
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 08 July 2021
Sec. Nephrology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.688060
This article is part of the Research TopicImmune Landscape of Kidney PathologyView all 23 articles
 Elena Cantero-Navarro1,2
Elena Cantero-Navarro1,2 Sandra Rayego-Mateos1,2*Macarena Orejudo3,4Lucía Tejedor-Santamaria1,2
Sandra Rayego-Mateos1,2*Macarena Orejudo3,4Lucía Tejedor-Santamaria1,2 Antonio Tejera-Muñoz1,2
Antonio Tejera-Muñoz1,2 Ana Belén Sanz2,5
Ana Belén Sanz2,5 Laura Marquez-Exposito1,2Vanessa Marchant1,2Laura Santos-Sanchez1,2
Laura Marquez-Exposito1,2Vanessa Marchant1,2Laura Santos-Sanchez1,2 Jesús Egido3,4
Jesús Egido3,4 Alberto Ortiz2,5
Alberto Ortiz2,5 Teresa Bellon6
Teresa Bellon6 Raúl R. Rodrigues-Diez1,2†
Raúl R. Rodrigues-Diez1,2† Marta Ruiz-Ortega1,2*†
Marta Ruiz-Ortega1,2*†Inflammation is a key characteristic of kidney disease, but this immune response is two-faced. In the acute phase of kidney injury, there is an activation of the immune cells to fight against the insult, contributing to kidney repair and regeneration. However, in chronic kidney diseases (CKD), immune cells that infiltrate the kidney play a deleterious role, actively participating in disease progression, and contributing to nephron loss and fibrosis. Importantly, CKD is a chronic inflammatory disease. In early CKD stages, patients present sub-clinical inflammation, activation of immune circulating cells and therefore, anti-inflammatory strategies have been proposed as a common therapeutic target for renal diseases. Recent studies have highlighted the plasticity of immune cells and the complexity of their functions. Among immune cells, monocytes/macrophages play an important role in all steps of kidney injury. However, the phenotype characterization between human and mice immune cells showed different markers; therefore the extrapolation of experimental studies in mice could not reflect human renal diseases. Here we will review the current information about the characteristics of different macrophage phenotypes, mainly focused on macrophage-related cytokines, with special attention to the chemokine CCL18, and its murine functional homolog CCL8, and the macrophage marker CD163, and their role in kidney pathology.
Renal inflammation arises as a protective response after kidney injury to fight against the initial insult and to establish tissue repair. However, if the reparative processes fail, this inflammatory response could be deleterious, participating in the kidney disease progression (1). This inflammatory response involves many different populations of immune infiltrating cells, including monocytes/macrophages, neutrophils, CD8+ and CD4+ lymphocytes, dendritic cells, mast cells and natural killer cells (2). Importantly, in chronic kidney disease (CKD), regardless of the underlying etiology, there is a persistent activation of the inflammatory response, characterized by immune cell recruitment throughout the kidney, leading to a local overproduction of growth factors and pro-fibrotic cytokines. This altered environment could activate different cellular and molecular processes, which cause progressive nephron loss and glomerular and interstitial fibrosis, leading to end-stage kidney disease and/or premature death (1–3). CKD is emerging as an important health problem due to the absence of early diagnostic biomarkers and effective treatments. Although many in vitro and experimental studies have extensively characterized the distinctive macrophage phenotypes in physiological and pathological conditions, the differences between human and murine macrophages complicate the extrapolation of preclinical results into human kidney disease, being a limitation on approaching macrophages as a therapeutic target to treat human CKD. Now, we review the macrophage characteristics and phenotypes, comparing human and mice data, and focused on novel macrophage-related cytokines as biomarkers or therapeutic targets for kidney disease. We will focus in two interesting candidates, the macrophage derived chemokine CCL18, as well as its murine functional homolog Ccl8, and the macrophage marker CD163.
Macrophages are innate immune cells and the main component of the mononuclear phagocyte system. Therefore, they are crucial in host defense against pathogens. Furthermore, macrophages are present in almost every organ in adult mammals, where they participate in multiple cellular processes and play a major role in the maintenance of tissue homeostasis (4). Macrophages are considered as tissue sentinels that maintain tissue integrity by eliminating senescent and dead cells, and debris (5). Moreover, they also participate in extracellular matrix (ECM) remodeling, mainly through matrix metalloproteinases (MMPs) release (6), and in the restoration of lost cells and intercellular matrices through the production of various regenerative growth factors (4).
Tissue resident macrophages (TRMs) can be classified into two different subtypes depending on their origin; one derived from circulating monocytes (7–9), and the other from embryonic precursors that are able to locally proliferate and self-renew (10, 11). Monocytes are limited to the blood compartment, the spleen and the bone marrow (12), but in response to inflammation, and guided by the cytokine milieu and/or interactions with other cells and microbial products, monocytes are quickly recruited into injured tissues and then differentiate into several specific macrophage phenotypes depending on micro-environmental signals (8, 13).
Plasticity is the hallmark feature of macrophages. The term “macrophage polarization” is used to refer to an estimate of macrophage activation status and phenotype (11). The M1/M2 polarization axis was originally defined in the 1990s (14) to describe the dichotomy in macrophage function regardless of cytokines: classically activated macrophages/M1 microbicidal macrophages and alternatively activated macrophages/M2 macrophages (Figure 1). M1 macrophages participate in the infection clearance and act as an initial defense barrier. The M1 phenotype is generated in response to pro-inflammatory stimuli, such as pathogen/danger-associated molecular patterns (PAMPs or DAMPs) in the presence of interferon gamma (IFN-γ) (14, 15). M1 cells are characterized by their ability to secrete significant amounts of pro-inflammatory cytokines including tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), interleukin-12 (IL-12), interleukin-15 (IL-15), interleukin-18 (IL-18), and interleukin-23 (IL-23) (16–18). M2 macrophages can be a “two-edged sword.” On one side, the anti-inflammatory phenotype is essential for adequate tissue repair; on the other side, it is a potential mediator of fibrosis and scarring (15). These M2 macrophages can release a different cytokine profile, such as transforming growth factor-β1 (TGF-β1), interleukin-10 (IL-10), C-C Motif Chemokine Ligand 17 (CCL17), C-C Motif Chemokine Ligand 18 (CCL18) and C-C Motif Chemokine Ligand 22 (CCL22) (Figure 2) (19–24). The macrophage population is currently thought to represent a continuous phenotype ranging between M1/M2 extremes and including other ill-defined populations, as commented above (Figure 1). Moreover, the M1/M2 classification is not representative of in vivo events, given that M1 and M2 stimuli do not exist alone in tissues. While a much broader spectrum of innate immune responses has been characterized, the M1/M2 axis remains the main macrophage polarization axis in vivo (25).
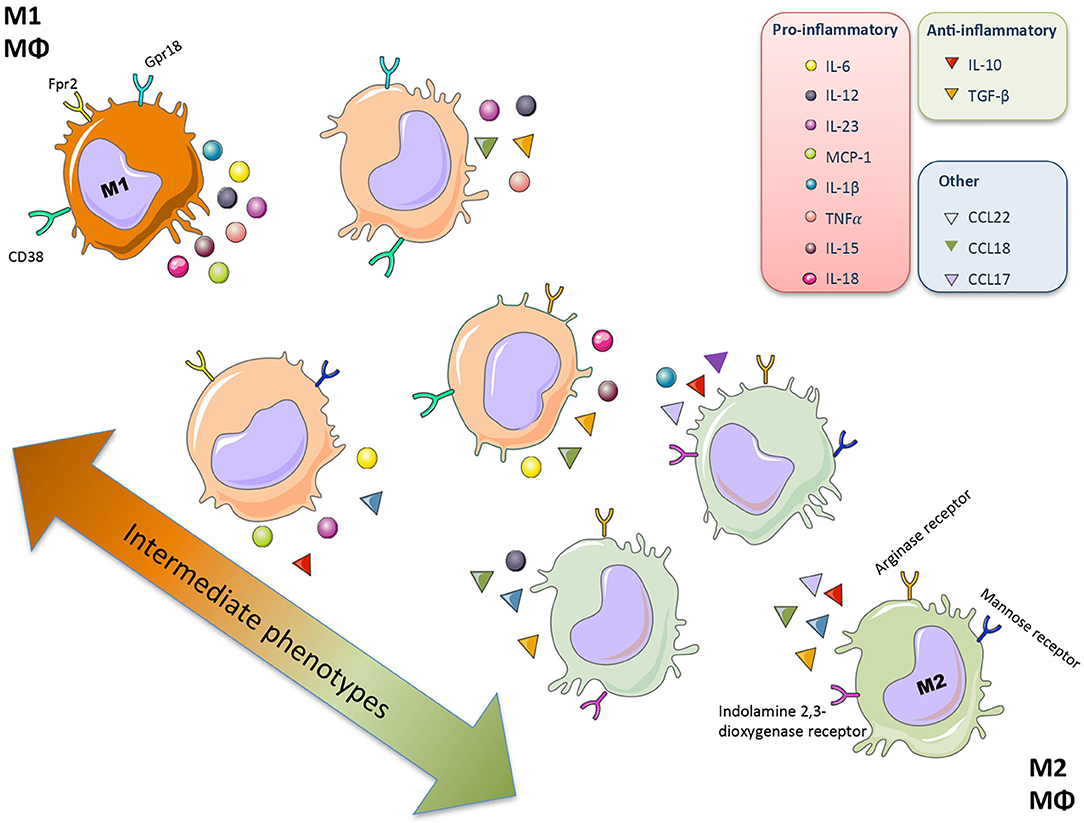
Figure 1. Macrophage polarization. Macrophages (Mθ) have a great plasticity and have different functional activation states and phenotypes that allow different specialized functions. The macrophage population represents a continuous phenotype ranging between M1/M2 extremes, characterized by a specific secretome (mainly cytokine and chemokines) and surface markers.
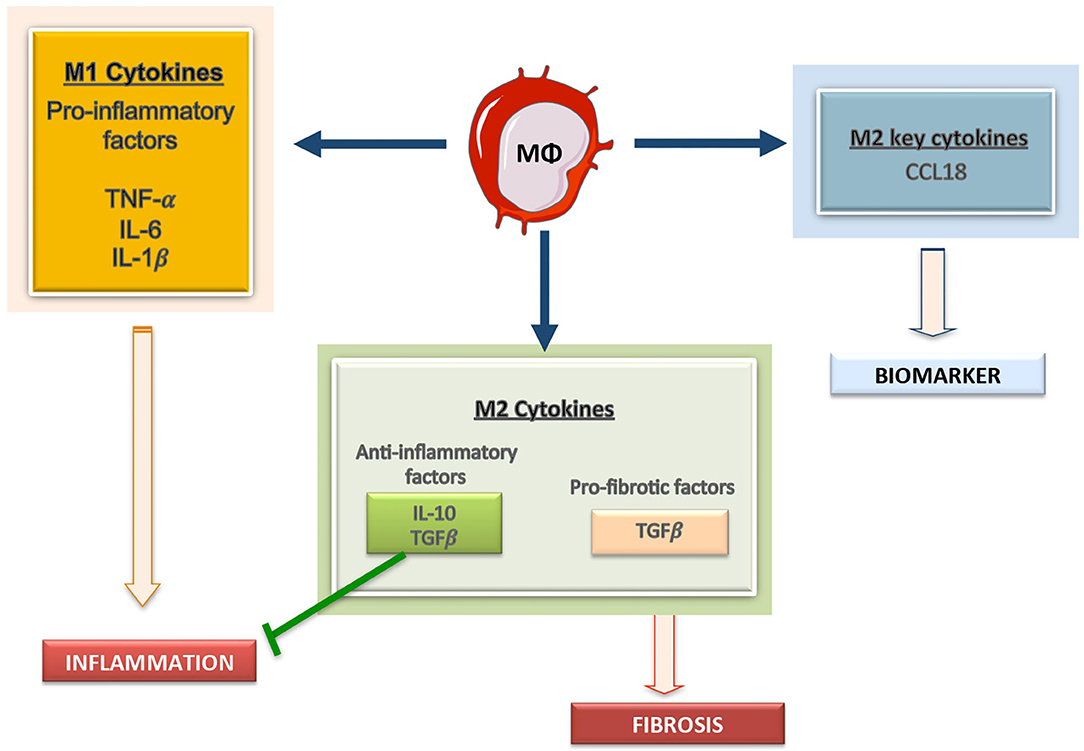
Figure 2. Macrophages derived cytokines can contribute to kidney damage. Macrophages can secrete a large array of molecules, including producing pro-inflammatory mediators, including IL1, Il6, and TNF, anti-inflammatory cytokines, such as IL-10, and profibrotic factors, like TGF-β. Among the M2 macrophage related factors CCL18 is a recent described cytokine and a potential biomarker of disease progression.
Most of the earliest studies on macrophage polarization were based on in vitro stimulation with different combination of cytokines. One of the most important regulators of macrophage differentiation is the colony-stimulating factor 1 receptor (CSF1R). This is a transmembrane tyrosine kinase receptor expressed on almost all mononuclear phagocytic cells (26). Csf1r gene deletion depleted macrophages in several tissues. Although this study demonstrates its key role in macrophages development, it also showed that some macrophages were still present, indicating the involvement of other growth factors in macrophage differentiation (27), including Granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-3 (IL-3), vascular endothelial growth factor (VEGF), and fms-like tyrosine kinase 3 ligand (Flt3L) (28–30). Macrophages respond in different ways to macrophage colony-stimulating factor (M-CSF) and GM-CSF with substantial polarizing effects. These factors have been used to study in vitro differentiation/polarization of macrophages (31–33). In this sense, GM-CSF activates the inflammatory program and extensive DNA methylation changes, while M-CSF-polarized cells (less differentiated state) show characteristics of anti-inflammatory repairing macrophages (33). Further research led to a sub-classification of M2 macrophages into M2a, M2b, and M2c described as wound-healing, immune complexes-related or regulatory macrophages, respectively (11, 16, 34). M2a activation induced by IL-4 or IL-13 was associated to changes in surface markers, including decreased CD14 expression, and upregulation of mannose receptor (CD206), CD209, and CD23. In M2a macrophages, pro-inflammatory cytokine expression is lower than in M1, but they release several factors, including CCL18 and vascular Endothelial Growth Factor (VEGF) (35). M2a can also secrete ECM components, like soluble fibronectin, and therefore their primary function is thought to be related to wound healing and tissue remodeling and repair (36). M2b cells are elicited in vitro by stimulation with LPS or IL-1β plus high concentrations of immune complexes. M2b macrophages are considered as functional converter of M1 cells with low IL-12 and high IL-10 production. Interestingly, they selectively produce the C-C Motif Chemokine Ligand (CCL1) (37). The M2c category includes cells stimulated with IL-10, TGF-β1 and glucocorticoids, being a heterogeneous group, but characterized by high expression of the surface marker CD163 (38). RNAseq studies have identified M2c-specific genes associated with angiogenesis, matrix remodeling, and phagocytosis, including CD163 (39). Accordingly, the analysis of the M2c macrophage-conditioned media revealed elevated production of MMPs (39). Due to the controversies in the field, a specific nomenclature for cytokine-induced macrophage polarization in vitro M (cytokine) have been proposed (40).
