 Sergi Codina
Sergi Codina Anna Manonelles
Anna Manonelles Maria Tormo
Maria Tormo Anna Sola
Anna Sola Josep M. Cruzado
Josep M. Cruzado
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 14 July 2021
Sec. Nephrology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.660334
This article is part of the Research Topic Management of Patients with Non-Dialysis Dependent Chronic Kidney Disease (ND-CKD) View all 22 articles
Chronic kidney disease (CKD) is increasing in most countries and kidney transplantation is the best option for those patients requiring renal replacement therapy. Therefore, there is a significant number of patients living with a functioning kidney allograft. However, progressive kidney allograft functional deterioration remains unchanged despite of major advances in the field. After the first post-transplant year, it has been estimated that this chronic allograft damage may cause a 5% graft loss per year. Most studies focused on mechanisms of kidney graft damage, especially on ischemia-reperfusion injury, alloimmunity, nephrotoxicity, infection and disease recurrence. Thus, therapeutic interventions focus on those modifiable factors associated with chronic kidney allograft disease (CKaD). There are strategies to reduce ischemia-reperfusion injury, to improve the immunologic risk stratification and monitoring, to reduce calcineurin-inhibitor exposure and to identify recurrence of primary renal disease early. On the other hand, control of risk factors for chronic disease progression are particularly relevant as kidney transplantation is inherently associated with renal mass reduction. However, despite progress in pathophysiology and interventions, clinical advances in terms of long-term kidney allograft survival have been subtle. New approaches are needed and probably a holistic view can help. Chronic kidney allograft deterioration is probably the consequence of damage from various etiologies but can be attenuated by kidney repair mechanisms. Thus, besides immunological and other mechanisms of damage, the intrinsic repair kidney graft capacity should be considered to generate new hypothesis and potential therapeutic targets. In this review, the critical risk factors that define CKaD will be discussed but also how the renal mechanisms of regeneration could contribute to a change chronic kidney allograft disease paradigm.
CKD is and will be one of the biggest threats for health systems worldwide due to its prevalence and cost, especially when referring to kidney replacement therapy (1). Nowadays, kidney transplantation is the treatment of choice since it has shown its superiority at improving survival and quality of life and reducing costs and comorbidity (2). The incidence of this technique has increased in recent years reaching a median rate of 33 pmp in Europe (3). Therefore, there is a significant number of patients living with a functioning kidney allograft, particularly in high-income countries. On the other side, progressive allograft function loss has become one of the most important causes of KRT requirement. Looking back, kidney allograft survival has undoubtedly improved. While in the 90's the median survival for kidney grafts was nearly 6.6 years, the current expected lifespan is about 8.8 years (4). However, this gain in allografts expected lifetime is mostly due to the improvement in short time graft survival (4). After the first post-transplant year, it has been estimated that chronic allograft damage may cause a 5% graft loss per year and this rate has seen very little decline in recent years (5). As a result, while short-term graft survival is relatively ensured, the tendency to improve long-term graft survival seems to have slowed down if we compare recent cohorts (3, 5).
From a clinical point of view, there is a wide spectrum of etiologies that explain graft damage (6). Currently, therapeutic interventions target those modifiable factors associated with kidney allograft attrition: reduce ischemia-reperfusion injury, minimize calcineurin-inhibitor exposure, control of classical risk factors or early identification of recurrence of primary renal disease. In addition, a lot of efforts are focused on improving immunologic risk stratification and on the development of new therapeutic strategies against kidney's rejection (7). Separately, all these etiologies can contribute in a major or minor form to the loss of renal mass that will end in chronic graft disease progression (8, 9).
Histologically, chronic graft damage means almost always fibrosis. Fibrosis develops in many patients during the first year after transplantation, especially in the first 3 months (10). Because most of the knowledge we have in this field is based on protocol biopsies, it is difficult to precisely titter the prevalence of fibrosis in allograft, but it varies from 17 to 66% according to the series (11, 12). However, fibrosis will continue to grow in the following years, especially in those patients with more fibrosis in former biopsies (12–14). This progression happens despite the absence or minimization of risk factors (13). Currently, alloimmunological damage is thought to be the main cause of kidney allograft fibrosis (15).
In short, a set of insults from different etiologies are capable of harming the kidney graft and altogether contribute to one common outcome. In this review, we will use the terminology chronic kidney allograft disease (CKaD) to refer to that global damage. We prefer the term CKaD in spite of other formulas (i.e., chronic allograft nephropathy) to emphasize CKaD as a global entity that summarizes the sum of all the damages of each etiology, not only immunological. In addition, we like the similarity with the term CKD as it helps to understand CKaD as a chronic, progressive, multifactorial disease, and above all, as a disease that requires a specific approach with nephroprotective strategies. In fact, despite the progress made in each area, clinical advances in terms of long-term kidney graft survival have been subtle (3) so new approaches are needed and the concept CKaD aims to provide a more holistic point of view. It is important to understand that chronic allograft deterioration is the consequence not only of the damage from various etiologies but also of the imbalance of kidney's own repair mechanisms (16). Based on recent investigations, it can be hypothesized that besides immunological and other mechanisms of damage, the graft's intrinsic capacity to repair could attenuate kidney damage and should be considered in new therapeutic approaches. In this review, the critical risk factors that define CKaD will be discussed but also how the knowledge on kidney mechanisms of repair could contribute to change the CKaD paradigm.
There are several mechanisms of kidney allograft damage and the most relevant will be described in this part (Table 1). Individually, all of them cause nephron loss on a solitary kidney and potentially activate the key mechanism of kidney disease progression by producing glomerular hypertension and hyperfiltration. Renin angiotensin blockade and SGTL-2 inhibitors could alleviate this common mechanism of CKD progression in native and transplanted kidneys. However, this topic is beyond the scope of this revision.
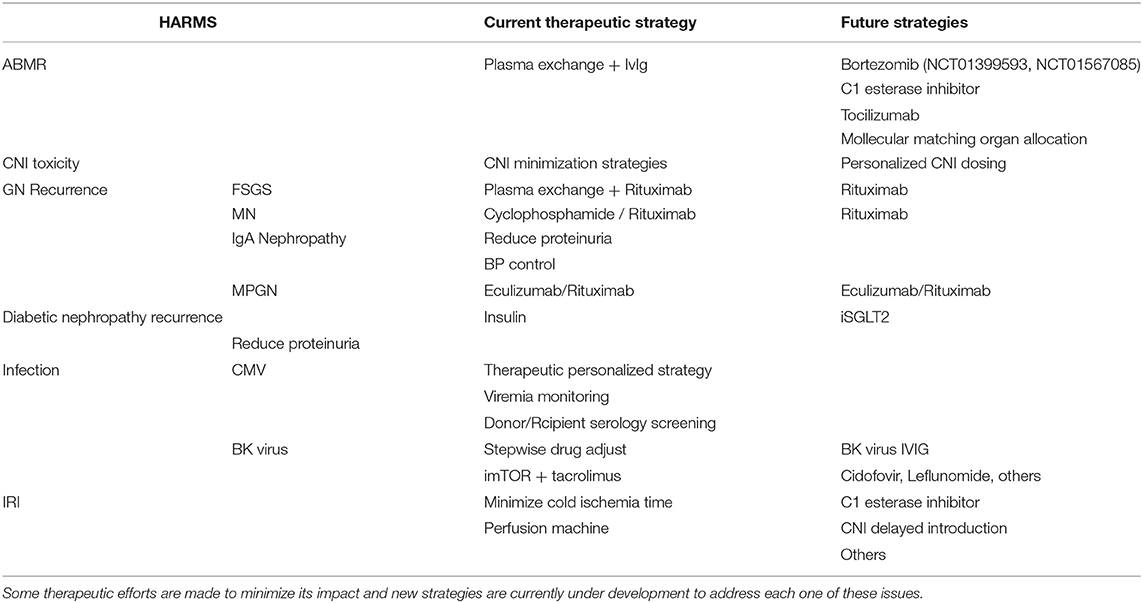
Table 1. Harms from different etiology contribute to CKaD.
Antibody-mediated rejection (ABMR) has been recognized as a major cause of organ-transplant failure during the past two decades (17). Nowadays, it is widely accepted that ABMR is the major risk factor for the development of CKaD and it can explain more than half of graft losses (18). Although there is some lack of knowledge about its natural history yet, understanding about ABMR has drastically increased in last years.
ABMR clinical impact increases poor graft and patient survival outcomes (19). In a large cohort of 885 kidney transplant recipients who underwent biopsies for graft dysfunction, patients with ABMR morphology showed an 8-year graft survival of 53% in C4d-positive and 66% in C4d-negative. This rate was significantly lower than the 81% seen in patients without any rejection features (20). To notice, the presence of complement-fixing antibodies, the extent of graft dysfunction at baseline and the presence of chronic lesions (capillary multilayering, arterial intimal fibrosis, glomerular basement membrane double contours) are associated with the poorest outcomes (21, 22). The main risk factor known for ABMR development is the number of HLA mismatches and poor adherence to medications.
Briefly, its pathogenesis is explained by the existence or de novo apparition of donor-specific antibodies (DSA). DSAs recognize some molecules expressed on the recipient's endothelium and this initiates an inflammatory cascade that leads to tissue damage. Among others, the classical pathway of the complement has been identified as an important way for antibody mediated damage (23). This simplistic explanation was the basis for the first definition of ABMR in the Banff classification from 1997 and allowed to stablish three standardized criteria for its diagnosis: First, identification of DSA. Second, evidence of endothelial damage. Third, proof of complement mediated damage through Cd4 staining.
Though, ABMR pathogenesis is more complex and further information has forced to readapt its definition until the actual one (24). Due to its predominant role in ABMR, DSA against HLA antigens are commonly screened in clinical practice, especially if rejection is suspected. However, DSA are not always targeted to HLA antigens but to other endothelial antigens (23). Thus, the current definition of ABMR accepts the diagnostic even without an identified DSA as long as evidence of antibody mediated damage exists (24). In addition, although complement activation is the most deleterious mechanism of allograft injury (25), antibody mediated harm also occurs through other pathways (26). Consequently, ABMR can also be diagnosed in C4d negative cases if DSA are present or in chronic ABMR.
No therapeutic strategy has received FDA approval for the treatment of antibody-mediated rejection. Based on ABMR pathogenesis, plasma exchange plus IVIG to remove circulant DSAs is considered the current standard of care despite low quality evidence (27, 28). Two trials were designed to test the effect of Rituximab plus plasmapheresis and/or IVIG on ABMR in order to decrease the production of DSA. Both trials ended with underpowered results showing no benefit on short graft survival nor on GFR loss prevention (29, 30). Furthermore, targeting plasmatic cells has not shown the desired effect. The BORTEJECT trial was based on the anti-proteosome drug Bortezomib. No differences in GFR attrition were observed but more gastrointestinal and hematological adverse events were observed in the Bortezomib group (31). Two more randomized clinical trials are currently active and will test Bortezomib (ClinicalTrials.gov numbers, NCT01399593 and NCT01567085). The aforementioned impact of the complement cascade in ABMR has been another therapeutic approach. To that end, the anti-C5 antibody Eculizumab, has been used both for ABMR treatment and for ABMR prevention in desensitization protocols. Despite some trends suggesting marginal beneficial effects (32), at this point of time Eculizumab has also failed to prevent graft damage secondary to ABMR (33, 34). Instead of targeting the end of the cascade, another valid strategy could be point to the top through C1 esterase inhibitors. In a phase II study with plasma derived C1 esterase inhibitor, Montgomery et al. (35) showed a secure profile of the treatment and described that no transplant glomerulopathy was found in the treatment group whereas transplant glomerulopathy were present in 3 of 7 patients in the control group. Another therapeutic approach with promising result is the use of the anti-IL-6 antibody Tocilizumab, which in a French cohort of KTR with refractory cABMR showed a graft survival of 80% at 6 years, with a reduction in glomerulitis, peritubular capilaritis and C4d deposition in allograft biopsies after treatment (36). RCTs are currently going on to confirm these therapeutic options. Other new strategies tested in ongoing registered trials are corticotropin or double filtration plasmapheresis (13). However, it should be pointed out that there is a surprising discrepancy between the theoretic high ABMR prevalence and the lack of power of interventional clinical trials on chronic ABMR due to the lack of patients fulfilling inclusion criteria.
