 Alice Drobny
Alice Drobny Phuong A. Ngo
Phuong A. Ngo Markus F. Neurath
Markus F. Neurath Friederike Zunke
Friederike Zunke Rocío López-Posadas
Rocío López-Posadas- 1Department of Molecular Neurology, University Hospital Erlangen, Friedrich-Alexander-University Erlangen-Nuremberg, Erlangen, Germany
- 2Medicine 1, University Hospital Erlangen, Erlangen, Germany
- 3Deutsches Zentrum Immuntherapie, Erlangen, Germany
Intestinal symptoms, such as nausea, vomiting, and constipation, are common in Parkinson's disease patients. These clinical signs normally appear years before the diagnosis of the neurodegenerative disease, preceding the occurrence of motor manifestations. Moreover, it is postulated that Parkinson's disease might originate in the gut, due to a response against the intestinal microbiota leading to alterations in alpha-synuclein in the intestinal autonomic nervous system. Transmission of this protein to the central nervous system is mediated potentially via the vagus nerve. Thus, deposition of aggregated alpha-synuclein in the gastrointestinal tract has been suggested as a potential prodromal diagnostic marker for Parkinson's disease. Interestingly, hallmarks of chronic intestinal inflammation in inflammatory bowel disease, such as dysbiosis and increased intestinal permeability, are also observed in Parkinson's disease patients. Additionally, alpha-synuclein accumulations were detected in the gut of Crohn's disease patients. Despite a solid association between neurodegenerative diseases and gut inflammation, it is not clear whether intestinal alterations represent cause or consequence of neuroinflammation in the central nervous system. In this review, we summarize the bidirectional communication between the brain and the gut in the context of Parkinson's disease and intestinal dysfunction/inflammation as present in inflammatory bowel disease. Further, we focus on the contribution of intestinal epithelium, the communication between intestinal epithelial cells, microbiota, immune and neuronal cells, as well as mechanisms causing alterations of epithelial integrity.
Introduction
Parkinson's Disease
Parkinson's disease (PD) is the second most common neurological disorder characterized by movement disabilities (1), but also by non-motor symptoms, including gastrointestinal dysfunction that often appears years before diagnosis of disease (2, 3). A neuropathological hallmark of PD is the aggregation of the synaptic protein alpha-synuclein (aSyn) within the central nervous system (CNS), leading to degeneration of dopaminergic neurons within the substantia nigra pars compacta (SNpc) of the midbrain (1). Moreover, research suggests that inflammatory responses within the CNS contribute to PD pathology. Hence, glial cell reactions and T cell infiltration result in increased levels of inflammatory cytokines within the CNS and are currently recognized as prominent features of PD (4, 5).
Intestinal Dysfunction and Inflammation Within PD
Interestingly, recent data indicate that intestinal inflammation contributes to the pathogenesis of PD (6), and increasing numbers of studies imply that PD may start in the gastrointestinal system years before any motor symptoms develop (7–9). An acute and chronic intestinal inflammation is a prominent feature of Inflammatory bowel disease (IBD) comprising the diseases Ulcerative colitis (UC) and Crohn's disease (CD). While UC mainly affects the colon and rectum, CD injures the entire GI tract (10). IBD is understood to be a result of gut microbiota dysbiosis and mucosal immune dysregulation (11). Also, intestinal inflammation in IBD is associated with intestinal epithelial cell (IEC) alterations and maintaining epithelial homeostasis helps in protecting against inflammation (12). Remarkably, PD and IBD share overlapping genetic factors found within a recent genome-wide-association study (GWAS) (13). The leucin-rich repeat kinase 2 (LRRK2) gene appears to be the most susceptibility-factor for both diseases (14, 15). Interestingly, LRRK2 is one of the genes most commonly associated with familial and sporadic PD (16). Recent studies show, that patients with IBD have a higher risk of developing PD as compared to non-IBD individuals (17, 18). It is well-established that IBD is characterized by chronic pro-inflammatory immune activity (11), which is now suggested to be a fundamental element of neurodegenerative disorders as well (5, 6). Furthermore, animal studies demonstrate that gut inflammation, similar to IBD, induces loss of dopaminergic neurons (19, 20). Additionally, chronic GI inflammation is likely to induce anxiety-like behavior and alter CNS biochemistry in mice (21). Interestingly, CD patients have been shown to accumulate aSyn in the gut (22).
Moreover, aSyn and its aggregated forms were also found in the enteric nervous system (ENS) of PD patients and symptoms outside the CNS were described including GI impairments (2, 23, 24). This gave rise to the hypothesis that PD pathology can spread from the gut to the brain and vice versa (23, 25, 26). This hypothesis is supported by recent animal studies, which recapitulated the transmission of aSyn pathology via the vagal nerve, connecting the central with the peripheral nervous system (27, 28). In this context, the discovery of aSyn expression in enteroendocrine cells (EECs) within the intestinal epithelium suggests these cells as sensors of luminal signals triggering the gut-neural circuit behind aSyn alteration (29, 30). This signal is then transmitted to the CNS, potentially via the vagus nerve. Thus, deposition of aggregated aSyn in the GI tract has been inferred as a potential diagnostic marker for prodromal PD.
This review focuses on overlapping disease pathologies and the molecular communication between the brain and the gut in the context of PD and gut inflammation, as present in IBD (Figure 1). We emphasis on the contribution of neurodegeneration and neuroinflammation in PD, gut-brain spreading of PD pathology, intestinal epithelium and the communication between IECs, microbiota and immune cells (Figure 2). Moreover, we discuss the mechanisms causing alterations of epithelial integrity and gastrointestinal (GI) dysfunction in PD.
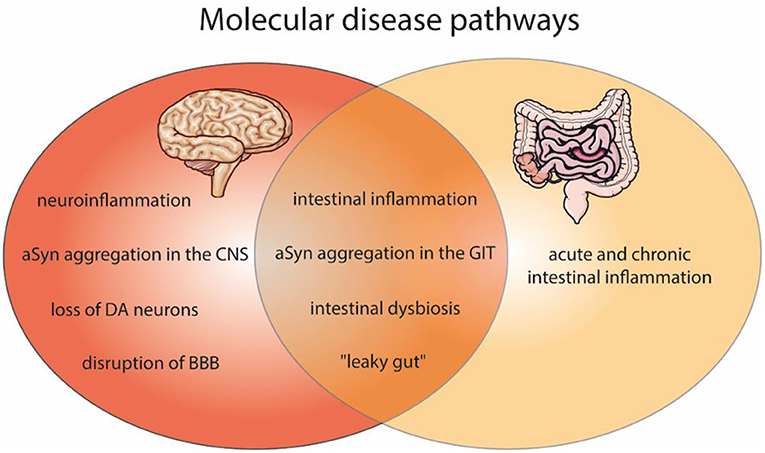
Figure 1. Shared molecular disease pathways of the brain and gut pathologies, as found in PD and IBD. Affected molecular features within PD (red) and gut inflammation (light orange) as well as in both disease (dark orange). Molecular pathways of PD include neuroinflammation, aSyn aggregation in the central nervous system (CNS), dopaminergic neurons (DA) degeneration, and the disruption of blood-brain barrier (BBB). In IBD an acute and chronic intestinal inflammation is described. Both diseases can comprise intestinal inflammation, aSyn aggregation in the gastrointestinal tract (GIT), intestinal dysbiosis and a “leaky gut.” The figure contains modified components of Servier Medical Art, licensed under the Creative Commons Attribution 3.0 Unported License (CC BY 3.0) https://smart.servier.com.
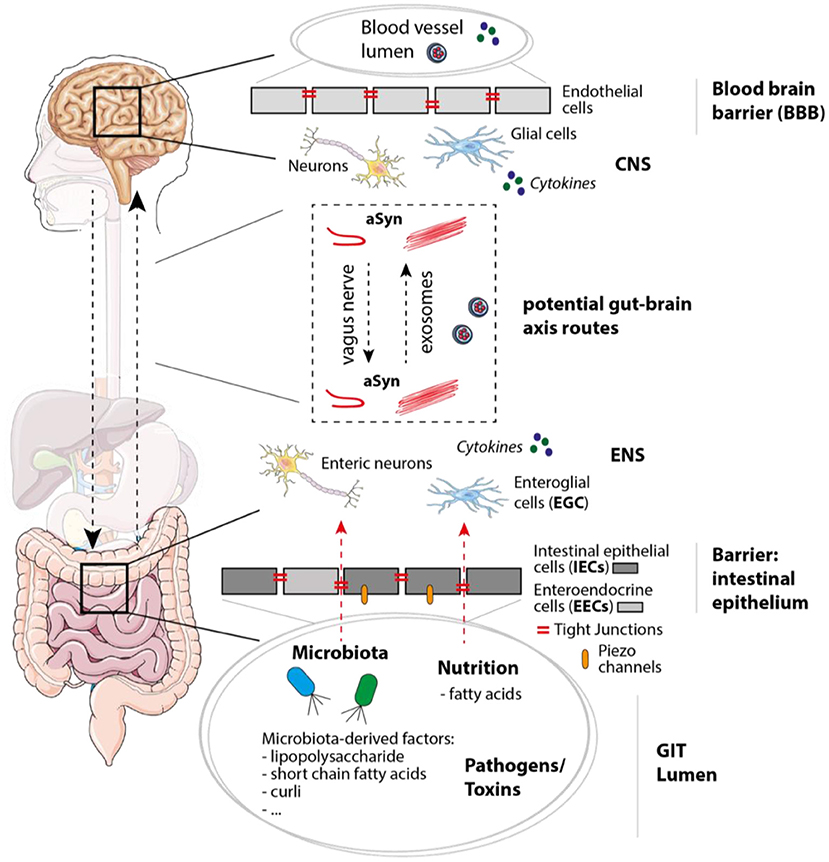
Figure 2. Mechanisms, molecules, and cell types involved in PD/IBD pathology and gut-brain communication. In the gastrointestinal tract (GIT) the intestinal epithelium functions as barrier and separates the GIT lumen from the surrounding enteric nervous system (ENS), which not only contains enteric neurons, but also enteroglial cells. Within the GIT lumen microbiota and microbiota-derived factors like polysaccharides, short chain fatty acids, and curli (bacterial amyloid protein) can be found, but also nutritions, possible pathogens and toxins. When integrity of the intestinal epithelium consisting of epithelial cells including enteroendocrine cells (IECs and EECs) is disturbed, molecules within the GIT lumen get in contact with cells of the ENS. Tight junctions and Piezo channels have been shown to play an important role in mechanosensation, peristalsis, and intestinal barrier function. Within the brain, the blood-brain barrier (BBB) separates the CNS (including neurons and glial cells) from the blood vessel lumen. Potential gut-brain axis routes on which aSyn and molecules like cytokines could be transferred are for instance the vagus nerve or exosomes [via the blood stream or cerebrospinal fluid (CSF)]. The figure contains modified components of Servier Medical Art, licensed under the Creative Commons Attribution 3.0 Unported License (CC BY 3.0) https://smart.servier.com.
Neuropathology in PD
Motor and Non-motor Manifestations of PD
PD is clinically characterized by classical motor symptoms including muscular rigidity, bradykinesia, rest tremor, and postural instability (1). Among several putative factors that may contribute to PD pathology, the most crucial indication of PD is the degeneration of neurons in the CNS. The loss of dopaminergic neurons within the SNpc is the most predominant feature during disease progression (31) and leads to excessive dopamine depletion within the basal ganglia, which results in the above mentioned parkinsonian motor characteristics (1). The administration of the amino acid precursor of dopamine, L-DOPA (L-3,4-dihydroxy-L-phenylalanine), has shown to be the most effective symptomatic treatment. However, if the motor symptoms occur in PD patients the continuous loss of neurons is already inexorable (32). Interestingly, PD manifests already >20 years before the motoric problems occur. This premotor or prodromal period of disease is defined by e.g., constipation, olfactory dysfunction, sleep disorder, cognitive impairment, autonomic dysfunction, pain and fatigue (1, 3). Altogether, this leads to the assumption that PD is a complex, multisystem disorder with both neurologic and systemic non-motor manifestations.
Lewy Body Pathology in the CNS
A neuropathological hallmark of PD is the formation of intracellular amyloid inclusions in neuronal bodies and neurites, known as Lewy bodies (LB) and Lewy neuritis (LN), respectively, consisting of aggregated aSyn (33). The appearance of these aSyn-carrying inclusions in patients is also collectively known as synucleinopathies, referring to PD, dementia with Lewy bodies (DLB) and multiple system atrophy (MSA) (33–35).
Physiologically, aSyn is natively unfolded and soluble with an amphipathic N-terminus, a hydrophobic central domain known as non-amyloid-β component (NAC) region, and an acidic C-terminus (36). Under pathological conditions, aSyn aggregates have been shown to exert cell toxic properties (37, 38). The aggregation mechanisms, by which soluble aSyn changes its structure to oligomers and ultimately to insoluble β-sheet rich fibrils, are still under debate (39–41). However, several factors have been described to induce structural changes of monomeric aSyn, involving the interactions with specific lipids (42, 43) and membranes (44). Further, posttranslational modifications such as phosphorylation (45), nitration and oxidation (46), ubiquitination (47), and sumoylation (48) have been shown to accelerate aSyn pathology. Multiple copies (e.g., duplications and triplications) (49, 50) and missense mutations (e.g., A53T, A30P, E46K, and H50Q) (51–53) of the gene encoding for aSyn (SNCA) foster protein aggregation. In addition, the cellular microenvironment of aSyn has been reported to play a role in aSyn conformation and solubility. For instance, aSyn aggregation behavior differs between neutral pH7.4 (e.g., cytosol) and acidic pH5 (e.g., lysosome) and aSyn purified from lysosomes was able to seed aggregation in a concentration-dependent manner (54, 55). Interestingly, dysfunction in lysosomal pathways have been linked to PD (56, 57). For degradation, aSyn is processed within lysosomes by specialized lysosomal enzymes (cathepsins) (58–60). Hence, deficiency within these lysosomal enzymes, important for lysosomal function and aSyn degradation, lead to its aggregation and pathology (61–64). Targeting lysosomal enzymes by boosting their activity has become a promising therapeutic approach, which might lower aSyn burden within neuronal cells and thus decrease the risk of pathological aSyn aggregation and neurotoxicity (65, 66). In a nutshell, intracellular accumulation of aSyn, due to inefficient clearance mechanisms, might drive further aggregation of the protein (67). In this regard, it was shown that toxic aSyn species can be released to the periphery from stressed and/or dying neurons and are subsequently taken up by surrounding cells, leading to spreading of pathology (68–70). Especially, aggregation intermediates, such as aSyn oligomers, exhibit highly cell toxic properties (43, 71, 72).
Neuroinflammation in PD
In recent years, evidence evolved that aSyn and inflammatory processes are extraordinarily connected. In that sense, chronic neuroinflammation is another characteristic indicator of PD pathophysiology and is considered to promote the progression of dopaminergic cell death (73, 74). In general, neuroinflammation is defined as the immune response of cells within the brain and plays an important role in maintenance of nervous tissue homeostasis (4). On the one hand, a moderate inflammation can protect neurons from damage (75); on the other hand, inflammatory factors do also affect neurons directly and convey neurodegeneration. In addition, neuronal cell death induces inflammatory mechanisms, and contributes to a vicious cycle of inflammation and progressive loss of neurons in the brain (76). The neuroinflammatory response is mediated by resident immune cells (microglia and astrocytes), which release cytokines and chemokines (4, 77).
Many neuroinflammatory circumstances at post-mortem stage have also been identified on a molecular basis in PD. For example, numerous proinflammatory cytokines and factors such as tumor necrosis factor (TNF)-α, β2-microglobulin, epidermal growth factor (EGF), transforming growth factor α (TGFα), TGFβ1, and interleukin (IL)-1β, IL-6, and IL-2 were found in the striatum of PD patients (78). Furthermore, TNF-α, IL-1β, and interferon (IF)-γ were also detected in the SNpc of PD patients (79). Interestingly, dopaminergic neurons express the receptors of these cytokines (80), that might explain the vulnerability of DA neurons to inflammatory processes inside the brain. In addition, increased levels of proinflammatory mediators, such as IL-1β, IL-2, TNF-α, and IL-6 are present in the serum and the cerebrospinal fluid (CSF) of PD patients (81–84). These results suggest the direct migration of immune cells from the periphery (blood stream) to the brain (or vice versa) during neurodegenerative process.
Microglia, the resident macrophages in the brain, and astrocytes, the most abundant glial subtype in the CNS, are considered to drive the inflammatory response in PD (85). Of relevance, microglia initiate the innate immune response in the brain, therefore representing key players upon inflammatory stimulus (86, 87). Under pathological conditions, activated microglia release proinflammatory cytokines and reactive oxygen species (ROS), which affect dopaminergic neuron viability (73, 88). Reactive microglia were found in various brain regions (89, 90) including the SNpc of PD patients (91). Besides microglia activation, reactive astrogliosis contributes to PD pathogenesis and progression (85). Astroglial cells secret the glial cell-line derived neurotrophic factor (GDNF), which promotes survival of dopaminergic neurons (92), and regulates the permeability of the blood-brain-barrier (BBB) (93, 94). Interestingly, in this regard the BBB is found to be defective in PD patients (95–97). The mechanism of an altered BBB function is still elusive, however, the increased levels of proinflammatory cytokines IL-6, IL-1β, and TNF-α have been associated with a disruption of trans-endothelial electrical resistance, indicating an increased BBB permeability (98). Recently, a study showed that aSyn-mediated release of proinflammatory cytokines and chemokines by pericytes induces disruption of BBB (99). Further, accumulations of aSyn in astrocytes are found in post-mortem analysis of PD patients (100). Reactive astrocytes manifest with PD progression by increased proinflammatory cytokine secretion such as IL-1β, TNF-α, and IFN-γ (101–103). A recent study indicates the close interplay between microglia and astrocytes showing induction of neurotoxic A1 astrocytes by microglial secretion of IL-1α, TNF-α and complement component 1q (C1q) (104). In this regard, it was shown that pathological aSyn inoculation in vitro and in vivo induces microglia to secrete cytokines and chemokines followed by astrocyte A1 activation that caused neuronal cell death in culture and neurodegeneration in mice (105).
Moreover, is has been reported that aSyn itself has an important role in the initiation and maintenance of inflammation in PD. Recent reports have suggested that aSyn acts as a damage-associated molecular pattern (DAMP), capable of modulating inflammatory cytokine production in microglia and inducing intracellular signaling cascades (106, 107). It has been demonstrated that extracellular oligomeric aSyn is a putative activator of toll-like receptor (TLR) 2 and promotes microglia-mediated inflammatory cytokine and ROS production (108, 109). The exact contribution of different aSyn conformations to TLR activation is currently unclear, however, there are strong indications that the activation seems to be conformation dependent. Specifically, TLR4 appears to be involved in the uptake of fibrillary aSyn (110). While the presence of monomeric aSyn seems to enhance phagocytic function, aggregated forms seem to inhibit this process (111).
Furthermore, lymphocyte infiltration might also play a role in inflammation processes inside the brain of PD patients. It was reported that cytotoxic T lymphocytes (CD8+) as well as CD4+ T helper (Th) cells were more abundant in the brains of PD patients compared to healthy individuals (112). In this regard, T cells, in particular Th17 cells, were increased in number in PD brain and blood. Furthermore, Th17 cells induced cell death in co-cultures autologous induced pluripotent stem cells (iPSC)-derived neurons from PD patients (113). Interestingly, aSyn is able to activate helper and cytotoxic T cell responses in PD patients, which suggests a possible role of autoimmune inflammation in PD (114).