Originally, the distinction between M1 and M2 phenotypes was also based on differences in arginine metabolism (14), but recent research has unraveled the metabolic differences associated with macrophage phenotype switching (41). As an overview, and based on the premise that M1 macrophages kill pathogens and M2 resolve inflammation and tissue repair, M1 are more likely to be involved in catabolic pathways, and M2 in anabolic ones (42). Therefore, M1 tend to rely on working aerobic glycolysis likewise fatty acid biosynthesis, because of the speed of producing ATP, but it also have impaired oxidative phosphorylation (43). Moreover, in M1 macrophages, nitric oxide synthase is highly expressed, playing an essential role in the M1 function of killing pathogens, being nitric oxide the source of reactive oxygen species (ROS) with antimicrobial properties (36, 44). On the other hand, M2 are more suitable to trigger fatty acid oxidation as energy-production pathway (42). These macrophages secrete insulin-like growth factor 1 (IGF-1) (45), TGF-β1, and VEGF (46), as well as use arginine to produce precursors of collagen (47), thereby dampening inflammation.
Though we are aware that it may represent an oversimplification, since most of the previous literature has used the M1/M2 classification nomenclature, this terminology has been maintained in some parts of this review.
A limitation in the field of macrophage research is the extrapolation of preclinical studies to humans. Macrophages play a key role in the inflammatory response, but the pathogen type that infects humans can be different from other species, including rodents, as well as the molecules released by macrophages to control infections.
The studies investigating M1/M2 macrophage polarization, some of them included in the previous section, were done using mouse or human macrophages of different cell sources; including isolated circulating monocytes, bone marrow-derived cells or peritoneal macrophages, as well as established cell lines (such as THPs or RAW 264.7). A macrophage comparative study evaluating different cell sources depicted similarities between mouse and human cytokine profiles stimulated with a specific combination of cytokines, such as CXCL-10 and CXCL-11 for M (LPS,IFN) and CCL17 and CCL22 for M (IL4, IL13)-induced macrophage polarization (48). Although murine M1/M2-polarized macrophage subsets can be distinguished on the basis of combinatorial gene expression profiles, the identification of equivalent subsets in humans is still unresolved. Another key point is the specific M1/M2 macrophage subsets markers. In humans, similarly to mice, three monocyte subsets have been described according to differential expression of CD14 and CD16 on HLA-DR+ cells (49). In human macrophages, CD68 is a general marker, whereas HLA-DR and CD163 are M1 and M2 markers, respectively (49, 50). Murine and human macrophages express the antigens CD68, CD11b, CSF1R, and CD163 (5). F4/80 is expressed on most tissue macrophages in the mouse and it has been extensively studied by immunohistochemistry, but has limited usefulness in humans as F4/80 is predominantly expressed on eosinophils (51). Recent research is focused on the search of additional markers. The early growth response protein 2 (Egr2) and c-Myc have been described in murine models to identify in vivo polarized M2 cells. One the other hand, some in vivo pro-inflammatory factors have been shown to be M1 macrophage markers, such as G-protein-coupled receptor 18 (Gpr18), formyl peptide receptor 2 (Fpr2), CD40, and CD38 (52, 53). Among them, CD38 could be also a marker of M1 pro-inflammatory macrophages in humans (54). Comparative biology, together with omics technologies, such as transcriptomics, metabolomics, proteomics, and epigenomics could potentially be employed to assess similarities between mouse and human macrophages (4). Recently, the single-cell RNA sequencing across multiple mammalian species has led to the identification of CD74 and CD81 as surface markers for kidney resident macrophages (55). Future studies in human biopsies of different pathologies using cutting edge technologies will reveal human macrophages characteristics in each state of the disease.
Macrophages participate in immune surveillance and in the regulation of kidney homeostasis. The macrophage response to kidney injury varies enormously depending on the nature and duration of the insult (23). Macrophages participate in the inflammatory response, both in acute kidney injury (AKI) and in CKD. They can promote kidney repair or contribute to the AKI-to-CKD transition and fibrosis, highlighting their remarkable plasticity (2, 56).
Macrophages can actively participate in all AKI-related processes, including cell death, resolution/regeneration phase or progression to kidney fibrosis (57). Importantly, macrophage phenotypes can change along AKI, depending on disease stage and evolution. Circulating classical monocytes (CD11b+ Ly6Chigh in the mouse or CD14highCD16low in humans) are recruited into the kidney, where they differentiate to pro-inflammatory M1 macrophages during the early phase of renal injury, in response to infection or cell damage (23). Many preclinical data suggest that M1 macrophages play a pathogenic role in the early phases of AKI. Studies done in the model of folic acid in mice showed an upregulation of cytokines and chemokines, such MCP-1/CCL2 (the main macrophage chemotactic factor) associated to the presence of M1 macrophages in the injured kidney at 48 h (58). Moreover, in ischemia-reperfusion injury (IRI) in mice M1 macrophages, identified by their high expression of iNOS, IL-12, IL-23, and Ly6C, were detected also at 48 h (59). Accordingly, depletion of kidney macrophages by liposomal clodronate (LC) at the early stages of IRI reduces AKI and improves renal repair. Furthermore, adoptive transfer of IFN-γ-stimulated macrophages in LC-treated IRI mice worsens AKI (49). Following AKI, once pathogens or injured cells are cleared up, a rapid polarization of macrophage phenotypes is necessary for tissue regeneration. During the regeneration process there is a restoration of the polarized tubular epithelium and basement membrane integrity, together with neovascularization, ensuring the recovery of tissue oxygenation by the injured microvasculature, leading to the reestablishment of the tubular cell functionality (60, 61). Therefore, a decrease in infiltrating M1 macrophages is essential to minimize injury of surrounding cells, since macrophages release cytotoxic compounds that do not distinguish self from exogenous pathogens (62, 63). Preclinical studies have described high levels of IL-4, IL-10, and IL-13, leading to a macrophage switch to M2 phenotype characterized by high expression of arginase-1 (Arg1), mannose receptor (MR, also termed CD206), chitinase-like protein (e.g., Ym1), resistin-like protein (Fizz1) and CD36 (fatty acid translocase), associated with down-regulated expression of proinflammatory markers (i.e., IL-12 and iNOS) (49). On the other hand, the prolonged presence of infiltrating macrophages in the kidney might be associated with persistent release of wound-healing growth factors, such as TGF-β1. This may turn the initial wound healing process to pathological one, resulting in further tissue damage, and contributing to the AKI-to-CKD progression, and irreversible fibrosis (64). Nevertheless, data on human macrophage phenotypes and related cytokines in healthy and AKI kidneys have been scarcely studied, limiting the translation of preclinical studies to humans. Moreover, the mechanisms enabling macrophage switch from the M1 to M2 subset remain unclear (49). Studies of human AKI biopsies have identified macrophages as the main cell type infiltrating the kidney that persist during tissue repair, being CD163 expressing -macrophages the predominant phenotype in the late phase of AKI (50, 61). Future studies in human AKI at different disease stages are needed.
Kidney infiltration by macrophages is common in human CKD. The magnitude of macrophage infiltration correlates with the severity of kidney injury suggesting an effector function of macrophages in CKD (19). However, the role of M1/M2 macrophages in human CKD progression is still poorly understood. In experimental progressive CKD, M1 macrophages are present in the early phases of inflammation (23) and, as this process progresses, M2 macrophages predominate to encourage repair and/or fibrosis (65), as described in unilateral ureteral obstruction (UUO) model (57, 66, 67). Some studies have suggested that the M1/M2 macrophage balance could influence CKD development (68, 69). Both M1 and M2 responses coexist during CKD (68). Indeed, M2 macrophages could originate from M1 macrophages or from proliferation/differentiation of monocytes (23, 65, 68). Some evidences suggest that macrophages can directly promote kidney fibrosis. The CD206+ subset of M2 macrophages is strongly associated with kidney fibrosis in both human and experimental diseases (23). Indeed, bone marrow-derived M2-type pro-fibrotic macrophages are highly proliferative, which may contribute to promote kidney fibrosis in experimental models such as UUO and the Adriamycin nephropathy (70–72).
In experimental hypertension, M2 macrophages expressing Mouse Chitinase 3-like 3(Chi3l3)/YM1were associated with renal fibrosis (73). Macrophages may promote hypertension through the generation of M1 cytokines, such as TNF-α, IL-6, and IL-1β, and reactive oxygen species (ROS) resulting in enhanced renal sodium retention and organ damage (74, 75). In this sense, recent studies suggest that the pro-inflammatory cytokine IL-17A, produced by CD4+/T and γδ-lymphocytes, but by not macrophages, plays a key role in the onset of hypertension and in hypertensive end-organ damage, such as the heart, vessels and kidneys (76, 77). However, the Th17 phenotype is sustained by interleukin-23 (IL-23), produced mainly by M1 macrophages (16), showing an interplay between macrophages and immune cells in hypertension.
Macrophages can release a wide array of cytokines, which varies depending on pathological conditions. One well-known product of macrophages is MCP-1/CCL2, a chemokine driving their recruitment into injured tissues (78). Some clinical data suggest that MCP-1/CCL2 could be a biomarker of kidney fibrosis and function decline (79, 80). As commented above, M1 macrophages can produce TNF-α, IL-1β, IL-6, IL-12, IL-15, IL-18, and IL-23 whereas M2 macrophages release TGF-β1, IL-10, CCL17, CCL18, and CCL22 (Figure 2). Therefore, macrophage-derived biomarkers in blood and other biological fluids can reflect the activation of macrophage populations in tissues. In this sense, IL-6 is now considered an important cardiovascular risk biomarker (81). Some of these macrophage-derived cytokines can be relevant as biomarkers of kidney disease progression. Additionally, in the AKI-to-CKD transition, the evaluation of M1/M2 macrophage markers or secretome-derived factors can be used to monitor disease progression and/or remission (23). We will now focus in two interesting candidates, the macrophage derived cytokine CCL18, and the macrophage marker CD163.
Chemokine (C-C motif) ligand 18 (CCL18) regulates several inflammatory and immunological processes, participating in cell recruitment (82, 83) and in phenotype transformations in cancer cells (84). This chemokine is constitutively expressed in the lung and in antigen presenting cells, such as dendritic cells and keratinocytes (83). CCL18 is one of the most highly expressed chemokines in human chronic inflammatory diseases, including allergies, fibrotic disorders and certain cancers (83). As commented before, M2 macrophages secrete high amounts of CCL18 (35). Moreover, stimulation of monocyte/macrophages with CCL18 induces an M2 spectrum macrophage phenotype (20).
In some diseases, CCL18 levels are used as a biomarker of disease progression, for instance in Gaucher disease (85) idiopathic pulmonary fibrosis (86), and chronic periaortitis (87), as well as in several proliferative disorders, including breast (88) and lung (89) cancer, glioblastoma (90), bladder cancer (91), osteosarcoma (92), and prostate cancer (93, 94). However, there is scarce information in kidney diseases. Serum CCL18 levels were proposed as a biomarker of disease activity in ANCA-associated crescentic glomerulonephritis (95). In patients with CKD undergoing peritoneal dialysis treatment, CCL18 levels in peritoneal effluent correlated with progressive ultrafiltration failure and peritoneal fibrosis, suggesting that CCL18 could also be a biomarker of peritoneal damage (96).
The human CC chemokine receptor 8 (CCR8) is a seven-transmembrane-spanning G protein–coupled receptor, whose canonical ligand is Chemokine (C-C motif) ligand 1 (CCL1/I-309) (97). CCL18 was recently discovered as another CCR8 agonist with less affinity than CCL1 (98). CCR8 is expressed mostly in monocytes and thymus and acts as chemoattractant receptor for Th2 cells (99). Moreover, this receptor was found in lymphocytes in human healthy skin for preserving tissue homeostasis (100). However, CCR8 is not expressed in human kidneys in normal or pathological conditions, such as renal transplant rejection (101). In contrast, CCR8 expression was detected mainly in tumor renal cells of human renal cell carcinoma (102). Accordingly, upregulated expression of CCR8 has been described in circulating cells of patients with bladder and renal carcinoma (103).
Human CCL18 and mouse chemokine (C-C motif) ligand 8 (CCL8), named here as mCCL8, have been proposed to be functional analogs (98). The CCL18 gene has only orthologs in primates (104), whereas mouse Ccl8 gene (also known as Mcp-2) lacks a human ortholog as showed by phylogenetic analysis and synteny mapping, but binds to CCR8 (105), and shares functions with CCL18 (98). Importantly, human CCL8/MCP2 is a different cytokine, exerting chemotactic actions for macrophages, and signaling through CCR1 and CCR5 (106). Therefore, to avoid confusion along the manuscript we have named mouse CCL8 here as mCcl8.
A microarray analysis of renal biopsy samples of patients with newly diagnosed ANCA-associated crescentic glomerulonephritis identified CCL18 as the most upregulated gene (95). Immunohistochemical analysis identified myeloid dendritic cells and CD68+ macrophages as CCL18-producing cells, and determined that the density of CCL18+ cells correlated with interstitial inflammation, crescent formation and impairment of renal function at the time of biopsy. Serum CCL18 levels also correlated with kidney disease activity, being lower in patients with immunosuppressive rescue therapy and higher in relapsing kidney disease (95). In accordance to these data, we found that CD163+/CCL18 expressing macrophages colocalized with Gremlin protein expression in another cohort of patients with ANCA-associated crescentic glomerulonephritis (107). In this study, we proposed urinary Gremlin levels as a potential biomarker of disease progression, showing a parallelism to CCL18. Despite these comprehensive studies, there are no published data on CCL18 in other human kidney diseases. In cultured human tubular cells, CCL18 increased the production of fibronectin in diabetic conditions (108). On the other hand, there is scarce information about the role of mCCL8 in preclinical kidney disease. In murine renal artery stenosis, kidney mCcl8 gene expression was higher than in control mice (109). However, in cultured murine tubular cells, we have found that these cells lack Ccr8 expression and were not responsive to mouse recombinant CCL8 protein (110). In experimental folic acid-induced AKI, kidney mCcl8 expression was unchanged in the acute phase (24–72 h). However, kidney mCcl8 expression was upregulated at 7 days, a time point associated to the AKI-to-CKD transition. In UUO, kidney mCcl8 expression was already increased at 5 days (111), and increased progressively over time (Figure 3). The evaluation of M1/M2 cytokine profiles showed that kidney mCcl8 expression correlates to M1-related cytokine downregulation and M2 cytokine overexpression. Therefore, these data suggest a potential role of mCcl8 in mouse macrophage polarization toward the M2 phenotype, helping to maintain chronic inflammation and favoring kidney fibrosis.
