 Elena Grossini1,2*
Elena Grossini1,2* Divya Praveen Garhwal1,2
Divya Praveen Garhwal1,2 Giuseppe Calamita3
Giuseppe Calamita3 Raffaele Romito4Cristina Rigamonti2,5
Raffaele Romito4Cristina Rigamonti2,5 Rosalba Minisini2,5Carlo Smirne2,5Daniela Surico2,6
Rosalba Minisini2,5Carlo Smirne2,5Daniela Surico2,6 Mattia Bellan2,5Mario Pirisi2,5
Mattia Bellan2,5Mario Pirisi2,5- 1Laboratory of Physiology, Department of Translational Medicine, University East Piedmont, Novara, Italy
- 2AGING Project, Department of Translational Medicine, University East Piedmont, Novara, Italy
- 3Laboratory of Cellular and Molecular Physiology and Pathophysiology, Department of Biosciences, Biotechnologies and Biopharmaceutics, University of Bari “Aldo Moro”, Bari, Italy
- 4General Surgery Unit, Azienda Ospedaliera Maggiore della Carità University Hospital, Novara, Italy
- 5Internal Medicine Unit, Department of Translational Medicine, University East Piedmont, Novara, Italy
- 6Obstetrics and Gynecology Unit, Department of Translational Medicine, University East Piedmont, Novara, Italy
Changes of lipidic storage, oxidative stress and mitochondrial dysfunction may be involved in the pathogenesis of non-alcoholic fatty liver disease (NAFLD). Although the knowledge of intracellular pathways has vastly expanded in recent years, the role and mechanisms of circulating triggering factor(s) are debated. Thus, we tested the hypothesis that factors circulating in the blood of NAFLD patients may influence processes underlying the disease. Huh7.5 cells/primary human hepatocytes were exposed to plasma from 12 NAFLD patients and 12 healthy subjects and specific assays were performed to examine viability, H2O2 and mitochondrial reactive oxygen species (ROS) release, mitochondrial membrane potential and triglycerides content. The involvement of NLRP3 inflammasome and of signaling related to peroxisome-proliferator-activating-ligand-receptor-γ (PPARγ), sterol-regulatory-element-binding-protein-1c (SREBP-1c), nuclear-factor-kappa-light-chain-enhancer of activated B cells (NF-kB), and NADPH oxidase 2 (NOX2) was evaluated by repeating the experiments in the presence of NLRP3 inflammasome blocker, MCC950, and through Western blot. The results obtained shown that plasma of NAFLD patients was able to reduce cell viability and mitochondrial membrane potential by about 48 and 24% (p < 0.05), and to increase H2O2, mitochondrial ROS, and triglycerides content by about 42, 19, and 16% (p < 0.05), respectively. An increased expression of SREBP-1c, PPARγ, NF-kB and NOX2 of about 51, 121, 63, and 46%, respectively, was observed (p < 0.05), as well. Those effects were reduced by the use of MCC950. Thus, in hepatocytes, exposure to plasma from NAFLD patients induces a NAFLD-like phenotype by interference with NLRP3-inflammasome pathways and the activation of intracellular signaling related to SREBP-1c, PPARγ, NF-kB and NOX2.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is currently the most common liver disease in the world and the second most common cause of liver transplantation in the United States (1, 2).
Our understanding of NAFLD pathogenesis and natural history has greatly expanded in the last decade, however, many issues remain unsolved. The liver plays a central role in regulating lipid homeostasis through processes that are strictly regulated by complex interactions between hormones, nuclear receptors, and transcription factors-related pathways. Their alteration may cause the retention of fat within the liver and the subsequent development of NAFLD (3, 4).
Another important player in the onset of NAFLD and its progression to non-alcoholic steatohepatitis (NASH) is mitochondrial dysfunction (5) that can alter the balance between prooxidants/antioxidants, leading to an increase of non-metabolized free fatty acids (FFAs) in the cytosol and the consequent induction of reactive oxygen species (ROS) production. A “primary” mitochondrial dysfunction has been proposed as one of the mechanisms of FFAs accumulation in the hepatocytes of NAFLD patients (6), however, an overload of FFAs into mitochondria may be able by itself to lead to dissipation of the membrane potential, loss of ATP synthesis capacity, and enhanced ROS generation (7). In addition, the accumulation of lipotoxic intermediates may promote inflammation and alter insulin signaling, facilitating the establishment of an insulin resistance state (5, 8).
Further topics of major interest regard the interference with pathways associated to recognition receptors including Toll-like receptors (TLRs) and NOD-like receptors (NLRs) (9), and downstream signaling involving peroxisome proliferator activating ligand receptors (PPAR), sterol regulatory element binding protein 1c (SREBP-1c), nuclear factor kappa-light-chain enhancer of activated B cells (NF-kB), and NADPH oxidase 2 (NOX2) (1, 10, 11).
In an “hormonocentric” view of NAFLD pathogenesis, all these processes may be triggered by circulating factors (12), that may represent diagnostic or prognostic biomarkers.
Based on these premises, we aimed to evaluate whether factors circulating in the blood of NAFLD patients may modulate one or more of the above-mentioned processes. Hence, in the present study we examined the effects of plasma of NAFLD patients on the viability and function of hepatocytes.
Materials and Methods
Patients and Controls
Experiments were conducted between October 2018 and March 2019 using plasma from 12 NAFLD patients and 12 healthy subjects. The use of excess plasma remaining from blood samples taken for the routinary clinical monitoring of NAFLD patients has been approved by the local Ethics Committee (Ethical Committee of the “Azienda Ospedaliera Maggiore della Carità” University Hospital in Novara). Patients and controls have given written informed consent for experimental use of pseudonymized clinical data and blood specimens. The work was carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki).
The clinical monitoring and plasma sampling of patients and controls were performed at Liver Clinic Unit, “Azienda Ospedaliera Maggiore della Carità” University Hospital in Novara.
Culture of Huh7.5 Cells
The human hepatocellular carcinoma cell line Huh7.5 (Apath L.L.C New York, USA) (13) was maintained in Dulbecco's modified Eagle's medium (DMEM; Sigma, Milan, Italy) supplemented with 10% fetal bovine plasma (FBS; Euroclone, Pero, Milan, Italy), 2 mM L-glutamine (Euroclone), 1% penicillin-streptomycin (P/S; Euroclone), at 37°C with 5% CO2 in incubator.
Isolation of Hepatocytes From Human Liver Biopsy Specimens
From October 2018 until November 2019, 10 liver samples (i.e., 50–100 g each) were obtained from patients undergoing surgery at General Surgery Unit, Azienda Ospedaliera Maggiore della Carità University Hospital, in Novara, due to primary or metastatic liver resectable tumor.
