation and maturation of mast cells but also enhances Th17 response during airway inflammation (47, 48). In macrophages, IL-33/ST2 signaling enhances their activation by upregulating Toll-like receptor 4 (TLR4), myeloid differentiation protein-2 (MD2), and MyD88 (49). In the dendritic cells, the administration of IL-33 not only increases the levels of CD80, CD40, and C-C motif chemokine receptor 7 (CCR7) but also increases the production of IL-5, C-C motif chemokine ligand 17 (CCL17), and tumor necrosis factor alpha (TNF-α) (50). Therefore, IL-33, as an alarmin and damage-associated molecular pattern (DAMP) molecule, can activate tissue cells and innate immune cells; upregulate costimulatory molecules, adhesion molecules, and chemokines; and initiate and maintain innate immunity (Figure 2).
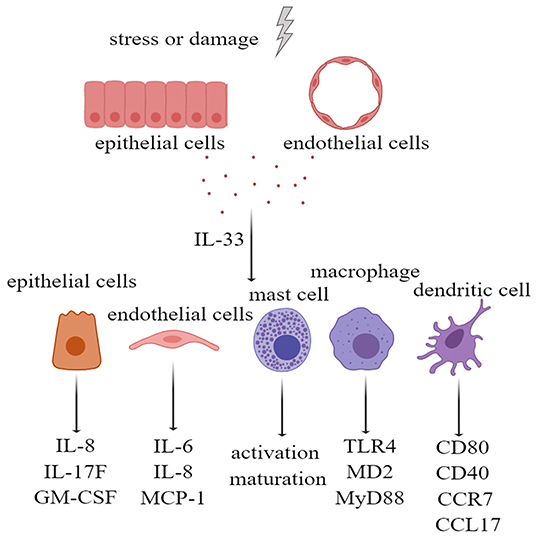
Figure 2. Interleukin-33 (IL-33), as alarmin, activates tissue cells and a variety of innate immune cells. Extracellular IL-33, acting on bronchial epithelial cells, led to upregulation of IL-8, IL-17F, and GM-CSF. IL-33 also upregulated IL-6, IL-8, and MCP-1 by stimulating pulmonary endothelial cells. IL-33 not only stimulated the activation and maturation of mast cells, and upregulation of TLR4, MD2, and MyD88 in macrophages, but also upregulated CD80, CD40, CCR7, and CCL17 in dendritic cells. GM-CSF, granulocyte-macrophage colony stimulating factor; MCP-1, monocyte chemoattractant protein-1; TLR-4, Toll-like receptor 4; MD2, myeloid differentiation protein-2; MyD88, myeloid differential protein-88; CCR7, cxc chemokine receptor 7; CCL17, chemokine (C-C motif) ligand 17.
IL-33 Indirectly Promotes the Production of IFN-γ
Interferon-gamma (IFN-γ) plays an important role in the development of rheumatic immune disease. IFN-γ can activate macrophages or other immune cells to aggravate tissue damage and can promote the ectopic expression of MHC class II antigens on tissue cells, which may contribute to the presentation of autoantigen. Several studies have proven that IL-33, in the presence of IL-12, can increase the secretion of IFN-γ by NK cells, natural killer T cells (NKT cells), ILC1 cells, and Th1 cells (51–54). Therefore, in the progression of rheumatic disease, IL-33 may increase the production of IFN-γ and may amplify the immune effects (Figure 3). In addition, IL-33 is also thought to increase antibody levels in the immune response.
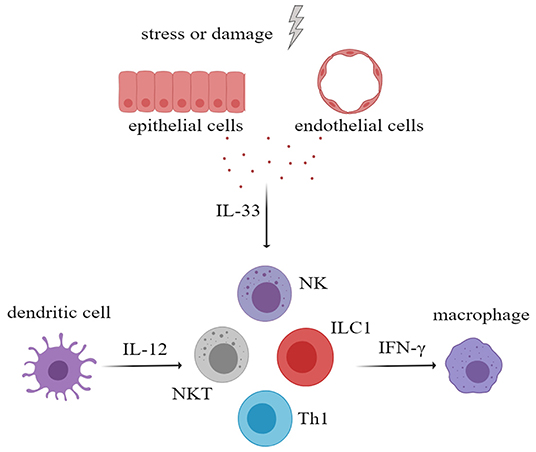
Figure 3. After activation of innate immune cells, especially myeloid dendritic cells, large amounts of interleukin-12 (IL-12) were secreted. IL-33 and IL-12 had a synergistic effect in stimulating NK cells, NKT cells, ILC1, and Th1 to secrete more IFN-γ, which will aggravate tissue damage. NK cells, natural killer cells; NKT cell, natural killer T cell; ILC, innate lymphoid cell.
IL-33 Promotes Tissue Repair and Fibrosis
Although IL-33 is an alarmin and is involved in inflammatory processes, there is evidence that IL-33 plays a role in wound healing and fibrosis. IL-33/ST2 signal enables M2 macrophages to promote the closure of damaged epidermis and angiogenesis, etc. In addition, M2 macrophages are also involved in tissue remodeling and fibrosis (55). In addition, IL-33 can act on eosinophils, mast cells, and ILC2 to increase their production of IL-13 and IL-5, which are closely related to fibrosis (56). More importantly, IL-33 can increase the number of ST2+ regulatory T cell (Treg), a pivotal type of cells in fibrogenesis and immunosuppression (57). IL-33 is also believed to affect the activity of matrix metalloproteinases and promote the deposition of extracellular matrix (56). All these indicate that IL-33 plays an important role in tissue repair and fibrosis (Figure 4).
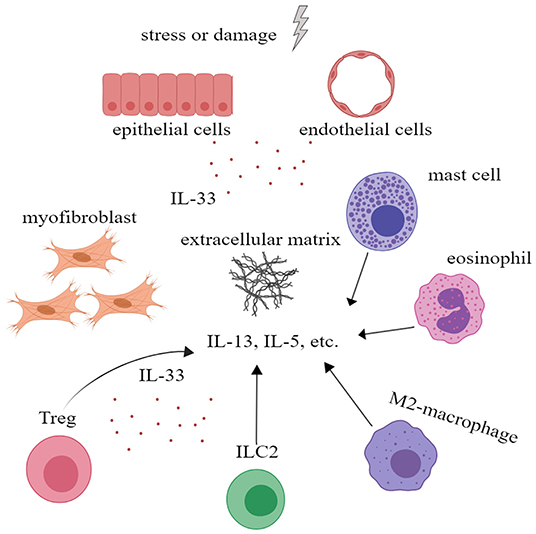
Figure 4. Interleukin-33 (IL-33) could promote tissue repair and fibrosis. IL-33 could promote the proliferation of Treg to further alleviate inflammation. IL-33 could also promote extracellular matrix deposition and tissue fibrosis by acting on mast cells, eosinophil, M2 macrophage, ILC2 cells, and Treg to increase the secretion of IL-5, IL-13, and other cytokines. ILC, innate lymphoid cell; Treg, regulatory T cell.
IL-33/ST2 Axis in Rheumatic Diseases
Rheumatic diseases are immune-mediated chronic inflammatory syndromes, which are characterized by the hyperactivity of effector Th1 cells and Th17 cells, dysfunction of Tregs, activation of autoreactive B cells, and production of autoantibodies (58). A growing number of studies have found that the level of IL-33 is associated with the severity of rheumatic disease, indicating that IL-33 and ST2 may be potential targets for predicting the development of disease and improving the clinical outcomes (59, 60). Next, we will discuss the role of the IL-33/ST2 axis in several common rheumatic diseases (Table 1, Figure 5).

Table 1. Expression and mechanisms of IL-33/ST2 in rheumatic diseases.