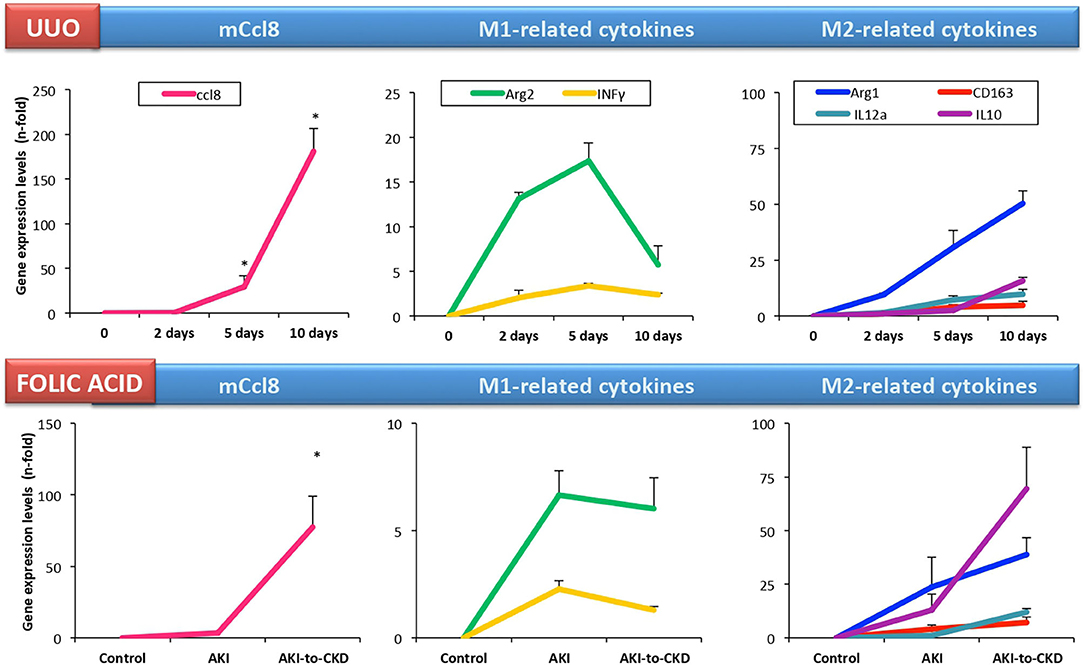
Figure 3. Kidney Ccl8 gene expression is associated to M2 cytokines in experimental kidney injury. Kidney Ccl8 expression and M1 and M2-related factors were evaluated in CKD models: unilateral ureteral obstruction (UUO) and the acute and chronic phase of folic acid (FA) nephropathy mice [data from Cantero-Navarro et al. (110)].
We previously showed that increased M2 macrophage numbers associated to peritoneal CCL18 effluent concentrations in patients with CKD undergoing peritoneal dialysis (96). Moreover, in a murine model of exposure to peritoneal dialysis fluids (112) we have found that mCcl8 expression was increased associated to macrophage infiltration (113), supporting the concept of mouse Ccl8 as a functional analog of human CCL18.
CD163 is a glycosylated membrane protein, member of the scavenger receptor cysteine-rich family, also known as the hemoglobin scavenger receptor (13, 114, 115). CD163 is expressed nearly exclusively by cells of monocytic origin (e.g., monocytes, macrophages, some dendritic cells, and some tumor cells) presenting high expression levels in macrophages (13, 114, 115). By ectodomain shedding, the extracellular portion of CD163 can be released from the cell surface into circulation, in a process regulated by ADAM17/TACE (tumor necrosis factor α-converting enzyme) (116). Soluble CD163 (sCD163) is stable and measured easily in serum, and has been investigated as potential biomarker of macrophage activation in various disease contexts (117–119).
CD163 functions have been extensively reviewed (120, 121). Early studies described that CD163 expression is a hallmark of the wound healing macrophage, related to the resolution phase of inflammation after cardiopulmonary bypass surgery (122) and in human inflammatory skin disease induced by Cantharidin (123). Some studies in animal models of renal damage pointed out the key role of CD163 in the beginning and progression of renal disease (124–126). In AKI induced by experimental rhabdomyolysis increased levels of M1 macrophages were observed in the early pathological stages. These macrophages suffer a partial differentiation to M2 phenotype characterized by CD163 overexpression via HO-1 activation and IL-10 release. Moreover, peritoneal macrophages stimulated by myoglobin can induce fibrosis through the regulation of profibrotic mediators, such as TGF-β and CTGF/CCN2 (124). In cisplatin-induced nephrotoxic damage in rats increased levels of CD163 M2 macrophages associated to fibrosis were found (126). Other study in Lupus nephritis (LN) in mice described an increased CD163+/CD68+cell ratio. In addition, Cd163 gene expression was increased in LN mice while Ho-1, levels were reduced, the last one associated to elevated Bach1 and Il-6 expression. The gene blockade of Bach1 in mice (Bach1-deficient MRL/lpr mice) improved the loss of renal function in experimental LN assessed by BUN levels (125).
In vitro studies in human macrophages found that CD163 overexpression induces a change on the cytokine profile secretion from pro-inflammatory M1-related cytokines to M2-cytokines (127). Importantly, CD163+/CD68+ macrophages may be involved in the pathogenesis of proliferative glomerular crescents, such as in ANCA-associated glomerulonephritis or active LN. CD163+/CD68+ macrophages were found in glomerular crescents and were correlated to proteinuria and to estimated glomerular filtration rate (eGFR) (positively and negatively, respectively) (128). Moreover, two different M2 macrophages populations, CD163+ and CD206+, were mainly expressed in fibrous crescents and were more common in Lupus nephritis (LN) and ANCA-associated vasculitis than in IgA nephropathy and Henoch Schönlein purpura glomerulonephritis (129, 130). Patients with early stage of idiopathic membranous nephropathy had higher levels of circulating CD14+/CD163+, CD14+/CD163+/CD206+, and CD14+/CD163+/CD206+/CD115+ macrophages in comparison with healthy controls (131). M2-macrophages were considered the dominant subpopulation in human LN and M2a subpopulations were associated with disease progression (132). In ANCA glomerulonephritis, urinary CD11b+ and CD163+ correlated with leukocyte recruitment in the kidney (133). Indeed, urinary sCD163 is a marker of human glomerulonephritis (118, 134) and active renal vasculitis (135, 136). On the other hand in LN (119), sCD163 in the urine is considered as a marker of disease activity and treatment response (137, 138). Interestingly, CD163+ cells in crescents of ANCA glomerulonephritis patients colocalized with CCL18 (95) and Gremlin (107). Both CCL18 and Gremlin levels have been independently proposed as potential biomarkers of disease progression (95, 107). These two independent studies suggest that the M2 macrophage secretome could be a source of biomarkers of kidney disease progression, mainly in crescentic glomerulonephritis. Accordingly, in peritoneal dialysis patients, CD163+ macrophages were present in peritoneal effluents and increased during peritonitis. Moreover, peritoneal CCL18 effluent concentrations correlated with decreased peritoneal function, showing the contribution of macrophage-derived cytokines to peritoneal damage and fibrosis (96). Despite there are few data about sCD163 in human diabetic nephropathy some studies described that sCD163 levels were also strongly associated with later development of type 2 diabetes in both lean and obese subjects, likely reflecting macrophage recruitment in the adipose tissue (117, 139–143) and in the liver (144–146), an effect associated to ADAM17/TACE-mediated shedding of TNF-α and sCD163 (116). Diabetic patients had higher numbers of circulating CD163+ monocytes (147). In addition, sCD163 was also identified as a good risk biomarker of diabetic nephropathy and/or diabetic retinopathy (148). Anti-inflammatory CD163+ macrophages were elevated in glomeruli from diabetic patients and were associated with pathological features such as tubular atrophy, interstitial fibrosis and glomerulosclerosis (149). In murine RAW264.7 macrophages preincubated with high glucose, calcitriol (1,25(OH)2D3) treatment blockaded M1 macrophage activation and M2 phenotype differentiation. The same result was observed in streptozotocin (STZ)-diabetic rats treated with calcitriol (150). Moreover, the uremic toxins that are accumulated in the last stage of renal disease have a role in M2 induction. A study in THP-1 cells showed that indoxil sulfate (IS) induced CD163 expression and transition to macrophages through AhR/Nrf2 activation (151). Finally, other study linked the increase of fat mass with elevated levels of sCD163, suggesting that adipose tissue macrophages play a key role in CKD proinflammatory state (140).
sCD163 was also been found in heme-related human kidney injury, such IgA nephropathy (152), intravascular hemolysis (e.g., paroxysmal nocturnal hemoglobinuria) (153), favism (154), and rhabdomyolysis AKI (124). In macrophages of patients with IgA nephropathy and macroscopic hematuria-related AKI, CD163 was associated with incomplete recovery of kidney function (152, 155) describing as the predominant subpopulations in kidney tissues, the M2a (CD206+/CD68+) and M2b (CD86+/CD68+) macrophages (130).
There have been multiple attempts to inhibit or modulate the inflammatory response to prevent or retard CKD progression. The most widely used approaches to modulate macrophage levels and/or phenotypes have been directed to M1-related cytokines, such as IL1, IL-6, and TNF-α. We now discuss preclinical studies targeting those inflammatory mediators and the key monocyte/macrophage chemotactic factor MCP-1/CCL2, and the potential translation to humans.
The earliest studies targeting macrophage related cytokines tested IL-1 strategies. Treatment with an IL-1 receptor (IL-1R) antagonist (IL-1Ra) ameliorated experimental anti-glomerular basement membrane antibody-associated glomerulonephritis (anti-GBM) in rats (156) as well as spontaneous IgA nephropathy in ddY mice (157). Gene deletion of IL-1 Type 1 Receptor (IL1R1) or IL-1β demonstrated that IL-1β but not IL-1α contributed to crescent formation and inflammatory cell recruitment in murine anti-GBM crescentic glomerulonephritis (158). Similarly, genetic IL1R1 deletion modestly improved survival and attenuated cyst volume in experimental murine ADPKD (159). The IL-1R1 antagonist anakinra prevented nephropathy in diabetic mice (160). Accordingly, anti-IL-1β antibody attenuated the progressive loss of kidney function and preserved podocytes in murine loss of kidney mass in diabetic db/db mice (161). Human studies also showed beneficial effects, including improved vascular endothelial function in patients with non-dialysis-dependent CKD after 12 weeks of treatment with the IL-1 inhibitor rilonacept, whereas no changes in kidney function were observed nor expected in such a short follow-up (162). A clinical trial of the canakinumab (neutralizing antibody against IL-1β) observed a reduction in the cardiovascular event rates in atherosclerosis patients with CKD without modifying kidney function (163). However, a clinical trial of gevokizumab (antibody against IL-1β) in diabetic kidney disease was terminated prematurely, based on company priorities (164).
There are multiple preclinical examples of kidney protection afforded by anti-TNF strategies. Administration of a pegylated form of the soluble TNF type 1 receptor (PEG-sTNFR1) reduced renal fibrosis in experimental rat CKD induced by renal mass reduction (165) and kidney inflammation and tubular cell apoptosis in rat UUO (166). Similarly, TNF-α gene deletion or silencing attenuated kidney injury induced by high fat diet in mice by reducing fibrosis and glomerulosclerosis (167). A neutralizing anti-TNF-α antibody reduced glomerular inflammation, crescent formation, and tubulointerstitial scarring, and preserved kidney function in rat anti-GBM crescentic glomerulonephritis (168). An anti-TNF-α antibody decreased albuminuria, plasma creatinine, histopathologic changes, and kidney macrophage recruitment in an experimental type I diabetes in B6-Ins2Akita/MatbJ mice (169). Moreover, macrophage-specific TNF-α-deficient mice (CD11bCre/TNF-αFlox/Flox; C57BL/6) presented the same beneficial effects after streptozotocin-induced diabetes (169). In high fat diet-induced kidney injury, TNF-α deletion reduced kidney fibrosis, glomerulosclerosis, oxidative stress, inflammation and apoptosis (167). Despite promising preclinical results, targeting TNF-α in human kidney disease for kidney protection is controversial. A clinical trial of infliximab in LN failed in the recruitment phase (NCT00368264). However, anti-TNF-α monoclonal antibodies are used routinely to treat rheumatoid arthritis (RA), ankylosing spondylitis (AS) or psoriasis. Thus, there is mixed information on such patients and kidney disease. Some reports observed that in RA patients with CKD, anti-TNF-α drugs (adalimumab, etanercept, or infliximab) had no deletereous effect on kidney function (170) or presented slower loss of renal function (171). However, cases of AKI, focal segmental glomerulosclerosis (FSGS) or IgA nephropathy have been reported in AS, RA, or inflammatory Bowel Disease patients treated with anti-TNF-α drugs (172–176).
Due to the key role of IL-6 in kidney diseases (177), the impact of targeting IL-6 has been studied. However, preclinical results differed for some kidney diseases. Neutralizing IL-6 (178), IL-6 receptor (IL-6R) (179) or genetic IL-6 deletion (180) decreased disease severity in different experimental models of LN in MRL-Faslpr and NZB/WF1 mice. However, anti-IL-R6 or anti-IL-6 strategies increased the severity of murine anti-GBM nephritis, while selective inhibition of IL-6 trans-signaling by sgp130Fc did not (181). Similarly, genetic IL-6 deletion did not decrease fibrosis in murine UUO (182), whereas treatment with Fc-gp130 reduced inflammation, immune cell infiltration and fibrosis in both murine UUO and IRI (65). These findings fit well with the concept that IL-6 classic signaling is anti-inflammatory and protective, whereas IL-6 trans-signaling is pro-inflammatory.