The specimens were acquired from the non-neoplastic portion of the liver parenchyma resected during the surgery. All tissue donors gave written informed consent for experimental use of pseudonymized clinical data and liver specimen prior to surgery. Then fresh samples were transferred on ice cold physiologic saline solution to Physiology laboratory, and hepatocytes were immediately isolated, as previously performed (14, 15). Briefly, liver specimens were minced with a scalpel and undergone a two steps collagenase procedure; the cell pellet was, then, centrifugated at 300 g for 5 min and the cell pellet was filtered through a nylon mesh. Thereafter, hepatocytes were pooled and seeded into plates coated with collagen-I (Sigma), and maintained in DMEM/HAM'S F-12 (Euroclone) supplemented with 10% FBS (Euroclone), 100 U/ml penicillin (Sigma), 0.1 mg/ml streptomycin (Sigma), and 2 mM L-glutamine (Sigma). Finally, they were transferred into 75-cm2 culture flasks (Euroclone) in incubator under standard conditions. The culturing was performed until passage 15 (16).
Experimental Protocol
To evaluate the effects of plasma samples taken from NAFLD patients and healthy subjects, on cell viability (MTT Assay), mitochondrial membrane potential (JC-1 Assay), H2O2 release (ROS-Glo H2O2,) mitochondrial ROS (mitoROS) release, triglycerides content (Triglyceride assay) and protein expression (Western Blot) in Huh7.5 cells/primary human hepatocytes, co-culture experiments were performed, by using specific Transwell inserts (Supplementary Figure 1A).
For the experiments, plasma samples from 12 NAFLD patients and 12 healthy subjects were plated in the apical compartment of the insert and left to act for 3 h, while, Huh7.5 cells/primary human hepatocytes were plated in the basal compartment. The 3 h time limit for stimulation was chosen based on preliminary time-course experiments during which Huh7.5 cells were exposed to plasma for 3, 12, and 24 h, showing that cell viability was drastically reduced already at 12 h. In addition, preliminary experiments were performed on both Huh7.5 cells and primary human hepatocytes with 5, 10, and 20% plasma calculated in relation to the total volume of each insert. In those experiments the effects of plasma from NAFLD patients and healthy subjects on cell viability and H2O2 release were measured (Supplementary Figure 1B). The results obtained allowed us to select the proper plasma concentration to be used for all next experiments performed on Huh7.5 cells. Furthermore, in some experiments, 1 nM inflammasome NLRP3 inhibitor (MCC950) was administrated to Huh7.5 cells for 30 min alone or before 200 pM TNFα (for 3 h) in co-stimulation with plasma. Some samples of Huh7.5 cells and primary human hepatocytes were not treated with plasma and were used as “control.” At the end of stimulations, various assays were performed (Supplementary Figure 1A). All experiments were conducted in triplicate and repeated at least five times.
1) Cell Viability
Cell viability was examined in Huh7.5 cells and primary human hepatocytes by using the 1% 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT; Life Technologies Italia, Monza, Italy; catalog number CT02) dye, as previously performed (17–21) and described in Supplementary Material. Cell viability was determined by measuring the absorbance through a spectrometer (VICTOR™ X Multilabel Plate Reader; PerkinElmer) with a wavelength of 570 nm and cell viability was calculated by setting control cells as 100%.
Mitochondrial Membrane Potential Measurement
Mitochondrial membrane potential measurement in Huh7.5 cells was performed with JC-1 assay (17, 19–22). The detailed description of methods is reported in Supplementary Material. The mitochondrial membrane potential was determined by measuring the red (excitation 550 nm/emission 600 nm) and green (excitation 485 nm/emission 535 nm) fluorescence through a spectrometer (VICTOR™ X Multilabel Plate Reader; PerkinElmer). The data were normalized vs. control cells.
2) Mitochondrial ROS Quantification
MitoROS production was determined through the Cayman's Mitochondrial ROS Detection Assay Kit (Cayman Chemical; catalog number 701600), as previously performed (23) and described in Supplementary Material. The MitoROS production was measured with an excitation and emission wavelength of 480 and 560 nm, respectively, by using a spectrophotometer (VICTOR™ X Multilabel Plate Reader; PerkinElmer). The data were normalized vs. control cells.
3) ROS-Glo H2O2 Quantification
H2O2 production was determined by the ROS-Glo H2O2 Assay, following the manufacturer's instructions (Promega Corporation; Padova, Italy; catalog number G8820) (24, 25). The detailed description of methods is reported in Supplementary Material. The H2O2 production was quantified as relative luminescence by using a spectrophotometer (VICTOR™ X Multilabel Plate Reader; PerkinElmer). The data were normalized vs. control cells.
4) Triglycerides Quantification
Triglycerides measurement was performed with a specific kit (Cayman Chemical; catalog number 10010303) and as described in Supplementary Material. The triglycerides content was detected following the manufacturer's instructions through a spectrometer (VICTOR™ X Multilabel Plate Reader; PerkinElmer) at excitation/emission wavelengths of 530–550 nm (22). The value of each sample was quantified in respect to triglycerides standard curve and expressed as triglycerides content (mg/dl).
5) Cell Lysates
For protein expression/activation, Huh7.5 cells were stimulated as described for various assays. For the experiments, 400000 Huh7.5 cells/insert in 6-Transwell plate were plated. At the end of stimulation, Huh7.5 cells were lysed in iced Ripa buffer supplemented with sodium orthovanadate (2 mM; Sigma) and protease inhibitors cocktail (1 mM; Thermo Fisher Scientific) and phenylmethanesulfonyl fluoride (1 mM; Sigma). The extracted proteins were quantified through bicinchoninic acid protein (BCA, Pierce) and used for electrophoresis and immunoblotting studies (16–22). Due to shortage of plasma specimens, experiments were conducted with plasma taken from three NAFLD patients and three healthy subjects and were repeated at least three times.
Western Blot Analysis
Cell lysates (30 μg protein each sample) were dissolved in 5 × Laemmli buffer, boiled for 5 min and resolved in 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis gels (Bio-Rad Laboratories). After electrophoresis they were transferred to polyvinylidene fluoride membranes (Bio-Rad Laboratories) and incubated overnight at 4°C with specific primary antibodies: anti Nf-kB (p-50; 1:1,000; Santa Cruz Biotechnology, catalog number sc-8414), anti gp91-phox (NOX2; 1:1,000; Santa Cruz Biotechnology, catalog number sc-130543), anti PPAR-γ (1:1,000; Santa Cruz Biotechnology, catalog number sc-271392), anti SREBP-1c (1:1,000; Santa Cruz Biotechnology, catalog number sc-13551). The membranes were washed and then incubated with horseradish peroxidase-coupled goat anti-rabbit IgG (Sigma), peroxidase-coupled rabbit anti-goat IgG and horseradish peroxidase-coupled goat anti-mouse IgG (Sigma) for 45 min and were developed through a non-radioactive method using Western Lightning Chemiluminescence (PerkinElmer Life and Analytical Sciences). Phosphorylated protein expression was calculated as a ratio toward β-actin (1:5,000; Santa Cruz Biotechnology; catalog number sc-47778) detection.
Statistical Analysis
Statistical analysis was performed using STATVIEW version 5.0.1 for Microsoft Windows (SAS Institute Inc., Cary NC, USA). Data were checked for normality before statistical analysis. All data are presented as means ± standard deviation (SD) of five independent experiments for each experimental protocol. Differences between groups were analyzed by one-way ANOVA and Bonferroni post hoc tests. The threshold for statistical significance was 0.05 (two-tails).