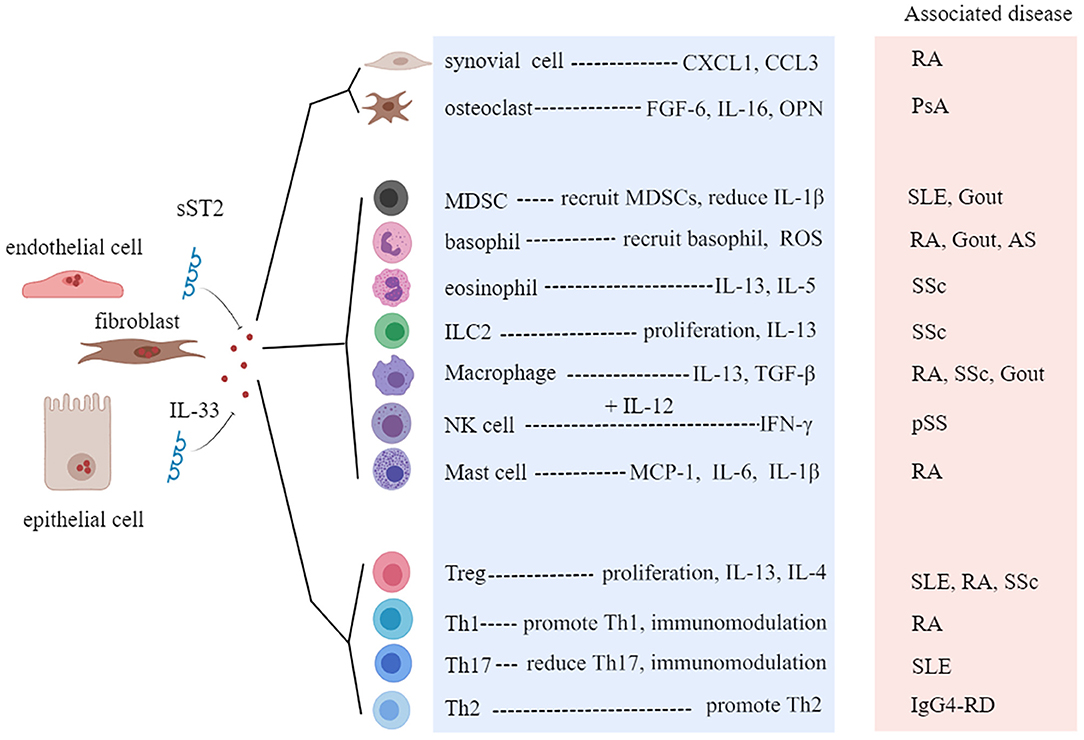
Figure 5. Roles of interleukin-33 (IL-33) in rheumatic diseases. sST2, soluble ST2; CXCL1, C-X-C motif chemokine ligand 1; FGF-6, fibroblast growth factor 6; OPN, osteopontin; MDSC, myeloid-derived suppressor cell; ROS, reactive oxygen species; ILC, innate lymphoid cell; Treg, regulatory T cell; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; pSS, primary Sjögren's syndrome; SSc, systemic sclerosis; PsA, psoriatic arthritis; IgG4-RD, IgG4-related disease; AS, ankylosing spondylitis.
Systemic Lupus Erythematosus
SLE is a chronic connective tissue disease of unknown etiology with multiple systemic involvements. The male-to-female ratio is about 1:9, and it is a major cause of death in young women with chronic inflammatory diseases (97). The prevalence of SLE is (30.13–70.41)/100,000 in China. The level of serum IL-33 was reported to be significantly higher in patients with SLE than that in healthy subjects and was positively correlated with erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), IgA, and Sjögren's syndrome antigen B (SSB) antibody levels (61). On the other hand, the serum soluble ST2 (sST2) level in patients was also significantly increased and was positively correlated with the level of anti-double-stranded DNA (dsDNA) antibodies and the disease activity index, while it was negatively correlated with the complement C3 (62). In addition, Guo et al. suggested that IL-33 and other pro-inflammatory cytokines might be involved in innate lymphoid cell disorder with a higher frequency of ILC1 in the peripheral blood of SLE patients (63). However, there were also other studies showing reduced or similar levels of serum IL-33 in SLE patients compared with healthy controls (62). This discrepancy is probably due to the detection efficacy of the enzyme-linked immunosorbent assay (ELISA) kit or the multiple roles of IL-33 plays in the disease (98). To further explore its effect, Li et al. found that the administration of neutralizing antibodies against IL-33 could reduce the mortality, serum anti-dsDNA level, and immune complex deposition in MRL/lpr lupus mice. The protective effect was associated with the increase of regulatory T cells and myeloid-derived suppressor cells and the reduction of Th17 cells and pro-inflammatory factors (64). These studies also indirectly supported the finding that two polymorphisms of the IL-33 gene (rs1929992-G and rs1891385-C) were associated with increased susceptibility to SLE (99–101). However, further studies are required to further explore the corresponding mechanism.
Rheumatoid Arthritis
RA is an autoimmune disease with erosive arthritis as the main manifestation and synovitis as the pathological basis (102). The male-to-female ratio is 1:4. In severe cases, patients with uncontrolled active RA may develop joint deformities and disabilities. The prevalence of RA in China is 0.28–0.41%. Immunopathology showed that the differentiation of Th1 lymphocytes increased and that the number of Treg decreased in RA patients (103). Elevated IL-33 levels were found in both serum and local joint synovial fluid in patients with RA (65, 66, 103). Serum sST2 levels were also elevated (68). Moreover, IL-33 levels were associated with RA severity parameters, such as rheumatoid factor (RF), anti-cyclic citrullinated peptide (anti-CCP) antibodies, IL-6, ESR, lung involvement, and bone erosion (68, 104–106). IL-33 was also expressed in synovial fibroblasts (69). Inflammatory factors (such as TNF-α) could stimulate synovial fibroblasts to produce IL-33; and IL-33 not only upregulated matrix metalloproteinase-3 (MMP-3), IL-8, and IL-6 but also upregulated B-cell lymphoma-2 (Bcl-2) to inhibit apoptosis and promote proliferation (107). Furthermore, the level of serum IL-33 also had a certain significance for the prediction of patient's response to the biological agents. For example, in patients with poor response to tumor necrosis factor inhibitor (TNFi), serum and synovial fluid IL-33 levels continued to rise (70). In the mouse model, IL-33 mRNA levels increased in the early stages of collagen-induced arthritis (CIA) (65). With the use of ST2 knockout mice and ST2 neutralizing antibodies in the CIA model, suppression of ST2 signaling alleviated arthritis symptoms and reduced levels of IL-17, TNF-α, and IFN-γ (65, 69). In contrast, the administration of IL-33 resulted in the exacerbation of arthritis (108). Interestingly, this effect was absent in mice lacking mast cells. Further studies showed that mast cells expressed high levels of ST2, which responded to IL-33 by producing various pro-inflammatory factors such as monocyte chemoattractant protein-1 (MCP-1), IL-6, and IL-1β (69). In addition, IL-33 could also recruit neutrophils by stimulating macrophages and synovial cells to release chemokines (such as CXCL1 and CCL3) (71). Although most studies support the deleterious effect of the IL-33/ST2 axis in the pathogenesis of RA, there are still some studies with opposite results. For example, repeated administration of IL-33 in the early and late stages of CIA mice models could relieve symptoms of arthritis. The protective mechanism might involve the regulation of immunity and the proliferation of Tregs (109). Despite the controversy, research from the gene polymorphism suggests that downregulating the expression of IL-33 shows resistance to disease. Further studies are needed to explore the specific role and molecular mechanisms of IL-33 in RA.