In addition to inflammation, IL-6 controls glucose metabolism and the hypothalamic-pituitary-adrenal axis among other processes which could result in deleterious effects when IL-6 is modulated (183). Therapies modulating IL-6 (olokizumab or clazakizumab) or the α subunit of its receptor (tocilizumab or sarilumab) have been used clinically to treat inflammatory diseases, such as RA, systemic lupus erythematous (SLE), diabetes and, more recently, coronavirus disease 2019 (COVID-19) (184, 185) that may develop kidney disease, but no clinical trial studied kidney function or renal injury as primary outcome. An ongoing trial is exploring the efficacy of clazakizumab to preserve kidney function in highly sensitized patients awaiting kidney transplantation (NCT03380962) or in kidney transplant receptors with late antibody-mediated rejection (NCT03444103).
MCP-1/CCL2 is the main factor driving monocyte recruitment and differentiation during inflammatory response. Therefore, MCP-1/CCL2 has been targeted in preclinical kidney disease (186). Both genetic deficiency of the MCP-1 receptor (CCR2) or MCP-1/CCL2 antagonism improved LN and vasculitis in MRL/lpr mice (187, 188). Likewise, MCP-1/CCL2 neutralizing antibody reduced glomerular macrophage infiltration and decreased crescent formation in experimental rat and murine crescentic glomerulonephritis (189, 190). Targeting MCP-1/CCL2/CCR2 was also protective in experimental diabetic nephropathy. A CCR2 antagonist (propagermanium) protected the kidneys in type 1 diabetic mice overexpressing type 2 nitric-oxide synthase (191), MCP-1/CCL2 gene deletion decreased glomerular and interstitial macrophage accumulation and fibrosis in murine STZ-induced diabetic nephropathy (192), and the MCP-1/CCL2 antagonistic Spiegelmer mNOX-E36 (emapticap pegol) reduced the number of glomerular macrophages and improved glomerular filtration rate (GFR) in uninephrectomized db/db mice (193) and in STZ- diabetic ApoE knockout mice (194). However, in experimental murine Alport nephropathy (Col4a3-deficient mice), mNOX-E36 administration reduced glomerular and interstitial macrophage recruitment, but did not improve glomerular or interstitial histopathology or survival (195).
In humans, phase 2 clinical trials were promising in diabetic kidney disease. A selective inhibitor of CCR2 (CCX140-B) added to standard care (196) and the Spielgelmer NOX-E36 showed evidence of kidney protection in patients with type 2 diabetes and kidney disease (197). However, there are no current ongoing phase 3 trials. In this regard, the standard of care has been changed by the efficacy of sodium-glucose cotransporter-2 (SGLT2) inhibitors and any future trial should test the efficacy of new drugs on top of SGLT2 inhibition and renin-angiotensin system (RAS) blockade (198). On the other hand, there are ongoing phase 2 trials of CCX140-B (Ilacirnon) for FSGS (NCT03536754, NCT03703908).
Inducing macrophage polarization has been also proposed as a potential approach to reduce the kidney inflammatory response (21, 199). Strategies to induce macrophage polarization, that include stimulation with cytokines, miRNAs regulation or genetic manipulation (199), have been extensively investigated in preclinical studies to reduce the inflammatory response in kidney diseases.
Cell stimulation with different cytokines has been used to induce macrophage phenotype polarization and polarized cells were subsequently administered to treat experimental kidney disease (199). Administration of spleen derived macrophages (SPDM) polarized to M2 phenotype by IL-4/IL-13 stimulation ex vivo decreased histological and functional kidney injury as compared to M1 macrophages in experimental adriamycin nephropathy (AN) in immunodeficient mice (200). Likewise, transference of M2 macrophages generated ex vivo by incubation of SPDM with IL-10/TGF-β1 decreased kidney inflammation, structural injury and functional decline in the same model (201). Similarly, the administration of M1 macrophages induced by incubation with INF-γ increased tubular injury in murine IRI while M2 macrophages induced by incubation with IL-4 did not (59). In STZ-induced diabetic mice, transfusion of SPDM polarized to M2 by incubation with IL-4/IL-13 decreased tubular atrophy, glomerular hypertrophy, interstitial expansion and kidney fibrosis (202). In murine nephrotoxic nephritis, the transfusion of bone marrow-derived macrophages (BMDM) polarized to M2 phenotype (CD206+) by ex vivo incubation with IL-4/IL-13 reduced kidney injury, proteinuria, and glomerular inflammatory cell infiltration (203). Treatment with both BMDM polarized ex vivo to M2 macrophages by IL-4/IL-13 as well as in vivo M2 polarization induced by IL-4/IL-13 injections, reduced renal crystal formation in murine experimental kidney stone disease (204). A more recent study corroborated the beneficial effects of M2 transplantation in AKI. Peritoneal macrophages isolated from mice under peritoneal dialysis were polarized to M2 by incubation with IL-4/IL-13 and then injected into the renal cortex of mice with experimental ischemia reperfusion injury (IRI). M2 macrophages administration improved kidney injury and decreased inflammation compared to those injected with non-activated (M0) macrophages (205). Finally, the intrarenal administration of an Elastin-like Polypeptide (ELP-VEGF) construct induced a clear polarization to M2 macrophage phenotype in CKD in pigs and improved renal hemodynamics and fibrosis, despite no differences in renal macrophage infiltration were found compared to control (206).
Macrophages have been used successfully in cell therapy to deliver targeted therapeutic genes in models of inflammatory kidney disease. The adoptive transfer of bone-marrow-derived macrophages (BMDM) genetically modified with adenoviral particles encoding the anti-inflammatory cytokine interleukin 1 receptor antagonist (IL-1Ra) reduced kidney injury in murine anti-GBM (207). Similar results were observed using the same approach in unilateral ureteral obstruction (UUO) mice: the transference of macrophages encoding IL-1Ra after ureter obstruction reduced kidney interstitial macrophage infiltration, overall inflammation and fibrosis (208). In the same way, rat alveolar macrophages (NR8383) transfected with an adenovirus encoding IL-4 reduced albuminuria and glomerular macrophage infiltration (209). The delivery of IL-10-expressing macrophages or a dominant-negative I-KB in rat nephrotoxic nephritis also reduced kidney injury (210, 211). In murine kidney ischemia reperfusion injury (IRI), the transference of adenoviral transduced BMDM encoding Heme oxygenase-1 (HO-1)preserved kidney function (212). Similar results were observed in a rat kidney IRI. Herein, transference with adenoviral-IL-10-transduced BMDM markedly reduced albuminuria, the number of pro-inflammatory macrophages and fibrosis (210).
MicroRNAs (miRNAs) are small, single-stranded non-coding RNAs (20–24 nucleotides) that regulate post-transcriptional gene expression (213). Since their discovery, multiple potential roles have been attributed to these molecules, including their participation in the inflammatory response by regulating multiple processes, including macrophage phenotype polarization (214). The miRNA expression profiles in M1 and M2 macrophages have been characterized both in human and in murine macrophages (215–217) (Table 1). A recent review highlighted that miR-9, miR-127, miR-155, and miR-125b promote M1 polarization while miR-124, miR-223, miR-34a, let-7c, miR-132, miR-146a, and miR-125a-5p induce the M2 phenotype in both species (214). The relevance of some of these miRNAs in the macrophage phenotype context has been also corroborated in kidney disease with different results. miR-146a regulated local chronic inflammation in autoimmune glomerulonephritis in B6.MRLc1 mice, suggesting that it is a potential therapeutic target (224). However, genetic miR-146a deletion increased proteinuria, renal macrophage infiltration, glomerular hypertrophy and fibrosis in murine streptozotocin-induced diabetes (225). The anti-inflammatory effect of miR-146 through regulation of IL-6 expression was also demonstrated in macrophages derived from cystic fibrosis patients (222). Similarly, a transwell assay using glomerular mesangial cells from LN patients stimulated with a miR-146 mimic showed a reduction in macrophage migration (223). In the same way, although miR-9 has been reported to promote pro-inflammatory macrophage phenotype, mice transfected with lentiviral vectors expressing miR-9-5p were protected from UUO-induced kidney fibrosis through decreased infiltrating monocytes/macrophages and induction of metabolic reprogramming (218).
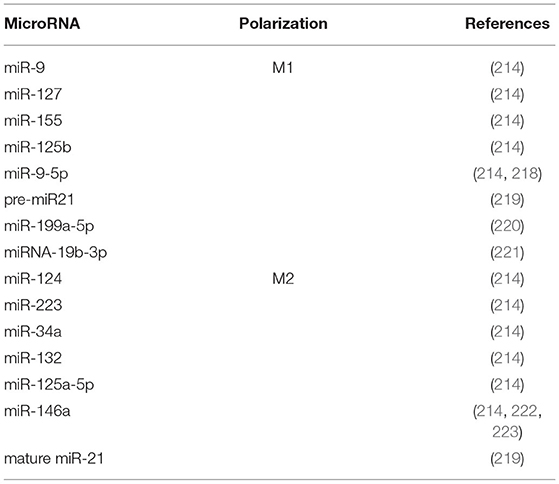
Table 1. MicroRNA and macrophage polarization.
miR-21 is a key mediator of the anti-inflammatory response in macrophages and has been proposed to exert a dual role. Pre-miR21 polarizes macrophages to the M1 phenotype while mature miR-21 polarizes to M2 and upregulates anti-inflammatory targets, such as IL-10 (219). However, although miR-21 has been widely studied in inflammatory conditions, its role in experimental kidney disease reported controversial results. Thus, while genetic deletion of miR-21 reduced kidney fibrosis in murine UUO, IRI, Alport syndrome (226, 227) and polycystic kidney disease (228), miR-21 was also reported to protect from murine kidney IRI (229). Lademirsen sodium (RG 012), an anti–miR-21, is currently undergoing phase 2 clinical trials for Alport syndrome (NCT02855268).
Apart from the miRNAs expressed directly in macrophages, miRNAs from extracellular vesicles also influence the macrophage polarization process. For example, miR-199a-5p present in extracellular vesicles from albumin-induced tubular epithelial cells promoted the M1 macrophage phenotype in mice with high fat diet (HFD)/STZ-induced diabetes (220). Similar results were obtained in another recent study demonstrating that miRNA-19b-3p from tubular epithelial-derived exosomes promotes M1 macrophage activation in a murine endotoxemia-induced AKI (221).
Macrophages actively participate in the initial phases on kidney injury, as well as in kidney damage resolution and progression. Current pharmacological treatments that present beneficial effects in human CKD diminish macrophages infiltration in the kidney (1), including the SGLT2 inhibitors (230). Future investigations targeting macrophage polarization, and macrophage-derived cytokines could provide novel therapeutic approaches to further reduce the inflammatory response in kidney diseases.
In spite of the intensive research done, the lack of consistent nomenclature, and reliable polarization markers that are conserved between species and between in vivo and in vitro models of macrophage polarization has delayed progress in the field and complicates the extrapolation of preclinical results into human kidney disease (53). Moreover, it is necessary to develop preclinical models that truly resemble the human situation, something specially relevant in human kidney diseases due to the complex etiology of CKD (1). Some studies have been focused on comparative biology approaches on macrophages to accelerate the translation into the clinic (231). Moreover, individual variation in cytokine production in the human population has been reported (232). Novel data on transcriptional machinery of different macrophage subtypes have increased the knowledge about macrophage functions (233, 234). However, the mechanisms of genetic regulation remains unraveled. Future studies using cutting-edge technologies, including single-cell RNA sequencing, genomics, and proteomics approaches, as well as spatial transcriptomics in human biopsies are needed to improve the understanding of cytokine profiles, macrophage phenotypes and functions in different human diseases, including AKI, AKI-to-CKD transition, and CKD progression.
LT-S and AT-M contributed to the design of the figures. MO, LM-E, LS-S, and EC-N participated in the development of mouse models and analysis of data of the unpublished data and to the draft of some parts of the manuscript. EC-N, SR-M, TB, AS, AT-M, VM, and RR-D contributed to the draft of some parts of the manuscript. JE and AO contributed to the critical review of the manuscript and the financial support of the work. MR-O contributed to the draft of the manuscript and financial support. All the authors have reviewed the manuscript and approved the final version.
This work was supported by grants from the Instituto de Salud Carlos III (ISCIII) and Fondos FEDER European Union (PI17/00119, PI17/01495, PI18/00718, PI19/0588, PI20/00140, PI20/00487, DTS20/00083, RTC-2017-6089-1, and DTS19/00093), and CIBERDEM to JE; Red de Investigación Renal REDINREN: RD16/0009 to MR-O, AO), SR-M, NOVELREN-CM: Enfermedad renal crónica: nuevas Estrategias para la prevención, Diagnóstico y tratamiento; B2017/BMD-3751, CIFRA2-CM B2017/BMD-3686, Convocatoria Dinamización Europa Investigación 2019 MINECO (EIN2019-103294 to MR-O and SR-M); Juan de la Cierva Incorporación del Ministerio de Economía, Industria y Competitividad (MINECO) para SR-M (IJC2018-035187-I); Innovation programme under the Marie Skłodowska-Curie grant of the European Union's Horizon 2020 (IMProve-PD ID: 812699) to MR-O. Fundación Conchita Rabago supports the salary of LT-S. ISCIII Miguel Servet to AS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Authors want to thank Rafael Selgas for his support during these years.
