
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Med. , 08 September 2021
Sec. Pathology
Volume 8 - 2021 | https://doi.org/10.3389/fmed.2021.655574
 Qiushi Wang1†
Qiushi Wang1† Chunlin Lu1†Minrui Jiang2†Mengxia Li3Xiao Yang3Lei Zhang4
Chunlin Lu1†Minrui Jiang2†Mengxia Li3Xiao Yang3Lei Zhang4 Yong He5Chengyi Mao1Ping Fu1
Yong He5Chengyi Mao1Ping Fu1 Ying Yang4
Ying Yang4 Hualiang Xiao1*
Hualiang Xiao1*Pulmonary Sclerosing Pneumocytoma (PSP) is considered as a benign tumor, although a few cases have been reported to have multiple lesions, recurrence, and even regional lymph nodes (LNs) metastasis. Here, we report a case of PSP with atypical histologic features and malignant biological behavior, and explore its molecular genetic changes. The 23-year-old male showed a 6.5-cm pulmonary nodule in the right middle lobe (RML) and enlarged media stinal LNs. He underwent thoracoscopic RML lobectomy, systematic LNs dissection, and mediastinal lymphadenectomy. The metastases to the cervical LNs and liver were detected in a short period and then resected. Postoperative pathological examination confirmed the diagnosis of PSP in all the lesions, based on the histological characteristics and immune phenotypes. Furthermore, whole-exome sequencing identified both AKT1 E17K somatic mutation and TP53 C176Y germline mutation in this case. Thus, we presented an extremely rare case of atypical PSP with rapid recurrence and multiply metastases, which can easily be misdiagnosed as primary lung cancer. In addition, PSP-specific AKT1 E17K somatic E17K somatic mutation accompanied with TP53 C176Y germline mutation may contribute to the malignant clinical course of this tumor.
Pulmonary sclerosing pneumocytoma (PSP), formerly known as pulmonary sclerosing hemangioma, is a rare primary lung tumor originated from incompletely differentiated type II pneumocytes. Although PSP has some clinical and imaging characteristics for differentiation from similar lesions, these are not specific for diagnosis (1, 2). At present, its diagnosis still depends on surgical pathology. Occasionally, PSP may manifest as multiple lesions, recurrence, regional lymph nodes (LNs), or single organ metastasis, but is not likely to affect the prognosis. Therefore, PSP is considered as a benign or a low-grade malignant potential primary lung tumor (3–8). Herein, we report an extremely rare PSP case with atypical histological features, rapid recurrence and multiply metastases, and try to elucidate the molecular mechanism of the malignant progression through clinic pathological, Immunohistochemistry (IHC), and whole-exome sequencing (WES) data.
A 23-year-old male was referred to Daping Hospital, Army Medical University for non-productive cough, chest distress, and fever for 1 month. He had no smoking and family disease history. Chest enhanced computed tomography (CT) scans confirmed a 6.5-cm pulmonary nodule in the right middle lobe (RML), with enlarged media stinal LN (Figures 1A,B). Positron-emission tomography (PET) showed increased uptake in the nodule and confirmed multiple media stinal LNs (Figures 1C,D), with no other areas of uptake. The blood examination showed that carcino embryonic antigen (CEA 8.4 ng/mL, the normal range 0.0–5.0 ng/mL) and CA153 (274.55 U/mL, the normal range 0.0–31.3 U/mL) were abnormal. The transthoracic radiology-guided biopsy considered epithelial tumor, preferred as adenocarcinoma. The patient underwent thoracoscopic RML lobectomy and systematic LNs dissection on July 27, 2018. Four months after the initial operation, the patient presented productive bloody cough. Chest enhanced CT scans showed an upper paratracheal LN measuring 3.2 × 2.2 cm. The second thoracoscopic media stinal lymphadenectomy was performed on November 20, 2018. On February 2019, 3 months after the second operation, neck-enhanced magnetic resonance imaging (MRI) showed an enlarged right supraclavicular LN measuring 5.4 × 3.8 cm (Figure 1E). The abdominal enhanced MRI showed a 1.5 × 1.2 cm nodule in the upper left lobe of the liver (Figure 1F). The patient underwent right neck mass resection on February 27, 2019, and laparoscopic liver mass resection on March 27, 2019.