Due to the lack of a solid therapeutic approach, prevention of ABMR occurrence may be crucial. Nowadays, to talk about ABMR prevention means improvement of compatibility. Scientific literature is full of evidence showing the correlation with more HLA matches and better graft outcomes (37). However, HLA incompatible transplantations cannot be avoided in current health systems. The study of these HLA miss-matched transplants has identified that some of these miss-matches confers more risk (Unacceptable miss-match) while others are better tolerated (acceptable mismatches) (38, 39). Differences in permissibility between HLA-mismatched combinations may be explained by a different impact of amino-acid polymorphisms on peptide-binding features. Some tools have been developed to calculate this binding-site or epitope miss-match to further compatibility stratification. In a Dutch multi-center study, 2,918 donor–recipient couples were retrospectively stratified using one of these tools (Predicted Indirectly ReCognizable HLA Epitopes presented by recipient HLA class II: PIRCHE-II) (40). For these donors–recipients couples, PIRCHE-II numbers were related to graft survival in univariate and multivariable analyses. Recently, DQ-antibody verified eplet miss-match has showed to be associated with an increased risk of dnDSA formation, kidney rejection, decline of graft function and graft failure (41). However, no study has reported benefits of these tools in a decision-making prospective protocol.
In short, ABMR is the main cause of long-term graft loss and so far, we have failed to dramatically improve its outcomes. In the absence of effective therapeutic approaches, prevention of ABMR occurrence plays a key role. Probably, molecular matching techniques will be included in organ allocation schemes in the next years. Early treatment and avoiding fibrosis could be essential to prevent long-term allograft attrition.
The importance of CNI toxicity on CKaD has drastically decreased over the decades (42). Putting on some perspective, CNI toxicity leading to chronic damage was pointed out after Myers et al. (43) published a case-control study of cardiac transplant recipients under treatment with Cyclosporine for at least 12 months. In this study, 2 patients developed ESKD and the median eGFR of patients treated with CNI presented an important attrition in comparison with controls. Since then, CNI related toxicity leading to ESKD has been observed in other solid organ transplantation cases. Nevertheless, more than three decades have passed and there have been some changes in CNI paradigm: first, the better knowledge of antibody mediated damage and the development of new diagnostic techniques has brought out the important role of alloimmunity in CKaD (see above). Second, CNI levels have been taken to the minimum necessary to ensure safety for both patient and graft survival. Third, Tacrolimus more than Cyclosporine is the current CNI of choice in many cases reducing associated adverse events (44, 45). After all these changes, CNI toxicity has been displaced as a cause of CKaD by some authors. In 2012 Sellarés et al. (18) tried to find a diagnostic explanation beyond “chronic graft nephropathy” for every graft loss in a cohort of 315 patients. In that cohort, no graft failure was attributed to CNI toxicity.
How can CNI toxicity as cause of CKaD have gone from all to nothing? Probably, there are a set of characteristics that make CNI toxicity something like Santa: “you can only see if you believe in it”:
a) On one hand, existence of CNI toxicity should be beyond doubt. Its damage mechanisms have been characterized, with an acute reversible damage related to an imbalance between vasodilators and vasoconstrictors and tubular vacuolization; and a chronic damage secondary to reduced glomerular blood flow with ischemic-reperfusion mediated injury (46). In the other hand, when large sparing CNI clinical trials were performed, differences in graft function seemed to be more related to the introduction of IL-2 antibody and MMF optimization than to CNI dosage (7, 47, 48).
b) CNI toxicity is dose-dependent (49, 50). However, the range of tacrolimus levels in those studies were between 5 and 25 ng/ml and the actual recommended target levels are much narrower, conferring less significant differences (51).
c) Histologically, chronic damage due to CNI toxicity shows arteriolar hyalinosis, interstitial fibrosis, tubular atrophy, juxtaglomerular apparatus hyperplasia, glomerular capsular fibrosis and global glomerulosclerosis. Unfortunately, none of these findings are pathognomonic, have not any specific marker and its presence can be explained by other common graft comorbidities.
d) Finally, some factors which are not measured in clinical practice could explain important differences in CNI susceptibility. For example, local concentration of Cyclosporine in kidney is associated with more susceptibility to toxicity and chronic damage (52). However, local concentration does not correlate with systemic blood levels and has a great interindividual variation due to genetic differences and drug interactions. Similarly, while some authors have described the presence of DNA SNPs that confers and increased risk of CNI toxicity (53), personalized CNI dosing according to genetic SNPs exists but is not globally extended.
In conclusion, CNI toxicity is an exclusion diagnosis without any specific marker. From a pragmatic point of view, while in other solid organ transplants chronic kidney damage could reasonably be attributed to CNI toxicity (54); in kidney transplantation is hard to find a situation in which graft loss occurs without any other comorbidity that could contribute to chronic renal damage. Thus, isolated chronic CNI toxicity diagnostic is always controversial and should be performed with caution. However, beyond a categorical diagnostic, CNI effects on renal function and chronic damage through fibrosis are probably always present, contributing to global graft attrition and therefore to CKaD.
GN recurrence accounts for a large amount of kidney graft recipients evolving to end stage kidney disease. Its exact proportion varies from 3 to 18% of graft losses according to the series (55, 56) but probably, GN recurrence is under diagnosed in transplant recipients (57). There are four main entities that explain the majority of cases of GN recurrence: focal segmental glomerulosclerosis (FSGS), membranous nephropathy (MN), membranoproliferative GN (MPGN), and IgA nephropathy (IgAN). In 2017, Cosio and Cattran (58) published an outstanding review on this topic. So, in this paper we will focused on the key points and actualize the issue with current evidence (Table 2).
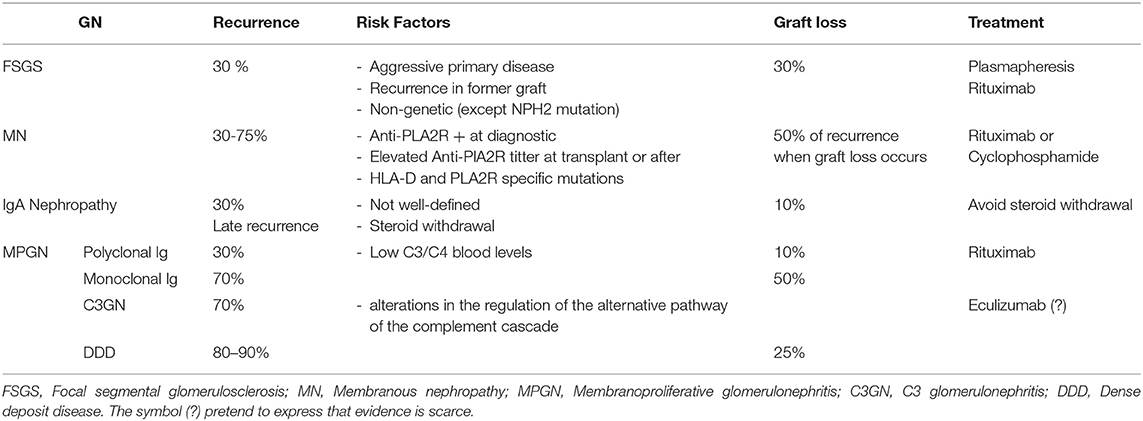
Table 2. Recurrence profile of the main Glomerulonephritis (GN).
FSGS is a common cause of recurrence reaching 30% in 3 years. Of these, nearly 30% will advance to graft loss (58). FSGS recurrence occurs early after transplantation. Genetic forms of FSGS are less probable to appear in the allograft except for podocin mutations (NPHS2) (59). Clinical predictors of GN recurrence are those signs of disease severity in the primary episode: evolution to dialysis in <3 years after the diagnosis and high levels of proteinuria after transplantation. Classically, young age is associated with more recurrence though some new evidence contradicts this idea (60). Unfortunately, these factors are imprecise and the most reliable predictor of GN recurrence is the recurrence itself in a former graft.
Although the pathogenesis of primary FSGS remains unknown, the clinical profile after transplantation hardly suggests the presence of a circulating factor (61). In a blind attempt to remove this factor, plasmapheresis is commonly used to treat GN recurrence. The TANGO project aims to create an international collaborative data sharing network about GN recurrence in order to generate stronger evidence on the field (62). As a result of this project, Uffing et al. (60) showed that in current clinical practice the therapeutic response after plasmapheresis was nearly 50%. The second most common pharmacological approach seen in this study was the use of Rituximab. Whether the beneficial effect of Rituximab is through targeting B lymphocyte or directly to podocyte SMPDL-3b protein is currently under discussion (63). Curiously, in the TANGO study there is an almost despicable use of Cyclosporine despite previous data suggested a potentially beneficial effect combined with Plasmapheresis (64).
Globally, recurrence of MN is about 50%. However, its range varies from 30 to 75%. This variation is in close relationship with anti-PLA2R titter profile. Thus, primary MN anti-PLA2R mediated has more risk of recurrence than no antibody mediated disease (58, 65, 66). In addition, the presence of persistent elevated titters of antibodies after transplantation carries a higher risk of recurrence. Also, genetic factors have been associated to MN risk of recurrence. For example, the presence of SNP mutations on HLA-D and PLA2R loci confer more risk of recurrence when presented by the donor (67). Most recipients with recurrent MN are under CNI treatment, an effective therapy in native kidneys. Another line of treatment is the use of alkylating agents such as cyclophosphamide, but it has a high risk of medullar toxicity specially if treatment with MMF is not stopped. Current evidence suggest that the use of Rituximab is an effective and safe strategy for these patients, with the additional advantage that do not precise the adjustment of the other immunosuppressors (66, 68) though further studies are needed.
Prevalence of recurrent IgA Nephropathy is 30% according to clinical reports (69). However, two major considerations must be made: first, unlike other primary diseases, IgA recurrence occurs later, even 10 years after transplantation, so some series may underestimate real prevalence due to short follow-up. Second, the histological immunofluorescence pattern characteristic of the disease appears in a very large proportion of patients much before clinical manifestation (70). Though, it should be noticed that some apparently normal donors (living or deceased) may have latent IgA deposits in the kidney (71). Currently, there is no way to know which latent IgA will evolve to clinical recurrence or will otherwise disappear. No robust evidence about other risk factors for IgA recurrence exists, although some authors suggest that primary disease's activity (presence of crescents, rapid evolution to ESKD) could be associated with higher rate of recurrence (72). IgA nephropathy impact on graft survival is mild and survival graft curves just differ from other entities beyond 10 years post transplantation (69). Histological Oxford classification could have prognostic value for allograft failure (73). No specific treatment is recommended for IgA recurrence. According to KDIGO guidelines, treatment should aim to reduce proteinuria, to optimize blood pressure and to reduce inflammation (74). Steroid withdrawal is associated with major incidence of recurrence and poorer graft outcomes (75). Studies from Japan reported favorable outcomes after tonsillectomy, but these results need to be confirmed (76).
Each type and subtype of MPGN has its particular evolution after transplantation (58). In general, MPGN due to Immunoglobulin (Ig) deposition has a lower risk of recurrence (30–70%) specially if the Ig are polyclonal. Instead, recurrence of MPGN due to complement deposition rises to 70–90%. Regarding MPGN with Ig deposition, polyclonal Ig MPGN usually appears after some years and presents a slow course (77). Instead, recurrence by monoclonal Ig occurs more often, earlier and is associated with a more aggressive course that often leads to graft failure (77). On the other hand, both DDD and G3GN are associated with a very high risk of recurrence. In addition, C3GN is associated with poor graft survival (78). Treatment of Ig deposition associated MPGN targets Ig production through Rituximab. Though the absence of CT some series has shown promising response rates (79, 80). As C3GN and DDD patients have presented alterations in alternative pathway of the complement cascade, there are several reports on the use of monoclonal antibodies that inhibit activation of the C5 component of complement (eculizumab) with distinct results (81, 82) but so far, no RCT has been done.