Results
NAFLD patients were seven males and five females, aged 51 ± 14 years, had a body mass index (BMI) of 31.1 ± 4.0 kg/m2, plasma alanine aminotransferase (ALT) concentration of 43 ± 29 U/L, plasma total cholesterol of 182 ± 30.4 mg/dl, liver stiffness and a controlled attenuation parameter measured by FibroScan® of 7.1 ± 1.9 kPa and 297 ± 45 dB/m, respectively. Healthy subjects were seven males and five females, aged 24.2 ± 0.4 year, had a BMI 22.9 ± 2.9 kg/m2, plasma ALT concentration of 23 ± 4 U/L, plasma total cholesterol of 167 ± 19 mg/dl, liver stiffness and a controlled attenuation parameter measured by FibroScan® of 5.4 ± 1 kPa and 186 ± 27 dB/m, respectively. Significant differences were observed in age, BMI, ALT, controlled attenuation parameter and liver stiffness measured between NAFLD and non-NAFLD patients (p < 0.05).
A dose response study was executed in order to choose the proper plasma concentration for the in vitro experiments. As shown in Supplementary Figure 2, plasma from NAFLD patients was able to reduce cell viability and increase H2O2 release in both Huh7.5 cells and primary human hepatocytes. In addition, a grading response was seen by using 5–20% plasma. Indeed, as regarding primary human hepatocytes viability, it amounted to 25, 34, and 43% about, in cells treated with 20, 10, and 5% NAFLD plasma, respectively. At the same time ROS release increased nearly by 100, 76, and by 56%. Moving on Huh7.5 cells, cell viability amounted to 34, 43, and 53% about, in cells treated with 20, 10, and 5% NAFLD plasma, respectively, whereas ROS release increased nearly by 80, 60, and 40%. Although the highest effects were observed after the treatment with the 20% plasma concentration, we decided to choose the 5% plasma concentration for all subsequent experiments in order to save plasma. As described in Figure 1, in Huh7.5 cells, plasma of NAFLD patients was able not only to reduce the cell viability but also mitochondrial membrane potential, by 47.25 ± 13.2% and 23.5 ± 0.8%, respectively. The finding of a reduction of mitochondrial membrane polarization would confirm the harmful effects elicited by NAFLD plasma. It is to note that those effects were accompanied by an increase not only of H2O2 release amounting to 45 ± 1.3%, but also of mitoROS release of 20.2 ± 0.7%. In addition, an increase of triglycerides content of 27.8 ± 2.1% was observed, as well.
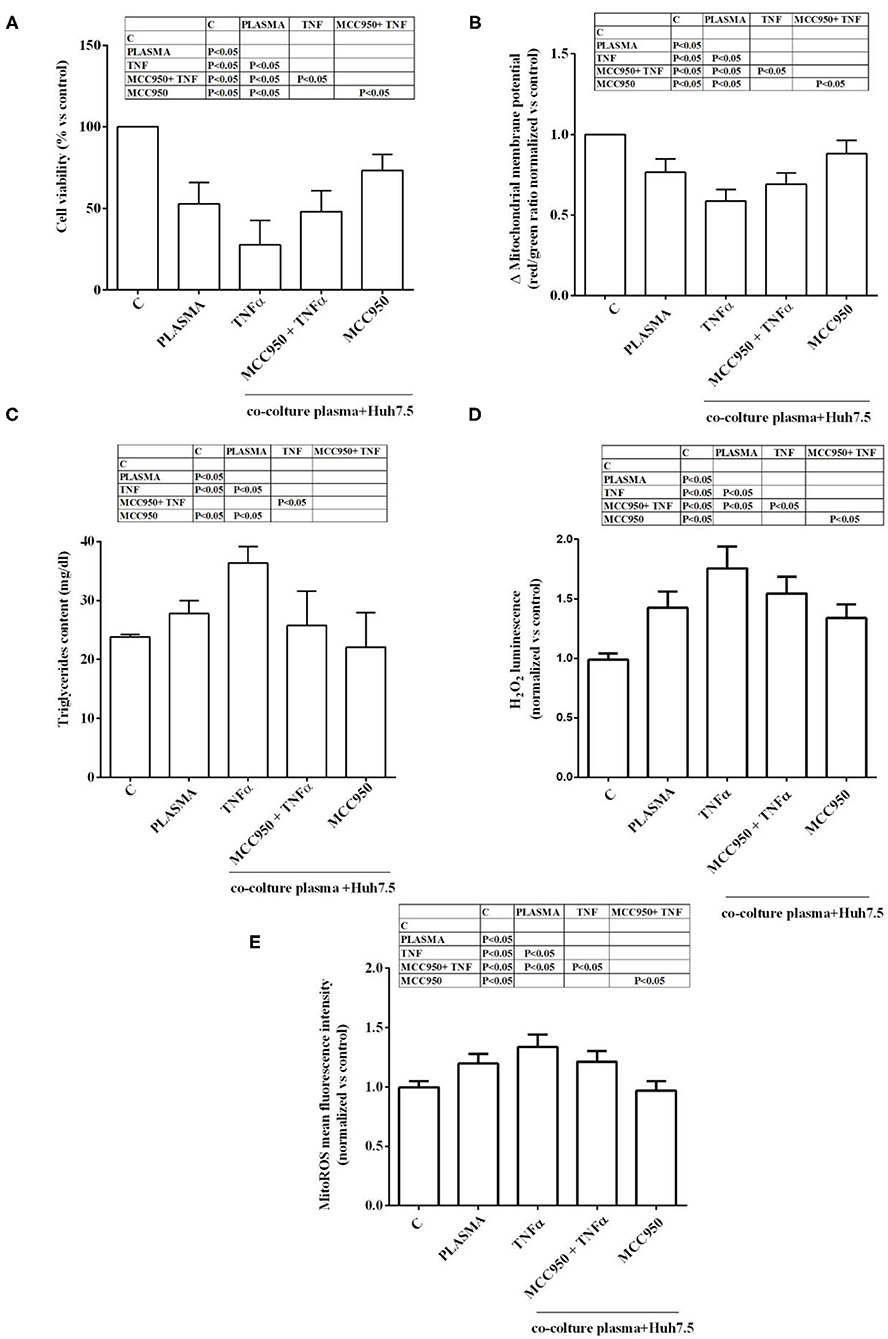
Figure 1. Effects of NAFLD plasma on Huh7.5 cell viability (A), mitochondrial membrane potential (B), triglycerides content (C), H2O2 release (D) and mitochondrial ROS (mitoROS, E). C=non-treated cells. MCC950 (NLRP3 inflammasome inhibitor, 1 nM for 30 min); TNFα (200 pM for 3 h). Reported data are means ± SD of five independent experiments for each experimental protocol.
In the presence of MCC950, the NLRP3 inflammasome inhibitor, the effects of NAFLD plasma on Huh7.5 cells were counteracted. Hence, cell viability and mitochondrial membrane potential were increased in comparison with findings obtained in Huh7.5 cells treated with NAFLD plasma alone (Figures 1A,B). Moreover, ROS and mitoROS release were reduced, as well as, the triglycerides content (Figures 1C–E).