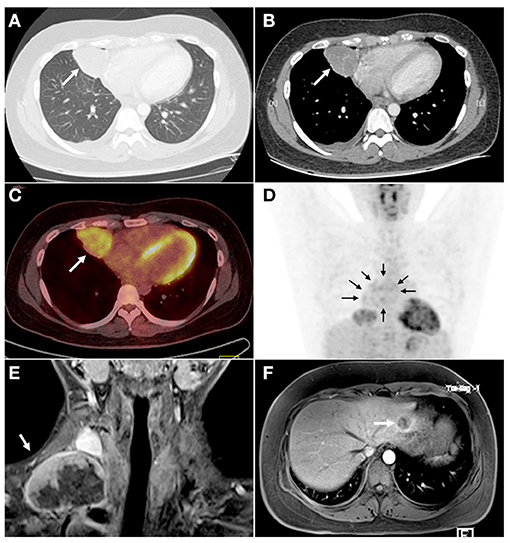
Figure 1. CT, PET, and MRI scans showed a primary lesion in the right lung and metastatic tumors in mediastinum, right neck, and right liver. (A) The lung window of CT examination showed a round-like mass (arrow) in the medial segment of the right middle lobe with a smooth margin. (B) The lesions (arrow) were slightly enhanced, and the tortuous small vessel shadow was seen inside. (C,D) PET examination showed that the radioactivity uptake of the right pulmonary pericardial lesions was increased (C, arrow), and the suspected annular radioactivity uptake in the mediastinum (D, arrows) was increased. No obvious abnormal radioactivity uptake was seen in other organs. (E) The neck MRI showed metastatic tumors (arrow) in the right neck after 5 months of the operation (thoracoscopic assisted resection of the right middle lung and systemic lymphadenectomy). The lesion was uneven enhancement and had abundant necrotic areas. (F) The abdomen MRI showed a lesion in the left lateral lobe of the liver (arrow). CT, chest enhanced computed tomography; PET, positron-emission tomography; MRI, magnetic resonance imaging.
A well-demarcated and circular mass, with grayish-white, slightly tough texture cut surface, was found in the RML (Figure 2A), 6 cm in diameter. Microscopically, the boundary between the tumor and the surrounding lung tissue was relatively clear except that a few minor infiltrative nodules were observed outside the main body of the tumor (Figure 2B). Solid cell area, papillary structure and sclerotic area composed of different proportion of surface epithelioid cells and polygonal stromal cells constituted the common mixed growth pattern of PSP (Figure 2C). Transition zone of solid and papillary pattern (Figure 2D), as well as the papillary structures in sclerosing background (Figure 2E) were observed, respectively. Focally in solid cell area, polygonal tumor cells distributed diffusely, with the increased cell density, and some pleomorphism and atypia appeared, but the mitosis was difficult to be found (Figure 2F). Focal tumor necrosis was noticed (Figure 2G). IHC analysis revealed that both the stromal cells and cuboidal surface cells showed positive for EMA and TTF-1 (Figure 2I), whereas only the surface cells expressed CK (Figure 2H), CEA, and Napsin A (Figure 2J). Most of the tumor cells were p53-positive (Figure 2K). The Ki-67 labeling index (LI) was about 5% (Figure 2L). PR was positive in polygonal stromal cells, while ER and Syn were positive in a few tumor cells. S-100, actin, desmin, HMB45, p63, and TFE3 were all negative. Double-labeling IHC showed CK7 and TTF-1 positive but PR negative in surface-lining epithelial cells, while CK7 negative but TTF-1 and PR positive in polygonal stromal cells (Figures 2M,N).