Taken together, these current data implicate that PD is an extraordinary complex disease with many pathophysiological processes driving disease progression. It becomes evident, that PD is rather a systemic disorder with a variety of pathological facets than ‘just' neurological degeneration.
Gut-Brain Axis in PD
Approximately 80% of PD patients suffer from GI manifestations (115), including constipation, which seems to be an important risk factor for PD (116). As mentioned above, intestinal symptoms may precede motor manifestations by several years, suggesting that PD might originate in the gut. This is in line with the estimation that 90% of idiopathic PD cases are due to oral ingestion of substances causing cell toxicity (oxidative stress, mitochondrial dysfunction), such as herbicides and pesticides (117). According to the hypothesis that PD originates from the gut (118), aggregates of aSyn were detected in the intestine of PD samples (119). Despite the evidence of gut to brain communication in the context of PD, there are still open questions regarding (A) the exact localization in the gut where PD might originate, (B) the dissemination pathways within the gut and to the brain, (C) the declutching event of proteinopathy in the gut, and (D) the role of the intestinal microbiota, as well as microbiota-epithelial-immune communication. All this lacking information is indispensable in order to develop potential PD diagnosis strategies based on GI premotor symptoms.
aSyn in the Gut and Its Propagation to the CNS
As already mentioned, LBs and different aSyn conformers were observed in variety of organs despite the brain. aSyn was reported to be present in the spinal cord and the peripheral nervous system (PNS) including the paravertebral sympathetic ganglia, vagus nerve, the GI tract and among others (120, 121). Indeed, phosphorylated aSyn, a pathological form of aSyn, has been detected in the GI tract up to 20 years before onset of PD motor symptoms (9). Also, Braak and colleagues hypothesized that synucleinopathy begins in the anterior olfactory nucleus and the dorsal motor nucleus of the vagus nerve (DMV) (dual-hit theory) (8, 23), favoring the idea that PD pathology invades the brain via retrograde axonal transport (25, 26). Braak and colleagues even suggest that a pathogen, a pathogen-derived component or other exposures are entering the nervous system through axons of the myenteric (Auerbach's) plexus and/or the submucosal (Meissner's) plexus via postganglionic neurons and may trigger aSyn conformation to aggregates and fibrils (8, 122). Thus, the microbiota has been suggested as a key player, since local immune activation can lead to systemic inflammation affecting the BBB, finally causing neuroinflammation and neurodegeneration (123) (Figure 2). Although it is still not clear whether microbiota changes are cause or consequence, dysbiosis is considered as a risk factors for PD development.
Detection of aSyn in the Gastrointestinal Tract
In the context of the ENS, aSyn was first identified in the esophagus and the colon (124), but it is still not clear where the deposition under pathological conditions initiates. Current literature demonstrates that aSyn can also be detected in salivary glands (125), pharyngeal sensory nerves (126), the esophagus (120), the stomach and the small intestine (127), the colon (123), and the appendix (128). Colonic aSyn has been detected even in premotor PD (119, 129). These observations postulate detection of intestinal aSyn as a diagnostic tool in PD, even in early phases of the disease. However, inconsistencies in the detection of aSyn conformers imply the need of alternative and more accurate methods for its detection (granular staining in the lamina propia, perivascular/vascular wall mucosa staining, lacy-granular pattern in the submucosa, or epithelial cell nuclear staining, 2D/3D electrophoresis) (24).
Monomeric aSyn expressed in gut neurons can be released in form of free protein or exosomes, which can be taken up by neighboring neurons via endocytosis (130). Most commonly, aSyn is transported directly from neuron to neuron (131, 132), which requires close cellular contacts and intact synaptic connections (133). In the gut, this is possible via the connection between submucosal/myenteric neurons to the preganglional vagal nerves, which allows aSyn propagation (134). Proteinopathy within the GI innervation might be due to a neurotropic pathogen/agent, which initiates Lewy pathology in the gut (8). Therefore, a connection between the ENS and the mentioned agent is necessary, since neurons/nerves do not reach the intestinal lumen. An attractive candidate in this context would be the intestinal epithelium, which is in direct contact with luminal content, and therefore acts as a physical and immunological barrier in the gut. On the other hand, disturbances of intestinal sealing in chronic intestinal inflammation leading to leaky gut might allow direct contact of the initiating factor and the ENS (Figure 2) (135–137).
Propagation of aSyn Between the Gut and the CNS
The next important question is how aSyn propagates from the ENS to the CNS. The connection between the ENS and the CNS, so called gut-brain axis, permits a mutual effect from the ENS to the CNS, and vice versa. This communication mainly occurs via the sympathetic system and the vagus nerve of the autonomic nervous system, and the spinal cord. Four levels of control have been defined (138): (1) ENS, including myenteric and submucosal ganglia, and enteroglial cells; (2) prevertebral ganglia (visceral reflex responses); (3) spinal tract the through tractus solitaires in the brain stem and the dorsal motor nucleus of the vagus nerve; and (4) cortical and basal ganglia neurons. Healthy individuals maintain intestinal functions, and patients with neurodegenerative disease suffer from GI problems, not only PD, but also Alzheimer's disease, transmissible spongiform encephalopathies, or amyotrophic lateral sclerosis; while GI disorders leads to CNS-related symptoms. The connection between the vagus nerve and the luminal content has been suggested to be mediated via EECs (139), which might produce metabolites acting on the vagus nerve, transmitting information from the nutrients toward the brain, in a glutamatergic neurotransmission (140) (Figure 2).
The vagus nerve, one of the largest nerves connecting the gut and brain, is considered to be the direct link between these two organs (141). Recent data from rodent models could evaluate a direct propagation of aSyn pathology from the gut to the brain via the vagal nerve (28, 142–144) (see section Animal Models of PD and GI Symptoms). Moreover, there has also been also research in alternative hypothesis of a brain-to-gut spread of aSyn pathology, showing that a vector-mediated overexpression of aSyn in the midbrain lead to accumulations of aSyn in enteric nerves and stomach walls (145). Further, a more recent study presents that a nigral overexpression of aSyn exerts significant alteration on the ENS followed by changes in the microbiome (146). Subsequently, loss of neuronal plexus and activation of glial cells in the gut impact on intestinal permeability, barrier function, inflammation, and GI motor functions. Taken together, this data suggests a bidirectional potential of aSyn to move both anterogradely and retrogradely within neurons (Figure 2). If the vagus nerve is the main route of bidirectional aSyn transmission, vagotomy could be protective against developing PD. Studies questioning whether a vagotomy leads to a reduced risk to develop PD could not find a strong association (147, 148). Only when the cases of a full truncal vagatomy were restricted >20 years after surgery a decreased risk for subsequent PD was observed (149). Overall, many studies support the idea of aSyn gut to brain and inversely brain to gut spread, however, there are still clinical studies missing that investigate the start and/or early development of PD progression, respectively.
Another way of aSyn transmission from the gut to the brain and vice-versa is thought to be possible through extracellular vesicles called exosomes, which are found in the blood serum and CSF of PD patients (Figure 2) (150–152). In fact, it was shown that exosomes derived from PD patients incorporate oligomeric aSyn and spread oligomerization of aSyn in a dose-dependent manner (130, 153, 154). An alternative gut-brain communication via the circulation has been also suggested in primates, where the damage of the CNS could be observed upon intestinal injection of aSyn without affecting the vagus nerve, but elevated aSyn levels in the circulation (155). Overall, this indicates that exosomes may function as intracellular cargo distributing aSyn pathology throughout the body (Figure 2).
Enteric Nervous System (ENS)
Being the largest and most complex part of the peripheral nervous system (PNS), the ENS controls crucial functions within the gastrointestinal tract, such as peristalsis, substance transport, or local blood supplies. The ENS innervates the whole GI tract, from the mouth to the rectum, including the salivary glands. Neuron networks in the gut wall formed ganglia, which are interconnected by dense fiber bundles. The nerve plexuses are organized in myenteric and submucosal plexuses, which are, in turn, interconnected. The myenteric plexus is localized between the longitudinal and circular muscle layers throughout the GI tract, and controls smooth muscle activity and motility. The submucosal plexus is located mainly in the small and large intestine, also in the stomach, but not in the esophagus (156).
ENS-mediated control of the GI function is independent from the CNS; therefore, the ENS allows complete sensory-motor reflexes, based on the existence of primary afferent neurons, interneurons and motor neurons. However, apart from this intrinsic innervation within the ENS, the gut is also innervated via the sympathetic and parasympathetic nervous system. More than 100 million entities from 20 different neuron subtypes (depending on the expression of neuropeptides) coexist with enteroglial cells (EGCs) in the ENS. EGCs express glial fibrillary acidic protein (GFAP), vimentin and S-100, but also receptors for cytokines, neuropeptides and neurotrophins, and therefore, have a dual function on the ENS. As astrocyte-like cells they also contribute to the function of the intestinal immune system. Moreover, ECGs participate in the structure of the ENS and contribute to the maintenance of mucosal barrier and tissue homeostasis (157). Interestingly, EGCs also serve as a communication tool between IECs and the ENS (158). Among ENS neurons, dopaminergic neurons are present in both plexus (159), and are more frequent in the proximal part of the GI tract; although the association between the loss of dopaminergic neurons and PD has been only demonstrated in the colon (160).
Microbiota
Seeing it as a super-organism, the human body is not only composed of human cells but also numerous microorganisms colonizing at mucosal surfaces, allowing various important body's functions such as maturation, education of host immune responses, protection against pathogen proliferation, and induction of responses to specific drugs. The human gut-microbiome carries millions of microorganisms and indeed, has been defined as the most complex ecosystem ever. It contributes greatly to intestinal immune function as a consequence of the continuous contact with gut lumen commensals and potentially harmful agents. A symbiotic relationship between the human body and these microorganisms permits the digestion of nutrients and pathogen colonization resistance. Thus, the intestinal microbiota modulates several functions of the gastrointestinal tract, such as permeability (161), mucosal immune function, motility (162), sensory nerve function and ENS activity (163). Interestingly, it is also associated with brain functions (164), such as response to stress (165), emotions (138), pain, digestive behavior (166), and brain biochemistry (167).
The balance between the human body and the microbiota (eubiosis) is challenged by several external factors, such as antibiotic treatment, various diseases, highly processed foods or lack of sleep. This can lead to microbiota alterations or dysbiosis, which in most cases is shown by variations in the composition and reduced diversity between different species. Dysbiosis has been associated with several pathological situations, including IBD (168) and PD (169) (Figure 1); although in many cases it is not clear whether its alteration represents cause or consequence of the subjacent pathology. The most accepted hypothesis for the pathogenesis of IBD claims that chronic intestinal inflammation occurs as an exacerbated immune response against components of the microbiota in genetically predisposed individuals. The first hint pointing to the association between the intestinal microbiota and IBD came from animal studies showing that experimental inflammation in a number of well-established animal models was abolished in germ-free mice (170). In addition, inflammation could be challenged upon colonization with caecal bacteria, while specific species were able to protect upon recolonization. Despite numerous efforts in order to identify a single specie capable of triggering chronic intestinal inflammation (171), nowadays IBD is considered as a polymicrobial disease, where dysregulation in the composition of the microbiota affects several species. In addition to activation of signals upon detection of the microbiota or derived-antigens, another important aspect is the release of metabolites derived from the microbiota. This has been identified using next-generation sequencing, metagenomics and metabolomics, allowing the description of the microbiome and its potential alterations (172, 173).
Based on the relevance of the intestinal microbiota, its modulation in order to restore eubiosis, appears as an attractive strategy for therapy purposes. In this context, fecal transplantation implies the transfer of microbiota from healthy donors to IBD patients. Fecal microbiota transplantation (FMT) has been tested in various pathological conditions, such as IBD, diabetes type 2 and even neurodegenerative disease. A recent study demonstrates the efficacy of this strategy in an experimental colitis model induced by adoptive transfer of naïve T cells, since transfer of healthy vs. IBD patient fecal content permits restoration of T cell responses (decreased Th17/Th2); and increased Treg/IFNγ and ameliorates thereby colitis (174). Despite limitations based on the donor testing, the limited duration of the treatment and the potential alterations upon antibiotic treatment, FMT it is approved for the treatment of other intestinal conditions, such as Clostridium difficile infections (175, 176). In addition to fecal transplantation, a recent review collects other therapy strategies based on the modulation of the microbiota via direct or indirect mechanisms, such as enteral nutrition; pre-, pro-, and post-biotics; inhibition of Adherent-invasive Escherichia coli (AIEC) adhesion and tungstate treatment (168). All these strategies to restore eubiosis are potentially valuable in diverse pathologies coursing with dysbiosis.
Microbiota in PD
Compared to GI homeostasis, more surprising is the association between microbiota and brain function, and the fact that the intestinal flora modulates immune, endocrine, and neuroendocrine maturation in nervous system sprouting. Colonization of the human gut upon birth is important for neonatal brain development, since it allows the synthesis of vitamins and fatty acids, regulation of BDNF (Brain-derived neurotrophic factor), synaptophysin and PSD-95 (177). Experimentally, sterile mice elicit decreased expression of BDNF in the cerebral cortex and hippocampus, and they show signs of anxiety and less activity performance (178); while another study shows that recolonization with healthy flora permitted production of different neurotransmitters (NTs) and the abolition of anxiety symptoms (179). An additional important aspect to be considered is the ability of the microbiota to directly produce inhibitory NT (GABA) or regulate their synthesis by the host (180, 181). Moreover, GABA signaling system (GAD and GABAAR) was detected in IECs and GABAAR stimulation played important role in regulating intestinal fluid secretion in rat (182). On the other hand, preventing the reuptake of NTs (for example, inhibiting 5-HT reuptake by fluoxetine) can regulate colonization in the gut (183). In addition, important to mention here is the production of short-chain fatty acids (SCFA) as microbiota-derived factors, which can affect the CNS thank to their passaging through the BBB via specific transporters. SCFAs in the brain regulated microglia homeostasis (184), have impact on G-protein coupled receptors (GPCRs) (185, 186) and maintain to the GPR41-mediated SNS activity (187). According to an association between brain function/development and colonization of the intestinal tract, the microbiota impacts then on social behavior, sleep cycle, mood disorders, and neurodegenerative disease including Alzheimer's disease and PD (188).
In the context of the gut-brain axis, components of the microbiota and its metabolites can act directly on neurons at the ENS, or signal through IECs (Figure 2) (189). Nowadays, several pieces of evidence demonstrate a correlation between dysbiosis and prodromal signs in PD (190–192). Importantly, changes affecting Firmicutes, Prevotella, Helicobater pylori (193), Bacteroides, or Bifidobacterium (194) as well as the imbalance between pro- and anti-inflammatory species, and the increased release of LPS should be mentioned (195). Based on a recent Metabolome wide association studies (MWAS) (196), the dysbiosis in PD patients is characterized by: increase of opportunistic pathogens (Porphyromonas, Corynebacterium, Prevotella, Porphyromonas, and Corynebacterium); reduction of SCFA-producing bacteria (Oscillospira, Lachnospiraceae_UCG-04, Lachnospiraceae_ND3007_group, Agathobacter, Butyricicoccus, Blautia, Faecalibacterium, Lachnospira, Fusicatenibacter, Roseburia); and elevated carbohydrate-metabolizing probiotics becoming immunogenic (Lactobacillus or Biffidoacerium). An independent meta-analysis of 223 PD vs. 137 control patients from America and Europe suggests elevation of Akkermansia, Catabacter genera, and Akkermansiaceae family together with reduction of general Roseburia and Faecalibacterium (197). Beyond alterations of the microbiota composition, related metabolic changes have also been observed in PD patients, such as reduced carbohydrates fermentation, butyrate synthesis, increased proteolytic fermentation, and amino acid metabolism (198). Interestingly, some of these metabolites play crucial roles for nervous system-related intestinal functions; for instance, SCFA contribute to 5-HT release and colon motility, proving again the gut-brain connection (199).
Interestingly, many of the PD-related microbiota alterations can also be linked to dysbiosis in IBD. Akkermansia muciniphila is a well-known actor in the context of IBD, since it can degrade the mucus layer and thereby impair the barrier function (200), which might favor the contact between the luminal content and the ENS. On the other hand, Roseburia and Faecalibacterium (197) possess an anti-inflammatory effect in IBD, due to their ability to produce SCFAs (201, 202); while decreased Prevotellaceae is associated with alterations of intestinal permeability via a similar mechanism (203). On its part, accumulation of Enterobacteriaceae leads to increased levels of LPS, explaining its correlation with disease progression and motor symptoms. Increased LPS levels can contribute to GI alterations by several mechanisms, such as causing epithelial leakage (204), inducing the production of cytokines and inflammation. Moreover, it can pass through the BBB (205), triggering direct destruction of the substantia nigra (206). Based on the neuroprotective effect of SCFA and ghrelin, reduced Lactobacillaceae can also affect intestinal inflammation, correlating with disease severity (207). Jointly, overlaps between dysbiosis profiles in IBD and PD might contribute to the associated barrier function alterations.
Changes in the gut microbiota composition might lead to aSyn accumulation in the gut, originating oxidative stress and mucosal inflammation. However, it is not clear whether changes in the microbiota composition, PD associated symptoms (constipation) or PD pharmacological treatment are a consequence of aSyn proteinopathy. Supporting a causative role of microbiota and/or microbiota-derived factors, a recent study shows induction of motor symptoms in mice upon fecal transplantation from human PD patients, due to aSyn pathology and neuroinflammation engendered by microbiota metabolites, such as SCFA. Furthermore, aSyn overexpressing mice (under the Thy1-promoter) show less motor symptoms in germ-free conditions, as well as upon antibiotic-treatment; while colonization with healthy or, in particular, PD patient-derived microbiota, lead to worsening of motor symptoms (192).
Beyond commensal bacteria, also pathogens in the lumen interact with the ENS, mostly via non-neuronal cells, such as EECs within the intestinal epithelium. On the other hand, local gut infections can impact on affective state and emotional responsiveness. This communication occurs via toxins promoting secretion and therefore, diarrhea, as observed in the case of Vibrio cholera, Clostriiodes difficile; or toxins promoting emesis, including Staphyloccoccus aureus or Bacillus cereus. However, not only bacteria, also viruses and parasites demonstrate an interplay with the ENS and CNS. The viremic hit hypothesis defends that PD occurs upon Influenza and HSV1 infections (dual-hit theory), leading to the aSyn aggregation in peripheral nervous tissues, and subsequently propagation to the brain (208–210). Interestingly, HIV targets the ENS, since it activates glial cells, which can then be propagated to the CNS. Furthermore, HIV Tat peptide can synergize with LPS by interfering with TLR4, inducing the release of cytokines, and promoting the proinflamamtory effect of LPS (211). ENS infection by HSV-1 leads to macrophage recruitment, releasing ROS and causing ENS neuroplasticity and destruction of enteric ganglia as well as GI dysmotility (212). Finally, parasites modulate 5-HT secretion, the release of enzymes degrading NTs, such as acetylcholinesterases (Anisakis or Schistosome), and NT secretion, while they are tightly linked to the immune system function (213).