Primary Sjögren's Syndrome
pSS is a chronic systemic rheumatic disease mainly involving salivary and lacrimal glands. The male-to-female ratio is 1:(9–20) (110). The prevalence of pSS in China is 0.28–0.41%. Multiple studies have described the pathogenic role of the IL-33/ST2 axis in patients with pSS (72, 73, 111–113). Compared with those in the control group, serum IL-33 and sST levels were elevated in pSS patients. Although serum IL-33 levels and EULAR Sjögren's syndrome disease activity index (ESSDAI) or lymphocyte infiltration were not correlated, serum sST2 levels were significantly correlated with ESSDAI, disease duration, and thrombocytopenia (72, 73). Immunohistochemical staining showed that IL-33 and its receptors (ST2 and IL-1RAcP) were expressed in the salivary glands. The expression of IL-33 in patients with pSS showed a dynamic pattern: IL-33 was significantly increased in salivary glands with Chisholm scores of 2 and 3 but was expressed at a lower level in salivary glands with Chisholm scores of 1 and 4. Its receptors (ST2 and IL-1RAcP) were expressed in a similar pattern (73). In addition, a recent study showed that the levels of IL-33 in the tears of patients with pSS were also significantly increased. Furthermore, IL-33 levels were correlated with the degree of ocular involvement and levels of IL-4 and IL-5 in tears (74). However, IL-33 alone did not lead to the release of pro-inflammatory factors. But when combined with IL-12 and/or IL-23, it promoted the release of IFN-γ by up to 10 times in NK and NKT cells. Moreover, TNF-α, IL-1β, and IFN-γ in the inflammatory environment could further increase the activation of IL-33, forming positive feedback (60). Therefore, the targeting IL-33/ST2 axis may be a promising treatment option for pSS.
Systemic Sclerosis
SSc is a rheumatic disease with unknown etiology characterized by the deposition of the extracellular matrix and diffuse skin thickening (114). The male-to-female ratio is 1:(3–7), and the lence rate in China is about 0.026%. It was reported that the serum IL-33 level in patients with early SSc was higher than that of healthy controls and patients with advanced SSc. This might be due to the activation of endothelial cells in the early stages of the disease, and elevated IL-33 level was positively correlated with skin lesions, degree of sclerosis, and degree of pulmonary fibrosis (75, 76). In skin biopsies of healthy subjects, ST2 was only expressed in fibroblasts and endothelial cells at a low level, while IL-33 was constitutively expressed in keratinocytes and endothelial cells (77). However, in the early stage of SSc, ST2 was highly expressed in endothelial cells, macrophages, T cells, B cells, and myofibroblasts of the affected organs, while the expression of IL-33 in tissues was not significantly increased until the late stage of SSc (77). In fact, the IL-33/ST2 axis could polarize M2 macrophages to promote the production of TGF-β1 and IL-13 and could also induce the proliferation of ILC2 to promote the accumulation of eosinophil granulocyte and expression of IL-13. Moreover, in vitro experiments have shown that IL-33 could induce the differentiation of Tregs into Th2-like cells, resulting in the production of IL-4 and IL-13 and local Treg dysfunction (77–79). The study in our laboratory also found that IL-33 could directly promote the proliferation of primary human skin fibroblasts and their expression of collagen. The administration of ST2 neutralizing antibody was able to effectively alleviate bleomycin-induced skin fibrosis in mice. Moreover, the polymorphism of IL-33 gene rs7044343 is associated with SSc-associated dyspnea in the Chinese population and SSc susceptibility in the Turkish population (115, 116). However, further research is needed to determine the therapeutic effect of IL-33/ST2 targeting therapy in human SSc.
Psoriatic Arthritis
PsA is a chronic, inflammatory, musculoskeletal disease affecting the skin, peripheral joint, spine, nails, and entheses (117). It was reported that up to 30% of patients with psoriasis might develop PsA (118). Several studies showed that IL-33 not only played a role in the development of psoriasis but also participated in the progress of PsA (119). One study detected elevated serum IL-33 in patients with PsA, but there was no correlation with osteoclastogenesis-related cytokines and PSA joint activity index (PSAJAI) (80). In another study, however, IL-33 was not detected in serum and synovial fluid from PsA patients but only in endothelial cells of the synovium and synovial fibroblast (120). Another study focused on the effects of skin inflammation on bone damage. IL-33 together with IL-17 increased the gene expression of the pro-osteoclastogenic factor, such as fibroblast growth factor (FGF-6), IL-16, and osteopontin (OPN). Moreover, IL-33, together with OPN, IL-17, and TNF-α, could also induce the release of bone contributing factor receptor activator of NF-κB ligand (RANKL) in the skin, thus inducing the differentiation of osteoclast precursor (OCP) into monocytes (81). IL-33 was also expressed in the synovium in a mouse model of PsA. But there was no difference between IL-33−/− and wild-type (WT) mice in frequencies of Treg, Th1, and Th17 cells in this model (121). In conclusion, the present studies suggest that IL-33 is involved in the development of human PsA, while studies in mouse models are limited. Further studies are needed to obtain more evidence.
Gout
Gout is an inflammatory disease characterized by the deposition of uric acid crystals in the joints (122). The male-to-female ratio is 15:1, and its incidence in China is 1% to 3%. It was reported that higher levels of IL-33 and neutrophil counts were detected in joint synovial fluid in patients with gout than those in osteoarthritis (82). Direct injection of uric acid crystals into the articular cavity could induce acute attacks of gout in mice. In this mice model, exogenous administration of IL-33 could aggravate the production of reactive oxygen species, recruitment of neutrophils, and hyperalgesia. Correspondingly, knocking out ST2 could significantly alleviate oxidative stress and reduce neutrophils' recruitment into the ankle joint. It was also found that macrophages in gout could produce IL-33 and increase CXCL1, CCL 3, and IL-1β through an autocrine pattern. These results supported the pathogenic role of the IL-33/ST2 axis in gout (83). Paradoxically, IL-33 was also believed to alleviate the mouse peritonitis model induced by uric acid crystals and to reduce neutrophil counts as well as the production of IL-1β and IL-6 (84). The mechanism was associated with the recruitment of bone marrow-derived suppressor cells by IL-33, which reduced the production of IL-1β in the peritoneal cavity (84). These results also precisely reflect the distinct action patterns of IL-33 in different sites and under different pathological conditions.
IgG4-Related Disease
The IgG4-related disease (IgG4-RD) is an idiopathic, fibroinflammatory disease characterized by elevated serum IgG4 levels, tumefaction, and tissue infiltration by IgG4-positive plasma cells (123). The ratio of male to female is ~(2–3):1. The current incidence in China is unknown. However, with the improvement in disease cognition and detection, the number of patients is gradually increasing. Furukawa et al. found that IL-33 could act as an inducer of Th2 response in IgG4-RD (85). Further studies found that in patients with IgG4-related autoimmune pancreatitis, plasmacytoid dendritic cells could produce IL-33 and interferon alpha (IFN-α), which were closely related to the fibrosis of the disease. The authors further validated these results in a mice model and demonstrated that depletion of plasmacytoid dendritic cells and blockade of signaling pathways related to type 1 interferon and IL-33 could prevent chronic fibrosis (124). Treatment with prednisolone was able to improve the swelling of the pancreas with a significant reduction of serum IFN-α and IL-33, but the serum IgG, IgG4, and IgE concentrations only slightly decreased. This suggested that the IFN-α/IL-33 axis may be a better biomarker reflecting the disease activity of IgG4-RD compared with serum levels of IgG, IgG4, and IgE (86, 125, 126). There is no correlation between serum IL-33 and serum IgG4 or IgG4:IgG ratio (126). Another study on IgG4-related sialadenitis demonstrated that TLR7-positive M2 macrophage was able to produce high levels of IL-33 in vitro, which activated the Th2 immune response and promoted tissue fibrosis in IgG4-RD (87). In conclusion, IL-33 plays an important role in the development of IgG4-related diseases as an important inducer of type 2 immunity and an important pro-fibrogenic factor. However, the specific mechanism is still unclear.