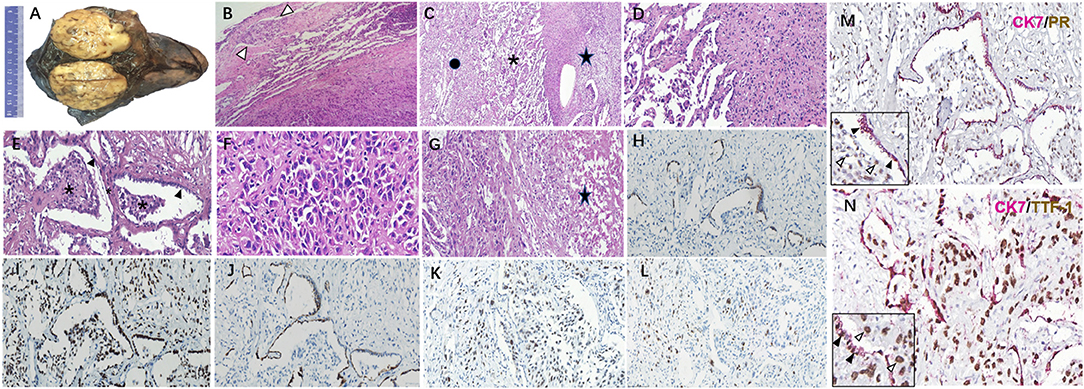
Figure 2. Histopathology of the primary pulmonary lesion in this unusual PSP case. (A) A large solid mass in the middle right lobe with relatively clear boundary and grayish-white, slightly tough texture cut surface. (B) The boundary between tumor and normal lung tissue was relatively clear, with the invasion of minor tumor nodules (arrow). (C) Solid cell area (star), papillary structure (asterisk) and sclerotic area (spot) in turn beneath normal lung tissue. (D) The migration of solid cell zone and papillary structures. (E) Papillary structures of different size (asterisk) covered with surface epithelial cells (arrow) appear in sclerotic background. (F) In the solid cell area, polygonal tumor cells were mainly diffusely arranged, certain atypia and giant tumor cells could be seen, but mitosis was hardly to be found. (G) Focal tumoral necrosis could be seen (star). CK (H), TTF-1 (I), and NapsinA (J) expressed in the same region as illustrated in this figure (E). P53 (K) and Ki-67 (L) expressed in the solid cell region and papillary structures as illustrated in this figure (F). (M) Double-labeling IHC showed the surface epithelial cells (black arrow) positive for CK7 and negative for PR, while the stromal-like cells (white arrow) in papilla's on the contrary. (N) Double labeling IHC showed CK7 and TTF-1 co-expressing in surface-lining cells (black arrow), while only TTF-1 positive in stromal-like cells (white arrow).Original magnification × 40 (B–E, G–M), original magnification×100 (F,N). PSP, pulmonary sclerosing pneumocytoma; IHC, immunohistochemistry.
The histopathological and IHC staining of the resected specimens confirmed multiple organ metastasis of hilar, mediastinum (Figures 3A–E), cervical LNs, and liver (Figures 3F–J). Strikingly, the normal structures of LNs and the liver were partially destroyed. The polygonal tumor cells showed diffuse or patchy infiltration, increased pleomorphism and atypia, with intra-nuclear inclusions and tumor giant cells. Interstitial vascular hyperplasia and focal necrosis were easily identified. The metastatic tumors showed same immuno-phenotypes as the stromal cells of the primary lung tumor: diffuse expression of EMA, TTF-1(Figures 3C,H), PR (Figures 3D,I) and p53 (Figure 3J), but negative for CK7 and Napsin A (Figure 3C, red box). The Ki-67 LI of mediastinum LNs (Figure 3E) and liver metastasis were higher than that of the primary lesion (25 vs. 5%).
