 Alina-Andreea Zimta1†
Alina-Andreea Zimta1† Ionut Hotea2,3†Melen Brinza4,5
Ionut Hotea2,3†Melen Brinza4,5 Cristina Blag6,7
Cristina Blag6,7 Sabina Iluta2Catalin Constantinescu2,8Atamyrat Bashimov1Elisabeth-Antonia Marchis-Hund1Alexandra Coudsy1Laetitia Muller-Mohnssen1
Sabina Iluta2Catalin Constantinescu2,8Atamyrat Bashimov1Elisabeth-Antonia Marchis-Hund1Alexandra Coudsy1Laetitia Muller-Mohnssen1 Noemi Dirzu1
Noemi Dirzu1 Diana Gulei1Delia Dima2Margit Serban9,10,11
Diana Gulei1Delia Dima2Margit Serban9,10,11 Daniel Coriu4,5
Daniel Coriu4,5 Ciprian Tomuleasa1,2,3*
Ciprian Tomuleasa1,2,3*- 1Medfuture Research Center for Advanced Medicine, Iuliu Hatieganu University of Medicine and Pharmacy, Cluj Napoca, Romania
- 2Department of Hematology, Iuliu Hatieganu University of Medicine and Pharmacy, Cluj Napoca, Romania
- 3Department of Hematology, Ion Chiricuta Clinical Cancer Center, Cluj Napoca, Romania
- 4Department of Hematology, Fundeni Clinical Institute, Bucharest, Romania
- 5Department of Hematology, Carol Davila University of Medicine and Pharmacy, Bucharest, Romania
- 6Department of Pediatrics, Iuliu Hatieganu University of Medicine and Pharmacy, Cluj Napoca, Romania
- 7Department of Hematology, Emergency Clinical Children's Hospital, Cluj Napoca, Romania
- 8Intensive Care Unit, Clinical Hospital for Infectious Diseases, Cluj Napoca, Romania
- 9Louis Turcanu Emergency Children's Hospital, Timisoara, Romania
- 10European Hemophilia Treatment Center, Timisoara, Romania
- 11Department of Pediatrics, Victor Babes University of Medicine and Pharmacy, Timisoara, Romania
Hemophilia type A (HA) is the most common type of blood coagulation disorder. While the vast majority of cases are inherited and caused by mutations in the F8 gene, recent data raises new questions regarding the non-heritability of this disease, as well as how other molecular mechanisms might lead to the development of HA or increase the severity of the disease. Some data suggest that miRNAs may affect the severity of HA, but for some patients, miRNA-based interference might cause HA, in the absence of an F8 mutation. A mechanism in HA installation that is also worth investigating and which could be identified in the future is the epigenetic silencing of the F8 gene that might be only temporarily. Acquired HA is increasingly reported and as more cases are identified, the description of the disease might become challenging, as cases without FVIII autoantibodies might be identified.
Introduction—“Classical” View of Hemophilia Type A and the Still Unanswered Questions
Hemophilia type A (HA) is a blood coagulation disorder described as an inherited condition caused by mutations in the F8 gene. Two mutations are found the most often in HA: intron 22 inversion (inv 22) and intron 1 inversion. Inv 22 is the most frequent mutation with different reported frequency, depending on the population: 35% (1), 45% of severe HA cases (2). Inv1 is the second most common mutation of HA in severe HA, with a different reported frequency of between 5 (3) and 7% (1). One large populational study from the US, the My Life, Our Future (MLOF) project concluded that the inv22 is found in 42% of severe HA cases and 3.7% of mild/moderate cases. Inv1 was found in 1.2% of severe HA cases and 0.2% of mild/moderate HA cases. In the majority of HA cases, which account for around 79.5% of patients, the most common type of mutation is the missense mutation (4).
Every year, new mutations in the F8 gene are discovered and the connection with severity of the disease (5–7) is reevaluated to gain more insight into HA and the reasons for different degrees of severity. Even though this continuing approach sustains patient stratification and risk evaluation, it does not offer a complete picture in terms of recent discoveries of FVIII repression at a transcriptional or translational level independent of the type of F8 mutation. More proof regarding the non-mutational influence of the circulating level of FVIII has been gathered from studies on healthy individuals, in whom FVIII levels increase with age (8).
Hematology presents these scenarios by analyzing the non-genetic influence of HA onset and severity. These may be related to the epigenetic repression of the F8 gene, non-coding RNA post-translational inhibition of the F8 mRNA, or direct repression of the FVIII protein. Through these analyses, new types of HA might be identified and monitored differently than in the case of “classical” HA.
MiRNAs Effects on Severity, Bleeding, and Complications—THE Need to Introduce More ncRNA Analysis in the Context of HA
The first line of evidence regarding the non-genetic causes of HA onset and progression comes from non-coding RNAs (ncRNAs), especially microRNAs (miRNAs). These RNAs can interact with the F8 mRNA and suppress its translation.
The evidence so far is scarce and done on a small number of patients, but it points to some missing details in the current overall view of HA. Jankowska et al. analyzed the miRNA able to bind to the F8 3′UTR through in silico data analysis and through RNA-affinity purification. The data was then compared to RNAseq results from 2 mild/moderate HA patients without F8 mutation and one severe HA patient with no known F8 mutation. In total, 22 up-regulated and 8 down-regulated miRNAs were found to be common with the miRNA targeting the F8 gene. MiR-19b-3p, miR-186-5p, miR-30c-5p, miR-93-3p, and miR-1246, all of which target F8 are also up-regulated in HA, and their expression increases with the severity of HA. At the same time, miR-93-5p, miR-196b-5p, let-7g-5p, miR-320a, and miR-221-3p showed inverse correlation with HA severity. The miR-19b-3p and miR-186-5p were experimentally demonstrated to target and down-regulate the F8 gene (9).