It is to note that all the effects of NAFLD plasma on Huh7.5 cells were higher than those caused by plasma from healthy subjects (Figure 2). Also, TNFα was able to reduce cell viability, mitochondrial membrane potential, to increase triglycerides content and promote hydrogen peroxide and mitoROS release in a stronger way in the presence of NAFLD plasma than healthy subjects' plasma. The NLRP3 inflammasome inhibitor, MCC950, was so effective in preventing the deleterious effects of NAFLD plasma that no differences could be observed between NAFLD plasma and healthy subjects' plasma, as regarding mitochondrial membrane potential, triglycerides content and mitoROS release.
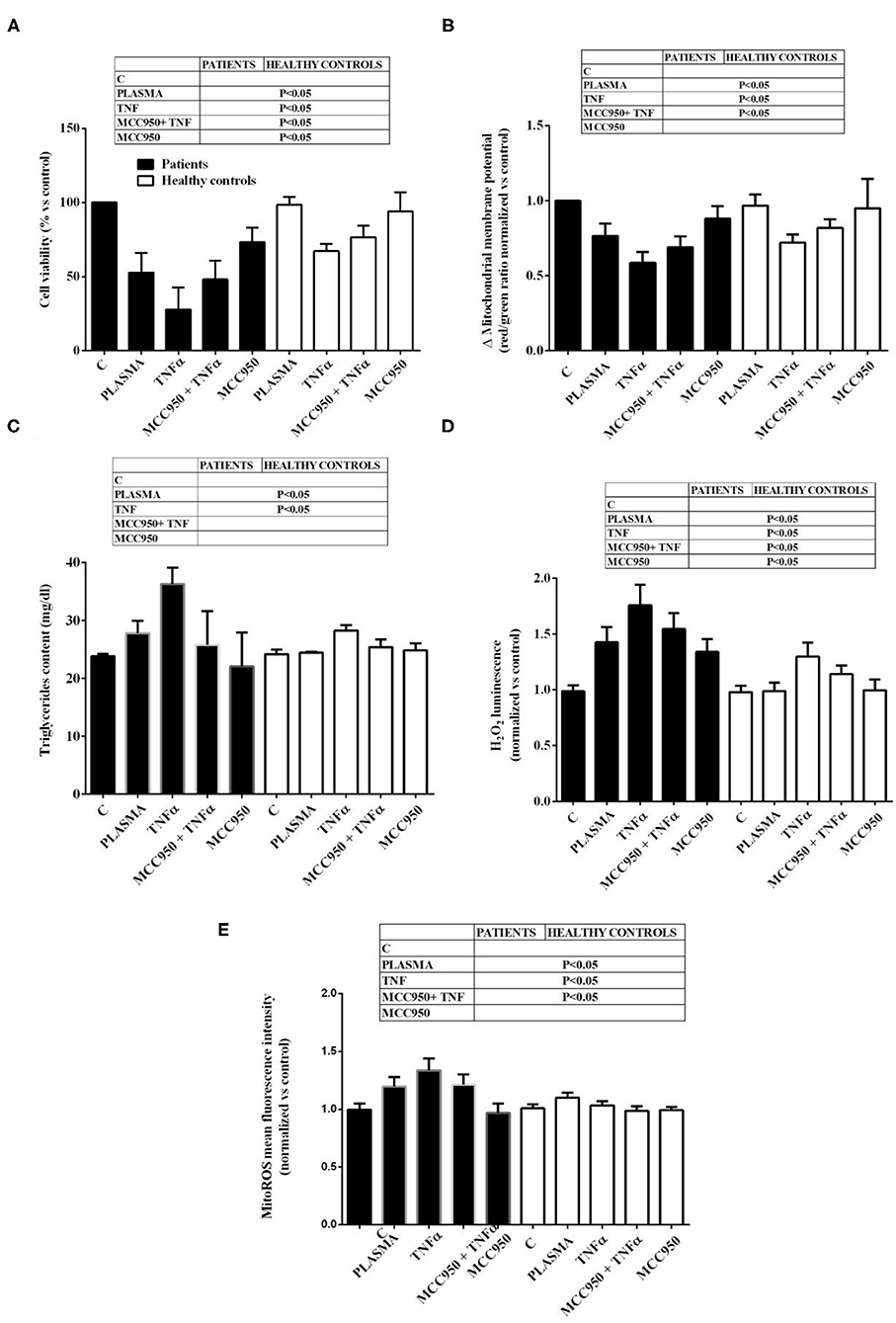
Figure 2. Comparison between the effects of NAFLD plasma and of plasma from healthy subjects on Huh7.5 cell viability (A), mitochondrial membrane potential (B), triglycerides content (C), H2O2 release (D) and mitochondrial ROS (mitoROS, E). Abbreviations are as described in Figure 1. Reported data are means ± SD of five independent experiments for each experimental protocol.
Western blot analysis performed on Huh7.5 cells treated with NAFLD plasma at 5% concentration showed the involvement inflammatory pathways and of an intracellular signaling related to lipid accumulation in liver during NAFLD. Indeed, in Huh7.5 cells treated with plasma of NAFLD patients, an increase in the expression of NF-kB, NOX2, PPARγ and SREBP-1c of 28.7 ±14.8%, 31,8 ± 16,1%, 55 ± 13%, 34,7 ± 13%, respectively, was observed (Figures 3, 4).