Diabetic nephropathy is not only the main cause of ESKD but is also associated with greater morbidity and mortality when it occurs in the kidney graft (83, 84). Post-transplant diabetic nephropathy (PTDN) shares pathophysiological and histological characteristics with primary diabetic nephropathy (85). However, the associated complications seem to develop at an accelerated rate (86). Older age, and obesity have shown to be the main risk factors for the development of PTDN (87). In addition, early low-grade proteinuria (<0.3 g/day) and hypertension (especially systolic blood pressure and elevated pulse pressure) have also been described as risk factors (88). Strategies with reduction or avoidance of steroids tend to decrease the incidence of PTDN (89). CNI are also associated with a high risk of developing PTDN. Among them, Cyclosporine seems to be less hyperglycemic according to the Diabetes Incidence after Renal Transplantation trial (DIRECT) (90). However, Tacrolimus continues to have a safer cardiovascular profile due superior lipid, blood pressure and kidney function effects (91).
The treatment of PTDN does not differ from native diabetic nephropathy. Screening for PTDM should be performed after starting treatment with glucocorticoids, sirolimus, or CNI (74). The choice of oral medication vs. insulin treatment must be done under the exact same rationale than in diabetic nephropathy. To mention, there's little evidence that basal insulin initiation to avoid hyperglycemic status immediately post transplantation can prevent further presentation of PTDN (92). In patients who develop PTDN with overt micro and macroalbuminuria, use of angiotensin inhibitors and statins are strongly recommended. This recommendation is an extrapolation from the effects observed in general population. However, in transplant population with proteinuria of any cause, treatment with angiotensin inhibitor has not demonstrated any benefit in long-term graft survival (93). Lately, iSGLT2 have shown remarkable results in decreasing cardiovascular risk and increasing survival both in general and in chronic kidney disease population (94–96). Given the enthusiasm that they have caused, it was predictable that some papers would appear defending that its use in kidney transplant recipients is safe and effective (97–100). Certainly, randomized trials will attempt this issue soon.
Beyond diabetic nephropathy, there are other risk factors of kidney disease progression but its impact in CKaD are not clearly defined. Briefly, hypertension is a common risk factor of renal disease progression, especially if it is associated with proteinuria. The use of RAAS blockade is one of the main strategies to reduce CKD progression. Hypothetically, the same mechanisms of damage occur on the kidney graft and may lead to CKaD but when it has been studied, results are not clear. While some retrospective cohorts associated the use of ACEI/ARB to better graft and patient survival (101, 102), prospective trials have not confirmed these association with graft survival (93, 103). The effect of obesity in graft outcomes is also controversial. Lafranca et al. (104) recently reviewed its effect, showing better outcomes for graft survival in patients with low BMI (<30). However, patient survival expressed in hazard ratios was in significant favor of high BMI recipients. Hyperlipidemia has also shown important outcomes related to patient survival but not related with CKAD. Though the anti-inflammatory effects of statins, associated with the inhibition of HMG CoA, have reported some good results in other solid organ transplantation (105) its use in kidney transplantation hasn't reported strong evidence in decreasing CKaD (106).
Infections are one of the most common complications after kidney transplantation due to immunosuppression (107). Besides of its impact in mortality, infections are a well-known risk factor for graft loss (108). Two different scenarios are especially important in relation to CKaD: first, the majority of infections are bacterial urinary tract infections (UTI). They occur more in the elderly due to immunosenescence, frailty, functional impairment and multiple comorbidities (109, 110). Female gender and obesity are also risk factors for the development of urinary infections. They usually occur within the first year after transplantation (111, 112). Despite screening of asymptomatic bacteriuria being common in Kidney Transplant Units (113), its treatment hasn't proved to reduce the incidence of acute pyelonephritis (114). Some series have demonstrated the association between the presence of UTI in the first year after transplant and poorer graft outcomes (108, 115). To explain this, we could extrapolate the effect of acute pyelonephritis on renal scarring and nephron mass loss observed in children (116). However, the association between UTI and loss of kidney functions has been observed also in patients with one or a few episodes of infection in which major renal mass loss is not expected (108). Another explanation is that infections and its clinical context could lead to an immunological imbalance triggering a rejection that would be the culprit for the dysfunction. Interestingly, in one of the few papers that has evaluated this issue, acute pyelonephritis was not independently associated with long term graft survival (117). Altogether, acute pyelonephritis is the most common complication in graft survival and has severe consequences on both patient's and graft's survival.
Second, viral infections play a relevant role in the development of CKaD. Cytomegalovirus (CMV) is the most infectious pathogen in KT recipients and it has been associated with both poor patient and graft survivals as it has been associated with cardiovascular mortality and an increased risk for acute rejection (118). Luckily, the better knowledge and prevention strategies have led to a drastic reduction of the prevalence of CMV infection from 40–100% to 0–37% (118). On the contrary, human polyomavirus has significantly increased its prevalence and it is associated with an important number of graft losses. BK virus nephropathy (BKVN) is an entity that occurs in up to 10% of renal transplant recipients and can result in graft loss in up to 50% of those affected (119). BK virus is a human polyomavirus of high prevalence and low morbidity with an estimated prevalence in adults of 80– 90% (120). After infection, BK virus may establish itself in a state of non-replicative asymptomatic infection in the renal epithelial and urothelial cells (121). In the host, BK virus can reactive itself in context of both immunosuppression and cellular injury (“two-hit hypothesis”). Three stages of the disease have been described: BKVN starts with viral cytopathic effects (stage A), then leads to an inflammation phase (stage B) and finally tubular atrophy and interstitial fibrosis (stage C) (122). The last stage would explain irreversible graft damage and then CKAD. The lack of strategies to prevent or treat BKVN explain the ominous prognosis of the entity with respect to graft survival. Since treatment options are limited and have poor results, strategies to prevent BKVN are crucial. In kidney transplant units, it is common to perform BK virus surveillance by monitoring BKV viremia post-transplant at different time points (123). This monitoring combined with stepwise drug adjustment is the strategy most commonly used and has become the gold standard. We must assume that lowering immunosuppression implies an increased risk of T-cell-mediated rejection (TCMR) and antibody-mediated rejection (ABMR) (124–126). Based on observations from DIRECT (92) and TRANSFORM (127) trials, treatment with ciclosporin and mTOR seemed to be better than Tacrolimus, although the combination of everolimus and tacrolimus was associated with lower incidence of BKVN than MMF with tacrolimus (128, 129). Some other interventions have been attempted with IVIG, cidofovir or Leflunomide among others. Some papers show 90% clearance of BK viremia and sustained graft function after 12 months with IVIG (130–132). Cidofovir has shown to stop polyomavirus replications in vitro, as well as good results on achieving BK clearance in a total of 11 cases in the literature (133–135). Conversion from MPA/MMF to Leflunomide has shown therapeutic response in some studies (136) but not in others (137).
After surgical removal of the organs for transplantation, kidneys are stored in a cold solution to preserve their viability. Cold ischemia time (CIT) is defined as the time that passes from surgical graft removal until the organ is warmed by recipient's blood supply after artery unclamping. CIT is a well-known risk factor for the development of delayed graft function (DGF) (138, 139) and acute rejection (AR) (140). This association implies both poorest graft and patient survival for those transplants with more CIT. Importantly, CIT itself does not seem to determine a decrease in long term graft survival (140–142).
Despite there are many definitions, DGF is the termed used in literature for those grafts that need at least one dialysis session in the first week after kidney transplantation. A longer CIT expanded criteria donors and terminal serum creatinine previous to donation are the main risk factors for DGF (142). The presence of DGF is detrimental for graft performance both in short and long-term. Interestingly, DGF is not only associated with a major rate of acute rejection episodes (143–145) but has also shown to be an independent factor for CKaD (143, 144).
Finally, IRI is a bimodal pathogenic way of tissue damage. Though cold storage and donor hypothermia try to minimize cell metabolism during CIT, some cells like renal tubular epithelial cells remain active in a state of hypoxia. In response to hypoxia, their mitochondria increase the production of reactive oxygen species and tend to develop intracellular acidosis. Thus, prolonged CIT will relentlessly lead to cell death and acute tubular necrosis: the ischemia damage. After reperfusion, the microvascular injury caused by ischemia enhances fluid filtration, with leukocyte plugging in capillaries and damaged endothelial cells secreting factors to favor inflammatory mediators and proteolytic enzymes (146). The global outcome of this ischemia-reperfusion damage is a harmful environment that through DAMPS and PAMPS enhances both innate and cellular immunity (147).
Thus, CIT is associated with a decrease in long term graft survival, but its impact is explained for the risk of developing an AR and DGF. Meanwhile, DGF is independently associated with both decreased long term graft survival and increased AR risk. IRI is a pathogenic phenomenon intimately related long CIT and explains DGF, chronic lesions and rejection risk (148).
Currently, no pharmacological intervention has proved to mitigate DGF neither in terms of duration nor its consequences. In clinical practice some strategies are used based on theoretical rationales without evidence. CNI delayed introduction to avoid toxicity and complement activation is one of these strategies although current evidence does not show significant differences (149). Combined to CNI delayed introduction, the use of Anti-Thymocyte Globulins (ATG) has been used to prevent AR and mitigate damage due to IRI. Even though ATG induction seems to have a better security profile in immunological high-risk patients, it is not clear that this grants any advantages in low-risk recipients compared with Basiliximab, not even in terms of reducing DGF (150). The results of PREDICT-DGF trial (NCT02056938, EudraCT #2014-000332-42) will provide more light about if ATG reduce DGF in comparison with Basiliximab in a selected group of patients at a high risk of developing DGF (151). The role of complement on IRI damage has also been tested as a therapeutic target. The use of C1 esterase inhibitor (C1INH) has shown good results in phase I/II studies, especially in those patients with more risk of DGF (152). Novel therapeutic options are also been tested like siRNA, mesenchymal stem cells or thrombin-targeted per-fluorocarbon nanoparticles (PFC-NP) (153, 154). However, all these strategies are far from being used in clinical practice.
Instead, the only strategy that has been proven effective to reduce CIT related DGF is the use of machine perfusion technology during kidney storage. Specifically, Tingle SJ et al. (155) have recently published a meta-analysis on this topic, showing that in deceased donors storage in hypotermic machine perfusion the incidence of DGF was reduced by 23% in comparison with static cold storage solution.
As reviewed, lots of efforts are currently being put into understanding the whole spectrum of kidney graft damages better. On each field, small strides are being made but with a global vision, CKaD is still worrying. A common limit for all therapeutic strategies is the presence of chronic lesions. These lesions, histologically characterized by fibrosis and extracellular matrix (ECM) deposition, are considered scars which can no longer be repaired. Yet, from another point of view, scars are the result of a wound healing mechanism designed to repair or regenerate kidney damage.
Schematically, when damage of any etiology happens there is a loss of mass of functioning cells and consequently an attrition in organ function. Therefore, the healing process has to (1) stop the damage, (2) refill the gaps left by cell loss, and (3) compensate the loss of organ function (Figure 1). Probably, the magnitude and duration of the damage, the age of the organism and the dialogue among the effectors of kidney regeneration will determine the balance between healing and regeneration processes. These are the main regeneration mechanisms for kidney regeneration (Table 3).