Intestinal Epithelium and “Leaky Gut”
The intestine is in charge of nutrition and water/ion absorption, but represents also a fundamental immunological organ, harboring the most extended immune cell population in the body. On its part, the intestinal epithelium together with the attached mucus constitute a physical and immunological barrier segregating the environment (intestinal lumen) and the human body. The gut epithelium consists of a monolayer of columnar epithelial cells allowing trans- and para-cellular transport required for nutrition, however, simultaneously impairing the invasion of potentially harmful pathogens. Thus, sealing of the epithelium has to be tightly maintained, in order to prevent transmucosal passage of microbiota-derived factors, which can then get in contact with the plethora of immune cells present in the sub-epithelial space. This is achieved via intercellular junctions (tight junctions, adherens junctions, and desmosomes) (214), as well as a tight regulation of cell architecture and polarity, mostly regulated by the function of the actin-myosin cytoskeleton (12, 215). Together, the intestinal epithelium accounts for the intestinal barrier function, which has been critically involved in the pathogenesis of intestinal disorders, such as chronic intestinal inflammation (including IBD) (216). This barrier function is challenged during the renewal or turnover of the epithelial layer. Lgr5+ stem cells located at the crypt bottom proliferate and give rise to pluripotent daughter cells located in the transient-amplifying area, which, in turn, differentiate into five IEC subtypes [enterocytes, goblet cells, paneth cells, EECs, and tuft cells (217)]. All differentiated IECs, except paneth cells which remain at the crypt, migrate upwards to the villus tip (small intestine) or the crypt surface (colon), where aged cells will be extruded to the lumen (cell shedding) and finally die. Temporary leakage occurring at the villus tip is tightly regulated by rearrangement of tight junctions and the so-called zipper effect of neighboring cells (218, 219), which allows resealing of the epithelium.
As mentioned above, the maintenance of epithelial integrity plays a fundamental role to keep tissue homeostasis in the gut, and therefore, avoid inflammation (220). Loss of epithelial sealing and leakage of the intestinal layer has been associated with chronic inflammatory disorders, such as IBD (221). Indeed, some observations claim that epithelial-intrinsic alterations can play a causative role in the disease. For instance, increased intestinal permeability appears in non-diagnosed relatives of IBD patients (222), and precedes flares in patients with an IBD diagnosis (223), suggesting that epithelial leakage heads the activation of the inflammatory response. Moreover, based on immune-epithelial communication in the gut, epithelial architecture and function can also be modified due to the effect of pro-inflammatory mediators present in the gut mucosa upon activation of an immune response, such as immune-cell derived cytokines (TNF, IL-6, IL-1β, IL-13, etc.). These cytokines affect mainly tight junction assembly (224), activation of different cell death pathways or cell shedding (225), as well as IEC damage (226). Altogether, via epithelial intrinsic and extrinsic mechanisms, epithelial barrier function is challenged in the context of IBD, and this correlates with pathogenesis of the disease. A proof of this association are recently introduced epithelial restoration therapy strategies, which indeed show promising results in the context of IBD pharmacological management (227, 228).
Beyond being a pure physical fence against components present in the lumen, the intestinal epithelium displays innate immune responses based on the expression of pattern-recognition receptors (PRRs), allowing them to recognize pathogen-associated molecular patterns (PAMPs) from diverse microorganisms in the lumen, amplify the initial immune response, and finally prime the adaptive immune system. PRRs also recognize endogenous molecules produced in stress conditions, so called DAMPs. Membrane-bound [TLRs and C-type lectin receptors (CLRs)] and cytoplasmic Nucleotide-binding oligomerization domain-like receptors or NOD-like receptors (NLRs), retinoic acid-inducible gene-I-like receptor (RLRs), absent-in-melanoma 2 (AIM2)-like receptors, and cyclic GMP-AMP synthase (cGAS) receptors act together in order to detect pathogens in multiple cellular compartments. Although TLRs are the best characterized PRRs, they are not unique in the context of IECs and IBD; others relevant receptors comprise CLRs (229) and NLRs (NOD2) (230, 231).
Focusing on the well-studied TLRs, deficiency of TLR2 is associated with aggravated colitis in DSS-treated mice (232) and multidrug resistance colitis (233). Similarly, poly(I:C)-mediated TLR3 activation protects epithelial barrier function and ameliorate DSS-induced colitis (234, 235). In contrast, several strategies based on TLR4 blocking show promising results in the context of epithelial restoration in IBD (not in the case of necrotizing enterocolitis), while constitutively activated TLR4 predisposes for DSS-colitis and colitis-induced neoplasia (236–238). Mechanistically, this is based on impaired NF-kB-mediated cytokine production and migration of epithelial cells. Although TLR5 was identified as one of the first IBD loci and its deletion triggers spontaneous colitis (239), controversial results regarding flagellin-mediated activation implies the need of future studies in this context (240). Another important candidate is TLR7, since its activation leads to production of antimicrobial peptides (AMPs) and protects against DSS (241) or TNBS colitis. Interestingly, as already mentioned in section Neuroinflammation in PD, recent studies suggest aSyn as a DAMP-activating TLRs on the surface of microglia (108, 109). This opens the hypothesis of a TLR-mediated recognition of aSyn in the gut, even via specific stimulation of intestinal epithelium or IEC subtypes. As mentioned above, it is important to consider potential specificity of TLR activation based on the conformation of the different aSyn aggregates in this context (242).
Intestinal Permeability in PD
PD pathogenesis is associated with “leaky gut” (Figure 1) and increased intestinal permeability (243), correlating with aSyn and LPS levels in the mucosa (190). Elevated intestinal permeability in turn promotes subsequent inflammation, and therefore, aSyn accumulation and aggregation in the ENS (192). In fact, increased expression of pro-inflammatory cytokines and glial markers, also in the gut, positively correlated with disease progression and severity. Mechanistically, recent studies have suggested that PD patients (123) and animal models of PD show altered expression and distribution of tight junction proteins, such as ZO-1, E-cadherin (244), and claudin-1 (245).
aSyn and Intestinal Epithelial Cells
Beyond the association between PD and decreased expression of tight junction proteins within the intestinal epithelium (123), the current knowledge about a potential interaction between aSyn and the intestinal epithelium is still scarce. The fact that EECs express aSyn make them attractive candidate players in this context [see chapter Enteroendocrine Cells (EECs)]. aSyn can be transmitted in a prion-like manner from epithelial cells to enteric neurons (30). Enteric glia is a crucial communication tool between the intestinal epithelium and the ENS. Thus, intestinal pathological conditions associated with alterations of epithelial permeability might trigger alterations of the EGCs as the declutching event for a local immune response and neuroinflammation affecting the ENS (see chapter Neuroinflammation in PD). In order to get in contact with IECs, aSyn should translocate across the mucus barrier protecting the monolayer of epithelial cells. A recent study has shown that, despite mucoadhesive properties, aSyn penetrates the mucus by inducing rearrangement of the mucin matrix (246). Other studies suggest that rather than aSyn itself, other phenomena associated with alterations of the microbiota, such as increased levels of LPS are responsible for epithelial alterations, including redistribution of ZO1 and E-cadherin (244). Enteric biofilms are produced by bacteria in the gut in order to promote their survival, and can in turn, activate local immune response, since some of their components act as DAMPs activating TLRs, for instance. Curli-containing biofilms in several experimental infection models caused alterations of the epithelial layer; the mechanism behind includes the fibrillization of aSyn (247). Undoubtedly, further research on the impact of aSyn on IECs, as well as other mechanisms explaining epithelial alterations in the context of PD pathogenesis are required.
The use of brain organoids derived from PD patient iPSCs has been extended in the last years. Recently, technical development in the field, such as co-culture of neuronal cells with astrocytes (248) and the use of assembloids have permitted modeling of cellular crosstalk between different areas of the brain (249). However, controversial opinions about the ability of these in vitro models to mimic complexity of the human brain still exist. In the context of PD, midbrain organoids containing dopamine-related neurons, astrocytes and oligodendrocytes (250) have demonstrated to recapitulate pathological hallmarks upon appropriate conditions (e.g., LRRK2 mutations), such as neurotoxic damage, endosomal phosphorylated aSyn, and increased mitophagy (27). Future advances regarding midbrain organoids may be the inclusion of other cell types, such as microglia, which enables to study the relevance of innate immunity in PD. Therefore, two strategies have been proposed: on one hand, the development of brain organoids including microglia (251); and on the other hand, exogenously add iPSC-derived microglia to brain organoids (252). Moreover, in order to model the BBB and the potential immune cell trafficking, neurovascular communication has been developed and implemented via organ-on-chip technology (endothelial-like cells, astrocytes, and neurons) (253).
The relevance of the gut-brain axis in PD opens the path for exploiting intestinal organoids as in vitro models of PD. Described in 2009, intestinal organoids or enteroids are 3D structures developed from intestinal stem cells cultures allowing the intricate differentiation of IECs, and mimicking the complex architecture of the intestinal epithelium (254). Although extremely useful in the context of of epithelial-intrinsic phenomenon, two aspects of intestinal organoids limit their use in studies dealing with microbial-epithelial communication. On one hand, the apical side of the polarized epithelium is projected toward the inside of the organoid (lumen) and makes microbial stimulation highly challenging; and on the other hand, culture conditions with high oxygen concentrations are not optimal for the growth of a vast majority of anaerobic intestinal microbiota. Moreover, some limitations also accounted in the case of co-culture settings with immune cells, for instance, the lack of nutrient support and mechanical constrains to immune cells mediated by blood flow and circulation. Thus, organ on a chip cultures mimicking the inter-organ communication and allowing the interaction with the microbiota as well, appear as suitable alternative. Highly challenging tissue engineering approaches combined with transplantation into mice have tried to implement in vitro systems including the ENS to co-cultures of intestinal organoids and smooth muscle cells; however, these strategies have not been successful until now, based on the lack of maturity of neuronal cells (255). More advances have been achieved in the context of immune-epithelial and microbiota-epithelial communication in organoid cultures. Addition of macrophages affected epithelial barrier function and maturity (256); while neutrophils in combination with pathological bacteria cause loss of epithelial integrity (257) and epithelial development and/or maturation is promoted by TNF-producing CD4+ T cells (258).
A step further in the field of PD research will be the combination of gut and brain organoids. Recent advances have focused on “patient-on-chip” models, such as the combination of separately developed multiorgan organoids (259); or the use of gut organ-chip models fluidically coupled to vascular endothelium lined channels (260), such as MINERVA (MIcroboita-Gut-BraiN EngineeRed platform to eVAluate intestinal microflora impact on brain functionality) (261). Experimental setups based on intestinal organoids and multiorgan organoids might provide important knowledge of the communication between the gut and the brain.
Enteroendocrine Cells (EECs)
Considered sensory cells within the secretory lineage of IECs, EECs represent the largest source of hormones in the body and play vital roles in many physiological processes like appetite control, sensing of gut microbiota, GI immunity, motility, barrier function, insulin and growth hormone secretion (262). Upon sensing of nutrients, EECs produce neuropeptides and hormones to the basal space. In the gut epithelium, enterochromaffin cells (ECs)—a subtype of EECs, react to mechanical forces during gut peristalsis by secreting 5-HT, accounting for 95% of body 5-HT (263). For decades, 5-HT is known as an important neurotransmitter signaling molecule, holding a key role in gut motility, secretion and pain sensation. Many studies indeed showed the link between abnormal regulation of 5-HT and GI disorders, such as IBD and irritable bowel syndrome (IBS) as well as in many CNS disorders (264, 265), suggesting a significant role of 5-HT in gut-brain-gut communication. Recently, EECs have been proposed as an alternative source for Notch ligands, supporting the stem cell population in Paneth-deficient mice (266). Therefore, it is predictable that many gut dysfunction diseases, including IBD, are associated with EECs alterations.
EECs possess a tightly organized apical brush border, and basal membrane projections (neuropods) allowing the intercellular communication with nerves and neurons (267). Interestingly, EECs show a certain overlapping expression profile with neuronal cells, such as neurotrophin receptors, pre- and post-synaptic proteins including aSyn, neurofilaments mimicking axons and their functions (neuropods), and dopamine synthesis machinery (268). Indeed, EECs not only synapse with enteric nerves (29) but also establish a direct contact with enteric glia (269). Thus, EECs can serve as a connection between the intestinal lumen and the ENS, and represents a key population in the context of gut-brain axis in neurodegenerative diseases (267). Besides direct cellular contact, EECs communicate with the ENS via the release of NT and hormones; or even act as the entry pathway for pathogens, which can then act on neurons in the gut. Most importantly, based on their neuron-like features, they can serve as niche for proteinopathy upon luminal signals, which is further supported by the expression of aSyn from these cells (30). Hence, the question arises, whether EECs may be the starting point or declutching event for aSyn pathology in the gut, which is then further transmitted to the CNS.
The fact that EECs express aSyn opens the path for the study of proteinopathy specifically in these cells. An important aspect in this context is the exposure of EECs to the lumen, which make them accessible via endoscopy, as a future early diagnostic tool of premotor PD (270). Interestingly, different TLRs (TLR1, 2, and 4) are expressed in EEC cell lines (271); while TLR4, -5, and -9 ligands induced secretion of EECs hormones in mice (272). On the other hand, Bacteroides thetaiotaomicron contributes to neurogenic colon activity via a TLR2- and EEC-dependent mechanism (273). Interestingly, TLR overstimulation has also been suggested in PD pathology (274). Another mechanism by which EECs contribute to the barrier function might be mediated by the expression of SCFA receptors, such as FFAR2 and FFAR3 (275–277).
Interestingly, qualitative and quantitative alterations of EECs have been associated with GI dysfunctions also observed in PD, such as constipation or alterations of transit times. Rotavirus infection courses with EEC-mediated 5-HT secretion, which activates the ENS and the extrinsic vagal afferent to the brain causing nausea, vomiting, and diarrhea (278). In contrast, increased 5-HT secretion protects intestinal barrier function due to the production of neutropic factors (279). Similar EECs-5-HT-dependent mechanisms operate also in diarrhea upon viral infections, such as Adenovirus infection (280) and even COVID-19 patients (281).
Mechanosensations in the Gut
Mechanosensation is vital for proper function of electrically excitable organs, those constantly exposed to and/or generating mechanical forces (heart, bladder, and GI). Physiologically, all cells in the gut epithelial layer are mechanosensitive, they need to sense the static forces (e.g., stretching, crowding) to adjust cell numbers and maintain epithelial integrity. Among them, so-called mechanosensitive cells, develop specific ion channels to sense acute mechanical forces (e.g., pressure from luminal food content); these cells are important to maintain gut functions like food digestion and peristalsis. Beyond peristalsis, mechanical issues are also crucial for maintenance of epithelial architecture. It is well-known that stem cell proliferation is important to maintain tissue homeostasis and avoid pathological conditions. Interestingly, in Drosophila, the strict regulation of stem cells is indeed associated with food digestion via gut epithelial stretching. Changes in mechanical properties upon ingestion (gut distension), lead to the decrease of misshapen (a Hippo pathway regulator) membrane association and phosphorylation, which then stimulates stem cell activity and contributes to control intestine adaptive growth (282). During epithelial turnover, aged or damaged cells are shed into the lumen in order to leave space for newly generated cells. This process must be tightly governed to maintain epithelial integrity, and therefore requires intercellular sensing communication between shedding and neighboring cells to finally extrude the dying cell. In general, little is known about biochemical pathways governing sensing and responses to mechanical forces.
Although several membrane ion channels have been revealed as important players in this context, the recently identified Piezo channels show their notable roles in many cellular mechanosensitive processes, from light-touch sensing, controlling red blood cell volume to muscular shear stress (283). In Drosophila midgut, the unique Piezo isoform is expressed in low division precursor cells differentiating into EECs. Adult Piezo mutant fly showed decreased number of EECs compared to WT fly. Moreover, Piezo overexpression or increasing Ca2+ level in fly intestinal stem cells induced both cell proliferation and EEC differentiation (284). In zebrafish, Piezo1 ion channel is reported to participate in live cell extrusion (285) and cell division (286), in response to crowding and stretching, respectively. Disturbing cell extrusion via Piezo1 channel lead to formation of cell masses, which hypothetically can lead to tumorigenesis. Gudipaty et al. have proposed a model on how Piezo1 acts as a regulator of epithelial cell number by shifting its localization between nuclear envelope and cytoplasm/ plasma membrane in order to control cell division and extrusion (286). Altogether, these studies suggested that investigating Piezo-mediated mechanosensations will give us insights into intracellular pathways regulating cell numbers and epithelial integrity, and therefore, be relevant in the context of intestinal inflammation and tumorigenesis.
Peristalsis
Peristalsis, or the impulsion of food based on muscle contraction and relaxation, is regulated by sensation of mechanical forces, but the molecular mechanism behind remains elusive. Generally, peristaltic waves in small intestine consist of weak and infrequent contractions around the bolus, while they continuous and gradually increased toward the anus in the colon. Under specific circumstances, for example diarrhea, an intense and powerful peristaltic wave is triggered in the whole small intestine, which quickly relieves mucosa irritation or unusual gut distension. In the small intestine, peristalsis helps driving food against intestinal wall for nutrient absorption and persistently push it toward the large intestine. In the large intestine, peristalsis is important for feces elimination and mechanical removal of gas and bacteria. At a cellular level, when food particles are formed, EECs are stimulated to secrete 5-HT, while mechanosensory neurons in circular and longitudinal muscles are activated to declutch gut motility (287).
The muscle contraction depends on signals received from ENS or CNS, such as substance P, neuropeptide Y or inhibitory neurotransmitters including nitric oxide (NO) and vasoactive intestinal polypeptide (VIP) (288). How the excitatory and inhibitory motor neurons are activated is still a controversy, however, a population of sensory neurons in the distal colon of guinea-pig are believed to be stretch-sensitive rather than muscle tone or contraction sensitive (289).
In order to respond to mechanical stimulations, the intestinal tract contains various mechanosensitive cell types carrying membrane mechanically gated ion channels such as ECs within the epithelial layer, smooth muscles, interstitial cells of Cajal or different types of sensory neurons in the lamina propria. They sense and respond to mechanical changes in different ways; for instance, by 5-HT secretion in the case of ECs. Even though the molecular mechanism behind mechanically induced 5-HT-release in ECs is unknown, recent evidence revealed that Piezo2 ion channel is specifically expressed in human and mouse 5-HT positive ECs, and Piezo2 activation by mechanical forces is necessary for 5-HT release and mucosal secretion (290). Another study suggests that Piezo2 is selectively expressed in a large number of NeuroD1+ cells—a subset of EC cell, and mechanical stimulation of NeuroD1+ cells leads to Piezo2-dependent, but not Piezo1-dependent Ca2+ increase inducing 5-HT production (291). Paradoxically, a newly published study showed that 5-HT release is crucially regulated upon detection of bacterial derived single-stranded RNA by Piezo1 channel in the gut epithelium, indicative of a new potential pathway for gut and bone disorder therapies. Even though the function of the Piezo family in EECs is not clear, Piezo1 was found to regulate gut peristalsis positively in vivo and the lack of Piezo1 in epithelial caused whole gut transit time delay (292). Considering mentioned evidences, Piezo1 and Piezo2 channels in gut epithelium could be possible key elements to uncover the mechanism behind EECs-related mechanisms operating behind constipation and altering transit time in PD (Figure 2). This knowledge might even elucidate the phenomena explaining misfolded aSyn-EECs and reveal the initiation of PD origin.