1. Ruiz-Ortega M, Rayego-Mateos S, Lamas S, Ortiz A, Rodrigues-Diez RR. Targeting the progression of chronic kidney disease. Nat Rev Nephrol. (2020) 16:269–88. doi: 10.1038/s41581-019-0248-y
2. Meng XM, Nikolic-Paterson DJ, Lan HY. Inflammatory processes in renal fibrosis. Nat Rev Nephrol. (2014) 10:493–503. doi: 10.1038/nrneph.2014.114
3. Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol. (2003) 35:881–900. doi: 10.1016/S1357-2725(02)00271-6
4. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. (2013) 496:445–55. doi: 10.1038/nature12034
5. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
6. Zhao H, Dong Y, Tian X, Tan TK, Liu Z, Zhao Y, et al. Matrix metalloproteinases contribute to kidney fibrosis in chronic kidney diseases. World J Nephrol. (2013) 6:84–9. doi: 10.5527/wjn.v2.i3.84
7. Hettinger J, Richards DM, Hansson J, Barra MM, Joschko AC, Krijgsveld J, et al. Origin of monocytes and macrophages in a committed progenitor. Nat Immunol. (2013) 14:821–30. doi: 10.1038/ni.2638
8. Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. (2014) 5:514. doi: 10.3389/fimmu.2014.00514
9. Bian Z, Gong Y, Huang T, Lee CZW, Bian L, Bai Z, et al. Deciphering human macrophage development at single-cell resolution. Nature. (2020) 582:571–6. doi: 10.1038/s41586-020-2316-7
10. Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, Van Rooijen N, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of T H2 inflammation. Science. (2011) 332:1284–8. doi: 10.1126/science.1204351
11. Funes SC, Rios M, Escobar-Vera J, Kalergis AM. Implications of macrophage polarization in autoimmunity. Immunology. (2018) 154:186–95. doi: 10.1111/imm.12910
12. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. (2009) 325:612–6. doi: 10.1126/science.1175202
13. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000 Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
14. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164:6166–173. doi: 10.4049/jimmunol.164.12.6166
15. Ley K. M1 means kill; M2 means heal. J Immunol. (2017) 199:2191–3. doi: 10.4049/jimmunol.1701135
16. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. (2008) 13:453–61. doi: 10.2741/2692
17. Wawrocki S, Druszczynska M, Kowalewicz-Kulbat M, Rudnicka W. Interleukin 18 (IL-18) as a target for immune intervention. Acta Biochim Pol. (2016) 63:59–63. doi: 10.18388/abp.2015_1153
18. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.26429
19. Wang Y, Harris DCH. Macrophages in renal disease. J Am Soc Nephrol. (2011) 22:21–7. doi: 10.1681/ASN.2010030269
20. Schraufstatter IU, Zhao M, Khaldoyanidi SK, Discipio RG. The chemokine CCL18 causes maturation of cultured monocytes to macrophages in the M2 spectrum. Immunology. (2012) 135:287–98. doi: 10.1111/j.1365-2567.2011.03541.x
21. Chen T, Cao Q, Wang Y, Harris DCH. M2 macrophages in kidney disease: biology, therapies, and perspectives. Kidney Int. (2019) 95:760–73. doi: 10.1016/j.kint.2018.10.041
22. Chen YT, Hsu H, Lin CC, Pan SY, Liu SY, Wu CF, et al. Inflammatory macrophages switch to CCL17-expressing phenotype and promote peritoneal fibrosis. J Pathol. (2020) 250:55–66. doi: 10.1002/path.5350
23. Tang PMK, Nikolic-Paterson DJ, Lan HY. Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol. (2019) 15:144–58. doi: 10.1038/s41581-019-0110-2
24. Tanita K, Fujimura T, Sato Y, Lyu C, Kambayashi Y, Ogata D, et al. Bexarotene reduces production of CCL22 from tumor-associated macrophages in cutaneous T-cell lymphoma. Front Oncol. (2019) 9:907. doi: 10.3389/fonc.2019.00907
25. Buscher K, Ehinger E, Gupta P, Pramod AB, Wolf D, Tweet G, et al. Natural variation of macrophage activation as disease-relevant phenotype predictive of inflammation and cancer survival. Nat Commun. (2017) 24:8:16041. doi: 10.1038/ncomms16041
26. Hume DA. Macrophages as APC and the dendritic cell myth. J Immunol. (2008) 181:5829–35. doi: 10.4049/jimmunol.181.9.5829
27. Satpathy AT, Wu X, Albring JC, Murphy KM. Re(de)fining the dendritic cell lineage. Nat Immunol. (2012) 13:1145–54. doi: 10.1038/ni.2467
28. Niida S, Kaku M, Amano H, Yoshida H, Kataoka H, Nishikawa S, et al. Vascular endothelial growth factor can substitute for macrophage colony- stimulating factor in the support of osteoclastic bone resorption. J Exp Med. (1999) 190:293–8. doi: 10.1084/jem.190.2.293
29. Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. (2009) 9:259–70. doi: 10.1038/nri2528
30. Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol. (2012) 13:888–99. doi: 10.1038/ni.2370
31. Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol. (2007) 178:5245–52. doi: 10.4049/jimmunol.178.8.5245
32. Lacey DC, Achuthan A, Fleetwood AJ, Dinh H, Roiniotis J, Scholz GM, et al. Defining GM-CSF– and macrophage-CSF–dependent macrophage responses by in vitro models. J Immunol. (2012) 188:5752–65. doi: 10.4049/jimmunol.1103426
33. Rodriguez RM, Suarez-Alvarez B, Lavín JL, Ascensión AM, Gonzalez M, Lozano JJ, et al. Signal integration and transcriptional regulation of the inflammatory response mediated by the GM-/M-CSF signaling axis in human monocytes. Cell Rep. (2019) 29:860–72.e5. doi: 10.1016/j.celrep.2019.09.035
34. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
35. Little AC, Pathanjeli P, Wu Z, Bao L, Goo LE, Yates JA, et al. IL-4/IL-13 stimulated macrophages enhance breast cancer invasion via rho-GTPase regulation of synergistic VEGF/CCL-18 signaling. Front Oncol. (2019) 9:456. doi: 10.3389/fonc.2019.00456
36. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
37. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
38. Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol. (2012) 189:3508–20. doi: 10.4049/jimmunol.1200662
39. Lurier EB, Dalton D, Dampier W, Raman P, Nassiri S, Ferraro NM, et al. Transcriptome analysis of IL-10-stimulated (M2c) macrophages by next-generation sequencing. Immunobiology. (2017) 222:847–56. doi: 10.1016/j.imbio.2017.02.006
40. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
41. Wang Y, Smith W, Hao D, He B, Kong L. M1 and M2 macrophage polarization and potentially therapeutic naturally occurring compounds. Int Immunopharmacol. (2019) 70:459–66. doi: 10.1016/j.intimp.2019.02.050
42. Batista-Gonzalez A, Vidal R, Criollo A, Carreño LJ. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front Immunol. (2020) 10:2993. doi: 10.3389/fimmu.2019.02993
43. Van den Bossche J, O'Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol. (2017) 38:395–406. doi: 10.1016/j.it.2017.03.001
44. Munder M, Eichmann K, Modolell M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol. (1998) 160:5347–54.
45. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. (2013) 229:176–85. doi: 10.1002/path.4133
46. Mills CD, Ley K. M1 and M2 macrophages: the chicken and the egg of immunity. J Innate Immun. (2014) 6:716–26. doi: 10.1159/000364945
47. Corraliza IM, Soler G, Eichmann K, Modolell M. Arginase induction by suppressors of nitric oxide synthesis (IL-4, IL-10 and PGE2) in murine bone-marrow-derived macrophages. Biochem Biophys Res Commun. (1995) 206:667–73. doi: 10.1006/bbrc.1995.1094
48. Spiller KL, Wrona EA, Romero-Torres S, Pallotta I, Graney PL, Witherel CE, et al. Differential gene expression in human, murine, and cell line-derived macrophages upon polarization. Exp Cell Res. (2016) 347:1–13. doi: 10.1016/j.yexcr.2015.10.017
49. Rossi M, Korpak K, Doerfler A, Boudjeltia KZ. Deciphering the role of heme oxygenase-1 (Ho-1) expressing macrophages in renal ischemia-reperfusion injury. Biomedicines. (2021) 9:306. doi: 10.3390/biomedicines9030306
50. Palmer MB, Vichot AA, Cantley LG, Moeckel GW. Quantification and localization of M2 macrophages in human kidneys with acute tubular injury. Int J Nephrol Renovasc Dis. (2014) 7:415–9. doi: 10.2147/IJNRD.S66936
51. Hamann J, Koning N, Pouwels W, Ulfman LH, van Eijk M, Stacey M, et al. EMR1, the human homolog of F4/80, is an eosinophil-specific receptor. Eur J Immunol. (2007) 37:2797–802. doi: 10.1002/eji.200737553
52. Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado JDD, Popovich PG, Partida-Sanchez S, et al. Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE. (2015) 10:e0145342. doi: 10.1371/journal.pone.0145342
53. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: Different gene signatures in M1(Lps+) vs. classically and M2(LPS-) vs. Alternatively activated macrophages. Front Immunol. (2019) 10:1084. doi: 10.3389/fimmu.2019.01084
54. Amici SA, Young NA, Narvaez-Miranda J, Jablonski KA, Arcos J, Rosas L, et al. CD38 is robustly induced in human macrophages and monocytes in inflammatory conditions. Front Immunol. (2018) 9:1593. doi: 10.3389/fimmu.2018.01593
55. Zimmerman KA, Bentley MR, Lever JM, Li Z, Crossman DK, Song CJ, et al. Single-cell RNA sequencing identifies candidate renal resident macrophage gene expression signatures across species. J Am Soc Nephrol. (2019) 30:767–81. doi: 10.1681/ASN.2018090931
56. Lee SB, Kalluri R. Mechanistic connection between inflammation and fibrosis. Kidney Int. (2010) 119:S22–6. doi: 10.1038/ki.2010.418
57. Andrade-Oliveira V, Foresto-Neto O, Watanabe IKM, Zatz R, Câmara NOS. Inflammation in renal diseases: new and old players. Front Pharmacol. (2019) 0:1192. doi: 10.3389/fphar.2019.01192
58. Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, Sanchez-Niño MD, et al. Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced AKI. J Am Soc Nephrol. (2017) 28:218–29. doi: 10.1681/ASN.2015121376
59. Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. (2011) 22:317–26. doi: 10.1681/ASN.2009060615
60. Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. (2011) 121:4210–21. doi: 10.1172/JCI45161
61. Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. (2011) 7:189–200. doi: 10.1038/nrneph.2011.16
62. Kroner A, Greenhalgh AD, Zarruk JG, PassosdosSantos R, Gaestel M, David S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron. (2014) 83:1098–116. doi: 10.1016/j.neuron.2014.07.027
63. Huen SC, Huynh L, Marlier A, Lee Y, Moeckel GW, Cantley LG. GM-CSF promotes macrophage alternative activation after renal ischemia/reperfusion injury. J Am Soc Nephrol. (2015) 26:1334–45. doi: 10.1681/ASN.2014060612
64. Ricardo SD, Van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. (2008) 118:3522–30. doi: 10.1172/JCI36150
65. Chen W, Yuan H, Cao W, Wang T, Chen WW, Yu H, et al. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics. (2019) 9:3980–91. doi: 10.7150/thno.32352
66. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. (2010) 30:245–57. doi: 10.1055/s-0030-1255354
67. Braga TT, Correa-Costa M, Azevedo H, Silva RC, Cruz MC, Almeida MES, et al. Early infiltration of p40IL12 + CCR7 + CD11b + cells is critical for fibrosis development. Immun Inflamm Dis. (2016) 4:300–14. doi: 10.1002/iid3.114
68. Han Y, Ma FY, Tesch GH, Manthey CL, Nikolic-Paterson DJ. Role of macrophages in the fibrotic phase of rat crescentic glomerulonephritis. Am J Physiol Ren Physiol. (2013) 304:1043–53. doi: 10.1152/ajprenal.00389.2012
69. Guiteras R, Flaquer M, Cruzado JM. Macrophage in chronic kidney disease. Clin Kidney J. (2016) 9:765–71. doi: 10.1093/ckj/sfw096
70. Cao Q, Wang Y, Zheng D, Sun Y, Wang C, Wang XM, et al. Failed renoprotection by alternatively activated bone marrow macrophages is due to a proliferation-dependent phenotype switch in vivo. Kidney Int. (2014) 85:794–806. doi: 10.1038/ki.2013.341
71. Engel DR, Krause TA, Snelgrove SL, Thiebes S, Hickey MJ, Boor P, et al. CX 3 CR1 reduces kidney fibrosis by inhibiting local proliferation of profibrotic macrophages. J Immunol. (2015) 194:1628–38. doi: 10.4049/jimmunol.1402149
72. Pan B, Liu G, Jiang Z, Zheng D. Regulation of renal fibrosis by macrophage polarization. Cell Physiol Biochem. (2015) 35:1062–9. doi: 10.1159/000373932
73. Cavalcante PAM, Alenina N, Budu A, Freitas-Lima LC, Alves-Silva T, Agudelo JSH, et al. Nephropathy in hypertensive animals is linked to M2 macrophages and increased expression of the YM1/Chi3l3 protein. Mediators Inflamm. (2019) 2019:9086758. doi: 10.1155/2019/9086758
74. Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. J Exp Med. (2018) 215:21–33. doi: 10.1084/jem.20171773
75. Wen Y, Crowley SD. Renal effects of cytokines in hypertension. In: Advances in Experimental Medicine and Biology. New York, NY: Springer (2019). p. 443–54. doi: 10.1007/978-981-13-8871-2_21
76. Orejudo M, Rodrigues-Diez RR, Rodrigues-Diez R, Garcia-Redondo A, Santos-Sánchez L, Rández-Garbayo J, et al. Interleukin 17A participates in renal inflammation associated to experimental and human hypertension. Front Pharmacol. (2019) 10:1015. doi: 10.3389/fphar.2019.01015
77. Orejudo M, García-Redondo AB, Rodrigues-Diez RR, Rodrigues-Díez R, Santos-Sanchez L, Tejera-Muñoz A, et al. Interleukin-17A induces vascular remodeling of small arteries and blood pressure elevation. Clin Sci. (2020) 134:513–27. doi: 10.1042/CS20190682
78. Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interface Cytokine Res. (2009) 29:313–25. doi: 10.1089/jir.2008.0027
79. Mansour SG, Puthumana J, Coca SG, Gentry M, Parikh CR. Biomarkers for the detection of renal fibrosis and prediction of renal outcomes: a systematic review. BMC Nephrol. (2017) 18:72. doi: 10.1186/s12882-017-0490-0
80. Satirapoj B. Tubulointerstitial biomarkers for diabetic nephropathy. J Diabetes Res. (2018) 2018:2852398. doi: 10.1155/2018/2852398
81. Chow SL, Maisel AS, Anand I, Bozkurt B, De Boer RA, Felker GM, et al. Role of biomarkers for the prevention, assessment, and management of heart failure: a scientific statement from the American Heart Association. Circulation. (2017) 135:e1054–91. doi: 10.1161/CIR.0000000000000490
82. Schutyser E. Involvement of CC chemokine ligand 18 (CCL18) in normal and pathological processes. J Leukoc Biol. (2005) 78:14–26. doi: 10.1189/jlb.1204712
83. Chenivesse C, Tsicopoulos A. CCL18 – beyond chemotaxis. Cytokine. (2018) 109:52–6. doi: 10.1016/j.cyto.2018.01.023
84. Wang H, Liang X, Li M, Tao X, Tai S, Fan Z, et al. Chemokine (CC motif) ligand 18 upregulates Slug expression to promote stem-cell like features by activating the mammalian target of rapamycin pathway in oral squamous cell carcinoma. Cancer Sci. (2017) 108:1584–93. doi: 10.1111/cas.13289
85. Deegan PB, Moran MT, McFarlane I, Schofield JP, Boot RG, Aerts MF, et al. Clinical evaluation of chemokine and enzymatic biomarkers of Gaucher disease. Blood Cells Mol. Dis. (2005) 35:259–67. doi: 10.1016/j.bcmd.2005.05.005
86. Prasse A, Probst C, Bargagli E, Zissel G, Toews GB, Flaherty KR, et al. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2009) 179:717–23. doi: 10.1164/rccm.200808-1201OC
87. Kollert F, Binder M, Probst C, Uhl M, Zirlik A, Kayser G, et al. CCL18 - Potential biomarker of fibroinflammatory activity in chronic periaortitis. J Rheumatol. (2012) 39:1407–12. doi: 10.3899/jrheum.111143
88. Lin L, Chen YS, Yao YD, Chen JQ, Chen JN, Huang SY, et al. CCL18 from tumor-associated macrophages promotes angio-genesis in breast cancer. Oncotarget. (2015) 6:34758–73. doi: 10.18632/oncotarget.5325
89. Huang H, Li J, Hu WJ, Chen C, Luo HQ, Tang XD, et al. The serum level of CC chemokine ligand 18 correlates with the prognosis of non-small cell lung cancer. Int J Biol Markers. (2019) 34:156–62. doi: 10.1177/1724600819829758
90. Ma L, Wang H, Li Z, Geng X, Li M. Chemokine (C-C motif) ligand 18 is highly expressed in glioma tissues and promotes invasion of glioblastoma cells. J Cancer Res Ther. (2019) 15:358–64. doi: 10.4103/jcrt.JCRT-360-17
91. Urquidi V, Kim J, Chang M, Dai Y, Rosser CJ, Goodison S. CCL18 in a multiplex urine-based assay for the detection of bladder cancer. PLoS ONE. (2012) 7:e37797. doi: 10.1371/journal.pone.0037797
92. Su Y, Zhou Y, Sun Y, jue, Wang YL, Yin J, et al. Macrophage-derived CCL18 promotes osteosarcoma proliferation and migration by upregulating the expression of UCA1. J Mol Med. (2019) 97:49–61. doi: 10.1007/s00109-018-1711-0
93. Chen G, Liang YX, Zhu JG, Fu X, Chen YF, Mo RJ, et al. CC chemokine ligand 18 correlates with malignant progression of prostate cancer. Biomed Res Int. (2014) 2014:230183. doi: 10.1155/2014/230183
94. Korbecki J, Olbromski M, Dziegiel P. Ccl18 in the progression of cancer. Int J Mol Sci. (2020) 21:1–24. doi: 10.3390/ijms21217955
95. Brix SR, Stege G, Disteldorf E, Hoxha E, Krebs C, Krohn S, et al. CC chemokine ligand 18 in ANCA-associated crescentic GN. J Am Soc Nephrol. (2015) 26:2105–17. doi: 10.1681/ASN.2014040407
96. Bellón T, Martínez V, Lucendo B, Del Peso G, Castro MJ, Aroeira LS, et al. Alternative activation of macrophages in human peritoneum: implications for peritoneal fibrosis. Nephrol Dial Transplant. (2011) 26:2995–3005. doi: 10.1093/ndt/gfq771
97. Roos RS, Loetscher M, Legler DF, Clark-Lewis I, Baggiolini M, Moser B. Identification of CCR8, the receptor for the human CC chemokine I-309. J Biol Chem. (1997) 272:17251–4. doi: 10.1074/jbc.272.28.17251
98. Islam SA, Ling MF, Leung J, Shreffler WG, Luster AD. Identification of human CCR8 as a CCL18 receptor. J Exp Med. (2013) 210:1889–98. doi: 10.1084/jem.20130240
99. Tiffany HL, Lautens LL, Gao JL, Pease J, Locati M, Combadiere C, et al. Identification of CCR8: a human monocyte and thymus receptor for the CC chemokine I-309. J Exp Med. (1997) 186:165–70. doi: 10.1084/jem.186.1.165
100. Ebert LM, Meuter S, Moser B. Homing and function of human skin γδ T cells and NK cells: relevance for tumor surveillance. J Immunol. (2006) 176:4331–6. doi: 10.4049/jimmunol.176.7.4331
101. Segerer S, Cui Y, Eitner F, Goodpaster T, Hudkins KL, Mack M, et al. Expression of chemokines and chemokine receptors during human renal transplant rejection. Am J Kidney Dis. (2001) 37:518–31. doi: 10.1053/ajkd.2001.22076
102. Fu Q, Chang Y, Zhou L, An H, Zhu Y, Xu L, et al. Positive intratumoral chemokine (C-C motif) receptor 8 expression predicts high recurrence risk of post-operation clear-cell renal cell carcinoma patients. Oncotarget. (2016) 7:8413–21. doi: 10.18632/oncotarget.6761
103. Eruslanov E, Stoffs T, Kim WJ, Daurkin I, Gilbert SM, Su LM, et al. Expansion of CCR8+ inflammatory myeloid cells in cancer patients with urothelial and renal carcinomas. Clin Cancer Res. (2013) 19:1670–80. doi: 10.1158/1078-0432.CCR-12-2091
104. Basu S, Schaefer TM, Ghosh M, Fuller CL, Reinhart TA. Molecular cloning and sequencing of 25 different rhesus macaque chemokine cDNAS reveals evolutionary conservation among C. CC, CXC, and CX3C families of chemokines. Cytokine. (2002) 18:140–8. doi: 10.1006/cyto.2002.0875
105. Islam SA, Chang DS, Colvin RA, Byrne MH, McCully ML, Moser B, et al. Mouse CCL8, a CCR8 agonist, promotes atopic dermatitis by recruiting IL-5+TH2 cells. Nat Immunol. (2011) 12:167–77. doi: 10.1038/ni.1984
106. Korbecki J, Kojder K, Simińska D, Bohatyrewicz R, Gutowska I, Chlubek D, et al. Cc chemokines in a tumor: A Review of pro-cancer and anti-cancer properties of the ligands of receptors ccr1, ccr2, ccr3, and ccr4. Int J Mol Sci. (2020) 21:1–29. doi: 10.3390/ijms21218412
107. Droguett A, Valderrama G, Burgos ME, Carpio D, Saka C, Egido J, et al. Gremlin, a potential urinary biomarker of anca-associated crescentic glomerulonephritis. Sci. Rep. (2019) 9:6867. doi: 10.1038/s41598-019-43358-5
108. Montero RM, Bhangal G, Pusey CD, Frankel AH, Tam FWK. CCL18 synergises with high concentrations of glucose in stimulating fibronectin production in human renal tubuloepithelial cells. BMC Nephrol. (2016) 17:1–7. doi: 10.1186/s12882-016-0352-1$
109. Kashyap S, Osman M, Ferguson CM, Nath MC, Roy B, Lien KR, et al. Ccl2 deficiency protects against chronic renal injury in murine renovascular hypertension. Sci Rep. (2018) 8:8598. doi: 10.1038/s41598-018-26870-y
110. Cantero-Navarro E, Orejudo M, Rodrigues-Diez RR, Bellón T, Selgas R, Ruiz-Ortega M. CCL8/MCP2, un nuevo marcador en inflamación renal. Hipertensión y Riesgo Vasc. (2019) 36:43−6. Available online at: https://revistas.inia.es/doi.org/10.1094/PD-66-491
111. Orejudo M, Rodrigues-Diez RR, García-Redondo AB, Alique M, Egido J, Selgas R, et al. MCP-2/CCR8 axis is activated in experimental renal and vascular inflammation. Nephrol Dial Transplant. (2015) 30 (Suppl 3): iii393–419. doi: 10.1093/ndt/gfv188.44
112. Rodrigues-Díez R, Aroeira LS, Orejudo M, Bajo MA, Heffernan JJ, Rodrigues-Díez RR, et al. IL-17A is a novel player in dialysis-induced peritoneal damage. Kidney Int. (2014) 86:303–15. doi: 10.1038/ki.2014.33
113. Orejudo M, Rodrigues-Diez RR, García-Redondo AB, Alique M, Bellón T, Egido J, et al. Investigación del papel de la quimioquina CCL-8 en el daño renal y vascular asociado a inflamación. Hipertensión y Riesgo Vasc. (2015) 32:1–116.
114. Zwadlo G, Voegeli R, Oslhoff KS, Sorg C. A monoclonal antibody to a novel differentiation antigen on human macrophages associated with the down-regulatory phase of the inflammatory process. Pathobiology. (1987) 55:295–304. doi: 10.1159/000163432
115. Ehrchen J, Steinmüller L, Barczyk K, Tenbrock K, Nacken W, Eisenacher M, et al. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood. (2007) 109:1265–74. doi: 10.1182/blood-2006-02-001115
116. Etzerodt A, Maniecki MB, Møller K, Møller HJ, Moestrup SK. Tumor necrosis factor α-converting enzyme (TACE/ADAM17) mediates ectodomain shedding of the scavenger receptor CD163. J Leukoc Biol. (2010) 88:1201–5. doi: 10.1189/jlb.0410235
117. Møller HJ. Soluble CD163. Scand J Clin Lab Invest. (2012) 72:1–13. doi: 10.3109/00365513.2011.626868
118. Aendekerk JP, Timmermans SAMEG, Busch MH, Potjewijd J, Heeringa P, Damoiseaux GMC, et al. Urinary soluble CD163 and disease activity in biopsy-proven ANCA-associated glomerulonephritis. Clin J Am Soc Nephrol. (2020) 15:1740–8. doi: 10.2215/CJN.07210520
119. Zhang T, Li H, Vanarsa K, Gidley G, Mok CC, Petri M, et al. Association of urine sCD163 with proliferative lupus nephritis, fibrinoid necrosis, cellular crescents and intrarenal M2 macrophages. Front Immunol. (2020) 11:671. doi: 10.3389/fimmu.2020.00671
120. Fabriek BO, Dijkstra CD, van den Berg TK. The macrophage scavenger receptor CD163. Immunobiology. (2005) 210:153–60. doi: 10.1016/j.imbio.2005.05.010
121. Etzerodt A, Moestrup SK. CD163 and inflammation: biological, diagnostic, therapeutic aspects. Antioxidants Redox Signal. (2013) 18:2352–63. doi: 10.1089/ars.2012.4834
122. Philippidis P, Mason JC, Evans BJ, Nadra I, Taylor KM, Haskard DO, et al. Hemoglobin scavenger receptor CD163 mediates interleukin-10 release and heme oxygenase-1 synthesis: antiinflammatory monocyte-macrophage responses in vitro, in resolving skin blisters in vivo, and after cardiopulmonary bypass surgery. Circ Res. (2004) 94:119–26. doi: 10.1161/01.RES.0000109414.78907.F9
123. Evans BJ, Haskard DO, Sempowksi G, Landis RC. Evolution of the macrophage CD163 phenotype and cytokine profiles in a human model of resolving inflammation. Int J Inflam. (2013) 2013:780502. doi: 10.1155/2013/780502
124. Rubio-Navarro A, Carril M, Padro D, Guerrero-Hue M, Tarín C, Samaniego R, et al. CD163-macrophages are involved in rhabdomyolysis-induced kidney injury and may be detected by MRI with targeted gold-coated iron oxide nanoparticles. Theranostics. (2016) 6:896–914. doi: 10.7150/thno.14915
125. Kishimoto D, Kirino Y, Tamura M, Takeno M, Kunishita Y, Takase-Minegishi K, et al. Dysregulated heme oxygenase-1 low M2-like macrophages augment lupus nephritis via Bach1 induced by type I interferons. Arthritis Res Ther. (2018) 20:64. doi: 10.1186/s13075-018-1568-1
126. Nakagawa M, Karim MR, Izawa T, Kuwamura M, Yamate J. Immunophenotypical characterization of M1/M2 macrophages and lymphocytes in cisplatin-induced rat progressive renal fibrosis. Cells. (2021) 10:257. doi: 10.3390/cells10020257
127. Alvarado-Vazquez PA, Bernal L, Paige CA, Grosick RL, Moracho Vilrriales C, Ferreira DW, et al. Macrophage-specific nanotechnology-driven CD163 overexpression in human macrophages results in an M2 phenotype under inflammatory conditions. Immunobiology. (2017) 222:900–912. doi: 10.1016/j.imbio.2017.05.011