Through in silico analysis, Rosset et al. concluded that miR-26a-5p and miR-26b-5p have a higher energetic score for binding to a mutated form of F8 mRNA (the c.8728 A>G variant) than in the case of the WT F8 gene. The F8 mutations are not disease-associated (10). Even if this data still needs experimental confirmation, it might show that even in the case of HA patients with known F8 mutation the microRNA completes the picture of this pathology and its severity by influencing the level of F8 gene variants, with no association with HA disease.
Throughout hemophilia progression, miRNAs also reflect acute episodes. It was proven that in hemophilia-induced arthropathy, the pro-inflammatory NF-kB pathway is activated and leads to the production of the pro-inflammatory cytokines interleukin (IL)-1β, IL-6, and tumor necrosis factor α (TNF α). MiRNAs that activate the NF-kB pathway, such as miR-155, miR-9, miR-16, and miR-181b, were also up-regulated. In the hemophilic joints, there is an increase in pro-angiogenic factors, such as the vascular endothelial growth factor α (VEGFα), hypoxia-inducible factor 1α (HIF1α), and matrix metalloproteinases (MMPs), that degrade the chondrocyte matrix. The miRNAs related to apoptosis were also mostly up-regulated, as is the case of miR-155, miR-186, miR-23a (11). In another study, published by Sen and Jayandharan in India, miR-15b expression was reported to specifically drop during a bleeding episode, as well as in the following 6 weeks. In an animal model, they later confirmed that the local induction of miR-15b overexpression leads to down-regulation of pro-angiogenic factors VEGF-α and HIF-2α, together with a reduction in the MMPs responsible for chondral degeneration (MMP1, MMP3, MMP9, and MMP14) (12). These results clearly show that miRNAs are capable of influencing HA progression in the context of different genetic backgrounds, thus the value of RNA analysis even in HA with known mutations might better aid the correct formulation of treatment. Pipe et al. considered missense mutations within the B domain, which were reported in patients with HA. They explored whether the B domain is dispensable for the secretion and function of F8, and hypothesized that these mutations should not cause HA in these patients. Plasmid vectors containing B domain missense mutations are reported to be linked to moderate and severe HA and have been analyzed for their effect on synthesis and secretion compared with F8 wild-type (WT) following transient transfection into COS-1 and CHO cells in vitro. The mutants were then expressed in vivo in a HA mouse model by hydrodynamic tail-vein injection. F8 activity and the antigen levels for all mutants expressed into the conditioned media of COS-1 and CHO cells were similar to FV8 WT. The plasma expression of these mutants was similar to F8 WT in HA mice. They concluded that most missense mutations within the FVIII B domain would be unlikely to lead to severe HA and that the majority of such missense mutations represent polymorphisms or non-pathologic mutations (13).
The RNASeq technology has proven its applications in identifying the molecular basis for allogenic inhibitor development in HA, analysis made from peripheral blood. The analysis reported that in the contexts of allogenic antibody development, there is an increase in the expression of inflammatory initiating molecules, namely NLRP3 and TLR8 (14). The next generation sequencing (NGS) of intronic regions describes several mechanisms for the non-coding RNA post-translational inhibition of F8 mRNA or direct repression of the FVIII protein, including the creation of splice sites resulting in the incorporation of intronic sequences into the mRNA and intronic mutations at the exon-intron boundaries, resulting in the excision of exons or deep intronic mutations occurring anywhere in the gene. This leads to sequences within the mRNA that can, for example, insert stop codons, resulting in premature truncations. Using NGS, Bach et al. identified 23 deep intronic candidate variants in several F8 introns, including six recurrent variants and three variants previously described. In each of the 15 patients analyzed, at least one deep intronic variant in the F8 gene was described to predict the alteration of F8 mRNA splicing. Reduced F8 mRNA levels or stability would thus be compatible with the patients' mild to moderate HA phenotypes (15). The same group also presented mRNA analysis of novel F8 mutations with possible impact on splicing in four HA patients with silent mutations and seven patients with intronic variants close to or within splice site consensus sequences. Seven of the 11 mutations examined in vitro were shown to affect F8 mRNA splicing and the results were compared to in silico predictions (16). Castaman et al. aimed to analyze F8 mRNA for mutations in five families with mild HA with no apparent genomic mutation and a reduced response to desmopressin. They concluded that even if rare, deep intronic variations may be responsible for mild HA where no other F8 mutations are identified and may be associated with a reduced biologic response to desmopressin (17). At the University of Bonn, Pezeshkpoor et al. determined F8 and vWF von Willebrand factor (vWF) activity and antigen levels and performed vWF-F8 binding and vWF-collagen binding assays, as well as vWF multimer analysis. vWF was completely sequenced to exclude mutations. The F8 locus, including the introns, was sequenced using overlapping long-range PCRs combined with an NGS approach. Moreover, the F8 mRNA was analyzed quantitatively and qualitatively by real-time PCR and overlapping reverse transcription PCRs, respectively (18). By systematically excluding all possible causes of HA, they concluded that deep intronic mutations in F8, although rare, cause abnormal mRNA splicing, leading to mild HA.