Constipation and PD
The abnormal defecation and reduced peristalsis can lead to constipation. Physically, constipation occurs when there is a decrease of bowel movement frequency, due to primary (idiopathic or functional) or secondary reasons (diet or medication). Approximately 52.48% PD patients experience constipation (293), making it the most common and distressing PD gastrointestinal symptoms (Figure 1). Indeed, a study with 551.324 volunteers in Taiwan showed that participants with mild to severe constipation symptoms tended to develop PD within 5.5 years and the constipation severity correlated with the risk of having PD (294).
Targeting the Gut for PD Treatment
Current pharmacological treatment for PD patients is based on the principle of escalating DA brain concentration, by (1) increasing/replacing DA levels; or (2) impairing its degradation. Since DA does not cross the BBB, the most commonly used drug is based on the action of Carbidopa/levodopa, a precursor of DA, which crosses the BBB and is believed to convert to DA in the brain. Other available medicines include DA agonists, monoamine oxidase type B (MAO B) inhibitors, catechol-O-methyltransferase (COMT) inhibitors, anticholinergics, Amantadine or Creatine (295). Pharmacological treatment can be also combined with surgery (deep brain stimulation) (296), gene therapy (297), immunotherapy (e.g., antibodies against aSyn) (298), or cell transplantation (299). However, none of the available therapeutic options is actually curative, nor able to stop disease progression (300, 301). Together, the need of alternative therapy strategies in PD is patent, which opens avenues for the identification of innovative strategies.
Considering gut-brain axis in the context of PD, nowadays it is suggested that PD can be, not only diagnosed based on GI manifestations, but even treated “from the gut.” This principle has been also exploited in the context of innovative strategies for levopoda therapy (302). For instance, currently used duopa therapy is based on the application of gels enabling the release of carbidopa/levodopa directly in the gut, allowing slow absorption and, therefore, impairing motion fluctuations and movement disorders. Tightly linked to intestinal function and microbiota, increasing attention has been paid to PD clinical management based on the diet, especially dietary fat. However, conflicting results do not permit drawing conclusive remarks in this context (303, 304); except for the fact that polyunsaturated fatty acid consumption has been associated with lower risk of PD (305). In accordance with the role of the microbiota in the pathogenesis of PD, several strategies modulating the microbiota demonstrated the potential in the context of PD. Antibiotics treatment ameliorate signs of PD, such as IL-1β and, TNF-α at the CNS and dopamine neuron loss (306, 307). Both pre- and pro-biotics have an effect on aSyn proteinopathy. Thus, butyrate activates aSyn autophagy and promotes barrier function of the intestinal epithelium (308). On the other hand, Bifidobacterium and Lactobacillus are able to reverse PD and PD-related constipation (309); while Lactobacillus promotes production of L-DOPA from L-tyrosine (310). The use of probiotics has been found to be beneficial in PD patients (311, 312) and experimental PD models (313, 314). Regarding fecal transplantation, there are controversial results; it is suggested that FMT not only improves GI symptoms (constipation) but also neuroinflammation in PD patients (315, 316); however, safety and efficacy are not clear. Experimentally, FMT lead to further decreased of Lachnospiraceae and Ruminococcaceae, and worsening of dyskinesia (191); while FMT from PD patients lead to worsening on motor symptoms in a PD model (192), but motor impairment was also observed in normal mice. FMT can impair TLR4 activation, improve gut dysbiosis, reduce activation of microglia, change NT secretion and the destruction of the substantia nigra (315). FMT can also ameliorate comorbidity in PD patients related to the GI tract, including ulcerative colitis (317).
Animal Models of PD and GI Symptoms
As mentioned above, the interplay between neurological and GI symptoms in PD is also nicely demonstrated in animal models. Thus, here we provide a summary of currently used experimental in vivo models of Parkinsonism, and the occurrence of GI pathological features, as well as aSyn propagation mechanisms supported by experimental observations using corresponding models.
Classical PD models are based on toxin-induced motor manifestations. Intragastric injection of rotenone causes Parkinsonism in mice, without increased systemic rotenone levels (133). The presence of aSyn in the GI tract or ENS depends on factors such as the administration route, dose or length of exposure. Thus, chronic exposure of rotenone involves non-motor GI symptoms (318), however, did not delay gastric emptying (319). Interestingly, rotenone toxicity is associated with changes in the microbiota composition, such as decreased Bifidobacterium and increased Rikenellaceae or Allobaculum (320); while severity of the symptoms is associated with decreased Lactobacillus and increased reactivity to LPS (245). In addition, cell toxicity is induced by the prodrug MPTP and the neurotoxin MPP+, causing dopaminergic neurons/tyrosine hydroxylase + (TH) neuron destruction in the brain and in the colonic ENS (321, 322). However, controversial data exists about the outcome of this GI affectation, and it is not clear if this is associated with increased intestinal motility (323) or constipation (322, 324). Strikingly, recent publications support a MPTP-mediated intestinal immune response, which might be provoked by activation of monocytes (325). Furthermore, direct brain injection of 6-OHDA induces PD-like constipation, delayed gastric emptying, and enteric inflammation (326, 327). Last but not least, paraquat injection into rats also demonstrated the relevance of the gut-brain axis, since it evokes reduced gastric motility tone and increased aSyn immunoreactivity in the DMV, which is blocked upon vagotomy (328, 329).
Most experimental genetic models are based on the induced expression of aSyn or mutations on the gene encoding for aSyn. These models recapitulate aSyn aggregation, similar to PD patients, however, require more time for pathological manifestations. In accordance with the gut-brain axis hypothesis, these models confirm that GI dysfunction and non-motor symptoms might represent early pathological features. The most commonly used model is the Thy1-aSyn overexpression model, which presents GI manifestations, delayed colon transit time and defecation accompanied by aSyn accumulation in colonic myenteric plexus (330, 331). CNS pathology in Thy1-aSyn mice is reduced upon microbial depletion, while FMT from PD causes worsening of the phenotype (192). However, a recent study claims that levels of LPS rather than microbiota alterations in Thy1-aSyn mice are responsible for colon intestinal permeability dysfunction and early motor manifestations (244). Other genetic models taking advantage of mutations on the aSyn gene, show aSyn accumulation in the olfactory bulb, myenteric plexus and adrenal neurons (aSyn-A53T) or accumulation of phospho-aSyn, slower transit time, abnormal stool and neuroinflammation at the ENS (PrP-A53T-aSyn) (332, 333). Even unique GI affectations, without motor dysfunction can be observed (BAC-A53T-aSyn) (334).
Mutations in PINK1 and PARK2 are associated with PD and activation of immune responses via modulation of mitophagy/autophagy (335). Interestingly, immune response in the context of PINK1 knockout mice is regulated by the microbiota, since colonization with bacteria leads to T cell mediated destruction of dopaminergic neurons in the periphery and the brain (336). On the other hand, the MitoPark model represents a noticeable example of experimental recapitulation of GI dysfunction and dysbiosis in PD Non-motor symptoms in this model include decreased motility and gradual progression of colon transit times, reduced fecal water content and activation of glial cells in the myenteric plexus. Disease progression in this model goes along with loss of TH+ neurons, reduction of central and intestinal DA levels, as well as changes in the microbiota composition (337).
As mentioned above, another important aspect within the gut-brain axis concept is the propagation route for aSyn. Thus, researchers in the field have concentrated on the development of experimental models based on the injection of aSyn. Therefore, pathological aSyn can be isolated from post-mortem human tissue; or recombinant aSyn preformed fibrils (PFF) are experimentally prepared. It has been demonstrated that the injection of patient-derived pathological aSyn directly into the gut leads to deposition of aSyn in myenteric neurons and intestinal inflammation in A53T transgenic mice (338). Intragastric aSyn can be transmitted to the brain in rats (142). Moreover, the injection of recombinant PFF in the olfactory bulb in WT mice caused the spread of aSyn to distant areas of the brain (339). While spreading of aSyn occurred only in aSyn transgenic mice upon injection into gastric wall and not in WT mice (340). Inoculation of PFFs in the duodenum of mice led to GI deficits and physiological changes of the ENS in addition to changes of aSyn histopathology in the midbrain and subsequent motor defects in elder, but not in young mice (28).
Conclusion
As outlined in our review, the disease mechanisms of PD are complex and exhibit a variety of pathological facets. GI manifestations are the most significant symptoms in the prodromal phase of PD (115), suggesting the direct communication of gut and brain. Recent studies have shown that pathogenic aSyn found within the GI system are able to spread and reach the CNS (28, 142, 339). In addition, the role of constipation in PD seems to support the hypothesis that the pathological pathway of PD spreads from the intestine to the brain. Besides, EECs were found to express aSyn and link directly to aSyn-containing nerves, creating neural circuit between the gut and nervous system. This raised an interesting hypothesis that the root of PD might start from misfolded aSyn in EECs, which is transmitted to the nervous system (30). Moreover, constipation is the most troublesome PD-gastrointestinal symptom and likely regulated by abnormal gut peristalsis (293). Accordingly, investigating the roles of EEC-mechanosensitive ion channels, which indeed was proven to be associated to peristalsis, could explain the reason why aSyn in ECs is misfolded, and reveal the mechanism behind PD origin.
Correspondingly, gut inflammation is a main pathological feature occurring in PD and IBD. Inflammatory processes and aSyn pathology appear to be extraordinarily linked to each other. In connection with inflammation, aSyn and its aggregated forms seem to mediate inflammatory responses by TLR activation (108, 109). This indicates the possibility of TLR-mediated release of proinflammatory cytokines in the gut by specific stimulation of IEC. Furthermore, IECs appear as key factor in inflammatory response, as they create a protective barrier against luminal antigens and microbes, helping to preserve gut homeostasis. IEC alterations, for example cytoskeletal rearrangement (12) or cell-to-cell adherens junction reorganization (341) could disturb the epithelial integrity and lead to intestinal permeability as seen in CD patients (342). In addition, PD pathogenesis is also associated with an increased intestinal permeability (243) along with impaired BBB function (97), promoting bidirectional inflammation cascades between the gut and the brain.
Many different routes for transmission between neuronal networks and intestinal cells are described to propagate aSyn pathology. Of interest, extracellular exosomes found in blood and CSF of PD patients have been described to spread pathology (151). Moreover, the vagus nerve is considered to be the most important bidirectional connection between these two organs (141). However, within this context, clinical studies investigating the origin of PD progression are still elusive.
Lastly, it is interesting that dysbiosis is a common feature in PD and IBD (168, 169). In order to affect ENS-specific pathways and spreading to the CNS, a connection between the GI lumen and the neurons/enteroglia is necessary. The intestinal epithelium is in direct contact with luminal content and therefore, acts as a physical and immunological barrier in the gut. Hence, a disturbance of the intestinal sealing allows direct contact of pathological factors and cells of the ENS. Interestingly, IBD (221) as well as PD (243) patients can suffer from intestinal inflammation concomitantly exhibiting a leaky gut. The disturbance of intestinal barrier function has been suggested to promote aSyn aggregation in the ENS, which is further able to spread to the CNS (30, 304), along the so-called gut-brain axis.
In recent years, numerous studies have been addressing the role of the gut-brain axis in neurodegenerative disorders, like PD. However, there are still open questions regarding the understanding about its impact in disease progression and regulation. Further studies and comparisons of disease mechanisms of PD and IBD, as presented in this review, might help to connect missing dots and shed light into the role of aSyn aggregation within the intestine as well as intestinal inflammation in PD. A detailed comprehension of the mechanisms and regulation of the gut-brain axis is essential to establish novel disease biomarkers, clinical read-outs and identify novel targets for (early) treatment strategies.
Author Contributions
AD, PN, FZ, and RL-P wrote the manuscript. MN read the manuscript and contributed to the finalized version. All authors approved the final version of the manuscript.
Funding
This research was funded by the Deutsche Forschungsgemeinschaft (DFG), grant numbers (TRR241-A07, SPP-1782, 125440785—SFB, and 877-B11) and supported by the Interdisciplinary Center for Clinical Research (IZKF) at the University Hospital of the University of Erlangen-Nuremberg (Jochen-Kalden funding programme N8).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank PD Dr. Wei Xiang and Dr. Alana Hoffmann (both Department of Molecular Neurology, University Hospital Erlangen) for proofreading the manuscript.
References
1. Kalia LV, Lang AE. Parkinson's disease. Lancet. (2015) 386:896–912. doi: 10.1016/S0140-6736(14)61393-3
2. Postuma RB, Aarsland D, Barone P, Burn DJ, Hawkes CH, Oertel W, et al. Identifying prodromal Parkinson's disease: pre-motor disorders in Parkinson's disease. Mov Disord. (2012) 27:617–26. doi: 10.1002/mds.24996
3. Postuma RB, Berg D. Prodromal Parkinson's disease: the decade past, the decade to come. Mov Disord. (2019) 34:665–75. doi: 10.1002/mds.27670
4. Hirsch EC, Vyas S, Hunot S. Neuroinflammation in Parkinson's disease. Parkinsonism Relat Disord. (2012) 18(Suppl. 1):S210–12. doi: 10.1016/S1353-8020(11)70065-7
5. Tansey MG, Romero-Ramos M. Immune system responses in Parkinson's disease: early and dynamic. Eur J Neurosci. (2019) 49:364–83. doi: 10.1111/ejn.14290
6. Devos D, Lebouvier T, Lardeux B, Biraud M, Rouaud T, Pouclet H, et al. Colonic inflammation in Parkinson's disease. Neurobiol Dis. (2013) 50:42–8. doi: 10.1016/j.nbd.2012.09.007
7. Braak H, Del Tredici K, Rüb U, Vos RI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. (2003) 24:197–211. doi: 10.1016/S0197-4580(02)00065-9
8. Braak H, Rüb U, Gai WP, Del Tredici K. Idiopathic Parkinson's disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. (2003) 110:517–36. doi: 10.1007/s00702-002-0808-2
9. Stokholm MG, Danielsen EH, Hamilton-Dutoit SJ, Borghammer P. Pathological α-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann Neurol. (2016) 79:940–9. doi: 10.1002/ana.24648
10. Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. (2007) 369:1627–40. doi: 10.1016/S0140-6736(07)60750-8
11. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. (2014) 14:329–42. doi: 10.1038/nri3661
12. Lopez-Posadas R, Sturzl M, Atreya I, Neurath MF, Britzen-Laurent N. Interplay of GTPases and cytoskeleton in cellular barrier defects during gut inflammation. Front Immunol. (2017) 8:1240. doi: 10.3389/fimmu.2017.01240
13. Grenn FP, Kim JJ, Makarious MB, Iwaki H, Illarionova A, Brolin K, et al. The Parkinson's disease genome-wide association study locus browser. Mov Disord. (2020) 35:2056–67. doi: 10.1002/mds.28197
14. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. (2008) 40:955–62. doi: 10.1038/ng.175
15. Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol. (2008) 7:583–90. doi: 10.1016/S1474-4422(08)70117-0