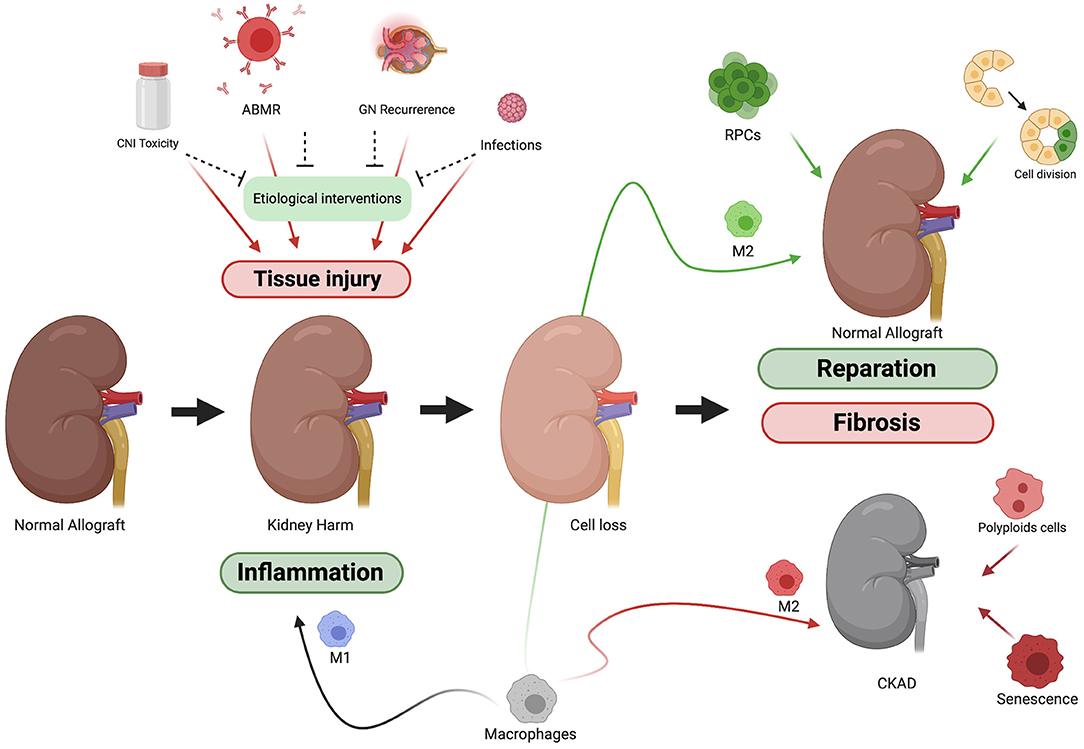
Figure 1. Schematic representation of kidney allograft injury and reparation process. Kidney allograft is exposed to many insults. Although there are mechanisms to protect the graft from each of them, tissue damage is often present at the end. Once the tissue is injured the kidney's owns mechanisms of reparation come into action. To stop the insult, M1 macrophages among others will trigger and inflammatory response. After that, the damage caused (cell loss) must be compensated. Factors like the type and duration of the damage or the age of the donor will determine whether the reparation process tends more toward reparation or scarring (fibrosis).
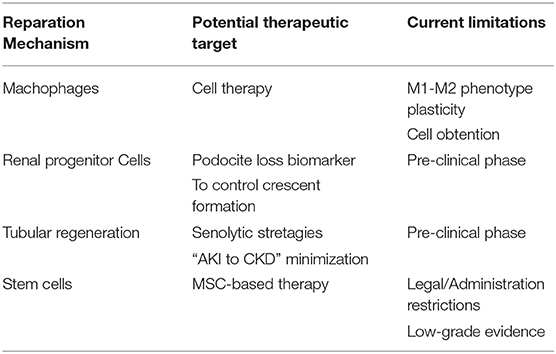
Table 3. Kidney regeneration mechanisms.
One of the clearest examples of why the reparations mechanisms are doubled-edged swords is the study of macrophages in CKaD. There is a considerable body of evidence that both circulating and, mainly resident kidney macrophages play a crucial role in kidney inflammation and healing. These hematopoietic cells derived from the yolk sac seem to remain in the kidney during embryogenesis forming niches and they are activated when the harm occurs (156).
After injury, the presence of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) is recognized via Toll-like receptors (TLR) or patterns recognition-receptors (PRR) and activate a subpopulation of macrophages (M1 or activated macrophage). These activated M1 macrophages are proinflammatory cells capable of secreting pro-inflammatory cytokines (IL-6, IL-1, and TNF-α), superoxide anions and oxygen and nitrogen radicals (157). The amplification of the inflammation cascade contributes to fight against the cause of the injury but as a side effect it does also kill host cells increasing tissue damage. These cells will also contribute to kidney fibrosis through secretion of MMP-9, which increases tubular cell ECM transition via the β-catenin pathway (158). In animal models, the depletion of M1 macrophages ameliorated kidney injury (159). These results were also seen in rat models of acute rejection (160).
In addition, there are other populations of macrophages with distinct function in the reparation process. The alternatively activated macrophages (AAM or M2) usually appear later and have an anti-inflammatory function. Up to three types of M2 macrophages can be differentiated: M2a, stimulated by IL-4 and/or IL-3, are capable of secreting ECM components and therefore they also participate in wound healing and tissue remodeling. M2b have an immunoregulatory profile, inducing IL-10 secretion, upregulating antigen presentation through MHC II and downregulating IL-12, IL-6 and TNF. Finally, M2c macrophages are induced by IL-10, TGF-ß and glucocorticoids and produce anti-inflammatory cytokines (161). In a model with depletion of M2 macrophages, a reduction in tubular cell proliferation and repair is observed (159). Altogether, the macrophage system seems to contribute to kidney regeneration in two differentiated phases. First, immediately after kidney injury M1 population contributes to fight the cause of the aggression through inflammation. Second, once the aggression is neutralized, M1 and M2a contribute to restore the damaged tissue by the production of ECM while M2b and M2c populations reduce inflammation to restore kidney homeostasis.
However, an imbalance in this system can contribute to aggravate kidney damage both in animals and humans (159). For example, macrophage depletion is a useful tool to reduce kidney damage in in vitro models, yet a selective depletion to manipulate the M1/M2 ratio has different effects. Moreover, the activation of M2 macrophages that usually results in a reduction of the damage can be the main effector of kidney fibrosis when chronic or constitutive damage occurs (162). Interestingly, Wang et al. demonstrated that after kidney rejection, fibrosis is associated with a constitutive activation of macrophages (mostly M2) especially in chronic active forms. In addition, they identified as etiologic factors of fibrosis not only the production of ECM components but also a macrophage to myofibroblasts transition (163).
Cell therapy using the macrophage system has been attempted in animal models to control kidney fibrosis. The phenotype plasticity of this cell type is one of the main limitations. Cao et al. (164) failed to protect kidney function after infusion of bone narrow derived M2 macrophages, mainly due to phenotypic changes of the infused cells. Spleen macrophages, on the contrary, seem to be more stable and have demonstrated a beneficial effect in model of Adriamycin induced nephropathy.
In 2006, a population of progenitor cells surrounding the Bowman's capsule was identified. These cells, which are characterized by the co-expression of CD133 and CD24 markers, display a multipotent capacity of evolving into kidney specific cells terminally differentiated (podocytes and tubular cells) (165). Through differentiation, these RPCs exhibit the capacity to ameliorate acute kidney damage (166). At the same time, under certain conditions, as a sustained injury, these cells could also contribute to crescent formation (167) or glomerulosclerosis (16, 168). Sicking et al. (169) observed how after podocyte damage caused by doxycycline, RPCs from Bowman's capsule tended to leave their position to replace podocytes. After, the remaining RPCs formed cellular extensions to cover the denuded Bowman's capsule surface (expressing de novo CD44). Throughout the observation period, the induced proliferation of RPCs persisted, resulting in the formation of typical cellular crescents with periglomerular infiltrate. Thus, it can be hypothesized that RPCs act as a physiological renal regenerative mechanism that when overcome tend to scar in order to prevent further damage. Importantly, urinary detection of RPCs is feasible and has been already used to perform functional and genotypic studies without the need of invasive procedures (170, 171).
Recently, Manonelles et al. (172) published the first and only experience of RPCs isolation in kidney transplant recipients. In this study, a cohort of stable kidney transplant recipients with 6 months protocol biopsy was divided into two groups depending on the presence or absence of urinary RPC. A total cohort of 66 patients were then followed for 2 years. Interestingly, at the beginning of the study both groups were identical considering clinical variables, alloimmune response, renal function, albuminuria and graft pathology. However, uRPC+ group showed increased podocyturia and a higher rate of proliferating RPCs along the Bowman's capsule, suggesting that RPCs were proliferating to compensate a podocyte loss. Consequently, 2 years follow up evidenced poorer outcomes in the uRPC+ group with worse renal function, increased albuminuria, wider mesangial expansion and more severe interstitial fibrosis. If these results are confirmed, the detection of urinary RPCs could act as a marker of current injury much before clinical, immunological or histological damage is detected. Whether a therapeutic intervention at this time could prevent function graft attrition must be proved in further studies.
Tubular epithelium is the kidney structure that most commonly suffers damage due to its high metabolic activity and decreased blood supply. Tubular regeneration model has great efficiency and is able to repair after an injury with no or little consequences. One of the important characteristics in this model is that tubular epithelial cells (TECs) are simpler than other kidney cells like podocytes. This feature allows TECs to easily dedifferentiate after an injury in order to do mitosis and repopulate the epithelia (173). This model for regeneration through dedifferentiation has been classically proposed for tubular regeneration but some objections have been done recently.
Lazzeri et al. suggested that the regeneration capacity of the tubule through dedifferentiation had largely been overestimated. Instead, they proposed a model of regeneration highly preserved in other human organs (174). This model is based on two main effectors: first, niches of progenitor cells can be found near the remaining tissue in a non-differentiated state. Second, after injury the remaining differentiated cells increase its content in DNA without undergoing mitosis through a specific cell cycle called endocycle. Thus, these cells can improve their performance reducing the loss of function of the organ while complete regeneration occurs. The same group has already demonstrated the presence of niches of cells that express progenitor markers next to the tubule and also the existence of endocycling cells, in both animals and humans (175).
In this model, the injury would cause a cell loss that implies decrease of organ of function of the organ. After that, progenitor cells would be mobilized from their niches and would finalize their differentiation process to repopulate the epithelia. In the meantime, to preserve organ function (or even survival) the remaining tubular cells would enter endocycle in order to duplicate its DNA content and keep the function of the organ. These cells though, will not be able to perform a normal cell cycle ever again; so, they are doomed to become a hyperfunctioning cell with a hypersecretory state of profibrotic mediators driving fibrogenesis, that is, cell senescence (176). These aberrant profibrotic cells would be responsible for fibrosis after AKI explaining, at least partially, the AKI-to-CKD transition. The balance between progenitors' repopulation and endocycling cells would then be crucial in a complete regeneration of the organ without later damage. Consistently, it has been observed that, the amount of progenitor cells in mammals decrease with aging and endocycling cells take a more important role in reparation processes, which would lead to greater fibrosis (177).
To involve the kidney's own repair processes as a therapeutic tool is a strategy that has been already tested with promising results through the infusion of stem-cells. In general, based on their therapeutic potential, mesenchymal stem cells (MSCs) from bone marrow or other tissues are considered one the most powerful tools for treating several human diseases. MSC action is based not only on the capacity to differentiate into terminal renal cells but they have also been associated with the release of pro-mitotic, anti-apoptotic, anti-inflammatory and immunomodulatory soluble factors as well as to the mitigation of metabolomic and oxidative stress imbalance (178). A number of clinical trials have been designed to evaluate the safety and efficacy of MSC-based therapy and some good results have been observed in acute kidney injury trials (179). Very recently, the TRITON study (180) has used the infusion of autologous MSCs in 29 kidney transplant recipients to withdraw CNI. After 24 weeks of follow up, no differences were observed in graft function, acute rejection, graft loss, major adverse events or in kidney fibrosis. In a post-hoc analysis of this study, a longer follow up (5-year) was performed observing a more preserved renal function in the MSC group. Although there are already many limitations and restrictions to cell therapy, this study shows the feasibility of these treatments and which could be a cornerstone in future kidney transplantation therapeutic regimens.
Long-term graft survival is a major concern in the transplant community due to its clinical impact. Until now, lots of efforts have been put into identifying and precisely mitigate the impact of every potential graft damage. Consequently, advances in the treatment of ABMR are expected to report greater outcomes than BK virus or pyelonephritis prevention. However, from a pragmatic point of view, all the aforementioned harmful situations will be present in every kidney graft contributing to the final outcome: Chronic Kidney Allograft Disease. CKaD needs to be addressed by a holistic strategy. A therapeutic approach that considers to abrogate the mechanisms of graft injury and to improve the intrinsic mechanisms of kidney repair could have a transversal impact and lead to a significant improvement in CKaD. Further studies are needed to address this issue in the coming years.
SC reviewed the literature and prepared the manuscript. AM, MT, and AS reviewed the manuscript. JC supervised and reviewed the manuscript. All authors approve this manuscript for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank CERCA Program/Generalitat de Catalunya and the ISCIII RETICS RedinRen RD16/0009/0003 for institutional support. We also want to thank the Fundació Catalunya la Pedrera that through the program Talents has granted Sergi Codina. We thank Pau Cruzado-Boix for his assistance with the English language revision.
1. Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, et al. Chronic kidney disease: global dimension and perspectives. Lancet. (2013). 382:260–72. doi: 10.1016/S0140-6736(13)60687-X
2. Wolfe RA, Ashby VB, Milford EL, Ojo AO, Ettenger RE, Agodoa LY, et al. Comparison of mortality in all patients on dialysis, patiens on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med. (1999) 341:1725–30 doi: 10.1056/NEJM199912023412303
3. Kramer A, Boenink R, Maarlies N, Bosdriesz JR, Stel VS, Beltran P, et al. The ERA-EDTA Registry Annual Report 2017: A Summary. (2020). CKJ 1-17.
4. Lamb KE, Lodhi S, Meier-Kriesche HU. Long-term renal allograft survival in the United States: A critical reappraisal. Am J Transplant. (2011) 11:450–62. doi: 10.1111/j.1600-6143.2010.03283.x
5. Wekerle T, Segev D, Lechler R, Oberbauer R. Strategies for long-term preservation of kidney graft function. Lancet. (2017) 389:2152–62. doi: 10.1016/S0140-6736(17)31283-7
6. Vanhove T, Goldschmeding R, Kuypers D. Kidney fibrosis: origins and interventions. Trasnplantation. (2017) 101:713–26. doi: 10.1097/TP.0000000000001608
7. Böhming GA, Eskandrary F, Doberer K, Halloran PF. The therapeutic challenge of late antibody-mediated kidney allograft rejection. Trasnpl Int. (2019) 8:775–88. doi: 10.1111/tri.13436
8. Galichon P, Xu-Dubois YC, Finianos S, Hertig A, Rondeau E. Clinical and histological predictors of long-term kidney graft survival. Nephrol. Dial Transplant. (2013) 6:1362–70. doi: 10.1093/ndt/gfs606
9. Novick AC, Gephardt G, Guz B, Steinmuller D, Tubbs RR. Long-term follow-up after partial removal of a solitary kidney. N Engl J Med. (1991) 325:1058–62. doi: 10.1056/NEJM199110103251502
10. Kuypers DR, Chapman JR, O'Connel PJ, Allen RD, Nankivell BJ. Predictors of renal transplant histology at three months. Transplantation. (1999) 67:1222–30. doi: 10.1097/00007890-199905150-00005
11. Nankinvell BJ, Borrows RJ, Fung C, O'Connell PJ, Allen RD, Chapman JR. The natural history of chronic allograft nephropathy. N Engl J Med. (2003) 349:2326–33. doi: 10.1056/NEJMoa020009
12. Stegall MD, Park WD, Larson TS, Gloor JM, Cornell LD, Sethi S, et al. The histology of solitary renal allografts at 1 and 5 years after transplantation. Am J Transplant. (2011) 11:698–707. doi: 10.1111/j.1600-6143.2010.03312.x
13. Cosio FG, Grande JP, Larson TS, Gloor JM, Velosa JA, Textor SC, et al. Kidney allograft fibrosis and atrophy early after living donor transplantation. Am J Transplant. (2005) 5:1130–6. doi: 10.1111/j.1600-6143.2005.00811.x
14. Servais A, Meas-Yedid V, Noël LH, Martinez F, Panterne C, Kreis H, et al. Interstitial fibrosis evolution on early sequential screening renal allograft biopsies using quantitative image analysis. Am J Transplant. (2011) 11:1456–63. doi: 10.1111/j.1600-6143.2011.03594.x
15. Schwarz C, Lawitschka A, Bohmig GA, Dauber EM, Greinix H, Kozakowski N, et al. Kidney transplantation with corticosteroids alone after haploidentical HSCT from the same donor. Transplantation. (2016) 100:2219–21. doi: 10.1097/TP.0000000000001213
16. Smeets B, Kuppe C, Sicking EM, Fuss A, Jirak P, van Kuppevelt TH, et al. Parietal epithelial cells participate in the formation of sclerotic lesions in focal segmental glomerulosclerosis. (2011) J Am Soc Nephrol. 22, 1262–74. doi: 10.1681/ASN.2010090970
17. Lefaucheur C, Loupy A. Antibody-mediated rejection of solid-organ allografts. N Engl J Med. (2018) 379:1150–60. doi: 10.1056/NEJMra1802677
18. Sellares J, de Freitas DG, Mengel M, Reeve J, Einecke G, Sis B, et al. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant. (2012) 12:388–99. doi: 10.1111/j.1600-6143.2011.03840.x
19. Kaplan B, Meier-Kriesche HU. Death after graft loss: an important late study endpoint in kidney transplantation. Am J Transplant. (2002) 2:970–4. doi: 10.1034/j.1600-6143.2002.21015.x
20. Dragun D, Müller DN, Bräsen JH, Fristsche L, Nieminen-Kelhä M, Dechend R, et al. Angiotensin II type 1–receptor activating antibodies in renal-allograft rejection. N Engl J Med. (2005) 352:558–6. doi: 10.1056/NEJMoa035717
21. Wiebe C, Gibson IW, Blydt-Hansen TD, Pochinco D, Birk PE, Ho J, et al. Rates and determinants of progression to graft failure in kidney allograft recipients with de novo donor-specific antibody. Am J Transplant. (2015) 15:2921–30. doi: 10.1111/ajt.13347
22. Cosio FG, Gloor JM, Sethi S, Stegall MD. Transplant glomerulopathy. Am J Transplant. (2008) 8:492–6. doi: 10.1111/j.1600-6143.2007.02104.x
23. Stites E, Le Quintrec M, Thurman JM. The complement system and antibody-mediated transplant rejection. J Immunol. (2015) 195:5525–3. doi: 10.4049/jimmunol.1501686
24. Haas M, Loupy A, Lefaucheur C, Roufosse C, Glotz D, Seron D. The Banff 2017 kidney meeting report: Revised diagnostic criteria for chronic active T cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am J Trasnplant. (2018) 18:293–307. doi: 10.1111/ajt.14625
25. Loupy A, Lefaucheur C, Vernerey D, Vernerey D, Prugger C, van Huyen JPD, et al. Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med. (2013) 369:1215–26. doi: 10.1056/NEJMoa1302506
26. Kikic Z, Kainz A, Kozakowski N, Oberbauer R, Rgele H, Bond G, et al. Capillary C4d and kidney allograft outcome in relation to morphologic lesions suggestive of antibody-mediated rejection. Clin J Am Soc Nephrol. (2015) 10:1435–43. doi: 10.2215/CJN.09901014
27. Montgomery RA, Zachary AA, Racusen LC, Leffell MS, King KE, Burdick J, et al. Plasmapheresis and intravenous immune globulin provides effective rescue therapy for refractory humoral rejection and allows kidneys to be successfully transplanted into cross-match-positive recipients. Transplantation. (2000) 70:887–95. doi: 10.1097/00007890-200009270-00006
28. Rocha PN, Butterly DW, Greenberg A, Reddan DN, Tuttle-Newhall J, Collins BH, et al. Beneficial effect of plasmapheresis and intravenous immunoglobulin on renal allograft survival of patients with acute humoral rejection. Transplantation. (2003) 75:1490–5. doi: 10.1097/01.TP.0000060252.57111.AC
29. Moreso F, Crespo M, Ruiz JC, Torres A, Gutierrez-Dalmau A, Osuna A, et al. Treatment of chronic antibody mediated rejection with intravenous immuno- globulins and rituximab: a multicenter, prospective, randomized, double-blind clinical trial. Am J Transplant. (2018) 18:927–35. doi: 10.1111/ajt.14520
30. Sautenet B, Blancho G, Büchler M, Morelon E, Toupance O, Barrou B, et al. One-year results of the effects of rituximab on acute antibody-mediated rejection in renal transplantation: RITUX ERAH, a multicenter double-blind randomized placebo-controlled trial. Transplantation. (2016) 100:391–9. doi: 10.1097/TP.0000000000000958
31. Eskandary F, Regele H, Baumann L, Bond G, Kozakowski N, Wahrmann M, et al. A randomized trial of Bortezomib in Late Antibody-Mediated Kidney Transplant Rejection. J Am Soc Nephrol. (2018) 29:591–605. doi: 10.1681/ASN.2017070818
32. Stegall MD, Diwan T, Raghavaiah S, Cornell LD, Burns J, Dean PG, et al. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am J Transplant. (2011) 11:2405–13. doi: 10.1111/j.1600-6143.2011.03757.x
33. Cornell LD, Schinstock CA, Gandhi MJ, Kremers WK, Stegall MD. Positive crossmatch kidney transplant recipients treated with eculizumab: outcomes be- yond 1 year. (2015) Am J Transplant.15:1293–302. doi: 10.1111/ajt.13168
34. Marks WH, Mamode N, Montgomery RA, Stegall MD, Ratner LE, Cornell LD, et al. Safety and efficacy of eculizumab in the prevention of antibody-mediated rejection in living-donor kidney transplant recipient requiring desensitization therapy; A randomized controlled trial. Am J Transplant. (2019) 19:2876–88. doi: 10.1111/ajt.15364
35. Montgomery RA, Orandi BJ, Racusen L, Jackson AM, Garonzik-Wang JM, Shah T, et al. Plasma-Derived C1 esterase inhibitor for acute antibody-mediated rejection following kidney transplantation: results of a randomized double-blind placebo-controlled pilot study. Am J Transplant. (2016) 16:3468–3478. doi: 10.1111/ajt.13871
36. Choi J, Aubert O, Vo A, Loupy A, Haas M, Puliyanda D. Assesment of Tocilizumab (Anti-Interleukin-6 Receptor Monoclonal) as a potential treatmentfor chronic antibody-mediated rejection and transplant glomerulopathy in HLA-sensitized renal allograft recipients. Am J Transplant. (2017) 17:2381–9. doi: 10.1111/ajt.14228
37. Opelz G, Dohler B. Effect of human leukocyte antigen compatibility on kidney graft survival: comparative analysis of two decades. Transplantation. (2007) 84:137–43. doi: 10.1097/01.tp.0000269725.74189.b9
38. Kawase T, Morishima Y, Matsuo K, Kashiwase K, Inoko H, Saji H, et al. High-risk HLA allele mismatch combinations responsible for severe acute graft-versus-host disease and implication for its molecular mechanism. Blood. (2007). 110:2235–41. doi: 10.1182/blood-2007-02-072405
39. Kawase T, Matsuo K, Kashiwase K, Inoko H, Saji H, Ogawa S, et al. HLA mismatch combinations associated with decreased risk of relapse: implications for the molecular mechanism. Blood. (2009) 113:2851–8. doi: 10.1182/blood-2008-08-171934
40. Geneugelijk K, Thus KA, Spierings E. Predicting alloreactivity in transplantation. J Immunol Res. (2014) 2014:159479. doi: 10.1155/2014/159479
41. Senev A, Coemans M, Lerut E, van Sandt V, Kerkhofs J, Daniëls L. Eplet Mismatch load and de novo occurrence of donor-specific anti-HLA antibodies, rejection and graft failure after kidney transplantation: An observational cohort study. J Am Soc Nephrol. (2020) 31:2193–204. doi: 10.1681/ASN.2020010019
42. Gaston RS. Our evolving understanding of late kidney allograft failure. Curr Opin Organ Transplant. (2011) 16:594–9. doi: 10.1097/MOT.0b013e32834c23a7
43. Myers BD, Ross J, Newton L, Luetscher J, Perlroth M. Cyclosporine-associated chronic nephropathy. N Engl J Med. (1984) 311:699–705. doi: 10.1056/NEJM198409133111103
44. Vincenti F, Jensik SC, Filo RS, Miller J, Pirsch J. A long-term comparison of tacrolimus (FK506) and cyclosporine in kidney transplantation: evidence for improved allograft survival at five years. Transplantation. (2002) 73:775–82. doi: 10.1097/00007890-200203150-00021
45. Nankivell BJ, Chapman JR, Bonovas G, Gruenewals S. Oral cyclosporine but not tacrolimus reduces renal transplant blood flow. Transplantation. (2004) 77:1457–9. doi: 10.1097/01.TP.0000121196.71904.E0
46. Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. J Am Soc Nephrol. (2009) 4:481–508. doi: 10.2215/CJN.04800908