miRNAs in the Prevention of Successful Gene Therapy in HA and How to Prevent Their Effect
Another way in which ncRNAs interfere in HA progression might be linked to their impairment of the efficacy for future treatments. Evidence of this comes from hemophilia type B (HB) but is still highly valuable in understanding the principle. Gene therapy is becoming a successful strategy for both HA and HB treatment. It is more efficient for HB according to clinical trials, which is why more evidence comes from HB (19–21). However, the therapy is often suppressed by the targeted inhibition of exogenous F8 or F9 mRNA through miRNAs (22–24). MiRNAs and other types of non-coding RNAs, such as small interfering RNAs (siRNAs), which target a factor from the coagulation cascade can impair the application of gene therapy for HA or HB. Other miRNAs or siRNAs have a therapeutic role in these pathologies. For instance, Fitusiran is a siRNA targeting antithrombin (AT) that increases the level of thrombin and increases blood coagulability in HA or HB (25). Some studies have proposed and successfully implemented the inclusion of miRNA target sites on the lentiviral vector, that would act as a miRNA-sponge. One such example was the use of the miR-142-3p sponge sequence in the lentiviral vectors, also containing the F9 gene. The vector transfected the extravascular hematopoietic lineage and managed to successfully induce FIX expression (26). In HA, this strategy was not as successful in the first trial as for HB. Besides the F8 gene, the vector contained a liver-restricted promoter and GP54 envelope glycoprotein and the target sites for miR-142. The expression of FVIII was restricted to the liver. Thus, more pronounced results were obtained in the case of transfection in mice neonates, as opposed to adults. This occurred because neonates did not have a fully developed immune system and the initial transfected cells expanded naturally throughout their lifetime (27).
Nourse et al. investigated the miRNA-mediated regulation of the hemostatic system and sed an integrative screening approach that combines functional aspects of miRNA silencing with biophysical miRNA interaction based on RNA pull-downs coupled to NGS. By assessing a panel of 27 hemostasis-associated gene 3'UTRs, they showed that the majority had substantial Dicer-dependent silencing capability, suggesting functional miRNA targeting. They identified 150 specific miRNA interactions with 14 3'UTRs, of which 52, involving 40 miRNAs, were functionally confirmed. This includes cooperative miRNA regulation of key hemostatic genes comprising procoagulant (F7, F8, F11, FGA, FGG, and KLKB1) and anticoagulant (SERPINA10, PROZ, SERPIND1, and SERPINC1) as well as fibrinolytic (PLG) components (28). In the Netherlands, Vossen et al. described 3'UTR variants in coagulation genes that influence coagulation factor levels and venous thrombosis risk. The 3'UTR of coagulation genes were sequenced in subjects with extremely high or low plasma levels of these factors in two case-control studies. MiR-544 caused a decrease of the luciferase activity not observed with an rs4253430 mutated vector. Thus, they stated that microRNAs are candidates that play a role both in hemostasis and thrombosis (29).
We believe that the non-specificity of microRNAs leads to these contradictory reports, since miRNA targets hundreds of other genes besides F8 mRNA, which might also contribute to the results. It could be helpful to look at different species of ncRNAs, some of which might be more specific. This could be done with the RNA Seq technique by gathering a number of patients and comparing them with healthy individuals, HA patients with F8 mutations, and HA patients without F8 mutation. This is a critically important point, and a key challenge that affected the studies mentioned previously, in investigating an ultra-rare subpopulation within a rare disease.
There are two long non-coding RNAs (lncRNAs), specifically NONHSAT139215 and NONHSAT139219, that are located in the F8 gene loci. Through molecular docking simulation of the interaction between the 3D structure of the aforementioned lncRNAs and the FVIII protein, it was proven that these two interact. Upon comparative RT-PCR analysis of the expression of these two ncRNAs, it was proven that they have a decreased level in HA patients compared to healthy controls. The severity of the decrease in expression was also correlated with the severity of hemophilia (30). MiR-1246 is markedly increased in HA patients compared to healthy controls, regardless of the co-present mutations in the F8 gene and its expression is high also in patients who have not developed anti-FVIII inhibitors. This microRNA binds in the 931–937 of F8 3′UTR (31).
NcRNAs as the Sole Cause of HA in the Absence of F8 Mutations-Possibly a New Type of HA
Besides the above-mentioned examples, the rare cases where the F8 gene is wild-type and there are no autoantibodies, but still the patients develop HA, are interesting uncommon scenarios for these rare bleeding disorders. Although there are some instances in the study of these cases, more detailed research is required as they may constitute a new type of HA that is non-inherited and that is different from acquired hemophilia, meaning the absence of auto-antibodies.
More precisely, it has been reported that 2–5% of HA patients do not have any mutations in the F8 gene, but still, the FVIII protein is absent from the blood and HA develops. Jankowska et al., from the United States Food and Drug Administration, did a miRNA Seq analysis on two cases of mild/moderate HA patients, with WT F8 gene, compared with healthy donors. This was a significant step forward from the genetic-orientated analysis of hemophilia toward a transcriptomic orientated one. From the list of miRNAs up-regulated in HA patients, miR-374b-5p and miR-30c-5b was also found to directly target F8 gene mRNAs and thus impair FVIII protein translation, according to in silico analysis (microRNA.org, TargetScan, miRDB) and an experimental validation done on HEK293T, HeLa, and MS1 cells. Moreover, miR-30c inhibition up-regulates the level of FVIII (9, 32). This was a huge change in perspective as classical HA can no longer be predicted in a family solely based on F8 mutation status and other mechanisms that are still being uncovered can lead to the development of HA.