16. Kumari U, Tan EK. LRRK2 in Parkinson's disease: genetic and clinical studies from patients. FEBS J. (2009) 276:6455–63. doi: 10.1111/j.1742-4658.2009.07344.x
17. Park S, Kim J, Chun J, Han K, Soh H, Kang EA, et al. Patients with inflammatory bowel disease are at an increased risk of parkinson's disease: a south Korean nationwide population-based study. J Clin Med. (2019) 8:1191. doi: 10.3390/jcm8081191
18. Villumsen M, Aznar S, Pakkenberg B, Jess T, Brudek T. Inflammatory bowel disease increases the risk of Parkinson's disease: a Danish nationwide cohort study 1977-2014. Gut. (2019) 68:18–24. doi: 10.1136/gutjnl-2017-315666
19. Villaran RF, Espinosa-Oliva AM, Sarmiento M, De Pablos RM, Arguelles S, Delgado-Cortes MJ, et al. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: potential risk factor in Parkinson‘s disease. J Neurochem. (2010) 114:1687–700. doi: 10.1111/j.1471-4159.2010.06879.x
20. Garrido-Gil P, Rodriguez-Perez AI, Dominguez-Meijide A, Guerra MJ, Labandeira-Garcia JL. Bidirectional neural interaction between central dopaminergic and gut lesions in Parkinson's disease models. Mol Neurobiol. (2018) 55:7297–316. doi: 10.1007/s12035-018-0937-8
21. Bercik P, Verdu EF, Foster JA, Macri J, Potter M, Huang X, et al. Chronic gastrointestinal inflammation induces anxiety-like behavior and alters central nervous system biochemistry in mice. Gastroenterology. (2010) 139:2102–12.e2101. doi: 10.1053/j.gastro.2010.06.063
22. Prigent A, Lionnet A, Durieu E, Chapelet G, Bourreille A, Neunlist M, et al. Enteric alpha-synuclein expression is increased in Crohn's disease. Acta Neuropathol. (2019) 137:359–61. doi: 10.1007/s00401-018-1943-7
23. Braak H, Vos RI, Bohl J, Del Tredici K. Gastric α-synuclein immunoreactive inclusions in Meissner's and Auerbach's plexuses in cases staged for Parkinson's disease-related brain pathology. Neurosci Lett. (2006) 396:67–72. doi: 10.1016/j.neulet.2005.11.012
24. Corbille AG, Neunlist M, Derkinderen P. Cross-linking for the analysis of α-synuclein in the enteric nervous system. J Neurochem. (2016) 139:839–47. doi: 10.1111/jnc.13845
25. Breen DP, Halliday GM, Lang AE. Gut-brain axis and the spread of α-synuclein pathology: vagal highway or dead end? Mov Disord. (2019) 34:307–16. doi: 10.1002/mds.27556
26. Leclair-Visonneau L, Neunlist M, Derkinderen P, Lebouvier T. The gut in Parkinson's disease: bottom-up, top-down, or neither? Neurogastroenterol Motil. (2020) 32:e13777. doi: 10.1111/nmo.13777
27. Kim H, Park HJ, Choi H, Chang Y, Park H, Shin J, et al. Modeling G2019S-LRRK2 sporadic Parkinson's disease in 3D midbrain organoids. Stem Cell Rep. (2019) 12:518–31. doi: 10.1016/j.stemcr.2019.01.020
28. Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, Hamilton AM, et al. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci. (2020) 23:327–36. doi: 10.1038/s41593-020-0589-7
29. Bohorquez DV, Shahid RA, Erdmann A, Kreger AM, Wang Y, Calakos N, et al. Neuroepithelial circuit formed by innervation of sensory enteroendocrine cells. J Clin Invest. (2015) 125:782–6. doi: 10.1172/JCI78361
30. Chandra R, Hiniker A, Kuo YM, Nussbaum RL, Liddle RA. α-Synuclein in gut endocrine cells and its implications for Parkinson's disease. JCI Insight. (2017) 2:e92295. doi: 10.1172/jci.insight.92295
31. Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci. (2017) 18:101–13. doi: 10.1038/nrn.2016.178
32. Fahn S. The history of dopamine and levodopa in the treatment of Parkinson's disease. Mov Disord. (2008) 23(Suppl. 3):S497–508. doi: 10.1002/mds.22028
33. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature. (1997) 388:839–40. doi: 10.1038/42166
34. Wakabayashi K, Tanji K, Mori F, Takahashi H. The Lewy body in Parkinson's disease: molecules implicated in the formation and degradation of α-synuclein aggregates. Neuropathology. (2007) 27:494–506. doi: 10.1111/j.1440-1789.2007.00803.x
35. Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. (2013) 9:13–24. doi: 10.1038/nrneurol.2012.242
36. Giasson BI, Murray IV, Trojanowski JQ, Lee VM. A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J Biol Chem. (2001) 276:2380–6. doi: 10.1074/jbc.M008919200
37. Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. (2013) 14:38–48. doi: 10.1038/nrn3406
38. Riederer P, Berg D, Casadei N, Cheng F, Classen J, Dresel C, et al. α-Synuclein in Parkinson's disease: causal or bystander? J Neural Transm. (2019) 126:815–40. doi: 10.1007/s00702-019-02025-9
39. Bartels T, Choi JG, Selkoe DJ. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. (2011) 477:107–10. doi: 10.1038/nature10324
40. Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. (2013) 501:45–51. doi: 10.1038/nature12481
41. Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat Med. (2017) 23:1–13. doi: 10.1038/nm.4269
42. Bosco DA, Fowler DM, Zhang Q, Nieva J, Powers ET, Wentworth P, et al. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate α-synuclein fibrilization. Nat Chem Biol. (2006) 2:249–53. doi: 10.1038/nchembio782
43. Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, et al. Reversible conformational conversion of α-synuclein into toxic assemblies by glucosylceramide. Neuron. (2018) 97:92–107.e110. doi: 10.1016/j.neuron.2017.12.012
44. Hogen T, Levin J, Schmidt F, Caruana M, Vassallo N, Kretzschmar H, et al. Two different binding modes of α-synuclein to lipid vesicles depending on its aggregation state. Biophys J. (2012) 102:1646–55. doi: 10.1016/j.bpj.2012.01.059
45. Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. (2002) 4:160–4. doi: 10.1038/ncb748
46. Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, et al. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science. (2000) 290:985–9. doi: 10.1126/science.290.5493.985
47. Liani E, Eyal A, Avraham E, Shemer R, Szargel R, Berg D, et al. Ubiquitylation of synphilin-1 and α-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson's disease. Proc Natl Acad Sci USA. (2004) 101:5500–5. doi: 10.1073/pnas.0401081101
48. Dorval V, Fraser PE. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and α-synuclein. J Biol Chem. (2006) 281:9919–24. doi: 10.1074/jbc.M510127200
49. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. α-Synuclein locus triplication causes Parkinson's disease. Science. (2003) 302:841. doi: 10.1126/science.1090278
50. Konno T, Ross OA, Puschmann A, Dickson DW, Wszolek ZK. Autosomal dominant Parkinson's disease caused by SNCA duplications. Parkinsonism Relat Disord. (2016) 22(Suppl. 1):S1–6. doi: 10.1016/j.parkreldis.2015.09.007
51. Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat Med. (1998) 4:1318–20. doi: 10.1038/3311
52. Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, et al. The E46K mutation in α-synuclein increases amyloid fibril formation. J Biol Chem. (2005) 280:7800–7. doi: 10.1074/jbc.M411638200
53. Ghosh D, Mondal M, Mohite GM, Singh PK, Ranjan P, Anoop A, et al. The Parkinson's disease-associated H50Q mutation accelerates α-Synuclein aggregation in vitro. Biochemistry. (2013) 52:6925–7. doi: 10.1021/bi400999d
54. Buell AK, Galvagnion C, Gaspar R, Sparr E, Vendruscolo M, Knowles TPJ, et al. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc Natl Acad Sci USA. (2014) 111:7671–6. doi: 10.1073/pnas.1315346111
55. Eymsh B, Drobny A, Heyn TR, Xiang W, Lucius R, Schwarz K, et al. Toxic metamorphosis-how changes from lysosomal to cytosolic pH modify the α-synuclein aggregation pattern. Biomacromolecules. (2020) 21:4673–4. doi: 10.1021/acs.biomac.0c00629
56. Klein AD, Mazzulli JR. Is Parkinson's disease a lysosomal disorder? Brain. (2018) 141:2255–62. doi: 10.1093/brain/awy147
57. Zunke F. The function of lysosomes and their role in Parkinson's disease. Neuroforum. (2020) 26:43–51. doi: 10.1515/nf-2019-0035
58. Sevlever D, Jiang P, Yen SH. Cathepsin D is the main lysosomal enzyme involved in the degradation of α-synuclein and generation of its carboxy-terminally truncated species. Biochemistry. (2008) 47:9678–87. doi: 10.1021/bi800699v
59. Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P, et al. Cathepsin D expression level affects α-synuclein processing, aggregation, and toxicity in vivo. Mol Brain. (2009) 2:5. doi: 10.1186/1756-6606-2-5
60. McGlinchey RP, Lee JC. Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc Natl Acad Sci USA. (2015) 112:9322–7. doi: 10.1073/pnas.1500937112
61. Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. (2011) 146:37–52. doi: 10.1016/j.cell.2011.06.001
62. Rothaug M, Zunke F, Mazzulli JR, Schweizer M, Altmeppen H, Lullmann-Rauch R, et al. LIMP-2 expression is critical for beta-glucocerebrosidase activity and α-synuclein clearance. Proc Natl Acad Sci USA. (2014) 111:15573–8. doi: 10.1073/pnas.1405700111
63. Robak LA, Jansen IE, Van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson's disease. Brain. (2017) 140:3191–203. doi: 10.1093/brain/awx285
64. Bunk J, Prieto Huarcaya S, Drobny A, Dobert JP, Walther L, Rose-John S, et al. Cathepsin D variants associated with neurodegenerative diseases show dysregulated functionality and modified α-synuclein degradation properties. Front Cell Dev Biol. (2021) 9:581805. doi: 10.3389/fcell.2021.581805
65. Mazzulli JR, Zunke F, Tsunemi T, Toker NJ, Jeon S, Burbulla LF, et al. Activation of beta-glucocerebrosidase reduces pathological α-synuclein and restores lysosomal function in Parkinson's patient midbrain neurons. J Neurosci. (2016) 36:7693–706. doi: 10.1523/JNEUROSCI.0628-16.2016
66. Zunke F, Andresen L, Wesseler S, Groth J, Arnold P, Rothaug M, et al. Characterization of the complex formed by beta-glucocerebrosidase and the lysosomal integral membrane protein type-2. Proc Natl Acad Sci USA. (2016) 113:3791–6. doi: 10.1073/pnas.1514005113
67. Lopes Da Fonseca T, Villar-Piqué A, Outeiro TF. The interplay between α-synuclein clearance and spreading. Biomolecules. (2015) 5:435–71. doi: 10.3390/biom5020435
68. Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL. α-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson's disease. J Biol Chem. (1999) 274:19509–12. doi: 10.1074/jbc.274.28.19509
69. Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. (2012)338:949–53. doi: 10.1126/science.1227157
70. Rey NL, Bousset L, George S, Madaj Z, Meyerdirk L, Schulz E, et al. α-Synuclein conformational strains spread, seed and target neuronal cells differentially after injection into the olfactory bulb. Acta Neuropathol Commun. (2019) 7:221. doi: 10.1186/s40478-019-0859-3
71. Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, et al. In vivo demonstration that α-synuclein oligomers are toxic. Proc Natl Acad Sci USA. (2011) 108:4194–9. doi: 10.1073/pnas.1100976108
72. Xiang W, Schlachetzki JC, Helling S, Bussmann JC, Berlinghof M, Schaffer TE, et al. Oxidative stress-induced posttranslational modifications of α-synuclein: specific modification of α-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol Cell Neurosci. (2013) 54:71–83. doi: 10.1016/j.mcn.2013.01.004
73. De Pablos RM, Herrera AJ, Espinosa-Oliva AM, Sarmiento M, Munoz MF, Machado A, et al. Chronic stress enhances microglia activation and exacerbates death of nigral dopaminergic neurons under conditions of inflammation. J Neuroinflammation. (2014) 11:34. doi: 10.1186/1742-2094-11-34
74. He J, Zhu G, Wang G, Zhang F. Oxidative stress and neuroinflammation potentiate each other to promote progression of dopamine neurodegeneration. Oxid Med Cell Longev. (2020) 2020:6137521. doi: 10.1155/2020/6137521
75. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. (2010) 140:918–34. doi: 10.1016/j.cell.2010.02.016
76. Rocha NP, Miranda AS, Teixeira AL. Insights into neuroinflammation in Parkinson's disease: from biomarkers to anti-inflammatory based therapies. Biomed Res Int. (2015) 2015:628192. doi: 10.1155/2015/628192
77. Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. (2016) 353:777–83. doi: 10.1126/science.aag2590
78. Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, et al. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. (1994) 180:147–50. doi: 10.1016/0304-3940(94)90508-8
79. Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debré P, et al. FcepsilonRII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci. (1999) 19:3440–7. doi: 10.1523/JNEUROSCI.19-09-03440.1999
80. Mogi M, Togari A, Kondo T, Mizuno Y, Komure O, Kuno S, et al. Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from parkinsonian brain. J Neural Transm.(2000) 107:335–41. doi: 10.1007/s007020050028
81. Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. (1994) 165:208–10. doi: 10.1016/0304-3940(94)90746-3
82. Stypuła G, Kunert-Radek J, Stepień H, Zylińska K, Pawlikowski M. Evaluation of interleukins, ACTH, cortisol and prolactin concentrations in the blood of patients with parkinson's disease. Neuroimmunomodulation. (1996) 3:131–4. doi: 10.1159/000097237
83. Dobbs RJ, Charlett A, Purkiss AG, Dobbs SM, Weller C, Peterson DW. Association of circulating TNF-α and IL-6 with ageing and parkinsonism. Acta Neurol Scand. (1999) 100:34–41. doi: 10.1111/j.1600-0404.1999.tb00721.x
84. Rentzos M, Nikolaou C, Andreadou E, Paraskevas GP, Rombos A, Zoga M, et al. Circulating interleukin-15 and RANTES chemokine in Parkinson's disease. Acta Neurol Scand. (2007) 116:374–9. doi: 10.1111/j.1600-0404.2007.00894.x
85. Hirsch EC, Hunot S. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol. (2009) 8:382–97. doi: 10.1016/S1474-4422(09)70062-6
86. Nayak D, Roth TL, McGavern DB. Microglia development and function. Annu Rev Immunol. (2014) 32:367–402. doi: 10.1146/annurev-immunol-032713-120240
87. Tay TL, Savage JC, Hui CW, Bisht K, Tremblay M-È. Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. J Physiol. (2017) 595:1929–45. doi: 10.1113/JP272134
88. Mosley RL, Benner EJ, Kadiu I, Thomas M, Boska MD, Hasan K, et al. Neuroinflammation, oxidative stress and the pathogenesis of Parkinson's disease. Clin Neurosci Res. (2006) 6:261–81. doi: 10.1016/j.cnr.2006.09.006
89. Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, et al. Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann Neurol. (2005) 57:168–75. doi: 10.1002/ana.20338
90. Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with 11C(R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis. (2006) 21:404–12. doi: 10.1016/j.nbd.2005.08.002
91. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. (1988) 38:1285–91. doi: 10.1212/WNL.38.8.1285
92. Cortes D, Carballo-Molina OA, Castellanos-Montiel MJ, Velasco I. The non-survival effects of glial cell line-derived neurotrophic factor on neural cells. Front Mol Neurosci. (2017) 10:258. doi: 10.3389/fnmol.2017.00258
93. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. (2010) 119:7–35. doi: 10.1007/s00401-009-0619-8
94. Cabezas R, Avila M, Gonzalez J, El-Bachá RS, Báez E, García-Segura LM, et al. Astrocytic modulation of blood brain barrier: Perspectives on Parkinson's disease. Front Cell Neurosci. (2014) 8:211. doi: 10.3389/fncel.2014.00211
95. Kortekaas R, Leenders KL, Van Oostrom JCH, Vaalburg W, Bart J, Willemsen ATM, et al. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol. (2005) 57:176–9. doi: 10.1002/ana.20369
96. Lee H, Pienaar IS. Disruption of the blood-brain barrier in Parkinson's disease: curse or route to a cure? Front Biosci. (2014) 19:272–80. doi: 10.2741/4206
97. Gray MT, Woulfe JM. Striatal blood-brain barrier permeability in Parkinson's disease. J Cerebr Blood Flow Metab. (2015) 35:747–50. doi: 10.1038/jcbfm.2015.32
98. Wong D, Dorovini-Zis K, Vincent SR. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp Neurol. (2004) 190:446–55. doi: 10.1016/j.expneurol.2004.08.008
99. Dohgu S, Takata F, Matsumoto J, Kimura I, Yamauchi A, Kataoka Y. Monomeric α-synuclein induces blood-brain barrier dysfunction through activated brain pericytes releasing inflammatory mediators in vitro. Microvasc Res. (2019) 124:61–6. doi: 10.1016/j.mvr.2019.03.005
100. Braak H, Sastre M, Del Tredici K. Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson's disease. Acta Neuropathol. (2007) 114:231–41. doi: 10.1007/s00401-007-0244-3
101. Lee HJ, Kim C, Lee SJ. α-synuclein stimulation of astrocytes: potential role for neuroinflammation and neuroprotection. Oxid Med Cell Longev. (2010) 3:283–7. doi: 10.4161/oxim.3.4.12809
102. Leal MC, Casabona JC, Puntel M, Pitossi FJ. Interleukin-1beta and tumor necrosis factor-alpha: reliable targets for protective therapies in Parkinson's Disease? Front Cell Neurosci. (2013) 7:53. doi: 10.3389/fncel.2013.00053
103. Sonninen TM, Hamalainen RH, Koskuvi M, Oksanen M, Shakirzyanova A, Wojciechowski S, et al. Metabolic alterations in Parkinson's disease astrocytes. Sci Rep. (2020) 10:14474. doi: 10.1038/s41598-020-71329-8
104. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
105. Yun SP, Kam T-I, Panicker N, Kim S, Oh Y, Park J-S, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson's disease. Nat Med. (2018) 24:931–8. doi: 10.1038/s41591-018-0051-5