128. Li J, Liu CH, Xu DL, Gao B. Significance of CD163-positive macrophages in proliferative glomerulonephritis. Am J Med Sci. (2015) 350:387–92. doi: 10.1097/MAJ.0000000000000569
129. Li J, Yu YF, Liu CH, Wang CM. Significance of M2 macrophages in glomerulonephritis with crescents. Pathol Res Pract. (2017) 213:1215–20. doi: 10.1016/j.prp.2017.04.011
130. Hu W, Lin J, Lian X, Yu F, Liu W, Wu Y, et al. M2a and M2b macrophages predominate in kidney tissues and M2 subpopulations were associated with the severity of disease of IgAN patients. Clin Immunol. (2019) 205:8–15. doi: 10.1016/j.clim.2019.05.005
131. Hou J, Zhang M, Ding Y, Wang X, Li T, Gao P, et al. Circulating CD14+CD163+CD206+ M2 monocytes are increased in patients with early stage of idiopathic membranous nephropathy. Mediators Inflamm. (2018) 21:2018:5270657. doi: 10.1155/2018/5270657
132. Olmes G, Büttner-Herold M, Ferrazzi F, Distel L, Amann K, Daniel C. CD163+ M2c-like macrophages predominate in renal biopsies from patients with lupus nephritis. Arthritis Res. Ther. (2016) 18:18:90. doi: 10.1186/s13075-016-0989-y
133. Yokoe Y, Tsuboi N, Imaizumi T, Kitagawa A, Karasawa M, Ozeki T, et al. Clinical impact of urinary CD11b and CD163 on the renal outcomes of anti-neutrophil cytoplasmic antibody-associated glomerulonephritis. Nephrol Dial Transplant. (2020) 8:gfaa097. doi: 10.1093/ndt/gfaa097
134. Nielsen AJ, Nielsen MC, Birn H, Ivarsen P, Møller HJ, Hviid CVB. Urine soluble CD163 (sCD163) as biomarker in glomerulonephritis: stability, reference interval and diagnostic performance. Clin Chem Lab Med. (2020) 19:cclm-2020-0466. doi: 10.1515/cclm-2020-0466
135. O'Reilly VP, Wong L, Kennedy C, Elliot LA, O'Meachair S, Coughlan AM, et al. Urinary soluble CD163 in active renal vasculitis. J Am Soc Nephrol. (2016) 27:2906–916. doi: 10.1681/ASN.2015050511
136. Hatemi G, Esatoglu SN, Yazici Y. Biomarkers in vasculitis. Curr Opin Rheumatol. (2018) 30:30–5. doi: 10.1097/BOR.0000000000000447
137. Endo N, Tsuboi N, Furuhashi K, Shi Y, Du Q, Abe T, et al. Urinary soluble CD163 level reflects glomerular inflammation in human lupus nephritis. Nephrol Dial Transplant. (2016) 31:2023–33. doi: 10.1093/ndt/gfw214
138. Mejia-Vilet JM, Zhang XL, Cruz C, Cano-Verduzco ML, Shapiro JP, Nagaraja HN, et al. Urinary soluble CD163: a novel noninvasive biomarker of activity for lupus nephritis. J Am Soc Nephrol. (2020) 31:1335–47. doi: 10.1681/ASN.2019121285
139. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. (2003) 112:1796–808. doi: 10.1172/jci19246
140. Axelsson J, Møller HJ, Witasp A, Qureshi AR, Carrero JJ, Heimbürger O, et al. Changes in fat mass correlate with changes in soluble sCD163, a marker of mature macrophages, in patients with CKD. Am J Kidney Dis. (2006) 48:916–25. doi: 10.1053/j.ajkd.2006.08.022
141. Levy AP, Purushothaman KR, Levy NS, Purushothaman M, Strauss M, Asleh R, et al. Downregulation of the hemoglobin scavenger receptor in individuals with diabetes and the Hp 2-2 genotype: implications for the response to intraplaque hemorrhage and plaque vulnerability. Circ Res. (2007) 101:106–10. doi: 10.1161/CIRCRESAHA.107.149435
142. Shakeri-Manesch S, Zeyda M, Huber J, Ludvik B, Prager G, Stulnig TM. Diminished upregulation of visceral adipose heme oxygenase-1 correlates with waist-to-hip ratio and insulin resistance. Int J Obes. (2009) 33:1257–64. doi: 10.1038/ijo.2009.160
143. Sporrer D, Weber M, Wanninger J, Weigert J, Neumeier M, Stögbauer F, et al. Adiponectin downregulates CD163 whose cellular and soluble forms are elevated in obesity. Eur J Clin Invest. (2009) 39:671–9. doi: 10.1111/j.1365-2362.2009.02170.x
144. Hiraoka A, Horiike N, Akbar SMF, Michitaka K, Matsuyama T, Onji M. Soluble CD163 in patients with liver diseases: very high levels of soluble CD163 in patients with fulminant hepatic failure. J Gastroenterol. (2005) 40:52–6. doi: 10.1007/s00535-004-1493-8
145. Møller HJ, Grønbæk H, Schiødt FV, Holland-Fischer P, Schilsky M, Munoz S, et al. Soluble CD163 from activated macrophages predicts mortality in acute liver failure. J Hepatol. (2007) 47:671–6. doi: 10.1016/j.jhep.2007.05.014
146. Holland-Fischer P, Grønbæk H, Sandahl TD, Moestrup SK, Riggio O, Ridola L, et al. Kupffer cells are activated in cirrhotic portal hypertension and not normalised by TIPS. Gut. (2011) 60:1389–93. doi: 10.1136/gut.2010.234542
147. Min D, Brooks B, Wong J, Aamidor S, Seehoo R, Sutanto S, et al. Monocyte CD163 is altered in association with diabetic complications: possible protective role. J Leukoc Biol. (2016) 100:1375–83. doi: 10.1189/jlb.3a1015-461rr
148. Samuelsson M, Dereke J, Svensson MK, Landin-Olsson M, Hillman M. Soluble plasma proteins ST2 and CD163 as early biomarkers of nephropathy in Swedish patients with diabetes, 15-34 years of age: a prospective cohort study. Diabetol Metab Syndr. (2017) 25:9:41. doi: 10.1186/s13098-017-0240-2
149. Klessens CQF, Zandbergen M, Wolterbeek R, Bruijn JA, Rabelink TJ, Bajema IM, et al. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol Dial Transplant. (2017) 32:1322–9. doi: 10.1093/ndt/gfw260
150. Zhang XL, Guo YF, Song ZX, Zhou M. Vitamin D prevents podocyte injury via regulation of macrophage M1/M2 phenotype in diabetic nephropathy rats. Endocrinology. (2014) 155:4939–50. doi: 10.1210/en.2014-1020
151. Barisione C, Garibaldi S, Furfaro AL, Nitti M, Palmieri D, Passalacqua M, et al. Moderate increase of indoxyl sulfate promotes monocyte transition into profibrotic macrophages. PLoS ONE. (2016) 11:e0149276.doi: 10.1371/journal.pone.0149276
152. Cleary CM, Moreno JA, Fernández B, Ortiz A, Parra EG, Gracia C, et al. Glomerular haematuria, renal interstitial haemorrhage and acute kidney injury. Nephrol Dial Transplant. (2010) 25:4103–6. doi: 10.1093/ndt/gfq493
153. Ballarín J, Arce Y, Torra Balcells R, Diaz Encarnación M, Manzarbeitia F, Ortiz A, et al. Acute renal failure associated to paroxysmal nocturnal haemoglobinuria leads to intratubular haemosiderin accumulation and CD163 expression. Nephrol Dial Transplant. (2011) 26:3408–11. doi: 10.1093/ndt/gfr391
154. García-Camín RM, Goma M, Osuna RG, Rubio-Navarro A, Buendía I, Ortiz A, et al. Molecular mediators of favism-induced acute kidney injury. Clin Nephrol. (2014) 81:203–9. doi: 10.5414/CN107520
155. Gutiérrez E, Egido J, Rubio-Navarro A, Buendía I, Blanco Colio LM, Toldos O, et al. Oxidative stress, macrophage infiltration and CD163 expression are determinants of long-term renal outcome in macrohematuria-induced acute kidney injury of igA nephropathy. Nephron Clin Pract. (2012) 121:c42–53. doi: 10.1159/000342385
156. Tang WW, Feng L, Vannice JL, Wilson CB. Interleukin-1 receptor antagonist ameliorates experimental anti-glomerular basement membrane antibody-associated glomerulonephritis. J Clin Invest. (1994) 93:273–9. doi: 10.1172/JCI116956
157. Chen A, Sheu LF, Chou WY, Tsai SC, Chang DM, Liang SC, et al. Interleukin-1 receptor antagonist modulates the progression of a spontaneously occurring IgA nephropathy in mice. Am J Kidney Dis. (1997) 30:693–702. doi: 10.1016/S0272-6386(97)90495-9
158. Timoshanko JR, Kitching AR, Iwakura Y, Holdsworth SR, Tipping PG. Contributions of IL-1β and IL-1α to crescentic glomerulonephritis in mice. J Am Soc Nephrol. (2004) 15:910–8. doi: 10.1097/01.ASN.0000115704.86897.F4
159. Yang B, Fu L, Privratsky JR, Lu X, Ren J, Mei C, et al. Interleukin-1 receptor activation aggravates autosomal dominant polycystic kidney disease by modulating regulated necrosis. Am J Physiol Ren Physiol. (2019) 317:F221–8. doi: 10.1152/ajprenal.00104.2019
160. Shahzad K, Bock F, Dong W, Wang H, Kopf S, Kohli S, et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. (2015) 87:74–84. doi: 10.1038/ki.2014.271
161. Lei Y, Devarapu SK, Motrapu M, Cohen CD, Lindenmeyer MT, Moll S, et al. Interleukin-1β inhibition for chronic kidney disease in obese mice with type 2 diabetes. Front Immunol. (2019) 10:1223. doi: 10.3389/fimmu.2019.01223
162. Nowak KL, Chonchol M, Ikizler TA, Farmer-Bailey H, Salas N, Chaudhry R, et al. IL-1 inhibition and vascular function in CKD. J Am Soc Nephrol. (2017) 28:971–980. doi: 10.1681/ASN.2016040453
163. Ridker PM, MacFadyen JG, Glynn RJ, Koenig W, Libby P, Everett BM, et al. Inhibition of interleukin-1β by canakinumab and cardiovascular outcomes in patients with chronic kidney disease. J Am Coll Cardiol. (2018) 71:2405–14. doi: 10.1016/j.jacc.2018.03.490
164. Perez-Gomez M, Sanchez-Niño M, Sanz A, Martín-Cleary C, Ruiz-Ortega M, Egido J, et al. Horizon 2020 in diabetic kidney disease: the clinical trial pipeline for add-on therapies on top of renin angiotensin system blockade. J Clin Med. (2015) 4:1325–47. doi: 10.3390/jcm4061325
165. Therrien FJ, Agharazii M, Lebel M, Larivire R. Neutralization of tumor necrosis factor-alpha reduces renal fibrosis and hypertension in rats with renal failure. Am J Nephrol. (2012) 36:151–61. doi: 10.1159/000340033
166. Meldrum KK, Metcalfe P, Leslie JA, Misseri R, Hile KL, Meldrum DR. TNF-α neutralization decreases nuclear factor-κB activation and apoptosis during renal obstruction. J Surg Res. (2006) 131:182–8. doi: 10.1016/j.jss.2005.11.581
167. Gai Z, Kullak-Ublick GA. TNF-α deficiency prevents renal inflammation and oxidative stress in obese mice. Kidney Blood Press Res. (2017) 42:416–27. doi: 10.1159/000478869
168. Khan SB, Cook HT, Bhangal G, Smith J, Tam FWK, Pusey CD. Antibody blockade of TNF-α reduces inflammation and scarring in experimental crescentic glomerulonephritis. Kidney Int. (2005) 67:1812–20. doi: 10.1111/j.1523-1755.2005.00279.x
169. Awad AS, You H, Gao T, Cooper TK, Nedospasov SA, Vacher J, et al. Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int. (2015) 88:722–33. doi: 10.1038/ki.2015.162
170. Hueber AJ, Tunc A, Schett G, Manger B. Anti-tumour necrosis factor alpha therapy in patients with impaired renal function. Ann Rheum Dis. (2007) 66:981–2. doi: 10.1136/ard.2006.069211
171. Kim HW, Lee CK, Cha HS, Choe JY, Park EJ, Kim J. Effect of anti-tumor necrosis factor alpha treatment of rheumatoid arthritis and chronic kidney disease. Rheumatol Int. (2015) 35:727–34. doi: 10.1007/s00296-014-3146-4
172. Stokes MB, Foster K, Markowitz GS, Ebrahimi F, Hines W, Kaufman D, et al. Development of glomerulonephritis during anti-TNF-alpha; therapy for rheumatoid arthritis. Nephrol Dial Transplant. (2005) 20:1400–6. doi: 10.1093/ndt/gfh832
173. Ávila PJ, Gracia Iguacel C, Ortiz Arduán A, Martín-Cleary C, Acuña Ramos M, Sainz Prestel V, et al. Fracaso renal agudo en paciente tratado con antifactor de necrosis tumoral-alfa. Nefrologia. (2011) 31:484–8. doi: 10.3265/Nefrologia.pre2011.May.10978
174. Yarkan Tugsal H, Zengin B, Kenar G, Can G. Ünlü M, Önen F, et al. Infliximab-associated focal segmental glomerulosclerosis in a patient with ankylosing spondylitis. Rheumatol Int. (2019) 39:561–7. doi: 10.1007/s00296-019-04241-8
175. PremuŽić V, Padjen I, Cerovec M, Corić M, Jelaković B, Anić B. The association of TNF-alpha inhibitors and development of IgA nephropathy in patients with rheumatoid arthritis and diabetes. Case Reports Nephrol. (2020) 2020:1–8. doi: 10.1155/2020/9480860
176. Strobel T, Ahmed W, De la Sancha C, Bohm M, Fischer M. IgA nephropathy in the setting of anti-TNF-α therapy for inflammatory bowel disease. ACG Case Reports J. (2020) 7:e00462. doi: 10.14309/crj.0000000000000462
177. Su H, Lei CT, Zhang C. Interleukin-6 signaling pathway and its role in kidney disease: an update. Front Immunol. (2017) 8:405. doi: 10.3389/fimmu.2017.00405
178. Liang B, Gardner DB, Griswold DE, Bugelski PJ, Song XYR. Anti-interleukin-6 monoclonal antibody inhibits autoimmune responses in a murine model of systemic lupus erythematosus. Immunology. (2006) 119:296–305. doi: 10.1111/j.1365-2567.2006.02433.x