As Salvajer and Pipe argue, the results from miRNA studies in F8 WT HA cases are contradictory and the definitive association between certain miRNAs and F8 mRNA levels is still too early to be assessed. They discuss another study analyzing miRNAs that targeted genes from the coagulation cascade that did not find the miR-374b-5p and miR-30c-5b as targets of the F8 gene. More detailed analysis on a larger cohort of HA patients with unmutated F8 gene needs to be conducted (33).
The Epigenetic Influence of HA Severity and a Possible Connection With HA Development in Some Cases
Besides the significant role of ncRNAs in inducing HA and influencing its severity, a lesser-known mechanism is the epigenetic silencing of the F8 gene, which in theory might be reversible. Proof of this influence comes from how existing mutations affect the epigenetic status of the F8 gene. The well-known inversions in the intron 1 or intron 22 of the F8 gene affect the methylation pattern of the surrounding sites (CpG island methylation), regardless of other confounding effects, such as age. Epigenetic changes can differentiate between intron 1 or intron 22 inversion (34). The role of epigenetics is revealed by the heritability of mutations in the F8 gene. An analysis of different members of the same family, all carriers of Xq28 deletions, which included the F8 gene, showed different degrees of severity of the disease. For instance, some members showed preferential epigenetic inactivation of the X chromosome with Xq28 deletion, while others did not show inactivation of the mutated X chromosome (35). The potential link between epigenetics and HA severity must be confirmed by further thorough studies as there is uncertainty regarding the methylation pattern of the F8 gene. Zimmermann et al. reported no difference in the methylation pattern of hemophilic patients and the controls (36). Genomic inversions in the F8 (Xq28) region are associated with detectable changes in methylation levels, thus inv1 and inv22 were associated with changes in the methylation pattern of CpG regions. However, this only applies in F8 inversions. Considering that the HA is caused by a great variety of mutations in the F8 genes, the epigenetic silencing of the F8 gene may be mutation specific and carry some F8 variants, and epigenetic disease-specific epigenetic changes might be completely absent.
AHA—A Recently Described HA With a Non-Mutational Background With Insufficiently Understood Epigenetic Influences
An increasingly well-known cause of HA that differs from the “classical” view is the recently described acquired hemophilia (AHA). AHA is a very rare, acquired bleeding disorder caused by F8 autoantibodies, which neutralize F8 activity. These inhibitors differ from alloantibodies against F8, which can occur in congenital HA after repeated exposure to plasma-derived or recombinant FVIII products. In most cases, the disease occurs suddenly in subjects without a personal or familiar history of bleeding, with symptoms that may be mild, moderate, or severe. AHA mostly affects female patients while HA is mainly caused by a mutation in F8 and affects male patients (37–39). The bleeding pattern of AHA is different from that of HA. Most patients with F8 autoantibodies have hemorrhages in the skin, muscles, or soft tissues and mucous membranes, whereas hemarthrosis, a typical feature of congenital F8 deficiency, is uncommon. This disease develops during a person's lifetime, usually in special conditions, such as pregnancy, after major surgery or very often linked to underlying malignancy, and it is described as an autoimmune disease (40, 41). In acquired hemophilia, the newly developed antibodies are different from the alloantibodies developed as a response to rF8 treatment in congenital HA (39) (Figure 1). These represent an acute response with more severe consequences. The disease may be idiopathic or present with severe symptoms. The paradigm of acquired hemophilia has also shifted over the last few years. It was shown that in two acquired hemophilia patients a point mutation was found (c.8899G>A) in the F8 gene (42). As follows, more evidence may come if acquired HA patients will also go through an analysis of the ncRNA or epigenetic mark-up.
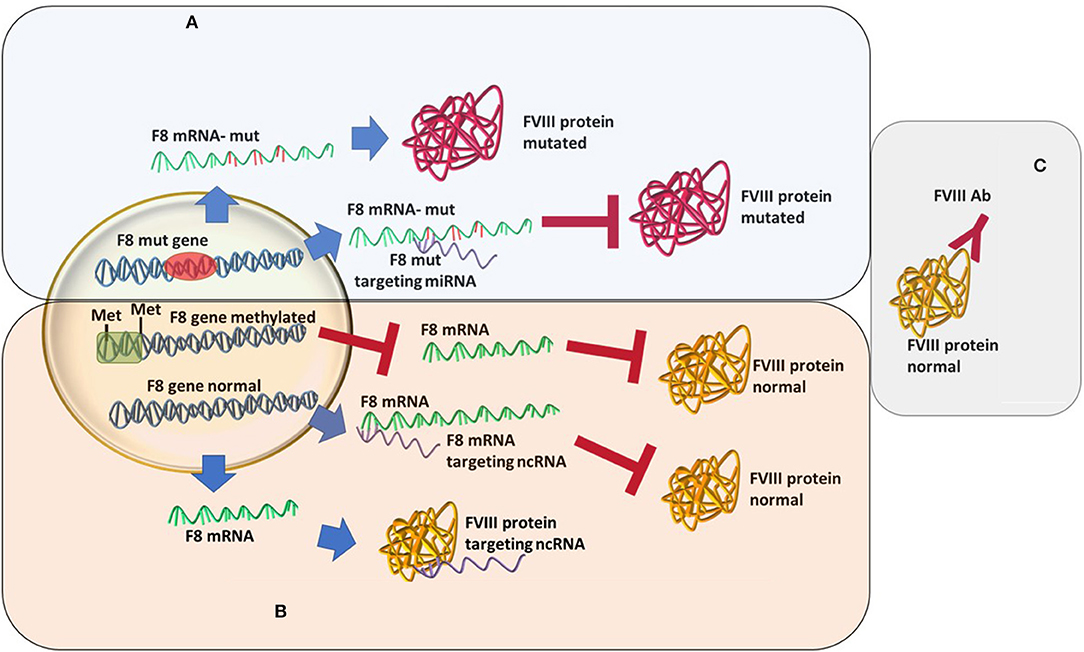
Figure 1. Different types of hemophilia type A and how epigenetic silencing of ncRNA repression might influence the FVIII level regardless of the mutation status. (A) Classic hemophilia-new mutations are found, miRNAs changes in expression increases severity. (B) Non-inherented rare hemophilia-still unclear or lesser known molecular mechanisms. (C) Acquired hemophilia-recently characterized.