106. Alvarez-Erviti L, Couch Y, Richardson J, Cooper JM, Wood MJ. α-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci Res. (2011) 69:337–42. doi: 10.1016/j.neures.2010.12.020
107. Beraud D, Hathaway HA, Trecki J, Chasovskikh S, Johnson DA, Johnson JA, et al. Microglial activation and antioxidant responses induced by the Parkinson's disease protein α-synuclein. J Neuroimmune Pharmacol. (2013) 8:94–117. doi: 10.1007/s11481-012-9401-0
108. Kim C, Ho D-H, Suk J-E, You S, Michael S, Kang J, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. (2013) 4:1562. doi: 10.1038/ncomms2534
109. Dzamko N, Gysbers A, Perera G, Bahar A, Shankar A, Gao J, et al. Toll-like receptor 2 is increased in neurons in Parkinson's disease brain and may contribute to α-synuclein pathology. Acta Neuropathol. (2017) 133:303–19. doi: 10.1007/s00401-016-1648-8
110. Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, et al. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia. (2013) 61:349–60. doi: 10.1002/glia.22437
111. Park J-Y, Paik SR, Jou I, Park SM. Microglial phagocytosis is enhanced by monomeric α-synuclein, not aggregated α-synuclein: implications for Parkinson's disease. Glia. (2008) 56:1215–23. doi: 10.1002/glia.20691
112. Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. (2009) 119:182–92. doi: 10.1172/JCI36470
113. Sommer A, Marxreiter F, Krach F, Fadler T, Grosch J, Maroni M, et al. Th17 Lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson's disease. Cell Stem Cell. (2018) 23:123–31.e126. doi: 10.1016/j.stem.2018.06.015
114. Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T cells from patients with Parkinson's disease recognize α-synuclein peptides. Nature. (2017) 546:656–61. doi: 10.1038/nature22815
115. Poirier AA, Aube B, Cote M, Morin N, Di Paolo T, Soulet D. Gastrointestinal dysfunctions in Parkinson's disease: symptoms and treatments. Parkinsons Dis. (2016) 2016:6762528. doi: 10.1155/2016/6762528
116. Frazzitta G, Ferrazzoli D, Folini A, Palamara G, Maestri R. Severe constipation in Parkinson's disease and in Parkinsonisms: prevalence and affecting factors. Front Neurol. (2019) 10:621. doi: 10.3389/fneur.2019.00621
117. Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, et al. Rotenone, paraquat, and Parkinson's disease. Environ Health Perspect. (2011) 119:866–72. doi: 10.1289/ehp.1002839
118. Hawkes CH, Del Tredici K, Braak H. Parkinson's disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol. (2007) 33:599–614. doi: 10.1111/j.1365-2990.2007.00874.x
119. Shannon KM, Keshavarzian A, Mutlu E, Dodiya HB, Daian D, Jaglin JA, et al. α-synuclein in colonic submucosa in early untreated Parkinson's disease. Mov Disord. (2012) 27:709–15. doi: 10.1002/mds.23838
120. Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White CL Iii, et al. Multi-organ distribution of phosphorylated α-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. (2010) 119:689–702. doi: 10.1007/s00401-010-0664-3
121. Gelpi E, Navarro-Otano J, Tolosa E, Gaig C, Compta Y, Rey MJ, et al. Multiple organ involvement by α-synuclein pathology in Lewy body disorders. Mov Disord. (2014) 29:1010–8. doi: 10.1002/mds.25776
122. Wakabayashi K, Takahashi H, Takeda S, Ohama E, Ikuta F. Parkinson's disease: the presence of Lewy bodies in Auerbach's and Meissner's plexuses. Acta Neuropathol. (1988) 76:217–21. doi: 10.1007/BF00687767
123. Clairembault T, Leclair-Visonneau L, Coron E, Bourreille A, Le Dily S, Vavasseur F, et al. Structural alterations of the intestinal epithelial barrier in Parkinson's disease. Acta Neuropathol Commun. (2015) 3:12. doi: 10.1186/s40478-015-0196-0
124. Qualman SJ, Haupt HM, Yang P, Hamilton SR. Esophageal Lewy bodies associated with ganglion cell loss in achalasia. Similarity to Parkinson's disease. Gastroenterology. (1984) 87:848–56. doi: 10.1016/0016-5085(84)90079-9
125. Cersosimo MG, Perandones C, Micheli FE, Raina GB, Beron AM, Nasswetter G, et al. α-synuclein immunoreactivity in minor salivary gland biopsies of Parkinson's disease patients. Mov Disord. (2011) 26:188–90. doi: 10.1002/mds.23344
126. Mu L, Sobotka S, Chen J, Su H, Sanders I, Adler CH, et al. α-synuclein pathology and axonal degeneration of the peripheral motor nerves innervating pharyngeal muscles in Parkinson disease. J Neuropathol Exp Neurol. (2013) 72:119–29. doi: 10.1097/NEN.0b013e3182801cde
127. Sanchez-Ferro A, Rabano A, Catalan MJ, Rodriguez-Valcarcel FC, Fernandez Diez S, Herreros-Rodriguez J, et al. In vivo gastric detection of α-synuclein inclusions in Parkinson's disease. Mov Disord. (2015) 30:517–24. doi: 10.1002/mds.25988
128. Killinger BA, Madaj Z, Sikora JW, Rey N, Haas AJ, Vepa Y, et al. The vermiform appendix impacts the risk of developing Parkinson's disease. Sci Transl Med. (2018) 10:eaar5280. doi: 10.1126/scitranslmed.aar5280
129. Schaeffer E, Kluge A, Bottner M, Zunke F, Cossais F, Berg D, et al. α synuclein connects the gut-brain axis in Parkinson's disease patients - A view on clinical aspects, cellular pathology and analytical methodology. Front Cell Dev Biol. (2020) 8:573696. doi: 10.3389/fcell.2020.573696
130. Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, et al. Exosomal cell-to-cell transmission of α synuclein oligomers. Mol Neurodegener. (2012) 7:42. doi: 10.1186/1750-1326-7-42
131. Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc Natl Acad Sci USA. (2009) 106:13010–5. doi: 10.1073/pnas.0903691106
132. Angot E, Steiner JA, Lema Tome CM, Ekstrom P, Mattsson B, Bjorklund A, et al. α-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS ONE. (2012) 7:e39465. doi: 10.1371/journal.pone.0039465
133. Pan-Montojo F, Anichtchik O, Dening Y, Knels L, Pursche S, Jung R, et al. Progression of Parkinson's disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS ONE. (2010) 5:e8762. doi: 10.1371/journal.pone.0008762
134. Pan-Montojo F, Schwarz M, Winkler C, Arnhold M, O'Sullivan GA, Pal A, et al. Environmental toxins trigger PD-like progression via increased α-synuclein release from enteric neurons in mice. Sci Rep. (2012) 2:898. doi: 10.1038/srep00898
135. Chorell E, Andersson E, Evans ML, Jain N, Gotheson A, Aden J, et al. Bacterial chaperones CsgE and CsgC differentially modulate human α-synuclein amyloid formation via transient contacts. PLoS ONE. (2015) 10:e0140194. doi: 10.1371/journal.pone.0140194
136. Bhattacharyya D, Mohite GM, Krishnamoorthy J, Gayen N, Mehra S, Navalkar A, et al. Lipopolysaccharide from gut microbiota modulates α-synuclein aggregation and alters its biological function. ACS Chem Neurosci. (2019) 10:2229–36. doi: 10.1021/acschemneuro.8b00733
137. Chen QQ, Haikal C, Li W, Li JY. Gut inflammation in association with pathogenesis of Parkinson's disease. Front Mol Neurosci. (2019) 12:218. doi: 10.3389/fnmol.2019.00218
138. Foster JA, McVey Neufeld KA. Gut-brain axis: how the microbiome influences anxiety and depression. Trends Neurosci. (2013) 36:305–12. doi: 10.1016/j.tins.2013.01.005
139. Sherwin E, Bordenstein SR, Quinn JL, Dinan TG, Cryan JF. Microbiota and the social brain. Science. (2019) 366:eaar2016. doi: 10.1126/science.aar2016
140. Bonaz B, Bazin T, Pellissier S. The vagus nerve at the interface of the microbiota-gut-brain axis. Front Neurosci. (2018) 12:49. doi: 10.3389/fnins.2018.00049
141. Bonaz B, Sinniger V, Pellissier S. Vagus Nerve Stimulation at the Interface of Brain-Gut Interactions. Cold Spring Harb Perspect Med. (2019) 9. doi: 10.1101/cshperspect.a034199
142. Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Bjorklund T, et al. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. (2014) 128:805–20. doi: 10.1007/s00401-014-1343-6
143. Ulusoy A, Musgrove RE, Rusconi R, Klinkenberg M, Helwig M, Schneider A, et al. Neuron-to-neuron α-synuclein propagation in vivo is independent of neuronal injury. Acta Neuropathol Commun. (2015) 3:13. doi: 10.1186/s40478-015-0198-y
144. Kim S, Kwon SH, Kam TI, Panicker N, Karuppagounder SS, Lee S, et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson's disease. Neuron. (2019) 103:627–41.e627. doi: 10.1016/j.neuron.2019.05.035
145. Ulusoy A, Phillips RJ, Helwig M, Klinkenberg M, Powley TL, Di Monte DA. Brain-to-stomach transfer of α-synuclein via vagal preganglionic projections. Acta Neuropathol. (2017) 133:381–93. doi: 10.1007/s00401-016-1661-y
146. O'Donovan SM, Crowley EK, Brown JR-M, O'Sullivan O, O'Leary OF, Timmons S. Nigral overexpression of α-synuclein in a rat Parkinson's disease model indicates alterations in the enteric nervous system and the gut microbiome. Neurogastroenterol Motil. (2020) 32:e13726. doi: 10.1111/nmo.13726
147. Tysnes O-B, Kenborg L, Herlofson K, Steding-Jessen M, Horn A, Olsen JH, et al. Does vagotomy reduce the risk of Parkinson's disease? Ann Neurol. (2015) 78:1011–2. doi: 10.1002/ana.24531
148. Liu B, Fang F, Pedersen NL, Tillander A, Ludvigsson JF, Ekbom A, et al. Vagotomy and Parkinson disease: a Swedish register-based matched-cohort study. Neurology. (2017) 88:1996–2002. doi: 10.1212/WNL.0000000000003961
149. Svensson E, Horváth-Puhó E, Thomsen RW, Djurhuus JC, Pedersen L, Borghammer P, et al. Does vagotomy reduce the risk of Parkinson's disease: the authors reply. Ann Neurol. (2015) 78:1012–3. doi: 10.1002/ana.24518
150. Fussi N, Höllerhage M, Chakroun T, Nykänen N-P, Rösler TW, Koeglsperger T, et al. Exosomal secretion of α-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis. (2018) 9:757. doi: 10.1038/s41419-018-0816-2
151. Parnetti L, Gaetani L, Eusebi P, Paciotti S, Hansson O, El-Agnaf O, et al. CSF and blood biomarkers for Parkinson's disease. Lancet Neurol. (2019) 18:573–86. doi: 10.1016/S1474-4422(19)30024-9
152. Jiang C, Hopfner F, Katsikoudi A, Hein R, Catli C, Evetts S, et al. Serum neuronal exosomes predict and differentiate Parkinson's disease from atypical parkinsonism. J Neurol Neurosurg Psychiatry. (2020) 91:720–9. doi: 10.1136/jnnp-2019-322588
153. Stuendl A, Kunadt M, Kruse N, Bartels C, Moebius W, Danzer KM, et al. Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson's disease and dementia with Lewy bodies. Brain. (2016) 139:481–94. doi: 10.1093/brain/awv346
154. Ngolab J, Trinh I, Rockenstein E, Mante M, Florio J, Trejo M, et al. Brain-derived exosomes from dementia with Lewy bodies propagate α-synuclein pathology. Acta Neuropathol Commun. (2017) 5:46. doi: 10.1186/s40478-017-0445-5
155. Arotcarena ML, Dovero S, Prigent A, Bourdenx M, Camus S, Porras G, et al. Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain. (2020) 143:1462–75. doi: 10.1093/brain/awaa096
156. Giuffre M, Moretti R, Campisciano G, Da Silveira ABM, Monda VM, Comar M, et al. You talking to me? Says the Enteric Nervous System (ENS) to the microbe how intestinal microbes interact with the ENS. J Clin Med. (2020) 9:3705. doi: 10.3390/jcm9113705
157. Christofi FL. TRPV1 Sensory neurons and enteric glia in ENS link tachykinins to neuroinflammation and nociception. Cell Mol Gastroenterol Hepatol. (2018) 6:354–5. doi: 10.1016/j.jcmgh.2018.06.011
158. Walsh KT, Zemper AE. The enteric nervous system for epithelial researchers: basic anatomy, techniques, and interactions with the epithelium. Cell Mol Gastroenterol Hepatol. (2019) 8:369–78. doi: 10.1016/j.jcmgh.2019.05.003
159. Anlauf M, Schafer MK, Eiden L, Weihe E. Chemical coding of the human gastrointestinal nervous system: cholinergic, VIPergic, and catecholaminergic phenotypes. J Comp Neurol. (2003) 459:90–111. doi: 10.1002/cne.10599
160. Singaram C, Ashraf W, Gaumnitz EA, Torbey C, Sengupta A, Pfeiffer R, et al. Dopaminergic defect of enteric nervous system in Parkinson's disease patients with chronic constipation. Lancet. (1995) 346:861–4. doi: 10.1016/S0140-6736(95)92707-7
161. Frazier TH, Dibaise JK, McClain CJ. Gut microbiota, intestinal permeability, obesity-induced inflammation, and liver injury. JPEN J Parenter Enteral Nutr. (2011) 35:14S−20S. doi: 10.1177/0148607111413772
162. Cani PD, Everard A, Duparc T. Gut microbiota, enteroendocrine functions and metabolism. Curr Opin Pharmacol. (2013) 13:935–40. doi: 10.1016/j.coph.2013.09.008
163. Valdez-Morales EE, Overington J, Guerrero-Alba R, Ochoa-Cortes F, Ibeakanma CO, Spreadbury I, et al. Sensitization of peripheral sensory nerves by mediators from colonic biopsies of diarrhea-predominant irritable bowel syndrome patients: a role for PAR2. Am J Gastroenterol. (2013) 108:1634–43. doi: 10.1038/ajg.2013.241
164. Forsythe P, Kunze WA. Voices from within: gut microbes and the CNS. Cell Mol Life Sci. (2013) 70:55–69. doi: 10.1007/s00018-012-1028-z
165. Dinan TG, Cryan JF. Regulation of the stress response by the gut microbiota: implications for psychoneuroendocrinology. Psychoneuroendocrinology. (2012) 37:1369–78. doi: 10.1016/j.psyneuen.2012.03.007
166. Finger BC, Dinan TG, Cryan JF. The temporal impact of chronic intermittent psychosocial stress on high-fat diet-induced alterations in body weight. Psychoneuroendocrinology. (2012) 37:729–41. doi: 10.1016/j.psyneuen.2011.06.015
167. Stilling RM, Dinan TG, Cryan JF. Microbial genes, brain and behaviour - epigenetic regulation of the gut-brain axis. Genes Brain Behav. (2014) 13:69–86. doi: 10.1111/gbb.12109
168. Schirmer M, Garner A, Vlamakis H, Xavier RJ. Microbial genes and pathways in inflammatory bowel disease. Nat Rev Microbiol. (2019) 17:497–511. doi: 10.1038/s41579-019-0213-6
169. Baizabal-Carvallo JF, Alonso-Juarez M. The link between gut dysbiosis and neuroinflammation in Parkinson's disease. Neuroscience. (2020) 432:160–73. doi: 10.1016/j.neuroscience.2020.02.030
170. Rath HC, Wilson KH, Sartor RB. Differential induction of colitis and gastritis in HLA-B27 transgenic rats selectively colonized with Bacteroides vulgatus or Escherichia coli. Infect Immun. (1999) 67:2969–74. doi: 10.1128/IAI.67.6.2969-2974.1999
171. Van Kruiningen HJ. Is Mycobacterium paratuberculosis or any other infectious agent involved in the pathogenesis of IBD? Inflamm Bowel Dis. (2008) 14 (Suppl 2):S88–89. doi: 10.1002/ibd.20724
172. Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. (2012) 13:R79. doi: 10.1186/gb-2012-13-9-r79
173. Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol. (2019) 4:293–305. doi: 10.1038/s41564-018-0306-4
174. Britton GJ, Contijoch EJ, Mogno I, Vennaro OH, Llewellyn SR, Ng R, et al. Microbiotas from humans with inflammatory bowel disease alter the balance of gut Th17 and RORgammat(+) regulatory T cells and exacerbate colitis in mice. Immunity. (2019) 50:212–24.e214. doi: 10.1016/j.immuni.2018.12.015
175. Roshan N, Clancy AK, Borody TJ. Faecal microbiota transplantation is effective for the initial treatment of clostridium difficile infection: a retrospective clinical review. Infect Dis Ther. (2020) 9:935–42. doi: 10.1007/s40121-020-00339-w
176. Tamilarasan AG, Krishnananthan T. Faecal microbiota transplantation: what's beyond Clostridium difficile infection? Eur J Gastroenterol Hepatol. (2020) 33:487–4. doi: 10.1097/MEG.0000000000001938
177. Codagnone MG, Stanton C, O'Mahony SM, Dinan TG, Cryan JF. Microbiota and neurodevelopmental trajectories: role of maternal and early-life nutrition. Ann Nutr Metab. (2019) 74(Suppl. 2):16–27. doi: 10.1159/000499144
178. Arentsen T, Raith H, Qian Y, Forssberg H, Diaz Heijtz R. Host microbiota modulates development of social preference in mice. Microb Ecol Health Dis. (2015) 26:29719. doi: 10.3402/mehd.v26.29719
179. De Theije CG, Wopereis H, Ramadan M, Van Eijndthoven T, Lambert J, Knol J, et al. Altered gut microbiota and activity in a murine model of autism spectrum disorders. Brain Behav Immun. (2014) 37:197–206. doi: 10.1016/j.bbi.2013.12.005
180. Barrett E, Ross RP, O'Toole PW, Fitzgerald GF, Stanton C. gamma-Aminobutyric acid production by culturable bacteria from the human intestine. J Appl Microbiol. (2012) 113:411–7. doi: 10.1111/j.1365-2672.2012.05344.x
181. Pokusaeva K, Johnson C, Luk B, Uribe G, Fu Y, Oezguen N, et al. GABA-producing Bifidobacterium dentium modulates visceral sensitivity in the intestine. Neurogastroenterol Motil. (2017) 29:e12904. doi: 10.1111/nmo.12904
182. Li Y, Xiang YY, Lu WY, Liu C, Li J. A novel role of intestine epithelial GABAergic signaling in regulating intestinal fluid secretion. Am J Physiol Gastrointest Liver Physiol. (2012) 303:G453–60. doi: 10.1152/ajpgi.00497.2011
183. Fung TC, Vuong HE, Luna CDG, Pronovost GN, Aleksandrova AA, Riley NG, et al. Intestinal serotonin and fluoxetine exposure modulate bacterial colonization in the gut. Nat Microbiol. (2019) 4:2064–73. doi: 10.1038/s41564-019-0540-4
184. Erny D, Hrabe De Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. (2015) 18:965–77. doi: 10.1038/nn.4030
185. Tazoe H, Otomo Y, Kaji I, Tanaka R, Karaki SI, Kuwahara A. Roles of short-chain fatty acids receptors, GPR41 and GPR43 on colonic functions. J Physiol Pharmacol. (2008) 59(Suppl. 2):251–62.