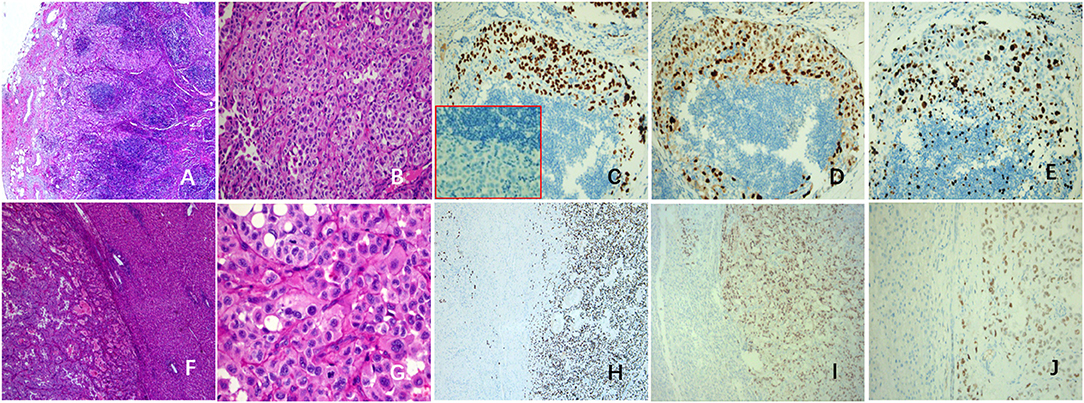
Figure 3. Histopathological changes in metastatic lesions of this case. Tumors cells of mediastinal LNs (A,B) were positive for TTF-1 (C) and PR (D), but negative for NapsinA (C, red box). The Ki-67 LI was about 25% (E). The tumor cells of liver metastasis (F,G) were like the polygonal stromal cells in the primary pulmonary lesion, with obvious pleomorphism and atypia, and the mitosis is easily to be found (G). The tumor cells were positive for TTF-1 (H), PR (I), and P53 (J). LNs, lymph nodes; LI, labeling index.
Herein, we conducted a comprehensive examination of genetic alterations (somatic mutations) in formalin fixed paraffin-embedded (FFPE) samples from the primary and three metastatic lesions with matched adjacent normal tissue. High frequencies of AKT1 E17K mutations was detected in primary lung tumor, media stinal LN, cervical LN, and liver metastatic lesions (16.14, 16.83, 34.51, and 21.88%, respectively). TP53 C176Y mutations were identified in the primary lung lesion, mediastinal LN, cervical LN, and liver metastatic samples, with the frequencies of 42.41, 68.63, 72.84, and 50.52%, respectively. Interestingly, TP53 C176Y mutations were also detected in the paired normal tissue adjacent to the primary lung tumor (frequency 33.02%), suggesting that TP53 C176Y was a germline mutation in this patient (Table 1). This phenotype was further confirmed by the identification of the same mutation in the peripheral blood mononuclear cell (PBMC) sample.
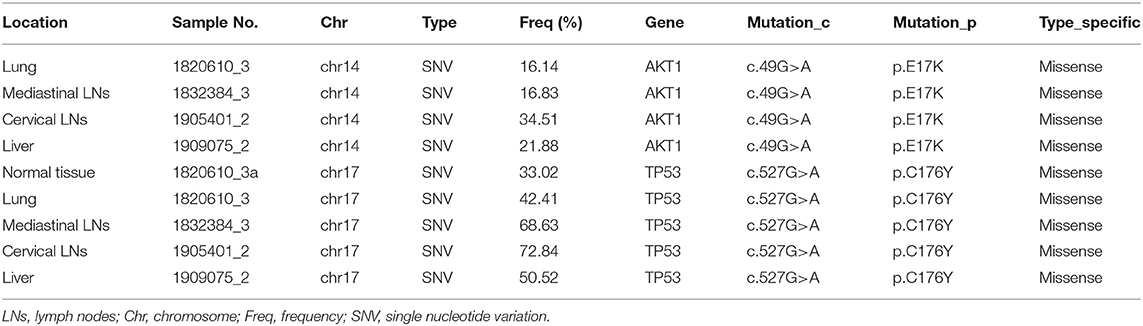
Table 1. Clinical data, AKT1, and TP53 gene mutations in this atypical pulmonary sclerosing pneumocytomacase.