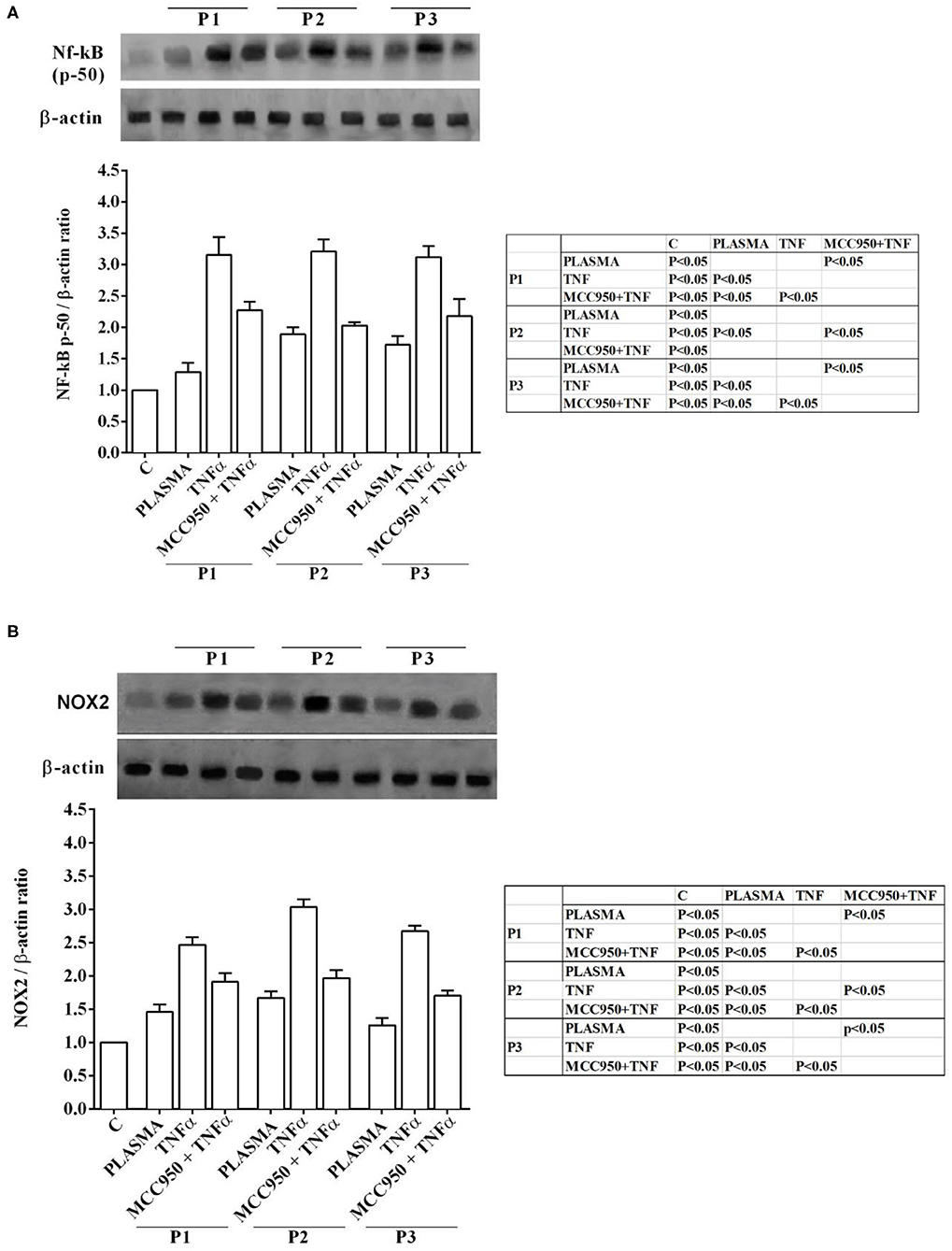
Figure 3. Effects of NAFLD plasma on NF-kB (A) and NOX2 (B) expression in Huh7.5 cells. P, patient; M, MCC950 (1 nM, for 30 min). Other abbreviations are as described in previous Figures. In the densitometric analysis, all values are normalized vs. control which is normalized, as well, and considered as 1. They are shown as fold changes vs. control. Reported data are means ± SD of five independent experiments for each experimental protocol.
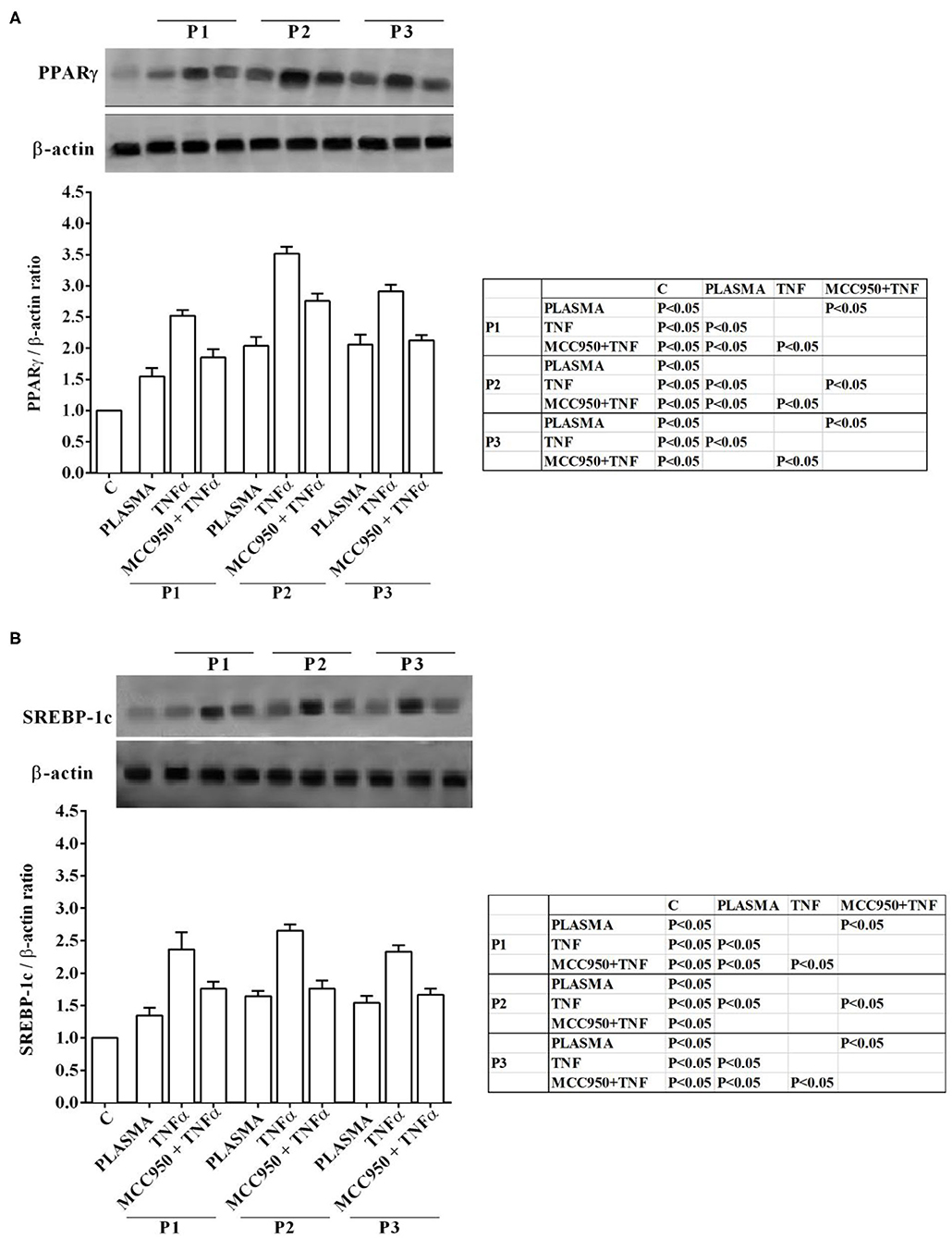
Figure 4. Effects of NAFLD plasma on PPARγ (A) and SREBP-1c (B) expression in Huh7.5 cells. P, patient; M, MCC950 (1 nM, for 30 min). Other abbreviations are as described in previous Figures. In the densitometric analysis, all values are normalized vs. control which is normalized, as well, and considered as 1. They are shown as fold changes vs. control. Reported data are means ± SD of five independent experiments for each experimental protocol.
In our experiments we used TNFα as positive “inflammatory” control to stimulate Huh7.5 cells treated with plasma of both NAFLD patients and healthy subjects. As observed in previous experiments, also in Western blot analysis all effects of plasma of NAFLD patients were potentiated by TNFα and reduced by MCC950 (Figures 1–4).