186. Van De Wouw M, Boehme M, Lyte JM, Wiley N, Strain C, O'Sullivan O, et al. Short-chain fatty acids: microbial metabolites that alleviate stress-induced brain-gut axis alterations. J Physiol. (2018) 596:4923–44. doi: 10.1113/JP276431
187. Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, et al. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc Natl Acad Sci USA. (2011) 108:8030–5. doi: 10.1073/pnas.1016088108
188. Ceppa FA, Izzo L, Sardelli L, Raimondi I, Tunesi M, Albani D, et al. Human gut-microbiota interaction in neurodegenerative disorders and current engineered tools for its modeling. Front Cell Infect Microbiol. (2020) 10:297. doi: 10.3389/fcimb.2020.00297
189. Lund ML, Egerod KL, Engelstoft MS, Dmytriyeva O, Theodorsson E, Patel BA, et al. Enterochromaffin 5-HT cells - A major target for GLP-1 and gut microbial metabolites. Mol Metab. (2018) 11:70–83. doi: 10.1016/j.molmet.2018.03.004
190. Hasegawa S, Goto S, Tsuji H, Okuno T, Asahara T, Nomoto K, et al. Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson's disease. PLoS ONE. (2015) 10:e0142164. doi: 10.1371/journal.pone.0142164
191. Keshavarzian A, Green SJ, Engen PA, Voigt RM, Naqib A, Forsyth CB, et al. Colonic bacterial composition in Parkinson's disease. Mov Disord. (2015) 30:1351–60. doi: 10.1002/mds.26307
192. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell. (2016) 167:1469–80 e1412. doi: 10.1016/j.cell.2016.11.018
193. Camci G, Oguz S. Association between Parkinson's disease and Helicobacter pylori. J Clin Neurol. (2016) 12:147–50. doi: 10.3988/jcn.2016.12.2.147
194. Minato T, Maeda T, Fujisawa Y, Tsuji H, Nomoto K, Ohno K, et al. Progression of Parkinson's disease is associated with gut dysbiosis: two-year follow-up study. PLoS ONE. (2017) 12:e0187307. doi: 10.1371/journal.pone.0187307
195. Pal GD, Shaikh M, Forsyth CB, Ouyang B, Keshavarzian A, Shannon KM. Abnormal lipopolysaccharide binding protein as marker of gastrointestinal inflammation in Parkinson disease. Front Neurosci. (2015) 9:306. doi: 10.3389/fnins.2015.00306
196. Wallen ZD, Appah M, Dean MN, Sesler CL, Factor SA, Molho E, et al. Characterizing dysbiosis of gut microbiome in PD: evidence for overabundance of opportunistic pathogens. NPJ Parkinsons Dis. (2020) 6:11. doi: 10.1038/s41531-020-0112-6
197. Nishiwaki H, Ito M, Ishida T, Hamaguchi T, Maeda T, Kashihara K, et al. Meta-analysis of gut dysbiosis in Parkinson's disease. Mov Disord. (2020) 35:1626–35. doi: 10.1002/mds.28119
198. Cirstea MS, Yu AC, Golz E, Sundvick K, Kliger D, Radisavljevic N, et al. Microbiota composition and metabolism are associated with gut function in Parkinson's disease. Mov Disord. (2020) 35:1208–17. doi: 10.1002/mds.28052
199. Fukumoto S, Tatewaki M, Yamada T, Fujimiya M, Mantyh C, Voss M, et al. Short-chain fatty acids stimulate colonic transit via intraluminal 5-HT release in rats. Am J Physiol Regul Integr Comp Physiol. (2003) 284:R1269–76. doi: 10.1152/ajpregu.00442.2002
200. Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell. (2016) 167:1339–53 e1321. doi: 10.1016/j.cell.2016.10.043
201. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. (2008) 105:16731–6. doi: 10.1073/pnas.0804812105
202. Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. (2014) 63:1275–83. doi: 10.1136/gutjnl-2013-304833
203. Scheperjans F, Pekkonen E, Kaakkola S, Auvinen P. Linking smoking, coffee, urate, and Parkinson's disease - A role for gut microbiota? J Parkinsons Dis. (2015) 5:255–62. doi: 10.3233/JPD-150557
204. Guo S, Al-Sadi R, Said HM, Ma TY. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am J Pathol. (2013) 182:375–87. doi: 10.1016/j.ajpath.2012.10.014
205. Rosenberg GA, Estrada EY, Mobashery S. Effect of synthetic matrix metalloproteinase inhibitors on lipopolysaccharide-induced blood-brain barrier opening in rodents: differences in response based on strains and solvents. Brain Res. (2007) 1133:186–92. doi: 10.1016/j.brainres.2006.11.041
206. Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. (2007) 55:453–62. doi: 10.1002/glia.20467
207. Torres-Fuentes C, Golubeva AV, Zhdanov AV, Wallace S, Arboleya S, Papkovsky DB, et al. Short-chain fatty acids and microbiota metabolites attenuate ghrelin receptor signaling. FASEB J. (2019) 33:13546–59. doi: 10.1096/fj.201901433R
208. Gesser RM, Koo SC. Oral inoculation with herpes simplex virus type 1 infects enteric neuron and mucosal nerve fibers within the gastrointestinal tract in mice. J Virol. (1996) 70:4097–102. doi: 10.1128/jvi.70.6.4097-4102.1996
209. Jang H, Boltz D, Sturm-Ramirez K, Shepherd KR, Jiao Y, Webster R, et al. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci USA. (2009) 106:14063–8. doi: 10.1073/pnas.0900096106
210. Mori I. Viremic attack explains the dual-hit theory of Parkinson's disease. Med Hypotheses. (2017) 101:33–6. doi: 10.1016/j.mehy.2017.02.007
211. Guedia J, Brun P, Bhave S, Fitting S, Kang M, Dewey WL, et al. HIV-1 Tat exacerbates lipopolysaccharide-induced cytokine release via TLR4 signaling in the enteric nervous system. Sci Rep. (2016) 6:31203. doi: 10.1038/srep31203
212. Brun P, Qesari M, Marconi PC, Kotsafti A, Porzionato A, Macchi V, et al. Herpes simplex virus type 1 infects enteric neurons and triggers gut dysfunction via macrophage recruitment. Front Cell Infect Microbiol. (2018) 8:74. doi: 10.3389/fcimb.2018.00074
213. Podolska M, Nadolna K. Acetylcholinesterase secreted by Anisakis simplex larvae (Nematoda: Anisakidae) parasitizing herring, Clupea harengus: an inverse relationship of enzyme activity in the host-parasite system. Parasitol Res. (2014) 113:2231–8. doi: 10.1007/s00436-014-3878-9
214. Koch S, Nusrat A. Dynamic regulation of epithelial cell fate and barrier function by intercellular junctions. Ann N Y Acad Sci. (2009) 1165:220–7. doi: 10.1111/j.1749-6632.2009.04025.x
215. Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol. (2010) 177:512–24. doi: 10.2353/ajpath.2010.100168
216. Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. (2009) 9:799–809. doi: 10.1038/nri2653
217. Van Der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. (2009) 71:241–60. doi: 10.1146/annurev.physiol.010908.163145
218. Watson AJ, Chu S, Sieck L, Gerasimenko O, Bullen T, Campbell F, et al. Epithelial barrier function in vivo is sustained despite gaps in epithelial layers. Gastroenterology. (2005) 129:902–12. doi: 10.1053/j.gastro.2005.06.015
219. Marchiando AM, Shen L, Graham WV, Edelblum KL, Duckworth CA, Guan Y, et al. The epithelial barrier is maintained by in vivo tight junction expansion during pathologic intestinal epithelial shedding. Gastroenterology. (2011) 140:1208–18 e1201–1202. doi: 10.1053/j.gastro.2011.01.004
220. Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front Immunol. (2013) 4:280. doi: 10.3389/fimmu.2013.00280
221. Lim LG, Neumann J, Hansen T, Goetz M, Hoffman A, Neurath MF, et al. Confocal endomicroscopy identifies loss of local barrier function in the duodenum of patients with Crohn's disease and ulcerative colitis. Inflamm Bowel Dis. (2014) 20:892–900. doi: 10.1097/MIB.0000000000000027
222. Munkholm P, Langholz E, Hollander D, Thornberg K, Orholm M, Katz KD, et al. Intestinal permeability in patients with Crohn's disease and ulcerative colitis and their first degree relatives. Gut. (1994) 35:68–72. doi: 10.1136/gut.35.1.68
223. Kiesslich R, Duckworth CA, Moussata D, Gloeckner A, Lim LG, Goetz M, et al. Local barrier dysfunction identified by confocal laser endomicroscopy predicts relapse in inflammatory bowel disease. Gut. (2012) 61:1146–53. doi: 10.1136/gutjnl-2011-300695
224. Suzuki T, Yoshinaga N, Tanabe S. Interleukin-6 (IL-6) regulates claudin-2 expression and tight junction permeability in intestinal epithelium. J Biol Chem. (2011) 286:31263–71. doi: 10.1074/jbc.M111.238147
225. Watson AJ, Hughes KR. TNF-α-induced intestinal epithelial cell shedding: implications for intestinal barrier function. Ann N Y Acad Sci. (2012) 1258:1–8. doi: 10.1111/j.1749-6632.2012.06523.x
226. Kawashima R, Kawamura YI, Oshio T, Son A, Yamazaki M, Hagiwara T, et al. Interleukin-13 damages intestinal mucosa via TWEAK and Fn14 in mice-a pathway associated with ulcerative colitis. Gastroenterology. (2011) 141:2119–29 e2118. doi: 10.1053/j.gastro.2011.08.040
227. Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. (2008) 118:534–44. doi: 10.1172/JCI33194
228. Zhao H, Zhang H, Wu H, Li H, Liu L, Guo J, et al. Protective role of 1,25(OH)2 vitamin D3 in the mucosal injury and epithelial barrier disruption in DSS-induced acute colitis in mice. BMC Gastroenterol. (2012) 12:57. doi: 10.1186/1471-230X-12-57
229. Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science. (2012) 336:1314–7. doi: 10.1126/science.1221789
230. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. (2001) 411:599–603. doi: 10.1038/35079107
231. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. (2001) 411:603–6. doi: 10.1038/35079114
232. Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology. (2007) 132:1359–74. doi: 10.1053/j.gastro.2007.02.056
233. Ey B, Eyking A, Klepak M, Salzman NH, Gothert JR, Runzi M, et al. Loss of TLR2 worsens spontaneous colitis in MDR1A deficiency through commensally induced pyroptosis. J Immunol. (2013) 190:5676–88. doi: 10.4049/jimmunol.1201592
234. Vijay-Kumar M, Wu H, Aitken J, Kolachala VL, Neish AS, Sitaraman SV, et al. Activation of toll-like receptor 3 protects against DSS-induced acute colitis. Inflamm Bowel Dis. (2007) 13:856–64. doi: 10.1002/ibd.20142
235. Zhao HW, Yue YH, Han H, Chen XL, Lu YG, Zheng JM, et al. Effect of toll-like receptor 3 agonist poly I:C on intestinal mucosa and epithelial barrier function in mouse models of acute colitis. World J Gastroenterol. (2017) 23:999–1009. doi: 10.3748/wjg.v23.i6.999
236. Fort MM, Mozaffarian A, Stover AG, Correia Jda S, Johnson DA, Crane RT, et al. A synthetic TLR4 antagonist has anti-inflammatory effects in two murine models of inflammatory bowel disease. J Immunol. (2005) 174:6416–23. doi: 10.4049/jimmunol.174.10.6416
237. Fukata M, Shang L, Santaolalla R, Sotolongo J, Pastorini C, Espana C, et al. Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflamm Bowel Dis. (2011) 17:1464–73. doi: 10.1002/ibd.21527
238. Rakhesh M, Cate M, Vijay R, Shrikant A, Shanjana A. A TLR4-interacting peptide inhibits lipopolysaccharide-stimulated inflammatory responses, migration and invasion of colon cancer SW480 cells. Oncoimmunology. (2012) 1:1495–506. doi: 10.4161/onci.22089
239. Gewirtz AT, Vijay-Kumar M, Brant SR, Duerr RH, Nicolae DL, Cho JH. Dominant-negative TLR5 polymorphism reduces adaptive immune response to flagellin and negatively associates with Crohn's disease. Am J Physiol Gastrointest Liver Physiol. (2006) 290:G1157–63. doi: 10.1152/ajpgi.00544.2005
240. Ivison SM, Himmel ME, Hardenberg G, Wark PA, Kifayet A, Levings MK, et al. TLR5 is not required for flagellin-mediated exacerbation of DSS colitis. Inflamm Bowel Dis. (2010) 16:401–9. doi: 10.1002/ibd.21097
241. Sainathan SK, Bishnupuri KS, Aden K, Luo Q, Houchen CW, Anant S, et al. Toll-like receptor-7 ligand Imiquimod induces type I interferon and antimicrobial peptides to ameliorate dextran sodium sulfate-induced acute colitis. Inflamm Bowel Dis. (2012) 18:955–67. doi: 10.1002/ibd.21867
242. Kumari M, Hanpude P, Maiti TK. α-Synuclein exhibits differential membrane perturbation, nucleation, and TLR2 binding through its secondary structure. ACS Chem Neurosci. (2020) 11:4203–14. doi: 10.1021/acschemneuro.0c00480
243. Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, et al. Increased intestinal permeability correlates with sigmoid mucosa α-synuclein staining and endotoxin exposure markers in early Parkinson's disease. PLoS ONE. (2011) 6:e28032. doi: 10.1371/journal.pone.0028032
244. Gorecki AM, Preskey L, Bakeberg MC, Kenna JE, Gildenhuys C, Macdougall G, et al. Altered gut microbiome in Parkinson's disease and the influence of lipopolysaccharide in a human α-synuclein over-expressing mouse model. Front Neurosci. (2019) 13:839. doi: 10.3389/fnins.2019.00839
245. Dodiya HB, Forsyth CB, Voigt RM, Engen PA, Patel J, Shaikh M, et al. Chronic stress-induced gut dysfunction exacerbates Parkinson's disease phenotype and pathology in a rotenone-induced mouse model of Parkinson's disease. Neurobiol Dis. (2020) 135:104352. doi: 10.1016/j.nbd.2018.12.012
246. Marczynski M, Rickert CA, Semerdzhiev SA, Van Dijk WR, Segers-Nolten IMJ, Claessens M, et al. α-Synuclein penetrates mucin hydrogels despite its mucoadhesive properties. Biomacromolecules. (2019) 20:4332–44. doi: 10.1021/acs.biomac.9b00905
247. Tursi SA, Tukel C. Curli-containing enteric biofilms inside and out: matrix composition, immune recognition, and disease implications. Microbiol Mol Biol Rev. (2018) 82:e00028–18. doi: 10.1128/MMBR.00028-18
248. Burke EE, Chenoweth JG, Shin JH, Collado-Torres L, Kim SK, Micali N, et al. Dissecting transcriptomic signatures of neuronal differentiation and maturation using iPSCs. Nat Commun. (2020) 11:462. doi: 10.1038/s41467-019-14266-z
249. Marton RM, Pasca SP. Organoid and assembloid technologies for investigating cellular crosstalk in human brain development and disease. Trends Cell Biol. (2020) 30:133–43. doi: 10.1016/j.tcb.2019.11.004
250. Kwak TH, Kang JH, Hali S, Kim J, Kim KP, Park C, et al. Generation of homogeneous midbrain organoids with in vivo-like cellular composition facilitates neurotoxin-based Parkinson's disease modeling. Stem Cells. (2020) 38:727–40. doi: 10.1002/stem.3163
251. Ormel PR, Vieira De Sa R, Van Bodegraven EJ, Karst H, Harschnitz O, Sneeboer M, et al. Microglia innately develop within cerebral organoids. Nat Commun. (2018) 9:4167. doi: 10.1038/s41467-018-06684-2
252. Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, et al. iPSC-derived human microglia-like cells to study neurological diseases. Neuron. (2017) 94:278–93.e279. doi: 10.1016/j.neuron.2017.03.042
253. Vatine GD, Barrile R, Workman MJ, Sances S, Barriga BK, Rahnama M, et al. Human iPSC-derived blood-brain barrier chips enable disease modeling and personalized medicine applications. Cell Stem Cell. (2019) 24:995–1005.e1006. doi: 10.1016/j.stem.2019.05.011
254. Sato T, Vries RG, Snippert HJ, Van De Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. (2009) 459:262–5. doi: 10.1038/nature07935
255. Workman MJ, Mahe MM, Trisno S, Poling HM, Watson CL, Sundaram N, et al. Engineered human pluripotent-stem-cell-derived intestinal tissues with a functional enteric nervous system. Nat Med. (2017) 23:49–59. doi: 10.1038/nm.4233
256. Noel G, Baetz NW, Staab JF, Donowitz M, Kovbasnjuk O, Pasetti MF, et al. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci Rep. (2017) 7:45270. doi: 10.1038/srep46790
257. Karve SS, Pradhan S, Ward DV, Weiss AA. Intestinal organoids model human responses to infection by commensal and Shiga toxin producing Escherichia coli. PLoS ONE. (2017) 12:e0178966. doi: 10.1371/journal.pone.0178966
258. Schreurs R, Baumdick ME, Sagebiel AF, Kaufmann M, Mokry M, Klarenbeek PL, et al. Human fetal TNF-α-cytokine-producing CD4(+) effector memory T cells promote intestinal development and mediate inflammation early in life. Immunity. (2019) 50:462–76.e468. doi: 10.1016/j.immuni.2018.12.010
259. Loskill P, Marcus SG, Mathur A, Reese WM, Healy KE. muOrgano: a Lego(R)-like plug and play system for modular multi-organ-chips. PLoS ONE. (2015) 10:e0139587. doi: 10.1371/journal.pone.0139587
260. Herland A, Maoz BM, Das D, Somayaji MR, Prantil-Baun R, Novak R, et al. Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips. Nat Biomed Eng. (2020) 4:421–36. doi: 10.1038/s41551-019-0498-9
261. Raimondi MT, Albani D, Giordano C. An organ-on-a-chip engineered platform to study the microbiota-gut-brain axis in neurodegeneration. Trends Mol Med. (2019) 25:737–40. doi: 10.1016/j.molmed.2019.07.006
262. Yu Y, Yang W, Li Y, Cong Y. Enteroendocrine cells: sensing gut microbiota and regulating inflammatory bowel diseases. Inflamm Bowel Dis. (2019) 26:11–20. doi: 10.1093/ibd/izz217
263. Banskota S, Ghia JE, Khan WI. Serotonin in the gut: blessing or a curse. Biochimie. (2019) 161:56–64. doi: 10.1016/j.biochi.2018.06.008
264. Crowell MD. Role of serotonin in the pathophysiology of the irritable bowel syndrome. Br J Pharmacol. (2004) 141:1285–93. doi: 10.1038/sj.bjp.0705762
265. Galligan JJ. 5-hydroxytryptamine, ulcerative colitis, and irritable bowel syndrome: molecular connections. Gastroenterology. (2004) 126:1897–9. doi: 10.1053/j.gastro.2004.04.028
266. Van Es JH, Wiebrands K, López-Iglesias C, Van De Wetering M, Zeinstra L, Van Den Born M, et al. Enteroendocrine and tuft cells support Lgr5 stem cells on Paneth cell depletion. Proc Natl Acad Sci USA. (2019) 116:26599–605. doi: 10.1073/pnas.1801888117
267. Liddle RA. Neuropods. Cell Mol Gastroenterol Hepatol. (2019) 7:739–47. doi: 10.1016/j.jcmgh.2019.01.006
268. Kaelberer MM, Rupprecht LE, Liu WW, Weng P, Bohorquez DV. Neuropod cells: the emerging biology of gut-brain sensory transduction. Annu Rev Neurosci. (2020) 43:337–53. doi: 10.1146/annurev-neuro-091619-022657
269. Bohorquez DV, Samsa LA, Roholt A, Medicetty S, Chandra R, Liddle RA. An enteroendocrine cell-enteric glia connection revealed by 3D electron microscopy. PLoS ONE. (2014) 9:e89881. doi: 10.1371/journal.pone.0089881
270. Ruffmann C, Parkkinen L. Gut feelings about α-Synuclein in gastrointestinal biopsies: biomarker in the making? Mov Disord. (2016) 31:193–202. doi: 10.1002/mds.26480
271. Bogunovic M, Dave SH, Tilstra JS, Chang DT, Harpaz N, Xiong H, et al. Enteroendocrine cells express functional Toll-like receptors. Am J Physiol Gastrointest Liver Physiol. (2007) 292:G1770–83. doi: 10.1152/ajpgi.00249.2006
272. Palazzo M, Balsari A, Rossini A, Selleri S, Calcaterra C, Gariboldi S, et al. Activation of enteroendocrine cells via TLRs induces hormone, chemokine, and defensin secretion. J Immunol. (2007) 178:4296–303. doi: 10.4049/jimmunol.178.7.4296
273. Aktar R, Parkar N, Stentz R, Baumard L, Parker A, Goldson A, et al. Human resident gut microbe Bacteroides thetaiotaomicron regulates colonic neuronal innervation and neurogenic function. Gut Microbes. (2020) 11:1745–57. doi: 10.1080/19490976.2020.1766936
274. Caputi V, Giron MC. Microbiome-gut-brain axis and toll-like receptors in Parkinson's disease. Int J Mol Sci. (2018) 19:1689. doi: 10.3390/ijms19061689
275. Kaji I, Karaki S, Tanaka R, Kuwahara A. Density distribution of free fatty acid receptor 2 (FFA2)-expressing and GLP-1-producing enteroendocrine L cells in human and rat lower intestine, and increased cell numbers after ingestion of fructo-oligosaccharide. J Mol Histol. (2011) 42:27–38. doi: 10.1007/s10735-010-9304-4
276. Nohr MK, Pedersen MH, Gille A, Egerod KL, Engelstoft MS, Husted AS, et al. GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology. (2013) 154:3552–64. doi: 10.1210/en.2013-1142
277. Psichas A, Sleeth ML, Murphy KG, Brooks L, Bewick GA, Hanyaloglu AC, et al. The short chain fatty acid propionate stimulates GLP-1 and PYY secretion via free fatty acid receptor 2 in rodents. Int J Obes. (2015) 39:424–9. doi: 10.1038/ijo.2014.153
278. Hagbom M, Istrate C, Engblom D, Karlsson T, Rodriguez-Diaz J, Buesa J, et al. Rotavirus stimulates release of serotonin (5-HT) from human enterochromaffin cells and activates brain structures involved in nausea and vomiting. PLoS Pathog. (2011) 7:e1002115. doi: 10.1371/journal.ppat.1002115
279. Hagbom M, De Faria FM, Winberg ME, Westerberg S, Nordgren J, Sharma S, et al. Neurotrophic factors protect the intestinal barrier from rotavirus insult in mice. MBio. (2020) 11:e02834–19. doi: 10.1128/mBio.02834-19
280. Westerberg S, Hagbom M, Rajan A, Loitto V, Persson BD, Allard A, et al. Interaction of human enterochromaffin cells with human enteric adenovirus 41 leads to serotonin release and subsequent activation of enteric glia cells. J Virol. (2018) 92:e00026–18. doi: 10.1128/JVI.00026-18
281. Ha S, Jin B, Clemmensen B, Park P, Mahboob S, Gladwill V, et al. Serotonin is elevated in COVID-19-associated diarrhoea. Gut. (2021). doi: 10.1136/gutjnl-2020-323542
282. Li Q, Nirala NK, Nie Y, Chen HJ, Ostroff G, Mao J, et al. Ingestion of food particles regulates the mechanosensing misshapen-yorkie pathway in drosophila intestinal growth. Dev Cell. (2018) 45:433–49.e436. doi: 10.1016/j.devcel.2018.04.014
283. Wu J, Lewis AH, Grandl J. Touch, tension, and transduction – The function and regulation of Piezo ion channels. Trends Biochem Sci. (2017) 42:57–71. doi: 10.1016/j.tibs.2016.09.004
284. He L, Si G, Huang J, Samuel ADT, Perrimon N. Mechanical regulation of stem-cell differentiation by the stretch-activated Piezo channel. Nature. (2018) 555:103–6. doi: 10.1038/nature25744
285. Eisenhoffer GT, Loftus PD, Yoshigi M, Otsuna H, Chien C-B, Morcos PA, et al. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature. (2012) 484:546–9. doi: 10.1038/nature10999
286. Gudipaty SA, Lindblom J, Loftus PD, Redd MJ, Edes K, Davey CF, et al. Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature. (2017) 543:118–21. doi: 10.1038/nature21407
287. Feher J. 8.3—Intestinal and colonic chemoreception and motility. In: Feher J, editor. Quantitative Human Physiology. 2nd ed. Boston, MA: Elsevier (2017). p. 796–809. doi: 10.1016/B978-0-12-800883-6.00079-3
288. Nezami BG, Srinivasan S. Enteric nervous system in the small intestine: pathophysiology and clinical implications. Curr Gastroenterol Rep. (2010) 12:358–65. doi: 10.1007/s11894-010-0129-9
289. Spencer NJ, Hennig GW, Smith TK. Stretch-activated neuronal pathways to longitudinal and circular muscle in guinea pig distal colon. Am J Physiol Gastrointest Liver Physiol. (2003) 284:G231–41. doi: 10.1152/ajpgi.00291.2002
290. Wang F, Knutson K, Alcaino C, Linden DR, Gibbons SJ, Kashyap P, et al. Mechanosensitive ion channel Piezo2 is important for enterochromaffin cell response to mechanical forces. J Physiol. (2017) 595:79–91. doi: 10.1113/JP272718
291. Alcaino C, Knutson KR, Treichel AJ, Yildiz G, Strege PR, Linden DR, et al. A population of gut epithelial enterochromaffin cells is mechanosensitive and requires Piezo2 to convert force into serotonin release. Proc Nat Acad Sci USA. (2018) 115:E7632–41. doi: 10.1073/pnas.1804938115
292. Sugisawa E, Takayama Y, Takemura N, Kondo T, Hatakeyama S, Kumagai Y, et al. RNA sensing by gut Piezo1 is essential for systemic serotonin synthesis. Cell. (2020) 182:609–24.e621. doi: 10.1016/j.cell.2020.06.022
293. Martinez-Martin P, Schapira AHV, Stocchi F, Sethi K, Odin P, Macphee G, et al. Prevalence of nonmotor symptoms in Parkinson's disease in an international setting; Study using nonmotor symptoms questionnaire in 545 patients. Mov Disord. (2007) 22:1623–9. doi: 10.1002/mds.21586
294. Lin C-H, Lin J-W, Liu Y-C, Chang C-H, Wu R-M. Risk of Parkinson's disease following severe constipation: a nationwide population-based cohort study. Parkinsonism Relat Disord. (2014) 20:1371–5. doi: 10.1016/j.parkreldis.2014.09.026
295. Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: a review. JAMA. (2014) 311:1670–83. doi: 10.1001/jama.2014.3654
296. Okun MS. Deep-brain stimulation for Parkinson's disease. N Engl J Med. (2012) 367:1529–38. doi: 10.1056/NEJMct1208070
297. Axelsen TM, Woldbye DPD. Gene therapy for Parkinson's disease, an update. J Parkinsons Dis. (2018) 8:195–215. doi: 10.3233/JPD-181331
298. Wang Z, Gao G, Duan C, Yang H. Progress of immunotherapy of anti-α-synuclein in Parkinson's disease. Biomed Pharmacother. (2019) 115:108843. doi: 10.1016/j.biopha.2019.108843
299. Lindvall O. Treatment of Parkinson's disease using cell transplantation. Philos Trans R Soc Lond B Biol Sci. (2015) 370:20140370. doi: 10.1098/rstb.2014.0370
300. Charvin D, Medori R, Hauser RA, Rascol O. Therapeutic strategies for Parkinson disease: beyond dopaminergic drugs. Nat Rev Drug Discov. (2018) 17:844. doi: 10.1038/nrd.2018.184
301. Iarkov A, Barreto GE, Grizzell JA, Echeverria V. Strategies for the treatment of Parkinson's disease: beyond dopamine. Front Aging Neurosci. (2020) 12:4. doi: 10.3389/fnagi.2020.00004
302. Lewitt PA. New levodopa therapeutic strategies. Parkinsonism Relat Disord. (2016) 22(Suppl. 1):S37–40. doi: 10.1016/j.parkreldis.2015.09.021
303. Powers KM, Smith-Weller T, Franklin GM, Longstreth WT Jr, Swanson PD, Checkoway H. Dietary fats, cholesterol and iron as risk factors for Parkinson's disease. Parkinsonism Relat Disord. (2009) 15:47–52. doi: 10.1016/j.parkreldis.2008.03.002
304. Miyake Y, Sasaki S, Tanaka K, Fukushima W, Kiyohara C, Tsuboi Y, et al. Dietary fat intake and risk of Parkinson's disease: a case-control study in Japan. J Neurol Sci. (2010) 288:117–22. doi: 10.1016/j.jns.2009.09.021
305. Kamel F, Goldman SM, Umbach DM, Chen H, Richardson G, Barber MR, et al. Dietary fat intake, pesticide use, and Parkinson's disease. Parkinsonism Relat Disord. (2014) 20:82–7. doi: 10.1016/j.parkreldis.2013.09.023
306. Pu Y, Chang L, Qu Y, Wang S, Zhang K, Hashimoto K. Antibiotic-induced microbiome depletion protects against MPTP-induced dopaminergic neurotoxicity in the brain. Aging. (2019) 11:6915–29. doi: 10.18632/aging.102221
307. Koutzoumis DN, Vergara M, Pino J, Buddendorff J, Khoshbouei H, Mandel RJ, et al. Alterations of the gut microbiota with antibiotics protects dopamine neuron loss and improve motor deficits in a pharmacological rodent model of Parkinson's disease. Exp Neurol. (2020) 325:113159. doi: 10.1016/j.expneurol.2019.113159
308. Qiao CM, Sun MF, Jia XB, Shi Y, Zhang BP, Zhou ZL, et al. Sodium butyrate causes α-synuclein degradation by an Atg5-dependent and PI3K/Akt/mTOR-related autophagy pathway. Exp Cell Res. (2020) 387:111772. doi: 10.1016/j.yexcr.2019.111772
309. Cassani E, Privitera G, Pezzoli G, Pusani C, Madio C, Iorio L, et al. Use of probiotics for the treatment of constipation in Parkinson's disease patients. Minerva Gastroenterol Dietol. (2011) 57:117–21.