Ankylosing Spondylitis
AS is a chronic, inflammatory rheumatic disease affecting the spine and sacroiliac joints. The main clinical features are inflammatory back pain, bone erosion, and syndesmophyte formation (127). In China, the prevalence of AS is 0.25~0.5%, and the ratio of male-to-female is about 4–1. Due to the lack of suitable animal models, all studies of IL-33 in AS have been conducted in humans. Serum IL-33 levels were reported to be elevated in patients with AS, especially in patients with active AS (88). In addition, IL-33 enhanced the production of TNF-α and IL-6 in peripheral blood mononuclear cells (PBMCs) and induced neutrophil migration when the dose of IL-33 exceeded 10 ng/ml (89). Another study explored the relationship between IL-33 gene polymorphisms and disease susceptibility and found that AS patients carrying the IL-33 rs16924159 AA genotype had higher disease activity and a worse response to anti-TNF therapy (90). But overall, IL-33 could be still used as a predictor of the therapeutic effect of infliximab in the treatment of AS (91). These results suggest that IL-33 is involved in the pathogenesis of AS and is a potential therapeutic target, but more studies are still needed.
Other Rheumatic Diseases
IL-33 also played a role in other rheumatic diseases such as idiopathic inflammatory myopathies (IIM), adult-onset Still's disease (AOSD), and Behcet's disease (BD).
IIM is a chronic rheumatic disease, which can lead to skin and internal organ involvement. IIM includes dermatomyositis (DM) and polymyositis (PM). It was reported that serum sST2 levels were significantly elevated in DM and PM patients and decreased after treatment. In addition, serum sST2 levels were correlated with CRP, creatine kinase (CK), and lactate dehydrogenase (LDH) (92). In another study, serum IL-33 could not be detected in the majority of IIM patients, but serum sST2 levels were elevated and even much higher in patients with anti-signal recognition particle (anti-SRP) antibodies (93). Considering the abnormal expression of sST2 and the short detection time window of IL-33, it can be speculated that IL-33 may be involved in the pathogenesis of IIM.
AOSD is a rare systemic inflammatory disorder, which is characterized by high spiking fever, an evanescent rash, polyarthralgia, arthritis, and hepatosplenomegaly. It was reported that serum levels of IL-33 and sST2 were elevated in patients with active AOSD; and serum IL-33 levels correlated with systemic score, ESR, ferritin levels, and aspartate transaminase levels, while serum soluble ST2 levels correlated only with ferritin levels (94). These results indicated that the IL-33/ST2 signaling pathway may play a role in the pathogenesis of AOSD.
BD is a multisystem inflammatory disease, characterized by recurrent oral ulceration, skin lesions, genital ulcerations, and uveitis. Serum and skin tissue of IL-33 and sST2 levels were reported to be elevated in patients with BD, and sST2 is associated with ESR and CRP (95). Another study reported that in BD patients of Iran, the expression of IL-33 mRNA in PBMCs was much higher than in healthy controls, and rs1342326 T/G polymorphism of the IL-33 gene might contribute to the genetic susceptibility to BD (96). These results suggest that IL-33 may play an important role in the pathogenesis of BD.
Discussion
In this review, we summarize the role of the IL-33/ST2 axis in rheumatic diseases by summarizing the evidence from clinical patients, mouse models, and in vitro cell culture. IL-33 is characterized as an alarmin, with ILC2, Th2, and Tregs as the main target cells in immune system. Because of the complexity and functional diversity of IL-33, it may play distinct roles in different stages of disease and different immune microenvironments. For example, administration of exogenous IL-33 with different treatment duration, different concentrations, or different stage of disease may result in different, even opposite, therapeutic effects. Nevertheless, previous studies have demonstrated a detrimental role of IL-33/ST2 axis in RA, scleroderma, SLE, psoriasis, and gout. The potential mechanisms may involve the immunomodulation, fibrogenesis, and tissue repair.
IL-33 is able to promote the polarization of macrophages, activate mast cells ILC2, and induce eosinophil activation. We and others have shown that some tissue cells including epithelial cells and fibroblasts also express IL-33 and its receptor ST2. IL-33 can also participate in the pathogenesis of rheumatic disease by interacting with ST2-expressing tissue cells, such as cardiomyocytes, oligodendrocytes, epithelial cells, and endothelial cells. The activation, dysfunction, and destruction of these cells are directly involved in the development of many rheumatic diseases. In general, IL-33/ST2 axis plays a detrimental role in both early and advanced stages of most rheumatic diseases. In the early stages of the disease, IL-33 can be released from damaged epithelial cells acting as an alarmin to activate other local tissue cells and recruit immune cells. In addition, IL-33 seems to interact with various cytokines such as IL-12 to produce more IFN-γ in the early inflammation environment. In contrast, during the advanced repair and fibrosis stages, IL-33 as an important pro-fibrogenic factor may play a protective role in maintaining the integrity of the barrier system. These characteristics determine the pleiotropy and complexity of IL-33.
Conclusion
In summary, the sources and targets of IL-33 involve a variety of cell types. Although great advances have been made in recent years, more evidence is needed to clarify the exact role of the IL-33/ST2 axis in rheumatic diseases.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grants from the National Natural Scientific Foundation of China (No. 81771754) and Tongji Hospital Clinical Research Flagship Program (No. 2019CR206).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Baekkevold ES, Roussigne M, Yamanaka T, Johansen FE, Jahnsen FL, Amalric F, et al. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am J Pathol. (2003) 163:69–79. doi: 10.1016/S0002-9440(10)63631-0
2. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
3. Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA. (2007) 104:282–7. doi: 10.1073/pnas.0606854104
4. Bertheloot D, Latz E. HMGB1, IL-1alpha, IL-33 and S100 proteins: dual-function alarmins. Cell Mol Immunol. (2017) 14:43–64. doi: 10.1038/cmi.2016.34
5. Cayrol C, Girard JP. Interleukin-33 (IL-33): a nuclear cytokine from the IL-1 family. Immunol Rev. (2018) 281:154–68. doi: 10.1111/imr.12619
6. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. (2016) 16:676–89. doi: 10.1038/nri.2016.95
7. Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity. (2015) 42:1005–19. doi: 10.1016/j.immuni.2015.06.006
8. Moussion C, Girard JP. Dendritic cells control lymphocyte entry to lymph nodes through high endothelial venules. Nature. (2011) 479:542–6. doi: 10.1038/nature10540
9. Girard JP, Moussion C, Forster R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat Rev Immunol. (2012) 12:762–73. doi: 10.1038/nri3298
10. Liu X, Xiao Y, Pan Y, Li H, Zheng SG, Su W. The role of the IL-33/ST2 axis in autoimmune disorders: friend or foe? Cytokine Growth Factor Rev. (2019) 50:60–74. doi: 10.1016/j.cytogfr.2019.04.004
11. Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin'? PLoS ONE. (2008) 3:e3331. doi: 10.1371/journal.pone.0003331
12. Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J Immunol. (2012) 188:3488–95. doi: 10.4049/jimmunol.1101977
13. Kakkar R, Hei H, Dobner S, Lee RT. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J Biol Chem. (2012) 287:6941–8. doi: 10.1074/jbc.M111.298703
14. Roussel L, Erard M, Cayrol C, Girard JP. Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A-H2B acidic pocket. Embo Rep. (2008) 9:1006–12. doi: 10.1038/embor.2008.145
15. Bessa J, Meyer CA, de Vera MM, Schlicht S, Smith SH, Iglesias A, et al. Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J Autoimmun. (2014) 55:33–41. doi: 10.1016/j.jaut.2014.02.012
16. Kurow O, Frey B, Schuster L, Schmitt V, Adam S, Hahn M, et al. Full length interleukin 33 aggravates radiation-induced skin reaction. Front Immunol. (2017) 8:722. doi: 10.3389/fimmu.2017.00722
17. Talabot-Ayer D, Lamacchia C, Gabay C, Palmer G. Interleukin-33 is biologically active independently of caspase-1 cleavage. J Biol Chem. (2009) 284:19420–6. doi: 10.1074/jbc.M901744200
18. Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA. (2009) 106:9021–6. doi: 10.1073/pnas.0812690106
19. Lefrancais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard JP, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA. (2012) 109:1673–8. doi: 10.1073/pnas.1115884109
20. Lefrancais E, Duval A, Mirey E, Roga S, Espinosa E, Cayrol C, et al. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci USA. (2014) 111:15502–7. doi: 10.1073/pnas.1410700111
21. Waern I, Lundequist A, Pejler G, Wernersson S. Mast cell chymase modulates IL-33 levels and controls allergic sensitization in dust-mite induced airway inflammation. Mucosal Immunol. (2013) 6:911–20. doi: 10.1038/mi.2012.129
22. Luthi AU Cullen SP McNeela EA Duriez PJ Afonina IS Sheridan C . Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. (2009) 31:84–98. doi: 10.1016/j.immuni.2009.05.007
23. Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, England E, et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun. (2015) 6:8327. doi: 10.1038/ncomms9327
24. Galand C, Leyva-Castillo JM, Yoon J, Han A, Lee MS, McKenzie A, et al. IL-33 promotes food anaphylaxis in epicutaneously sensitized mice by targeting mast cells. J Allergy Clin Immunol. (2016) 138:1356–66. doi: 10.1016/j.jaci.2016.03.056
25. Imai Y, Yasuda K, Sakaguchi Y, Futatsugi-Yumikura S, Yoshimoto T, Nakanishi K, et al. Immediate-type contact hypersensitivity is reduced in interleukin-33 knockout mice. J Dermatol Sci. (2014) 74:159–61. doi: 10.1016/j.jdermsci.2014.01.009
26. Sundnes O, Pietka W, Loos T, Sponheim J, Rankin AL, Pflanz S, et al. Epidermal expression and regulation of interleukin-33 during homeostasis and inflammation: strong species differences. J Invest Dermatol. (2015) 135:1771–80. doi: 10.1038/jid.2015.85
27. Kuchler AM, Pollheimer J, Balogh J, Sponheim J, Manley L, Sorensen DR, et al. Nuclear interleukin-33 is generally expressed in resting endothelium but rapidly lost upon angiogenic or proinflammatory activation. Am J Pathol. (2008) 173:1229–42. doi: 10.2353/ajpath.2008.080014
28. Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. (2011) 12:631–8. doi: 10.1038/ni.2045
29. Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, et al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol. (2012) 129:216–7.e1–6. doi: 10.1016/j.jaci.2011.10.036
30. Wills-Karp M, Rani R, Dienger K, Lewkowich I, Fox JG, Perkins C, et al. Trefoil factor 2 rapidly induces interleukin 33 to promote type 2 immunity during allergic asthma and hookworm infection. J Exp Med. (2012) 209:607–22. doi: 10.1084/jem.20110079
31. Fock V, Mairhofer M, Otti GR, Hiden U, Spittler A, Zeisler H, et al. Macrophage-derived IL-33 is a critical factor for placental growth. J Immunol. (2013) 191:3734–43. doi: 10.4049/jimmunol.1300490
32. Hsu CL, Neilsen CV, Bryce PJ. IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS ONE. (2010) 5:e11944. doi: 10.1371/journal.pone.0011944
33. Tung HY, Plunkett B, Huang SK, Zhou Y. Murine mast cells secrete and respond to interleukin-33. J Interferon Cytokine Res. (2014) 34:141–7. doi: 10.1089/jir.2012.0066
34. Shimokawa C Kanaya T Hachisuka induction of group 2 innate lymphoid cells and clearance of helminth infections. M, Ishiwata K, Hisaeda H, Kurashima Y, et al. Mast cells are crucial for Immunity. (2017) 46:863–74.e4. doi: 10.1016/j.immuni.2017.04.017
35. Toki S, Goleniewska K, Reiss S, Zhou W, Newcomb DC, Bloodworth MH, et al. The histone deacetylase inhibitor trichostatin A suppresses murine innate allergic inflammation by blocking group 2 innate lymphoid cell (ILC2) activation. Thorax. (2016) 71:633–45. doi: 10.1136/thoraxjnl-2015-207728
36. Griesenauer B, Paczesny S. The ST2/IL-33 axis in immune cells during inflammatory diseases. Front Immunol. (2017) 8:475. doi: 10.3389/fimmu.2017.00475