47. Ekberg H, Tedesco-Silva H, Demirbas A, Vítko S, Nasham B, Gürkan A, et al. Reduced exposure to calcineurin inhbitors in renal transplantation. N Engl. J Med. (2007) 357:2562–75. doi: 10.1056/NEJMoa067411
48. Ekberg H, Grinyó J, Nashan B, Vanrenterghem Y, Vicenti F, Voulgari A, et al. Cyclosporine sparing with mycophenolate mofetil, Daclizumab and corticosteroids in renal allograft recipients: The CAESAR Study. Am J Transplant. (2007) 7:560–70. doi: 10.1111/j.1600-6143.2006.01645.x
49. Kershner RP, Fitzsimmons WE. Relationship of FK506 whole blood concentrations and efficacy and toxicity after liver and kidney transplantation. Transplantation. (1996) 62:920–6. doi: 10.1097/00007890-199610150-00009
50. Vincenti F, Laskow DA, Neylan JF, Mendez R, Matas AJ. One-year follow-up of an open-label trial of FK506 for primary kidney transplantation. A report of the U.S. Multicenter FK506 kidney transplant group. Transplantation. (1996) 61:1576–81. doi: 10.1097/00007890-199606150-00005
51. Girerd S, Schikowski J, Girerd N, Duarte K, Busby H, Gambier N, et al. Impact of reduced exposure to calcineurin inhibitors on the development of de novo DSA: a cohort of non-immunized first kidney graft recipients between 2007 and 2014. BMC Nephrology. (2018) 19:232. doi: 10.1186/s12882-018-1014-2
52. Anglicheau D, Pallet N, Rabant M, Marquet P, Cassinat B, Meria P, et al. Role of P-glycoprotein in cyclosporine cytotoxicity in the cyclosporine-sirolimus interaction. Kidney Int. (2006) 70:1019–25. doi: 10.1038/sj.ki.5001649
53. Jacobson PA, Schladt D, Israni A, Oetting WS, Lin YC. Genetic and clinical determinants of early, acute calcineurin inhibitor-realated nephrotoxicity: results from a kidney transplant consortium. Transplantation. (2012) 93:624–31. doi: 10.1097/TP.0b013e3182461288
54. Schwarz A, Haller H, Schmitt R, Schiffer M, Koenecke C, Strassburg C, et al. Biopsy-diagnosed renal disease in patients after transplantation of other organs and tissues. Am J Transplant. (2010) 10:2017–25. doi: 10.1111/j.1600-6143.2010.03224.x
55. Briganti EM, Russ GR, McNeil JJ, Atkins RC, Chadban SJ. Risk of renal allograft loss from recurrent glomerulonephritis. N Engl J Med. (2002) 347:103–9. doi: 10.1056/NEJMoa013036
56. Cosio FG, El Ters M, Cornell LD, Schinstock CA, Stegall MD. Changing kidney allograft histology early posttransplant: prognostic implications of 1-year protocol biopsies. Am J Transplant. (2016). 16:194–203. doi: 10.1111/ajt.13423
57. Rodriguez EF, Cosio FG, Nasr SH, Sethi S, Fidler ME, Stegall MD, et al. The pathology and clinical features of early recurrent membranous glomerulonephritis. Am J Transplant. (2012) 12:1029–38. doi: 10.1111/j.1600-6143.2011.03903.x
58. Cosio FG, Cattran DC. Recent advances in our understanding of recurrent primary glomerulonephritis after kidney transplantation. Kidney Int. (2017) 91:304–14. doi: 10.1016/j.kint.2016.08.030
59. Bertelli R, Ginevri F, Caridi G, Dagnino M, Sandrini S, Di Duca M, et al. Recurrence of focal segmental glomerulosclerosis after renal transplantation in patients with mutations of podocin. Am J Kidney Dis. (2003) 41:1314–21. doi: 10.1016/S0272-6386(03)00364-0
60. Uffing A, Perez-Saez MJ, Mazzali M, Manfro RC, Bauer AC, Drumond FS, et al. Recurrence of FSGS after kidney transplantation in adults. Clin J Am Soc Nephrol. (2020). 15:247–56. doi: 10.2215/CJN.08970719
61. Shuai T, Pei Jing Y, Huang Q, Xiong H, Liu J, Zhu L, et al. Serum soluble urokinase type plasminogen activated receptor and focal segmental glomerulosclerosis: a systematic review and meta-analysis. BMJ Open. (2019) 9:e031812. doi: 10.1136/bmjopen-2019-031812
62. Uffing A, Perez-Saez MJ, La Manna G, Comai G, Fischman C, Farouk S, et al. A large, international study on post-transplant glomerular diseases: the TANGO project. BMC Nephrology. (2018) 19:229. doi: 10.1186/s12882-018-1025-z
63. Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, et al. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med. (2011) 3:85ra46. doi: 10.1126/scitranslmed.3002231
64. Canaud G, Zuber J, Sberro R, Royale V, Anglicheau D, Snanoudj R, et al. Intensive and prolonged treatment of focal and segmental glomerulosclerosis recurrence in adult kidney transplant recipients: a pilot study. Am J Transplant. (2009) 9:1081–6. doi: 10.1111/j.1600-6143.2009.02580.x
65. Quintana LF, Blasco M, Seras M, Perez NS, Lopez-Hoyos M, Villaroel P, et al. Antiphospholipase A2 receptor antibody levels predict the risk of posttransplantation recurrence of membranous nephropathy. Transplantation. (2015) 99:1709–14. doi: 10.1097/TP.0000000000000630
66. Grupper A, Cornell LD, Fervenza FC, Beck LH, Lorenz E, Cosio FG. Recurrent Membranous nephropathy after kidney transplantation: treatment and long-term implications. Transplantation. (2016) 100:2710–6. doi: 10.1097/TP.0000000000001056
67. Berchtold L, Letouzé E, Alexander MP, Canaud G, Logt AEvd, Hamilton P, et al. HLA-D and PLA2R1 risk alleles associate with recurrent primary membranous nephropathy in kidney transplant recipients. Kidney Int. (2020) 99:671–85. doi: 10.1016/j.kint.2020.08.007
68. Sprangers B, Lefkowitz GI, Cohen SD, Stoked MB, Valeri A, Appel GB, et al. Beneficial effect of rituximab in the treatment of recurrent idiopathic membranous. Clin J A Soc Nephrol. (2010) 5:790–7. doi: 10.2215/CJN.04120609
69. Moroni G, Longhi S, Quaglini S, Gallelli B, Banfi G, Montagnino G, et al. The long-term outcom of renal transplantation of IgA nephropathy and the impact of recurrence on graft survival. Nephrol Dial Transplant. (2013) 28:1305–14. doi: 10.1093/ndt/gfs472
70. Ortiz F, Gelpi R, Koskinen P, Manonelles A, Räisänen-Sokolowski A, Carrera M. IgA nephropathy recurs early in the graft when assessed by protocol biopsy. Nephrol Dial Transplant. (2012) 27:2553–8. doi: 10.1093/ndt/gfr664
71. Suzuki K, Honda K, Tanabe K, Toma H, Nihei H, Yamaguchi Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int. (2003) 63:2286–94. doi: 10.1046/j.1523-1755.63.6s.2.x
72. Soler MJ, Mir M, Rodriguez E, Orfila A, Munne A, Vazquez S, et al. Recurrence of IgA nephropathy and Henoch-Schönlein purpura after kidney transplantation: risk factors and graft survival. Transplant Proc. (2005) 37:3705–9. doi: 10.1016/j.transproceed.2005.09.172
73. Park S, Go H, Baek CH, Kim YH, Kim YC, Yang SH, et al. Clinical importance of the updated Oxford classification in allograft IgA nephropathy. Am J Transplant. (2019) 19:2855–64. doi: 10.1111/ajt.15400
74. Kidney Disease. Improving Global Outcomes (KDIGO) Transplant Work Group. KDIGO clinical practice guideline for the care of kidney transplant recipients. Am J Transplant. (2009). 9:S1–155. doi: 10.1111/j.1600-6143.2009.02834.x
75. Clayton P, McDonald S, Chadban S. Steroid and recurrent IgA nephropathy after kidney transplantation. Am J Transplant. (2011) 11:1645–9. doi: 10.1111/j.1600-6143.2011.03667.x
76. Liu LL, Wang L, Jiang Y, Yao L, Dong L, Li X. Tonsillectomy for IgA Nephropathy: A Meta-analysis. Am J Kidney Dis. (2015) 65:80–87. doi: 10.1053/j.ajkd.2014.06.036
77. Lorenz EC, Sethi S, Leung N, Dispenzieri A, Fervenza FC, Cosio FG. Recurrent membranoproliferative glomerulonephritis after kidney transplantation. Kidney Int. (2010) 77:721–8. doi: 10.1038/ki.2010.1
78. Braun MC, Stablein DM, Hamiwka LA, Bell L, Bartosh MS, Striffe CF, et al. Recurrence of membranoproliferative glomerulonephritis type II in renal allografts: the North American Pediatric Renal Transplant Cooperative Study experience. J Am Soc Nephrol. (2005) 16:2225–33. doi: 10.1681/ASN.2005020175
79. Guiard E, Karras A, Plaisier E, van Huyen JPD, Fakhouri F, Rougier JP, et al. Patterns of noncryoglobulinemic glomerulonephritis with monoclonal Ig deposits: correlation with IgG subclass and response to rituximab. Clin J Am Soc Nephrol. (2011) 6:1609–16. doi: 10.2215/CJN.10611110
80. Buxeda A, Said SM, Nasr SH, Leung N, El Ters M, Cosio FG. Recurrent proliferative glomerulonephritis with monoclonal immunoglobulin deposits in kidney allografts treated with anti-CD20 antibodies. Transplantation. (2019) 103:1477–85. doi: 10.1097/TP.0000000000002577
81. Zuber J, Fakhouri F, Roumenina LT, Loirat C, Frémeauz-Bacchi V. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. (2012) 8:643–57. doi: 10.1038/nrneph.2012.214
82. Le Quintrec M, Lionet A, Kandel C, Bourdon F, Gnemmi V, Colombat M, et al. Eculizumab for treatment of rapidily progressive C3 glomerulopathy. Am J Kidney Dis. (2015) 65:484–9. doi: 10.1053/j.ajkd.2014.09.025
83. Cooper L, Oz N, Fishman G, Shohat T, Rahaminov R, Mor E, et al. New onset diabetes after kidney transplantation is associated with increased mortality. A retrospective cohort study. Diabetes Metab Res Rev. (2017) 33. doi: 10.1002/dmrr.2920
84. Sharif A, Baboolal K. Complications associated with new-onset diabetes after kidney transplantation. Nat Rev Nephrol. (2011) 8:34–42. doi: 10.1038/nrneph.2011.174
85. Bhalla V, Nast CC, Stollenwerk N, Tran S, Barba L, Kamil ES, et al. Recurrent and de novo diabetic nephropathy in renal allografts. Transplantation. (2003) 75:66–71. doi: 10.1097/00007890-200301150-00012
86. Burroughs TE, Swindle J, Takemoto S, Lentine KL, Machnicki G, Irish WD, et al. Diabetic complications associated with new-onset diabetes mellitus in renal transplant recipients. Transplantation. (2007) 83:1027–34. doi: 10.1097/01.tp.0000259617.21741.95
87. Roland M, Gataut P, Doute C, Büchler M, Al-Najjar A, Barbet C, et al. Immunosuppressive medications, clinical and metabolic parameters in new-onset diabetes mellitus after kidney transplantation. Transpl Int. (2008) 21:523–30. doi: 10.1111/j.1432-2277.2008.00640.x
88. Roland M, Gataut P, Doute C, Büchler M, Al-Najjar A, Barbet C, et al. Early pulse pressure and low-grade proteinuria as independent long-term risk factors for new-onset diabetes mellitus after kidney transplantation. Am J Transplant. (2008) 8:1719–28. doi: 10.1111/j.1600-6143.2008.02308.x
89. Luan FL, Steffick DE, Ojo AO. New-onset diabetes mellitus in kidney transplant recipients discharged on steroid-free immunosuppresion. Transplantation. (2011) 91:334–41. doi: 10.1097/TP.0b013e318203c25f
90. Vicenti F, Friman S, Scheuermann E, Rostaing L, Jenssen T, Campistol JM, et al. Results of an international, randomized trial comparing glucose metabolism disorders and outcome with cyclosporine versus tacrolimus. Am J Transplant. (2007) 7:1506–14. doi: 10.1111/j.1600-6143.2007.01749.x