310. Surwase SN, Jadhav JP. Bioconversion of L-tyrosine to L-DOPA by a novel bacterium Bacillus sp. JPJ Amino Acids. (2011) 41:495–506. doi: 10.1007/s00726-010-0768-z
311. Tamtaji OR, Taghizadeh M, Daneshvar Kakhaki R, Kouchaki E, Bahmani F, Borzabadi S. Clinical and metabolic response to probiotic administration in people with Parkinson's disease: a randomized, double-blind, placebo-controlled trial. Clin Nutr. (2019) 38:1031–5. doi: 10.1016/j.clnu.2018.05.018
312. Van Kessel SP, Frye AK, El-Gendy AO, Castejon M, Keshavarzian A, Van Dijk G, et al. Gut bacterial tyrosine decarboxylases restrict levels of levodopa in the treatment of Parkinson's disease. Nat Commun. (2019) 10:310. doi: 10.1038/s41467-019-08294-y
313. Srivastav S, Neupane S, Bhurtel S, Katila N, Maharjan S, Choi H, et al. Probiotics mixture increases butyrate, and subsequently rescues the nigral dopaminergic neurons from MPTP and rotenone-induced neurotoxicity. J Nutr Biochem. (2019) 69:73–86. doi: 10.1016/j.jnutbio.2019.03.021
314. Hsieh TH, Kuo CW, Hsieh KH, Shieh MJ, Peng CW, Chen YC, et al. Probiotics alleviate the progressive deterioration of motor functions in a mouse model of Parkinson's disease. Brain Sci. (2020) 10:206. doi: 10.3390/brainsci10040206
315. Sun MF, Zhu YL, Zhou ZL, Jia XB, Xu YD, Yang Q, et al. Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson's disease mice: Gut microbiota, glial reaction and TLR4/TNF-α signaling pathway. Brain Behav Immun. (2018) 70:48–60. doi: 10.1016/j.bbi.2018.02.005
316. Huang H, Xu H, Luo Q, He J, Li M, Chen H, et al. Fecal microbiota transplantation to treat Parkinson's disease with constipation: a case report. Medicine. (2019) 98:e16163. doi: 10.1097/MD.0000000000016163
317. Choi HH, Cho YS. Fecal microbiota transplantation: current applications, effectiveness, and future perspectives. Clin Endosc. (2016) 49:257–65. doi: 10.5946/ce.2015.117
318. Drolet RE, Cannon JR, Montero L, Greenamyre JT. Chronic rotenone exposure reproduces Parkinson's disease gastrointestinal neuropathology. Neurobiol Dis. (2009) 36:96–102. doi: 10.1016/j.nbd.2009.06.017
319. Tasselli M, Chaumette T, Paillusson S, Monnet Y, Lafoux A, Huchet-Cadiou C, et al. Effects of oral administration of rotenone on gastrointestinal functions in mice. Neurogastroenterol Motil. (2013) 25:e183–93. doi: 10.1111/nmo.12070
320. Perez-Pardo P, Dodiya HB, Engen PA, Naqib A, Forsyth CB, Green SJ, et al. Gut bacterial composition in a mouse model of Parkinson's disease. Benef Microbes. (2018) 9:799–814. doi: 10.3920/BM2017.0202
321. Chaumette T, Lebouvier T, Aubert P, Lardeux B, Qin C, Li Q, et al. Neurochemical plasticity in the enteric nervous system of a primate animal model of experimental Parkinsonism. Neurogastroenterol Motil. (2009) 21:215–22. doi: 10.1111/j.1365-2982.2008.01226.x
322. Natale G, Kastsiushenka O, Fulceri F, Ruggieri S, Paparelli A, Fornai F. MPTP-induced parkinsonism extends to a subclass of TH-positive neurons in the gut. Brain Res. (2010) 1355:195–206. doi: 10.1016/j.brainres.2010.07.076
323. Anderson G, Noorian AR, Taylor G, Anitha M, Bernhard D, Srinivasan S, et al. Loss of enteric dopaminergic neurons and associated changes in colon motility in an MPTP mouse model of Parkinson's disease. Exp Neurol. (2007) 207:4–12. doi: 10.1016/j.expneurol.2007.05.010
324. Ellett LJ, Hung LW, Munckton R, Sherratt NA, Culvenor J, Grubman A, et al. Restoration of intestinal function in an MPTP model of Parkinson's Disease. Sci Rep. (2016) 6:30269. doi: 10.1038/srep30269
325. Cote M, Poirier AA, Aube B, Jobin C, Lacroix S, Soulet D. Partial depletion of the proinflammatory monocyte population is neuroprotective in the myenteric plexus but not in the basal ganglia in a MPTP mouse model of Parkinson's disease. Brain Behav Immun. (2015) 46:154–67. doi: 10.1016/j.bbi.2015.01.009
326. Blandini F, Balestra B, Levandis G, Cervio M, Greco R, Tassorelli C, et al. Functional and neurochemical changes of the gastrointestinal tract in a rodent model of Parkinson's disease. Neurosci Lett. (2009) 467:203–7. doi: 10.1016/j.neulet.2009.10.035
327. Pellegrini C, Fornai M, Colucci R, Tirotta E, Blandini F, Levandis G, et al. Alteration of colonic excitatory tachykininergic motility and enteric inflammation following dopaminergic nigrostriatal neurodegeneration. J Neuroinflammation. (2016) 13:146. doi: 10.1186/s12974-016-0608-5
328. Anselmi L, Toti L, Bove C, Hampton J, Travagli RA. A nigro-vagal pathway controls gastric motility and is affected in a rat model of Parkinsonism. Gastroenterology. (2017) 153:1581–93. doi: 10.1053/j.gastro.2017.08.069
329. Anselmi L, Bove C, Coleman FH, Le K, Subramanian MP, Venkiteswaran K, et al. Ingestion of subthreshold doses of environmental toxins induces ascending Parkinsonism in the rat. NPJ Parkinsons Dis. (2018) 4:30. doi: 10.1038/s41531-018-0066-0
330. Wang L, Fleming SM, Chesselet MF, Tache Y. Abnormal colonic motility in mice overexpressing human wild-type α-synuclein. Neuroreport. (2008) 19:873–6. doi: 10.1097/WNR.0b013e3282ffda5e
331. Wang L, Magen I, Yuan PQ, Subramaniam SR, Richter F, Chesselet MF, et al. Mice overexpressing wild-type human α-synuclein display alterations in colonic myenteric ganglia and defecation. Neurogastroenterol Motil. (2012) 24:e425–36. doi: 10.1111/j.1365-2982.2012.01974.x
332. Naudet N, Antier E, Gaillard D, Morignat E, Lakhdar L, Baron T, et al. Oral exposure to paraquat triggers earlier expression of phosphorylated alpha-synuclein in the enteric nervous system of A53T mutant human alpha-synuclein transgenic mice. J Neuropathol Exp Neurol. (2017) 76:1046–57. doi: 10.1093/jnen/nlx092
333. Rota L, Pellegrini C, Benvenuti L, Antonioli L, Fornai M, Blandizzi C, et al. Constipation, deficit in colon contractions and alpha-synuclein inclusions within the colon precede motor abnormalities and neurodegeneration in the central nervous system in a mouse model of alpha-synucleinopathy. Transl Neurodegener. (2019) 8:5. doi: 10.1186/s40035-019-0146-z
334. Taguchi T, Ikuno M, Hondo M, Parajuli LK, Taguchi K, Ueda J, et al. α-Synuclein BAC transgenic mice exhibit RBD-like behaviour and hyposmia: a prodromal Parkinson's disease model. Brain. (2020) 143:249–65. doi: 10.1093/brain/awz380
335. Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, et al. Parkinson's disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell. (2016) 166:314–27. doi: 10.1016/j.cell.2016.05.039
336. Matheoud D, Cannon T, Voisin A, Penttinen AM, Ramet L, Fahmy AM, et al. Intestinal infection triggers Parkinson's disease-like symptoms in Pink1(-/-) mice. Nature. (2019) 571:565–9. doi: 10.1038/s41586-019-1405-y
337. Ghaisas S, Langley MR, Palanisamy BN, Dutta S, Narayanaswamy K, Plummer PJ, et al. MitoPark transgenic mouse model recapitulates the gastrointestinal dysfunction and gut-microbiome changes of Parkinson's disease. Neurotoxicology. (2019) 75:186–99. doi: 10.1016/j.neuro.2019.09.004
338. Lee HJ, Suk JE, Lee KW, Park SH, Blumbergs PC, Gai WP, et al. Transmission of synucleinopathies in the enteric nervous system of A53T α-synuclein transgenic mice. Exp Neurobiol. (2011) 20:181–8. doi: 10.5607/en.2011.20.4.181
339. Rey NL, George S, Steiner JA, Madaj Z, Luk KC, Trojanowski JQ, et al. Spread of aggregates after olfactory bulb injection of α-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol. (2018) 135:65–83. doi: 10.1007/s00401-017-1792-9
340. Uemura N, Yagi H, Uemura MT, Yamakado H, Takahashi R. Limited spread of pathology within the brainstem of α-synuclein BAC transgenic mice inoculated with preformed fibrils into the gastrointestinal tract. Neurosci Lett. (2020) 716:134651. doi: 10.1016/j.neulet.2019.134651
341. Mehta S, Nijhuis A, Kumagai T, Lindsay J, Silver A. Defects in the adherens junction complex (E-cadherin/ β-catenin) in inflammatory bowel disease. Cell Tissue Res. (2015) 360:749–60. doi: 10.1007/s00441-014-1994-6
342. Breslin NP, Nash C, Hilsden RJ, Hershfield NB, Price LM, Meddings JB, et al. Intestinal permeability is increased in a proportion of spouses of patients with Crohn's disease. Am J Gastroenterol. (2001) 96:2934–8. doi: 10.1111/j.1572-0241.2001.04684.x
Glossary
6-OHDA, 6-Hydroxydopamine; AIEC, Adherent invasive Escherichia coli; AMPs, Antimicrobial peptides; aSyn, alpha-Synuclein; BBB, Blood-brain barrier; BDNF, Brain-derived neurotrophic factor; CD, Crohn's disease; cGAS, cyclic GMP-AMP synthase; CLR, C-type lectin receptor; CNS, Central nervous system; COMT, Catechol-O-methyltransferase; CSF, Cerebrospinal fluid; CXCL, Chemokine C-X-X- motif ligand; DA, Dopamine; DAMP, Damage-associated molecular pattern; DLB, Dementia with Lewy Bodies; DMV, Dorsal motor nucleus of the vagus nerve; DSS, Dextran sodium sulfate; EC, Enterochromaffin cells; EEC, Enteroendocrine cells; EGC, Enteroglial cells; EGF, Epidermal growth factor; ENS, Enteric nervous system; FFAR, Free fatty acid receptor; FMT, Fecal Microbiota Transplantation; GABA, γ-Amicobutyric acid; GDNF, Glial cell-line derived neurotrophic factor; GFAP, Glial fibrillary acidic protein; GI, gastrointestinal; GMP-AMP, Guanosin monophosphate-adenosine monophosphate; GPCRS, G-Protein coupled receptors; GWAS, Genome-Wide-Association studies; HSV1, Herpes simplex virus type 1; IBD, Inflammatory bowel disease; IEC, Intestinal epithelial cells; IF, Interferon; IL, Interleukin; iPSC, Induced pluripotent stem cells; LB, Lewy Bodies; L-DOPA, L-3,4-dihydroxy-L-phenylalanine; LN, Lewy neuritis; LRRK2, Leucin-rich repeat kinase 2; LP, Lamina propria; LPS, Lipopolysaccharide; MAO-B, Monoamine oxidase type B; MINERVA, MIcroboita-gut-braiN EngineeRed platform to eVAluate intestinal microflora impact on brain functionality; MPP, 1-Methyl-4-phenylpyridinium; MPTP, 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridin; MSA, Multiple system atrophy; MWAS, Metagenome-wide association studies; NAC, Non-amyloid-β component; NF-kB, Nuclear factor “kappa-light-chain-enhancer” of activated B-cells; NLR, NOD-like receptor; NO, Nitric oxide; NOD, Nucleotide-binding oligomerization domain; NT, Neurotransmitter; PAMPs, Pathogen-associated molecular patterns; PD, Parkinson's disease; PFF, Preformed fibrils; PNS, Peripheral nervous system; PRRs, Pattern-recognition receptors; PSD-95, postsynaptic density protein 95; RLR, Retinoic acid-inducible gene-like receptor; SCFA, Short-chain fatty acids; SNpc, Substantia nigra pars compacta; TGFα, Transforming growth factor α; TH, Tyrosine hydroxylase; Th, T helper cells; TLR, Toll-like receptor; TNBS, Trinitrobenzene sulfate; TNF, Tumor necrosis factor; UC, Ulcerative colitis; VIP, Vasoactive intestinal polypeptide; ZO, Zonula occludens.
Keywords: Parkinson's disease, gut-brain axis, enteroendocrine cells, alpha-synuclein, intestinal inflammation, inflammatory bowel diseases
Citation: Drobny A, Ngo PA, Neurath MF, Zunke F and López-Posadas R (2021) Molecular Communication Between Neuronal Networks and Intestinal Epithelial Cells in Gut Inflammation and Parkinson's Disease. Front. Med. 8:655123. doi: 10.3389/fmed.2021.655123
Received: 18 January 2021; Accepted: 14 June 2021;
Published: 22 July 2021.
Edited by:
Xingshun Qi, General Hospital of Shenyang Military Command, ChinaReviewed by:
Pandu R. Gangula, Meharry Medical College, United StatesMarta Castro, University of Zaragoza, Spain
Ling Yang, Huazhong University of Science and Technology, China
Copyright © 2021 Drobny, Ngo, Neurath, Zunke and López-Posadas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rocío López-Posadas, Um9jaW8ubG9wZXotcG9zYWRhc0B1ay1lcmxhbmdlbi5kZQ==; Friederike Zunke, ZnJpZWRlcmlrZS56dW5rZUBmYXUuZGU=
†These authors share first authorship