The patient received anti-estrogen therapy with medroxyprogesterone after the fourth operation, but the disease progressed rapidly. In May 2019, chest and abdominal enhanced CT scans showed multiple nodules in the right lung and multiple enlarged LNs in neck, jaw and right supraclavicular fossa. In September 2019, the patient felt that the neck mass was significantly larger than before. Chest and abdominal enhanced CT scans showed an enlarged mass in both lung hilum (5.8 × 5.4 cm) and right supraclavicular fossa (5.7 cm in maximum dimension), multiple nodules in the liver and left kidney, and thoracic 8th and 11th vertebral osteolytic bone destruction. The patient was then administered pembrolizumab treatment at 2 mg/kg intravenously every 3 weeks, combined with apatinib at 250 mg/day orally since October 2, 2019. As of January 10, 2021, after multiple cycles of combined treatment, the metastatic lesions were obviously reduced or in a stable state. The patient was in good mental state and had no significant change in weight.
Due to the lack of specificity in clinical and imaging features, the diagnosis of PSP mainly depends on postoperative pathological examination. The diagnosis of typical PSP, characterized by two different cell components, four typical histological types, and immune phenotypes with specific features, is not difficult for pathologists (9). However, some atypical cases may exhibit either different clinical behaviors from typical PSP, such as recurrence or metastasis, or confused histological characteristics, such as limited typical patterns, cytological atypia, and focal necrosis. These atypical cases could be misdiagnosed as papillary or solid subtype of lung adenocarcinoma and neuroendocrine tumor, especially in the case of needle biopsy or intraoperative frozen diagnosis (10). In this case, the delayed diagnosis at biopsy was mainly attributed to the atypical phenomenon, including multiple LN metastases, papillary and solid growth pattern, cellular atypia, tumoral necrotic foci, and higher Ki-67 LI than that of ordinary PSP cases. Interestingly, only interstitial round cells, no surface cells, in LN and liver metastases of this case were observed. Consistently with other reports of the literature (3, 5, 11, 12), this morphology indicated that the stromal cells might play a critical role in malignant progression of PSP. Therefore, for accurate conclusions, we should comprehensively analyze the results of imaging, gross specimen performance, histopathology, IHC, and molecular detection, if necessary.
There are several points worth emphasizing on the pathological diagnosis. Firstly, PSP usually presents as an isolated, solid, and well-defined mass, which is different from the general changes of invasive adenocarcinoma. Secondly, we should deduce the basic structure for the diagnosis of PSP by observing the sections carefully and comprehensively: the papillary growth pattern and its diffuse distribution of interstitial cell components, as well as other histological patterns, such as solid cell area, intra-alveolar hemorrhage, and sclerotic changes. Thirdly, although there is no specific single antibody for diagnosis, establishing the diagnosis by the appropriate combination of antibodies and observing the obvious difference of immunophenotypes between epithelioid cells and stromal cells is imperative. Finally, for the atypical cases, the molecular pathologic findings are helpful for diagnosis. As in this case, we identified the AKT1 E17K point mutation through WES analysis, which is a relatively specific molecular feature of PSP (13, 14), but no other common driving gene mutations related to lung cancer were found, which played an important role in strengthening our confidence in the diagnosis of this atypical disease.
PSPs were mostly diagnosed in female patients (83.34%), aged 38–61 years (15). The malignant progression and metastasis of PSP were extremely rare. The reported PSP patients with LN (3, 7, 8, 16) or organ metastases (5, 11) were mostly females, and only one male patient suffered from mediastinal LN metastasis (17). Herein, we reported an extremely atypical case: a young male patient had been suffering from an aggressive PSP with multiple LN and organ metastases during 7 months after the resection of the primary lung lesion.