91. Artz MA, Boots JM, Ligtenberg G, Roodnat JI, Christiaans MH, Vos PF, et al. Improved cardiovascular risk profile and renal function in renal transplant patients after randomized conversion from cyclosporine to tacrolimus. J Am Soc Nephrol. (2003) 14:1880–8. doi: 10.1097/01.ASN.0000071515.27754.67
92. Hecking M, Haidinger M, Döller D, Werzowa J, Tura A, Zhang J, et al. Early basal insulin therapy decreases new-onset diabetes after renal transplantation. J Am Soc Nephrol. (2012) 23:739–49. doi: 10.1681/ASN.2011080835
93. Knoll GA, Fergusson D, Chassé M, Hebert P, Wells G, Tibbles LA, et al. Ramipril versus placebo in kidney transplant recipients with proteinuria: a multicentre, double-blind randomized controlled trial. Lancet Diabetes Endocrinol. (2016) 4:318–26. doi: 10.1016/S2213-8587(15)00368-X
94. Perkovic V, Jardine M, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canaglifozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. (2019) 380:2295–306. doi: 10.1056/NEJMoa1811744
95. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapaglifozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2018) 380:347–57. doi: 10.1056/NEJMoa1812389
96. Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, et al. Empaglifozin and progression of kidney disease in type 2 diabetes. N Engl J Med. (2016). 375:323–34. doi: 10.1056/NEJMoa1515920
97. Alkindi F, Al-Omary HL, Hussain Q, Al Hakim M, Chaaban A, Boobes Y. Outocmes of SGLT2 inhibitors use in diabetic renal transplant patients. Trasnplant Proc. (2020) 52:175–8. doi: 10.1016/j.transproceed.2019.11.007
98. Strom Halden TA, Kvitne KE, Midtvedt K, Rajakumar L, Robertsen I, Brox J. Efficacy and safety of empaglifozin in renal transplant recipients with posttransplant diabetes mellitus. Diabetes Care. (2019) 42:1067–74. doi: 10.2337/dc19-0093
99. Mahling M, Schork A, Nadalin S, Fritsche A, Heyne N, Guthoff M. Sodium-glucose cotransporter 2 (SGLT2) inhibition in kidney transplant recipients with diabetes mellitus. Kidney Blood Press Res. (2019) 44:984–92. doi: 10.1159/000501854
100. Shah M, Virani Z, Rajput P, Shah B. Efficacy and safety of Canaglifozin in kidney transplant recipients. Indian J Nephrol. (2019) 29:278–81. doi: 10.4103/ijn.IJN_2_18
101. Heinze G, Mitterbauer C, Regele H, Kramar R, Winkelmayer WC, Curhan GC, et al. Angiotensin-converting enzyme inhibitor or angiotensin II type 1 receptor antagonist therapy is associated with prolonged patient and graft survival after renal transplantation. J Am Soc Nephrol. (2006) 17:889–99. doi: 10.1681/ASN.2005090955
102. Hillebrand U, Suwelack B, Loley K, Lang D, Reuter S, Amler S, et al. Blood pressure, antihypertensive treatment, and graft survival in kidney transplant patietns. Transpl Int. (2009) 22:1073–80. doi: 10.1111/j.1432-2277.2009.00922.x
103. Hernández D, Muriel A, Abraira V, Pérez G, Porrini E, Marrero D. Renin-angiotensin system blockade and kidney transplantation: a longitudinal cohort study. Nephrol Dial Trasnplant. (2012) 27:417–22. doi: 10.1093/ndt/gfr276
104. Lafranca JA, Ijerman JNM, Betjes MGH, Dor FJM. Body mass index and outcome in renal transplant recipients: a systematic review and meta-analysis. BMC Med. (2015) 13:111. doi: 10.1186/s12916-015-0340-5
105. Vallakati A, Reddy S, Dunlap ME, Taylor DO. Impact of statin use after heart transplantation: a meta-analysis. Circ Heart Fail. (2016) 9:e003265. doi: 10.1161/CIRCHEARTFAILURE.116.003265
106. Soveri I, Abedini S, Holdaas H, Jardine A, Eriksson N, Fellström B. Graft loss risk in renal transplant recipients with metabolic syndrome: subgroup analyses of the ALERT trial. J Neprhol. (2012) 25:245–54. doi: 10.5301/JN.2011.8450
107. Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med. (2007) 357:2601–14. doi: 10.1056/NEJMra064928
108. Naik AS, Dharnidharka VR, Schnitzler MA, Brennan DC, Segev DL, Axelrod D, et al. Clinical and economic consequences of first-year urinary tract infections, sepsis, and pneumonia in contemporary kidney transplantation practice. Transpl Int. (2016) doi: 10.1111/tri.12711
109. Kobashigawa J, Dadhania D, Bhorade S, Adey D, Berger J, Bhat G, et al. Report from the American Society of Transplantation on frailty in solid organ transplantation. Am J Transplant. (2018) 19:984–94. doi: 10.1111/ajt.15198
110. Hemmersbach-Miller M, Alexander BD, Sudan DL, Pieper C, Schmader K. Infections after kidney transplantation. Does age matter? Clin Transpl. (2019) 33:e13516. doi: 10.1111/ctr.13516
111. Hollyer I, Ison MG. The challenge of urinary tract infections in renal transplant recipients. Transpl Infect Dis. (2018) 20:e12828. doi: 10.1111/tid.12828
112. Rice JC, Safdar N. Urinary tract infections in solid organ transplant recipients. Am J Transplan. (2009) 9(Suppl. 4):S267–72. doi: 10.1111/j.1600-6143.2009.02919.x
113. Coussement J, Maggiore U, Manuel O, Scemla A, Lopez-Medrano F, Nagler EV, et al. Diagnosis and management of asymptomatic bacteriuria in kidney transplant recipients: a survey of current practice in Europe. Nephrol Dial Transplant. (2018) 33:1661–8. doi: 10.1002/14651858.CD011357.pub2
114. Sabe N, Oriol I, Melilli E, Manonelles A, Bestard O, Polo C, et al. Antibiotic treatment versus no treatment for asymptomatic bacteriuria in kidney transplant recipients: A multicenter randomized trial. Open Forum Infect Dis. (2019) 6:ofz243. doi: 10.1093/ofid/ofz243
115. Abbot KC, Swanson SJ, Richter ER, Bohen EM, Agodoa LY, Peters TG. Late urinary tract infections after renal transplantation in the United States. Am J Kidney Dis. (2004) 44:353–62. doi: 10.1053/j.ajkd.2004.04.040
116. Rushton HG. The evaluation of acute pyelonephritis and renal scarring with technetium 99-mdimercaptosuccinic acid renal scintigraphy: evolving concepts and future directions. Pediatr Nephrol. (1997) 11:108–20. doi: 10.1007/s004670050243
117. Giral M, Pascuariello G, Karam G, Hourmant M, Cantarovich D, Dantal J, et al. Acute graft pyelonephritis and long-term kidney allograft outcome. Kidnet Int. (2002) 61:1880–6. doi: 10.1046/j.1523-1755.2002.00323.x
118. De Keyzer K, Van Laecke S, Peeters P, Vanholder R. Human cytomegalovirus and kidney transplantation: a clinician's update. Am J Kidney Dis. (2011) 58:118–26. doi: 10.1053/j.ajkd.2011.04.010
119. Jamboti J. BK virus nephropathy in renal transplant recipients. Nephrology. (2016) 21:647–54. doi: 10.1111/nep.12728
120. Hariharan S. BK virus nephritis after renal transplantation. Kidney Int. (2006) 69:655–62. doi: 10.1038/sj.ki.5000040
121. Hirsch HH. BK virus: opportunity makes a pathogen. Clin Infect Dis. (2005) 41:354–60. doi: 10.1086/431488
122. Sis B, Mengel M, Haas M, Colvin RB, Halloran PF, Racusen LC, et al. Banff '09 meeting report: antibody mediated graft deterioration and implementation of Banff working groups. Am J Transplant. (2010) 10:464–71. doi: 10.1111/j.1600-6143.2009.02987.x
123. Hirsch HH, Randhawa PS. BK polyomavirus in solid organ transplantation- Guidelines from the American Society of transplantation infectious diseases community of practice. Clin Transpl. (2019) 33:e13528. doi: 10.1111/ctr.13528
124. Bischof N, Hirsch HH, Wehmeier C, Amico P, Dickenmann M, Hirt-Minkowski P, et al. Reducing calcineurin inhibitor-first for treating BK polyomavirus replication after kidney transplantation: long-term outcomes. Nephrol Dial Transplant. (2018) 34:1240–50. doi: 10.1093/ndt/gfy346
125. Cheungpasitporn W, Kremers WK, Lorenz E, Amer H, Cosio FG, Stegall MD, et al. De novo donor-specific antibody following BK nephropathy: The incidence and association with antibody-mediated rejection. Clin Transplant. (2018) 32:e13194. doi: 10.1111/ctr.13194
126. Gard L, van Doesum W, Niesters HGM, van Son WJ, Diepstra A, Stegeman CA, et al. A delicate balance between rejection and BK polyomavirus associated nephropathy; a retrospective cohort study in renal transplant recipients. PLoS ONE. (2017) 12:e0178801. doi: 10.1371/journal.pone.0178801
127. Pascual J, Berger SP, Witzke O, Tedesco H, Mulgaonkar S, Qazi Y, et al. Everolimus with reduced calcineurin inhibitor exposure in renal transplantation. J Am Soc Nephrol. (2018) 29:1979–91. doi: 10.1681/ASN.2018010009
128. Mallat SG, Tanios BY, Itani HS, Lofti T, McMullan C, Gabardi S, et al. CMV and BKPyV infections in renal transplant recipients receiving an mTOR inhibitor-based regimen versus a CNI-based regimen: a systematic review and meta-analysis of ran- domized, controlled trials. Clin J Am Soc Nephrol. (2017) 12:1321–36. doi: 10.2215/CJN.13221216
129. Bussalino E, Marsano L, Parodi A, Russo R, Massarino F, Ravera M, et al. Everolimus for BVN nephropathy in kidney transplant recipients: a prospective, controlled study. J Neprhol. (2020) 34:531–8. doi: 10.1007/s40620-020-00777-2
130. Vu D, Shah T, Ansari J, Naraghi R, in D. Efficacy of intravenous immunoglobulin in the treatment of persistent BK viremia and BK virus nephropathy in renal transplant recipients. Transplant Proc. (2015) 47:394–8. doi: 10.1016/j.transproceed.2015.01.012
131. Sener A, House AA, Jevnikar AM, Boudville N, McAlister VC, Muirhead N, et al. Intravenous immunoglobulin as a treatment for BK virus associated nephropathy: one-year follow-up of renal allograft recipients. Transplantation. (2006) 81:117–20. doi: 10.1097/01.tp.0000181096.14257.c2
132. Moon J, Chang Y, Shah T, Min D. Effects of intravenous immunoglobulin therapy and Fc gamma receptor polymorphisms on BK virus nephropathy in kidney transplant recipients. Transpl Infect Dis. (2020) 22:e13300. doi: 10.1111/tid.13300
133. Keller LS, Peh CA, Nolan J, et al. BK transplant nephropathy successfully treated with cidofovir. Nephrol Dial Transplant. (2003) 18:1013–14. doi: 10.1093/ndt/gfg061
134. Kuypers DR, Vandooren AK, Lerut E, Evenepoel P, Claes K, Snoeck R, et al. Adjuvant low-dose cidofovir therapy for BK polyomavirus interstitial nephritis in renal transplant recipients. Am J Transplant. (2015) 5:1997–2004. doi: 10.1111/j.1600-6143.2005.00980.x
135. Kadambi PV, Josephson MA, Williams J, Corey L, Jerome KR, Meehan SM, et al. Treatment of refractory BK virus-associated nephropathy with cidofovir. Am J Transplant. (2003) 3:186–91. doi: 10.1034/j.1600-6143.2003.30202.x
136. Josephson MA, Gillen D, Javaid B, Kadambi P, Meehan S, Foster P, et al. Treatment of renal allograft polyoma BK virus infection with leflunomide. Transplantation. (2006) 81:704–10. doi: 10.1097/01.tp.0000181149.76113.50