Non-mutation induced HA is defined as a sudden abnormal hemorrhage diagnosed in a patient, not on anticoagulation, without personal or family bleeding history, with an isolated prolonged aPTT and a mixing study consistent with an inhibitor. The abnormal prolongation of aPTT before surgery must always be investigated. Still, for 30% of cases, only laboratory alterations occur. A diagnosis must be confirmed by a hematology department with expertise in coagulation disorders, as well as in the management of inhibitors against coagulation factors. Clinical management recommends avoiding invasive procedures. If necessary, procedures must be performed in a hemophilia center or after consultation with it. Physicians are advised to investigate the underlying cause of the disease as soon as the diagnosis of acquired HA (AHA) has been made. Should the underlying cause be identified, therapy should be initiated.
The many medical conditions that trigger AHA, such as autoimmune disorders, malignancy, drug reaction, or even idiopathic, can affect the patient to such extent that they may require complex observation or treatment in a High-Dependency Unit (HDU) or the intensive care unit (ICU) (43, 44). To correctly manage such a patient before admission and during the first minutes, doctors should follow a focused and straightforward approach. The complications that in the end lead to the death of the patient are those of a delayed diagnosis (mostly because of a lack of experience), difficulty achieving hemostasis, other associated diseases that further decompensate and lead to organ failure and complications of treatment (such as venous thrombosis). The clinical problems that these patients pose are: how do we control the bleeding and how do we get rid of the autoantibodies. The ABC approach is the most commonly used worldwide, but during hemorrhage situations, the correct one is CABC (45). Before assessing the airway, try to find the source of bleeding (if there is an evident one) and apply hemostasis, if it is possible. Continue with the airway (A) and breathing (B) check, where it is mandatory to maintain a permeable airway and give oxygen if the SpO2 is <94% in atmospheric air, to fully saturate the hemoglobin remaining in the circulation and achieve optimal delivery of oxygen to the tissues. Returning to the (C)-circulation step, adequate intravenous access [at least two 18 gauge (G) intravenous catheters] are available and if a shock state is suspected, one should begin fluid infusion (crystalloid or colloid) or blood-derived products. During bleeding situations make sure the patient is connected to a monitor where there is a constant evaluation of clinical signs, such as ECG, SpO2, and blood pressure. Daily clinical examination and laboratory studies are mandatory. Pay attention to the need for invasive procedures and avoidance of antiplatelet, anticoagulant, or other therapies that may cause further bleeding. Most patients present with bleeding symptoms and require the initiation of antihemorrhagic treatment. By knowing the antibodies titers the correct medication may be chosen. If the inhibitor titer is ≤ 5 BU human FVIII concentrates or DDAVP can be used as first-line treatment. For the cases with high-titer (>5 BU), bypassing agents can be used, such as activated prothrombin complex concentrate (aPCC), which contains activated FII, FVII, FIX, and FX (dependent vitamin-K factors) and recombinant activated FVII (rFVIIa), achieving hemostasis by generating thrombin (in the absence of FVIII) at the site of bleeding (46). The concomitant use of both agents is not recommended, only in life-threatening situations, where hemostasis could not be achieved by the use of one agent (47). In the situations of mucosal bleeding, an antifibrinolytic drug can be used (epsilon-aminocaproic acid or tranexamic acid) (48). After the hemostasis has been achieved, ongoing treatment should be continued (as prophylaxis) to prevent recurrence of bleeding. Furthermore, the treatment is oriented on the possibility of elimination of the antibodies. The recommended first-line treatment for autoantibody eradication is an immunosuppressive regimen mainly consisting of prednisone (1 mg/kg/day) alone or in combination with oral cyclophosphamide (50–100 mg/day), with the target being an undetectable inhibitor (<0.6 BU) and normal FVIII levels (>50%) (49). Rituximab, calcineurin inhibitors cyclosporine, tacrolimus) and mycophenolate mofetil are alternatives to rituximab in patients who do not respond to first-line treatment (50–52).
At times, because the management of these patients is complex, and there is difficulty in achieving hemostasis, liaison with hemophilia specialists and treatment centers is highly recommended.
Conclusion
HA is a disease far more complex than initially thought. In the future, there might be more varieties of the disease than originally thought, as more studies focus on the epigenetic and transcriptomic background of the disease. These might not only explain the differences in the severity of the disease but also offer answers to the cases with no mutations and no autoantibodies. Based on the research literature and critical analysis, we propose that there are at least two more types of uncharacterized HA: the epigenetic one and one caused by ncRNA. Thus, future research should focus on deciphering the role of both epigenetic events and non-coding RNAs in the pathophysiology of both “classical HA,” as well as AHA.