37. Kumar S, Minnich MD, Young PR. ST2/T1 protein functionally binds to two secreted proteins from Balb/c 3T3 and human umbilical vein endothelial cells but does not bind interleukin 1. J Biol Chem. (1995) 270:27905–13. doi: 10.1074/jbc.270.46.27905
38. Kumar S, Tzimas MN, Griswold DE, Young PR. Expression of ST2, an interleukin-1 receptor homologue, is induced by proinflammatory stimuli. Biochem Biophys Res Commun. (1997) 235:474–8. doi: 10.1006/bbrc.1997.6810
39. Yanagisawa K, Takagi T, Tsukamoto T, Tetsuka T, Tominaga S. Presence of a novel primary response gene ST2L, encoding a product highly similar to the interleukin 1 receptor type 1. Febs Lett. (1993) 318:83–7. doi: 10.1016/0014-5793(93)81333-U
40. Tominaga S, Kuroiwa K, Tago K, Iwahana H, Yanagisawa K, Komatsu N. Presence and expression of a novel variant form of ST2 gene product in human leukemic cell line UT-7/GM. Biochem Biophys Res Commun. (1999) 264:14–8. doi: 10.1006/bbrc.1999.1469
41. Palmer G, Lipsky BP, Smithgall MD, Meininger D, Siu S, Talabot-Ayer D, et al. The IL-1 receptor accessory protein (AcP) is required for IL-33 signaling and soluble AcP enhances the ability of soluble ST2 to inhibit IL-33. Cytokine. (2008) 42:358–64. doi: 10.1016/j.cyto.2008.03.008
42. Kakkar R, Lee RT. The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov. (2008) 7:827–40. doi: 10.1038/nrd2660
43. Gupta RK, Gupta K, Dwivedi PD. Pathophysiology of IL-33 and IL-17 in allergic disorders. Cytokine Growth Factor Rev. (2017) 38:22–36. doi: 10.1016/j.cytogfr.2017.09.005
44. De la Fuente M, MacDonald TT, Hermoso MA. The IL-33/ST2 axis: Role in health and disease. Cytokine Growth Factor Rev. (2015) 26:615–23. doi: 10.1016/j.cytogfr.2015.07.017
45. Garlanda C, Anders HJ, Mantovani A. TIR8/SIGIRR: an IL-1R/TLR family member with regulatory functions in inflammation and T cell polarization. Trends Immunol. (2009) 30:439–46. doi: 10.1016/j.it.2009.06.001
46. Drake LY, Prakash YS. Contributions of IL-33 in non-hematopoietic lung cells to obstructive lung disease. Front Immunol. (2020) 11:1798. doi: 10.3389/fimmu.2020.01798
47. Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol. (2007) 179:2051–4. doi: 10.4049/jimmunol.179.4.2051
48. Cho KA, Suh JW, Sohn JH, Park JW, Lee H, Kang JL, et al. IL-33 induces Th17-mediated airway inflammation via mast cells in ovalbumin-challenged mice. Am J Physiol Lung Cell Mol Physiol. (2012) 302:L429–40. doi: 10.1152/ajplung.00252.2011
49. Espinassous Q, Garcia-de-Paco E, Garcia-Verdugo I, Synguelakis M, von Aulock S, Sallenave JM, et al. IL-33 enhances lipopolysaccharide-induced inflammatory cytokine production from mouse macrophages by regulating lipopolysaccharide receptor complex. J Immunol. (2009) 183:1446–55. doi: 10.4049/jimmunol.0803067
50. Su Z, Lin J, Lu F, Zhang X, Zhang L, Gandhi NB, et al. Potential autocrine regulation of interleukin-33/ST2 signaling of dendritic cells in allergic inflammation. Mucosal Immunol. (2013) 6:921–30. doi: 10.1038/mi.2012.130
51. Ochayon DE, Ali A, Alarcon PC, Krishnamurthy D, Kottyan LC, Borchers MT, et al. IL-33 promotes type 1 cytokine expression via p38 MAPK in human NK cells. J Leukoc Biol. (2020) 107:663–71. doi: 10.1002/JLB.3A0120-379RR
52. Clark JT, Christian DA, Gullicksrud JA, Perry JA, Park J, Jacquet M, et al. IL-33 promotes innate lymphoid cell-dependent IFN-gamma production required for innate immunity to Toxoplasma gondii. Elife. (2021) 10:e65614. doi: 10.7554/eLife.65614
53. Seltmann J, Werfel T, Wittmann M. Evidence for a regulatory loop between IFN-gamma and IL-33 in skin inflammation. Exp Dermatol. (2013) 22:102–7. doi: 10.1111/exd.12076
54. Ferhat MH, Robin A, Barbier L, Thierry A, Gombert JM, Barbarin A, et al. The impact of invariant NKT cells in sterile inflammation: the possible contribution of the alarmin/cytokine IL-33. Front Immunol. (2018) 9:2308. doi: 10.3389/fimmu.2018.02308
55. Lee JS, Seppanen E, Patel J, Rodero MP, Khosrotehrani K. ST2 receptor invalidation maintains wound inflammation, delays healing and increases fibrosis. Exp Dermatol. (2016) 25:71–4. doi: 10.1111/exd.12833
56. Kotsiou OS, Gourgoulianis KI, Zarogiannis SG. IL-33/ST2 axis in organ fibrosis. Front Immunol. (2018) 9:2432. doi: 10.3389/fimmu.2018.02432
57. Huang E, Peng N, Xiao F, Hu D, Wang X, Lu L. The roles of immune cells in the pathogenesis of fibrosis. Int J Mol Sci. (2020) 21:5303. doi: 10.3390/ijms21155203
58. Wang L, Wang FS, Gershwin ME. Human autoimmune diseases: a comprehensive update. J Intern Med. (2015) 278:369–95. doi: 10.1111/joim.12395
59. Xu D, Barbour M, Jiang HR, Mu R. Role of IL-33/ST2 signaling pathway in systemic sclerosis and other fibrotic diseases. Clin Exp Rheumatol. (2019) 37(Suppl. 119):141–6.
60. Soyfoo MS, Nicaise C. Pathophysiologic role of Interleukin-33/ST2 in Sjogren's syndrome. Autoimmun Rev. (2021):102756. doi: 10.1016/j.autrev.2021.102756
61. Yang Z, Liang Y, Xi W, Li C, Zhong R. Association of increased serum IL-33 levels with clinical and laboratory characteristics of systemic lupus erythematosus in Chinese population. Clin Exp Med. (2011) 11:75–80. doi: 10.1007/s10238-010-0115-4
62. Mok MY, Huang FP, Ip WK, Lo Y, Wong FY, Chan EY, et al. Serum levels of IL-33 and soluble ST2 and their association with disease activity in systemic lupus erythematosus. Rheumatology. (2010) 49:520–7. doi: 10.1093/rheumatology/kep402
63. Guo C, Zhou M, Zhao S, Huang Y, Wang S, Fu R, et al. Innate lymphoid cell disturbance with increase in ILC1 in systemic lupus erythematosus. Clin Immunol. (2019) 202:49–58. doi: 10.1016/j.clim.2019.03.008
64. Li P, Lin W, Zheng X. IL-33 neutralization suppresses lupus disease in lupus-prone mice. Inflammation. (2014) 37:824–32. doi: 10.1007/s10753-013-9802-0
65. Palmer G, Talabot-Ayer D, Lamacchia C, Toy D, Seemayer CA, Viatte S, et al. Inhibition of interleukin-33 signaling attenuates the severity of experimental arthritis. Arthritis Rheum. (2009) 60:738–49. doi: 10.1002/art.24305
66. Hong YS, Moon SJ, Joo YB, Jeon CH, Cho ML, Ju JH, et al. Measurement of interleukin-33 (IL-33) and IL-33 receptors (sST2 and ST2L) in patients with rheumatoid arthritis. J Korean Med Sci. (2011) 26:1132–9. doi: 10.3346/jkms.2011.26.9.1132