AKT1 E17K mutation, identified from all primary and metastatic lesions in this PSP case, were localized to the pleckstrin homology domain (PH domain), which is crucial for membrane localization and downstream activation of AKT1 (18) and is known to promote growth factor-independent cell proliferation (19, 20). However, though more than 40 PSP cases have been reported with AKT1 E17K mutation (13, 14), malignant progression has been rarely reported, which indicated that single E17K mutation on the PH domain of the AKT1 gene might not be sufficient to initiate the malignant transformation of the tumor. A 17-year-old girl suffering from multiple nodules in the right lung lobe diagnosed as PSP with both AKT1 E17K and BRAF V600E mutations (21). The other PSP case with diffusely scattered nodules in the right lung, harbored AKT1 E17K and other 14 somatic gene mutations (22). These indicated that the combination of AKT1 mutations with other oncogenes might accelerate the malignant progression of benign PSPs. For the first time, we reported a TP53 C176Y germline mutation, a likely pathogenic mutation according to the ClinVar and 1000Genomes database, in this extremely aggressive PSP case. Which consistent with the positive expression of the P53 mutant protein in the IHC assay. Therefore, rapid malignant progression leading to multiple metastases of this PSP case might be partially attributed to the combination of somatic AKT1 E17K mutation and germ line TP53 C176Y mutation. Germ line mutations of TP53 gene have been identified in 80% of patients with Li-Fraumeni syndrome (LFS), a cancer predisposition syndrome associated with high risks for a diverse spectrum of childhood- and adult-onset malignancies (23). However, neither first- nor second-degree relatives had been diagnosed with any cancer or sarcoma, this patient does not meet the classic LFS diagnosis criteria (24).
We report an extremely rare case of PSP, which showed obvious atypical features in histopathology and malignant biological behaviors, such as rapid recurrence and multiple metastases. Based on the results of WES, we speculated that the somatic AKT1 E17K and germline TP53 C176Y mutations might account for this malignant progression.
All data sets generated for this study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Clinical Ethics Committee of Daping Hospital, Army Medical University (Chongqing, China). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
QW and HX designed the study. MJ and XY investigated and provided the clinical data. QW, CL, LZ, and YY performed the molecular experiments. CM, CL, and PF performed the pathologic slides. QW and HX wrote the manuscript. ML, YH, and HX revised and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by The National Natural Science Foundation of China (grant no. 81802781).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Shin SY, Kim MY, Oh SY, Lee HJ, Hong SA, Jang SJ, et al. Pulmonary sclerosing pneumocytoma of the lung: CT characteristics in a large series of a tertiary referral center. Medicine. (2015) 94:e498. doi: 10.1097/MD.0000000000000498
2. Zhu J. Analysis of the clinical differentiation of pulmonary sclerosing pneumocytoma and lung cancer. J Thorac Dis. (2017) 9:2974–81. doi: 10.21037/jtd.2017.08.07
3. Pokharel S, Dhillon SS, Ylagan L, George S, Yendamuri S. Sclerosing pneumocytoma with lymph node metastasis. J Thorac Oncol. (2016) 11:1802–4. doi: 10.1016/j.jtho.2016.06.005
4. Arumugam VG, Joseph LD, Thangavel P, Swaminathan R, Sunderaj RR. Sclerosing pneumocytoma of the lung: a case report. J Clin Diagn Res. (2017) 11:ED12–4. doi: 10.7860/JCDR/2017/22279.9271
5. Kim MK, Jang SJ, Kim YH, Kim SW. Bone metastasis in pulmonary sclerosing hemangioma. Kor J Intern Med. (2015) 30:928–30. doi: 10.3904/kjim.2015.30.6.928
6. Xu HM, Zhang G. A rare case of pulmonary sclerosing hemagioma with lymph node metastasis and review of the literature. Int J Clin Exp Pathol. (2015) 8:8619–23.