Author Contributions
All authors contributed in the design and writing of the manuscript.
Funding
IH was funded by an internal grant from the Iuliu Hatieganu University-School of Doctoral Studies (PCD 2018-2021). This paper was published under the frame of European Social Found, Human Capital Operational Programme 2014–2020, Project No. POCU/380/6/13/125171. This work was also supported by an international grant awarded by the Novo Nordisk Haemophilia Foundation 2020–2021, aiming to Strengthen haemophilia care in the north and establish a national registry in Romania awarded to the Romanian Society of Hematology (grant director Ciprian Tomuleasa). It was funded by grants from the Romanian Ministry of Research and Innovation, CCCDI-UEFISCDI, Project No. PN-III-P4-ID-PCCF-2016-0112 within PNCDI III, for Young Research Teams 2020–2022 (Grant No. PN-III-P1-1.1-TE-2019-0271, grant director Ciprian Tomuleasa).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Antonarakis SE, Rossiter JP, Young M, Horst J, de Moerloose P, Sommer SS, et al. Factor VIII gene inversions in severe hemophilia A: results of an international consortium study. Blood. (1995) 86:2206–12. doi: 10.1182/blood.V86.6.2206.bloodjournal8662206
2. Lakich D, Kazazian HHJ, Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat Genet. (1993) 5:236–41. doi: 10.1038/ng1193-236
3. Bagnall RD, Waseem N, Green PM, Giannelli F. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood. (2002) 99:168–74. doi: 10.1182/blood.V99.1.168
4. Johnsen JM, Fletcher SN, Huston H, Roberge S, Martin BK, Kircher M, et al. Novel approach to genetic analysis and results in 3000 hemophilia patients enrolled in the my life, our future initiative. Blood Adv. (2017) 1:824–34. doi: 10.1182/bloodadvances.2016002923
5. Roualdes O, Nougier C, Fretigny M, Talagrand E, Durand B, Negrier C, et al. Usefulness of an in vitro cellular expression model for haemophilia a carrier diagnosis: illustration with five novel mutations in the F8 gene in women with isolated factor VIII:C deficiency. Haemophilia. (2015) 21:e202–9. doi: 10.1111/hae.12651
6. Riccardi F, Tagliaferri A, Martorana D, Rivolta GF, Valdrè L, Rodorigo G, et al. Spectrum of F8 gene mutations in haemophilia a patients from a region of Italy: identification of 23 new mutations: F8 mutation in North Italian patients. Haemophilia. (2010) 16:791–800. doi: 10.1111/j.1365-2516.2010.02228.x
7. Nissen SK, Laursen AL, Poulsen LH, Mogensen TH. Identification of a novel mutation in the factor VIII gene causing severe haemophilia A. BMC Hematol. (2018) 18:17. doi: 10.1186/s12878-018-0113-4
8. Biguzzi E, Castelli F, Lijfering WM, Cannegieter SC, Eikenboom J, Rosendaal FR, et al. Rise of levels of von Willebrand factor and factor VIII with age: role of genetic and acquired risk factors. Thromb Res. (2021) 197:172–8. doi: 10.1016/j.thromres.2020.11.016
9. Jankowska KI, McGill J, Pezeshkpoor B, Oldenburg J, Sauna ZE, Atreya CD. Further evidence that microRNAs can play a role in hemophilia a disease manifestation: F8 gene downregulation by MiR-19b-3p and MiR-186-5p. Front Cell Dev Biol. (2020) 8:669. doi: 10.3389/fcell.2020.00669
10. Rosset C, Vieira IA, Salzano FM, Bandinelli EA. Germline variant affects putative MiRNA-binding sites at the F8 3′UTR and acts as a potential haemophilia A phenotype modifier in Southern Brazilian patients. Haemophilia. (2016) 22:e327–9. doi: 10.1111/hae.12953
11. Sen D, Chapla A, Walter N, Daniel V, Srivastava A, Jayandharan GR. Nuclear factor (NF)-κB and its associated pathways are major molecular regulators of blood-induced joint damage in a murine model of hemophilia. J Thromb Haemost. (2013) 11:293–306. doi: 10.1111/jth.12101
12. Sen D, Jayandharan G. MicroRNA-15b modulates molecular mediators of blood induced arthropathy in hemophilia mice. IJMS. (2016) 17:492. doi: 10.3390/ijms17040492
13. Ogata K, Selvaraj SR, Miao HZ, Pipe SW. Most factor VIII B domain missense mutations are unlikely to be causative mutations for severe hemophilia a: implications for genotyping. J Thromb Haemost. (2011) 9:1183–90. doi: 10.1111/j.1538-7836.2011.04268.x
14. Karim AF, Soltis AR, Sukumar G, Königs C, Ewing NP, Dalgard CL, et al. Hemophilia A inhibitor subjects show unique PBMC gene expression profiles that include up-regulated innate immune modulators. Front Immunol. (2020) 11:1219. doi: 10.3389/fimmu.2020.01219
15. Bach JE, Wolf B, Oldenburg J, Müller CR, Rost S. Identification of deep intronic variants in 15 haemophilia A patients by next generation sequencing of the whole factor VIII gene. Thromb Haemost. (2015) 114:757–67. doi: 10.1160/TH14-12-1011