67. Matsuyama Y, Okazaki H, Tamemoto H, Kimura H, Kamata Y, Nagatani K, et al. Increased levels of interleukin 33 in sera and synovial fluid from patients with active rheumatoid arthritis. J Rheumatol. (2010) 37:18–25. doi: 10.3899/jrheum.090492
68. Wang Y, Chen Z, Huang Y, Yafei L, Tu S. Prognostic significance of serum interleukins and soluble ST2 in Traditional Chinese Medicine (TCM) syndrome-differentiated rheumatoid arthritis. Med Sci Monit. (2018) 24:3472–3478. doi: 10.12659/MSM.907540
69. Xu D, Jiang HR, Kewin P, Li Y, Mu R, Fraser AR, et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc Natl Acad Sci USA. (2008) 105:10913–8. doi: 10.1073/pnas.0801898105
70. Matsuyama Y, Okazaki H, Hoshino M, Onishi S, Kamata Y, Nagatani K, et al. Sustained elevation of interleukin-33 in sera and synovial fluids from patients with rheumatoid arthritis non-responsive to anti-tumor necrosis factor: possible association with persistent IL-1beta signaling and a poor clinical response. Rheumatol Int. (2012) 32:1397–401. doi: 10.1007/s00296-011-1854-6
71. Verri WJ, Souto FO, Vieira SM, Almeida SC, Fukada SY, Xu D, et al. IL-33 induces neutrophil migration in rheumatoid arthritis and is a target of anti-TNF therapy. Ann Rheum Dis. (2010) 69:1697–703. doi: 10.1136/ard.2009.122655
72. Jung SM, Lee J, Baek SY, Lee JH, Lee J, Park KS, et al. The Interleukin 33/ST2 axis in patients with primary Sjogren syndrome: expression in serum and salivary glands, and the clinical association. J Rheumatol. (2015) 42:264–71. doi: 10.3899/jrheum.140234
73. Awada A, Nicaise C, Ena S, Schandene L, Rasschaert J, Popescu I, et al. Potential involvement of the IL-33-ST2 axis in the pathogenesis of primary Sjogren's syndrome. Ann Rheum Dis. (2014) 73:1259–63. doi: 10.1136/annrheumdis-2012-203187
74. Luo G, Xin Y, Qin D, Yan A, Zhou Z, Liu Z. Correlation of interleukin-33 with Th cytokines and clinical severity of dry eye disease. Indian J Ophthalmol. (2018) 66:39–43. doi: 10.4103/ijo.IJO_405_17
75. Vettori S, Cuomo G, Iudici M, D'Abrosca V, Giacco V, Barra G, et al. Early systemic sclerosis: serum profiling of factors involved in endothelial, T-cell, and fibroblast interplay is marked by elevated interleukin-33 levels. J Clin Immunol. (2014) 34:663–8. doi: 10.1007/s10875-014-0037-0
76. Wagner A, Kohm M, Nordin A, Svenungsson E, Pfeilschifter JM, Radeke HH. Increased serum levels of the IL-33 neutralizing sST2 in limited cutaneous systemic sclerosis. Scand J Immunol. (2015) 82:269–74. doi: 10.1111/sji.12317
77. Manetti M, Ibba-Manneschi L, Liakouli V, Guiducci S, Milia AF, Benelli G, et al. The IL1-like cytokine IL33 and its receptor ST2 are abnormally expressed in the affected skin and visceral organs of patients with systemic sclerosis. Ann Rheum Dis. (2010) 69:598–605. doi: 10.1136/ard.2009.119321
78. Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. (2009) 183:6469–77. doi: 10.4049/jimmunol.0901575
79. MacDonald KG, Dawson N, Huang Q, Dunne JV, Levings MK, Broady R. Regulatory T cells produce profibrotic cytokines in the skin of patients with systemic sclerosis. J Allergy Clin Immunol. (2015) 135: 946–955.e9. doi: 10.1016/j.jaci.2014.12.1932
80. Li J, Liu L, Rui W, Li X, Xuan D, Zheng S, et al. new interleukins in psoriasis and psoriatic arthritis patients: the possible roles of interleukin-33 to interleukin-38 in disease activities and bone erosions. Dermatology. (2017) 233:37–46. doi: 10.1159/000471798
81. Raimondo A, Lembo S, Di Caprio R, Donnarumma G, Monfrecola G, Balato N, et al. Psoriatic cutaneous inflammation promotes human monocyte differentiation into active osteoclasts, facilitating bone damage. Eur J Immunol. (2017) 47:1062–74. doi: 10.1002/eji.201646774
82. Fattori V, Staurengo-Ferrari L, Zaninelli TH, Casagrande R, Oliveira RD, Louzada-Junior P, et al. IL-33 enhances macrophage release of IL-1beta and promotes pain and inflammation in gouty arthritis. Inflamm Res. (2020) 69:1271–82. doi: 10.1007/s00011-020-01399-x
83. Yin C, Liu B, Li Y, Li X, Wang J, Chen R, et al. IL-33/ST2 induces neutrophil-dependent reactive oxygen species production and mediates gout pain. Theranostics. (2020) 10:12189–203. doi: 10.7150/thno.48028
84. Shang K, Wei Y, Su Q, Yu B, Tao Y, He Y, et al. IL-33 ameliorates the development of MSU-induced inflammation through expanding MDSCs-like cells. Front Endocrinol. (2019) 10:36. doi: 10.3389/fendo.2019.00036
85. Furukawa S, Moriyama M, Miyake K, Nakashima H, Tanaka A, Maehara T, et al. Interleukin-33 produced by M2 macrophages and other immune cells contributes to Th2 immune reaction of IgG4-related disease. Sci Rep. (2017) 7:42413. doi: 10.1038/srep42413
86. Minaga K, Watanabe T, Hara A, Kamata K, Omoto S, Nakai A, et al. Identification of serum IFN-alpha and IL-33 as novel biomarkers for type 1 autoimmune pancreatitis and IgG4-related disease. Sci Rep. (2020) 10:14879. doi: 10.1038/s41598-020-71848-4
87. Ishiguro N, Moriyama M, Furusho K, Furukawa S, Shibata T, Murakami Y, et al. Activated M2 macrophages contribute to the pathogenesis of IgG4-related disease via toll-like receptor 7/interleukin-33 signaling. Arthritis Rheumatol. (2020) 72:166–78. doi: 10.1002/art.41052
88. Han GW, Zeng LW, Liang CX, Cheng BL, Yu BS, Li HM, et al. Serum levels of IL-33 is increased in patients with ankylosing spondylitis. Clin Rheumatol. (2011) 30:1583–8. doi: 10.1007/s10067-011-1843-x
89. Li GX, Wang S, Duan ZH, Zeng Z, Pan FM. Serum levels of IL-33 and its receptor ST2 are elevated in patients with ankylosing spondylitis. Scand J Rheumatol. (2013) 42:226–31. doi: 10.3109/03009742.2012.735700
90. Iwaszko M, Wielinska J, Swierkot J, Kolossa K, Sokolik R, Bugaj B, et al. IL-33 gene polymorphisms as potential biomarkers of disease susceptibility and response to TNF inhibitors in rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis patients. Front Immunol. (2021) 12:631603. doi: 10.3389/fimmu.2021.631603
91. Li L, Chen B, Zhao H, Wang G. Bone changes and curative effect of infliximab in patients with ankylosing spondylitis. J Musculoskelet Neuronal Interact. (2020) 20:437–43.
92. Yuan L, Yao L, Zhao L, Xia L, Shen H, Lu J. Serum levels of soluble ST2 and interleukin-33 in patients with dermatomyositis and polymyositis. Clin Exp Rheumatol. (2013) 31:428–32.