In addition, all the above effects observed in Huh7.5 cells treated with plasma of NAFLD patients were accompanied by an increase in the expression of NF-kB, NOX2, PPARγ and SREBP-1c of 28.7 ±14.8%, 31,8 ± 16,1%, 55 ± 13%, 34,7 ± 13%, respectively (Figures 3, 4). All effects of plasma of NAFLD patients were potentiated by TNFα and reduced by MCC950 (Figures 1–4).
Discussion
The present paper documents that exposure of hepatocytes to plasma from NAFLD patients was able to affect cell viability and to modulate intracellular pathways in vitro in the direction predicted by current hypotheses on NAFLD pathogenesis.
In recent years, several in vitro model of NAFLD of increasing complexity have been proposed, from culture of a single cell type to 3D cocultures and body-on-a-chip systems (26). Human hepatoma cell lines are easy to maintain and may be apt for multiparametric evaluation of steatosis (27), though their enzymatic profile may differ significantly from that of normal hepatocytes. On these premises, we decided to set a system in which human hepatoma cells could be maintained at ease varying conditions in which they were grown by using specific stimulant and inhibitor molecules. It should be of particular interest and represent a touch of originality the use of plasma for treating hepatocytes, Hence, almost all previous studies have been performed by simulating an in vitro condition of NAFLD milieu, using mixed FFAs solutions. Here, we aimed to examining the role of any circulating factors playing a pathophysiological role in the onset of NAFLD. The Huh7.5 cell line that we employed in our experiments has gained popularity for being highly permissive for hepatitis C virus replication in vitro (28). Moreover, some experiments have been performed in human primary hepatocytes, as well, to set up the optimal experimental conditions and to confirm the results obtained in Huh7.5 cells.
It is to note that the method employed for hepatocytes isolation is the one that was reported to result in hepatocytes populations of high purity with retained physiological activity in vitro (14).
In both Huh7.5 cells and human primary hepatocytes we tested 5, 10, and 20% NAFLD and non-NAFLD plasma samples on cell viability and H2O2 release, so that we could select the proper plasma concentration to be used in all other experiments performed on Huh7.5 cells only. To culture medium of Huh7.5 cells we also added TNFα, to reproduce in vitro conditions mimicking the steatohepatitis milieu (29), where TNFα plays a major role (30).
Furthermore, experiments were conducted in the presence or absence of the potent and selective inhibitor MCC950, which targets directly the NLRP3 ATP-hydrolysis motif for inflammasome inhibition (31).
Regarding the loss of cell viability that we observed after exposure to NAFLD plasma, it was presumably due to the differences in triglycerides content and oxidants release observed in Huh7.5 cells. It is to note that H2O2 and mitoROS have widely been considered as main contributors to liver injury and disease progression in NAFLD (32–35).
These effects were amplified in the presence of TNFα and reduced by the pretreatment with MCC950. It is noteworthy that in similar in vitro models, TNFα appears to mediate an increase of mitochondrial Ca2+ leading to production of ATP and ROS, with the shedding of TNF receptor 1 (which acts as a decoy) limiting the propagation of the inflammatory response (36). Our results would indicate the potentiation by TNFα of the harmful effects elicited by not yet clarified “inflammatory” factors circulating in the plasma of NAFLD patients, a mechanism that was counteracted by the NLRP3 inflammasome inhibition.
Following the “multiple-hit” hypothesis, hepatic lipid overload would induce the overproduction of oxidants that could be detrimental for DNA, lipids and proteins and lead to the accumulation of the so called “damage-associated molecular pattern” (DAMPS), which would induce liver injury. In addition, the impairment of the electrons transfer chain and the fall of mitochondrial membrane potential could be followed by cell death (35).
Mitochondrial membrane potential is essential for cells, enabling them to store energy: a long-lasting drop of mitochondrial potential is deleterious to cell viability, possibly more because of interference by products of ATP hydrolysis than by ATP in itself. About this issue it is to note that the increased mitoROS release we have observed was accompanied by the fall of mitochondrial membrane potential in Huh7.5 cells.
Interestingly, we also found an increase of SREBP-1c and PPARγ expression, which are involved in the intracellular triglycerides accumulation (1) and an increase of NF-kB and NOX2. As concerning SREBP-1c, it has been found to be involved in mitochondrial oxidants release through AQP8 upregulation (36). NF-kB is responsible for the transcription of pro-inflammatory molecules; among other stimuli like SREBP-1c (37). Also, NOX enzymes, especially NOX1, NOX2, and NOX4, have been associated with liver injuries in NAFLD both in vivo and in vitro, through the increased ROS release and the reciprocal interaction between different NOX enzymes and mitochondria (38).
The data we have obtained with MCC950 highlight the involvement of NLRP3 inflammasome in eliciting the harmful effects of NAFLD plasma in Huh7.5 cells. Our findings are in agreement with previous observations about the role of NOD-like receptors in NAFLD pathogenesis. In fact, the release by dying hepatocytes of DAMPS can be detected by members of NOD-like receptors, such as the NLRP3 inflammasome. Its activation is followed by cleavage of pro-caspase-1 into active caspase-1, which, in turn, cleaves pro-IL-1β into mature IL-1β (39, 40). Overall, the activation of NLRP3 inflammasome may potentiate a proinflammatory condition, resulting into stellate cells activation and transformation in a myofibroblastic phenotype. The fact however, that the only some effects of NAFLD plasma were abolished by MCC950, whereas others were just reduced, would suggest, on the one side, the important role played by NLRP3 inflammasome, and on the other side, the existence of other mechanisms which could trigger the damage. As also previously reported and confirmed by our results, intracellular pathways related to SREBP-1c and PPARγ could be involved. In addition, the modulation of inflammatory/anti-inflammatory cytokines, like interleukin 6 or adiponectin, could play a role and could be object of future studies.
Interestingly, the present data suggest the existence of an association between the activation of the NLRP3 inflammasome and that of intracellular pathways associated with SREBP-1c, PPARγ, NOX2 and NF-kB. In the presence of the inhibitor MCC950, the expression of all the above proteins was reduced in Huh7.5 cells exposed to NAFLD plasma. Data about SREBP-1c are also in agreement with previous findings in HepRG cells (41). Thus, it could be hypothesized a role for the NLRP3 inflammasome in the regulation of triglycerides accumulation via involvement of SREBP-1c and PPARγ, and in the onset of oxidative stress. Concomitantly or alternatively, the NLRP3 inflammasome may be responsible for oxidants release through signaling involving NOX2 and NF-kB. Future studies could be organized aimed at the analysis of other intracellular pathways related to all the above pathways and their relationships. In this context, it would be of interest to examine the role of AMP-activated protein kinase (AMPK), which is crucial for the regulation of fat mebabolism in liver.