16. Zimmermann MA, Gehrig A, Oldenburg J, Müller CR, Rost S. Analysis of F8 MRNA in haemophilia A patients with silent mutations or presumptive splice site mutations. Haemophilia. (2013) 19:310–7. doi: 10.1111/hae.12039
17. Castaman G, Giacomelli SH, Mancuso ME, D'Andrea G, Santacroce R, Sanna S, et al. Deep intronic variations may cause mild hemophilia A. J Thromb Haemost. (2011) 9:1541–8. doi: 10.1111/j.1538-7836.2011.04408.x
18. Pezeshkpoor B, Zimmer N, Marquardt N, Nanda I, Haaf T, Budde U, et al. Deep intronic “mutations” cause hemophilia A: application of next generation sequencing in patients without detectable mutation in F8 CDNA. J Thromb Haemost. (2013) 11:1679–87. doi: 10.1111/jth.12339
19. Croteau SE, Wang M, Wheeler AP. 2021 clinical trials update: innovations in hemophilia therapy. Am J Hematol. (2021) 96:128–44. doi: 10.1002/ajh.26018
20. Mancuso ME, Mahlangu JN, Pipe SW. The changing treatment landscape in haemophilia: from standard half-life clotting factor concentrates to gene editing. Lancet. (2021) 397:630–40. doi: 10.1016/S0140-6736(20)32722-7
21. Sidonio RF, Pipe SW, Callaghan MU, Valentino LA, Monahan PE, Croteau SE. Discussing investigational AAV gene therapy with hemophilia patients: a guide. Blood Rev. (2020) 100759. doi: 10.1016/j.blre.2020.100759
22. Spadarella G, Di Minno A, Milan G, Franco N, Polimeno M, Castaldo F, et al. Paradigm shift for the treatment of hereditary haemophilia: towards precision medicine. Blood Rev. (2020) 39:100618. doi: 10.1016/j.blre.2019.100618
23. Ling G, Nathwani AC, Tuddenham EGD. Recent advances in developing specific therapies for haemophilia. Br J Haematol. (2018) 181:161–72. doi: 10.1111/bjh.15084
24. Korte W, Graf L. The potential close future of hemophilia treatment—gene therapy, TFPI inhibition, antithrombin silencing, and mimicking factor VIII with an engineered antibody. Transfus Med Hemother. (2018) 45:92–6. doi: 10.1159/000488152
25. Machin N, Ragni MV. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J Blood Med. (2018) 9:135–40. doi: 10.2147/JBM.S159297
26. Brown BD, Venneri MA, Zingale A, Sergi LS, Naldini L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat Med. (2006) 12:585–91. doi: 10.1038/nm1398
27. Matsui H, Hegadorn C, Ozelo M, Burnett E, Tuttle A, Labelle A, et al. A microRNA-regulated and GP64-pseudotyped lentiviral vector mediates stable expression of FVIII in a murine model of hemophilia A. Mol Ther. (2011) 19:723–30. doi: 10.1038/mt.2010.290
28. Nourse J, Braun J, Lackner K, Hüttelmaier S, Danckwardt S. Large-scale identification of functional microRNA targeting reveals cooperative regulation of the hemostatic system. J Thromb Haemost. (2018) 16:2233–45. doi: 10.1111/jth.14290
29. Vossen CY, van Hylckama Vlieg A, Teruel-Montoya R, Salloum-Asfar S, de Haan H, Corral J, et al. Identification of coagulation gene 3'UTR variants that are potentially regulated by microRNAs. Br J Haematol. (2017) 177:782–90. doi: 10.1111/bjh.14629
30. Naderi N, Namvar A, Amani N, Nasoohi N, Bolhassani A. Analysis of long non-coding RNA expression in hemophilia A patients. Hematology. (2019) 24:255–62. doi: 10.1080/16078454.2018.1560934
31. Sarachana T, Dahiya N, Simhadri VL, Pandey GS, Saini S, Guelcher C, et al. Small NcRNA expression-profiling of blood from hemophilia A patients identifies MiR-1246 as a potential regulator of factor 8 gene. PLoS ONE. (2015) 10:e0132433. doi: 10.1371/journal.pone.0132433
32. Jankowska KI, McGill J, Pezeshkpoor B, Oldenburg J, Atreya CD, Sauna ZE. Clinical manifestation of hemophilia A in the absence of mutations in the F8 gene that encodes FVIII: role of microRNAs. Transfusion. (2020) 60:401–13. doi: 10.1111/trf.15605
33. Selvaraj SR, Pipe SW. Not in the genotype: can unexplained hemophilia A result from “micro(RNA) management”? Transfusion. (2020) 60:227–8. doi: 10.1111/trf.15668
34. Jamil MA, Sharma A, Nuesgen N, Pezeshkpoor B, Heimbach A, Pavlova A, et al. F8 inversions at Xq28 causing hemophilia A are associated with specific methylation changes: implication for molecular epigenetic diagnosis. Front Genet. (2019) 10:508. doi: 10.3389/fgene.2019.00508
35. Janczar S, Kosinska J, Ploski R, Pastorczak A, Wegner O, Zalewska-Szewczyk B, et al. Haemophilia A and cardiovascular morbidity in a female SHAM syndrome carrier due to skewed X chromosome inactivation. Eur J Med Genet. (2016) 59:43–7. doi: 10.1016/j.ejmg.2015.12.004
36. Zimmermann MA, Hansmann T, Haaf T, Oldenburg J, Müller CR, Rost S. Methylation analysis of the promoter region and intron 1 of the factor VIII gene in haemophilia A patients. Hamostaseologie. (2013) 33(Suppl. 1):S46–9. doi: 10.1055/s-0037-1619797