7. Soo IX, Sittampalam K, Lim CH. Pulmonary sclerosing pneumocytoma with mediastinal lymph node metastasis. Asian Cardiovasc Thorac Ann. (2017) 25:547–9. doi: 10.1177/0218492317727668
8. Wang X, Zhang L, Wang Y, Jia X, Wang J, Zhang H. Sclerosing pneumocytoma with metastasis to the mediastinal and regional lymph nodes. Indian J Pathol Microbiol. (2018) 60:407–9. doi: 10.4103/IJPM.IJPM_98_17
9. Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. (2015) 10:1243–60. doi: 10.1097/JTO.0000000000000630
10. Yang CH, Lee LY. Pulmonary sclerosing pneumocytoma remains a diagnostic challenge using frozen sections: a clinicopathological analysis of 59 cases. Histopathology. (2018) 72:500–8. doi: 10.1111/his.13391
11. Bae YS, Ro JY, Shim HS, Hong SW, Yoon SO. Pulmonary sclerosing haemangioma with metastatic spread to stomach. Histopathology. (2012) 60:1162–4. doi: 10.1111/j.1365-2559.2012.04213.x
12. Gao Q, Zhou J, Zheng Y, Cui J, Teng X. Clinical and histopathological features of pulmonary sclerosing pneumocytoma with dense spindle stromal cells and lymph node metastasis. Histopathology. (2020) 77:718–27. doi: 10.1111/his.14159
13. Jung SH, Kim MS, Lee SH, Park HC, Choi HJ, Maeng L, et al. Whole-exome sequencing identifies recurrent AKT1 mutations in sclerosing hemangioma of lung. Proc Natl Acad Sci USA. (2016) 113:10672–7. doi: 10.1073/pnas.1606946113
14. Yeh YC, Ho HL, Wu YC, Pan CC, Wang YC, Chou TY. AKT1 internal tandem duplications and point mutations are the genetic hallmarks of sclerosing pneumocytoma. Mod Pathol. (2020) 33:391–403. doi: 10.1038/s41379-019-0357-y
15. Lovrenski A, Vasilijević M, Panjković M, Tegeltija D, Vučković D, Baroš I, et al. Sclerosing pneumocytoma: a ten-year experience at a Western Balkan University Hospital. Medicina. (2019) 55:27. doi: 10.3390/medicina55020027
16. Adachi Y, Tsuta K, Hirano R, Tanaka J, Minamino K, Shimo T, et al. Pulmonary sclerosing hemangioma with lymph node metastasis: a case report and literature review. Oncol Lett. (2014) 7:997–1000. doi: 10.3892/ol.2014.1831
17. Katakura H, Sato M, Tanaka F, Sakai H, Bando T, Hasegawa S, et al. Pulmonary sclerosing hemangioma with metastasis to the mediastinal lymph node. Ann Thorac Surg. (2005) 80:2351–3. doi: 10.1016/j.athoracsur.2004.06.099
18. Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. (2002) 2:489–501. doi: 10.1038/nrc839
19. Parikh C, Janakiraman V, Wu WI, Foo CK, Kljavin NM, Chaudhuri S, et al. Disruption of PH-kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc Natl Acad Sci USA. (2012) 109:19368–73. doi: 10.1073/pnas.1204384109
20. Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. (2007) 448:439–44. doi: 10.1038/nature05933
21. Jiang G, Zhang M, Tan Q, Lin S, Zeng Y, Liu C, et al. Identification of the BRAF V600E mutation in a patient with sclerosing pneumocytoma: a case report. Lung Cancer. (2019) 137:52–5. doi: 10.1016/j.lungcan.2019.09.004
22. Fan X, Lin L, Wang J, Wang Y, Feng A, Nie L, et al. Genome profile in aextremely rare case of pulmonary sclerosing pneumocytoma presenting with diffusely-scattered nodules in the right lung. Cancer Biol Ther. (2018) 19:13–9. doi: 10.1080/15384047.2017.1360443
23. Villani A, Shore A, Wasserman JD, Stephens D, Kim RH, Druker H, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol. (2016) 17:1295–305. doi: 10.1016/S1470-2045(16)30249-2
Keywords: pulmonary sclerosing pneumocytoma, metastasis, AKT1, TP53, whole-exome sequencing
Citation: Wang Q, Lu C, Jiang M, Li M, Yang X, Zhang L, He Y, Mao C, Fu P, Yang Y and Xiao H (2021) Case Report and Literature Review: Pulmonary Sclerosing Pneumocytoma With Multiple Metastases Harboring AKT1 E17K Somatic Mutation and TP53 C176Y Germline Mutation. Front. Med. 8:655574. doi: 10.3389/fmed.2021.655574
Received: 19 January 2021; Accepted: 03 August 2021;
Published: 08 September 2021.
Edited by:
Mattia Barbareschi, Azienda Provinciale per i Servizi Sanitari (APSS), ItalyReviewed by:
Alberto Cavazza, Azienda USL/IRCCS Reggio Emilia, ItalyCopyright © 2021 Wang, Lu, Jiang, Li, Yang, Zhang, He, Mao, Fu, Yang and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hualiang Xiao, ZHBibF94aGxAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.