One limitation of the present study is that NAFLD patients and healthy subjects were different as regarding BMI and age. This bias is somewhat inevitable, since NAFLD and NAFLD progression are strongly related to BMI and age. Moreover, sample size could be increased. The hepatoma cell line we used is not fully comparable to those of normal hepatocytes, though preliminary experiments performed on primary human hepatocytes (data not shown) confirmed the results obtained regarding cell viability and oxidant release. One may also argue that the model does not allow the identification of any specific pathogenic factor(s) and/or of putative biomarker(s); on the other hand, it has the advantage of being free of bias on the kind of substances involved. As said, our aim was to examine the role of any circulating factor possibly involved in the onset of NAFLD without venturing in the attempt of identifying all possibleculprits. Indeed, this is the first study performed by using NAFLD plasma on Huh7.5 cells and aimed at the analysis of cell viability, mitochondrial function and oxidants release. In addition, the role of main pathways involved in NAFLD onset has been investigated by performing experiments in the presence of an inhibitor and through Western Blot. The findings of harmful effects elicited by NAFLD plasma on hepatocytes would represent the starting point for the execution of future studies aimed at deepening the aforementioned issues. In particular, the application of proteomic and metabolomic profiling technologies to NAFLD plasma might contribute to identify any circulating factor(s) (microRNA, extracellular vesicles), whereas, in vitro experiments could be organized to increase the knowledge about the intracellular mechanisms (SREBP-1c, PPARγ, inflammasome molecules, AMPK).
Conclusions
In summary, we have established an in vitro model in which exposure to NAFLD plasma, in agreement with what predicted by “hormonocentric” theories on NAFLD pathogenesis (12, 42) resulted in loss of cell viability and activation of pathways known to be involved in inflammation and tissue damage. Substances present in NAFLD plasma could have affected lipid metabolism of Huh7.5 cells: triglycerides accumulation activated as downstream signaling involving SREBP-1c and PPARγ may have acted as a starting point. Hence, accumulation of triglycerides may have hampered mitochondrial function either directly or through increased oxidants release, NOX2 and NLRP3 inflammasome-related pathways.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethical Committee of the Azienda Ospedaliera Maggiore della Carità University Hospital in Novara. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
EG and MP: concenptualization and funding acquisition. DG, RM, RR, CR, CS, and MB: methodology and investigation. DS, MB, and MP: resources. DG, GC, CR, CS, and MB: formal analysis. EG, DG, GC, RR, CR, RM, CS, DS, MB, and MP: supervision, validation, visualization, writing/review, and editing. EG, GC, and MP: writing/original draft preparation. All authors involved in editing the paper and had final approval of the submitted and published versions.
Funding
This study was (partially) funded by FAR-2017 Università del Piemonte Orientale.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge the friendly contribution of personnel in the General Pathology Unit of the Azienda Ospedaliera Maggiore della Carità, University Hospital in collecting and processing blood specimens from NAFLD patients.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2021.693997/full#supplementary-material
References
1. Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci. (2018) 75:3313–27. doi: 10.1007/s00018-018-2860-6
2. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of non-alcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. (2016) 64:73–84. doi: 10.1002/hep.28431
3. Nguyen P, Leray V, Diez M, Serisier S, Le Bloc'h J, Siliart B, et al. Liver lipid metabolism. J Anim Physiol Anim Nutr. (2008) 92:272–83. doi: 10.1111/j.1439-0396.2007.00752.x
4. Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol. (2012) 56:952–64. doi: 10.1016/j.jhep.2011.08.025
5. Masarone M, Rosato V, Dallio M, Gravina AG, Aglitti A, Loguercio C, et al. Role of oxidative stress in pathophysiology of non-alcoholic fatty liver disease. Oxid Med Cell Longev. (2018) 11:9547613. doi: 10.1155/2018/9547613
6. Silva MF, Aires CC, Luis PB, Ruiter JP, IJlst L, Duran M, et al. Valproic acid metabolism and its effects on mitochondrial fatty acid oxidation: a review. J Inherit Metab Dis. (2008) 31:205–16. doi: 10.1007/s10545-008-0841-x
7. Grattagliano I, De Bari O, Bernardo TC, Oliveira PJ, Wang DQ, Portincasa P. Role of mitochondria in non-alcoholic fatty liver disease-from origin to propagation. Clin Biochem. (2012) 45:610–8. doi: 10.1016/j.clinbiochem.2012.03.024
8. Patterson RE, Kalavalapalli S, Williams CM, Nautiyal M, Mathew JT, Martinez J, et al. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am J Physiol Endocrinol Metab. (2016) 310:E484–94. doi: 10.1152/ajpendo.00492.2015
9. Del Campo JA, Gallego P, Grande L. Role of inflammatory response in liver diseases: therapeutic strategies. World J. Hepatol. (2018) 10:1–7. doi: 10.4254/wjh.v10.i1.1
10. Del Ben M, Polimeni L, Carnevale R, Bartimoccia S, Nocella C, Baratta F, et al. NOX2-generated oxidative stress is associated with severity of ultrasound liver steatosis in patients with non-alcoholic fatty liver disease. BMC Gastroenterol. (2014) 14:81. doi: 10.1186/1471-230X-14-81
11. Sarkar S, Kimono D, Albadrani M, Seth RK, Busbee P, Alghetaa H, et al. Environmental microcystin targets the microbiome and increases the risk of intestinal inflammatory pathology via NOX2 in underlying murine model of non-alcoholic fatty liver disease. Sci Rep. (2019) 9:8742. doi: 10.1038/s41598-019-45009-1
12. Lonardo A, Carani C, Carulli N, Loria P. 'Endocrine NAFLD' a hormonocentric perspective of non-alcoholic fatty liver disease pathogenesis. J Hepatol. (2006) 44:1196–207. doi: 10.1016/j.jhep.2006.03.005
13. Kholodenko IV, Kholodenko RV, Manukyan GV, Burunova VV, Yarygin KN. Mesenchymal-epithelial transition in culture of stromal progenitor cells isolated from the liver of a patient with alcoholic cirrhosis. Bull Exp Biol Med. (2016) 162:115–9. doi: 10.1007/s10517-016-3559-z
14. Werner M, Driftmann S, Kleinehr K, Kaiser GM, Mathé Z, Treckmann JW, et al. All-in-one: advanced preparation of human parenchymal and non-parenchymal liver cells. PloS ONE. (2015) 10:e0138655. doi: 10.1371/journal.pone.0138655