37. Shetty S, Bhave M, Ghosh K. Acquired hemophilia a: diagnosis, aetiology, clinical spectrum and treatment options. Autoimmun Rev. (2011) 10:311–6. doi: 10.1016/j.autrev.2010.11.005
38. Holme PA, Brosstad F, Tjønnfjord GE. Acquired haemophilia: management of bleeds and immune therapy to eradicate autoantibodies. Haemophilia. (2005) 11:510–5. doi: 10.1111/j.1365-2516.2005.01136.x
39. Mazzucconi MG, Baldacci E, Ferretti A, Santoro C. Acquired haemophilia A: an intriguing disease. Mediterr J Hematol Infect Dis. (2020) 12:e2020045. doi: 10.4084/mjhid.2020.045
40. Yousphi AS, Bakhtiar A, Cheema MA, Nasim S, Ullah W. Acquired hemophilia A: a rare but potentially fatal bleeding disorder. Cureus. (2019) 11:e5442. doi: 10.7759/cureus.5442
41. Tiede A, Collins P, Knoebl P, Teitel J, Kessler C, Shima M, et al. International recommendations on the diagnosis and treatment of acquired hemophilia A. Haematologica. (2020) 105:1791–801. doi: 10.3324/haematol.2019.230771
42. Hwang SH, Lim JA, Kim HC, Lee HW, Kim HS. Identification of a shared F8 mutation in the Korean patients with acquired hemophilia A. Korean J Hematol. (2011) 46:49. doi: 10.5045/kjh.2011.46.1.49
43. Knoebl P, Marco P, Baudo F, Collins P, Huth-Kühne A, Nemes L, et al. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost. (2012) 10:622–31. doi: 10.1111/j.1538-7836.2012.04654.x
44. Janbain M, Leissinger CA, Kruse-Jarres R. Acquired hemophilia A: emerging treatment options. J Blood Med. (2015) 6:143–50. doi: 10.2147/JBM.S77332
45. Constantinescu C, Bodolea C, Pasca S, Teodorescu P, Dima D, Rus I, et al. Clinical approach to the patient in critical state following immunotherapy and/or stem cell transplantation: guideline for the on-call physician. J Clin Med. (2019) 8:884. doi: 10.3390/jcm8060884
46. Collins P, Baudo F, Huth-Kühne A, Ingerslev J, Kessler CM, Castellano MEM, et al. Consensus recommendations for the diagnosis and treatment of acquired hemophilia A. BMC Res Notes. (2010) 3:161. doi: 10.1186/1756-0500-3-161
47. Schneiderman J, Rubin E, Nugent DJ, Young G. Sequential therapy with activated prothrombin complex concentrates and recombinant FVIIa in patients with severe haemophilia and inhibitors: update of our previous experience. Haemophilia. (2007) 13:244–8. doi: 10.1111/j.1365-2516.2007.01451.x
48. Holmström M, Tran HTT, Holme PA. Combined treatment with APCC (FEIBA®) and tranexamic acid in patients with haemophilia A with inhibitors and in patients with acquired haemophilia A—a two-centre experience. Haemophilia. (2012) 18:544–9. doi: 10.1111/j.1365-2516.2012.02748.x
49. Huth-Kühne A, Baudo F, Collins P, Ingerslev J, Kessler CM, Lévesque H, et al. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica. (2009) 94:566–75. doi: 10.3324/haematol.2008.001743
50. Collins P, Baudo F, Knoebl P, Lévesque H, Nemes L, Pellegrini F, et al. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood. (2012) 120:47–55. doi: 10.1182/blood-2012-02-409185
51. Collins PW, Chalmers E, Hart D, Jennings I, Liesner R, Rangarajan S, et al. Diagnosis and management of acquired coagulation inhibitors: a guideline from UKHCDO. Br J Haematol. (2013) 162:758–73. doi: 10.1111/bjh.12463
Keywords: hemophilia, non-coding RNAs, epigenetics, hypothesis, acquired bleeding disorder
Citation: Zimta A-A, Hotea I, Brinza M, Blag C, Iluta S, Constantinescu C, Bashimov A, Marchis-Hund E-A, Coudsy A, Muller-Mohnssen L, Dirzu N, Gulei D, Dima D, Serban M, Coriu D and Tomuleasa C (2021) The Possible Non-Mutational Causes of FVIII Deficiency: Non-Coding RNAs and Acquired Hemophilia A. Front. Med. 8:654197. doi: 10.3389/fmed.2021.654197
Received: 15 January 2021; Accepted: 11 March 2021;
Published: 15 April 2021.
Edited by:
Robert Campbell, The University of Utah, United StatesReviewed by:
Bipin P. Kulkarni, Indian Council of Medical Research, IndiaZuben E. Sauna, United States Food and Drug Administration, United States
Chava Kimchi-sarfaty, United States Food and Drug Administration, United States
Katarzyna Jankowska, Center for Biologics Evaluation and Research (CBER), FDA, United States, in collaboration with reviewer CK-s
Copyright © 2021 Zimta, Hotea, Brinza, Blag, Iluta, Constantinescu, Bashimov, Marchis-Hund, Coudsy, Muller-Mohnssen, Dirzu, Gulei, Dima, Serban, Coriu and Tomuleasa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ciprian Tomuleasa, Y2lwcmlhbi50b211bGVhc2FAdW1mY2x1ai5ybw==
†These authors have contributed equally to this work and share first authorship