137. Faguer S, Hirsch HH, Kamar N, Guilbeau-Frugier C, Ribes D, Guitard J, et al. Leflunomide treatment for polyomavirus BK-associated nephropaty after kidney transplantation. Transpl Int. (2007) 20:962–9. doi: 10.1111/j.1432-2277.2007.00523.x
138. Debout A, Foucher Y, Trébern-Launay K, Legendre C, Kreis H, Mourad G, et al. Each additional hour of cold ischemia time significantly increases risk of graft and mortality following renal transplantation. Kidney Int. (2015) 87:343–9. doi: 10.1038/ki.2014.304
139. Doshi MD, Garg N, Reese PP, Parikh CR. Recipient risk factors associated with delayed graft function: a paired kidney analysis. Transplantation. (2011) 91:666–71. doi: 10.1097/TP.0b013e318209f22b
140. Milkhalski D, Wissing KM, Ghisdal L, Broeders N, Touly M, Hoang AD, et al. Cold ischemia is a major determinant of acutre rejection and renal graft survival in the modern era of immunosuppression. Transplantation. (2008) 85(7 Suppl):S3–9. doi: 10.1097/TP.0b013e318169c29e
141. Butala NM, Reese PP, Doshi MD, Parikh CR. Is delayed graft function causally associated with long-term outcomes after kidney transplantation? Instrumental variable analysis. Transplantation. (2013) 95:1008–14. doi: 10.1097/TP.0b013e3182855544
142. Mellih KV, Boynuegri B, Mustafa C, Nilgun A. Incidence, risk factors, and outcomes of delayed graft function in deceased donor kidney transplantation. Transplant Proc. (2019) 51:1096–100. doi: 10.1016/j.transproceed.2019.02.013
143. Yarlagadda SG, Coca SG, Formica RN, Poggio ED, Parikh CR. Association betweem delayed graft function and allograft and patient survival: a systematic review and meta-analysis. Nephrol Dial Transplant. (2009) 24:1039–47. doi: 10.1093/ndt/gfn667
144. Wu WK, Famure O, Li Y, Lim SJ. Delayed graft function and the risk of acutre rejection in the modern era of kidney transplantation. Kidney Int. (2015) 88:851–8. doi: 10.1038/ki.2015.190
145. Weber S, Dienemann T, Jacobi J, Eckardt KU, Weidemann A. Delayed graft function is associated with an increased rate of renal allograft rejection: a retrospective single center analysis. PLoS ONE. (2018) 13:e0199445. doi: 10.1371/journal.pone.0199445
146. Ponticelli C. The impact of cold ischemia time on renal transplant outcome. Kidney Int. (2015) 87:272–5. doi: 10.1038/ki.2014.359
147. Kreimann K, Jang MS, Rong S, Greite R, von Vietingshoff S, Schmitt R, et al. Ischemia reperfusion injury triggers CXCL13 release and B-cell recruitment after allogenic kidney transplantation. Front Immunol. (2020) 11:1204. doi: 10.3389/fimmu.2020.01204
148. Jain S, Plenter R, Nydam R, Jani A. Injury pathways that lead to AKI in a mouse kidney transplant model. Transplantation. (2020) 104:1832–41. doi: 10.1097/TP.0000000000003127
149. Ghadimi M, Dahsti-Khavidaki S, Khatami MR, Mahdavi-Mazdeh M, Gatmiri M, Minoo FS, et al. Comparing the effect of immediate versus delayed initiation of Tacrolimus on delayed graft function in kidney transplant recipients: a randomized open-label clinical trial. J Res Pharm Pract. (2018) 7:69–76. doi: 10.4103/jrpp.JRPP_17_90
150. Ulrich F, Niedzwiecki S, Pascher A, Kohler S, Weiss S, Fikatas P, et al. Long-term outcome of ATG vs. Basiliximab induction. Eur J Clin Invest. (2011) 41:971–8. doi: 10.1111/j.1365-2362.2011.02490.x
151. Chapal M, Foucher Y, Marguerite M, Neau K, Papuchon E, Daguin P, et al. PREventing delayed graft function by driving immunosuppressive infuCtion Treatment (PREDICT-DGF): study protocol for a randomized controlled trial. Trials. (2015) 16:282. doi: 10.1186/s13063-015-0807-x
152. Jordan SC, Choi J, Aubert O, Haas M, Loupy A, Huang E, et al. A phase I/II, double-bling, placebo-controlled study assessing safety and efficacy of C1 esterase inhibitor for prevention of delayed graft function in deceases donor kidney transplant recipients. Am J Transplan. (2018) 18:2955–64. doi: 10.1111/ajt.14767
153. Sun Q, Huang Z, Han F, Zhao M, Cao R, Zhao D, et al. Allogeneic mesenchymal stem cell as induction therapy are safe and feasible in renal allografts: pilot results of a multicenter randomized controlled trial. J Transl Med. (2018) 16:52. doi: 10.1186/s12967-018-1422-x
154. Vemuri C, Upadhya GA, Arif B, Jia J, Lin Y, Gaut JP, et al. Antithrombin perfluorocarbon nanoparticles improve renal allograft function in a murine deceased criteria donor model. Transplant Direc. (2018) 4:e384. doi: 10.1097/TXD.0000000000000817
155. Tingle SJ, Figueiredo RS, Moir JAG, Goodfellow M, Talbot D, Wilson CH. Machine perfusion preservation versus static cold storage for deceased donor kidney transplantation. Cochrane Database Syst Rev. (2019) 3:CD011671. doi: 10.1002/14651858.CD011671.pub2
156. Davies L, Jenkins S, Allen J, Taylor PR. Tissue-resident macrophages. Nat Immunol. (2014) 14:986–95. doi: 10.1038/ni.2705
157. Guiteras R, Flaquer M, Cruzado JM. Macrophage in chronic kidney disease. Clin Kidney J. (2016) 9:765–71. doi: 10.1093/ckj/sfw096
158. Cao Q, Harris DCH, Wang Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology. (2015) 30:183–94. doi: 10.1152/physiol.00046.2014
159. Lee S, Huen S, Nishio H, Nishio S, Lee HK, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. (2011) 22:317–326. doi: 10.1681/ASN.2009060615
160. Wyburn K, Wu H, Yin J, Jose M, Eris J, Chadban S. Macrophage-derived interleukin-18 in experimental renal allograft rejection. Nephrol Dial Transplant. (2005) 20:699–706. doi: 10.1097/00007890-200407271-01496
161. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. (2014) 6:1–13. doi: 10.12703/P6-13
162. Ricardo SD, Goor H, Van Eddy AA. Science in medicine Macrophage diversity in renal injury and repair. J Clin Invest. (2008) 118:3522–30. doi: 10.1172/JCI36150
163. Wang YY, Jiang H, Pan J, Huang XR, Wang YC, Huang HF, et al. Macrophage-to-myofibroblast transition contributes to interstitial fibrosis in chronic renal allograft injury. J Am Soc Nephrol. (2017) 28:2053–67. doi: 10.1681/ASN.2016050573
164. Wang Y, Wang YP, Zheng G, Lee VWS, Ouyang L, Chang D, et al. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. (2007) 72:290–9. doi: 10.1038/sj.ki.5002275
165. Sagrinati C, Netti GS, Mazzinghi B, Lazzeri E, Liotta F, Frosali F, et al. Isolation and characterization of multipotent progenitor cells from the Bowman's capsule of adult human kidneys. J Am Soc Nephrol. (2006) 17:2443–56. doi: 10.1681/ASN.2006010089
166. Lasagni L, Angelotti ML, Ronconi E. Podocyte regeneration driven by renal progenitors determine glomerular disease remission and can be pharmacologically enhanced. Stem Cell Rep. (2015) 5:248–63. doi: 10.1016/j.stemcr.2015.07.003
167. Sicking EM, Fuss A, Uhlig S, Jirak P, Dijkman H, Wetzels J, et al. Subtotal ablation of parietal epithelial cells induces crescent formation. J Am Soc Nephrol. (2012). 23:629–40. doi: 10.1681/ASN.2011050449
168. Naito S, Pippin JW, Shankland SJ. The glomerular parietal epithelial cell's responses are influenced by SM22 alpha levels. BMC Nephrol. (2014) 15:174. doi: 10.1186/1471-2369-15-174
169. Achenbach J, Mengel M, Tossidou I, Peters I, Park JK, Haubitz M, et al. Parietal epithelia cells in the urine as a marker of disease activity in glomerular diseases. Nephrol Dial Transplant. (2008) 23:3138–45. doi: 10.1093/ndt/gfn235
170. Lazzeri E, Ronconi E, Angelotti ML, Peired A, Mazzinghi B, Becherucci F, et al. Human urine-derived renal progenitors for personalized modeling of genetic kidney disorders. J Am Soc Nephrol. (2015) 26:1961–74. doi: 10.1681/ASN.2014010057
171. Arcolino FO, Zia S, Held K, Papadimitriou E, Theunis K, Bussolati B, et al. Urine of preterm neonates as a novel source of kidney progenitor cells. J Am Soc Nephrol. (2016) 2762–70. (2016). doi: 10.1681/ASN.2015060664
172. Manonelles A, Guiteras R, Melilli E, Lazzeri E, Goma M, Crespo E, et al. The presence of urinary renal progenitor cells in stable kidney transplant recipients anticipates allograft deterioration. Front Physiol. (2018) 9:1412. doi: 10.3389/fphys.2018.01412
173. Chang-Panesso M, Humphreys BD. Cellular plasticity in kidney injury and repair. Nat Rev Nephrol. (2017) 13:39–46. doi: 10.1038/nrneph.2016.169
174. Lazzeri E, Angelotti ML, Conte C, Anders HJ, Romagnani P. Surviving Acute Organ Failure: Cell Polyploidization and Progenitor Proliferation. Trends Mol Med. (2019) 25:366–81. doi: 10.1016/j.molmed.2019.02.006
175. Lazzeri E, Angelotti ML, Peired A, Conte C, Marschner JA, Maggi L, et al. Endocycle-related tubular cell hypertrophy and progenitor proliferation recover renal function after acute kidney injury. Nat Commun. (2018) 9:1–18. doi: 10.1038/s41467-018-03753-4
176. Velicky P, Meinhardt G, Plessl K, Vondra S, Weiss T, Haslinger P, et al. Genome amplification and cellular senescence are hallmarks of human placenta development. PLoS Genet. (2018) 14:e1007698. doi: 10.1371/journal.pgen.1007698
177. Sereti KI, Nguyen NB, Kamran P, Zhao P, Ranjbarvaziri S, Par S, et al. Analysis of cardiomyocyte clonal expansion during mouse heart development and injury. Nat Commun. (2018) 9:754. doi: 10.1038/s41467-018-02891-z
178. Monsel A, Zhu YG, GEnnai S, Hao Q, Liu J, Lee JW. Cell-based threapy for acute organ injury: preclinicial evidence and ongoing clinical trials using mesenchymal stem cells. Anesthesiology. (2014) 1212:1099–121. doi: 10.1097/ALN.0000000000000446
179. Swaminathan M, Stafford-Smith M, Chertow GM, Warnock DG, Paragamian V, Brenner RM, et al. Allogenic mesenchymal stem cells for treatment of aki after cardiac surgery. J Am Soc Nephrol. (2018) 29:260–67. doi: 10.1681/ASN.2016101150
180. Reinders MEJ, Groeneweg KE, Hendricks SH, Bank JR, Dreyer GJ, De Vries APJ, et al. Autologous bone marrow-derived mesenchymal stromal cell therapy with early tacrolimus withdrawal: the randomized prospective, single-center, open-label TRITON study. Am J Trasnplant. (2021). doi: 10.1111/ajt.16528. [Epub ahead of print].
Keywords: review, transplantation, kidney disease, graft survival, regeneration
Citation: Codina S, Manonelles A, Tormo M, Sola A and Cruzado JM (2021) Chronic Kidney Allograft Disease: New Concepts and Opportunities. Front. Med. 8:660334. doi: 10.3389/fmed.2021.660334
Received: 29 January 2021; Accepted: 10 June 2021;
Published: 14 July 2021.
Edited by:
Michele Provenzano, University of Catanzaro, ItalyReviewed by:
Pasquale Esposito, University of Genoa, ItalyCopyright © 2021 Codina, Manonelles, Tormo, Sola and Cruzado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Manonelles, YW1hbm9uZWxsZXNAYmVsbHZpdGdlaG9zcGl0YWwuY2F0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.