179. Kiberd BA. Interleukin-6 receptor blockage ameliorates murine lupus nephritis. J Am Soc Nephrol. (1993) 4:58–61.
180. Cash H, Relle M, Menke J, Brochhausen C, Jones SA, Topley N, et al. Interleukin 6 (IL-6) deficiency delays lupus nephritis in MRL-Fas lpr mice: the IL-6 pathway as a new therapeutic target in treatment of autoimmune kidney disease in systemic lupus erythematosus. J Rheumatol. (2010) 37:60–70. doi: 10.3899/jrheum.090194
181. Luig M, Kluger MA, Goerke B, Meyer M, Nosko A, Yan I, et al. Inflammation-induced IL-6 functions as a natural brake on macrophages and limits GN. J Am Soc Nephrol. (2015) 26:1597–607. doi: 10.1681/ASN.2014060620
182. Yang J, Chen J, Yan J, Zhang L, Chen G, He L, et al. Effect of interleukin 6 deficiency on renal interstitial fibrosis. PLoS ONE. (2012) 7:e52415. doi: 10.1371/journal.pone.0052415
183. Jones SA, Fraser DJ, Fielding CA, Jones GW. Interleukin-6 in renal disease and therapy. Nephrol Dial Transplant. (2015) 30:564–74. doi: 10.1093/ndt/gfu233
184. Kang S, Tanaka T, Narazaki M, Kishimoto T. Targeting interleukin-6 signaling in clinic. Immunity. (2019) 50:1007–23. doi: 10.1016/j.immuni.2019.03.026
185. Rosas IO, Bräu N, Waters M, Go RC, Hunter BD, Bhagani S, et al. Tocilizumab in hospitalized patients with severe covid-19 pneumonia. N Engl J Med. (2021) 25:NEJMoa2028700. doi: 10.1056/NEJMoa2028700
186. Haller H, Bertram A, Nadrowitz F, Menne J. Monocyte chemoattractant protein-1 and the kidney. Curr Opin Nephrol Hypertens. (2016) 25:42–9. doi: 10.1097/MNH.0000000000000186
187. Hasegawa H, Kohno M, Sasaki M, Inoue A, Ito MR, Terada M, et al. Antagonist of monocyte chemoattractant protein 1 ameliorates the initiation and progression of lupus nephritis and renal vasculitis in MRL/lpr mice. Arthritis Rheum. (2003) 48:2555–66. doi: 10.1002/art.11231
188. De Lema GP, Maier H, Franz TJ, Escribese M, Chilla S, Segerer S, et al. Chemokine receptor Ccr2 deficiency reduces renal disease and prolongs survival in MRL/lpr lupus-prone mice. J Am Soc Nephrol. (2005) 16:3592–601. doi: 10.1681/ASN.2005040426
189. Wada T, Yokoyama H, Furuichi K, Kobayashi K, Harada K, Naruto M, et al. Intervention of crescentic glomerulonephritis by antibodies to monocyte chemotactic and activating factor (MCAF/MCP-1). FASEB J. (1996) 10:1418–25. doi: 10.1096/fasebj.10.12.8903512
190. Lloyd CM, Minto AW, Dorf ME, Proudfoot A, Wells TNC, Salant DJ, et al. RANTES and monocyte chemoattractant protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J Exp Med. (1997) 185:1371–80. doi: 10.1084/jem.185.7.1371
191. Kanamori H, Matsubara T, Mima A, Sumi E, Nagai K, Takahashi T, et al. Inhibition of MCP-1/CCR2 pathway ameliorates the development of diabetic nephropathy. Biochem Biophys Res Commun. (2007) 360:772–7. doi: 10.1016/j.bbrc.2007.06.148
192. Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Rollin BJ, Tesch GH. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. (2006) 69:73–80. doi: 10.1038/sj.ki.5000014
193. Ninichuk V, Clauss S, Kulkarni O, Schmid H, Segerer S, Radomska E, et al. Late onset of Ccl2 blockade with the Spiegelmer mNOX-E36-3′PEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am J Pathol. (2008) 172:628–37. doi: 10.2353/ajpath.2008.070601
194. Boels MGS, Koudijs A, Avramut MC, Sol WMPJ, Wang G, van Oeveren-Rietdijk AM, et al. Systemic monocyte chemotactic protein-1 inhibition modifies renal macrophages and restores glomerular endothelial glycocalyx and barrier function in diabetic nephropathy. Am J Pathol. (2017) 187:2430–40. doi: 10.1016/j.ajpath.2017.07.020
195. Clauss S, Gross O, Kulkarni O, Avila-Ferrufino A, Radomska E, Segerer S, et al. Ccl2/Mcp-I blockade reduces glomerular and interstitial macrophages but does not ameliorate renal pathology in co//agen4A3-deficient mice with autosomal recessive alport nephropathy. J Pathol. (2009) 218:40–7. doi: 10.1002/path.2505
196. De Zeeuw D, Bekker P, Henkel E, Hasslacher C, Gouni-Berthold I, Mehling H, et al. The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: a randomised trial. Lancet Diabetes Endocrinol. (2015) 3:687–96. doi: 10.1016/S2213-8587(15)00261-2
197. Menne J, Eulberg D, Beyer D, Baumann M, Saudek F, Valkusz Z, et al. C-C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol Dial Transplant. (2017) 32:307–15. doi: 10.1093/ndt/gfv459
198. Fernandez-Fernandez B, Sarafidis P, Kanbay M, Navarro-González JF, Soler MJ, Górriz JL, et al. SGLT2 inhibitors for non-diabetic kidney disease: drugs to treat CKD that also improve glycaemia. Clin Kidney J. (2020) 13:728–33. doi: 10.1093/ckj/sfaa198
199. Engel JE, Chade AR. Macrophage polarization in chronic kidney disease: a balancing act between renal recovery and decline? Am J Physiol Ren Physiol. (2019) 317:F1409–13. doi: 10.1152/ajprenal.00380.2019
200. Wang Y, Wang YP, Zheng G, Lee VWS, Ouyang L, Chang DHH, et al. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. (2007) 72:290–9. doi: 10.1038/sj.ki.5002275
201. Cao Q, Wang Y, Zheng D, Sun Y, Wang Y, Lee VWS, et al. IL-10/TGF-β-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J Am Soc Nephrol. (2010) 21:933–42. doi: 10.1681/ASN.2009060592
202. Zheng D, Wang Y, Cao Q, Lee VWS, Zheng G, Sun Y, et al. Transfused macrophages ameliorate pancreatic and renal injury in murine diabetes mellitus. Nephron Exp Nephrol. (2011) 118:e87–99. doi: 10.1159/000321034
203. Du Q, Tsuboi N, Shi Y, Ito S, Sugiyama Y, Furuhashi K, et al. Transfusion of CD206+ M2 macrophages ameliorates antibody-mediated glomerulonephritis in mice. Am J Pathol. (2016) 186:3176–88. doi: 10.1016/j.ajpath.2016.08.012
204. Taguchi K, Okada A, Hamamoto S, Unno R, Moritoki Y, Ando R, et al. M1/M2-macrophage phenotypes regulate renal calcium oxalate crystal development. Sci Rep. (2016) 6:35167. doi: 10.1038/srep35167
205. Mao R, Wang C, Zhang F, Zhao M, Liu S, Liao G, et al. Peritoneal M2 macrophage transplantation as a potential cell therapy for enhancing renal repair in acute kidney injury. J Cell Mol Med. (2020) 24:3314–27. doi: 10.1111/jcmm.15005
206. Engel JE, Williams E, Williams ML, Bidwell GL, Chade AR. Targeted VEGF (Vascular Endothelial Growth Factor) therapy induces long-term renal recovery in chronic kidney disease via macrophage polarization. Hypertension. (2019) 74:1113–23. doi: 10.1161/HYPERTENSIONAHA.119.13469
207. Yokoo T, Ohashi T, Utsunomiya Y, Kojima H, Imasawa T, Kogure T, et al. Prophylaxis of antibody-induced acute glomerulonephritis with genetically modified bone marrow-derived vehicle cells. Hum Gene Ther. (1999) 10:2673–8. doi: 10.1089/10430349950016717
208. Yamagishi H, Yokoo T, Imasawa T, Mitarai T, Kawamura T, Utsunomiya Y. Genetically modified bone marrow-derived vehicle cells site specifically deliver an anti-inflammatory cytokine to inflamed interstitium of obstructive nephropathy. J Immunol. (2001) 166:609–16. doi: 10.4049/jimmunol.166.1.609
209. Kluth DC, Ainslie CV, Pearce WP, Finlay S, Clarke D, Anegon I, et al. Macrophages transfected with adenovirus to express IL-4 reduce inflammation in experimental glomerulonephritis. J Immunol. (2001) 166:4728–36. doi: 10.4049/jimmunol.166.7.4728
210. Wilson HM, Stewart KN, Brown PAJ, Anegon I, Chettibi S, Rees AJ, et al. Bone-marrow-derived macrophages genetically modified to produce IL-10 reduce injury in experimental glomerulonephritis. Mol Ther. (2002) 6:710–7. doi: 10.1006/mthe.2002.0802
211. Wilson HM, Chettibi S, Jobin C, Walbaum D, Rees AJ, Kluth DC. Inhibition of macrophage nuclear factor-κB leads to a dominant anti-inflammatory phenotype that attenuates glomerular inflammation in vivo. Am J Pathol. (2005) 167:27–37. doi: 10.1016/S0002-9440(10)62950-1
212. Ferenbach DA, Ramdas V, Spencer N, Marson L, Anegon I, Hughes J, et al. Macrophages expressing heme oxygenase-1 improve renal function in ischemia/reperfusion injury. Mol Ther. (2010) 18:1706–13. doi: 10.1038/mt.2010.100
213. Ren GL, Zhu J, Li J, Meng XM. Noncoding RNAs in acute kidney injury. J Cell Physiol. (2019) 234:2266–76. doi: 10.1002/jcp.27203
214. Essandoh K, Li Y, Huo J, Fan GC. MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock. (2016) 46:122–31. doi: 10.1097/SHK.0000000000000604
215. Graff JW, Dickson AM, Clay G, McCaffrey AP, Wilson ME. Identifying functional microRNAs in macrophages with polarized phenotypes. J Biol Chem. (2012) 287:21816–25. doi: 10.1074/jbc.M111.327031
216. Zhang Y, Zhang M, Zhong M, Suo Q, Lv K. Expression profiles of miRNAs in polarized macrophages. Int J Mol Med. (2013) 31:797–802. doi: 10.3892/ijmm.2013.1260
217. Cobos Jiménez V, Bradley EJ, Willemsen AM, van Kampen AHC, Baas F, Kootstra NA. Next-generation sequencing of microRNAs uncovers expression signatures in polarized macrophages. Physiol Genomics. (2014) 46:91–103. doi: 10.1152/physiolgenomics.00140.2013
218. Fierro-Fernández M, Miguel V, Márquez-Expósito L, Nuevo-Tapioles C, Herrero JI, Blanco-Ruiz E, et al. MiR-9-5p protects from kidney fibrosis by metabolic reprogramming. FASEB J. (2020) 34:410–31. doi: 10.1096/fj.201901599RR
219. Sheedy FJ. Turning 21: induction of miR-21 as a key switch in the inflammatory response. Front Immunol. (2015) 6:19. doi: 10.3389/fimmu.2015.00019
220. Jia Y, Zheng Z, Xue M, Zhang S, Hu F, Li Y, et al. Extracellular vesicles from albumin-induced tubular epithelial cells promote the m1 macrophage phenotype by targeting klotho. Mol Ther. (2019) 27:1452–66. doi: 10.1016/j.ymthe.2019.05.019
221. Lv LL, Feng Y, Wu M, Wang B, Li ZL, Zhong X, et al. Exosomal miRNA-19b-3p of tubular epithelial cells promotes M1 macrophage activation in kidney injury. Cell Death Differ. (2020) 27:210–26. doi: 10.1038/s41418-019-0349-y
222. Luly FR, Lévêque M, Licursi V, Cimino G, Martin-Chouly C, Théret N, et al. MiR-146a is over-expressed and controls IL-6 production in cystic fibrosis macrophages. Sci Rep. (2019) 9:16259. doi: 10.1038/s41598-019-52770-w
223. Zheng CZ, Shu YB, Luo YL, Luo J. The role of miR-146a in modulating TRAF6-induced inflammation during lupus nephritis. Eur Rev Med Pharmacol Sci. (2017) 21:1041–8.
224. Ichii O, Otsuka S, Sasaki N, Namiki Y, Hashimoto Y, Kon Y. Altered expression of microRNA miR-146a correlates with the development of chronic renal inflammation. Kidney Int. (2012) 81:280–92. doi: 10.1038/ki.2011.345
225. Bhatt K, Lanting LL, Jia Y, Yadav S, Reddy MA, Magilnick N, et al. Anti-inflammatory role of microrna-146a in the pathogenesis of diabetic nephropathy. J Am Soc Nephrol. (2016) 27:2277–88. doi: 10.1681/ASN.2015010111
226. Chau BN, Xin C, Hartner J, Ren S, Castano AP, Linn G, et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med. (2012) 4:121ra18. doi: 10.1126/scitranslmed.3003205
227. Gomez IG, MacKenna DA, Johnson BG, Kaimal V, Roach AM, Ren S, et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Invest. (2015) 125:141–56. doi: 10.1172/JCI75852
228. Lakhia R, Hajarnis S, Williams D, Aboudehen K, Yheskel M, Xing C, et al. MicroRNA-21 aggravates cyst growth in a model of polycystic kidney disease. J Am Soc Nephrol. (2016) 27:2319–30. doi: 10.1681/ASN.2015060634
229. Song N, Zhang T, Xu XL, Lu Z, Yu X, Fang Y, et al. miR-21 protects against ischemia/reperfusion-induced acute kidney injury by preventing epithelial cell apoptosis and inhibiting dendritic cell maturation. Front Physiol. (2018) 9:790. doi: 10.3389/fphys.2018.00790
230. Rayego-Mateos S, Morgado-Pascual JL, Opazo-Ríos L, Guerrero-Hue M, García-Caballero C, Vázquez-Carballo C, et al. Pathogenic pathways and therapeutic approaches targeting inflammation in diabetic nephropathy. Int J Mol Sci. (2020) 21:3798. doi: 10.3390/ijms21113798
231. Reynolds G, Haniffa M. Human and mouse mononuclear phagocyte networks: a tale of two species? Front Immunol. (2015) 6:330. doi: 10.3389/fimmu.2015.00330
232. Li Y, Oosting M, Deelen P, Ricaño-Ponce I, Smeekens S, Jaeger M, et al. Inter-individual variability and genetic influences on cytokine responses to bacteria and fungi. Nat Med. (2016) 22:952–60. doi: 10.1038/nm.4139
233. Das A, Yang CS, Arifuzzaman S, Kim S, Kim SY, Jung KH, et al. High-resolution mapping and dynamics of the transcriptome, transcription factors, and transcription co-factor networks in classically and alternatively activated macrophages. Front Immunol. (2018) 9:22. doi: 10.3389/fimmu.2018.00022
Keywords: macrophages, cytokines, kidney disease, CCL18, inflammation, CCL8
Citation: Cantero-Navarro E, Rayego-Mateos S, Orejudo M, Tejedor-Santamaria L, Tejera-Muñoz A, Sanz AB, Marquez-Exposito L, Marchant V, Santos-Sanchez L, Egido J, Ortiz A, Bellon T, Rodrigues-Diez RR and Ruiz-Ortega M (2021) Role of Macrophages and Related Cytokines in Kidney Disease. Front. Med. 8:688060. doi: 10.3389/fmed.2021.688060
Received: 30 March 2021; Accepted: 11 June 2021;
Published: 08 July 2021.
Edited by:
David Nikolic-Paterson, Monash University, AustraliaReviewed by:
Hee-Seong Jang, Icahn School of Medicine at Mount Sinai, United StatesCopyright © 2021 Cantero-Navarro, Rayego-Mateos, Orejudo, Tejedor-Santamaria, Tejera-Muñoz, Sanz, Marquez-Exposito, Marchant, Santos-Sanchez, Egido, Ortiz, Bellon, Rodrigues-Diez and Ruiz-Ortega. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Rayego-Mateos, U3JheWVnb0BmamQuZXM=; Marta Ruiz-Ortega, bXJ1aXpvQGZqZC5lcw==
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.