15. Del Campo JA, Valdecasas MG, Gil-Gómez A, Rojas A, Gallego P, Ampuero J, et al. Simvastatin and metformin inhibit cell growth in hepatitis C virus infected cells via mTOR increasing PTEN and autophagy. PloS ONE. (2018) 13:e0191805. doi: 10.1371/journal.pone.0191805
16. Farruggio S, Cocomazzi G, Marotta P, Romito R, Surico D, Calamita G, et al. Genistein and 17β-estradiol protect hepatocytes from fatty degeneration by mechanisms involving mitochondria, inflammasome and kinases activation. Cell Physiol Biochem. (2020) 54:401–16. doi: 10.33594/000000227
17. De Cillà S, Farruggio S, Vujosevic S, Raina G, Filippini D, Gatti V, et al. Anti-vascular endothelial growth factors protect retinal pigment epithelium cells against oxidation by modulating nitric oxide release and autophagy. Cell Physiol Biochem. (2017) 42:1725–38. doi: 10.1159/000479441
18. Surico D, Bordino V, Cantaluppi V, Mary D, Gentilli S, Oldani A, et al. Preeclampsia and intrauterine growth restriction: role of human umbilical cord mesenchymal stem cells-trophoblast cross-talk. PLoS One. (2019) 14:e0218437. doi: 10.1371/journal.pone.0218437
19. Savoia P, Raina G, Camillo L, Farruggio S, Mary D, Veronese F, et al. Anti-oxidative effects of 17 β-estradiol and genistein in human skin fibroblasts and keratinocytes. J Dermatol Sci. (2018) 92:62–77. doi: 10.1016/j.jdermsci.2018.07.007
20. Huang XY, Li D, Chen ZX, Huang YH, Gao WY, Zheng BY, et al. Hepatitis B virus X protein elevates Parkin-mediated mitophagy through lon peptidase in starvation. Exp Cell Res. (2018) 368:75–83. doi: 10.1016/j.yexcr.2018.04.016
21. Surico D, Ercoli A, Farruggio S, Raina G, Filippini D, Mary D, et al. Modulation of oxidative stress by 17 β-estradiol and genistein in human hepatic cell lines in vitro. Cell Physiol Biochem. (2017) 42:1051–62. doi: 10.1159/000478752
22. Grossini E, Farruggio S, Raina G, Mary D, Deiro G, Gentilli S. Effects of genistein on differentiation and viability of human visceral adipocytes. Nutrients. (2018) 10:pii: E978. doi: 10.3390/nu10080978
23. Noëmi JR, Urs D, Jamal B, Krähenbühl S. The uricosuric benzbromarone disturbs the mitochondrial redox homeostasis and activates the NRF2 signaling pathway in HepG2 cells. Free Radic Biol Med. (2020) 152:216–26. doi: 10.1016/j.freeradbiomed.2020.03.009
24. Wittmann C, Chockley P, Singh SK, Pase L, Lieschke GJ, Grabher C. Hydrogen peroxide in inflammation: messenger, guide, and assassin. Adv Hematol. (2012) 2012:541471. doi: 10.1155/2012/541471
25. Murata H, Takamatsu H, Liu S, Kataoka K, Huh NH, Sakaguchi M. NRF2 regulates PINK1 expression under oxidative stress conditions. PLoS ONE. (2015) 10:e0142438. doi: 10.1371/journal.pone.0142438
26. Müller FA, Sturla SJ. Human in vitro models of non-alcholic fatty liver disease. Cur Opin Toxicol. (2019) 16:9–16. doi: 10.1016/j.cotox.2019.03.001
27. Donato MT, Tolosa L, Jiménez N, Castell JV, Gómez-Lechón MJ. High-content imaging technology for the evaluation of drug-induced steatosis using a multiparametric cell-based assay. J Biomol Screen. (2012) 17:394–400. doi: 10.1177/1087057111427586
28. Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. (2002) 76:13001–14. doi: 10.1128/JVI.76.24.13001-13014.2002
29. Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C, et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. (2014) 59:886–97. doi: 10.1002/hep.26749
30. Dludla PV, Nkambule BB, Mazibuko-Mbeje SE, Nyambuya TM, Marcheggiani F, Cirilli I, et al. N-acetyl cysteine targets hepatic lipid accumulation to curb oxidative stress and inflammation in NAFLD: a comprehensive analysis of the literature. Antioxidants. (2020) 9:1283. doi: 10.3390/antiox9121283
31. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nature Chem Biol. (2019) 15:556–9. doi: 10.1038/s41589-019-0277-7
32. Kanda T, Matsuoka S, Yamazaki M, Shibata T, Nirei K, Takahashi H, et al. Apoptosis and non-alcoholic fatty liver diseases. World J Gastroenterol. (2018) 24:2661–72. doi: 10.3748/wjg.v24.i25.2661
33. Feng R, Luo C, Li C, Du S, Okekunle AP, Li Y, et al. Free fatty acids profile among lean, overweight and obese non-alcoholic fatty liver disease patients: a case—control study. Lipids Health Dis. (2017) 16:165. doi: 10.1186/s12944-017-0551-1
34. Feldstein AE, Werneburg NW, Canbay A, Guicciardi ME, Bronk SF, Rydzewski R, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. (2004) 40:185–94. doi: 10.1002/hep.20283
35. Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of non-alcoholic fatty liver disease. Free Radic Biol Med. (2020) 152:116–41. doi: 10.1016/j.freeradbiomed.2020.02.025
36. Tamma G, Valenti G, Grossini E, Donnini S, Marino A, Marinelli RA, et al. Aquaporin membrane channels in oxidative stress, cell signaling, and aging: recent advances and research trends. Oxid Med Cell Longev. (2018) 2018:1501847. doi: 10.1155/2018/1501847
37. Li X, Huang W, Gu J, Du X, Lei L, Yuan X, et al. SREBP-1c overactivates ROS-mediated hepatic NF-κB inflammatory pathway in dairy cows with fatty liver. Cell Signal. (2015) 27:2099–109. doi: 10.1016/j.cellsig.2015.07.011
38. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. (2014) 94:909–50. doi: 10.1152/physrev.00026.2013
39. Numann K, Schiller B, Tiegs G. NLRP3 inflammasome and IL-33: novel players in sterile liver inflammation. Int J Mol Sci. (2018) 19:pii:E2732. doi: 10.3390/ijms19092732
40. Wan X, Xu C, Yu C, Li Y. Role of NLRP3 inflammasome in the progression of NAFLD to NASH. Can J Gastroenterol Hepatol. (2016) 2016:6489012. doi: 10.1155/2016/6489012
41. Liu B, Mao X, Huang D, Li F, Dong N. Novel role of NLRP3-inflammasome in regulation of lipogenesis in fasting-induced hepatic steatosis. Diabetes Metab Syndr Obes. (2019) 12:801–11. doi: 10.2147/DMSO.S206558
Keywords: biomarker, inflammasome, mitochondria, NAFLD, oxidative stress
Citation: Grossini E, Garhwal DP, Calamita G, Romito R, Rigamonti C, Minisini R, Smirne C, Surico D, Bellan M and Pirisi M (2021) Exposure to Plasma From Non-alcoholic Fatty Liver Disease Patients Affects Hepatocyte Viability, Generates Mitochondrial Dysfunction, and Modulates Pathways Involved in Fat Accumulation and Inflammation. Front. Med. 8:693997. doi: 10.3389/fmed.2021.693997
Received: 13 April 2021; Accepted: 07 June 2021;
Published: 02 July 2021.
Edited by:
Carmen Peralta Uroz, Institut de Recerca Biomèdica August Pi i Sunyer (IDIBAPS), SpainReviewed by:
Hassen Ben Abdennebi, University of Monastir, TunisiaAnabel Fernández-Iglesias, Institut de Recerca Biomèdica August Pi i Sunyer (IDIBAPS), Spain
Copyright © 2021 Grossini, Garhwal, Calamita, Romito, Rigamonti, Minisini, Smirne, Surico, Bellan and Pirisi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Grossini, ZWxlbmEuZ3Jvc3NpbmlAbWVkLnVuaXVwby5pdA==