93. Opinc A, Sarnik J, Brzezinska O, Makowski M, Lewandowska-Polak A, Makowska J. Interleukin-33/suppression of tumorigenicity 2 (IL-33/ST2) axis in idiopathic inflammatory myopathies and its association with laboratory and clinical parameters: a pilot study. Rheumatol Int. (2020) 40:1133–41. doi: 10.1007/s00296-020-04554-z
94. Han JH, Suh CH, Jung JY, Ahn MH, Kwon JE, Yim H, et al. serum levels of interleukin 33 and soluble ST2 are associated with the extent of disease activity and cutaneous manifestations in patients with active adult-onset still's disease. J Rheumatol. (2017) 44:740–7. doi: 10.3899/jrheum.170020
95. Kim DJ, Baek SY, Park MK, Park KS, Lee JH, Park SH, et al. Serum level of interleukin-33 and soluble ST2 and their association with disease activity in patients with Behcet's disease. J Korean Med Sci. (2013) 28:1145–53. doi: 10.3346/jkms.2013.28.8.1145
96. Talei M, Abdi A, Shanebandi D, Jadidi-Niaragh F, Khabazi A, Babaie F, et al. Interleukin-33 gene expression and rs1342326 polymorphism in Behcet's disease. Immunol Lett. (2019) 212:120–4. doi: 10.1016/j.imlet.2018.11.005
97. Kiriakidou M, Ching CL. Systemic Lupus Erythematosus. Ann Intern Med. (2020) 172:ITC81-ITC96. doi: 10.7326/AITC202006020
98. Riviere E, Ly B, Boudaoud S, Chavez H, Nocturne G, Chanson P, et al. Pitfalls for detecting interleukin-33 by ELISA in the serum of patients with primary Sjogren syndrome: comparison of different kits. Ann Rheum Dis. (2016) 75:633–5. doi: 10.1136/annrheumdis-2015-208557
99. Xu W, Liu Y, Ye D. Association between IL-33 gene polymorphisms (rs1929992, rs7044343) and systemic lupus erythematosus in a Chinese Han population. Immunol Invest. (2016) 45:575–83. doi: 10.1080/08820139.2016.1193868
100. Guo J, Xiang Y, Peng YF, Huang HT, Lan Y, Wei YS. The association of novel IL-33 polymorphisms with sIL-33 and risk of systemic lupus erythematosus. Mol Immunol. (2016) 77:1–7. doi: 10.1016/j.molimm.2016.07.001
101. Zhu X, Xie L, Qin H, Liang J, Yang Y, Xu J, et al. Interaction between IL-33 gene polymorphisms and current smoking with susceptibility to systemic lupus erythematosus. J Immunol Res. (2019) 2019:1547578. doi: 10.1155/2019/1547578
102. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. (2010) 376:1094–108. doi: 10.1016/S0140-6736(10)60826-4
103. Boissier MC. Cell and cytokine imbalances in rheumatoid synovitis. Joint Bone Spine. (2011) 78:230–4. doi: 10.1016/j.jbspin.2010.08.017
104. Mu R, Huang HQ, Li YH, Li C, Ye H, Li ZG. Elevated serum interleukin 33 is associated with autoantibody production in patients with rheumatoid arthritis. J Rheumatol. (2010) 37:2006–13. doi: 10.3899/jrheum.100184
105. Xiangyang Z, Lutian Y, Lin Z, Liping X, Hui S, Jing L. Increased levels of interleukin-33 associated with bone erosion and interstitial lung diseases in patients with rheumatoid arthritis. Cytokine. (2012) 58:6–9. doi: 10.1016/j.cyto.2011.12.010
106. Kageyama Y, Torikai E, Tsujimura K, Kobayashi M. Involvement of IL-33 in the pathogenesis of rheumatoid arthritis: the effect of etanercept on the serum levels of IL-33. Mod Rheumatol. (2012) 22:89–93. doi: 10.3109/s10165-011-0480-1
107. Kunisch E, Chakilam S, Gandesiri M, Kinne RW. IL-33 regulates TNF-alpha dependent effects in synovial fibroblasts. Int J Mol Med. (2012) 29:530–40. doi: 10.3892/ijmm.2012.883
108. Tang S, Huang H, Hu F, Zhou W, Guo J, Jiang H, et al. Increased IL-33 in synovial fluid and paired serum is associated with disease activity and autoantibodies in rheumatoid arthritis. Clin Dev Immunol. (2013) 2013:985301. doi: 10.1155/2013/985301
109. Biton J, Khaleghparast AS, Thiolat A, Santinon F, Lemeiter D, Herve R, et al. In vivo expansion of activated Foxp3+ regulatory T cells and establishment of a type 2 immune response upon IL-33 treatment protect against experimental arthritis. J Immunol. (2016) 197:1708–19. doi: 10.4049/jimmunol.1502124
110. Brito-Zeron P, Baldini C, Bootsma H, Bowman SJ, Jonsson R, Mariette X, et al. Sjogren syndrome. Nat Rev Dis Primers. (2016) 2:16047. doi: 10.1038/nrdp.2016.47
111. Margiotta DP, Navarini L, Vadacca M, Lo VM, Pignataro F, Basta F, et al. The IL33/ST2 axis in Sjogren syndrome in relation to disease activity. Eur Rev Med Pharmacol Sci. (2016) 20:1295–9.
112. Olsson P, Skogstrand K, Nilsson A, Turesson C, Jacobsson L, Theander E, et al. Smoking, disease characteristics and serum cytokine levels in patients with primary Sjogren's syndrome. Rheumatol Int. (2018) 38:1503–10. doi: 10.1007/s00296-018-4063-8
113. Zhao L, Yao L, Yuan L, Xia L, Shen H, Lu J. Potential contribution of interleukin-33 to the development of interstitial lung disease in patients with primary Sjogren's syndrome. Cytokine. (2013) 64:22–4. doi: 10.1016/j.cyto.2013.07.006
114. Denton CP, Khanna D. Systemic sclerosis. Lancet. (2017) 390:1685–99. doi: 10.1016/S0140-6736(17)30933-9
115. Koca SS, Pehlivan Y, Kara M, Alibaz-Oner F, Oztuzcu S, Yilmaz N, et al. The IL-33 gene is related to increased susceptibility to systemic sclerosis. Rheumatol Int. (2016) 36:579–84. doi: 10.1007/s00296-015-3417-8
116. Huang XL, Wu GC, Wang YJ, Yang XK, Yang GJ, Tao JH, et al. Association of interleukin-1 family cytokines single nucleotide polymorphisms with susceptibility to systemic sclerosis: an independent case-control study and a meta-analysis. Immunol Res. (2016) 64:1041–52. doi: 10.1007/s12026-016-8797-7
117. Orbai AM, de Wit M, Mease P, Shea JA, Gossec L, Leung YY, et al. International patient and physician consensus on a psoriatic arthritis core outcome set for clinical trials. Ann Rheum Dis. (2017) 76:673–680. doi: 10.1136/annrheumdis-2016-210242
118. Ogdie A, Coates LC, Gladman DD. Treatment guidelines in psoriatic arthritis. Rheumatology. (2020) 59:i37–46. doi: 10.1093/rheumatology/kez383
119. Cannavo SP, Bertino L, Di Salvo E, Papaianni V, Ventura-Spagnolo E, Gangemi S. Possible roles of IL-33 in the innate-adaptive immune crosstalk of psoriasis pathogenesis. Mediators Inflamm. (2019) 2019:7158014. doi: 10.1155/2019/7158014
120. Talabot-Ayer D, McKee T, Gindre P, Bas S, Baeten DL, Gabay C, et al. Distinct serum and synovial fluid interleukin (IL)-33 levels in rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Joint Bone Spine. (2012) 79:32–7. doi: 10.1016/j.jbspin.2011.02.011
121. Athari SK, Poirier E, Biton J, Semerano L, Herve R, Raffaillac A, et al. Collagen-induced arthritis and imiquimod-induced psoriasis develop independently of interleukin-33. Arthritis Res Ther. (2016) 18:143. doi: 10.1186/s13075-016-1042-x
123. Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet. (2015) 385:1460–71. doi: 10.1016/S0140-6736(14)60720-0
124. Watanabe T, Yamashita K, Arai Y, Minaga K, Kamata K, Nagai T, et al. Chronic fibro-inflammatory responses in autoimmune pancreatitis depend on IFN-alpha and IL-33 produced by plasmacytoid dendritic cells. J Immunol. (2017) 198:3886–96. doi: 10.4049/jimmunol.1700060
125. Minaga K, Watanabe T, Kamata K, Takenaka M, Yasukawa S, Kudo M. The IFN-alpha-IL-33 axis as possible biomarkers in IgG4-related disease. Am J Gastroenterol. (2019) 114:1002–3. doi: 10.14309/ajg.0000000000000245
126. Akiyama M, Suzuki K, Yamaoka K, Yasuoka H, Takeshita M, Kaneko Y, et al. Number of circulating follicular helper 2 T cells correlates with IgG4 and interleukin-4 levels and plasmablast numbers in IgG4-related disease. Arthritis Rheumatol. (2015) 67:2476–81. doi: 10.1002/art.39209
Keywords: IL-33, alarmin, ST2, autoimmune, rheumatic disease
Citation: Dong Y, Zhong J and Dong L (2021) IL-33 in Rheumatic Diseases. Front. Med. 8:739489. doi: 10.3389/fmed.2021.739489
Received: 11 July 2021; Accepted: 13 August 2021;
Published: 13 September 2021.
Edited by:
João Eurico Fonseca, University of Lisbon, PortugalReviewed by:
Rita A. Moura, Faculdade de Medicina da Universidade de Lisboa, PortugalYi Liu, Sichuan University, China
Copyright © 2021 Dong, Zhong and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingli Dong, dGpoZG9uZ2xsQDE2My5jb20=; Jixin Zhong, emhvbmdqaXhpbjYyMEAxNjMuY29t